

Abstract
Methylation-mediated pathways play important roles in the progression of various diseases. Thus, targeting methyltransferases has proven to be a promising strategy for developing novel therapies. Nicotinamide N-methyltransferase (NNMT) is a major metabolic enzyme involved in epigenetic regulation through catalysis of methyl transfer from the cofactor S-adenosyl-L-methionine onto nicotinamide and other pyridines. Accumulating evidence infers that NNMT is a novel therapeutic target for a variety of diseases such as cancer, diabetes, obesity, cardiovascular and neurodegenerative diseases. Therefore, there is an urgent need to discover potent and specific inhibitors for NNMT to assess its therapeutical potential. Herein, we reported the design and synthesis of a fluorescent probe II138, exhibiting a Kd value of 369 ± 14 nM for NNMT. We also established a fluorescence polarization (FP)-based competition assay for evaluation of NNMT inhibitors. Importantly, the unique feature of this FP competition assay is its capability to identify inhibitors that interfere with the interaction of the NNMT active site directly or allosterically. In addition, this assay performane is robust with a Z factor of 0.76, indicating its applicability in high-throughput screening for NNMT inhibitors.
Keywords: protein methyltransferase, Nicotinamide N-methyltransferase, fluorescence polarization, inhibitor, high-throughput screening
Graphical Abstract
Introduction
Nicotinamide N-methyltransferase (NNMT) is a metabolic enzyme that catalyzes the transfer of a methyl group from the cofactor S-adenosyl-L-methionine (AdoMet) to nicotinamide (NAM) and other pyridines to produce S-adenosyl-L-homocysteine (AdoHcy) and respective methylated products (Fig. 1A). NNMT is predominantly expressed in the liver and adipose tissue [1]. NAM, the most studied endogenous substrate of NNMT, is converted to N1-methylnicotinamide (MNAM) after methylation. NAM is a form of vitamin B3 and also a precursor of salvage pathways to produce NAD+, an important coenzyme involved in redox metabolism and sirtuins-catalyzed deacetylation reaction [2,3]. Besides NAM, NNMT also methylates 4-phenylpyridine to generate the neurotoxin 1-methyl-4-phenylpyridinium ion, which interferes with complex I and is implicated in idiopathic Parkinson’s disease [4].
Fig. 1.
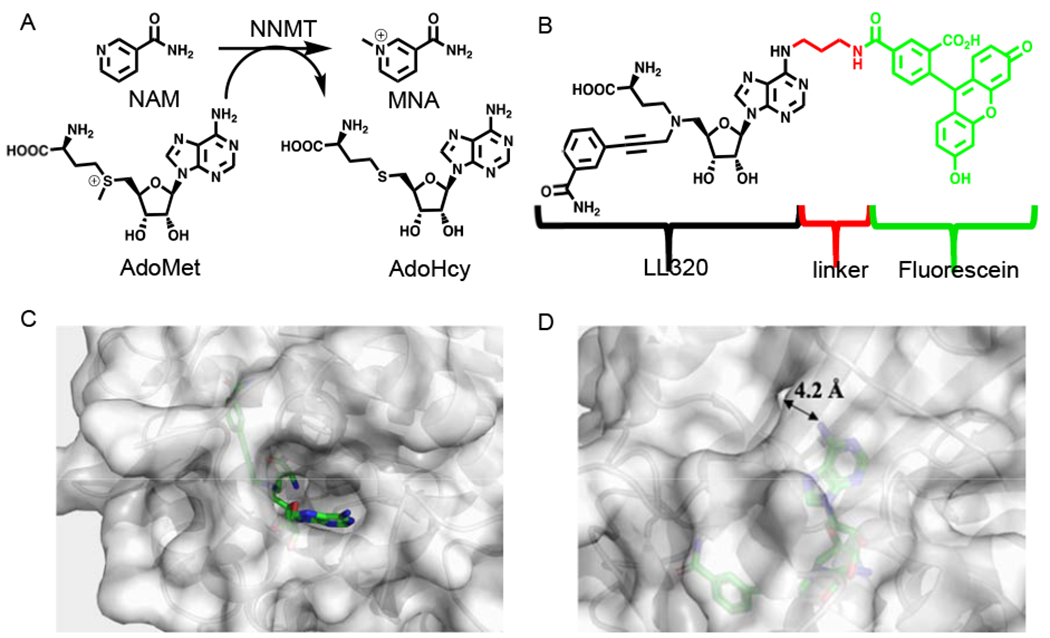
Design of FP probe II138. A. Methylation of nicotinamide (NAM) by NNMT. B. Structure of the fluorescent probe II138 comprised of LL320 (black), the linker (red), and fluorescein (green). C. Crystal structure of hNNMT-LL320 complex (PDB: 6PVS). hNNMT is colored gray and LL320 is shown in green stick with N6 adenosine pointing towards the solvent. B. The distance between N6 and the protein surface is 4.2 Å.
The dual functions of NNMT in the metabolism of both NAM and AdoMet manifest its significance in cellular metabolism and epigenetic pathways. Not surprisingly, upregulation of NNMT has been implicated in cancers, obesity, diabetes, cardiovascular and neurodegenerative diseases [3–10]. Genetic studies have shown that the loss of NNMT inhibits cancer cell growth and migration [11]. In addition, knockdown of NNMT increased metabolism and prevented weight gain [3]. The emerging importance of NNMT imposes the need to develop selective and cell-potent inhibitors. Despite recent progress in the development of potent NNMT bisubstrate inhibitors with low nM inhibitory activity, limited cell permeability restrains their applications in cell-based studies [12–14]. Meanwhile, reported small molecule NNMT inhibitors only bear moderate activity at μM levels [15,16]. Hence, new cell-potent NNMT inhibitors are in demand in order to delineate the physiological and pathological functions of NNMT.
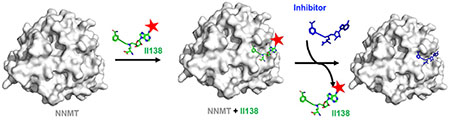
To expedite the discovery of NNMT inhibitors, it is important to have a convenient and efficient assay to screen and evaluate NNMT inhibitors. Currently, a widely used assay is an enzymatic assay to indirectly monitor the production of AdoHcy, either through an AdoHcy hydrolase (SAHH)-coupled fluorescence or a bioluminescent MTase-Glo™ assay (Promega, Inc). Both assays require additional coupling enzymes and reagents to produce a detectable signal [17,18]. The MTase-Glo™ assay needs several coupling enzymes to convert AdoHcy to ADP and produce a luciferase signal [17,19]. Direct assay to measure the methylation product like MNAM has been developed through the use of high-pressure liquid chromatography (HPLC), which is not amenable for high throughput screening (HTS) applications [20]. Additionally, a fluorescence assay to directly detect the inherent fluorescence of the methylated product methylquinolinium has been developed by using quinoline as the NNMT substrate, but this assay has a low signal-to-noise ratio [21,22]. Although biophysical methods like isothermal titration calorimetry and surface plasmon resonance are useful for direct determination of binding affinities of inhibitors to the target, the requirements of expensive instruments and protein stability have limited their applications as a primary HTS method. On the other hand, fluorescence polarization (FP) is a HTS-amenable method to characterize the binding of a fluorescent ligand to the target or any compound that interferes with the aforementioned interaction [23]. In this work, we report the design and synthesis of the first fluorescent probe II138 for NNMT to date. Furthermore, we have established and optimized a FP-based competition assay in an HTS-compatible format, allowing for identification and evaluation of NNMT inhibitors that perturb the binding to the active site either directly or allosterically.
2. Materials and methods
2.1. Materials and instruments
The reagents and solvents were purchased from commercial sources (Fisher and Sigma-Aldrich) and used directly. Analytical thin-layer chromatography was performed on ready-to-use plates with silica gel 60 (Merck, F254). Flash column chromatography was performed over silica gel (grade 60, 230–400 mesh) on Teledyne Isco CombiFlash purification system. Final compounds were purified on preparative reversed phase high-pressure liquid chromatography (RP-HPLC) and was performed on Agilent 1260 Series system with Agilent 1260 Infinity II Variable Wavelength Detector (G7114A, UV = 254 nM) and a Waters BEH C18 (130Å, 5 μm, 10 mm X 250 mm) column. Final compounds were eluted using a gradient 95% water (with 0.1% formic acid) and 5% acetonitrile (with 0.1% formic acid) at a flow rate of 4 mL/min over 30 min.
NMR spectra were acquired on a Bruker AV500 instrument (500 MHz for 1H-NMR, 126 MHz for 13C-NMR). TLC-MS were acquired using Advion CMS-L MS. The Agilent 1260 Infinity II Variable Wavelength Detector (G7114A, UV = 254 nM) and an Agilent ZORBAX RR SB-C18 (80Å, 3.5 μm, 4.6 x 150 mm) at a flow rate of 1 mL/min using a solvent system of 100% water with 0.1% TFA to 40 or 60% methanol over 20min were used to assess purity of final compounds. All the purity of target compounds showed >95% in RP-HPLC. FP was monitored on a BMG CLARIOstar microplate reader.
2.2. Protein Expression and Purification.
Expression and purification of full-length human NNMT wild type was performed as previously described [24]. Briefly, full-length hNNMT (amino acids 1-270) was codon optimized, synthesized, and cloned into pET28a(+)-TEV vector (GenScript). Protein was expressed in E. coli BL21-CodonPlus(DE3)-RIPL competent cells in LB media with kanamycin and induced by 0.3 mM isopropyl-D-1-thiogalactopyranoside at 16 °C for 20 hours. Harvested cells were lysed through sonication (Qsonica Q55 cell disruptor) on ice in 10 volumes of 50 mM KH2PO4/K2HPO4 (pH = 7.4) containing 500 mM NaCl, 25 mM imidazole, 5 mM ß-mercaptoethanol, and 100 μM PMSF. The cell lysate was centrifuged at 25,000 × g for 30 minutes at 4 °C. Then the supernatant was loaded to the HiTrap FF Ni-NTA column on a GE AKTA Prime purification system and eluted with a step gradient of imidazole (0 to 0.5 M), 50 mM KH2PO4/K2HPO4 (pH = 7.4), 500 mM NaCl, and 0.5 mM TCEP. The peak fractions were verified by SDS-PAGE analysis, combined to dialyze in the dialysis buffer (25 mM Tris, pH = 7.5, 150 mM NaCl, 50 mM KCl), and concentrated to 1.5 mg/mL.
2.3. FP measurement, binding and competition assay
All FP measurements were performed on a BMG CLARIOstar microplate reader in black opaque 384-well microplates (Corning #3820) with excitation 482 nm and emission 530 nm. All experiments were performed in triplicates in a volume of 20 μL per well in 25 mM Tris, 50 mM KCl, 0.01% Tween pH 7.5. For direct binding assay, different concentrations of NNMT were titrated against the probe at the concentration of 5 nM. The polarization (mP) was measured after both 30 min and 60 min incubation at room temperature. The dissociation constant (Kdapp) was obtained by fitting the fluorescence polarization values and the corresponding protein concentrations into a nonlinear regression model in GraphPad Prism 8. For the competition binding assay, 0.5 μM NNMT was incubated with the inhibitor at different concentrations at room temperature for 30 mins. Then FP probe was added to the above mixture with a final concentration of 5 nM. After incubation for 30 min, polarization was measured. The FP values were plotted against the log of inhibitor concentrations into a nonlinear regression model. The Ki were calculated from Binding-Competitive model in GraphPad Prism [25].
2.4. Z ’-factor determination and data analysis
The fluorescence polarization signals of 100 wells of negative control (no inhibitors added to the competition assay) and 100 wells of positive control samples (5 μM of bisubstrate inhibitor LL320, Figure 1) were measured in a single 384-well plate after 60 mins incubation at room temperature. The statistical parameter Z-factor was calculated based on the following equation [26]:
3. Results and Discussion
Probe Design
The inherent feature of the NNMT active site imposes a challenge on the discovery of a potent and specific inhibitor by solely targeting NAM or AdoMet binding site because of the relatively small binding site of NAM and conserved binding site of AdoMet. We have recently reported the development of a potent and selective propargyl-linked bisubstrate inhibitor LL320 (Ki = 1.6 ± 0.3 nM) for NNMT, displaying over 1,000-fold selectivity for a panel of MTases [13]. Inspired by the high potency and selectivity of LL320, we hypothesize that a fluorescent derivative of LL320 would enable the development of a FP assay for the identification of potent and specific NNMT inhibitors. Because LL320 occupies both NAM and AdoMet binding sites, the advantage of this FP assay is to allow the discovery of NNMT inhibitors directly or allosterically targeting either NAM or AdoMet binding site, or both. Our designed FP probe (II138) is comprised of three parts: LL320, linker, and the fluorescent dye (Fig. 1B). We chose fluorescein as the dye because of its high quantum yield and availability. The position, flexibility, and size of the linker of the FP probe is crucial to retain its binding affinity for NNMT. Examination of the co-crystal structure of NNMT-LL320 (PDB ID: 6PVS) suggests that the N6 of adenosine is a favorable site for tethering because it is exposed to the solvent (Fig. 1C). Since the distance between the N6 and the surface of NNMT is about 4.2 Å (Fig. 1D), we hypothesize that a 3-carbon linker will be enough to tether LL320 to fluorescein and retain the interaction with NNMT. Therefore, a propyl group was chosen to connect the fluorescein with LL320 (Figure 1B).
Synthesis of FP probe
The FP probe was synthesized from commercially available (2R,3R,4S,5R)-2-(6-chloro-9H-purin-9-yl)-5-(hydroxymethyl)tetrahydrofuran-3,4-diol 1 (Scheme 1). First, the diol was protected by acidic treatment to afford the acetal 2, which was subjected to substitution with propane-1,3-diamine and followed by treatment with ethyl trifluoroacetate to yield 3 [27]. The subsequent Mitsunobu reaction with isoindoline-1,3-dione afforded the masked amine 4, which was deprotected by hydrazine to produce 5. Reductive amination of 5 with tert-butyl (S)-2-((tert-butoxycarbonyl)amino)-4-oxobutanoate produced 6, which was subjected to a second reductive amination with 3-(3-oxoprop-1-yn-1-yl)benzonitrile to yield 7. The cyano group of 7 was oxidized by hydrogen peroxide to generate the primary amide 8, which was subjected to ammonia solution to provide 9. To synthesize the FP probe II138, compound 9 was subjected to the amidation reaction with 5-carboxyfluorescein to attach the fluorescein followed by global deprotection. Meanwhile, TFA treatment of 9 resulted in simultaneous removal of the acetal, Boc and tert-butoxyl protecting groups to yield 10, which was used as a control compound to investigate the effect of the propane-1,3-diamine linker.
Scheme 1.
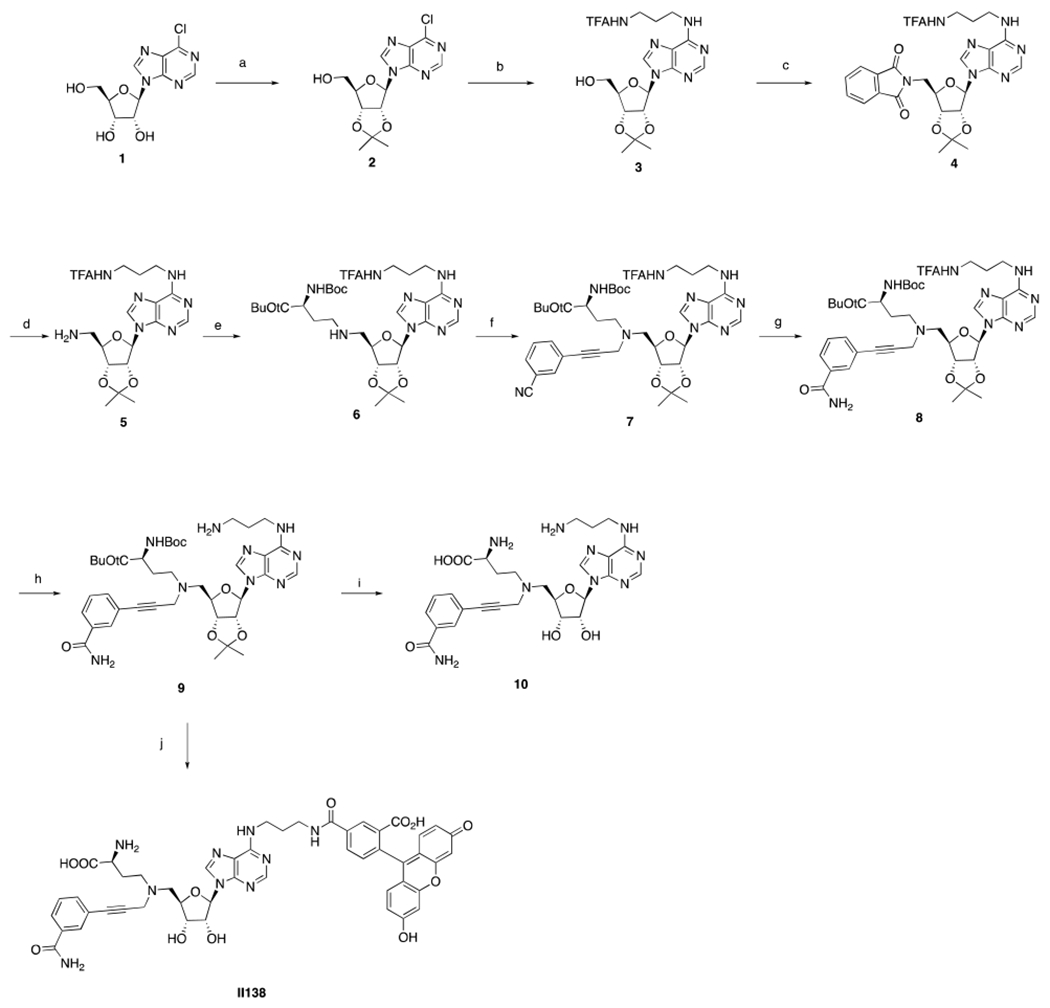
Synthesis of FP probea
aReagents and conditions: (a) pTsOH, acetone, 3 h, quant. yield; (b) propane-1,3-diamine, TEA, EtOH, 60 °C, 2 h then; CF3CO2Et, TEA, MeOH, 12 h, 89 %; (c) isoindoline-1,3-dione, PPh3, DIAD, THF, 2 h, 90 %; (d) NH2NH2, MeOH, 8 h, 63 %; (e) tert-butyl (S)-2-((tert-butoxycarbonyl)amino)-4-oxobutanoate, NaBH3CN, AcOH, MeOH, 2 h, 67 %; (f) 3-(3-oxoprop-1-yn-1-yl)benzonitrile, NaBH3CN, AcOH, MeOH, 2 h, 79 %; (g) K2CO3, H2O2, DMSO, quant. yield; (h) NH4OH, MeOH, 77 %; (i) TFA, H2O, 8 h, 37 %; (j) fluorescein, DIC, HOBt, DIPEA, DMF; 12 h then TFA, H2O, 8 h, 29 %.
Evaluation of the linker effect
In order to determine the effect of the linker, the Ki of compound 10 was determined by the SAHH-coupled fluorescence assay to be 0.21 ± 0.02 μM, reflecting about 100-fold loss in comparison with LL320 (Ki = 1.6 ± 0.3 nM) [13]. This is probably due to the loss of one hydrogen bond donor of the N6 amine in adenosine. Nevertheless, this decreased Ki value is acceptable for the FP probe that is intended for the FP assay development.
Assay optimization
Both Tris and HEPES buffers were investigated to evaluate their effects on the sensitivity (detection limit) in this assay. In Tris buffer, 5 nM concentration of the probe showed over 10-fold higher total fluorescence intensity compared to the blank, but in HEPES buffer, a probe concentration of >40 nM was required to display 10-fold higher total fluorescence over the blank. (Fig. 2A, B). To determine the linearity and the optimal concentration of II138, the fluorescence intensity of II138 was measured at various concentrations ranging from 0 to 1 μM in the Tris buffer. We chose the concentration of II138 at 5 nM because it gave about 10-fold higher intensity than the blank (Fig. 2A). The direct binding of the FP probe II138 to NNMT was determined by titrating NNMT (0-25 μM, 2-fold dilution) against 5 nM of II138. The Kd value of II138 was determined to be 369 ±14 nM, which is comparable to the Ki of parent compound 10 (210 ± 20 nM) (Fig. 2C).
Fig. 2.

Optimization of FP assay condition. (A) Linearity of the total fluorescence intensity of fluorescent probe II138 versus its concentration in Tris buffer (25 mM, 50 mM KCl, PH 7.5). (B) Linearity of the total fluorescence intensity of II138 versus its concentration in HEPES buffer (25 mM, 150 mM NaCl, pH 7.0). (C) NNMT binding curve with NNMT (0-25 μM, 2-fold dilution) titrated to II138 (5 nM). The dotted line indicated 0.5 μM NNMT reaching nearly 60% change in polarization (ΔmP). (D) Effect of different incubation time on the binding affinity of II138 to NNMT. All experiments were performed in duplicates (n=2) except NNMT binding in triplicates (n=3).
Next, we investigated the stability of II138 for the FP binding assay by varying the incubation time and temperature. The reaction mixture was incubated and analyzed at room temperature every 30 minutes for 4 hours. The system reached equilibrium within 30 min and the binding affinity remained stable for the entire 4 h duration of the experiment, indicating that NNMT was stable for the entire incubation time. The assay window was largely unchanged during the entire 4 h period of incubation (Fig. 2D). The stability was slightly worse at higher temperatures (30 °C and 37 °C), especially after 3 h (Fig. S1). Thus, we proceeded with the measurement for 30 minutes of incubation at room temperature because the Kd reached equilibrium and remained stable.
Competition FP assay.
Competition FP assay was performed to evaluate the displacement of the bound probe with reported inhibitors. To ensure sufficient signal, the fraction of the bound probe should be greater or equal to 50% [28]. According to NNMT titration results (Fig. 2C), we used 0.5 μM NNMT and 5 nM II138 because 0.5 μM NNMT produced nearly 60% increase in FP in comparison to the probe alone. Compound 10, the unlabeled analog of the probe II138, was employed in a “gold standard” assay. The Ki of the competitor (10) was determined to be 0.31 ± 0.7 μM from the FP assay (Fig. 3), which was less than 2-fold in comparison to the Ki value of 0.21 ± 0.02 μM in the SAHH-coupled fluorescence assay. The minor difference in both Ki values could be due to the different assay conditions, particularly the enzyme concentration [25]. Nevertheless, the close agreement between the Ki values obtained by FP and coupled assay establish the confidence of the FP probe to evaluate NNMT inhibitors.
Fig. 3.
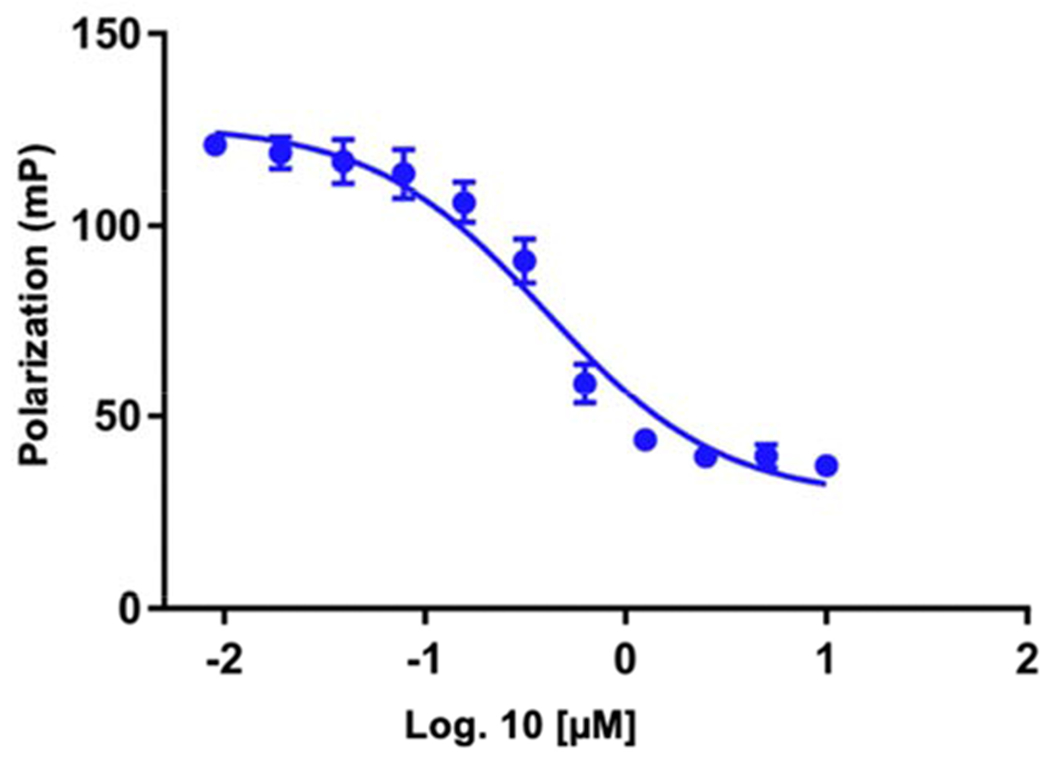
Gold Standard Assay. Competitive binding assay of the unlabeled probe. Compound 10 was titrated against a constant concentration of FP probe (5 nM) in the presence of 0.5 μM NNMT. The averages of two independent experiments done in duplicate are shown (n=2).
Assay performance assessment
In order to further validate potential application of this FP assay, we assessed a series of NNMT ligands (Table 1). In comparison with literature values from other assay formats, our results showed comparable Ki values in our FP assay for inhibitors that displayed a submicromolar to millimolar inhibitory activity. For instance, the cofactor AdoMet (Km of 24.0 ± 6.8 μM) was determined to have a Ki of 20.3 ± 2.9 μM [17]. The substrates NAM and 1 methyl quinoline (1MQ) exhibited over 1 mM Ki in our FP assay, which were in alignment with reported values. For AdoHcy, its Ki from our FP assay was 82.9 ± 14.3 μM, while its IC50 was reported to be 75.4 ± 6.3 μM from the UHPLC-MS assay [29]. Meanwhile, two previously-reported bisubstrate inhibitors VH45 and MS2756 displayed a Ki value of 59.2 ± 8.1 μM and 11.3 ± 1.8 μM in FP assay versus 29.2 ± 4.0 μM and 10.0 ± 0.35 μM in the SAHH coupled assay, respectively [13]. Likewise, LL335 exhibited a Ki value of 5.6 ± 1.2 μM in the FP assay and 3.0 ± 0.83 μM in the SAHH-coupled assay, respectively [13]. However, there is a discrepancy for tight-binding inhibitors LL319 and LL320, demonstrating a Ki value of 0.41 ± 0.05 μM and 0.22 ± 0.04 μM in the FP assay, respectively. There is nearly 5- and 130-fold difference for LL319 and LL320 in comparison with the values in the SAHH coupled assay, respectively [13]. This difference results from the sensitivity limitation that is imposed by the binding affinity (Kd = 369 ± 14 nM) of the probe II138 to NNMT. The Kd of a probe sets the sensitivity limitation that highly potent inhibitors (<500 nM) are not distinguished, which can be readily addressed with an orthogonal follow-up assay [30].
Table 1.
Comparison of Ki values from FP with literature values
| Compound | Structure | Literature value Ki or Km (μM) | Determined Ki from FP Assay (μM) |
|---|---|---|---|
| AdoHcy | 75.4 ± 6.3a | 82.9 ± 14.3 | |
| AdoMet | 24.0 ± 6.8 | 20.3 ± 2.9 | |
| VH45 | 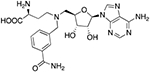 |
29.2 ± 4.0a | 59.2 ± 8.1 |
| MS2756 | 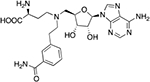 |
10.0 ± 0.3b | 11.3 ± 1.8 |
| LL335 | 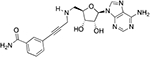 |
3.0 ± 0.8b | 5.6 ± 1.2 |
| LL319 | 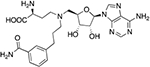 |
0.083 ± 0.007b | 0.41 ± 0.05 |
| LL320 |  |
0.0016 ± 0.0003b | 0.22 ± 0.04 |
| 10 | 0.21 ± 0.02b | 0.31 ± 0.07 | |
| NAM |  |
> 100b | > 1 mM |
| 1-MQ |  |
> 100b | > 1 mM |
Note:
determined by UHPLC-MS assay.
determined by SAHH coupled assay
Optimization of DMSO compatibility and Z-factor.
Next, we investigated the effects of DMSO on the FP assay because DMSO is a commonly used solvent to prepare stock solutions of small molecules. The Kd value of the FP probe was examined in the presence of DMSO up to 10% (Fig. 4A). The effects of DMSO on the binding affinity of the probe were negligible until DMSO reached 2.5% in the solution. The Kd value remained consistent in the presence of DMSO from 0 to 2.5%, displaying 0.34 μM and 0.38 μM at 0.5% DMSO and 2.5% DMSO, respectively. Then Kd increased proportionally to the percentage of DMSO when it increased from 5% - 10%. The assay window (ΔmP) remained relatively stable in the presence of DMSO up to 5%, but it gradually decreased from 5% to 10% DMSO. This reduction is likely due to the instability of protein at higher percentage of DMSO. Nonetheless, the tolerance to 2.5% DMSO makes this FP assay amenable for HTS of small molecules.
Fig. 4.
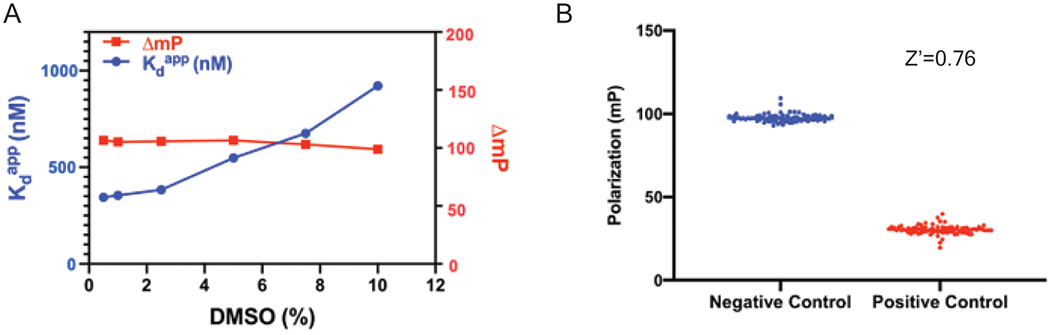
DMSO tolerance and Z’ factor determination (A) Effect of different DMSO concentrations on the binding affinity of probe to NNMT. (B) Z’ factor determination from blue dots (100 negative controls); red dots (100 positive controls).
The statistical parameter Z-factor (Z’) has been used to evaluate the assay performance quality of any given HTS [26]. Typically, a Z’ value of greater than 0.5 is considered to be acceptable. The invariance of the positive and negative control groups resulted in low standard deviation (Fig. 4B). The Z’ factor of our developed FP assay was calculated to be 0.76, indicating a robust assay performance for potential application in HTS.
Conclusion
In summary, we have described the development of a HTS-amenable FP competition assay for the discovery of NNMT inhibitors. We designed and synthesized a fluorescent probe II138 (Kdapp value of 369 ±14 nM) based on the known inhibitor LL320 by tethering the fluorescein on the N6 position of the nucleoside of LL320 via a propyl linker. The FP-based competition assay was performed to characterize a series of known ligands, demonstrating great agreement with reported values except for highly potent compounds (<500 nM). Notably, sensitivity of the assay is limited by the affinity of the probe for the enzyme, which does not allow this assay to readily differentiate those tight binding compounds. Because II138 occupies both the substrate and cofactor binding sites of NNMT, this FP competition assay can detect any molecules that interact with the NNMT active site either directly or indirectly. However, this FP assay will not detect inhibitors that require SAM binding such as suicide inhibitors. The requirement for small amount of the probe makes this FP competition assay not only an economical but valuable tool for identification and evaluation of NNMT inhibitors. Moreover, we also demonstrate the robustness of this assay with a Z’value of 0.76 and compatibility with DMSO, indicating the readiness of this FP competition assay for HTS to expedite the discovery of novel NNMT inhibitors.
EXPERIMENTAL SECTION
Chemistry General Procedures.
The reagents and solvents were purchased from commercial sources (Fisher and Sigma-Aldrich) and used directly. Analytical thin-layer chromatography was performed on ready-to-use plates with silica gel 60 (Merck, F254). Flash column chromatography was performed over silica gel (grade 60, 230–400 mesh) on Teledyne Isco CombiFlash purification system. Final compounds were purified on preparative reversed phase high-pressure liquid chromatography (RP-HPLC) was performed on Agilent 1260 Series system. Systems were run with 0-50% methanol/water gradient with 0.1% TFA. NMR spectra were acquired on a Bruker AV500 instrument (500 MHz for 1H-NMR, 126 MHz for 13C-NMR).
General procedure A (Reductive amination).
To a solution of NAM or 6 (0.06 mmol) in 0.5 mL MeOH was added the aldehyde (0.08 mmol) followed by 2 drops of AcOH. The resulting mixture was stirred for 30 min before NaBH3CN (0.1 mmol) was added. After stirring for 2 h at room temperature, the reaction was quenched with saturated NaHCO3 and extracted with DCM (3X). The combined organic layers were dried over Na2SO4, filtered, and concentrated. The residue was purified with silica gel column to provide desired product.
General procedure B (Deprotection of bisubstrate inhibitors).
The protected inhibitor was dissolved in DCM (0.5 mL). The resulting solution was cooled to 0 °C and treated with TFA (0.5 mL). The reaction mixture was warmed to rt and stirred for 6 h. The solution was concentrated under reduced pressure, dissolved in water and purified by reverse HPLC to get the corresponding bisubstrate compound.
((3aR,4R,6R,6aR)-6-(6-chloro-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methanol (2).
To a suspension of 1 (1.0 g, 3.4 mmol) in dry acetone (200 mL) was added p-TsOH monohydrate (6.6 g, 34.0 mmol) in one portion. The mixture was stirred at room temperature for 3 h. After completion of the reaction, ice-cold saturated NaHCO3 solution was added to the above mixture with stirring over 5 minutes. The volatiles were removed under reduced pressure and the crude was extracted with ethyl acetate (3X). The organic layers were combined, dried with sodium sulfate and concentrated to afford the crude product which was purified by column chromatography (dichloromethane: methanol 20:1) to afford 2 (1.1g, quant. yield) as a white solid.
2,2,2-trifluoro-N-(3-((9-((3aR,4R,6R,6aR)-6-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-9H-purin-6-yl)amino)propyl)acetamide (3).
To a solution of propane-1,3-diamine (450 mg, 6.1 mmol) and TEA (0.85 mL, 6.1 mmol) in EtOH (2 mL) was added slowly a solution of 2 (500 mg, 1.5 mmol) in EtOH (20 mL) and the reaction mixture was stirred at 60°C for 2 h. The solvent and excess of reagents were removed under reduced pressure to give the crude nucleoside. The crude nucleoside was dissolved in dry MeOH (20 mL) and TEA (0.42 mL, 3.1 mmol) and trifluoroacetic acid ethyl ester (1.1 mL, 9.1 mmol). The reaction mixture was stirred at room temperature for 12 h. The solvent was removed under reduced pressure and the crude product was purified by column chromatography (ethyl acetate 100 %) to give the protected nucleoside 3 (625 mg, 89 %) as a white solid. 1H NMR (500 MHz, CDCl3) δ 8.86 (s, 1H), 8.28 (s, 1H), 7.82 (s, 1H), 6.43 (t, J = 11.1 Hz, 2H), 5.85 (d, J = 4.9 Hz, 1H), 5.26 – 5.15 (m, 1H), 5.15 – 5.06 (m, 1H), 4.53 (s, 1H), 4.00 – 3.91 (m, 1H), 3.83 – 3.71 (m, 2H), 3.48 – 3.31 (m, 2H), 2.25 (s, 1H), 1.63 (s, 3H), 1.36 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 157.48, 157.19, 155.80, 152.44, 147.39, 140.01, 117.75, 115.59, 114.12, 94.33, 86.12, 83.03, 81.67, 63.37, 37.67, 35.56, 29.41, 27.64, 25.23. LCMS (ESI) m/z: [M + H]+ 461.2
N-(3-((9-((3aR,4R,6R,6aR)-6-((1,3-dioxoisoindolin-2-yl)methyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-9H-purin-6-yl)amino)propyl)-2,2,2-trifluoroacetamide (4).
To a solution of 3 (650 mg, 1.4 mmol) in dry THF (10 mL) was added phthalimide (249 mg, 1.7 mmol), triphenyl phosphine (740 mg, 2.8 mmol) and DIAD (566 mg, 2.8 mmol) at room temperature. The reaction mixture was stirred for 2 h. 10 mL of MeOH was added and stirring was continued for 15 min after which the crude was directly absorbed on silica gel and purified by column chromatography (ethyl acetate 100 %) to afford 4 (734 mg, 90 %) as a white solid. 1H NMR (500 MHz, CDCl3) δ 9.23 (s, 1H), 8.00 (s, 1H), 7.85 (s, 1H), 7.79 – 7.73 (m, 2H), 7.67 (dd, J = 5.5, 3.0 Hz, 2H), 6.36 (s, 1H), 6.08 – 5.96 (m, 1H), 5.50 (d, J = 6.3 Hz, 1H), 5.23 (dd, J = 6.4, 3.5 Hz, 1H), 4.54 (q, J = 5.3 Hz, 1H), 4.06 – 3.91 (m, 2H), 3.72 (s, 1H), 3.39 (s, 2H), 1.85 (s, 2H), 1.56 (s, 3H), 1.36 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 168.23, 157.48, 155.47, 152.58, 140.27, 134.12, 132.01, 123.39, 117.49, 115.20, 114.66, 90.86, 85.14, 84.18, 82.50, 39.54, 37.01, 35.50, 29.73, 27.19, 25.48. LCMS (ESI) m/z: [M + H]+ 590.3
N-(3-((9-((3aR,4R,6R,6aR)-6-(aminomethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-9H-purin-6-yl)amino)propyl)-2,2,2-trifluoroacetamide (5).
To a solution of 4 (200 mg, 0.34 mmol) in ethanol was added hydrazine (34 mg, 0.67 mmol). The reaction was stirred at room temperature overnight. The reaction mixture was absorbed on silica gel and purified by column chromatography (DCM: MeOH 5:1) to afford 5 (98 mg, 63 %) as a white solid. 1H NMR (500 MHz, MeOD) δ 8.26 (s, 1H), 8.22 – 8.19 (m, 1H), 7.83 (dd, J = 7.9, 3.3 Hz, 1H), 6.21 – 6.10 (m, 1H), 5.47 – 5.38 (m, 1H), 5.13 – 5.03 (m, 1H), 4.40 – 4.26 (m, 1H), 3.67 (d, J = 38.2 Hz, 2H), 3.43 – 3.36 (m, 1H), 3.19 – 3.04 (m, 2H), 3.04 – 2.95 (m, 1H), 2.05 – 1.96 (m, 1H), 1.96 – 1.84 (m, 2H), 1.59 (s, 3H), 1.37 (s, 3H). 13C NMR (126 MHz, MeOD) δ 159.15, 153.18, 141.70, 133.49, 129.53, 126.72, 115.87, 91.54, 86.36, 85.17, 83.25, 43.50, 38.17, 27.45, 25.50. LCMS (ESI) m/z: [M + H]+ 460.3
tert-butyl ((S)-1-((((3aR,4R,6R,6aR)-2,2-dimethyl-6-(6-((3-(2,2,2-trifluoroacetamido)propyl)amino)-9H-purin-9-yl)tetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)amino)-5,5-dimethyl-4-oxohexan-3-yl)carbamate (6).
Was prepared according to general procedure for reductive amination with 5 (200 mg, 0.4 mmol) and tert-butyl (S)-2-((tert-butoxycarbonyl)amino)-4-oxobutanoate (143 mg, 0.45 mmol) to afford 6 (209 mg, 67 %) as a white solid. Ή NMR (500 MHz, MeOD) δ 8.27 (s, 1H), 8.21 (s, 1H), 8.19 – 8.15 (m, 1H), 6.16 (d, J = 2.9 Hz, 1H), 5.44 (dd, J = 6.4, 3.0 Hz, 1H), 5.09 (dd, J = 6.5, 3.4 Hz, 1H), 4.42 – 4.34 (m, 1H), 4.06 (dd, J = 8.9, 4.8 Hz, 1H), 3.69 – 3.56 (m, 2H), 3.40 (t, J = 6.8 Hz, 2H), 3.10 (d, J = 5.8 Hz, 2H), 2.84 – 2.71 (m, 2H), 1.98 – 1.89 (m, 3H), 1.85 – 1.74 (m, 1H), 1.59 (s, 3H), 1.43 (s, 9H), 1.39 (s, 9H), 1.37 (s, 3H). 13C NMR (126 MHz, MeOD) δ 172.85, 159.09 – 158.51(m), 158.02, 156.30, 153.97, 141.56, 133.65, 126.62, 115.79, 91.85, 85.60, 84.88, 83.64, 82.90, 80.62, 53.87, 51.79, 46.90, 38.18, 31.37, 28.67, 28.22, 27.50, 25.60. LCMS (ESI) m/z: [M + H]+ 717.4
tert-butyl ((S)-1-((3-(3-cyanophenyl)prop-2-yn-1-yl)(((3aR,4R,6R,6aR)-2,2-dimethyl-6-(6-((3-(2,2,2-trifluoroacetamido)propyl)amino)-9H-purin-9-yl)tetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)amino)-5,5-dimethyl-4-oxohexan-3-yl)carbamate (7).
Was prepared according to general procedure for reductive amination with 6 (170 mg, 0.24 mmol) and 3-(3-oxoprop-1-yn-1-yl)benzonitrile (44 mg, 0.28 mmol) to afford 6 (160 mg, 79 %) as a white solid. 1H NMR (500 MHz, MeOD) δ 8.24 (d, J = 2.7 Hz, 2H), 7.71 – 7.63 (m, 2H), 7.59 (d, J = 7.8 Hz, 1H), 7.48 (dd, J = 9.4, 6.2 Hz, 1H), 6.18 (d, J = 2.4 Hz, 1H), 5.49 (dd, J = 6.5, 2.5 Hz, 1H), 5.10 (dd, J = 6.4, 3.4 Hz, 1H), 4.43 – 4.35 (m, 1H), 4.15 – 4.02 (m, 1H), 3.69 – 3.58 (m, 3H), 3.39 (t, J = 6.9 Hz, 2H), 3.36 – 3.34 (m, 1H), 2.92 (dd, J = 13.5, 5.4 Hz, 1H), 2.84 (dd, J = 13.7, 7.5 Hz, 1H), 2.66 (t, J = 6.8 Hz, 2H), 1.98 – 1.87 (m, 3H), 1.59 (s, 3H), 1.43 (d, J = 2.4 Hz, 9H), 1.41 (s, 9H), 1.39 (s, 4H). 13C NMR (126 MHz, MeOD) δ 173.52, 159.11 –158.82 (m), 157.99, 156.20, 153.92, 141.53, 137.00, 135.89, 133.86, 132.60, 130.71, 126.56, 125.80, 118.97, 115.49, 113.79, 91.55, 87.90, 87.28, 85.04, 84.60, 84.17, 82.61, 80.45, 56.71, 54.16, 52.14, 44.33, 38.19, 30.27, 28.74, 28.28, 27.49, 25.65. LCMS (ESI) m/z: [M + H]+ 856.5
tert-butyl ((S)-1-((3-(3-carbamoylphenyl)prop-2-yn-1-yl)(((3aR,4R,6R,6aR)-2,2-dimethyl-6-(6-((3-(2,2,2-trifluoroacetamido)propyl)amino)-9H-purin-9-yl)tetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)amino)-5,5-dimethyl-4-oxohexan-3-yl)carbamate (8).
A solution of 7 (160 mg, 0.18 mmol) and K2CO3 (103 mg, 0.75 mmol) in DMSO (3 mL) was cooled to 0 °C and then treated with hydrogen peroxide (38 mg, 1.12 mmol). The reaction mixture was warmed to room temperature and stirred for 3 h. The reaction mixture was diluted with water and extracted with ethyl acetate (3X). The organic layers were combined, dried with sodium sulfate and concentrated to afford the crude product which was purified by column chromatography (DCM: MeOH 10:1) to afford 8 (163 mg, quant. yield) as a white solid. 1H NMR (500 MHz, CDCl3) δ 9.08 (s, 1H), 8.30 (s, 1H), 7.99 (s, 1H), 7.88 – 7.73 (m, 2H), 7.46 (d, J = 7.7 Hz, 1H), 7.35 (t, J = 7.8 Hz, 1H), 6.85 (s, 1H), 6.40 (t, J = 58.7 Hz, 2H), 6.10 (s, 1H), 5.59 (t, J = 17.6 Hz, 1H), 5.47 (d, J = 6.3 Hz, 1H), 5.07 – 4.82 (m, 1H), 4.50 – 4.36 (m, 1H), 3.81 – 3.54 (m, 4H), 3.44 – 3.27 (m, 2H), 2.82 (p, J = 8.9, 7.2 Hz, 2H), 2.64 (dd, J = 16.3, 9.0 Hz, 2H), 2.02 – 1.74 (m, 4H), 1.61 (s, 3H), 1.42 (s, 9H), 1.40 (s, 9H), 1.39 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 171.85, 169.12, 157.54 –157.25 (m), 155.68, 155.53, 153.02, 139.79, 134.89, 133.70, 130.80, 128.74, 127.37, 123.48, 117.48, 115.19, 114.62, 90.97, 86.38, 85.25, 84.64, 84.07, 83.37, 81.99, 79.74, 55.84, 52.94, 50.87, 43.71, 30.06, 29.42, 28.44, 28.08, 27.24, 25.52. LCMS (ESI) m/z: [M + H]+ 874.4
tert-butyl ((S)-1-((((3aR,4R,6R,6aR)-6-(6-((3-aminopropyl)amino)-9H-purin-9-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)(3-(3-carbamoylphenyl)prop-2-yn-1-yl)amino)-5,5-dimethyl-4-oxohexan-3-yl)carbamate (9).
Nucleoside 8 (160 mg, 0.18 mmol) was dissolved in a mixture of MeOH (6 mL) and aqueous NH3 solution (6 mL, 33%) and stirred at room temperature for 12 h. The solvents were removed in vacuo, the crude product was dissolved in DCM (10 mL) and the organic layer washed with aqueous TEA solution. Removal of the solvent yielded the protected AdoHcy analog 9 (123 mg, 77%) as a clear oil. 1H NMR (500 MHz, CDCl3) δ 8.32 (s, 1H), 7.93 (s, 1H), 7.82 (d, J = 9.7 Hz, 2H), 7.47 – 7.42 (m, 1H), 7.34 (t, J = 7.7 Hz, 1H), 6.08 (d, J = 1.9 Hz, 1H), 5.59 (d, J = 8.1 Hz, 1H), 5.46 (d, J = 6.5 Hz, 1H), 5.03 – 4.94 (m, 1H), 4.44 – 4.35 (m, 1H), 4.29 – 4.17 (m, 1H), 3.78 – 3.52 (m, 4H), 2.88 – 2.77 (m, 4H), 2.67 – 2.56 (m, 2H), 1.86 – 1.73 (m, 4H), 1.59 (s, 4H), 1.42 (s, 9H), 1.40 (s, 9H), 1.38 (s, 3H). 13C NMR (126 MHz, CDCls) δ 171.84, 169.02, 155.64, 155.06, 153.37, 139.28, 134.79, 133.77, 130.68, 128.73, 127.68, 123.32, 120.42, 114.45, 90.93, 86.71, 85.25, 84.62, 84.12, 83.47, 81.93, 79.68, 55.82, 52.92, 50.93, 43.86, 39.82, 30.01, 28.45, 28.10, 27.23, 25.54. LCMS (ESI) m/z: [M + H]+ 778.5
(S)-amino-4-((((2R,3S,4R,5R)-5-(6-((3-aminopropyl)amino)-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methy)(3-3-carbamoylphenyl)prop-2-yn-1-yl)amino)butanoic acid (10).
Was prepared according to general procedure for deprotection of bisubstrate inhibitor 9 (20 mg, 0.026 mmol) to afford 10 (6 mg, 37 %) as a white solid. 1H NMR (500 MHz, Deuterium Oxide) δ 8.37 (s, 1H), 8.23 (d, J = 3.5 Hz, 1H), 7.87 (d, J = 7.4 Hz, 1H), 7.59 (s, 1H), 7.52 – 7.41 (m, 2H), 6.19 (d, J = 3.5 Hz, 1H), 4.51 (dd, J = 16.9, 3.4 Hz, 3H), 4.36 (dd, J = 17.0, 3.5 Hz, 2H), 4.06 – 3.98 (m, 2H), 3.94 – 3.89 (m, 1H), 3.85 – 3.79 (m, 1H), 3.63 (d, J = 7.2 Hz, 2H), 3.48 (s, 2H), 3.13 – 3.05 (m, 2H), 2.49 – 2.37 (m, 1H), 2.27 (d, J = 12.6 Hz, 1H), 2.10 – 1.99 (m, 2H). HRMS m/z calcd for C27H36N9O6 [M + H]+: 582.2783; found: 582.2783 found:582.2783
(S)-2-amino-4-((3-(3-carbamoylphenyl)prop-2-yn-1-yl)(((2R,3S,4R,5R)-5-(6-((3-(3′,6′-dihydroxy-3-oxo-3H-spiro[isobenzofuran-1,9′-xanthene]-5-carboxamido)propyl)amino)-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)amino)butanoic acid (11)
To a solution of 9 (30 mg, 0.038 mmol) in DMF (1 mL) was added DIC (10 mg, 0.077 mmol) and the reaction was stirred for 30 mins before the addition of HOBt (7.8 mg, 0.058 mmol), 5-Carboxyfluorescein (17 mg, 0.046 mmol) and DIPEA (10 mg, 0.077 mmol). The reaction was stirred at room temperature overnight. The reaction mixture was quenched with water and extracted with ethyl acetate (3X). The organic layers were combined, washed with brine, dried with sodium sulfate and concentrated to afford the crude product which was then subjected to the general procedure for deprotection of bisubstrate inhibitor to afford 11 (12 mg, 29 %) as a yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 10.26 (s, 1H), 8.91 (s, 1H), 8.45 (s, 1H), 8.37 (s, 1H), 8.24 (s, 2H), 8.08 (s, 1H), 7.94 (s, 1H), 7.85 (d, J = 7.3 Hz, 1H), 7.53 (s, 1H), 7.44 (s, 1H), 7.37 (d, J = 7.6 Hz, 1H), 6.71 (s, 2H), 6.55 (d, J = 14.1 Hz, 4H), 5.91 (s, 1H), 5.54 (s, 1H), 4.66 (s, 1H), 4.15 (d, J = 22.8 Hz, 2H), 3.94 (s, 1H), 2.95 (s, 2H), 2.83 (s, 2H), 2.02 (s, 1H), 1.91 (d, J = 38.7 Hz, 3H), 1.20 (d, J = 17.7 Hz, 1H). 13C NMR (126 MHz, DMSO-d6) δ 170.82, 168.19, 167.02, 164.68, 159.61, 154.61, 152.61, 151.76, 139.69, 136.31, 134.64, 134.53, 134.04, 130.40,129.07, 128.67, 127.51, 126.41, 124.23, 123.20, 122.25, 119.59, 112.68, 108.98, 102.27, 87.80, 84.87, 83.29, 81.88, 72.72, 71.82, 55.41, 51.09, 49.88, 42.74, 37.54, 37.06, 28.92, 27.14. HRMS m/z calcd for C48H46N9O12 [M + H]+: 940.3260; found: 940.3259
Supplementary Material
Acknowledgements
We appreciate Dr. Dongxing Chen for his assistance with the enzymatic activity assay and Krystal Diaz for her feedback. The authors acknowledge the support from NIH grants R01GM117275 (R.H.), U01CA214649 (R.H.), and P30 CA023168 (Purdue University Center for Cancer Research). We also thank supports from the Department of Medicinal Chemistry and Molecular Pharmacology (R.H.) at Purdue University.
1. Abbreviations used
- AdoHcy
S-adenosylhomocysteine
- AdoMet
S-adenosylmethionine
- SAHH
S-adenosylhomocysteine hydrolase
- NAM
nicotinamide
- MNAM
N1-methylnicotinamide
- NNMT
nicotinamide N- methyltransferase
- HPLC
High-Pressure Liquid chromatography
- FP
fluorescence polarization
- HTS
high-throughput screening
- TFA
trifluoroacetic acid
- NMR
Nuclear magnetic resonance
- Ki
inhibitory constant
- Kd
dissociation constant
- DMSO
dimethyl sulfoxide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no competing financial interest.
Supplementary data
Supplementary data associated with this article can be found, in the online version.
Development of a fluorescence polarization (FP)-based competition assay with a novel fluorescent probe II138 for evaluation of NNMT inhibitors that interfere with the interaction of the NNMT active site directly or allosterically.
References
- [1].Aksoy S, Szumlanski CL, Weinshilboum RM, Human liver nicotinamide N-methyltransferase. cDNA cloning, expression, and biochemical characterization, J. Biol. Chem 269 (1994) 14835–14840. [PubMed] [Google Scholar]
- [2].Pissios P, Nicotinamide N-methyltransferase: more than a vitamin B3 clearance enzyme, Trends Endocrinol. Metab 28 (2017) 340–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kraus D, Yang Q, Kong D, Banks AS, Zhang L, Rodgers JT, Pirinen E, Pulinilkunnil TC, Gong F, Wang YC, Cen Y, Sauve AA, Asara JM, Peroni OD, Monia BP, Bhanot S, Alhonen L, Puigserver P, Kahn BB, Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity, Nature. 508 (2014) 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Parsons RB, Smith SW, Waring RH, Williams AC, Ramsden DB, High expression of nicotinamide N-methyltransferase in patients with idiopathic Parkinson’s disease, Neurosci. Lett 342 (2003) 13–16. [DOI] [PubMed] [Google Scholar]
- [5].Xie X, Yu H, Wang Y, Zhou Y, Li G, Ruan Z, Li F, Wang X, Liu H, Zhang J, Nicotinamide N-methyltransferase enhances the capacity of tumorigenesis associated with the promotion of cell cycle progression in human colorectal cancer cells, Arch. Biochem. Biophys 564 (2014) 52–66. [DOI] [PubMed] [Google Scholar]
- [6].Markert JM, Fuller CM, Gillespie GY, Bubien JK, McLean LA, Hong RL, Lee K, Gullans SR, Mapstone TB, Benos DJ, Differential gene expression profiling in human brain tumors, Physiol. Genomics 5 (2001) 21–33. [DOI] [PubMed] [Google Scholar]
- [7].Xu J, Moatamed F, Caldwell JS, Walker JR, Kraiem Z, Taki K, Brent GA, Hershman JM, Enhanced expression of nicotinamide N-methyltransferase in human papillary thyroid carcinoma cells, J. Clin. Endocrinol. Metab 88 (2003) 4990–4996. [DOI] [PubMed] [Google Scholar]
- [8].Roessler M, Rollinger W, Palme S, Hagmann ML, Berndt P, Engel AM, Schneidinger B, Pfeffer M, Andres H, Karl J, Bodenmuller H, Ruschoff J, Henkel T, Rohr G, Rossol S, Rosch W, Langen H, Zolg W, Tacke M, Identification of nicotinamide N-methyltransferase as a novel serum tumor marker for colorectal cancer, Clin. Cancer Res 11 (2005) 6550–6557. [DOI] [PubMed] [Google Scholar]
- [9].Brenner C, Targeting a fat-accumulation gene, Nature. 508 (2014) 194–195. [DOI] [PubMed] [Google Scholar]
- [10].Domagala TB, Szeffler A, Dobrucki LW, Dropinski J, Polanski S, Leszczynska-Wiloch M, Kotula-Horowitz K, Wojciechowski J, Wojnowski L, Szczeklik A, Kalinowski L, Nitric oxide production and endothelium-dependent vasorelaxation ameliorated by N1-methylnicotinamide in human blood vessels, Hypertension. 59 (2012) 825–832. [DOI] [PubMed] [Google Scholar]
- [11].Pozzi V, Sartini D, Morganti S, Giuliante R, Di Ruscio G, Santarelli A, Rocchetti R, Rubini C, Tomasetti M, Giannatempo G, Orlando F, Provinciali M, Lo Muzio L, Emanuelli M, RNA-Mediated gene silencing of nicotinamide N-methyltransferase is associated with decreased tumorigenicity in human oral carcinoma cells, PLoS One. 8 (2013)e71272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gao Y, Van Haren MJ, Moret EE, Rood JJM, Sartini D, Salvucci A, Emanuelli M, Craveur P, Babault N, Jin J, Martin NI, Bisubstrate inhibitors of nicotinamide N-methyltransferase (NNMT) with enhanced activity, J. Med. Chem 62 (2019) 6597–6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chen D, Li L, Diaz K, Iyamu ID, Yadav R, Noinaj N, Huang R, Novel propargyl-linked bisubstrate analogues as tight-binding inhibitors for nicotinamide N-methyltransferase, J. Med. Chem 62 (2019) 10783–10797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Policarpo RL, Decultot L, May E, Kuzmic P, Carlson S, Huang D, Chu V, Wright BA, Dhakshinamoorthy S, Kannt A, Rani S, Dittakavi S, Panarese JD, Gaudet R, Shair MD, High-affinity alkynyl bisubstrate inhibitors of nicotinamide N-methyltransferase (NNMT), J. Med. Chem 62 (2019) 9837–9873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Neelakantan H, Wang HY, Vance V, Hommel JD, McHardy SF, Watowich SJ, Structure-Activity Relationship for small molecule inhibitors of nicotinamide N-methyltransferase, J. Med. Chem 60 (2017) 5015–5028. [DOI] [PubMed] [Google Scholar]
- [16].Kannt A, Rajagopal S, Kadnur SV, Suresh J, Bhamidipati RK, Swaminathan S, Hallur MS, Kristam R, Elvert R, Czech J, Pfenninger A, Rudolph C, Schreuder H, Chandrasekar DV, Mane VS, Birudukota S, Shaik S, Zope BR, Burri RR, Anand NN, Thakur MK, Singh M, Parveen R, Kandan S, Mullangi R, Yura T, Gosu R, Ruf S, Dhakshinamoorthy S, A small molecule inhibitor of nicotinamide N-methyltransferase for the treatment of metabolic disorders, Sci. Rep 8 (2018) 3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Babault N, Allali-Hassani A, Li F, Fan J, Yue A, Ju K, Liu F, Vedadi M, Liu J, Jin J, Discovery of bisubstrate inhibitors of nicotinamide N-methyltransferase (NNMT), J. Med. Chem 61 (2018) 1541–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Richardson SL, Mao Y, Zhang G, Hanjra P, Peterson DL, Huang R, Kinetic mechanism of protein N-terminal methyltransferase 1, J. Biol. Chem 290 (2015) 11601–11610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Collazo E, Couture JF, Bulfer S, Trievel RC, A coupled fluorescent assay for histone methyltransferases, Anal. Biochem 342 (2005) 86–92. [DOI] [PubMed] [Google Scholar]
- [20].Van Haren MJ, Sastre Torano J, Sartini D, Emanuelli M, Parsons RB, Martin NI, A rapid and efficient assay for the characterization of substrates and inhibitors of nicotinamide N-methyltransferase, Biochemistry. 55 (2016) 5307–5315. [DOI] [PubMed] [Google Scholar]
- [21].Neelakantan H, Vance V, Wang HYL, McHardy SF, Watowich SJ, Noncoupled fluorescent assay for direct real-time monitoring of nicotinamide N-methyltransferase activity, Biochemistry. 56 (2017) 824–832. [DOI] [PubMed] [Google Scholar]
- [22].Loring HS, Thompson PR, Kinetic mechanism of nicotinamide N-methyltransferase, Biochemistry. 57 (2018) 5524–5532. [DOI] [PubMed] [Google Scholar]
- [23].Moerke NJ, Fluorescence Polarization (FP) assays for monitoring peptide-protein or nucleic acid-protein binding, Curr. Protoc. Chem. Biol 1 (2009) 1–15. [DOI] [PubMed] [Google Scholar]
- [24].Peng Y, Sartini D, Pozzi V, Wilk D, Emanuelli M, Yee VC, Structural basis of substrate recognition in human nicotinamide N-methyltransferase, Biochemistry. 50 (2011) 7800–7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yung-Chi C, Prusoff WH, Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction, Biochem. Pharmacol 22 (1973) 3099–3108. [DOI] [PubMed] [Google Scholar]
- [26].Zhang JH, Chung TDY, Oldenburg KR, A simple statistical parameter for use in evaluation and validation of high throughput screening assays, J. Biomol. Screen 4 (1999) 67–73. [DOI] [PubMed] [Google Scholar]
- [27].Dalhoff C, Huben M, Lenz T, Poot P, Nordhoff E, Koster H, Weinhold E, Synthesis of S-adenosyl-L-homocysteine capture compounds for selective photoinduced isolation of methyltransferases, ChemBioChem. 11 (2010) 256–265. [DOI] [PubMed] [Google Scholar]
- [28].Roehrl MHA, Wang JY, Wagner G, Discovery of small-molecule inhibitors of the NFAT-calcineurin interaction by competitive high-throughput fluorescence polarization screening, Biochemistry. 43 (2004) 16067–16075. [DOI] [PubMed] [Google Scholar]
- [29].Van Haren MJ, Taig R, Kuppens J, Sastre Torano J, Moret EE, Parsons RB, Sartini D, Emanuelli M, Martin NI, Inhibitors of nicotinamide: N-methyltransferase designed to mimic the methylation reaction transition state, Org. Biomol. Chem 15 (2017) 6656–6667. [DOI] [PubMed] [Google Scholar]
- [30].Huang X, Fluorescence polarization competition assay: The range of resolvable inhibitor potency is limited by the affinity of the fluorescent ligand, J. Biomol. Screen 8 (2003) 34–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.