

Abstract
Prostate stem/progenitor cells (PrSCs) are responsible for adult prostate tissue homeostasis and regeneration. However, the related regulatory mechanisms are not completely understood. In this study, we examined the role of heparan sulfate (HS) in PrSC self-renewal and prostate regeneration. Using an in vitro prostate sphere formation assay, we found that deletion of the glycosyltransferase exostosin 1 (Ext1) abolished HS expression in PrSCs and disrupted their ability to self-renew. In associated studies, we observed that HS loss inhibited p63 and CK5 expression, reduced the number of p63+- or CK5+-expressing stem/progenitor cells, elevated CK8+ expression and the number of differentiated CK8+ luminal cells and arrested the spheroid cells in the G1/G0 phase of cell cycle. Mechanistically, HS expressed by PrSCs (in cis) or by neighboring cells (in trans) could maintain sphere formation. Furthermore, HS deficiency upregulated transforming growth factor β (TGFβ) signaling and inhibiting TGFβ signaling partially restored the sphere-formation activity of the HS-deficient PrSCs. In an in vivo prostate regeneration assay, simultaneous loss of HS in both epithelial cell and stromal cell compartments attenuated prostate tissue regeneration, whereas the retention of HS expression in either of the two cellular compartments was sufficient to sustain prostate tissue regeneration. We conclude that HS preserves self-renewal of adult PrSCs by inhibiting TGFβ signaling and functions both in cis and in trans to maintain prostate homeostasis and to support prostate regeneration.
Keywords: heparan sulfate, prostate, stem cell, self-renewal, TGFβ
Introduction
The prostate is a complex tubuloalveolar exocrine gland of the male reproductive system in mammals. The gland contains two major epithelial cell types: secretory luminal cells, which contribute to the bulk of seminal fluid, and basal cells that line the basement membrane (Wang et al. 2001). A small population of neuroendocrine cells also exists within the basal cell layer and regulates the activity of the two aforementioned epithelial lineages (Aumuller et al. 2001). The prostate epithelial cells (PrECs) are in an intimate contact with the surrounding stroma, which includes fibroblasts, smooth muscle, nerves and lymphatics. Interactions between the stromal and the epithelial compartment have been shown to mediate multiple growth factor signaling pathways that are involved not only in the growth and development of the prostate but also in the prostate cancer (Josson et al. 2010). The adult prostate is capable of undergoing multiple cycles of atrophy and regeneration following castration and readministration of androgen. This atrophy results mostly from apoptosis of the differentiated luminal cells, thus attesting to the presence of prostate stem/progenitor cells (PrSCs) in the androgen-independent basal cell layer (English et al. 1987; Kyprianou and Isaacs 1988). Intensive investigations have been taken to delineate the molecular mechanisms that regulate PrSCs fate determination. In this regards, some achievements have been made (Strand and Goldstein 2015). For example, autocrine TGFβ signaling has been shown to induce rat PrSCs to differentiate into luminal cells (Danielpour 1999) and maintains dormancy of PrSCs in the proximal region of prostate ducts (Salm et al. 2005), highlighting the critical role of TGFβ signaling on PrSC fate decision.
Heparan sulfate (HS) is a linear anionic polysaccharide expressed ubiquitously on the cell surface and in extracellular matrix (ECM), where it is covalently linked to core proteins to form HS proteoglycans (Wang et al. 2005; Fuster and Wang 2010; Sarrazin et al. 2011; Zhang et al. 2014; Qiu et al. 2018). The biosynthesis of HS chains begins in the golgi through the sequential activities of various HS biosynthetic enzymes (Bernfield et al. 1999; Esko and Selleck 2002). HS biosynthesis is initiated by adding the first N-acetylglucosamine (GlcNAc) residue to a tetrasaccharide linker that consists of glucuronic acid (GlcA)-galactose-galactose-xylose-O-(serine). A heterodimer of two glycosyltransferases, composed of exostosin 1 (Ext1) and Ext2, then polymerizes and extends the HS chain backbone by alternative addition of and GlcA and GlcNAc residues. As the chain is being elongated, a number of modifications take place resulting in N-deacetylation, N-sulfation of GlcNAc and epimerization of GlcA to iduronic acid followed by addition of 2-O, 6-O and 3-O sulfation modifications (Bernfield et al. 1999; Esko and Selleck 2002; Wang et al. 2005; Qiu et al. 2013, 2018; Zhang et al. 2014; Talsma et al. 2018). These modification reactions are incomplete, which results in a highly heterogeneous HS structure. The synthesized HS chains can be subjected to postsynthetic modification by 6-O-sulfatases (Sulf) at the cell surface and in the ECM to remove 6-O sulfates. The biosynthetic modifications and the postsynthetic remodeling work together to generate unique binding sites for various protein ligands, including growth factors and morphogens such as FGF, Wnt, Hedgehog, TGF and bone morphogenetic proteins, which are known to critically modulate organ development (Lyon et al. 1997; Ornitz 2000; Baeg et al. 2001; Kuo et al. 2010; Ortmann et al. 2015). Intriguingly, HS structures have been shown to be tissue, cell-type and developmental stage specific, implicating that HS plays cell-specific and spatiotemporal regulatory functions via interactions with a selection of protein ligands (Sarrazin et al. 2011).
We and others have previously reported that HS essentially regulates self-renewal and differentiation of mouse embryonic stem cells via facilitation of FGF and/or BMP signaling (Johnson et al. 2007; Baldwin et al. 2008; Sasaki et al. 2008; Kraushaar et al. 2010, 2012, 2013; Dejima et al. 2011; Saez et al. 2014; Levings et al. 2016). Other studies have emerged that HS may also critically modulate adult stem cell fate in various tissues such as the Drosophila germline niche and mouse bone marrow, appearing via distinct molecular mechanisms (Nurcombe and Cool 2007; Hayashi et al. 2009; Helledie et al. 2012; Saez et al. 2014; Watson et al. 2014; Levings et al. 2016). However, the HS function and underlying molecular mechanisms of HS in adult stem cells in many other tissue/organs remain largely unexplored. In this study, we show that HS is required to sustain self-renewal of adult PrSCs by inhibiting TGFβ signaling and it functions both in cis and in trans to maintain PrSc homeostasis as well as in facilitating prostate regeneration.
Results
Loss of HS diminishes self-renewal activity of adult PrSCs
HS is known to be ubiquitously expressed on the cell surface and in the ECM. Intriguingly, in adult mouse prostates, immunohistochemical staining with anti-HS antibody 10E4 reveals that HS is highly enriched at the junction of basal-stromal cells (Figure 1A), where the p63+ and/or CK5+ PrSCs are enriched, suggesting that HS may play important roles in maintaining PrSC fate.
Fig. 1.
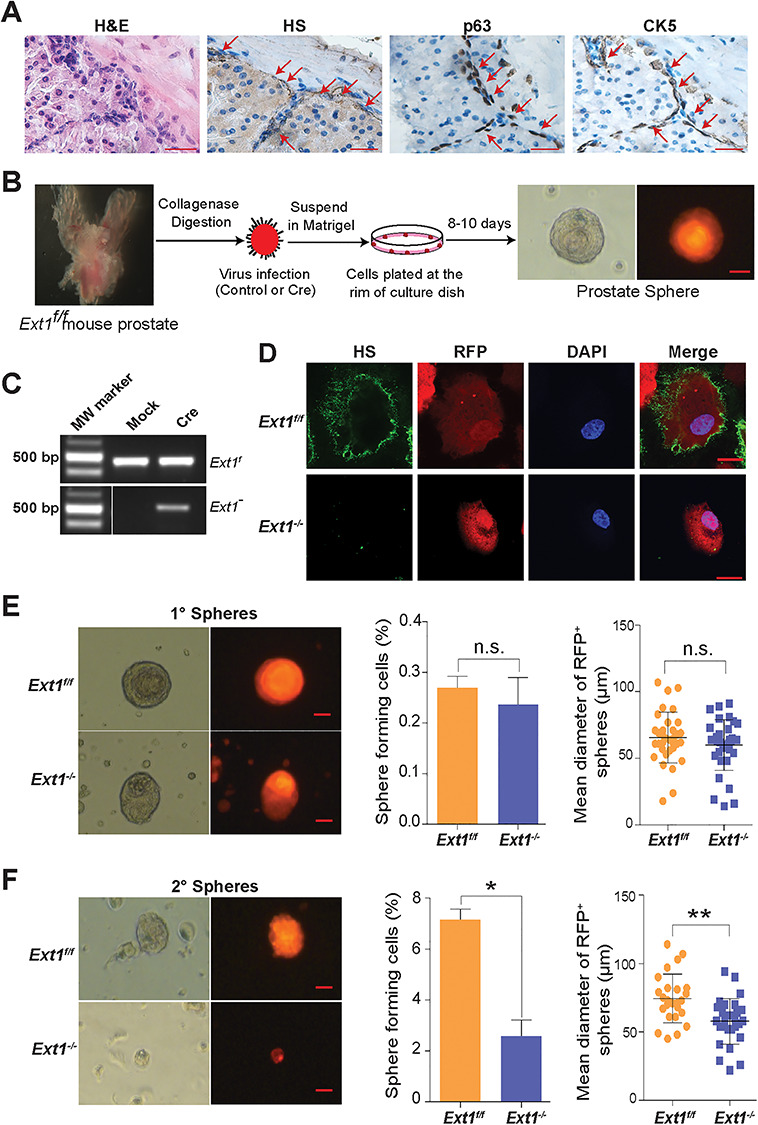
Loss of HS expression diminishes self-renewal activity of adult PrSCs. (A) H&E and immunohistochemical staining of cellularity, HS, p63, and CK5 in adult mouse prostate (scale = 20 μm). (B) Schematic representation of PrSC sphere forming assay. The Ext1f/f PrECs were transduced with control or Cre by lentiviral infection (with the RFP marker). The transduced cells were mixed with matrigel and plated at the rim of a petri dish. After 8–10 days incubation, prostate spheroids were formed. RFP expression indicates a sphere was transduced with Cre or control gene. Scale = 25 μm. (C) PCR analysis of genomic DNA isolated from control or Cre transduced prostate spheres. Recombination of conditional-targeting Ext1 allele (Ext1f) by Cre transduction led to generation of a mutant Ext1 allele (Ext1−) band. (D) Ext1f/f PrECs lost HS expression after transducing with Cre expressing virus. Primary prostate spheres were dissociated and sorted based on RFP expression, cultured as a monolayer culture and stained for HS epitopes (10E4 antibody). The Cre-transduced cells lost HS expression. Scale = 25 μm. (E, F) Phase and fluorescence images, and the size distribution of the spheres derived from control (Ext1f/f) or Cre (Ext1−/−) transduced groups in primary sphere (1°, E) and secondary (2°, F) sphere assays. Scale = 25 μm. The percentage of primary (1°, E) and secondary (2°, F) spheres formed from the control or Cre transduced PrEC preparations are shown too. The data represent mean ± SD from triplicate experiments. (*P < 0.05; **P < 0.005).
Prostate spheres clonally arise from PrSCs when cultured within matrigel (Reynolds and Weiss 1996; Dontu et al. 2003). The serial passaging of the sphere formation allows to assess the self-renewal potential of PrSCs (Xin et al. 2007). To determine if HS is functionally essential for PrSC self-renewal, PrECs from conditionally targeted Ext1 (Ext1f/f) adult mice were transduced with Cre-recombinase or control lentivirus and grew in matrigel to form spheres (Figure 1B). Ablation of Ext1 expression was confirmed by PCR analysis of the genomic DNA (Figures 1C and S1) (Kraushaar et al. 2010, 2012; Bianco et al. 2013), while deficiency of HS expression was confirmed by cell surface anti-HS antibody (10E4) staining (Figure 1D) (Wang et al. 2005; Wijelath et al. 2010). Both the control (Ext1f/f) and Cre-transduced (Ext1−/−) PrECs, after 8–10 days in culture, formed primary spheres (1° spheres) with comparable efficiency (Figure 1E). The size of Ext1−/− spheres appeared smaller, but the difference did not reach a statistical significance. When PrECs dissociated from primary spheres were reseeded to generate secondary spheres (2° spheres), a dramatically reduced number of 2° spheres were formed in the Ext1−/− group with many of the plated PrECs persisting as single cells, compared to the PrECs from the control-infected (Ext1f/f) primary spheres which retained a high sphere formation activity (Figure 1F). This observation illustrates that HS is required for PrSCs to maintain their self-renewal activity.
HS functions in trans and in cis to sustain PrSC self-renewal
HS expressed on cell surfaces can function in cis (on its own cell surface) or in trans (on the cell surface of a neighboring cell) (Jakobsson et al. 2006; Nakato and Li 2016). The diminished 2° sphere formation capacity of the Ext1−/− cells highlighted the essential requirement of HS to maintain the self-renewal activity of PrSCs and also revealed that HS functions in cis to mediate the biological function. However, we did not observe this dramatic loss of sphere formation of Ext1f/f PrSCs after Cre-lentiviral infection in the 1° sphere-forming assay. It is plausible that this may result from incomplete transduction of Cre-expressing lentivirus. In consequence, HS expressed by the residual Ext1f/f PrECs may function in trans to sustain the self-renewal/sphere formation of Ext1−/− PrSCs. To assess this possibility, we employed Rosa26mT/mG mouse, a double-fluorescent Cre reporter that expresses membrane-targeted tandem dimer Tomato (tdTomato) prior to Cre-mediated excision and membrane-targeted GFP (mG) expression after the Cre-mediated excision (Muzumdar et al. 2007), and generated Rosa26mT/mGExt1f/f mouse, which allows monitoring Cre-mediated Ext1 ablation in cells (Figure 2A). PrECs isolated from Rosa26mT/mGExt1f/f mice were transduced with Cre by lentiviral infection and cultured in matrigel to form 1° spheres. Interestingly, a majority of Cre transduced primary spheres were shown to be chimeras with both tdTomato and EGFP expression (tdTomato+EGFP+), indicating that Ext1−/− and Ext1f/f cells coexisted in most of the Cre-lentivirus transduced 1° spheroids (Figure 2B, top panel, and Figure 2C). A very low percentage of primary spheres showed complete loss of Tdtomato fluorescence (Tdtomato−EGFP+) and exhibited a dramatic decrease in size (Figure 2B, lower panel, and Figure 2D). These observations strongly suggest that HS expressed by neighboring cells functions in trans to sustain the self-renewal capacity of Ext1−/− PrSCs. To more directly determine the cis and trans function of HS in maintaining PrSC self-renewal in primary sphere assay, the control or Cre recombinase (with RFP reporter) transduced Ext1f/f PrECs forming 1° spheres were sorted (Figure S1) as soon as RFP expression was observed (~at day 4) and seeded for 2° sphere formation. The control cells formed abundant 2° spheres, whereas very few spheres were formed from Cre-expressing PrECs (Figure 2E). Taken together, these studies clearly show that HS functions both in trans and in cis to maintain self-renewal activity of PrSCs.
Fig. 2.
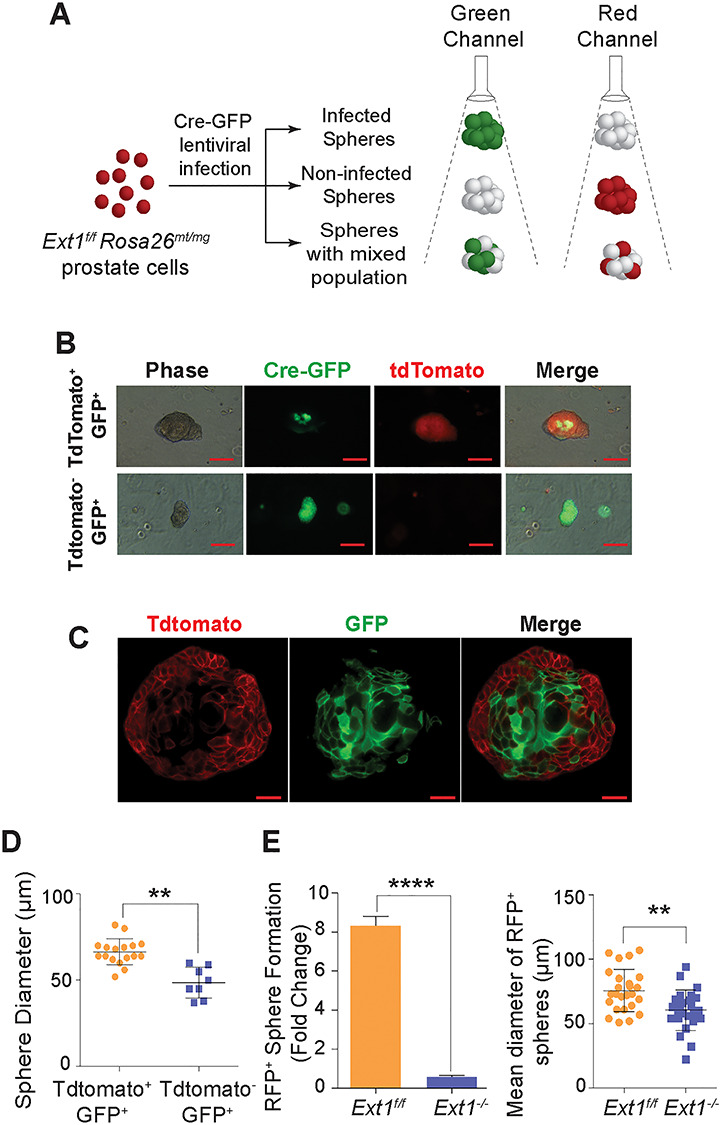
HS functions in trans and in cis to sustain self-renewal potential of PrSCs. (A) Schematic representation of the strategy employed in determining Cre expression in prostate spheres derived from Ext1f/f Rosa26mt/mg mouse PrECs. Due to incomplete Cre-lentiviral infection, the primary PrECs from Ext1f/f Rosa26mt/mg mice forming primary sphere exhibit mosaic pattern of Cre expression. While expression of Cre correlated with gain of GFP expression (green fluorescence) with concomitant loss of TdTomato expression (TdT, red fluorescence), non-Cre expressing cells retained Tdtomato expression. (B) Fluorescence image of Cre-infected primary (1°) prostate spheres from Ext1f/f Rosa26mt/mg PrECs. Scale = 50 μm. The top panel showed the incomplete Cre-lentiviral infection with mosaic expression of green and red color in the sphere. The bottom panel showed a uniform Cre expression, which all cells were green in a sphere. (C) Confocal image of Cre-infected mosaic primary (1°) prostate sphere from Ext1f/f Rosa26mt/mg PrECs. Scale = 25 μm. Only part of the spheroid cells had expression of Cre (GFP+), and the rest remained Ext1f/f (RFP+). The spheres with mosaic pattern of Cre expression formed normally. (D) The size distribution of the primary (1°) spheres with mosaic or complete Cre-expression. The size of sphere with GFP+RFP− (uniformed expression of Cre) was smaller than the ones with GFP+RFP+ (mosaic expression of Cre). Results show means ± SD representative from three independent experiments. Statistical significance was assessed by Student’s t-test. **P < 0.01. (E) Secondary (2°) sphere formation efficiency and size distribution of spheres derived from control or Cre transduced PrECs that were sorted from day 4 primary spheres. The data are represented as mean ± SD from three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001.
Loss of HS inhibits p63/CK5 expression and promotes PrSC differentiation
Prostate spheres are primarily generated from p63+ basal cells, which differentiate into cytokeratin 5 expressing (CK5+) prostate progenitor cells and further into fully differentiated CK8+ PrECs (Huang, Hamana, Liu, Wang, An, You, Chang, Xu, McKeehan et al. 2015). To further determine if loss of HS alters expressions of p63, CK5 and CK8 in prostate spheres, Cre-transduced Ext1f/f PrECs were isolated from 1° spheres (Figures 3A and S2) and analyzed for PrSC-related gene expression by qPCR analysis. Surprisingly, Ext1−/− cells showed significantly reduced expressions of p63 and CK5, and an increased expression of CK8 (Figure 3B). In agreement with these findings, IHC staining showed that, in the control 1° spheres, p63+ cells were abundant and present within the outer layers with uniform expression of CK5 and minimal CK8 expression, whereas the number of p63+ cells was dramatically reduced with reduced CK5+ cells and increased CK8+ cells in the Cre transduced sphere (Figure 3C). Collectively, these data demonstrate loss of HS expression diminishes PrSC homeostasis and induces PrSC differentiation.
Fig. 3.
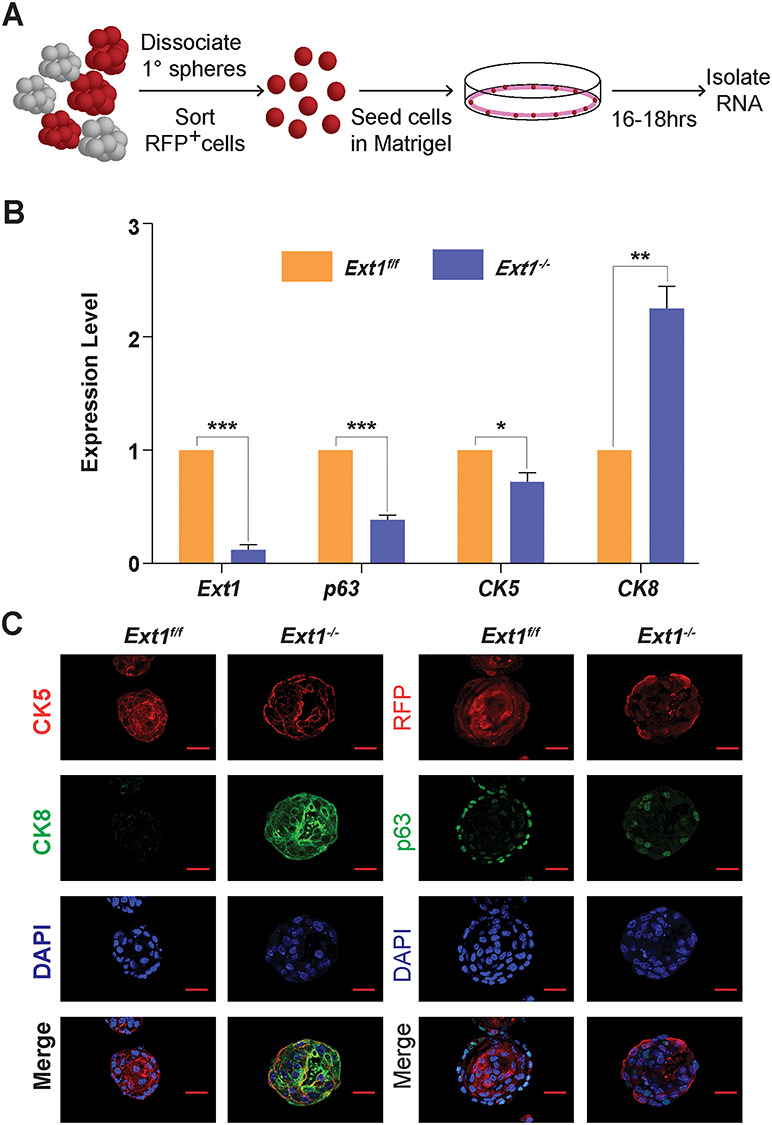
Deficiency of HS expression inhibits p63 and CK5 expression and promotes PrSC differentiation. (A) Schematic representation of mRNA isolation from secondary spheres. Dissociated primary PrECs were transduced with Cre (with RFP marker) by lentiviral infection. After 10 days, the primary spheres were dissociated into single cells. The RFP+ cells were collected by cell sorting, and reinoculated into the matrigel for the recovery of enriched cell. Total RNA was isolated after 16–18 h inoculation. (B) qRT-PCR analysis for expression levels of Ext1 and lineage markers (p63, CK5 and CK8) of the above short-cultured RFP+ cells. Fold Change was normalized to Gapdh. Results represent the mean ± SD of three independent experiments. Statistical significance was assessed by Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0 .001. (C) Immuno-histochemical staining for CK5, CK8 and p63 expression in Ext1f/f and Ext1−/− primary spheres. Scale = 25 μm.
Loss of HS arrests PrEC cycle progression
HS is known to interact with many growth factors and cytokines to modulate cell proliferation (Bishop et al. 2007). We further examined if loss of self-renewal activity of the Ext1−/− PrSCs was caused by altering cell cycle. Cells isolated from control or Cre-expressing Ext1f/f 1° spheres were analyzed for cell cycle progression (Figure S3). The percentage of cells in the G1/G0-phase was significantly higher in the Ext1−/− cells (Figure 4A). Furthermore, qRT-PCR analysis of the gene expression profile of cell cycle regulators, including cyclins, cyclin-dependent kinase (CDKs) and cyclin-dependent kinase inhibitors (CKIs), showed decreased expression levels of CDK4, cyclins A2, B1 and D1, and elevated expression of p21, a CKI, in the Ext1−/− spheroid cells (Figure 4B–D). No altered expression of apoptosis-related gene, including Bcl2, Bax and Caspase-3, was detected in the same cells (Figure 4E). These data suggest that loss of HS attenuates cell proliferation, not apoptosis, to contribute to the self-renewal defect of the Ext1−/− PrSCs.
Fig. 4.
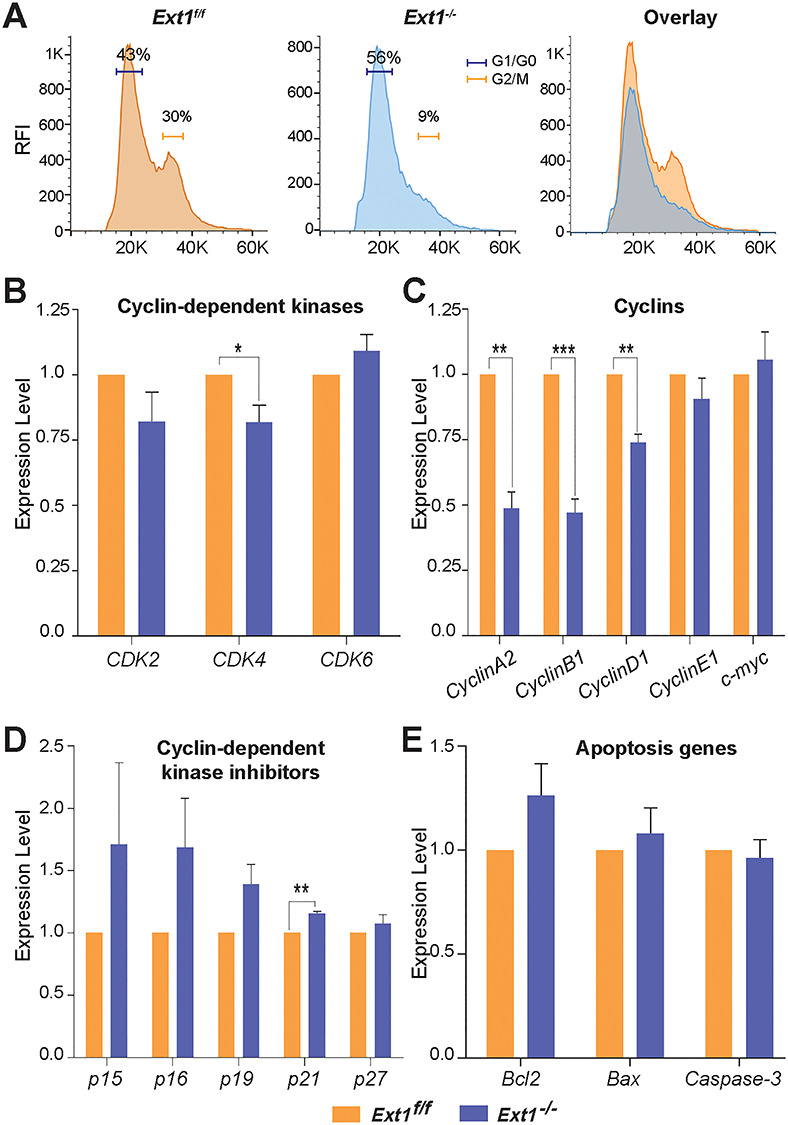
Loss of HS expression arrests cell cycle progression. (A) The primary prostate spheres derived from Ext1f/f or Ext1−/− PrECs were dissociated and subjected to cell cycle analysis after Hoechst 33342 staining. (B–E) qRT-PCR analysis of cell cycle regulators including CDKs (B), Cyclins (C), CKIs (D) and apoptotic genes (E) of the above cells. Fold change was normalized to Gapdh. Results represent mean ± SD of three independent experiments. Statistical significance was assessed by Student’s t-test. *P < 0.05, **P < 0.01 and ***P < 0.001.
Inhibiting TGFβ signaling partially restores self-renewal activity of Ext1−/− PrSCs
The homeostasis of adult PrSCs is the outcome of a variety of signaling networks including growth factors, morphogens and adhesion molecules (Witte 2009; Valdez et al. 2012). Within this signaling network, TGFβ signaling is one of the crucial regulators and functions to maintain dormancy of adult PrSCs (Danielpour 1999; Salm et al. 2005). HS is known to regulate TGFβ signaling in various cell types (Eickelberg et al. 2002; Qiu et al. 2013). We investigated if HS regulates TGFβ signaling to maintain self-renewal activity of PrSCs and analyzed expressions of TGFβ receptors (TGFβRs), ligands and signaling-related transcription factors and target genes. We observed that, while TGFβR2 and TGFβ2 expression levels were significantly increased in the Ext1−/− sphere cells (Figure 5A–C), the expression of TGFβRIII (betaglycan), a transcriptional repressor of TGFβ signals (Lopez-Casillas et al. 2003; Hempel et al. 2008), was reduced. Additionally, the expression of a number of TGFβ-responsive transcription factors was altered including the upregulation of 4E-BP1 and downregulation of E2F1. The expression levels of TGFβ target genes were also altered, including a reduction of Id1 and Id3 in the Ext1−/− sphere cells. In parallel, pSmad2 level was also examined. Upon TGFβ1 stimulation, nuclear pSmad2 level was significantly elevated in the Cre-transduced Ext1f/f PrECs (Ext1−/−) in comparison with the control-transduced cells (Ext1f/f) (Figure 5D). Additionally, the level of pSmad3 was also elevated in Ext1−/− cells in comparison with Ext1f/f cells under the induction of TGFβ1 in western blot analysis (Figures 5E and S4). To determine if the inhibition of pSmad levels upon TGFβ1 stimulation directly depends on HS, we also treated primary human PrECs with heparinases I–III prior to TGFβ stimulation. As shown in Figure 5F, the heparinase treatment efficiently degraded cell surface HS as evidenced by diminishing the top two Syndecan bands, which sat at 90–120 kd range in western blot. The heparinase-treated cells also showed elevated phosphorylation of Smad1, Smad2 and Smad 3 upon TGFβ1 stimulation. Collectively, these observations showed that loss of HS upregulates TGFβ-Smad1/2/3 signaling in PrECs.
Fig. 5.
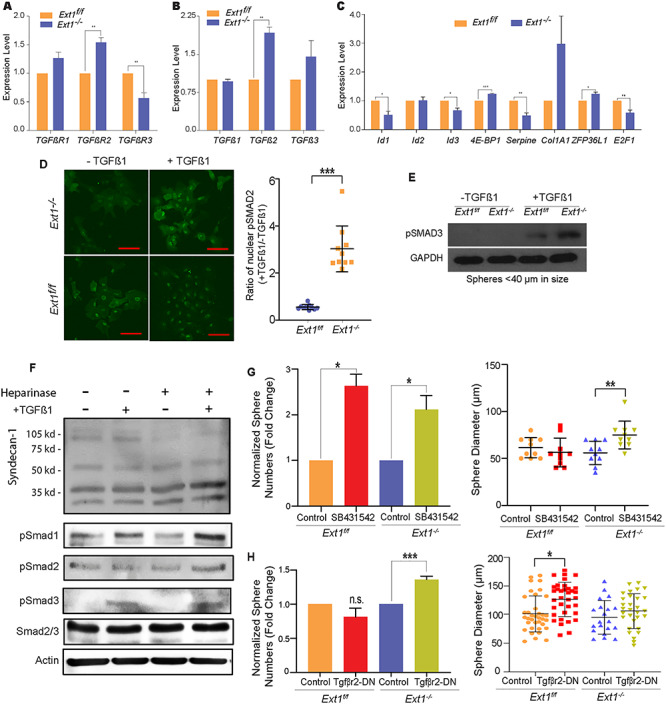
Inhibition of TGFβ signaling restores sphere formation activity of Ext1−/− PrSCs. (A–C) qRT-PCR analysis to assess expression levels of TGFβR1/2/3 (A), TGFβ1/2/3 (B), and TGFβ signaling downstream target genes including ld1/2/3, 4E-BP1, Serpine, Col1A1, ZFP36L1, and E2F1 in Ext1f/f or Ext1−/− prostate spheroid cells (C). (D) Expression levels of nuclear pSmad2 under the stimulation TGFβ1 in Ext1f/f and Ext1−/− PrECs. PrECs were sorted from primary prostate spheres transduced with control vector- or Cre gene (based on RFP reporter). The sorted cells were cultured as monolayer in PrEGM medium. After stimulation with TGFβ1 (1 ng/mL) for 10 min, the cells were fixed and stained with p-Smad2 antibody. Expression levels of p-Smad2 in the nuclear were quantified. The ratio of nuclear p-Smad2 with/without TGFβ1 were calculated in Ext1f/f or Ext1−/− cells. (E) Expression levels of pSmad3 under the stimulation TGFβ1 in Ext1f/f and Ext1−/− PrECs. Primary prostate spheres (<40 μm in diameter) transduced with control vector- or Cre gene (based on RFP marker) were collected and dissociated into single cells. The dissociated cells were cultured in PrEGM medium. After stimulation with TGFβ1 (1 ng/mL) for 10 min, cell lysates were extracted. Expression levels of p-Smad3 and GAPDH were examined by western blot analysis. The data are representative of 2–3 independent experiments. (F) Heparinase-treated human primary PrECs show enhanced Smad phosphorylation upon TGFβ1stimulation. Primary human PrECs at 80% confluence were treated with heparinases I, II and III (100 mU/mL) in serum-free medium for 3 h, and then stimulated with TGFβ1 (5 ng/mL) for 30 min. Following, the cells were lysed and analyzed for syndecan-1, pSmad1–3, Smad2/3 and actin in western blot. Heparinase treatment diminished the glycol-form (two bands at about 90–12 kd), and heparinase-treated cells showed increased phosphorylation of Smad1–3, although untreated cells had unchanged pSmad2 level upon TGFβ1treatment. The data shown are representative of two times of the experiment. (G, H) Primary sphere from wild type or Ext1−/− PrECs were dissociated into single cells. The dissociated cells were replated into matrigel and treated with SB43154 (F), or overexpressed with dominant negative form of TGFβR2 (TGFβR2-DN) (G) to form the secondary spheres. The number and size (>40 μm in diameter) of the secondary spheres were recorded. Results show means ± SD and is representative from three independent experiments. Statistical significance was assessed by Student’s t-test (*P < .05, **P < .01 and ***P < .001).
We further examined if the upregulated TGFβ signaling is responsible for the loss of self-renewal activity of the Ext1−/− PrSCs. Prostate spheroid cells derived from primary spheres were treated with SB-431542, a specific TGFβR1 inhibitor (Droguett et al. 2010). SB-431542 treatment increased the number and size (>40 μm) of spheres derived from Ext1−/− PrEC cells (Figure 5G). The treatment also increased the number, though not the size of Ext1f/f sphere (Figures 5G and S5A). Additionally, over-expression of TGFβR2-DN (Figure S6), a dominant negative TGFβR2 (Siegel et al. 2003), also increased Ext1−/− sphere number, but not size (>40 μm) (Figure 5H), and the slightly increased the size of control sphere by TGFβR2-DN (Figures 5H and S5B), which, in together, suggest that TGFβR1 and TGFβR2 signaling might function some differently in regulation of PrSC self-renewal, but both support that upregulated TGFβ signaling contribute to the loss of self-renewal phenotype of the Ext1−/− PrSCs. Collectively, the pSmad1/2/3 activation and self-renewal rescue data indicate that HS deficiency enhances TGFβ signaling which, in turn, inhibits self-renewal activity of PrSCs.
Simultaneous loss of HS in both epithelial and stromal cell compartments disrupts prostate regeneration in vivo
PrSCs are responsible for prostate regeneration, a highly orchestrated process that requires intimate regulatory crosstalk between the epithelial and the stromal cell compartments (Xin et al. 2003; Josson et al. 2010). To test if HS is required for PrSCs to regenerate prostate tissue, we carried out the in vivo prostate regeneration assay (Figure 6A) (Xin et al. 2003; Cai et al. 2011; Wu et al. 2016). The primary PrECs or urogenital sinus mesenchyme cells (UGSM) derived from Ext1f/f mice were transduced with the control or Cre recombinase. The transduced PrECs and UGSMs were mixed together in a collagen gel to form grafts, wherein HS was depleted in PrEC compartment (Ext1−/−-PrEC/Ext1f/f-UGSM), UGSM cell compartment (Ext1f/f-PrEC/Ext1−/−-UGSM) or both (Ext1−/−-PrEC/Ext1−/−-UGSM). The grafts were implanted under the kidney capsule in the severe combined immunodeficient (SCID) mice. Eight weeks after the implantation, the regenerated prostate tissues were harvested for analysis. The size, weight and number of prostatic tubules of the regenerated prostate tissues were comparable among the Ext1f/f-PrEC/Ext1f/f-UGSM (control), the Ext1−/−-PrEC/Ext1f/f-UGSM and Ext1f/f-PrEC/Ext1−/−-UGSM groups, but were significantly reduced in the Ext1−/−-PrEC/Ext1−/−-UGSM group (Figures 6B–D and S7). These observations indicate that expression of HS in PrSCs or stromal cells is sufficient to maintain the regeneration activity of PrSCs, whereas simultaneous loss of HS expression in both epithelial and stromal cells diminishes the potential for prostate regeneration. These observations illustrate that HS is essentially required for PrSCs to regenerate prostate and that HS expressed by PrECs and UGSMs can function in cis and in trans, respectively, to sustain PrSC regeneration capacity in vivo.
Fig. 6.
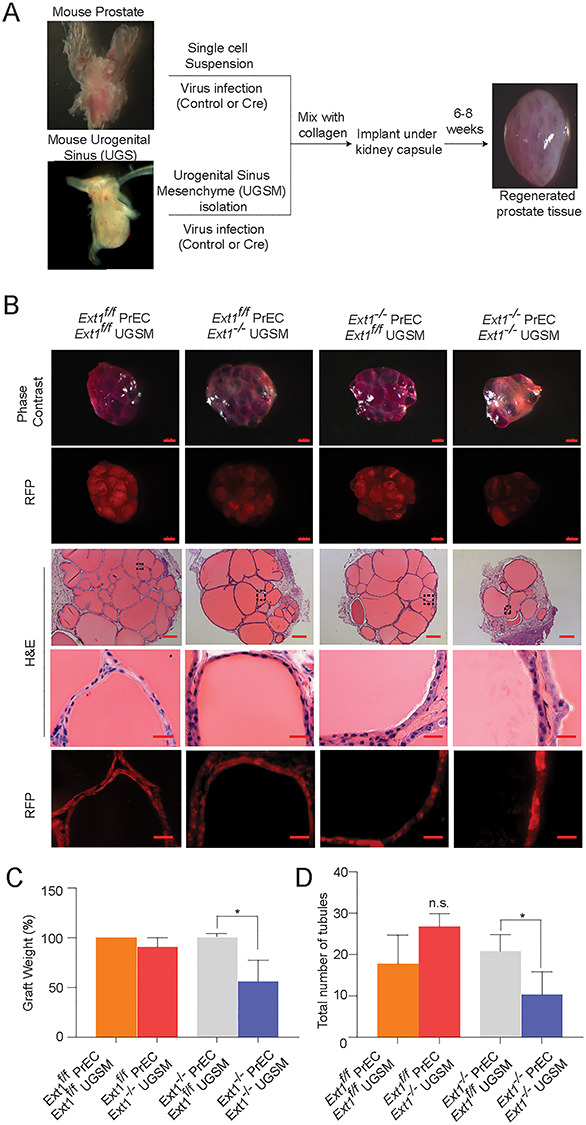
Loss of HS expression diminishes the activity of PrSCs to regenerate prostate in vivo. (A) Schematic representation of in vivo prostate regeneration assay. (B) The global view of regenerated prostate tissue. Phase contrast and RFP expression in grafts (scale = 0.5 mm) and H&E staining sections of the grafts (scale = 200 μm). (C, D) Percentage weight of grafts and total number of formed tubules. Results represent mean ± SD from three independent experiments. Statistical significance was assessed by Student’s t-test (P < 0.05).
Discussion
HS has been established to be essential in regulating embryonic stem cell fate (Johnson et al. 2007; Sasaki et al. 2008; Kraushaar et al. 2010, 2012, 2013; Lanner et al. 2010). Recent studies have proceeded to investigate the roles of HS in tissue-specific stem cells, and have reported that HS is essential for self-renewal or homeostasis of stem cells in skeletal muscle, epidermis, neural tissue, bone marrow, salivary gland, testis and intestine (Buono et al. 2010; Helledie et al. 2012; Patel et al. 2014; Saez et al. 2014; Dos Santos et al. 2016; Levings et al. 2016; Pawlikowski et al. 2017; Takemura and Nakato 2017). In present study, we provide in vitro and in vivo evidences demonstrating that HS is a crucial regulator of PrSC self-renewal and maintenance in prostate gland and is required to fulfill PrSCs capacity to regenerate prostate tissue. Our cell lineage marker analysis revealed that HS also regulates PrSC differentiation.
The prostate develops from the urogenital sinus during late embryonic development. Crosstalk between UGSM and the urogenital sinus epithelium are required for proper prostate development. It is well documented that UGSMs provide supportive as well as inductive signals in prostate gland development (Cunha and Lung 1979). In tissue recombination experiments, UGSM cells instruct PrSCs that are isolated from adult prostate to regenerate prostate tissue (Xin et al. 2003). Using in vitro sphere formation and in vivo tissue recombinant prostate regeneration assays, we provided first evidence, to our best knowledge, showing that HS functions in cis and in trans to sustain PrSC self-renewal activity and prostate regeneration (Figure 7), and demonstrating that HS is indispensable for PrSC homeostasis thereby prostate homeostatsis and regeneration.
Fig. 7.
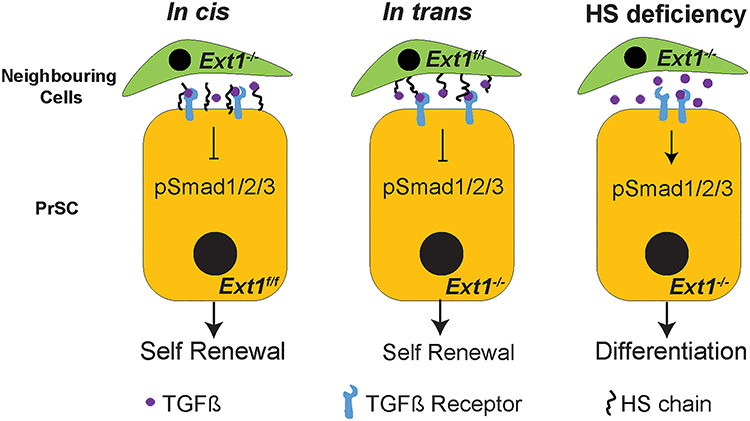
Schematic model of HS dependent regulation of TGFβ signaling in PrSCs. HS chains expressed on the surface of PrSCs (in cis) or on the surrounding cells (neighboring epithelial or stromal cells, in trans) interacts with TGFβ presumably resulting in decreased ratio of TGFβ binding to TGFβR-II and TGFβR-I (not shown), thereby suppressing TGFβ signals to maintain self-renewal activity of PrSCs. In the complete absence of HS, the inhibitory role of HS is lost leading to increased TGFβ signals, which results in attenuated self-renewal activity and enhanced differentiation of PrSCs to luminal cells.
In our study, the in trans function of HS-expressing cells has helped to interpret the normal primary sphere formation with HS-deficient PrSCs. We noticed that, due to the nature of the experimental system, the Cre recombinase expression becomes evident only around day 4 after Cre transduction. It is reasonable to believe it could take more time to completely ablate HS biosynthesis and to have the pre-existing HS metabolized, and our claimed “HS-deficient” PrECs should still express HS during the days 4–6 in the primary sphere formation assay. This may provide another possible reason to explain why complete loss of self-renewal activity in HS deficient PrSCs was observed only in the second sphere formation assay. Further time course studies by staining HS expression at different time points of the primary sphere formation assay may help to clearly address this issue.
Chondroitin sulfate is another glycosaminoglycan, which is structurally close to HS and sometimes functionally overlaps with HS. In previous studies, we observed that Ext1 deletion led to a ~2-fold upregulated chondroitin sulfate expression in mouse embryonic stem cells (29). Interestingly, we did not observed obvious altered chondroitin sulfate expression in the Cre-transduced PrECs (Figure S8). This suggests that alteration of chondroitin sulfate expression upon Ext1 deletion may be cell-type dependent.
Paracrine TGFβ signaling is required for UGSM to induce endoderm-derived epithelia and stem cells to form prostate (Li et al. 2009). An early study examined androgen dependence of HS biosynthesis in prostate following castration and subsequent androgen-supplementation-induced regeneration (Terry and Clark 1996). HS content was significantly decreased following castration and increased after androgen replacement, highlighting that HS may participate to mediate androgen-primed prostate regeneration. In the regeneration experiment, we provided the first evidence showing that HS indeed is essential for proper prostate regeneration. We further delineated that HS sustains self-renewal activity of PrSCs by inhibiting TGFβ signaling. Our results are consistent with the reports showing that TGFβ signaling promotes differentiation of PrSCs toward luminal cell fate (Danielpour 1999; Salm et al. 2005). Furthermore, increase in the levels of TGFβ2, TGFβ3 and TGFβR2, indicating elevated TGF signaling, in the rat PrEC line NRP-152 initiates luminal differentiation (Danielpour 1999). These reported studies supports our conclusions that HS mitigates TGFβ signaling to maintain PrSC self-renewal and homeostasis.
HS has been reported to positively or negatively modulate TGFβ signaling in various cellular or tissue contexts via different modes of action. For example, in mouse periocular mesenchyme, HS deficiency specifically inhibits TGFβ2 signaling manifested by diminished phosphorylation of Smad2 (Iwao et al. 2009). Studies of CHO cells show that TGFβ1 induces transcriptional activation of plasminogen activator inhibitor-1 and inhibits HS-deficient CHO cell (CHO-677 cell) more potently than wildtype CHO-K1 cells, indicating that HS inhibits TGFβ1 signaling (Chen et al. 2006). Currently, three possible modes of action for HS to modulate TGFβ signaling have been proposed. In the first mode, HS modulates the diffusion and the gradient of TGFβ within the local environment. A good example of this action mode is the Drosophila TGFβ homolog, Dpp, which moves along the cell surface via HS to restrict extracellular diffusion in order to maintain proper TGFβ signaling (Eickelberg et al. 2002; Chen et al. 2006). In the second mode of action, HS functions as a coreceptor to facilitate the interaction between TGFβ and the receptors on the cell surface (Kuo et al. 2010). In the third mode of action, HS decreases the ratio of TGFβ binding to TGFβR1 and TGFβR2 on cell surfaces, and facilitates caveolae/lipid-raft-mediated endocytosis and rapid degradation of TGFβ to attenuate TGFβ signaling (Chen et al. 2006). Using these three modes of action, HS can act to enhance or inhibit TGFβ signaling depending on the cellular and tissue context. In our study, we observed that PrECs express both TGFβR2 and TGFβR1, and HS mitigates TGFβ signaling to sustain self-renewal activity of PrSCs, suggesting HS acts in the third mode to regulate TGFβ signaling in order to maintain PrSC self-renewal activity and homeostasis. We postulate that HS functions both in cis and in trans to decrease the ratio of TGFβ binding to TGFβR2 and TGFβR1 on the PrSC surface, thereby mitigating TGFβ signaling to maintain PrSC self-renewal activity and homeostasis in adult prostate (Figure 7).
Our studies have noticed that inhibition of TGFβR1 using SB-431542 and TGFβR2 via overexpressing TGFBR2-DN did not lead to the same rescue effects of Ext1−/− PrECs. This may be explained by their different inhibitory spectrum in TGFβ signaling. SB-431542 inhibits TGFβR1–activin-like kinase 5 (ALK5) pathway as well as ALK4 and ALK7. Knockout of ALK4, ALK5 or ALK7 displays unique phenotypes indicating distinct cellular or contextual roles for these ALKs (Jornvall et al. 2004; Itoh et al. 2009; Vogt et al. 2011). The phenotype observed in Figure 5G is the collective inhibition of these ALKs. The over-expression of TGFβR2-DN only inhibits signals emanating from the receptor complex composed of TGFβR1 and TGFβR2 receptor, and this genetic manipulation is expected to have signaling outcome distinct from the SB-431542 treatment, leading to the rescue effect some different from the SB-431542. Since both treatments partially rescue the Ext1−/− PrSC phenotype, the both treatments serve to alternatively support that HS inhibits TGFβ signaling to maintain PrSC self-renewal.
We show that inhibition of TGFβ signaling only partially rescued the self-renewal activity of the HS-deficient PrSCs, suggesting the possibility that other HS-dependent signaling(s) is required to acquire full self-renewal activity. A recent study has shown that type 2 FGFR signaling preserves stemness and prevents differentiation of PrSCs from the basal compartment (Huang, Hamana, Liu, Wang, An, You, Chang, Xu, Jin et al. 2015). FGF requires HS as a coreceptor for proper signaling (Donjacour et al. 2003; Izvolsky et al. 2003). Therefore, it is possible that the loss of HS may also impair FGF signaling attributing to the self-renewal defect in Ext1−/− PrSCs. Additionally, several other paracrine growth factors that essentially regulate prostate development, including BMP4 and BMP7, also bind HS (Lamm et al. 2001; Grishina et al. 2005; Huang et al. 2005; Prins et al. 2006; Lin et al. 2007; Zhang et al. 2008; Abler et al. 2011). A recent study reported that exogenous BMP4 and BMP7 induce Sulf1 expression in the UGMS in in vitro organ culture, decrease epithelial HS 6-O-sulfation, and reduce intracellular signaling of urogenital sinus epithelium in response to FGF10 stimulation, revealing a pivotal role HS plays in regulating BMP and FGF10 signaling in prostate development (Buresh-Stiemke et al. 2012). Therefore, it is possible that HS also modulates BMP4/7-FGF10 signaling pathways to maintain self-renewal activity and the homeostasis of adult PrSCs and this will be examined in our further studies.
In conclusion, our studies demonstrate that HS is a crucial regulator of PrSC self-renewal and prostate regeneration. Our mechanistic studies revealed that HS inhibits TGFβ signaling and functions both in cis and in trans to sustain adult PrSC self-renewal and prostate homeostasis, and to facilitate prostate regeneration.
Experimental procedures
Mice
All animals received humane care in compliance with the protocol approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Georgia. The conditional Ext1f/f mice were kindly provided by Dr. Yu Yamaguchi (Sanford Burnham Prebys Medical Discovery Institute, La Jolla, CA, USA) (Inatani et al. 2003). The Rosa26mT/mG mice were obtained from the Jackson Laboratory (Bar Harbor, ME) (Muzumdar et al. 2007). Ext1f/f female mice were crossed with Rosa26mT/mG male mice to generate Rosa26mT/mGExt1f/f mice. Mice were genotyped by PCR using mouse genomic DNA from tail biopsy specimens. The sequences of genotyping primers and the expected PCR product sizes are listed in Table SI. PCR products were separated on 2% agarose gels. The C57BL/6 and CB.17SCID/SCID mice were obtained from Charles River (Wilmington, MA).
Lentivirus generation
Codon-optimized Cre recombinase (iCre) was cloned using the XbaI site of a FU-CRW vector or FU-CGW (Xin et al. 2006), generating the FU-Cre-CRW and FU-Cre-CGW vector. In this vector, iCre is driven by a human Ubiquitin promoter and is followed by a CMV promoter driving the expression of a monomeric red RFP or enhanced GFP, respectively. The TGFBR2-DN-expressing construct was derived from pCMV5 HA-TBRII (delta Cyt) that was purchased from Addgene (plasmid # 14051) (Siegel et al. 2003). The encoded gene was subcloned into the EcoRI and XbaI sites of the FU-CRW lentiviral vector (primers listed in Table SII). Lentivirus production and titration were performed as described previously (Cai et al. 2012). All procedures followed the safety guidelines and regulations of the University of Georgia.
Prostate primary and secondary sphere formation assay
Prostate tissue derived from 8-week-old male mice were dissected, minced into small pieces with a steel blade, and digested with collagenase I (190 units/mL, GIBCO) in 10 mL of DMEM 10% FBS (GIBCO) at 37°C for 90 min. PrECs were pelleted, washed once PBS without Ca2+ and Mg2+ and trypsinized with 0.05% Trypsin/EDTA for 5 min at 37°C. Trypsin is inactivated by addition of equal volume of media containing 10% FBS. The cells were then passed 4–5 times through a 21G needle, followed by a 251/2 G needle, and filtered with 40 μm nylon mesh (BD Biosciences, San Jose, CA) to obtain single cell suspension. Cells were washed twice with DMEM 10% FBS and resuspended in 1 mL of DMEM 10% FBS. Dissociated prostate cells were infected with control/Cre/TGFBR2-DN-expressing lentiviruses at a multiplicity of infection of 10–20 using the spinoculation method at 750 g for 120 min at 25°C (Xin et al. 2003).
For primary prostate sphere formation, the infected cells were collected, washed and finally resuspended in 50 μL prostate epithelial growth medium (PrEGM) (#CC-3166, Lonza, Walkersville, MD). About 50 μL of cell suspension was then mixed with 50 μL of matrigel (#356234, Corning) and plated around the rim of a well in a 12-well plate. After the cell–matrigel mixture solidified at 37°C for 45 min, 1 mL of PrEGM was added. Prostate spheres were formed after 8–10 days of incubation. Additionally, Cre (with RFP/GFP marker) was expressed due to viral integration. Prostate spheres were defined as a spheroid with diameter ≥ 40 μm after 8 days of culture. For secondary prostate spheres, dispase (final concentration of 1 mg/mL) was added to digest the matrigel matrix after the RFP/GFP expression was confirmed. Spheres were collected by passing through a 40 μm cell strainer to remove single cells while the spheres were retained. The spheres were washed once with PBS without Ca2+ and Mg2+ digested with collagenase I (190 units/mL, GIBCO) in 1 mL of DMEM 10% FBS (GIBCO) at 37°C for 90 min. The collected spheres were washed once with PBS without Ca2+ and Mg2+ and trypsinized for 10 min at RT with 0.05% trypsin-EDTA. Spheres were dissociated by passing through a 211/2 G needle thrice and filtered by a 40 μm cell strainer. Cells were then resuspended in PrEGM medium. After being counted, the dissociated primary spheroid cells were mixed with matrigel, and plated in the rim of a well in a 12-well plate. The secondary spheres were formed after 8–10 days incubation. In some cases, dissociated primary spheroid cells were sorted based on fluorescence before seeding them in matrigel for secondary sphere formation.
To exam if TGFβ signaling is required for sphere formation, SB431542 (Woburn, MA) was dissolved in DMSO and added at optimized concentrations in second sphere formation assay (Shahi et al. 2011; Zhang et al. 2011; Valdez et al. 2012). The SB431524-supplemented culture media was replaced every 24 h.
FACS
FACS analyses and sorting of RFP/GFP-expressing cells were performed using the Bio-Rad S3 cell sorter (Bio-rad, Hercules, CA) (Figure S1). For cell cycle analysis, dissociated primary sphere cells were seeded in matrigel for 16–20 h. They were then harvested, dissociated to single cells and stained with Hoechst 33342 ready flow reagent (Thermo Fischer Scientific) for 1 h at 37°C and analyzed on a HyperCyAn analyzer (Beckman Coulter, Indianapolis, IN) (Figure S2). The cell cycle profile was analyzed using the ModFit software (Verity software house).
In vivo prostate tissue regeneration assay
As described above, primary prostate cells were isolated from the Ext1f/f mice. PrECs (1 × 105 cells/graft) were transduced with control or iCre by lentiviral infection. In addition, Ext1f/f-UGSM was isolated from 16.5-day embryos of Ext1f/f mice. The UGSM cells were transduced with control or iCre gene by lentiviral infection. The control or Cre-transduced PrEC cells were combined with the control or Cre-transduced Ext1f/f-UGSM (1 × 105 cells/graft). The cell mixture was resuspended with 20 μL of collagen type I (neutral pH, #354236, Corning). After overnight incubation, grafts were implanted under the kidney capsule in SCID mice by survival surgery with simultaneous implantation of a subcutaneous testosterone pellet under the skin (12.5 mg of androgen per pellet, 90-day release; Innovative Research of America). Eight weeks after the plantation, the grafts were taken out for analysis. All animals were maintained and examined according to the surgical and experimental procedures approved by the University of Georgia IACUC.
RNA isolation and qRT-PCR analysis
Total RNA was isolated from cells using the RNeasy Plus Mini kit (Qiagen, Valencia, CA). Reverse transcription was performed using the high capacity cDNA reverse transcription kit (Thermo-Fischer Scientific). qRT-PCR was performed using the PerfeCTa SYBR Green FastMix (Quanta Bio, Beverly, MA) on an ABI Step one plus real-time PCR system (Applied Biosystems, Foster City, CA). Primer sequences for qPCR are listed in Table SIII.
Primary human prostate epithelial cell culture, heparinase treatment and TGF-β stimulation
Primary human prostate epithelial cells (HPrECs) were obtained from American Type Culture Collection (ATCC) (Rockville, MD). HPrECs were maintained in Prostate Epithelial Cell Basal Medium (ATCC- PCS-440-030) supplemented with Prostate Epithelial Cell Growth Kit (ATCC PCS-440-040). To delete HS expressed on the cells, HPrECs cells were cultured in 60 mm dishes to reach approximately 80% confluent. To remove expressed HS, the cells were washed with twice with serum-free medium and incubated in a medium with 0.1% serum with or without a mixture of heparinases I, II and III (100 milliunits/mL, Sigma-Aldrich) at RT for 3 h. Then, cells were washed twice with DPBS and treated with vehicle (0.025% TGF-β reconstitution solution including 0.1% BSA) and TGF-β (5 ng/mL) (PeproTech, Rocky Hill, NJ) for 30 min, and then the cells were processed for western blotting analysis.
Western Blotting
Prostate spheres or primary HPrECs were lysed in RIPA buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate and 1 mM Na3VO4) with protease inhibitors (Roche Applied Science) and phosphatase inhibitors 2 and 3 (Sigma). Protein concentrations were determined using a Bradford Assay kit (Bio-Rad). Protein was separated in 10% SDS/PAGE and transferred onto a 0.2 μm nitrocellulose membrane (Amersham Biosciences, Arlington Heights, IL). The membrane was blocked with 5% skim milk, and subsequently incubated with primary antibodies listed in Table SIV at 4°C O/N followed by incubation with peroxidase-conjugated goat antimouse IgG or goat antirabbit IgG (CST, Danvers, MA), and developed with Amersham ECL reagent (GE Healthcare Ltd., Buckinghamshire, UK).
Histology and immunostaining, immunohistochemistry
H&E and immunofluorescence staining were performed using standard protocols on 5 μm paraffin sections. Primary antibodies and dilutions used are listed in Table SIV. Slides were blocked with 3% BSA (Gold Bio, St. Louis, MO) and incubated with primary antibodies diluted in 3% BSA overnight at 4°C. Slides were washed and incubated with secondary antibodies (diluted 1:500 in 0.05% Tween 20 in phosphate-buffered saline), and labeled with Alexa Fluor 488 or 594 (Invitrogen/Molecular Probes). Sections were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich). Immunofluorescence staining was imaged using a Carl Zeiss Axio Observer A1 fluorescence microscope or a Nikon A1 confocal microscope.
For cell surface chondroitin sulfate staining, the primary prostate spheres, transduced with control or Cre lentivirus, were sorted and seeded in eight well chamber slide (Nunc Lab Tek) in PrEGM media. The cells were fixed with 4% PFA for 30 min at RT. The excess PFA was discarded and cells washed three times with PBS for 5 min each. The cells were then blocked with 1% BSA for 1 h followed by overnight incubation with Anti-CS antibody (CS-56, Sigma, 1:200) at 4°C. The cells were washed with PBS thrice and incubated with rabbit antimouse IgM antibody (1 μg/μL, diluted 1:100 in 1% BSA/PBS, Thermo Fischer Scientific) at RT for 1 h. Excess solution was removed from the chambers, the cells were washed three times and incubated with antimouse IgM Alexa Fluor 488 antibody (Thermo Fischer Scientific) for 1 h at RT. The cells were washed again with PBS, mounting media added (Vectashield) and imaged. Antibodies for Immunohistochemistry and western blot are listed in Table SIV.
Supplementary Material
Acknowledgements
We thank Ms. Karen Howard for here English revision of the manuscript. We thank Julie Nelson for cell sorting and flow cytometry analysis in the CTEGD Flow Cytometry Facility at the University of Georgia. We also thank James Barber in the CVM Cytometry Core Facility at the University of Georgia for helping with confocal microscopy.
Funding
National Institute of Health grants (R01HL093339 and P41GM103390 to LW, R01CA172495 to HC, and U01CA22574 to both LW and HC) and a Department of Defense (DOD) grant (W81XWH-15-1-0507 to HC).
Conflict of interest statement
The authors declare no conflicts of interest.
Author contributions
SR, OAA and HY conducted the experiments, acquisition and analysis of the results, and wrote the paper. SR, LW and HC designed the experiments and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Abbreviations
BMP, bone morphogenetic protein; CDK, cyclin-dependent kinase; CKI, cyclin-dependent kinase inhibitor; GFP, green fluorescent protein; Ext1, exostosin 1; GlcA, glucuronic acid; GlcNAc, N-acetyl glucosamine; HS, heparan sulfate; PrEC, prostate epithelial cell; PrEGM, prostate epithelial growth medium; PrSCs, prostate stem/progenitor cells; RFP, red fluorescent protein; Sulf, 6-O-sulfatase; TGFβRII, TGFβ receptor II; TGFBR2-DN, a dominant negative TGFBR2; UGSM, urogenital sinus mesenchyme
References
- Abler LL, Keil KP, Mehta V, Joshi PS, Schmitz CT, Vezina CM. 2011. A high-resolution molecular atlas of the fetal mouse lower urogenital tract. Dev Dyn. 240:2364–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aumuller G, Leonhardt M, Renneberg H, von Rahden B, Bjartell A, Abrahamsson PA. 2001. Semiquantitative morphology of human prostatic development and regional distribution of prostatic neuroendocrine cells. Prostate. 46:108–115. [DOI] [PubMed] [Google Scholar]
- Baeg GH, Lin X, Khare N, Baumgartner S, Perrimon N. 2001. Heparan sulfate proteoglycans are critical for the organization of the extracellular distribution of wingless. Development. 128:87–94. [DOI] [PubMed] [Google Scholar]
- Baldwin RJ, ten GB, van Kuppevelt TH, Lacaud G, Gallagher JT, Kouskoff V, Merry CLR. 2008. A developmentally regulated heparan sulfate epitope defines a subpopulation with increased blood potential during mesodermal differentiation. Stem Cells. 26:3108–3118. [DOI] [PubMed] [Google Scholar]
- Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. 1999. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 68:729–777. [DOI] [PubMed] [Google Scholar]
- Bianco P, Cao X, Frenette PS, Mao JJ, Robey PG, Simmons PJ, Wang CY. 2013. The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat Med. 19:35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop JR, Schuksz M, Esko JD. 2007. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 446:1030–1037. [DOI] [PubMed] [Google Scholar]
- Buono M, Visigalli I, Bergamasco R, Biffi A, Cosma MP. 2010. Sulfatase modifying factor 1-mediated fibroblast growth factor signaling primes hematopoietic multilineage development. J Exp Med. 207:1647–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buresh-Stiemke RA, Malinowski RL, Keil KP, Vezina CM, Oosterhof A, Kuppevelt TH, Marker PC. 2012. Distinct expression patterns of Sulf1 and Hs6st1 spatially regulate heparan sulfate sulfation during prostate development. Dev Dyn. 241:2005–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Memarzadeh S, Stoyanova T, Beharry Z, Kraft AS, Witte ON. 2012. Collaboration of Kras and androgen receptor signaling stimulates EZH2 expression and tumor-propagating cells in prostate cancer. Cancer Res. 72:4672–4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Smith DA, Memarzadeh S, Lowell CA, Cooper JA, Witte ON. 2011. Differential transformation capacity of Src family kinases during the initiation of prostate cancer. Proc Natl Acad Sci U S A. 108:6579–6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CL, Huang SS, Huang JS. 2006. Cellular heparan sulfate negatively modulates transforming growth factor-beta1 (TGF-beta1) responsiveness in epithelial cells. J Biol Chem. 281:11506–11514. [DOI] [PubMed] [Google Scholar]
- Cunha GR, Lung B. 1979. The importance of stroma in morphogenesis and functional activity of urogenital epithelium. In Vitro. 15:50–71. [DOI] [PubMed] [Google Scholar]
- Danielpour D. 1999. Transdifferentiation of NRP-152 rat prostatic basal epithelial cells toward a luminal phenotype: Regulation by glucocorticoid, insulin-like growth factor-I and transforming growth factor-beta. J Cell Sci. 112( Pt 2):169–179. [DOI] [PubMed] [Google Scholar]
- Dejima K, Kanai MI, Akiyama T, Levings DC, Nakato H. 2011. Novel contact-dependent bone morphogenetic protein (BMP) signaling mediated by heparan sulfate proteoglycans. J Biol Chem. 286:17103–17111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donjacour AA, Thomson AA, Cunha GR. 2003. FGF-10 plays an essential role in the growth of the fetal prostate. Dev Biol. 261:39–54. [DOI] [PubMed] [Google Scholar]
- Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, Wicha MS. 2003. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 17:1253–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Santos M, Michopoulou A, Andre-Frei V, Boulesteix S, Guicher C, Dayan G, Whitelock J, Damour O, Rousselle P. 2016. Perlecan expression influences the keratin 15-positive cell population fate in the epidermis of aging skin. Aging (Albany NY). 8:751–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Droguett R, Cabello-Verrugio C, Santander C, Brandan E. 2010. TGF-beta receptors, in a Smad-independent manner, are required for terminal skeletal muscle differentiation. Exp Cell Res. 316:2487–2503. [DOI] [PubMed] [Google Scholar]
- Eickelberg O, Centrella M, Reiss M, Kashgarian M, Wells RG. 2002. Betaglycan inhibits TGF-beta signaling by preventing type I-type II receptor complex formation. Glycosaminoglycan modifications alter betaglycan function. J Biol Chem. 277:823–829. [DOI] [PubMed] [Google Scholar]
- English HF, Santen RJ, Isaacs JT. 1987. Response of glandular versus basal rat ventral prostatic epithelial cells to androgen withdrawal and replacement. Prostate. 11:229–242. [DOI] [PubMed] [Google Scholar]
- Esko JD, Selleck SB. 2002. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem. 71:435–471. [DOI] [PubMed] [Google Scholar]
- Fuster MM, Wang L. 2010. Endothelial heparan sulfate in angiogenesis. Prog Mol Biol Transl Sci. 93:179–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishina IB, Kim SY, Ferrara C, Makarenkova HP, Walden PD. 2005. BMP7 inhibits branching morphogenesis in the prostate gland and interferes with notch signaling. Dev Biol. 288:334–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Kobayashi S, Nakato H. 2009. Drosophila glypicans regulate the germline stem cell niche. J Cell Biol. 187:473–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helledie T, Dombrowski C, Rai B, Lim ZX, Hin IL, Rider DA, Stein GS, Hong W, van Wijnen AJ, Hui JH et al. . 2012. Heparan sulfate enhances the self-renewal and therapeutic potential of mesenchymal stem cells from human adult bone marrow. Stem Cells Dev. 21:1897–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hempel N, How T, Cooper SJ, Green TR, Dong M, Copland JA, Wood CG, Blobe GC. 2008. Expression of the type III TGF-beta receptor is negatively regulated by TGF-beta. Carcinogenesis. 29:905–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Pu Y, Alam S, Birch L, Prins GS. 2005. The role of Fgf10 signaling in branching morphogenesis and gene expression of the rat prostate gland: Lobe-specific suppression by neonatal estrogens. Dev Biol. 278:396–414. [DOI] [PubMed] [Google Scholar]
- Huang Y, Hamana T, Liu J, Wang C, An L, You P, Chang JY, Xu J, Jin C, Zhang Z et al. . 2015a. Type 2 fibroblast growth factor receptor signaling preserves stemness and prevents differentiation of prostate stem cells from the basal compartment. J Biol Chem. 290:17753–17761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Hamana T, Liu J, Wang C, An L, You P, Chang JY, Xu J, McKeehan WL, Wang F. 2015. Prostate sphere-forming stem cells are derived from the P63-expressing basal compartment. J Biol Chem. 290:17745–17752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inatani M, Irie F, Plump AS, Tessier-Lavigne M, Yamaguchi Y. 2003. Mammalian brain morphogenesis and midline axon guidance require heparan sulfate. Science. 302:1044–1046. [DOI] [PubMed] [Google Scholar]
- Itoh F, Itoh S, Carvalho RL, Adachi T, Ema M, Goumans MJ, Larsson J, Karlsson S, Takahashi S, Mummery CL et al. . 2009. Poor vessel formation in embryos from knock-in mice expressing ALK5 with L45 loop mutation defective in Smad activation. Lab Investig. 89:800–810. [DOI] [PubMed] [Google Scholar]
- Iwao K, Inatani M, Matsumoto Y, Ogata-Iwao M, Takihara Y, Irie F, Yamaguchi Y, Okinami S, Tanihara H. 2009. Heparan sulfate deficiency leads to Peters anomaly in mice by disturbing neural crest TGF-beta2 signaling. J Clin Invest. 119:1997–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izvolsky KI, Shoykhet D, Yang Y, Yu Q, Nugent MA, Cardoso WV. 2003. Heparan sulfate-FGF10 interactions during lung morphogenesis. Dev Biol. 258:185–200. [DOI] [PubMed] [Google Scholar]
- Jakobsson L, Kreuger J, Holmborn K, Lundin L, Eriksson I, Kjellén L, Claesson-Welsh L. 2006. Heparan sulfate in trans potentiates VEGFR-mediated angiogenesis. Dev Cell. 10:625–634. [DOI] [PubMed] [Google Scholar]
- Johnson CE, Crawford BE, Stavridis M, ten Dam G, Wat AL, Rushton G, Ward CM, Wilson V, van Kuppevelt TH, Esko JD et al. . 2007. Essential alterations of heparan sulfate during the differentiation of embryonic stem cells to Sox1-enhanced green fluorescent protein-expressing neural progenitor cells. Stem Cells. 25:1913–1923. [DOI] [PubMed] [Google Scholar]
- Jornvall H, Reissmann E, Andersson O, Mehrkash M, Ibanez CF. 2004. ALK7, a receptor for nodal, is dispensable for embryogenesis and left-right patterning in the mouse. Mol Cell Biol. 24:9383–9389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josson S, Matsuoka Y, Chung LWK, Zhau HE, Wang R. 2010. Tumor–stroma co-evolution in prostate cancer progression and metastasis. Semin Cell Dev Biol. 21:26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraushaar DC, Dalton S, Wang L. 2013. Heparan sulfate: a key regulator of embryonic stem cell fate. Biol Chem. 394:741–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraushaar DC, Rai S, Condac E, Nairn A, Zhang S, Yamaguchi Y, Moremen K, Dalton S, Wang L. 2012. Heparan sulfate facilitates FGF and BMP signaling to drive mesoderm differentiation of mouse embryonic stem cells. J Biol Chem. 287:22691–22700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraushaar DC, Yamaguchi Y, Wang L. 2010. Heparan sulfate is required for embryonic stem cells to exit from self-renewal. J Biol Chem. 285:5907–5916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo WJ, Digman MA, Lander AD. 2010. Heparan sulfate acts as a bone morphogenetic protein coreceptor by facilitating ligand-induced receptor hetero-oligomerization. Mol Biol Cell. 21:4028–4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyprianou N, Isaacs JT. 1988. Activation of programmed cell death in the rat ventral prostate after castration. Endocrinology. 122:552–562. [DOI] [PubMed] [Google Scholar]
- Lamm ML, Podlasek CA, Barnett DH, Lee J, Clemens JQ, Hebner CM, Bushman W. 2001. Mesenchymal factor bone morphogenetic protein 4 restricts ductal budding and branching morphogenesis in the developing prostate. Dev Biol. 232:301–314. [DOI] [PubMed] [Google Scholar]
- Lanner F, Lee KL, Sohl M, Holmborn K, Yang H, Wilbertz J, Poellinger L, Rossant J, Farnebo F. 2010. Heparan sulfation-dependent fibroblast growth factor signaling maintains embryonic stem cells primed for differentiation in a heterogeneous state. Stem Cells. 28:191–200. [DOI] [PubMed] [Google Scholar]
- Levings DC, Arashiro T, Nakato H. 2016. Heparan sulfate regulates the number and centrosome positioning of drosophila male germline stem cells. Mol Biol Cell. 27:888–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang Y, Sharif-Afshar AR, Uwamariya C, Yi A, Ishii K, Hayward SW, Matusik RJ, Bhowmick NA. 2009. Urothelial transdifferentiation to prostate epithelia is mediated by paracrine TGF-beta signaling. Differentiation. 77:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Liu G, Zhang Y, Hu YP, Yu K, Lin C, McKeehan K, Xuan JW, Ornitz DM, Shen MM et al. . 2007. Fibroblast growth factor receptor 2 tyrosine kinase is required for prostatic morphogenesis and the acquisition of strict androgen dependency for adult tissue homeostasis. Development. 134:723–734. [DOI] [PubMed] [Google Scholar]
- Lopez-Casillas F, Riquelme C, Perez-Kato Y, Ponce-Castaneda MV, Osses N, Esparza-Lopez J, Gonzalez-Nunez G, Cabello-Verrugio C, Mendoza V, Troncoso V et al. . 2003. Betaglycan expression is transcriptionally up-regulated during skeletal muscle differentiation. Cloning of murine betaglycan gene promoter and its modulation by MyoD, retinoic acid, and transforming growth factor-beta. J Biol Chem. 278:382–390. [DOI] [PubMed] [Google Scholar]
- Lyon M, Rushton G, Gallagher JT. 1997. The interaction of the transforming growth factor-betas with heparin/heparan sulfate is isoform-specific. J Biol Chem. 272:18000–18006. [DOI] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. 2007. A global double-fluorescent Cre reporter mouse. Genesis. 45:593–605. [DOI] [PubMed] [Google Scholar]
- Nakato H, Li JP. 2016. Functions of heparan sulfate proteoglycans in development: Insights from drosophila models. Int Rev Cell Mol Biol. 325:275–293. [DOI] [PubMed] [Google Scholar]
- Nurcombe V, Cool SM. 2007. Heparan sulfate control of proliferation and differentiation in the stem cell niche. Crit Rev Eukaryot Gene Expr. 17:159–171. [DOI] [PubMed] [Google Scholar]
- Ornitz DM. 2000. FGFs, heparan sulfate and FGFRs: Complex interactions essential for development. Bioessays. 22:108–112. [DOI] [PubMed] [Google Scholar]
- Ortmann C, Pickhinke U, Exner S, Ohlig S, Lawrence R, Jboor H, Dreier R, Grobe K. 2015. Sonic hedgehog processing and release are regulated by glypican heparan sulfate proteoglycans. J Cell Sci. 128:2374–2385. [DOI] [PubMed] [Google Scholar]
- Patel VN, Lombaert IM, Cowherd SN, Shworak NW, Xu Y, Liu J, Hoffman MP. 2014. Hs3st3-modified heparan sulfate controls KIT+ progenitor expansion by regulating 3-O-sulfotransferases. Dev Cell. 29:662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlikowski B, Vogler TO, Gadek K, Olwin BB. 2017. Regulation of skeletal muscle stem cells by fibroblast growth factors. Dev Dyn. 246:359–367. [DOI] [PubMed] [Google Scholar]
- Prins GS, Huang L, Birch L, Pu Y. 2006. The role of estrogens in normal and abnormal development of the prostate gland. Ann N Y Acad Sci. 1089:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu H, Jiang JL, Liu M, Huang X, Ding SJ, Wang L. 2013. Quantitative phosphoproteomics analysis reveals broad regulatory role of heparan sulfate on endothelial signaling. Mol Cell Proteomics. 12:2160–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu H, Shi S, Yue J, Xin M, Nairn AV, Lin L, Liu X, Li G, Archer-Hartmann SA, Dela Rosa M et al. . 2018. A mutant-cell library for systematic analysis of heparan sulfate structure-function relationships. Nat Methods. 15:889–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds BA, Weiss S. 1996. Clonal and population analyses demonstrate that an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Dev Biol. 175:1–13. [DOI] [PubMed] [Google Scholar]
- Saez B, Ferraro F, Yusuf RZ, Cook CM, Yu VW, Pardo-Saganta A, Sykes SM, Palchaudhuri R, Schajnovitz A, Lotinun S et al. . 2014. Inhibiting stromal cell heparan sulfate synthesis improves stem cell mobilization and enables engraftment without cytotoxic conditioning. Blood. 124(19):2937047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salm SN, Burger PE, Coetzee S, Goto K, Moscatelli D, Wilson EL. 2005. TGF-{beta} maintains dormancy of prostatic stem cells in the proximal region of ducts. J Cell Biol. 170:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrazin S, Lamanna WC, Esko JD. 2011. Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol. 3(7). pii:a004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki N, Okishio K, Ui-Tei K, Saigo K, Kinoshita-Toyoda A, Toyoda H, Nishimura T, Suda Y, Hayasaka M, Hanaoka K et al. . 2008. Heparan sulfate regulates self-renewal and pluripotency of embryonic stem cells. J Biol Chem. 283:3594–3606. [DOI] [PubMed] [Google Scholar]
- Shahi P, Seethammagari MR, Valdez JM, Xin L, Spencer DM. 2011. Wnt and notch pathways have interrelated opposing roles on prostate progenitor cell proliferation and differentiation. Stem Cells. 29:678–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel PM, Shu W, Cardiff RD, Muller WJ, Massague J. 2003. Transforming growth factor beta signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc Natl Acad Sci U S A. 100:8430–8435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand DW, Goldstein AS. 2015. The many ways to make a luminal cell and a prostate cancer cell. Endocr Relat Cancer. 22:T187–T197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemura M, Nakato H. 2017. Drosophila Sulf1 is required for the termination of intestinal stem cell division during regeneration. J Cell Sci. 130:332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talsma DT, Katta K, Ettema MAB, Kel B, Kusche-Gullberg M, Daha MR, Stegeman CA, van den Born J, Wang L. 2018. Endothelial heparan sulfate deficiency reduces inflammation and fibrosis in murine diabetic nephropathy. Lab Investig. 98:427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry DE, Clark AF. 1996. Glycosaminoglycans in the three lobes of the rat prostate following castration and testosterone treatment. Biochem Cell Biol. 74:653–658. [DOI] [PubMed] [Google Scholar]
- Valdez JM, Zhang L, Su Q, Dakhova O, Zhang Y, Shahi P, Spencer DM, Creighton CJ, Ittmann MM, Xin L. 2012. Notch and TGFbeta form a reciprocal positive regulatory loop that suppresses murine prostate basal stem/progenitor cell activity. Cell Stem Cell. 11:676–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt J, Traynor R, Sapkota GP. 2011. The specificities of small molecule inhibitors of the TGFss and BMP pathways. Cell Signal. 23:1831–1842. [DOI] [PubMed] [Google Scholar]
- Wang L, Fuster M, Sriramarao P, Esko JD. 2005. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat Immunol. 6:902–910. [DOI] [PubMed] [Google Scholar]
- Wang Y, Hayward S, Cao M, Thayer K, Cunha G. 2001. Cell differentiation lineage in the prostate. Differentiation. 68:270–279. [DOI] [PubMed] [Google Scholar]
- Watson HA, Holley RJ, Langford-Smith KJ, Wilkinson FL, van Kuppevelt TH, Wynn RF, Wraith JE, Merry CL, Bigger BW. 2014. Heparan sulfate inhibits hematopoietic stem and progenitor cell migration and engraftment in mucopolysaccharidosis I. J Biol Chem. 289:36194–36203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijelath E, Namekata M, Murray J, Furuyashiki M, Zhang S, Coan D, Wakao M, Harris RB, Suda Y, Wang L et al. . 2010. Multiple mechanisms for exogenous heparin modulation of vascular endothelial growth factor activity. J Cell Biochem. 111:461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte JS. 2009. Prostate cancer genomics: Towards a new understanding. Nat Rev Genet. 10:77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Ingram L, Tolosa EJ, Vera RE, Li Q, Kim S, Ma Y, Spyropoulos DD, Beharry Z, Huang J et al. . 2016. Gli transcription factors mediate the oncogenic transformation of prostate basal cells induced by a Kras-androgen receptor Axis. J Biol Chem. 291:25749–25760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin L, Ide H, Kim Y, Dubey P, Witte ON. 2003. In vivo regeneration of murine prostate from dissociated cell populations of postnatal epithelia and urogenital sinus mesenchyme. Proc Natl Acad Sci U S A. 100:11896–11903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin L, Lukacs RU, Lawson DA, Cheng D, Witte ON. 2007. Self-renewal and multilineage differentiation in vitro from murine prostate stem cells. Stem Cells. 25:2760–2769. [DOI] [PubMed] [Google Scholar]
- Xin L, Teitell MA, Lawson DA, Kwon A, Mellinghoff IK, Witte ON. 2006. Progression of prostate cancer by synergy of AKT with genotropic and nongenotropic actions of the androgen receptor. Proc Natl Acad Sci U S A. 103:7789–7794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Xiao W, Qiu H, Zhang F, Moniz HA, Jaworski A, Condac E, Gutierrez-Sanchez G, Heiss C, Clugston RD et al. . 2014. Heparan sulfate deficiency disrupts developmental angiogenesis and causes congenital diaphragmatic hernia. J Clin Invest. 124:209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Valdez JM, Zhang B, Wei L, Chang J, Xin L. 2011. ROCK inhibitor Y-27632 suppresses dissociation-induced apoptosis of murine prostate stem/progenitor cells and increases their cloning efficiency. PLoS One. 6:e18271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang J, Lin Y, Lan Y, Lin C, Xuan JW, Shen MM, McKeehan WL, Greenberg NM, Wang F. 2008. Role of epithelial cell fibroblast growth factor receptor substrate 2alpha in prostate development, regeneration and tumorigenesis. Development. 135:775–784. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.