

Abstract
Fecal microbial community changes are associated with numerous disease states, including cardiovascular disease (CVD). However, such data are merely associative. A causal contribution for gut microbiota in CVD has been further supported by a multitude of more direct experimental evidence. Indeed, gut microbiota transplantation studies, specific gut microbiota-dependent pathways, and down-stream metabolites have all been shown to influence host metabolism and CVD, sometimes through specific identified host receptors. Multiple metaorganismal pathways (involving both microbe and host) both impact CVD in animal models and show striking clinical associations in human studies. For example, trimethylamine N-oxide (TMAO) and more recently, phenylacetyl glutamine (PAG), are gut microbiota-dependent metabolites whose blood levels are associated with incident CVD risks in large scale clinical studies. Importantly, a causal link to CVD for these and other specific gut microbial metabolites/pathways have been shown through numerous mechanistic animal model studies. PAG, for example, was recently shown to promote adverse cardiovascular phenotypes in the host via interaction with multiple adrenergic receptors, a class of key receptors that regulate cardiovascular homeostasis. In this review, we summarize recent advances of microbiome research in CVD and related cardiometabolic phenotypes that have helped to move the field forward from associative to causative results. We focus on microbiota and metaorganismal compounds/pathways, with specific attention paid to short chain fatty acids, secondary bile acids, TMAO, and PAG. We also discuss novel therapeutic strategies for directly targeting the gut microbiome to improve cardiovascular outcomes.
Keywords: Cardiovascular Disease; Gut Microbiome; Atherosclerosis; Thrombosis; Vascular Disease; Inflammation, Metabolism
Introduction
The collection of microbes living in the human intestinal tract (gut microbiota) and their combined genetic capacities (gut microbiome) have influence far beyond digestion. Indeed, gut microbiota generate biologically active metabolites that impact many aspects of host physiology, and collectively are widely considered the body’s largest endocrine organ. While gut microbiota facilitate many necessary and beneficial physiological processes, like the digestion of macronutrients and synthesis of some vitamins, numerous lines of evidence show gut microbiota can play a role in the development of adverse phenotypes. In particular, distinct changes in the microbial community structure and function are associated with multiple disease states, including cardiovascular disease (CVD)1.
Early work in the gut microbiome field demonstrated that alterations in fecal microbial community composition are associated with the development of obesity and insulin resistance, and that microbial transplantation could transmit heightened adiposity in the host2–4. Subsequently, it was discovered that disruptions to the microbiome early in life can promote heightened adiposity5. Early sequencing studies by Koren et al. suggested microbiota may be linked to atherosclerosis, because human atherosclerotic plaques were noted to contain bacterial DNA, though whether or not the DNA was derived from live bacteria within the artery wall was not determined6. The first studies revealing a potential causal link between the gut microbiome and CVD focused on trimethylamine N-oxide (TMAO), a metaorganismal metabolite formed following ingestion of dietary nutrients abundant in a Western diet (e.g. lecithin, choline, carnitine)7–9. The microbiome field has since rapidly expanded to involve many previously disparate areas of research, demonstrating the far-reaching effects of gut microbiota on human health and disease. Figure 1 illustrates several of the major pathways identified linking gut microbiota to CVD, numerous related phenotypes, and known molecular participants, including some of the identified host receptors. We will use Figure 1 as a template for organizing this review, first providing an overview of the field, and then focusing on several areas of more recent advancement. Finally, we will discuss therapeutic targeting of the gut microbiome as a potential treatment or prevention strategy for cardiovascular and metabolic diseases, highlighting recent advances in development of non-lethal small molecule inhibitors of specific microbial pathways as a novel approach to improve CVD outcomes.
Figure 1:
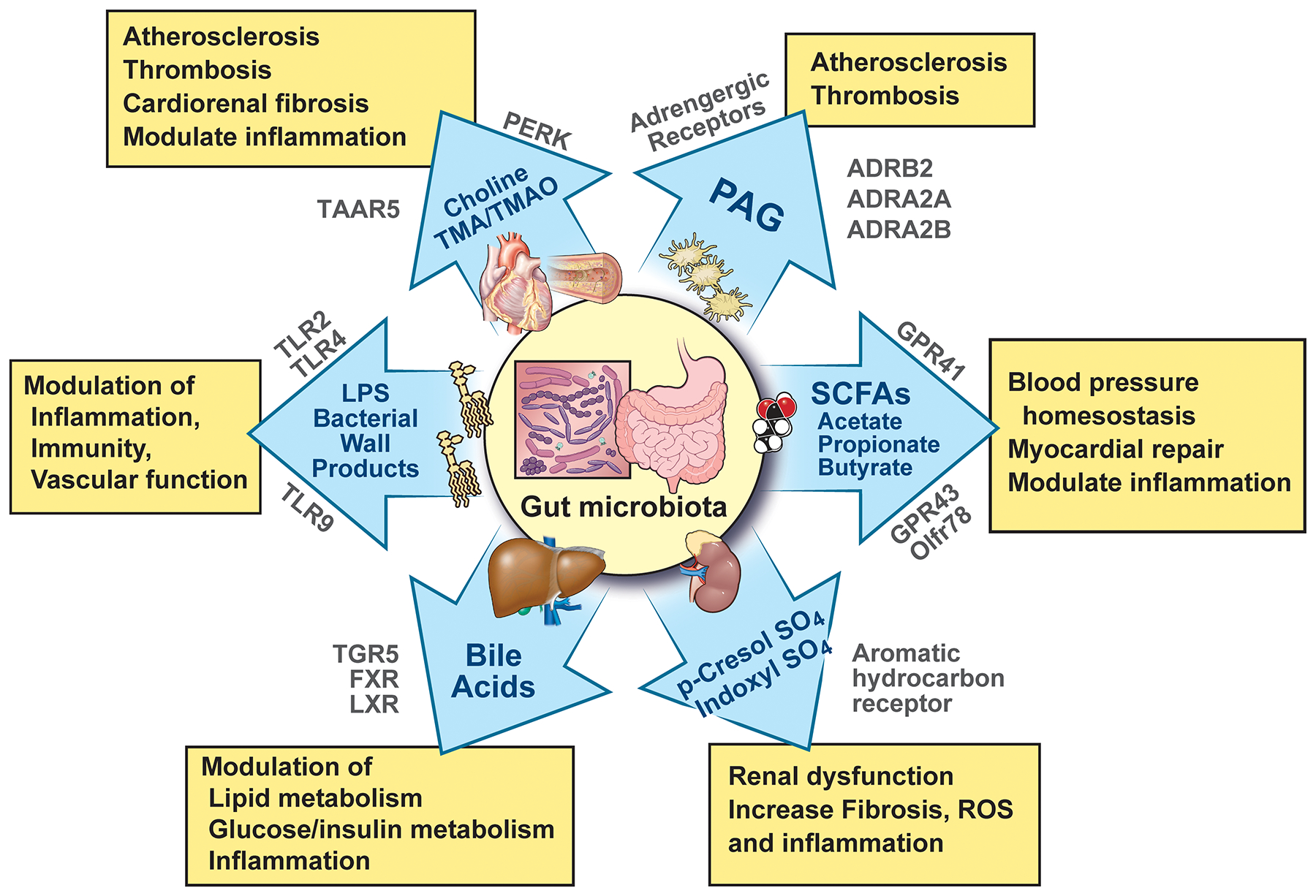
Molecular pathways and host receptors that link gut microbiota-derived products and metabolites with cardiovascular and cardiometabolic disease phenotypes. ADRA, adrenergic receptor alpha; ADRB, adrenergic receptor beta; GPR, G-protein–coupled receptor; FXR, farnesol X receptor; LPS, lipopolysaccharide; LXR, liver X receptor; Olfr, olfactory receptor; PAG, phenylacetylglutamine; PERK, protein kinase R-like endoplasmic reticulum kinase; ROS, reactive oxygen species; SCFA, short chain fatty acid; TGR, takeda G protein-coupled receptor; TLR, toll-like receptor; TMA, trimethylamine; TMAO, trimethylamine N-oxide; and TAAR, trace amine-associated receptor.
Gut bacterial community compositional changes and association with CVD
Due to advances in culture-independent sequencing technologies and bioinformatics, the vast majority of gut microbiome related studies assess gut (often fecal) microbial community compositional changes associated with various disease states. This has led to a wealth of associative data, which, while helpful, is limited in terms of investigating causality. While it is difficult to define a truly pathogenic bacterial community, the term dysbiosis has been used to describe an imbalance of intestinal microbiota composition within a disease state or phenotype. Many investigators have reported an association between CVD phenotypes and changes in the relative abundance of specific microbial taxa, or gut bacterial richness or diversity. For example, in early studies bacterial DNA was detected in atherosclerotic plaques with signatures that match taxa associated with disease states6, and microbial compositional changes have been reported in patients with numerous CVD risk factors including hypertension, dyslipidemia, insulin resistance, and other metabolic phenotypes10, 11. Changes in microbiota composition, diversity, and richness are associative, making it difficult to determine whether microbial changes are driving disease states or rather being driven by them. Moreover, typically-employed sequencing techniques often do not reach species or strain level resolution, and analyses often exclude less abundant microbes. Studies focusing primarily on abundance can thus neglect gain-of-function microbial pathways that contribute to disease, despite arising from microbes that can represent only a small proportion of the microbial community. A recent illustrative example of this is shown in a series of microbial transplantation studies designed to demonstrate a direct role of gut microbial cutC (a major microbial gene responsible for choline→TMA transformation12, 13) in host thrombosis potential14 (Figure 2). These studies used germ-free (GF) mice colonized with a synthetic polymicrobial community lacking choline→TMA functional capacity, and the addition of a human commensal (C. sporogenes) genetically engineered to either possess or lack a functional microbial cutC gene. While only present at a very low abundance (only ~ 0.1%) within the large intestine gut microbial community, the gain-of-function C. sporogenes mutant elevated circulating TMA and TMAO levels within the host, and both enhanced rate of thrombus formation and reduced time to cessation of blood flow following arterial injury in vivo14.
Figure 2:
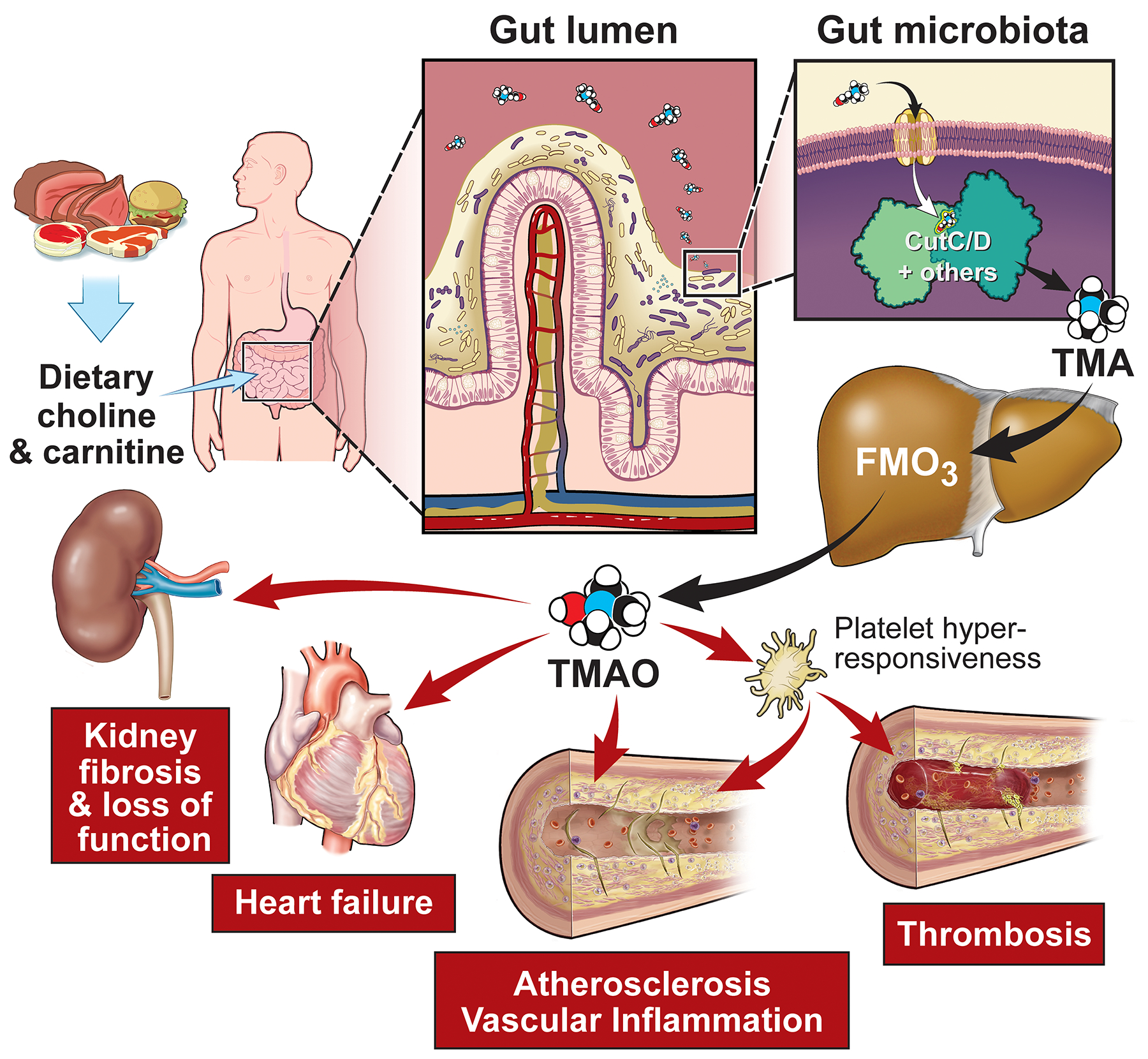
Dietary precursors, such as choline and carnitine, are metabolized into trimethylamine (TMA) by gut microbiota via specific genes, including members of the choline utilization gene cluster (Cut)C/D. Host hepatic flavin monooxygenases (FMOs) oxidize TMA into TMAO, which promotes metabolic and functional changes in the host including cardiovascular and renal end-organ damage.
Whole genome sequencing coupled with bioinformatics analyses can also be used to infer potential functional capacity of gut microbial communities. For example, deep sequencing and systematic analyses of biosynthetic gene clusters identified the presence of common antibiotic genes in human commensals15. But just like studies in vertebrate animals, the mere presence of a gene within bacteria does not provide information about its expression and activity. What’s more, the majority of microbial genes have unknown functions. Further, microbial protein expression is highly dynamic, as bacterial gene regulation is greatly influenced by environmental cues. This is demonstrated in recent work showing that dietary addition of a single type of fiber can cause dramatic changes in the expression of genes required for bacterial metabolism of fiber glycans16. So while studies and approaches that focus on microbial gene abundance and inference of potential functional capacity can be valuable tools for hypothesis generation, they need to be experimentally validated17.
Gut leakiness as a potential portal for gut microbiota-derived products and inflammation
In its healthy state, intestinal barrier function is maintained by physical factors including tight junctions between epithelial cells, mucus production, and mucosal immunity. And in heart failure patients, we often observe bowel wall edema and impaired barrier function18, 19. Following the leaky gut concept, impaired gut barrier function leads to translocation of bacterial products into host circulation, which can result in a pro-inflammatory state. Multiple studies show patients with heart failure have alterations in intestinal integrity, and that elevated levels of pro-inflammatory cytokines in the blood correlate with symptom severity and poorer outcomes20, 21. In the presence of heart failure, venous fluid overload, adaptive sympathetic activation resulting in redistribution of the systemic circulation, and low cardiac output contribute to bowel wall edema and reduced mucosal perfusion22. Intestinal hypoperfusion in heart failure alters mucosal function, as evidenced by increased paracellular permeability and augmented intestinal bacterial biofilm formation23. When the gut barrier is impaired, lipopolysaccharide (LPS) originating from Gram-negative bacteria can enter the host circulation, where it is mainly recognized by toll-like receptor (TLR)s on the surface of immune cells24. Upon binding of bacterial ligands, TLR-signaling induces release of pro-inflammatory cytokines that orchestrate a pro-inflammatory state in the host.
Enhanced levels of LPS and other bacterial wall products, presumably derived from gut microbiota, have been mechanistically linked to modulation of inflammation, immunity, and vascular function (Figure 1). Patients with decompensated heart failure have higher endotoxin levels in the blood compared to stable patients25. Translocation of LPS from the bowel in this setting is supported by higher endotoxin concentrations in the hepatic vei,n as compared to blood directly sampled from the ventricles26. The detection of gut microbiota and metaorganismal metabolites at heightened levels in subjects with CVD, or risk for incident adverse CVD events, may in part reflect alterations of the host barrier function (Figure 1). In a recent observational study, circulating LPS concentrations were predictive of MACE in a cohort of patients with atrial fibrillation, suggesting that endotoxin translocation impacts CVD complications27. It is worth noting that a Mediterranean diet was negatively associated with endotoxemia in this study, suggesting the involvement of the gut microbiota in gut barrier function of the host. Indeed, in preclinical models, gavage of western diet–fed Apoe−/− mice with live Akkermansia muciniphila decreased intestinal permeability and lowered fecal and circulating LPS levels, which was associated with reduced aortic atherosclerosis independent of lipid metabolism28. While the mechanism of Akkermansia-induced changes in barrier function needs to be explored, a recent proof-of-concept study in humans found that administration of pasteurized (but not live) Akkermansia muciniphila for 3 months led to reduction in plasma LPS in obese individuals with metabolic syndrome29. Although the intervention did not change body weight, the authors observed improvement in insulin sensitivity and dyslipidemia in the Akkermansia-treated group. Interestingly, structural differences of LPS subtypes from different gut microbial species have been associated with altered TLR recognition and their effects on host innate immunity30. For instance, gavage with the gram-negative Bacteroides (vulgatus and dorei) that produce penta- and tetra-acylated lipid A – in contrast to the hexa-acylated lipid A of e. coli, reduced colonic inflammation, endotoxemia, and atherosclerosis in ApoE−/− mice31.
It should be noted that factors beyond gut leakiness contribute to CVD. There are many disease conditions where enhanced “gut leakiness” is present, but not all such diseases show heightened associations with CVD risks. For example, while the presence of heart failure-induced bowel wall edema has been linked to endotoxemia and CVD progression, gut barrier defects caused by colitis and inflammatory bowel diseases are not classically associated with heightened CVD risks. These observations point to a more complex relationship between bowel wall integrity, changes in gut microbial communities, and the relationship between host systemic inflammation and altered susceptibility for development of CVD. We speculate that different mechanisms of “gut leakiness” (e.g. inflammatory, bowel wall edema, etc.) may differentially impact gut microbiome and pathophysiological processes that modulate susceptibility for development of CVD. Improved understanding of the role of gut bacterial pro-inflammatory factors in triggering systemic inflammatory cascades may help to provide novel therapeutic strategies to improve care and risk stratification among patients with CVD.
Numerous lines of evidence link multiple different facets of inflammation to heightened risks for cardiovascular disease32, 33. The role of inflammatory pathways in CVD has recently been reaffirmed by the CANTOS clinical trial. Administration of canakinumab, an antibody against Interleukin(IL)-1β, showed that inhibition of the IL-1β pathway reduces cardiovascular event risk independent of lipid level lowering34. Thus, treatment and prevention of CVD with immunomodulatory therapies seems promising. But targeting inflammatory pathways bears the risk for opportunistic infections, which may limit its use in patients with multiple comorbidities, as is often observed in CVD patients. Identification of gut microbiota that elicit host immune responses that participate in CVD pathogenesis may provide a therapeutic avenue to ameliorate inflammation-driven CVD phenotypes.
Bile acids are predominantly gut microbiota derived, and serve as modulators of host metabolism
A major function historically attributed to bile acids (BAs) has been to facilitate emulsification and adsorption of fat-soluble dietary nutrients. However, BAs are composed of a diverse array of structurally specific species whose concentrations differ by many orders of magnitude. More recent studies have shown structurally specific and distinct BAs also play additional roles, including but not limited to modulation of host lipid metabolism, glucose/insulin metabolism, and inflammation (Figure 1).
Initially synthesized from cholesterol in the host liver, primary BAs, which only represent a small fraction of the total BA pool, are then secreted into the intestinal (duodenum) lumen, where subsequent gut microbiota dependent modifications participate in the generation of a remarkably large array of BA species. The body maintains a large pool of hydrophobic BAs through reuptake in the ileum and through negative feedback. These negative feedback mechanisms are triggered by activation of the farnesoid x receptor (FXR) and cholesterol 7α hydroxylase35, or by expression of intestinal bile transporters36. BAs modulate gut microbial composition via potent antimicrobial properties, immune responses37 and FXR38. And bile obstruction can lead to bacterial overgrowth syndromes39. Gut microbiota modify primary BAs via bile salt hydrolysis and bile acid 7α dehydroxylation, yielding secondary BAs, many of which have hormone-like functions. Following systemic adsorption, some secondary BAs in turn impact host physiology though interaction with a variety of host nuclear receptors including FXR, liver X receptor (LXR), pregnane X receptor (PXR), and specific G-protein coupled receptors like TGR5 (Figure 1)40–44. Perturbations to the dynamic interactions among diet, gut microbiota, and specific BAs may thus contribute to cardiometabolic phenotypes and disease susceptibly. For example, altered levels of BAs in plasma are associated with insulin resistance in type 2 diabetes45, 46, and modulation of BA signaling may contribute to metabolic improvements during some anti-diabetic treatments47. As such, measuring systemic levels of BAs may aid in the assessment of potential gut microbiota contributions to cardiometabolic diseases46. A better understanding of how perturbation in bile acid profiles are associated with future development of disease states, or responses to therapy. Functional studies to determine if candidate bile acids are mechanistically linked to these processes are promising areas of future investigation.
Short chain fatty acids (SCFAs) and blood pressure in the host
SCFAs (fatty acids with 5 carbons or less) can be products of host metabolism (e.g. acetate48). However, they are also produced in large quantities by gut microbiota through anaerobic fermentation of dietary fiber49, 50. The most common SCFAs include acetate, propionate, and butyrate, which have been linked to alterations in host blood pressure homeostasis, myocardial repair and inflammation (Figure 1). The idea that circulating SCFAs are produced in large part by gut commensals is supported by studies showing that free SCFA levels are virtually undetectable in plasma recovered from germ-free animals51. Besides acting as an energy source for large intestine gut epithelial cells (e.g. colonocytes), SCFAs are absorbed into the portal blood and participate in various processes of the host, including lipid metabolism, glucose homeostasis, gut inflammation, and neurogenesis52. An association between SCFAs and adiposity early in life has been shown by Cho et al5. Exposure to antibiotics during weaning changed gut microbial communities with increasing metabolic capacity to produce acetate, propionate, and butyrate in C57BL/6J mice. Influx of these SCFAs to the liver resulted in substantial changes in the regulation of hepatic lipid metabolism and obese phenotype5. Moreover, antibiotic exposure early in life has also been associated with changes in microbiota diversity53, lasting effects on the host immunity54, and cardiometabolic diseases, such as diabetes55, 56. Interestingly, transfer of antibiotic-perturbed microbiota to the next generation of mice led to loss of microbial richness and changes in metagenomic gene expression and susceptibility for colitis57.
Initial clinical intervention studies reported that fiber intake is associated with a decrease in blood pressure58, and support the idea that SCFAs are involved in the regulation of blood pressure. Early mechanistic studies by Pluznick and colleagues substantiated this idea by demonstrating the G-protein coupled SCFA receptors olfactory receptor 78 (Olfr78) and G-protein receptor 41 (Gpr41) participate in host blood pressure regulation59–61. Being expressed in vascular smooth muscle cells as well as the juxtaglomerular apparatus, Olfr78 mediates renin release and changes in vascular resistance, contributing to hypertension60. By contrast, Gpr41 is expressed in the vascular endothelium and promotes reduction in blood pressure61. Propionate administration in the absence of Gpr41 tended to increase blood pressure, while it caused a pronounced drop in blood pressure in Olfr78−/− mice, suggesting a differential function of both receptors in SCFA-dependent regulation of blood pressure60. Interestingly, when the SCFA pool was depleted by antibiotics in Olfr78−/− mice, blood pressure went up, further corroborating a protective role of microbial SCFA generation to balance Olfr78 signaling. However, the overall hypotensive effects in wildtype animals might be explained by both a decline in cardiac output and the loss of vascular resistance exhibited by SCFAs62. While microbiota suppression with anti-microbials (i.e. poorly-absorbed antibiotics; Figure 3) can serve as a valuable tool for demonstrating involvement of the gut microbiome in a host phenotype, it is not a viable approach for therapeutically targeting the gut microbiome to achieve a desired long term outcome in the host, due to the selection of gut microbial communities with antibiotic resistance.
Figure 3:
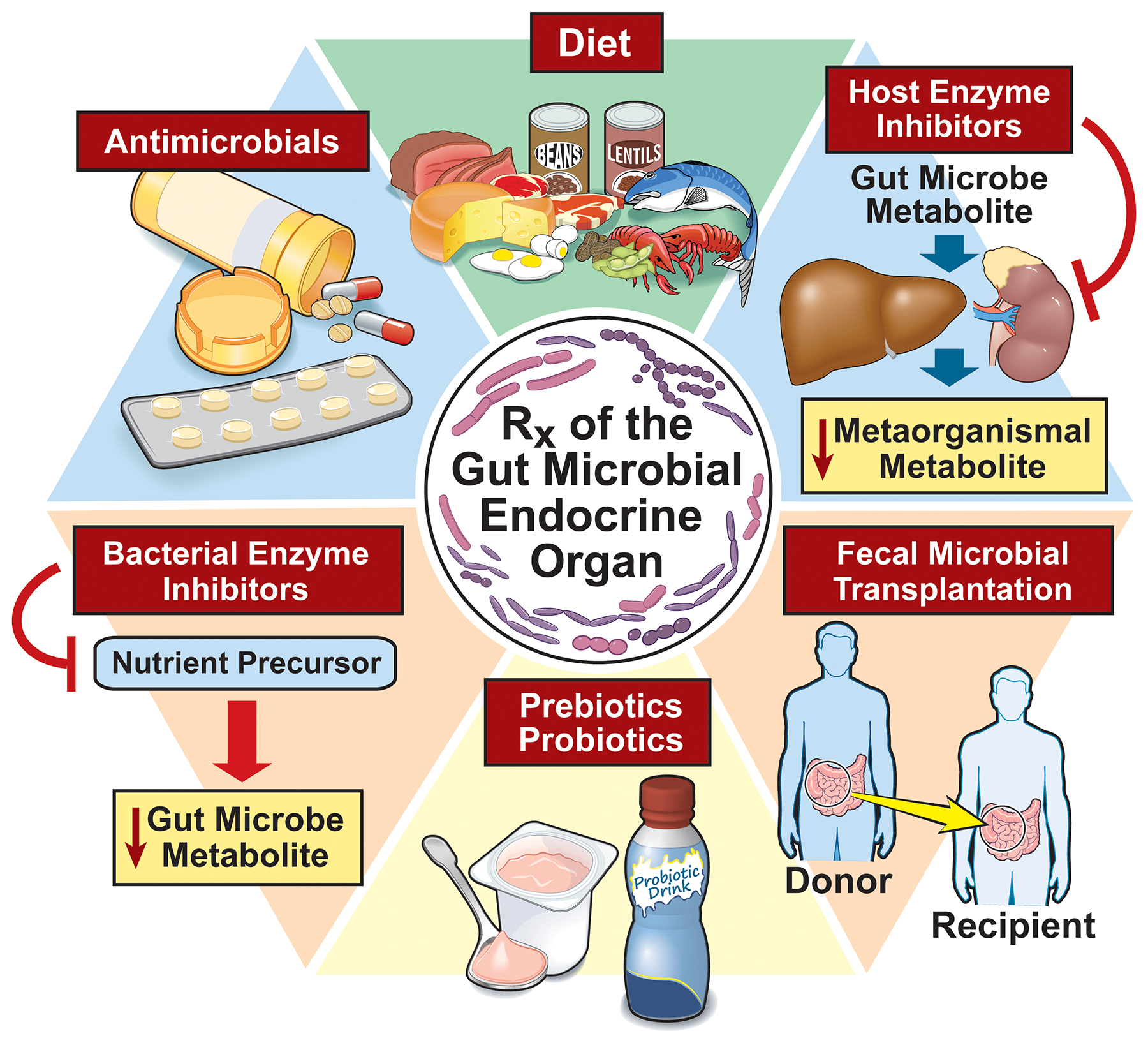
Illustration of current strategies to improve cardiovascular disease by manipulating gut microbiota include dietary interventions, targeting host enzymes involved in generation of metaorganismal metabolites, fecal microbial transplantation, pre/probiotics, bacterial enzyme inhibitors, and antimicrobials.
Many additional studies have supported a role for gut microbiota generation of SCFAs in modulation of blood pressure in the host. For example, fecal transfer from human hypertensive (versus normotensive) donors into germ-free mice revealed transmission of heightened blood pressure63. But in another study, transplantation of feces from normotensive Dahl salt-resistant rats into hypertensive Dahl salt-sensitive rats in fact exacerbated hypertension in the recipients, suggesting that additional host genetic variables may interact with microbial factors to modulate blood pressure control64. A role for SCFAs in hypertensive end-organ damage in angiotensin II-infused mice has also been suggested65. Thus, numerous lines of evidence show that the gut microbial community can impact blood pressure regulation in the host, and that SCFAs represent at least one of the microbial mediators that contribute to vasomotor tone and blood pressure. Recent studies provided further evidence that SCFAs are involved in other CVD processes, such as ischemia reperfusion injury, cardiac repair following myocardial infarction, and impaired arterial compliance66, 67.
SCFAs represent a readout of saccharidic metabolism by the whole microbial community, and often serve as terminal end products of (poly)microbial catabolic pathways. Therefore, their levels may reflect a convergence of multiple microbial participants and competing pathways. Further understanding of the factors that contribute to individual molecular species of SCFAs and the host receptors sensing them is an area of future investigation. Of particular interest, recent studies employing untargeted metabolomics suggest additional gut microbiota-derived plasma metabolites beyond SCFA may also contribute to host blood pressure regulation68. These studies found multiple structurally specific compounds including some “uremic toxins” that had previously been reported to activate the renin–angiotensin–aldosterone system (RAAS), and have been linked to heightened kidney injury in model systems69–71. A growing number of microbiota-dependent products, including uremic toxins like p-cresol sulfate, indoxyl sulfate, and a variety of aromatic amino acid metabolites, are thought to potentially alter host metabolism via specific receptors, including the aromatic hydrocarbon receptor Figure 3)72, 73.
Trimethylamine N-oxide – a metaorganismal metabolite causally linked to CVD and metabolic disease
Almost a decade ago, Wang et al. first causally linked gut microbiota with oral intake of nutrient precursors, TMAO production, and CVD risk (Figures 1 and 2)9. A combination of untargeted metabolomics and mechanistic animal model studies was used to uncover small molecules whose levels in blood associate with CVD risk in humans, and impact CVD relevant phenotypes in animal models. Several analytes linked to phosphatidylcholine metabolism, including TMAO, were identified, and TMAO was shown to both predict CVD risks in multiple clinical cohorts, and facilitate accelerated atherosclerosis (as did nutrient precursors with an intact host gut microbiome) in animal models9. Through these and other studies, generation of TMAO in humans and mice has been shown to occur via a multistep, metaorganismal pathway starting with the dietary precursors choline9, phosphatidylcholine8, 9, and carnitine7. These are most abundant in foods found in a Western diet, including red meat, egg yolks, and other animal products (Figure 2). Notably, plasma or serum levels of every additional TMA nutrient precursor identified (i.e. shown to generate TMA and TMAO in hosts via a gut microbiota dependent fashion), including betaine9, γ-butyrobetaine74, and trimethyllysine (TML)75 have all similarly shown associations with incident CVD risks in large scale clinical studies, and these associations appear be mediated by TMAO (since their clinical prognostic value becomes attenuated with TMAO in the model).
Gut microbial metabolism of TMA-containing nutrient precursors begins with specific microbial TMA lyases that generate TMA, an odorous gas, as a product. The major microbial choline TMA lyase is thought to be encoded by the microbial cutC/D genes (choline utilization gene cluster genes C (catalytic) and D)12. The TMA produced is then transported to liver via the portal vein and readily metabolized by host hepatic flavin monooxygenases (FMOs) (mainly FMO376) into TMAO9. In the circulation, TMA levels are typically negligible. When radiolabeled TMA or TMAO were orally administered to human volunteers, 95% of the dose label was excreted via the kidneys with the majority being TMAO77. TMAO has been shown to enhance atherosclerosis in most, but not all, mechanistic and animal model studies7, 9, 78–82. It has also been shown to promote platelet reactivity and thrombosis potential14, 83–85, vascular inflammation and inflammasome activation86–88, heightened heart failure89–91 and CKD92–95 related phenotypes in animal models (Figure 2).
Circulating levels of TMAO have been shown to associate with CVD and predict outcomes in the presence of multiple CVD phenotypes, including peripheral artery disease (PAD)96, coronary artery disease (CAD)97, acute coronary syndrome (ACS)97–99, and heart failure100–104. Notably, the prognostic value of TMAO withstands adjustment for traditional risk factors, highlighting its potential as a biomarker for risk stratification beyond what has been considered tradition risk. While not all studies have observed the relationship between heightened TMAO levels and incident CVD risks, examination of extant clinical studies with TMAO in multiple meta-analyses have concluded that a strong relationship indeed exists between elevated circulating TMAO levels and both CVD risk and mortality in multiple cohorts on different continents105–107. In many studies, a plasma TMAO cut off value of approximately > 6μM predicted heightened risk of adverse cardiac events108. And in one recent meta-analysis comprising >25000 subjects, a 7.6% increase in all-cause mortality was noted for each 10μmol/L increment of TMAO105.
Several factors impact circulating TMAO levels within subjects19, 108. First, the gut microbial community composition is critical since there is an initial obligatory role of gut microbes in TMA(O) generation7, 9. Second, renal functional decline leads to less efficient excretion and thus heightened levels of TMA(O)95. But elevated levels are also frequently observed among subjects with normal kidney function105. In all subjects, choline is a major and continuous nutrient precursor, since beyond diet, choline content in bile is remarkably high, and thus baths gut microbes in omnivore and vegans/vegetarians alike (the origins of the word choline are in the Greek word kholḗ, for “bile”, since choline was first isolated by Adolph Strecker from pig and ox bile in 1862)109. However, carnitine, which is found in high levels in red meat (and some energy drinks and over the counter supplements), also serves as a nutrient precursor, and can account for significant elevation in TMAO levels, particularly in some omnivores7, 110, 111. Large-scale clinical observational studies show that subjects with heightened circulating carnitine levels have higher risk for incident CVD events (heart attack, stroke, and death). The prognostic value for plasma carnitine levels, like other TMA precursors, seems to be mediated by TMAO, as inclusion of TMAO in statistical models attenuates the prognostic value of carnitine (but TMAO remains a robust predictor), and carnitine supplementation accelerates atherosclerosis development in animal models7, 112.
In a recent human dietary intervention study examining protein source (red meat vs white meat vs non-meat), substantially higher levels of circulating TMAO were observed when subjects consumed a red meat diet (equivalent to 8oz of steak daily for 1 month)111. Although there is a modest increase in choline content in omnivorous vs vegan diets, as noted above, a substantial amount of choline is introduced into the gut in the form of bile (choline in the form of phosphatidylcholine is a major component of bile). Thus, there is far more modest overall difference in choline exposure to the gut microbial community of vegan/vegetarian versus omnivore. By contrast, omnivorous diets show markedly higher carnitine content since vegan/vegetarian diets are virtually devoid of carnitine. In line with this, a major source of the observed elevation in plasma TMAO levels in subjects following 1 month of either a red meat rich diet versus white meat or non-meat diet (predominantly vegetarian protein source), was shown to arise from carnitine. In addition to having enhanced nutrient density of TMA(O) precursors (including carnitine), isotope tracer studies showed the red meat rich diet induced functional remodeling of the gut microbial community to enhance carnitine→→TMA transformation, but not choline→TMA generation111. Interestingly, the chronic exposure to a red meat rich diet also induced a functional change in the kidneys, with reduction in the fractional renal excretion rate for TMAO, despite no change in glomerular filtration rate111. It is also notable that numerous studies support a dose-dependent association between meat consumption and CVD risks and mortality113.
Trimethylamine N-oxide, a newly-recognized participant in atherosclerosis, thrombosis, and vascular inflammation
A causal contribution of gut microbiota to atherosclerosis susceptibility was first demonstrated with the discovery of TMAO as a gut microbiota-derived factor, and the initial functional studies demonstrating both direct provision of TMAO accelerated atherosclerosis in murine models, and that suppression of gut microbiota dependent conversion of nutrient precursors (choline) into TMA and TMAO (with antimicrobial/antibiotics) blocked choline diet dependent enhancement in atherosclerosis (Figure 2 and 3)9. In atherosclerosis-prone ApoE−/− mice, dietary supplementation with choline led to augmented atherosclerotic lesion burden, higher aortic expression of scavenger receptors (CD36 and scavenger receptor A), increased cholesterol laden macrophage foam cell formation9, and impaired in vivo reverse cholesterol transport7. In addition, TMAO suppressed bile acid pool size and therefore cholesterol clearance in the host7. Consistent and complementary to these findings, FMO3 knock down has been shown to impair TMA transformation into TMAO, thus reducing plasma TMAO levels and concomitantly, restoring cholesterol balance114. Early microbial transplantation studies of cecal microbial communities from high TMA-producing inbred C57BL/6J mice into atherosclerosis-resistant NZW/LacJ recipients demonstrated the transmissibility of dietary choline-induced TMA and TMAO generation, and atherosclerosis115. Not all TMAO precursor feeding studies, however, have shown similar results, supporting the notion that differences in the microbial communities present in the host can impact the final phenotypes observed116.
The striking association between circulating TMAO levels in subjects and thrombotic event risks, such as heart attack and stroke, has been witnessed across numerous large-scale clinical cohorts105, 117. This has prompted mechanistic studies in both humans and mice to explore the role of TMAO in thrombosis. Zhu et al. found that TMAO alters human platelet calcium signaling, heightening their responsiveness to sub-maximal stimulation by agonists (e.g. thrombin, ADP, collagen). Consequently, heightened thrombosis potential has been observed in both whole blood and in vivo arterial injury models14, 83–85, 118. In a subsequent human feeding study, healthy volunteers (both omnivore and vegan/vegetarian) that were orally supplemented with choline exhibited higher levels of TMAO and concomitant enhanced platelet responsiveness and aggregation77, 118. Importantly, higher TMAO levels were dose-dependently associated with increased platelet aggregation responsiveness, even in subjects on low-dose aspirin. This suggests that in subjects with elevated TMAO, the anti-platelet effects of aspirin may be attenuated, highlighting the possible involvement of TMAO in on-treatment platelet reactivity and so-called “aspirin resistance”.
The mechanistic involvement of the metaorganismal TMAO pathway in platelet function and in vivo thrombosis potential has also been examined through both genetic gain and loss of function manipulations, including to the host gene FMO3 (Figure 2). Multiple studies have confirmed through both genetic gain (as global FMO3 transgene) and loss (via anti-sense oligonucleotide to FMO3, and via global FMO3 knock out) of function studies in mice that manipulation of TMA and TMAO levels in vivo alters platelet responsiveness, rate of clot generation, and thrombosis potential83, 84, 119. Moreover, cecal microbial transplantation experiments confirmed that the prothrombotic phenotype mediated by a choline-rich diet was a transmissible trait83. Early studies by Cracium et al. first identified the choline utilization (cut) gene cluster in human commensals encoding the catalytic and regulatory gene products CutC and CutD12. The presence of the microbial cutC/D genes in human microbiota are associated with the ability to generate TMA from choline120, and with subsequent TMAO accumulation121. Importantly, studies using germ-free mice colonized with synthetic microbial communities +/- a genetically engineered gain or loss of function cutC mutant human commensal confirmed that a functional microbial cutC gene is sufficient to transmit TMA and TMAO generation, as well as in vivo thrombosis potential. Microbial cutC may thus represent a therapeutic target for preventing thromboembolic complications14.
Recent studies have shown that beyond impacting platelet function, TMAO induces expression of tissue factor (TF), the initiator of the extrinsic clotting, in endothelial cells in vitro122. Vascular TF promotes thrombosis and vascular inflammation123, particularl y in patients with type 2 diabetes who have higher levels of circulating TMAO124–126. Animal model studies are still needed to validate a contribution of gut microbiota and TMAO generation to alterations in TF pathway in vivo, and to altered thrombosis potential. A recent study reported that the absence of microbiota was associated with reduced hepatic von Willebrand factor (VWF) synthesis and reduced thrombus growth after carotid artery injury in C57BL/6 germfree mice as compared to conventionally raised littler mates127. However, the role of metaorganismal metabolites, in particular TMAO, in the VWF-depending thrombosis has yet to be determined.
Vascular inflammation is critically involved in the pathogenesis of atherosclerosis and thrombotic complications. Seldin et al. found that acute infusion of physiological levels of TMAO in mice heightens vascular inflammation (Figure 2), as supported by aortic endothelial cell activation (recovered by laser microdissection), including activation of mitogen-activated protein kinase (MAPK) signaling and nuclear factor (NF) kB nuclear translocation, leading to subsequent pro-inflammatory gene expression86. Complementary findings (TMAO stimulated vascular inflammation; Figure 2) have been observed in vitro using primary human aortic endothelial cells and vascular smooth muscle cells86. After an acute injection, mouse aortas also showed increased expression of vascular adhesion molecules, such as E selectin or intercellular cell adhesion molecule (ICAM)-1, even when TMAO had been cleared from circulation, implying sustained vascular inflammation. Further, TMAO was reported to increase oxidative stress and vascular senescence, which was characterized by impaired cell proliferation and migration in human umbilical vein endothelial cells128. In other animal model studies, gut microbiota suppression with oral poorly absorbed antibiotics was associated with decreased TMAO levels, improved endothelial function, reduced arterial stiffness, and lower oxidative stress129. Recent investigations have suggested TMAO can impact inflammation via priming and activation of the NLRP3 inflammasome in endothelial cells, as well as the arterial vascular wall in mice, involving mitochondrial reactive oxygen species (ROS) production, thioredoxin interacting protein (TXNIP) and lysosomal destabilization87, 130, 131. However, the exact mechanisms by which TMAO induces inflammasome activity have yet to be explored.
The receptor for TMA was identified long ago as a highly sensitive olfactory receptor called trace amine-associated receptor 5 (TAAR5)132. TAAR5 shows high specificity for TMA, and does not recognize TMAO. While TMA alone may contribute to pro-inflammatory signaling in the vasculature86, the role of TAAR5 in CVD requires further investigation. Recently, a receptor for TMAO has been reported and shown to participate in TMAO-dependent effects on glucose and insulin metabolism133. Chen et al. showed that TMAO directly binds to protein kinase R-like endoplasmic reticulum kinase (PERK), a main component of the unfolded protein response – a signaling pathway that adapts the cell to ER stress133 (Figure 1). Dietary supplementation of TMAO in C57BL/6J wildtype animals induced hepatic PERK expression, accompanied by increased Fox01, a key transcription factor in insulin singling and impaired glycemic control. Genetic manipulation demonstrated that the absence of hepatic PERK blunted the TMAO-induced increases in Fox01 expression and improved glycemic indices. Whether or not PERK plays a role in TMAO mediated effects on atherosclerosis or thrombosis remains unknown, and is an important area for further exploration. It is also interesting to note that ER stress is implicated in the pathogenesis of many CVD phenotypes. Whether TMAO and PERK participate in these associations also remains to be explored.
Theoretical benefits of TMAO
Due to its small size and combination of hydrophilic and hydrophobic properties, TMAO behaves as a chaotropic agent, with the ability to alter protein conformation and potentially serve as an allosteric modulator to proteins134. These features may have important physiological functions in the host (e.g. impacting the protein unfolding or ER stress response within cells). In some aquatic animals, including a subset of deep sea fish, TMAO is reported to act as an osmolyte, and to protect against pressure-induced protein destabilization135, 136. Indeed, some bony fish can use large amounts of TMAO for osmoregulation, and TMAO plasma levels within the fish increase with depth of the habitat, reaching levels up to 400mmol/kg in snailfish that were caught at 7000m depth in the South Pacific Ocean137. TMAO has been shown to stabilize proteins to elevated hydrostatic pressure, and is thought to both impact protein conformational changes, like those that occur with allosteric regulation, and modulate intracellular molecular crowding effects136, 138, 139. Thus, TMAO accumulation both in deep sea creatures and in mammals may represent an adaptive mechanism to impact protein stability and intracellular signaling processes.
Other studies have reported that TMAO is involved in the adaptive freeze avoidance response to extreme cold – in other words, it acts as antifreeze – such as within Newfoundland smelt140. These fish have the ability to elevate plasma osmolarity by seasonal accumulation of TMAO and other organic solutes, which depresses the freezing point of body fluids and allows them to survive at sub-freezing temperatures. It has recently been hypothesized that TMAO-induced protein stabilization may play a role to protect cardiomyocytes from hydrostatic pressure fluctuations during heart failure141. By contrast, high concentrations of TMAO may at times also impair function, as was reported for the activity of the actomyosin motor142.
TMAO may also be involved in tissue osmolality in vertebrates. When measuring concentrations in various mouse tissues, we observed that TMAO levels in kidney tissue largely exceeded those observed in corresponding paired plasma samples collected at the same time from the same animals (Hazen, unpublished data). In the kidneys, an osmotic gradient arising from the cortico-medullary boundary to the inner medullary tip is normally maintained as a mechanism that allows reabsorption of water and concentration of the urine, a process also called countercurrent multiplication143. It seems plausible that high levels of TMAO in the kidneys may function analogously to urea, playing a role in osmoregulation and renal function. Understanding the molecular participants involved in TMAO secretion and cellular transport in the kidneys and other tissues is an important potential area of future investigation, as it may reveal novel targets for therapeutic intervention. For example, one could theorize a small molecule drug that targeted renal TMAO transport/secretion might functioned as a diuretic, facilitating TMAO urinary excretion and both reduced blood pressure and cardiovascular disease risks.
Many studies have shown that high TMAO levels are associated with risk for thrombotic complications. However, these same properties may theoretically confer benefits to the host during situations with high bleeding risk, such as the delivery of a baby. It is thus interesting that one study reported TMAO levels may increase during pregnancy144. Studies by Bennett et al. found that human liver samples from female subjects showed higher FMO1/3 expression, and therefore an increased capacity for TMAO generation, than those from males. Likewise, gonadectomized male mice treated with testosterone show reduced hepatic FMO3 expression, while ovariectomized female mice treated with estrogen exhibit increases in hepatic FMO3 expression76. These results suggest that high levels of TMAO in the terminal stages of pregnancy may in theory better equip women to avoid severe blood loss during delivery. In addition, beyond FMO3 expression, microbial community alterations are known to occur during pregnancy145.
While purely speculative, evolutionary drive may have selected for metabolic changes in hosts that lead to harboring of gut commensal communities with the ability to produce TMA or other gut microbiota-generated pro-thrombotic metabolites, thus enabling hosts to better cope with environmental stressors that lead to hemorrhage (such as parturition). However, these features may have become detrimental for individuals living in modern western societies associated with a CVD-prone environment, and less need to survive traumatic injuries. Rodents show a significant sexual dimorphism with respect to FMO3 expression, and TMAO levels, with females showing higher levels (and greater atherosclerosis capacity9, 76). Although gene expression studies in humans have suggested gender differences in hepatic FMO3 expression, plasma levels of TMAO thus far reported have failed to show sex differences, and the prognostic value of TMAO appears to be similar in both men and women105. However, it should also be noted that virtually all clinical TMAO studies reported involve cohorts that are of postmenopausal age. A full exploration of sex specific differences TMAO levels in younger women warrants further investigation.
The metaorganismal metabolite phenylacetylglutamine (PAG) is both linked to CVD and acts via adrenergic receptors
The pathogenesis of type 2 diabetes goes beyond glycemic control, and traditional risk factors, including level of glucose control, poorly stratify CVD risk among diabetics. To investigate this, Nemet, Saha, Gupta, et al. used untargeted metabolomics to identify novel metabolites that associate with incident risk for MACE, are increased in type 2 diabetes subjects, and do not significantly correlate with glycemic control146. The candidate analyte showing the strongest association with MACE (m/z 265.1188) was subsequently identified as phenylacetylglutamine (PAG), a phenylalanine-derived metabolite (Figure 4). The association of PAGln with incident risk for major adverse cardiac events like heart attack, stroke and death was further validated in an independent and non-overlapping cohort comprised of 4000 stable cardiovascular subjects, and shown to be independent of traditional CVD risk factors in both diabetics and non-diabetics alike146. Additional functional studies revealed that the association of PAG with incident CVD risks likely occurs because the metabolite impacts host physiology, and fosters CVD related phenotypes. Moreover, PAG was shown to be generated via gut microbes during metabolism of phenylalanine, as illustrated in Figure 4. Genetic engineering studies in microbes coupled with transplantation into germ free mice confirmed gut microbial (and some of the gut microbial genes) contribution to host platelet reactivity and in vivo thrombosis potential. And through multiple gain of function and loss of function genetic and pharmacological studies, PAG was shown to interact with G protein-coupled receptors (GPCRs), including the α- and β-adrenergic receptors (ADRs)146. Adrenergic receptors are crucially involved in heart disease147 and platelet function148. However, until the discovery of PAG, ADR signaling had not yet been implicated in gut microbiota-derived factors driving CVD. The new studies by Nemet et al. also showed that adverse CVD related phenotypes observed with PAG administration at physiological levels was attenuated with the β-blocker carvedilol146.
Figure 4:
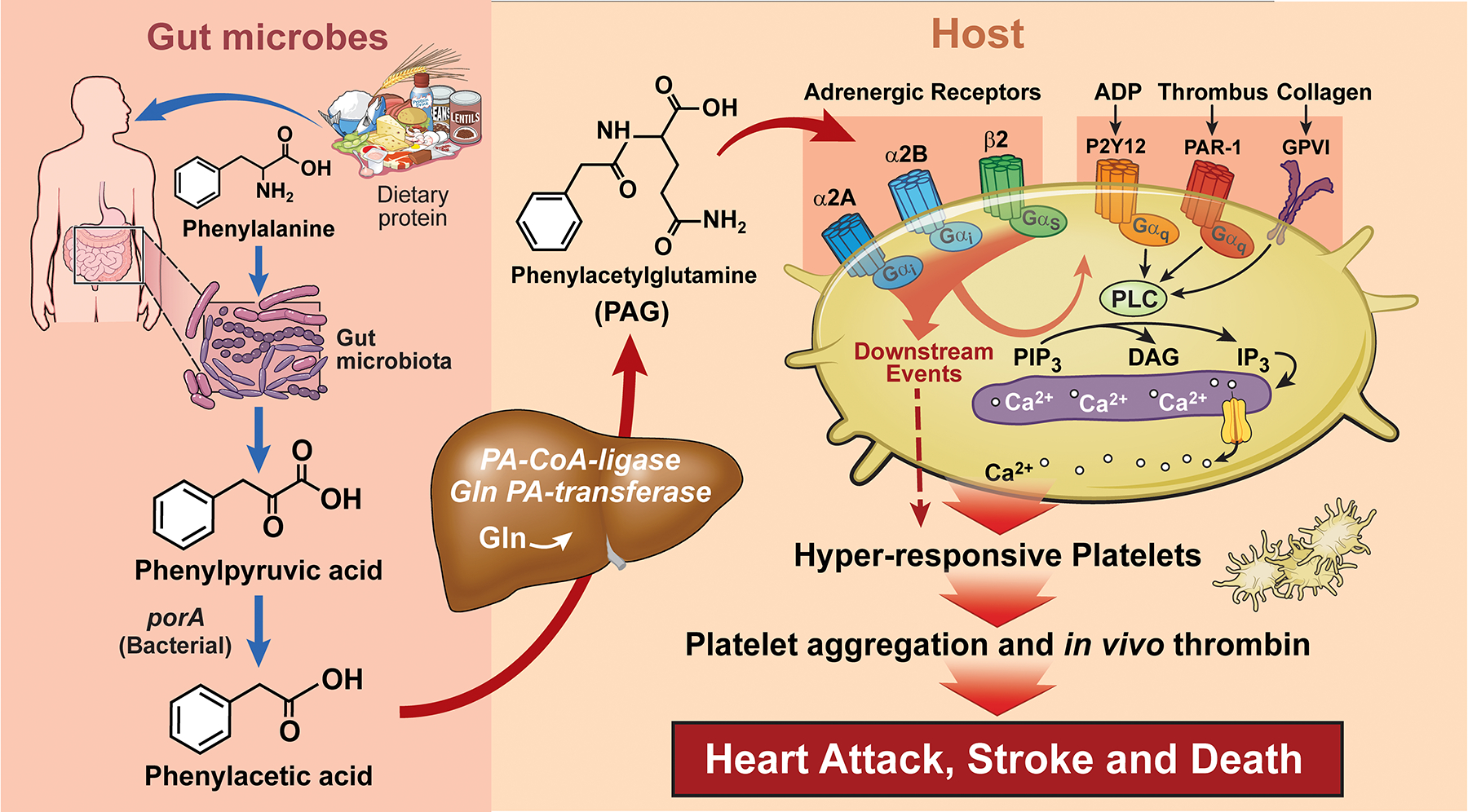
The metaorganismal metabolite phenylacetylglumatine (PAG) is derived from microbial metabolism of phenylalanine, and is involved in enhancement of platelet thrombotic potential via adrenergic receptors. ADP, adenosine diphosphate; α2A, α2A adrenergic receptor; α2B, α2B adrenergic receptor; β2, β2 adrenergic receptor; GPVI, Glycoprotein VI; IP3, inositol 1,4,5-triphosphate; P2Y12, purinergic receptor P2Y12,; PAR-1, Protease-activated receptor 1; PIP3, phosphatidylinositol trisphosphate, and PLC, phospholipase C.
It is striking that gut microbiota appear to elicit adverse cardiovascular phenotypes in the host via modulation of ADRs. Such a finding may help explain some of the beneficial effects of clinical beta blocker treatment. Interestingly, ADRs may also in turn modulate microbial abundance as observed in beta-adrenergic receptor knockout mice149. The novel discovery of a microbiota-ADR signaling axis is particularly interesting considering the widespread implication of ADRs in cardiovascular physiology and metabolism. There is no reason to assume that the subset of ADRs identified in this study are the only receptors modulated by PAG. It remains to be determined if other members of the ADR gene family, some of which are also known to participate in the regulation of cardiovascular homeostasis, are similarly modulated by PAG. It is intriguing to speculate that some phenotypes exhibited by microbiota are directly or indirectly mediated via ADRs. This is indirectly supported by the fact that absence of microbiota alters cardiovascular homeostasis, such as blood pressure regulation150, myocardial repair following post-infarction cardiac repair66, or thrombosis growth127 – functions that all involve ADRs. More studies are needed to further characterize PAG-mediated functions in the host. PAG also appears to represent another potential gut microbiome pharmacological target for future efforts151.
Drugging the microbiome
In response to accumulating evidence that the microbiota affects susceptibility for CVD and cardiometabolic diseases, researchers have begun to develop microbiota-directed interventions to improve clinical outcomes (Figure 3). The microbiome-host axis comprises many different layers, including dietary precursors, microbial communities, and metaorganismal pathways that generate bioactive metabolites recognized by host receptors – all of which represent potential therapeutic targets to modulate community output and host phenotype. Figure 3 illustrates several different therapeutic approaches for targeting the gut microbiome to exert a beneficial effect in the host. We have already mentioned the use of anti-microbial agents such as poorly absorbed antibiotics as a valuable tool for demonstrating involvement of gut microbiota in both animal models and humans9, but as a poor choice as a long-term therapeutic due to the development of antibiotic resistance. While many studies have reported associations between atherosclerotic plaque and the presence of pathogens such as cytomegalovirus, Chlamydia, and Helicobactor pylori152–155, multiple prospective randomized trials with antibiotics have thus far failed to demonstrate clinical benefit156–158. Further, the impact of antibiotic treatment on microbial communities is hard to predict, often because the microbial community that recolonizes after cessation of antibiotics can be variable, depending upon many factors, including the microbes one is exposed to as the antibiotics are metabolized and excreted. Currently, there is no clear evidence that antibiotics have efficacy in the treatment of CVD in humans. Use of antibiotics thus seems better suited to eradicating true pathogens, than as a chronic long-term preventive intervention.
In a recent study that screened over 1000 commonly prescribed non-antibiotic drugs for their impact on a broad selection of human gut commensals, nearly a quarter of the tested medications demonstrated antibiotic-like activity, significantly inhibiting the growth of at least one human commensal159. These results suggest that commonly prescribed medications may impact human gut microbial communities, thereby impacting host phenotypes though indirect effects mediated by changes in the gut microbiome. One well studied example of this is the widely-used anti-diabetic drug metformin. Recent studies by Bäckhed and colleagues show that some of the anti-diabetic effects of metformin therapy appear to be mediated by alterations in gut microbiota composition, since microbial transplantation studies reveal transmission of anti-diabetic effects in recipient germ free mice following fecal transfer from metformin treated donors160. In addition to the potential for common medications to impact gut microbial composition and function, thus potentially contributing to drug effects in the host, it is becoming increasingly clear that there is broad diversity in gut microbial metabolism of medications, leading to potential altered responsiveness to drugs. The diverse drug-microbiome interactions that vary between individuals are another area where further studies are needed, and shows promise for possible use in drug development efforts and personalized medicine161.
Defined microbial compositions (probiotics) and non-microbial substances that may alter microbial community structure (prebiotics) have also been proposed to improve CVD (Figure 3). Indeed, there are many preclinical and some clinical intervention studies using either prebiotics or probiotics that have shown promising results. However, these studies tend to be relatively small in size, and their adoption in clinical practice would require further study. The use of probiotics and prebiotics has recently been extensively reviewed, including discussions of the many promises and potential challenges of their development and use162–167. While a complete review of this topic is beyond the scope of the present review, below we discuss several preclinical studies involving probiotics, particularly where a mechanistic role for alteration in gut microbiome related processes to improved host phenotype were reported. One example includes a recent rodent hypertension study in which either a high-fiber diet (a prebiotic) or acetic acid alone (a gut microbial product) resulted in reduced blood pressure and adverse cardiac remodeling168. In a different mouse model involving Apoe E−/− mice on a n-3 polyunsaturated fatty acid (PUFA)-depleted diet for 12 weeks, supplementation of dietary inulin-type fructans were shown to reverse endothelial dysfunction in carotid arteries via activation of the nitric oxide (NO) synthase/NO pathway169. Beyond changes in vascular function, probiotic use in animal studies of heart failure have also shown promise. For example, administration of Lactobacillus rhamnosus GR-1 in rodents improved systolic and diastolic left ventricular function following coronary artery ligation170. This study was of interest because it was shown that while the GR-1 strain did not colonize to distal intestines where most anaerobes reside, beneficial cardiac remodeling was none-the-less observed. In another recent study, provision of Akkermansia muciniphila was associated with reduced aortic atherosclerosis in the hypercholesterolemic Apoe−/− mouse model28. This probiotic is of interest because of its use in human clinical investigations. For example, provision of Akkermansia muciniphila in a randomized placebo controlled double blinded interventional exploratory study was reported to promote a reduction in plasma LPS in obese individuals with metabolic syndrome29.
While prebiotic and probiotic interventions have shown ability to favorably alter metabolic profiles in some human studies, results are highly variable, and animal model findings have not yet been translated to evidence of clinical efficacy. It is still unclear whether most of the used microbes survive the acidity of the stomach, if they colonize the colon where most of the gut microbiota reside, and whether the beneficial effects are mediated by the ingested microbes, caused by shifts in the community structure, or possibly even caused by secondary effects on host immune education and function. One difficulty with probiotic studies where secondary effects on microbial communities likely plays a role in beneficial effects if observed is the vastness of the gut microbial community and the inter-individual differences in community structure. These factors are thought to give rise to variable responses to probiotics or prebiotics in general171. Thus, results of probiotic or prebiotic administration have been difficult to predict. Moreover, it is worth noting that the current selection of probiotics seems to be primarily driven by abundance-based analyses of microbiota composition, wherein microbial community members whose proportions are highly associated with beneficial phenotypes are the focus of interest. However, the keystone commensal organizing a community architecture, or providing a key gain of function as discussed above, can be a very low abundance component, and is often not easily detected by current conventional sequencing depths of analyses.
Perhaps the most obvious potential therapeutic intervention for targeting the gut microbiome is diet (Figure 3). The TMAO pathway is an excellent example of this, given the nutrient precursors are more abundant in a Western diet, and diets rich in phosphatidylcholine and carnitine are associated with heightened levels of TMAO, whereas vegetarian or vegan diets have reduced nutrient precursors111, 118. Notably, however, TMAO levels appear to be driven more so by the gut microbial community composition than by the dietary intervention, and significant variation in TMAO production among individuals on a given diet is observed14, 111. While a low choline or carnitine containing diet can be generated using a primarily vegetarian or vegan selection of food, and are rational recommendations for subjects with high TMAO levels, such dietary recommendations are harder to envision with other gut microbiota generated metabolites, where either the nutrient precursors are numerous (e.g. SCFA) or an essential amino acid (e.g. phenylalanine and PAG), so cannot be easily avoided. However, even with phenylalanine, there are dietary choices that can be made to reduce intake. For example, sufferers of the inborn error of metabolism called phenylketonuria (PKU) have a host enzyme deficiency that makes eating foods abundant in phenylalanine harmful (i.e. proteins). Consequently, throughout life, a phenylketonuric diet is highly recommended, which has an overall low phenylalanine content172. The impact of adopting a PKU diet in subjects with high PAG levels (e.g. subjects with diabetes, renal disease, etc) has yet to be examined, but is of considerable interest.
Recent work has examined the selective non-lethal targeting of gut microbial enzymes for TMA generation as a therapeutic approach for the treatment or prevention of CVD (Figure 3). Key to this approach is the development of a small molecule inhibitor that is “non-lethal” to the microbe, and thus does not trigger as great a selective pressure as an antibiotic for development of resistance. The first study of this type targeting the gut microbiome for the treatment of CVD used a choline structural analogue, 1,3 dimethylbutanol (DMB). Through a series of studies, DMB was shown to serve as a non-lethal microbial enzyme inhibitor of choline→TMA transformation, and to reduce TMAO production in vivo without affecting microbial fitness78. When fed to animals, DMB inhibited choline diet-dependent TMAO generation, reduced macrophage foam cell formation, and inhibited aortic atherosclerotic plaque development78. Next-generation choline TMA lyase inhibitors have since been developed that selectively target and accumulate in microbiota, thereby limiting systemic exposure in the host. The choline TMA lyase suicide substrate inhibitor fluoromethylcholine (FMC) was shown to be over 10,000 fold more potent an inhibitor of cutC than DMB, and markedly blocks microbial choline catabolism85. Interestingly, owing to their highly polar nature, FMC and related halomethylcholines were shown to be poorly absorbed into the host, limiting systemic exposure and thus chances of side effects. In addition, cutC inhibition by FMC and iodomethylcholine (IMC) was shown to result in microbial cytosolic choline elevation, which appears to be sensed as an abundant fuel source by the microbe. This then induces expression of the entire choline utilization gene cluster, including both cutC and the microbial choline transporter. This leads to active microbial uptake of FMC and the substrate of cutC, choline. A positive feedback loop is thus created, whereby the greater microbial catabolism of choline into TMA is inhibited, the greater the elevation in cytosolic choline within the gut microbe (sensed as an abundant fuel source), and the greater the sequestering of choline from the intestinal lumen into the inhibited gut microbe. As intestinal luminal choline is depleted, microbial community TMA production is globally inhibited, even from neighboring community members who might otherwise not be potently inhibited85.
Although human clinical studies with choline TMA lyase inhibitors have not yet been reported, numerous efforts are ongoing in this area. Like any drug, microbiota targeting non-lethal small molecule inhibitors will require the same sort of safety testing as any other drug. But with compounds that have reduced systemic absorption, there is a theoretical potential benefit of limiting adverse side effects from off target inhibitory activities in the host. Thus, pharmacological interventions aimed at “drugging the microbiome” with non-lethal small molecule inhibitors represents a novel therapeutic approach for both the treatment and prevention of cardiometabolic diseases that will need to be validated in clinical intervention studies.
Conclusions
Although our knowledge about how microbiota impact CVD is still rudimentary, the rate at which new discoveries are emerging is impressive. As outlined, there is overwhelming evidence that gut microbiota-derived processes in general are linked to numerous CVD relevant phenotypes, including but not limited to atherosclerosis, platelet reactivity and thrombosis potential, blood pressure, lipid metabolism, adiposity, glucose homeostasis, and vascular inflammation. Investigative approaches have included a wide array of microbiota transplantation studies, both animal and human dietary interventions in colonized versus antibiotic suppressed states (or germ-free mice), direct provision (dietary or via infusion) of specific gut microbiota metabolites, and both genetic and pharmacological studies that have targeted multiple components of metaorganismal pathways (including gut microbial genes, and both host transformational enzymes and end organ receptors). New therapeutic approaches that target gut microbes for the treatment and prevention of cardiovascular diseases represent exciting areas of investigation. The development of nonlethal microbial inhibitors that target specific pathways, yet show limited systemic exposure in the host, are just one of the new and potentially promising therapeutic approaches. Yet others include but are not limited to dietary interventions, probiotics, and/or prebiotics that hopefully can be used to someday “terraform” the microbial community to alter its functional output to the betterment of the host. As with any therapeutic, large prospective interventional studies will be needed to validate novel gut microbiome targeted therapeutics.
Supplementary Material
Acknowledgements
This work is supported by grants from the NIH and Office of Dietary Supplements (P01HL147823, R01HL103866), the German Research Foundation (WI 5229/1–1), and the Foundation Leducq. SLH reports being partially supported by a gift from the Krieger Fund.
Non-standard Abbreviations and Acronyms
- ACS
acute coronary syndrome
- ADP
adenosine diphosphate
- ADR
adrenergic receptor
- BA
bile acids
- CAD
coronary artery disease
- CKD
chronic kidney disease
- CVD
cardiovascular disease
- cut
choline utilization cluster
- DMB
dimethylbutanol
- FMC
fluoromethylcholine
- FMO
flavin monooxygenases
- FXR
farnesoid x receptor
- GPCR
G protein-coupled receptors
- GRP41
G-protein receptor 41
- ICAM
intercellular cell adhesion molecule
- IMC
iodomethylcholine
- LPS
lipopolysaccharide
- LXR
liver X receptor
- MACE
major adverse cardiac events
- MAPK
mitogen-activated protein kinase
- NFkB
nuclear factor NFkB
- NO
nitric oxide
- Olfr78
olfactory receptor 78
- PAD
peripheral artery disease
- PAG
phenylacetylglutamine
- PERK
protein kinase R-like endoplasmic reticulum kinase
- PKU
phenylketonuria
- PXR
pregnane X receptor
- RAAS
renin–angiotensin–aldosterone system
- ROS
reactive oxygen species
- SCFA
short chain fatty acids
- TAAR5
trace amine-associated receptor 5
- TF
tissue factor
- TLR
toll-like receptor
- TMA
trimethylamine
- TMAO
trimethylamine N-oxide
- TML
trimethyllysine
- VWF
von Willebrand factor
Footnotes
Conflict of Interest: SLH reports being named as co-inventor on pending and issued patents held by the Cleveland Clinic relating to cardiovascular diagnostics and therapeutics, being a paid consultant for P&G, having received research funds from P&G, and Roche Diagnostics, and being eligible to receive royalty payments for inventions or discoveries related to cardiovascular diagnostics or therapeutics from Cleveland HeartLab, Quest Diagnostics and P&G. The other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
References
- 1.Brown JM, Hazen SL. The gut microbial endocrine organ: Bacterially derived signals driving cardiometabolic diseases. Annu Rev Med. 2015;66:343–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;101:15718–15723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102:11070–11075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031 [DOI] [PubMed] [Google Scholar]
- 5.Cho I, Yamanishi S, Cox L, Methe BA, Zavadil J, Li K, Gao Z, Mahana D, Raju K, Teitler I, Li H, Alekseyenko AV, Blaser MJ. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature. 2012;488:621–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koren O, Spor A, Felin J, Fak F, Stombaugh J, Tremaroli V, Behre CJ, Knight R, Fagerberg B, Ley RE, Backhed F. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc Natl Acad Sci U S A. 2011;108 Suppl 1:4592–4598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, Smith JD, DiDonato JA, Chen J, Li H, Wu GD, Lewis JD, Warrier M, Brown JM, Krauss RM, Tang WH, Bushman FD, Lusis AJ, Hazen SL. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, Hazen SL. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368:1575–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, Wu Y, Schauer P, Smith JD, Allayee H, Tang WH, DiDonato JA, Lusis AJ, Hazen SL. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang T, Santisteban MM, Rodriguez V, Li E, Ahmari N, Carvajal JM, Zadeh M, Gong M, Qi Y, Zubcevic J, Sahay B, Pepine CJ, Raizada MK, Mohamadzadeh M. Gut dysbiosis is linked to hypertension. Hypertension. 2015;65:1331–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto JM, Kennedy S, Leonard P, Li J, Burgdorf K, Grarup N, Jorgensen T, Brandslund I, Nielsen HB, Juncker AS, Bertalan M, Levenez F, Pons N, Rasmussen S, Sunagawa S, Tap J, Tims S, Zoetendal EG, Brunak S, Clement K, Dore J, Kleerebezem M, Kristiansen K, Renault P, Sicheritz-Ponten T, de Vos WM, Zucker JD, Raes J, Hansen T, Bork P, Wang J, Ehrlich SD, Pedersen O. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500:541–546 [DOI] [PubMed] [Google Scholar]
- 12.Craciun S, Balskus EP. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc Natl Acad Sci U S A. 2012;109:21307–21312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Craciun S, Marks JA, Balskus EP. Characterization of choline trimethylamine-lyase expands the chemistry of glycyl radical enzymes. ACS Chem Biol. 2014;9:1408–1413 [DOI] [PubMed] [Google Scholar]
- 14.Skye SM, Zhu W, Romano KA, Guo CJ, Wang Z, Jia X, Kirsop J, Haag B, Lang JM, DiDonato JA, Tang WHW, Lusis AJ, Rey FE, Fischbach MA, Hazen SL. Microbial transplantation with human gut commensals containing cutc is sufficient to transmit enhanced platelet reactivity and thrombosis potential. Circ Res. 2018;123:1164–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Donia MS, Cimermancic P, Schulze CJ, Wieland Brown LC, Martin J, Mitreva M, Clardy J, Linington RG, Fischbach MA. A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell. 2014;158:1402–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patnode ML, Beller ZW, Han ND, Cheng J, Peters SL, Terrapon N, Henrissat B, Le Gall S, Saulnier L, Hayashi DK, Meynier A, Vinoy S, Giannone RJ, Hettich RL, Gordon JI. Interspecies competition impacts targeted manipulation of human gut bacteria by fiber-derived glycans. Cell. 2019;179:59–73.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hooks KB, O’Malley MA. Dysbiosis and its discontents. mBio 2017;8:e01492–01417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Polsinelli VB, Sinha A, Shah SJ. Visceral congestion in heart failure: Right ventricular dysfunction, splanchnic hemodynamics, and the intestinal microenvironment. Curr Heart Fail Rep. 2017;14:519–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang WHW, Li DY, Hazen SL. Dietary metabolism, the gut microbiome, and heart failure. Nat Rev Cardiol. 2019;16:137–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Munger MA, Johnson B, Amber IJ, Callahan KS, Gilbert EM. Circulating concentrations of proinflammatory cytokines in mild or moderate heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol. 1996;77:723–727 [DOI] [PubMed] [Google Scholar]
- 21.Rauchhaus M, Doehner W, Francis DP, Davos C, Kemp M, Liebenthal C, Niebauer J, Hooper J, Volk HD, Coats AJ, Anker SD. Plasma cytokine parameters and mortality in patients with chronic heart failure. Circulation. 2000;102:3060–3067 [DOI] [PubMed] [Google Scholar]
- 22.Fudim M, Hernandez AF, Felker GM. Role of volume redistribution in the congestion of heart failure. J Am Heart Assoc. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sandek A, Bauditz J, Swidsinski A, Buhner S, Weber-Eibel J, von Haehling S, Schroedl W, Karhausen T, Doehner W, Rauchhaus M, Poole-Wilson P, Volk HD, Lochs H, Anker SD. Altered intestinal function in patients with chronic heart failure. J Am Coll Cardiol. 2007;50:1561–1569 [DOI] [PubMed] [Google Scholar]
- 24.Hug H, Mohajeri MH, La Fata G. Toll-like receptors: Regulators of the immune response in the human gut. Nutrients. 2018;10:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niebauer J, Volk HD, Kemp M, Dominguez M, Schumann RR, Rauchhaus M, Poole-Wilson PA, Coats AJ, Anker SD. Endotoxin and immune activation in chronic heart failure: A prospective cohort study. Lancet. 1999;353:1838–1842 [DOI] [PubMed] [Google Scholar]
- 26.Peschel T, Schonauer M, Thiele H, Anker SD, Schuler G, Niebauer J. Invasive assessment of bacterial endotoxin and inflammatory cytokines in patients with acute heart failure. Eur J Heart Fail. 2003;5:609–614 [DOI] [PubMed] [Google Scholar]
- 27.Pastori D, Carnevale R, Nocella C, Novo M, Santulli M, Cammisotto V, Menichelli D, Pignatelli P, Violi F. Gut-derived serum lipopolysaccharide is associated with enhanced risk of major adverse cardiovascular events in atrial fibrillation: Effect of adherence to Mediterranean diet. J Am Heart Assoc. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J, Lin S, Vanhoutte PM, Woo CW, Xu A. Akkermansia muciniphila protects against atherosclerosis by preventing metabolic endotoxemia-induced inflammation in apoe−/− mice. Circulation. 2016;133:2434–2446 [DOI] [PubMed] [Google Scholar]
- 29.Depommier C, Everard A, Druart C, Plovier H, Van Hul M, Vieira-Silva S, Falony G, Raes J, Maiter D, Delzenne NM, de Barsy M, Loumaye A, Hermans MP, Thissen JP, de Vos WM, Cani PD. Supplementation with akkermansia muciniphila in overweight and obese human volunteers: A proof-of-concept exploratory study. Nat Med. 2019;25:1096–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vatanen T, Kostic AD, d’Hennezel E, Siljander H, Franzosa EA, Yassour M, Kolde R, Vlamakis H, Arthur TD, Hämäläinen AM, Peet A, Tillmann V, Uibo R, Mokurov S, Dorshakova N, Ilonen J, Virtanen SM, Szabo SJ, Porter JA, Lähdesmäki H, Huttenhower C, Gevers D, Cullen TW, Knip M, Xavier RJ. Variation in microbiome lps immunogenicity contributes to autoimmunity in humans. Cell. 2016;165:842–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoshida N, Emoto T, Yamashita T, Watanabe H, Hayashi T, Tabata T, Hoshi N, Hatano N, Ozawa G, Sasaki N, Mizoguchi T, Amin HZ, Hirota Y, Ogawa W, Yamada T, Hirata KI. Bacteroides vulgatus and bacteroides dorei reduce gut microbial lipopolysaccharide production and inhibit atherosclerosis. Circulation. 2018;138:2486–2498 [DOI] [PubMed] [Google Scholar]
- 32.Lawler PR, Bhatt DL, Godoy LC, Luscher TF, Bonow RO, Verma S, Ridker PM. Targeting cardiovascular inflammation: Next steps in clinical translation. Eur Heart J. 2020 [DOI] [PubMed] [Google Scholar]
- 33.Geovanini GR, Libby P. Atherosclerosis and inflammation: Overview and updates. Clin Sci (Lond) 2018;132:1243–1252 [DOI] [PubMed] [Google Scholar]
- 34.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131 [DOI] [PubMed] [Google Scholar]
- 35.Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Bile acids and the gut microbiome. Curr Opin Gastroenterol. 2014;30:332–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kemis JH, Linke V, Barrett KL, Boehm FJ, Traeger LL, Keller MP, Rabaglia ME, Schueler KL, Stapleton DS, Gatti DM, Churchill GA, Amador-Noguez D, Russell JD, Yandell BS, Broman KW, Coon JJ, Attie AD, Rey FE. Genetic determinants of gut microbiota composition and bile acid profiles in mice. PLoS Genetics. 2019;15:e1008073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, Yu RT, Shelton JM, Richardson JA, Repa JJ, Mangelsdorf DJ, Kliewer SA. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A. 2006;103:3920–3925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parseus A, Sommer N, Sommer F, Caesar R, Molinaro A, Stahlman M, Greiner TU, Perkins R, Backhed F. Microbiota-induced obesity requires farnesoid x receptor. Gut. 2017;66:429–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clements WD, Parks R, Erwin P, Halliday MI, Barr J, Rowlands BJ. Role of the gut in the pathophysiology of extrahepatic biliary obstruction. Gut. 1996;39:587–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hylemon PB, Zhou H, Pandak WM, Ren S, Gil G, Dent P. Bile acids as regulatory molecules. J Lipid Res. 2009;50:1509–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang B, Tontonoz P. Liver x receptors in lipid signalling and membrane homeostasis. Nat Rev Endocrinol. 2018;14:452–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou H, Hylemon PB. Bile acids are nutrient signaling hormones. Steroids. 2014;86:62–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chavez-Talavera O, Tailleux A, Lefebvre P, Staels B. Bile acid control of metabolism and inflammation in obesity, type 2 diabetes, dyslipidemia, and nonalcoholic fatty liver disease. Gastroenterology. 2017;152:1679–1694.e1673 [DOI] [PubMed] [Google Scholar]
- 44.Keitel V, Stindt J, Haussinger D. Bile acid-activated receptors: Gpbar1 (tgr5) and other g protein-coupled receptors. Handb Exp Pharmacol. 2019;256:19–49 [DOI] [PubMed] [Google Scholar]
- 45.Haeusler RA, Astiarraga B, Camastra S, Accili D, Ferrannini E. Human insulin resistance is associated with increased plasma levels of 12alpha-hydroxylated bile acids. Diabetes. 2013;62:4184–4191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choucair I, Nemet I, Li L, Cole MA, Skye SM, Kirsop JD, Fischbach MA, Gogonea V, Brown JM, Tang WHW, Hazen SL. Quantification of bile acids: A mass spectrometry platform for studying gut microbe connection to metabolic diseases. J Lipid Res. 2020;61:159–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gu Y, Wang X, Li J, Zhang Y, Zhong H, Liu R, Zhang D, Feng Q, Xie X, Hong J, Ren H, Liu W, Ma J, Su Q, Zhang H, Yang J, Wang X, Zhao X, Gu W, Bi Y, Peng Y, Xu X, Xia H, Li F, Xu X, Yang H, Xu G, Madsen L, Kristiansen K, Ning G, Wang W. Analyses of gut microbiota and plasma bile acids enable stratification of patients for antidiabetic treatment. Nat Commun. 2017;8:1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bose S, Ramesh V, Locasale JW. Acetate metabolism in physiology, cancer, and beyond. Trends Cell Biol. 2019;29:695–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morrison DJ, Preston T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes. 2016;7:189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deehan EC, Yang C, Perez-Munoz ME, Nguyen NK, Cheng CC, Triador L, Zhang Z, Bakal JA, Walter J. Precision microbiome modulation with discrete dietary fiber structures directs short-chain fatty acid production. Cell Host Microbe. 2020;27:389–404.e386 [DOI] [PubMed] [Google Scholar]
- 51.Perry RJ, Peng L, Barry NA, Cline GW, Zhang D, Cardone RL, Petersen KF, Kibbey RG, Goodman AL, Shulman GI. Acetate mediates a microbiome-brain-beta-cell axis to promote metabolic syndrome. Nature. 2016;534:213–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koh A, De Vadder F, Kovatcheva-Datchary P, Backhed F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell. 2016;165:1332–1345 [DOI] [PubMed] [Google Scholar]
- 53.Nobel YR, Cox LM, Kirigin FF, Bokulich NA, Yamanishi S, Teitler I, Chung J, Sohn J, Barber CM, Goldfarb DS, Raju K, Abubucker S, Zhou Y, Ruiz VE, Li H, Mitreva M, Alekseyenko AV, Weinstock GM, Sodergren E, Blaser MJ. Metabolic and metagenomic outcomes from early-life pulsed antibiotic treatment. Nat Commun. 2015;6:7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ruiz VE, Battaglia T, Kurtz ZD, Bijnens L, Ou A, Engstrand I, Zheng X, Iizumi T, Mullins BJ, Müller CL, Cadwell K, Bonneau R, Perez-Perez GI, Blaser MJ. A single early-in-life macrolide course has lasting effects on murine microbial network topology and immunity. Nat Commun. 2017;8:518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang XS, Li J, Krautkramer KA, Badri M, Battaglia T, Borbet TC, Koh H, Ng S, Sibley RA, Li Y, Pathmasiri W, Jindal S, Shields-Cutler RR, Hillmann B, Al-Ghalith GA, Ruiz VE, Livanos A, van ‘t Wout AB, Nagalingam N, Rogers AB, Sumner SJ, Knights D, Denu JM, Li H, Ruggles KV, Bonneau R, Williamson RA, Rauch M, Blaser MJ. Antibiotic-induced acceleration of type 1 diabetes alters maturation of innate intestinal immunity. Elife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Livanos AE, Greiner TU, Vangay P, Pathmasiri W, Stewart D, McRitchie S, Li H, Chung J, Sohn J, Kim S, Gao Z, Barber C, Kim J, Ng S, Rogers AB, Sumner S, Zhang XS, Cadwell K, Knights D, Alekseyenko A, Bäckhed F, Blaser MJ. Antibiotic-mediated gut microbiome perturbation accelerates development of type 1 diabetes in mice. Nat Microbiol. 2016;1:16140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schulfer AF, Battaglia T, Alvarez Y, Bijnens L, Ruiz VE, Ho M, Robinson S, Ward T, Cox LM, Rogers AB, Knights D, Sartor RB, Blaser MJ. Intergenerational transfer of antibiotic-perturbed microbiota enhances colitis in susceptible mice. Nat Microbiol. 2018;3:234–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Whelton SP, Hyre AD, Pedersen B, Yi Y, Whelton PK, He J. Effect of dietary fiber intake on blood pressure: A meta-analysis of randomized, controlled clinical trials. J Hypertens. 2005;23:475–481 [DOI] [PubMed] [Google Scholar]
- 59.Pluznick J A novel scfa receptor, the microbiota, and blood pressure regulation. Gut Microbes. 2014;5:202–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pluznick JL, Protzko RJ, Gevorgyan H, Peterlin Z, Sipos A, Han J, Brunet I, Wan LX, Rey F, Wang T, Firestein SJ, Yanagisawa M, Gordon JI, Eichmann A, Peti-Peterdi J, Caplan MJ. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci U S A. 2013;110:4410–4415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Natarajan N, Hori D, Flavahan S, Steppan J, Flavahan NA, Berkowitz DE, Pluznick JL. Microbial short chain fatty acid metabolites lower blood pressure via endothelial g protein-coupled receptor 41. Physiol Genomics. 2016;48:826–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Poll B, Steppan J, Lester L, Berkowitz D, Pluznick J. A short chain fatty acid produced by the gut microbiota plays a role in blood pressure regulation and cardiac contractility. FASEB J. 2019;33:569.519–569.519 [Google Scholar]
- 63.Li J, Zhao F, Wang Y, Chen J, Tao J, Tian G, Wu S, Liu W, Cui Q, Geng B, Zhang W, Weldon R, Auguste K, Yang L, Liu X, Chen L, Yang X, Zhu B, Cai J. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome. 2017;5:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mell B, Jala VR, Mathew AV, Byun J, Waghulde H, Zhang Y, Haribabu B, Vijay-Kumar M, Pennathur S, Joe B. Evidence for a link between gut microbiota and hypertension in the dahl rat. Physiol Genomics. 2015;47:187–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bartolomaeus H, Balogh A, Yakoub M, Homann S, Marko L, Hoges S, Tsvetkov D, Krannich A, Wundersitz S, Avery EG, Haase N, Kraker K, Hering L, Maase M, Kusche-Vihrog K, Grandoch M, Fielitz J, Kempa S, Gollasch M, Zhumadilov Z, Kozhakhmetov S, Kushugulova A, Eckardt KU, Dechend R, Rump LC, Forslund SK, Muller DN, Stegbauer J, Wilck N. Short-chain fatty acid propionate protects from hypertensive cardiovascular damage. Circulation. 2019;139:1407–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tang TWH, Chen HC, Chen CY, Yen CYT, Lin CJ, Prajnamitra RP, Chen LL, Ruan SC, Lin JH, Lin PJ, Lu HH, Kuo CW, Chang CM, Hall AD, Vivas EI, Shui JW, Chen P, Hacker TA, Rey FE, Kamp TJ, Hsieh PCH. Loss of gut microbiota alters immune system composition and cripples postinfarction cardiac repair. Circulation. 2019;139:647–659 [DOI] [PubMed] [Google Scholar]
- 67.Battson ML, Lee DM, Li Puma LC, Ecton KE, Thomas KN, Febvre HP, Chicco AJ, Weir TL, Gentile CL. Gut microbiota regulates cardiac ischemic tolerance and aortic stiffness in obesity. Am J Physiol Heart Circ Physiol. 2019;317:H1210–h1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cheema MU, Pluznick JL. Gut microbiota plays a central role to modulate the plasma and fecal metabolomes in response to angiotensin ii. Hypertension. 2019;74:184–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sun C-Y, Chang S-C, Wu M-S. Uremic toxins induce kidney fibrosis by activating intrarenal renin-angiotensin-aldosterone system associated epithelial-to-mesenchymal transition. PloS One. 2012;7:e34026–e34026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Itoh Y, Ezawa A, Kikuchi K, Tsuruta Y, Niwa T. Protein-bound uremic toxins in hemodialysis patients measured by liquid chromatography/tandem mass spectrometry and their effects on endothelial ros production. Anal Bioanal Chem. 2012;403:1841–1850 [DOI] [PubMed] [Google Scholar]
- 71.Gryp T, Vanholder R, Vaneechoutte M, Glorieux G. P-cresyl sulfate. Toxins (Basel). 2017;9:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sallée M, Dou L, Cerini C, Poitevin S, Brunet P, Burtey S. The aryl hydrocarbon receptor-activating effect of uremic toxins from tryptophan metabolism: A new concept to understand cardiovascular complications of chronic kidney disease. Toxins (Basel). 2014;6:934–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rothhammer V, Mascanfroni ID, Bunse L, Takenaka MC, Kenison JE, Mayo L, Chao CC, Patel B, Yan R, Blain M, Alvarez JI, Kébir H, Anandasabapathy N, Izquierdo G, Jung S, Obholzer N, Pochet N, Clish CB, Prinz M, Prat A, Antel J, Quintana FJ. Type i interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med. 2016;22:586–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Koeth RA, Levison BS, Culley MK, Buffa JA, Wang Z, Gregory JC, Org E, Wu Y, Li L, Smith JD, Tang WHW, DiDonato JA, Lusis AJ, Hazen SL. Gamma-butyrobetaine is a proatherogenic intermediate in gut microbial metabolism of l-carnitine to tmao. Cell Metab. 2014;20:799–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li XS, Wang Z, Cajka T, Buffa JA, Nemet I, Hurd AG, Gu X, Skye SM, Roberts AB, Wu Y, Li L, Shahen CJ, Wagner MA, Hartiala JA, Kerby RL, Romano KA, Han Y, Obeid S, Luscher TF, Allayee H, Rey FE, DiDonato JA, Fiehn O, Tang WHW, Hazen SL. Untargeted metabolomics identifies trimethyllysine, a tmao-producing nutrient precursor, as a predictor of incident cardiovascular disease risk. JCI Insight. 2018;3:e99096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bennett BJ, de Aguiar Vallim TQ, Wang Z, Shih DM, Meng Y, Gregory J, Allayee H, Lee R, Graham M, Crooke R, Edwards PA, Hazen SL, Lusis AJ. Trimethylamine-n-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013;17:49–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Al-Waiz M, Mitchell SC, Idle JR, Smith RL. The metabolism of 14c-labelled trimethylamine and its n-oxide in man. Xenobiotica. 1987;17:551–558 [DOI] [PubMed] [Google Scholar]
- 78.Wang Z, Roberts AB, Buffa JA, Levison BS, Zhu W, Org E, Gu X, Huang Y, FZamanian-Daryoush M, Culley MK, DiDonato AJ, Fu X, Hazen JE, Krajcik D, DiDonato JA, Lusis AJ, Hazen SL. Non-lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis. Cell. 2015;163:1585–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wu P, Chen J, Chen J, Tao J, Wu S, Xu G, Wang Z, Wei D, Yin W. Trimethylamine n-oxide promotes apoe(−/−) mice atherosclerosis by inducing vascular endothelial cell pyroptosis via the sdhb/ros pathway. J Cell Physiol. 2020; Online ahead of print [DOI] [PubMed] [Google Scholar]
- 80.Ding L, Chang M, Guo Y, Zhang L, Xue C, Yanagita T, Zhang T, Wang Y. Trimethylamine-n-oxide (tmao)-induced atherosclerosis is associated with bile acid metabolism. Lipids Health Dis. 2018;17:286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aldana-Hernandez P, Leonard KA, Zhao YY, Curtis JM, Field CJ, Jacobs RL. Dietary choline or trimethylamine n-oxide supplementation does not influence atherosclerosis development in ldlr−/− and apoe−/− male mice. J Nutr. 2020;150:249–255 [DOI] [PubMed] [Google Scholar]
- 82.Xue J, Zhou D, Poulsen O, Imamura T, Hsiao YH, Smith TH, Malhotra A, Dorrestein P, Knight R, Haddad GG. Intermittent hypoxia and hypercapnia accelerate atherosclerosis, partially via trimethylamine-oxide. Am J Respir Cell Mol Biol. 2017;57:581–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, Li L, Fu X, Wu Y, Mehrabian M, Sartor RB, McIntyre TM, Silverstein RL, Tang WHW, DiDonato JA, Brown JM, Lusis AJ, Hazen SL. Gut microbial metabolite tmao enhances platelet hyperreactivity and thrombosis risk. Cell. 2016;165:111–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhu W, Buffa JA, Wang Z, Warrier M, Schugar R, Shih DM, Gupta N, Gregory JC, Org E, Fu X, Li L, DiDonato JA, Lusis AJ, Brown JM, Hazen SL. Flavin monooxygenase 3, the host hepatic enzyme in the metaorganismal trimethylamine n-oxide-generating pathway, modulates platelet responsiveness and thrombosis risk. J Thromb Haemost. 2018;16:1857–1872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Roberts AB, Gu X, Buffa JA, Hurd AG, Wang Z, Zhu W, Gupta N, Skye SM, Cody DB, Levison BS, Barrington WT, Russell MW, Reed JM, Duzan A, Lang JM, Fu X, Li L, Myers AJ, Rachakonda S, DiDonato JA, Brown JM, Gogonea V, Lusis AJ, Garcia-Garcia JC, Hazen SL. Development of a gut microbe-targeted nonlethal therapeutic to inhibit thrombosis potential. Nat Med. 2018;24:1407–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Seldin MM, Meng Y, Qi H, Zhu W, Wang Z, Hazen SL, Lusis AJ, Shih DM. Trimethylamine n-oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor-κB. J Am Heart Assoc. 2016;5:e002767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen ML, Zhu XH, Ran L, Lang HD, Yi L, Mi MT. Trimethylamine-N-oxide induces vascular inflammation by activating the NLRP3 inflammasome through the SIRT3-SOD2-mtROS signaling pathway. J Am Heart Assoc. 2017;6:e002238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang X, Li Y, Yang P, Liu X, Lu L, Chen Y, Zhong X, Li Z, Liu H, Ou C, Yan J, Chen M. Trimethylamine-N-oxide promotes vascular calcification through activation of NLRP3 (nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3) inflammasome and NF-κB (nuclear factor kappab) signals. Arterioscler Thromb Vasc Biol. 2020;40:751–765 [DOI] [PubMed] [Google Scholar]
- 89.Wang G, Kong B, Shuai W, Fu H, Jiang X, Huang H. 3,3-dimethyl-1-butanol attenuates cardiac remodeling in pressure-overload-induced heart failure mice. J Nutr Biochem. 2020;78:108341. [DOI] [PubMed] [Google Scholar]
- 90.Jin B, Ji F, Zuo A, Liu H, Qi L, He Y, Wang Q, Zhao P. Destructive role of TMAO in t-tubule and excitation-contraction coupling in the adult cardiomyocytes. Int Heart J. 2020;61:355–363 [DOI] [PubMed] [Google Scholar]
- 91.Organ CL, Otsuka H, Bhushan S, Wang Z, Bradley J, Trivedi R, Polhemus DJ, Tang WH, Wu Y, Hazen SL, Lefer DJ. Choline diet and its gut microbe-derived metabolite, trimethylamine n-oxide, exacerbate pressure overload-induced heart failure. Circ Heart Fail. 2016;9:e002314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gupta N, Buffa JA, Roberts AB, Sangwan N, Skye SM, Li L, Ho KJ, Varga J, DiDonato JA, Tang WHW, Hazen SL. Targeted inhibition of gut microbial tmao production reduces renal tubulointerstitial fibrosis and functional impairment in a murine model of chronic kidney disease. Arterioscler Thromb Vasc Biol. 2020;40:1239–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nanto-Hara F, Kanemitsu Y, Fukuda S, Kikuchi K, Asaji K, Saigusa D, Iwasaki T, Ho HJ, Mishima E, Suzuki T, Suzuki C, Tsukimi T, Matsuhashi T, Oikawa Y, Akiyama Y, Kure S, Owada Y, Tomioka Y, Soga T, Ito S, Abe T. The guanylate cyclase c agonist linaclotide ameliorates the gut-cardio-renal axis in an adenine-induced mouse model of chronic kidney disease. Nephrol Dial Transplant. 2020;35:250–264 [DOI] [PubMed] [Google Scholar]
- 94.Sun G, Yin Z, Liu N, Bian X, Yu R, Su X, Zhang B, Wang Y. Gut microbial metabolite tmao contributes to renal dysfunction in a mouse model of diet-induced obesity. Biochem Biophys Res Commun. 2017;493:964–970 [DOI] [PubMed] [Google Scholar]
- 95.Tang WH, Wang Z, Kennedy DJ, Wu Y, Buffa JA, Agatisa-Boyle B, Li XS, Levison BS, Hazen SL. Gut microbiota-dependent trimethylamine n-oxide (tmao) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res. 2015;116:448–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Senthong V, Wang Z, Fan Y, Wu Y, Hazen SL, Tang WH. Trimethylamine N-oxide and mortality risk in patients with peripheral artery disease. J Am Heart Assoc. 2016;5:e004237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Senthong V, Li XS, Hudec T, Coughlin J, Wu Y, Levison B, Wang Z, Hazen SL, Tang WH. Plasma trimethylamine n-oxide, a gut microbe-generated phosphatidylcholine metabolite, is associated with atherosclerotic burden. J Am Coll Cardiol. 2016;67:2620–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tan Y, Sheng Z, Zhou P, Liu C, Zhao H, Song L, Li J, Zhou J, Chen Y, Wang L, Qian H, Sun Z, Qiao S, Xu B, Gao R, Yan H. Plasma trimethylamine N-oxide as a novel biomarker for plaque rupture in patients with ST-segment-elevation myocardial infarction. Circ Cardiovasc Interv. 2019;12:e007281. [DOI] [PubMed] [Google Scholar]
- 99.Li XS, Obeid S, Klingenberg R, Gencer B, Mach F, Raber L, Windecker S, Rodondi N, Nanchen D, Muller O, Miranda MX, Matter CM, Wu Y, Li L, Wang Z, Alamri HS, Gogonea V, Chung YM, Tang WH, Hazen SL, Luscher TF. Gut microbiota-dependent trimethylamine N-oxide in acute coronary syndromes: A prognostic marker for incident cardiovascular events beyond traditional risk factors. Eur Heart J. 2017;38:814–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lever M, George PM, Slow S, Bellamy D, Young JM, Ho M, McEntyre CJ, Elmslie JL, Atkinson W, Molyneux SL, Troughton RW, Frampton CM, Richards AM, Chambers ST. Betaine and trimethylamine-N-oxide as predictors of cardiovascular outcomes show different patterns in diabetes mellitus: An observational study. PLoS One. 2014;9:e114969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Suzuki T, Heaney LM, Bhandari SS, Jones DJ, Ng LL. Trimethylamine n-oxide and prognosis in acute heart failure. Heart. 2016;102:841–848 [DOI] [PubMed] [Google Scholar]
- 102.Troseid M, Ueland T, Hov JR, Svardal A, Gregersen I, Dahl CP, Aakhus S, Gude E, Bjorndal B, Halvorsen B, Karlsen TH, Aukrust P, Gullestad L, Berge RK, Yndestad A. Microbiota-dependent metabolite trimethylamine-n-oxide is associated with disease severity and survival of patients with chronic heart failure. J Intern Med. 2015;277:717–726 [DOI] [PubMed] [Google Scholar]
- 103.Tang WH, Wang Z, Shrestha K, Borowski AG, Wu Y, Troughton RW, Klein AL, Hazen SL. Intestinal microbiota-dependent phosphatidylcholine metabolites, diastolic dysfunction, and adverse clinical outcomes in chronic systolic heart failure. J Card Fail. 2015;21:91–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tang WH, Wang Z, Fan Y, Levison B, Hazen JE, Donahue LM, Wu Y, Hazen SL. Prognostic value of elevated levels of intestinal microbe-generated metabolite trimethylamine-n-oxide in patients with heart failure: Refining the gut hypothesis. J Am Coll Cardiol. 2014;64:1908–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Schiattarella GG, Sannino A, Toscano E, Giugliano G, Gargiulo G, Franzone A, Trimarco B, Esposito G, Perrino C. Gut microbe-generated metabolite trimethylamine-n-oxide as cardiovascular risk biomarker: A systematic review and dose-response meta-analysis. Eur Heart J. 2017;38:2948–2956 [DOI] [PubMed] [Google Scholar]
- 106.Qi J, You T, Li J, Pan T, Xiang L, Han Y, Zhu L. Circulating trimethylamine n-oxide and the risk of cardiovascular diseases: A systematic review and meta-analysis of 11 prospective cohort studies. J Cell Mol Med. 2018;22:185–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Heianza Y, Ma W, Manson JE, Rexrode KM, Qi L. Gut microbiota metabolites and risk of major adverse cardiovascular disease events and death: A systematic review and meta-analysis of prospective studies. J Am Heart Assoc. 2017;6:e004947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tang WHW, Backhed F, Landmesser U, Hazen SL. Intestinal microbiota in cardiovascular health and disease: Jacc state-of-the-art review. J Am Coll Cardiol. 2019;73:2089–2105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zempleni J, Suttie JW, Gregory JF 3rd, Stover PJ, editors. Handbook of vitamins. CRC Press; 2013. p. 492. [Google Scholar]
- 110.Koeth RA, Lam-Galvez BR, Kirsop J, Wang Z, Levison BS, Gu X, Copeland MF, Bartlett D, Cody DB, Dai HJ, Culley MK, Li XS, Fu X, Wu Y, Li L, DiDonato JA, Tang WHW, Garcia-Garcia JC, Hazen SL. L-carnitine in omnivorous diets induces an atherogenic gut microbial pathway in humans. J Clin Invest. 2019;129:373–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang Z, Bergeron N, Levison BS, Li XS, Chiu S, Jia X, Koeth RA, Li L, Wu Y, Tang WHW, Krauss RM, Hazen SL. Impact of chronic dietary red meat, white meat, or non-meat protein on trimethylamine n-oxide metabolism and renal excretion in healthy men and women. Eur Heart J. 2019;40:583–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sinha A, Ma Y, Scherzer R, Rahalkar S, Neilan BD, Crane H, Drozd D, Martin J, Deeks SG, Hunt P, Hsue PY. Carnitine is associated with atherosclerotic risk and myocardial infarction in hiv -infected adults. J Am Heart Assoc. 2019;8:e011037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Abete I, Romaguera D, Vieira AR, Lopez de Munain A, Norat T. Association between total, processed, red and white meat consumption and all-cause, cvd and ihd mortality: A meta-analysis of cohort studies. Br J Nutr. 2014;112:762–775 [DOI] [PubMed] [Google Scholar]
- 114.Warrier M, Shih DM, Burrows AC, Ferguson D, Gromovsky AD, Brown AL, Marshall S, McDaniel A, Schugar RC, Wang Z, Sacks J, Rong X, Vallim TA, Chou J, Ivanova PT, Myers DS, Brown HA, Lee RG, Crooke RM, Graham MJ, Liu X, Parini P, Tontonoz P, Lusis AJ, Hazen SL, Temel RE, Brown JM. The tmao-generating enzyme flavin monooxygenase 3 is a central regulator of cholesterol balance. Cell Rep. 2015;10:326–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gregory JC, Buffa JA, Org E, Wang Z, Levison BS, Zhu W, Wagner MA, Bennett BJ, Li L, DiDonato JA, Lusis AJ, Hazen SL. Transmission of atherosclerosis susceptibility with gut microbial transplantation. J Biol Chem. 2015;290:5647–5660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lindskog Jonsson A, Caesar R, Akrami R, Reinhardt C, Fak Hallenius F, Boren J, Backhed F. Impact of gut microbiota and diet on the development of atherosclerosis in apoe(−/−) mice. Arterioscler Thromb Vasc Biol. 2018;38:2318–2326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Haghikia A, Li XS, Liman TG, Bledau N, Schmidt D, Zimmermann F, Krankel N, Widera C, Sonnenschein K, Haghikia A, Weissenborn K, Fraccarollo D, Heimesaat MM, Bauersachs J, Wang Z, Zhu W, Bavendiek U, Hazen SL, Endres M, Landmesser U. Gut microbiota-dependent trimethylamine n-oxide predicts risk of cardiovascular events in patients with stroke and is related to proinflammatory monocytes. Arterioscler Thromb Vasc Biol. 2018;38:2225–2235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhu W, Wang Z, Tang WHW, Hazen SL. Gut microbe-generated trimethylamine n-oxide from dietary choline is prothrombotic in subjects. Circulation. 2017;135:1671–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shih DM, Zhu W, Schugar RC, Meng Y, Jia X, Miikeda A, Wang Z, Zieger M, Lee R, Graham M, Allayee H, Cantor RM, Mueller C, Brown JM, Hazen SL, Lusis AJ. Genetic deficiency of flavin-containing monooxygenase 3 ( fmo3) protects against thrombosis but has only a minor effect on plasma lipid levels-brief report. Arterioscler Thromb Vasc Biol. 2019;39:1045–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Romano KA, Vivas EI, Amador-Noguez D, Rey FE. Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine-n-oxide. mBio. 2015;6:e02481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Romano KA, Martinez-Del Campo A, Kasahara K, Chittim CL, Vivas EI, Amador-Noguez D, Balskus EP, Rey FE. Metabolic, epigenetic, and transgenerational effects of gut bacterial choline consumption. Cell Host Microbe. 2017;22:279–290.e277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cheng X, Qiu X, Liu Y, Yuan C, Yang X. Trimethylamine n-oxide promotes tissue factor expression and activity in vascular endothelial cells: A new link between trimethylamine n-oxide and atherosclerotic thrombosis. Thromb Res. 2019;177:110–116 [DOI] [PubMed] [Google Scholar]
- 123.Witkowski M, Landmesser U, Rauch U. Tissue factor as a link between inflammation and coagulation. Trends Cardiovasc Med. 2016;26:297–303 [DOI] [PubMed] [Google Scholar]
- 124.Witkowski M, Witkowski M, Saffarzadeh M, Friebel J, Tabaraie T, Ta Bao L, Chakraborty A, Dorner A, Stratmann B, Tschoepe D, Winter SJ, Krueger A, Ruf W, Landmesser U, Rauch U. Vascular miR-181b controls tissue factor-dependent thrombogenicity and inflammation in type 2 diabetes. Cardiovasc Diabetol. 2020;19:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Witkowski M, Weithauser A, Tabaraie T, Steffens D, Kränkel N, Witkowski M, Stratmann B, Tschoepe D, Landmesser U, Rauch-Kroehnert U. Micro-RNA-126 reduces the blood thrombogenicity in diabetes mellitus via targeting of tissue factor. Arterioscler Thromb Vasc Biol. 2016;36:1263–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Dambrova M, Latkovskis G, Kuka J, Strele I, Konrade I, Grinberga S, Hartmane D, Pugovics O, Erglis A, Liepinsh E. Diabetes is associated with higher trimethylamine N-oxide plasma levels. Exp Clin Endocrinol Diabetes. 2016;124:251–256 [DOI] [PubMed] [Google Scholar]
- 127.Jäckel S, Kiouptsi K, Lillich M, Hendrikx T, Khandagale A, Kollar B, Hörmann N, Reiss C, Subramaniam S, Wilms E, Ebner K, Brühl MV, Rausch P, Baines JF, Haberichter S, Lämmle B, Binder CJ, Jurk K, Ruggeri ZM, Massberg S, Walter U, Ruf W, Reinhardt C. Gut microbiota regulate hepatic von willebrand factor synthesis and arterial thrombus formation via toll-like receptor-2. Blood. 2017;130:542–553 [DOI] [PubMed] [Google Scholar]
- 128.Ke Y, Li D, Zhao M, Liu C, Liu J, Zeng A, Shi X, Cheng S, Pan B, Zheng L, Hong H. Gut flora-dependent metabolite trimethylamine-n-oxide accelerates endothelial cell senescence and vascular aging through oxidative stress. Free Radic Biol Med. 2018;116:88–100 [DOI] [PubMed] [Google Scholar]
- 129.Brunt VE, Gioscia-Ryan RA, Richey JJ, Zigler MC, Cuevas LM, Gonzalez A, Vazquez-Baeza Y, Battson ML, Smithson AT, Gilley AD, Ackermann G, Neilson AP, Weir T, Davy KP, Knight R, Seals DR. Suppression of the gut microbiome ameliorates age-related arterial dysfunction and oxidative stress in mice. J Physiol. 2019;597:2361–2378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Boini KM, Hussain T, Li P-L, Koka S. Trimethylamine-n-oxide instigates nlrp3 inflammasome activation and endothelial dysfunction. Cell Physiol Biochem. 2017;44:152–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sun X, Jiao X, Ma Y, Liu Y, Zhang L, He Y, Chen Y. Trimethylamine n-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via activating ros-txnip-nlrp3 inflammasome. Biochem Biophys Res Commun. 2016;481:63–70 [DOI] [PubMed] [Google Scholar]
- 132.Wallrabenstein I, Kuklan J, Weber L, Zborala S, Werner M, Altmüller J, Becker C, Schmidt A, Hatt H, Hummel T, Gisselmann G. Human trace amine-associated receptor taar5 can be activated by trimethylamine. PloS One. 2013;8:e54950–e54950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chen S, Henderson A, Petriello MC, Romano KA, Gearing M, Miao J, Schell M, Sandoval-Espinola WJ, Tao J, Sha B, Graham M, Crooke R, Kleinridders A, Balskus EP, Rey FE, Morris AJ, Biddinger SB. Trimethylamine n-oxide binds and activates perk to promote metabolic dysfunction. Cell Metab. 2019;30:1141–1151.e1145 [DOI] [PubMed] [Google Scholar]
- 134.Schneck E, Horinek D, Netz RR. Insight into the molecular mechanisms of protein stabilizing osmolytes from global force-field variations. J Phys Chem B. 2013;117:8310–8321 [DOI] [PubMed] [Google Scholar]
- 135.Kelly RH, Yancey PH. High contents of trimethylamine oxide correlating with depth in deep-sea teleost fishes, skates, and decapod crustaceans. Biol Bull. 1999;196:18–25 [DOI] [PubMed] [Google Scholar]
- 136.Yancey PH, Rhea MD, Kemp KM, Bailey DM. Trimethylamine oxide, betaine and other osmolytes in deep-sea animals: Depth trends and effects on enzymes under hydrostatic pressure. Cell Mol Biol (Noisy-le-grand). 2004;50:371–376 [PubMed] [Google Scholar]
- 137.Yancey PH, Gerringer ME, Drazen JC, Rowden AA, Jamieson A. Marine fish may be biochemically constrained from inhabiting the deepest ocean depths. Proc Natl Acad Sci USA. 2014;111:4461–4465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yancey PH, Siebenaller JF. Trimethylamine oxide stabilizes teleost and mammalian lactate dehydrogenases against inactivation by hydrostatic pressure and trypsinolysis. J Exp Biol. 1999;202:3597–3603 [DOI] [PubMed] [Google Scholar]
- 139.Miller AL, Elam WA, Johnson BH, Khan SH, Kumar R, Thompson EB. Restored mutant receptor:Corticoid binding in chaperone complexes by trimethylamine n-oxide. PLoS One. 2017;12:e0174183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Treberg JR, Wilson CE, Richards RC, Ewart KV, Driedzic WR. The freeze-avoidance response of smelt osmerus mordax: Initiation and subsequent suppression of glycerol, trimethylamine oxide and urea accumulation. J Exp Biol. 2002;205:1419–1427 [DOI] [PubMed] [Google Scholar]
- 141.Gawrys-Kopczynska M, Konop M, Maksymiuk K, Kraszewska K, Derzsi L, Sozanski K, Holyst R, Pilz M, Samborowska E, Dobrowolski L, Jaworska K, Mogilnicka I, Ufnal M. TMAO, a seafood-derived molecule, produces diuresis and reduces mortality in heart failure rats. Elife. 2020;9:e57028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kumemoto R, Yusa K, Shibayama T, Hatori K. Trimethylamine N-oxide suppresses the activity of the actomyosin motor. Biochim Biophys Acta. 2012;1820:1597–1604 [DOI] [PubMed] [Google Scholar]
- 143.Sands JM, Layton HE. The physiology of urinary concentration: An update. Semin Nephrol. 2009;29:178–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Visentin CE, Masih S, Plumptre L, Malysheva O, Nielsen DE, Sohn KJ, Ly A, Lausman AY, Berger H, Croxford R, El-Sohemy A, Caudill MA, O’Connor DL, Kim YI. Maternal choline status, but not fetal genotype, influences cord plasma choline metabolite concentrations. J Nutr. 2015;145:1491–1497 [DOI] [PubMed] [Google Scholar]
- 145.Gohir W, Whelan FJ, Surette MG, Moore C, Schertzer JD, Sloboda DM. Pregnancy-related changes in the maternal gut microbiota are dependent upon the mother’s periconceptional diet. Gut Microbes. 2015;6:310–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Nemet I, Saha PP, Gupta N, Zhu W, Romano KA, Skye SM, Cajka T, Mohan ML, Li L, Wu Y, Funabashi M, Ramer-Tait AE, Naga Prasad SV, Fiehn O, Rey FE, Tang WHW, Fischbach MA, DiDonato JA, Hazen SL. A cardiovascular disease-linked gut microbial metabolite acts via adrenergic receptors. Cell. 2020;180:862–877.e822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wang J, Gareri C, Rockman HA. G-protein-coupled receptors in heart disease. Circ Res. 2018;123:716–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Offermanns S Activation of platelet function through g protein-coupled receptors. Circ Res. 2006;99:1293–1304 [DOI] [PubMed] [Google Scholar]
- 149.Bartley A, Yang T, Arocha R, Malphurs WL, Larkin R, Magee KL, Vickroy TW, Zubcevic J. Increased abundance of lactobacillales in the colon of beta-adrenergic receptor knock out mouse is associated with increased gut bacterial production of short chain fatty acids and reduced IL17 expression in circulating CD4(+) immune cells. Front Physiol. 2018;9:1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Honour J The possible involvement of intestinal bacteria in steroidal hypertension. Endocrinology. 1982;110:285–287 [DOI] [PubMed] [Google Scholar]
- 151.Kim J, Padanilam BJ. Renal nerves drive interstitial fibrogenesis in obstructive nephropathy. J Am Soc Nephrol. 2013;24:229–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Epstein SE, Speir E, Zhou YF, Guetta E, Leon M, Finkel T. The role of infection in restenosis and atherosclerosis: Focus on cytomegalovirus. Lancet. 1996;348 Suppl 1:S13–17 [DOI] [PubMed] [Google Scholar]
- 153.Patel P, Mendall MA, Carrington D, Strachan DP, Leatham E, Molineaux N, Levy J, Blakeston C, Seymour CA, Camm AJ, et al. Association of helicobacter pylori and chlamydia pneumoniae infections with coronary heart disease and cardiovascular risk factors. BMJ. 1995;311:711–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Danesh J, Collins R, Peto R. Chronic infections and coronary heart disease: Is there a link? Lancet. 1997;350:430–436 [DOI] [PubMed] [Google Scholar]
- 155.Saikku P, Leinonen M, Mattila K, Ekman MR, Nieminen MS, Makela PH, Huttunen JK, Valtonen V. Serological evidence of an association of a novel chlamydia, TWAR, with chronic coronary heart disease and acute myocardial infarction. Lancet. 1988;2:983–986 [DOI] [PubMed] [Google Scholar]
- 156.O’Connor CM, Dunne MW, Pfeffer MA, Muhlestein JB, Yao L, Gupta S, Benner RJ, Fisher MR, Cook TD. Azithromycin for the secondary prevention of coronary heart disease events: The WIZARD study: A randomized controlled trial. JAMA. 2003;290:1459–1466 [DOI] [PubMed] [Google Scholar]
- 157.Cannon CP, Braunwald E, McCabe CH, Grayston JT, Muhlestein B, Giugliano RP, Cairns R, Skene AM. Antibiotic treatment of chlamydia pneumoniae after acute coronary syndrome. N Engl J Med. 2005;352:1646–1654 [DOI] [PubMed] [Google Scholar]
- 158.Andraws R, Berger JS, Brown DL. Effects of antibiotic therapy on outcomes of patients with coronary artery disease: A meta-analysis of randomized controlled trials. JAMA. 2005;293:2641–2647 [DOI] [PubMed] [Google Scholar]
- 159.Maier L, Pruteanu M, Kuhn M, Zeller G, Telzerow A, Anderson EE, Brochado AR, Fernandez KC, Dose H, Mori H, Patil KR, Bork P, Typas A. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature. 2018;555:623–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wu H, Esteve E, Tremaroli V, Khan MT, Caesar R, Mannerås-Holm L, Ståhlman M, Olsson LM, Serino M, Planas-Fèlix M, Xifra G, Mercader JM, Torrents D, Burcelin R, Ricart W, Perkins R, Fernàndez-Real JM, Bäckhed F. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med. 2017;23:850–858 [DOI] [PubMed] [Google Scholar]
- 161.Javdan B, Lopez JG, Chankhamjon P, Lee YJ, Hull R, Wu Q, Wang X, Chatterjee S, Donia MS. Personalized mapping of drug metabolism by the human gut microbiome. Cell. 2020; Online ahead of print [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Companys J, Pedret A, Valls RM, Solà R, Pascual V. Fermented dairy foods rich in probiotics and cardiometabolic risk factors: A narrative review from prospective cohort studies. Crit Rev Food Sci Nutr. 2020:1–10 [DOI] [PubMed] [Google Scholar]
- 163.Companys J, Pla-Pagà L, Calderón-Pérez L, Llauradó E, Solà R, Pedret A, Valls RM. Fermented dairy products, probiotic supplementation, and cardiometabolic diseases: A systematic review and meta-analysis. Adv Nutr. 2020; Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zhou Z, Chen X, Sheng H, Shen X, Sun X, Yan Y, Wang J, Yuan Q. Engineering probiotics as living diagnostics and therapeutics for improving human health. Microb Cell Fact. 2020;19:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Rondanelli M, Faliva MA, Perna S, Giacosa A, Peroni G, Castellazzi AM. Using probiotics in clinical practice: Where are we now? A review of existing meta-analyses. Gut Microbes. 2017;8:521–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Suez J, Zmora N, Segal E, Elinav E. The pros, cons, and many unknowns of probiotics. Nat Med. 2019;25:716–729 [DOI] [PubMed] [Google Scholar]
- 167.Moludi J, Maleki V, Jafari-Vayghyan H, Vaghef-Mehrabany E, Alizadeh M. Metabolic endotoxemia and cardiovascular disease: A systematic review about potential roles of prebiotics and probiotics. Clin Exp Pharmacol Physiol. 2020;47:927–939 [DOI] [PubMed] [Google Scholar]
- 168.Marques FZ, Nelson E, Chu PY, Horlock D, Fiedler A, Ziemann M, Tan JK, Kuruppu S, Rajapakse NW, El-Osta A, Mackay CR, Kaye DM. High-fiber diet and acetate supplementation change the gut microbiota and prevent the development of hypertension and heart failure in hypertensive mice. Circulation. 2017;135:964–977 [DOI] [PubMed] [Google Scholar]
- 169.Catry E, Bindels LB, Tailleux A, Lestavel S, Neyrinck AM, Goossens JF, Lobysheva I, Plovier H, Essaghir A, Demoulin JB, Bouzin C, Pachikian BD, Cani PD, Staels B, Dessy C, Delzenne NM. Targeting the gut microbiota with inulin-type fructans: Preclinical demonstration of a novel approach in the management of endothelial dysfunction. Gut. 2018;67:271–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Gan XT, Ettinger G, Huang CX, Burton JP, Haist JV, Rajapurohitam V, Sidaway JE, Martin G, Gloor GB, Swann JR, Reid G, Karmazyn M. Probiotic administration attenuates myocardial hypertrophy and heart failure after myocardial infarction in the rat. Circ Heart Fail. 2014;7:491–499 [DOI] [PubMed] [Google Scholar]
- 171.Salonen A, Lahti L, Salojarvi J, Holtrop G, Korpela K, Duncan SH, Date P, Farquharson F, Johnstone AM, Lobley GE, Louis P, Flint HJ, de Vos WM. Impact of diet and individual variation on intestinal microbiota composition and fermentation products in obese men. ISME J. 2014;8:2218–2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Trefz F, Maillot F, Motzfeldt K, Schwarz M. Adult phenylketonuria outcome and management. Mol Genet Metab. 2011;104 Suppl:S26–30 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.