

Abstract
A hexanucleotide repeat expansion in C90RF72 is the most common genetic variant contributing to Amyotrophic lateral sclerosis (ALS) and Frontotemporal dementia (FTD)1,2. The C90RF72 mutation acts through gain and loss of function mechanisms to induce pathways implicated in neural degeneration3–9. The expansion is transcribed into a long repetitive RNA, which negatively sequesters RNA binding proteins5 prior to its non-canonical translation into neural-toxic di-peptide proteins3,4. Failure of RNA-polymerase to read through the mutation also reduces abundance of the endogenous C90RF72 gene product, which functions in endo-lysosomal pathways and suppresses systemic and neural inflammation6–9. Notably, effects of the repeat expansion act with incomplete penetrance in ALS/FTD families, indicating that either genetic or environmental factors modify each individual’s risk of disease. Identifying disease modifiers is of significant translational interest, as it could suggest strategies that diminish the risk of developing ALS/FTD, or that slow progression. Here, we report that an environment with reduced abundance of immune-stimulating bacteria10,11 protects C9orf72 mutant mice from premature mortality and significantly ameliorates their underlying systemic inflammation and autoimmunity. Consistent with C9orf72 functioning to prevent microbiota from inducing a pathological inflammatory response, we found that reducing microbial burden in mutants with broad spectrum antibiotics, as well as transplanting gut microflora from a protective environment attenuated inflammatory phenotypes, even after their onset. Our studies provide further evidence that the microbial constituency of our gut plays an important role in brain health and can interact in surprising ways with well-known genetic risk factors for nervous system disorders.
To understand the consequences of the long-term reduction in C90RF72 activity found in patients, we and others previously studied mice harboring loss of function (LOF) mutations in the orthologous gene (C9orf72)6,7,12,13. We reported7 and others corroborated that reduced C9orf72 function led to age-dependent inflammation, characterized by cytokine storm7,14, neutrophilia6,7,14, pseudothrombocytopenia7, autoimmunity7,14, splenomegaly6,7,13,14, and neuroinflammation6,7. Informed by these observations and validating their importance, it was subsequently found that C90RF72 ALS/FTD patients were significantly more likely to have been diagnosed with autoimmune disease prior to their neurological diagnosis15,16.
However, long-term survival of C9orf72 LOF mutant mice varied dramatically between reports, despite many groups studying the same allele on a similar genetic background. We7 and others12 found that loss of one (+/−) or both (−/−) alleles of C9orf72 increased risk of premature mortality, while some13 noted reduced survival of C9orf72 −/− but not +/− animals and another group6 reported no survival differences between controls and mutants (Extended Data Fig. 1). These findings suggested the environment in which animals were reared might be a significant modifier of survival when C9orf72 levels are reduced. To test this hypothesis, we aseptically re-derived C9orf72 mutant animals into a new facility at the Broad Institute (C9orf72Broad), while continuing our colony at the Harvard BRI facility (C9orf72Harvard) (Fig. 1a). To assess the reproducibility of our original findings at Harvard, we aged an independent cohort of C9orf72Harvard animals (+/+ n=55, +/− n=114, −/− n=62) and again found that +/− animals (P = 0.0460) and −/− animals (P < 0.0001) were at increased risk of premature mortality (Fig. 1b). The causes of death in these animals, which included cervical lymphadenopathy, wasting, and severe ataxia, were indistinguishable from those we observed previously7 and closely tied to their underlying autoimmune condition (Extended Data Fig. 2a–b). In remarkable contrast, we observed no early mortality or motor behavior deficit in either heterozygous or homozygous mutant animals at the Broad Institute (C9orf72Broad +/+ n=22, +/− n=36, −/− n=23) (Fig. 1c and Extended Data Fig 2c). As a result, C9orf72 −/− mice were significantly more likely to die prematurely when reared at Harvard than at Broad (P = 0.0179). We therefore conclude that signals from the environment can be significant modifiers of lifespan when C9orf72 function declines.
Fig. 1 |.
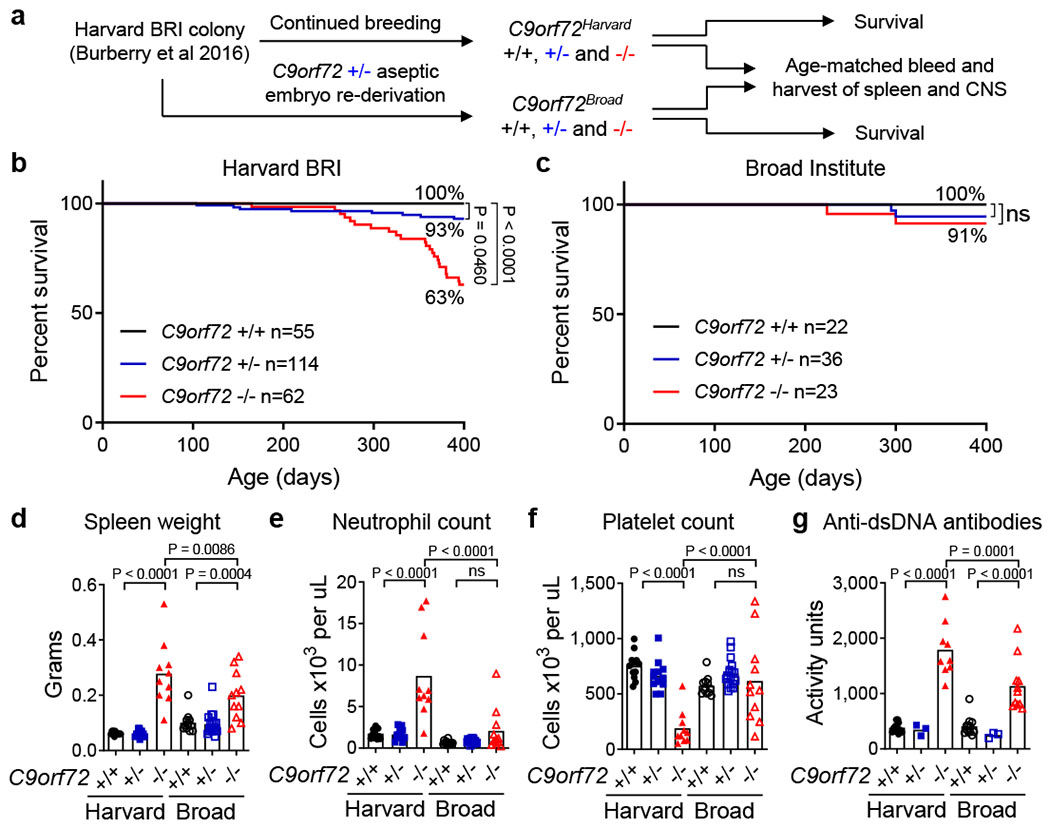
Environment governs survival, inflammation and autoimmunity in C9orf72 LOF mice.
a, Aseptic embryo transfer of C9orf72 neo deleted allele from Harvard BRI to Broad Institute. Males and females were aged for survival or tissue harvest. Survival of mice at b, Harvard BRI (C9orf72 +/+ n=55; +/− n=114; −/− n=62) or c, Broad Institute (C9orf72 +/+ n=22; +/− n=36; −/− n=23) (Gehan-Breslow-Wilcoxon). ns not significant Age-matched (48-week-old) mice reared at Harvard BRI (C9orf72 +/+ n=12; +/− n=13; −/− n=10) or Broad Institute (C9orf72 +/+ n=12; +/− n=18; −/− n=11) were assessed for d, spleen weight, e, blood neutrophil count, f, blood platelet count measured at 0ºC and g, plasma anti-double stranded (ds)DNA antibody activity, d-g, One way ANOVA with Sidak multiple comparisons. Each dot represents one animal.
To determine whether the improved survival we observed in C9orf72Broad mice was associated with a diminution of inflammatory and autoimmune endophenotypes7, we jointly analyzed age-matched animals reared at each facility (Fig. 1d–g and Extended Data Fig. 2d–e). As we previously reported, C9orf72Harvard animals exhibited autoimmune and inflammatory phenotypes including significantly elevated levels of IL-23, IL-10, IL-22, G-csf, IL-17a, Tnfα, Ifnγ, IL-1β and IL-12p70 (P < 0.05)(Extended data Fig. 1e) as well as splenomegaly (P < 0.0001)(Fig. 1d), neutrophilia (P < 0.0001) (Fig. 1e), pseudothrombocytopenia (P < 0.0001) (Fig. 1f and Extended Data Fig. 2f–g), and development of auto-antibodies (P < 0.0001) (Fig. 1g). Strikingly, and in every case, these inflammatory phenotypes were significantly reduced in C9orf72Broad −/− animals relative to their C9orf72Harvard mutant counterparts (Fig. 1d–g). In fact, the reduction of inflammation in the pro-survival Broad Institute environment was sufficiently reduced that many inflammatory phenotypes we routinely observed in mutant animals at Harvard were no longer significantly different between −/− and +/+ animals at Broad. It is notable that the few phenotypes that remained significantly different between C9orf72Broad −/− and C9orf72Broad +/+ animals, such as modest splenomegaly (P = 0.0004), were those most widely reported in the past6,13,14. Thus, an environment that improved survival also ameliorated the underlying inflammatory and autoimmune disease found in C9orf72 mutants.
Antibiotics prevent inflammation
We next considered variables between the two environments that might have contributed to such dramatic differences in the severity of mutant phenotypes. We found that diet, light-cycle and many other features of the two environments were similar. However, a review of microbial screening reports from the two facilities indicated that murine norovirus (P = 0.0140), Helicobacter spp. (P < 0.0001), Pasteurella pneumotropica (P = 0.0070) and Tritrichomonas muris (P < 0.0001) were significantly more common in C9orf72Harvard animals than in C9orf72Broad mice (Supplementary Table 1). It is important to note, that differences between the two colonies were well within norms for Assessment and Accreditation of Laboratory Animal Care (AAALAC) processes. The differential components of the microflora we found at Harvard are not generally considered pathogenic, consistent with the normal health and lifespan of control animals in that environment (Fig. 1b and ref7). However, Helicobacter spp. have been suggested to have immune-stimulating properties10, raising the possibility that changes in gut microflora between the environments might underlie the increased rate of mortality and inflammatory phenotypes we found in C9orf72Harvard mutant animals.
To learn whether resident microflora contributed to the inflammation and autoimmunity seen in C9orf72Harvard mutants, we weaned new C9orf72Harvard animals (+/+ n=14, −/− n=22) and administered either vehicle or broad-spectrum antibiotics prior to onset of inflammatory disease (Day 30), then monitored related phenotypes for 200 days (Fig. 2a). As expected, antibiotics significantly reduced the abundance and diversity of bacterial species including Helicobacter spp., without affecting levels of murine norovirus. The guts of vehicle treated controls were largely unaltered (Fig. 2b and Extended data Fig 3a). We found that vehicle had no effect on development of either inflammatory or autoimmune phenotypes in C9orf72Harvard −/− mice, including cytokine storm (Extended Data Fig. 3b), neutrophilia (Fig. 2c), pseudothrombocytopenia (Fig. 2d), autoimmunity (Fig. 2e) and splenomegaly (Fig. 2f and Extended data Fig 3c). In contrast, providing lifelong antibiotics treatment to C9orf72Harvard −/− mice completely suppressed the emergence of all of these phenotypes (Fig. 2c–f and Extended data Fig 3b–i). Thus, our experiments suggest that signals derived from gut bacteria promote inflammation and autoimmunity when C9orf72 function is diminished. However, we found that chronic antibiotics administration resulted in previously reported health consequences including hepatoxicity17, which prevented us from assessing behavioral and survival outcomes.
Fig. 2 |.
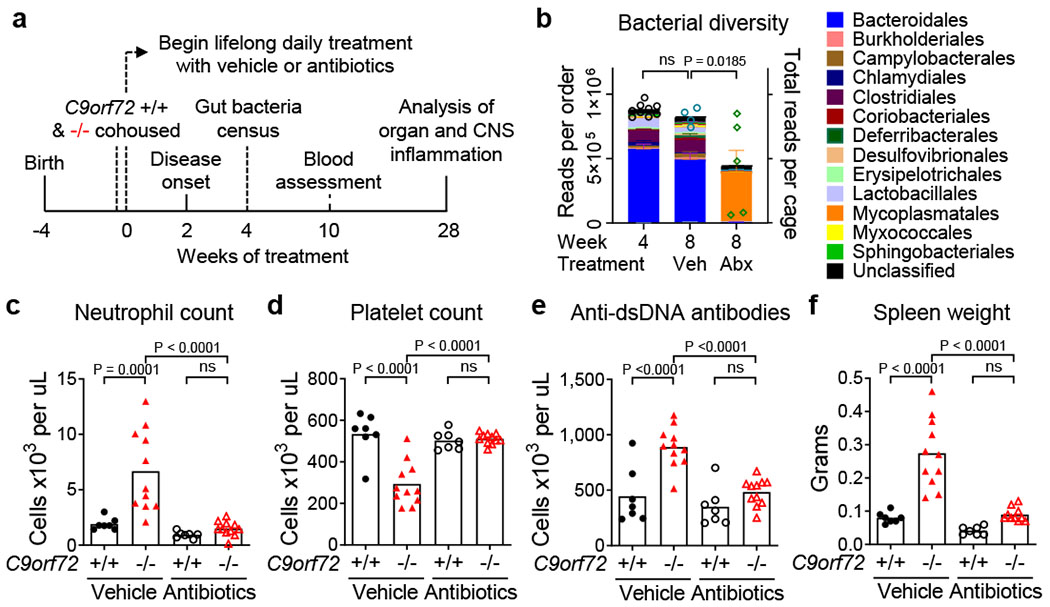
Lifelong suppression of gut microflora prevents inflammation and autoimmunity in C9orf72 LOF mice
a, Male and female C9orf72Harvard +/+ and −/− neo deleted mice of weaning age were cohoused by treatment group, then administered vehicle (+/+ n=7; −/− n=11) or antibiotics (+/+ n=7; −/− n=11) daily for life. Mice assessed for gut microbial composition (b, 4 weeks), blood measures (c-e, 8 weeks) and sacrificed for organ and CNS assessment (f, 28 weeks). b, 16S rDNA sequencing of bacteria diversity in feces. Each dot represents total sequencing reads per cage. One-way ANOVA with Dunnett multiple comparison. c, Blood neutrophil count. d, Blood platelet count measured at 0ºC. e, Plasma anti-dsDNA antibody activity. f, Spleen weight. c-f, One way ANOVA with Sidak multiple comparisons. Each dot represents one animal.
We next asked whether acute suppression of gut microbiota could ameliorate inflammatory and autoimmune phenotypes after their establishment in C9orf72Harvard mutant mice. To this end, we obtained another independent cohort of C9orf72Harvard mice (+/+ n=25, −/− n=24; Day 250), demonstrated these animals displayed the expected inflammatory phenotypes relative to controls and showed that they exhibited poor performance on the accelerating rotarod (Fig. 3a–d and Extended Data Fig 4a). Then we began acute administration of broad-spectrum antibiotics and monitored associated phenotypes over the course of 60 days. We found that this treatment significantly reduced each of the inflammatory and autoimmune phenotypes in mutant animals (Fig 3b–d), including splenomegaly (P = 0.0002) (Extended Data Fig. 4c–d) and improved rotarod performance (P = 0.0398) (Extended Data Fig 4a). In contrast, treatment with vehicle had no impact on these measures (Fig 3b–d and Extended Data Fig 4a–d).
Fig. 3 |.
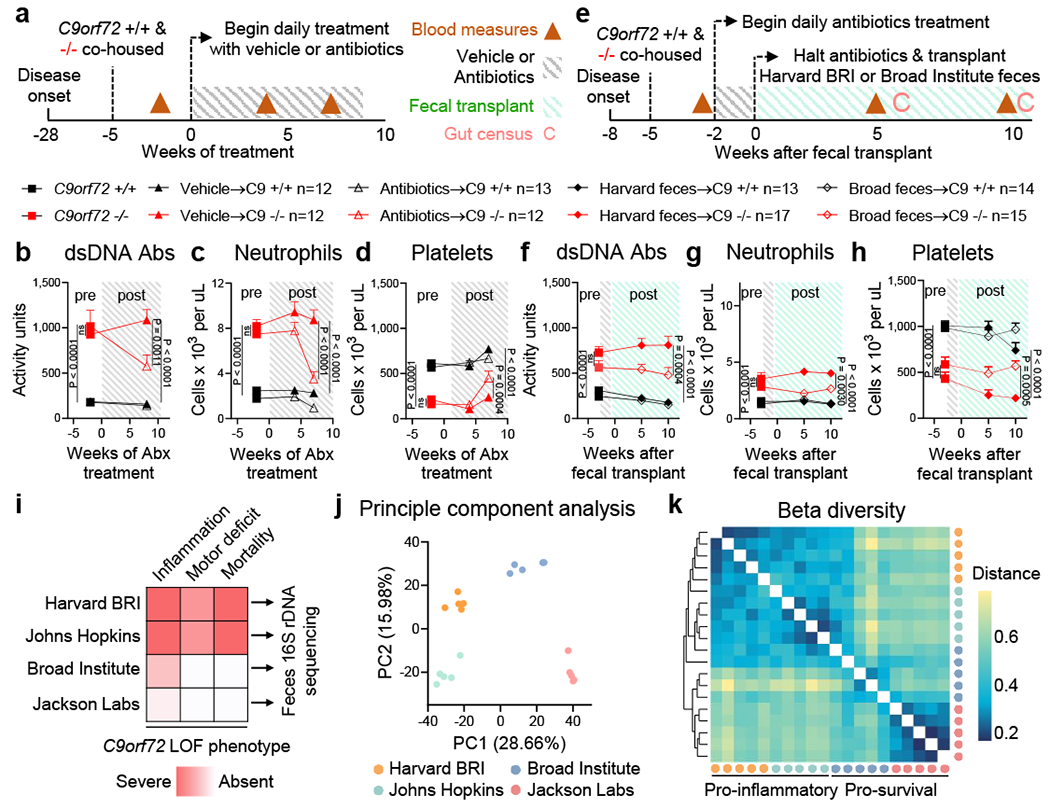
Gut bacteria propagates inflammation and autoimmunity in C9orf72 LOF mice
a, Age matched (36-week-old) female C9orf72Harvard +/+ and −/− neo deleted mice were cohoused by treatment group, then administered vehicle (+/+ n=12; −/− n=12) or antibiotics (+/+ n=13; −/− n=12) daily and assessed for b, plasma anti-dsDNA antibody activity, c, blood neutrophil count and d, blood platelet count measured at 0ºC. b-d and f-h, One way ANOVA with Sidak multiple comparisons. Each dot represents one animal. e, Age matched (13-week-old) female C9orf72Harvard +/+ and −/− neo deleted mice were cohoused by treatment group, administered antibiotics for two weeks, then gavaged Harvard BRI feces (+/+ n=13; −/− n=17) or Broad feces (+/+ n=14; −/− n=15) and assessed for f, plasma anti-dsDNA antibody activity, g, blood neutrophil count and h, blood platelet count measured at 0ºC. i, Fecal pellets (n=5 each) from two pro-inflammatory environments (Harvard BRI/Johns Hopkins) and two pro-survival environments (Broad Institute/Jackson Labs) were subjected to 16S rDNA sequencing and assessed by j, principle component analysis and k, Bray-Curtis dissimilarity matrix of beta diversity.
Fecal transplants mitigate inflammation
To more directly probe whether phenotypic improvements were due to microbial communities of the gut, rather than unrelated consequences of antibiotics treatment, we performed fecal transplant experiments. We produced another cohort of C9orf72Harvard mice (+/+ n=27, −/− n=32; Day 100) and demonstrated these animals displayed the expected inflammatory phenotypes relative to controls. We then suppressed these animals’ gut microflora with transient antibiotic treatment and transplanted with feces from either the pro-inflammatory (Harvard BRI) or pro-survival (Broad Institute) environment (Fig 3e). Transplantation of pro-survival gut microflora significantly improved each of the inflammatory and autoimmune phenotypes (Fig 3f–h and Extended Data Fig 4e). In contrast, transplant with microflora from the pro-inflammatory facility did not improve these measures, suggesting the benefits we observed when transplanting feces from the protective environment was not merely due to the brief antibiotic treatment that enabled microbial engraftment. Therefore, our studies establish that the inflammatory and autoimmune disease which underlies premature mortality in C9orf72Harvard mutant mice can be therapeutically prevented and that signals from certain gut microbiota help to maintain it.
Profiling gut bacteria
To identify bacterial constituents of the gut associated with severe phenotypes in C9orf72Harvard mutant mice, we surveyed the composition of feces from two pro-inflammatory environments where mutant mice perished7,12 as well as two pro-survival environments6, this report (Fig 3i). Principle components analysis readily separated samples from the four environments with the largest principle component (PC1; 28.7% of variance), separating the two pro-inflammatory environments from the two pro-survival environments (Fig 3j). Deeper investigation of this axis of variance revealed a shared significant decrease in alpha diversity in the two pro-inflammatory environments while unsupervised hierarchical clustering demonstrated that samples from the pro-inflammatory environments showed disparate beta diversity from that in pro-survival environments (Fig 3k and Extended data Fig 9f). Exemplifying these considerable differences in community structure, 62 of 301 bacterial species we identified (20.6%) were significantly altered in their abundance when jointly comparing the two pro-survival environments to the two pro-inflammatory environments (p < 0.0002)(Extended Data Fig 5a–e). Consistent with initial observation (Supplementary Table 1), Helicobacter spp. was found in both pro-inflammatory environments (Extended data Fig 5g–h and Supplementary Images) but absent in pro-survival environments.
We next characterized the extent of microbial reconstitution in our fecal transplant recipients (Extended data Fig 6a–f). Hierarchical clustering of beta diversity revealed that the microbial constituencies of mice receiving Broad transplants were more similar to Broad Institute feces than to feces from either animals housed at Harvard BRI or feces from Harvard transplant recipients (Extended data Fig 6d). Analysis of individual bacteria similarly supported the success of our transplants, with 85% (199/234) of bacterial species identified in Harvard BRI feces detected in Harvard fecal recipients and 75% (178/236) of bacterial species identified in Broad Institute feces detected in Broad fecal recipients (Extended Data Fig 6f). Quantitative PCR for Helicobacter spp. rDNA further confirmed the reconstitution of Harvard specific microbes in Harvard recipients and their elimination from Broad fecal recipients (Extended data Fig 5i and Supplementary Images).
Gut components regulate myeloid cytokines
To mechanistically explore how varied fecal components in separate environments alter cytokine burden and autoimmunity in C9orf72 −/− mice, we stimulated bone marrow derived macrophages (BMDMs) from C9orf72Harvard animals with chemical analogs of microbial components and found that both +/− and −/− C9orf72Harvard BMDMs released higher levels of several pro-inflammatory cytokines than +/+ control cells in response to bacterial lipopeptide, single stranded (ss)RNA and ssDNA (Extended data Fig 7a–c). Given these findings, we next asked whether fecal material from Harvard BRI housed animals contained higher levels of innate immune stimulating factors than feces from mice at the Broad Institute. To this end, we individually administered normalized concentrations of fecal eubacteria from both institutions to C9orf72Harvard −/− and +/+ BMDMs. Indeed, we found that C9orf72 −/− BMDMs produced significantly higher levels of TNF-alpha when exposed to Harvard BRI fecal material than when exposed to Broad feces (Extended data Fig 7d). In addition, serial dilutions revealed that a combination of Harvard BRI feces and a C9orf72 −/− genotype lead to TNF-alpha release at the lowest fecal concentrations (Extended data Fig 7d).
Environment governs neuroinflammation
Neuroinflammation is a pathological hallmark of C90RF72 ALS and FTD18,19, with substantial infiltration of peripheral immune cells noted in the spinal cord of ALS patients20,21. We used the pan-hematopoietic marker CD45 to distinguish CD45mid resident microglia from peripherally derived CD45hi cells22 and found that infiltrating cells were present at sites of focal inflammation within the spinal cord parenchyma of C9orf72Harvard −/− mice (Figure 4a and Extended data Fig 8a–h and Supplementary Videos 1–2). Mass Cytometry analysis revealed the CD45hi cells that infiltrated the spinal cord were mostly CD11b+ Ly6C+ Ly6G+ CD39− neutrophils and CD3e+ T cells (Extended Data Fig. 8a–f and Supplementary Gating Strategy). Strikingly, lifelong suppression of gut microflora with antibiotics prevented the accumulation of infiltrating myeloid cells within the spinal cord of C9orf72Harvard −/− mice (Figure 4b–c).
Fig. 4 |.
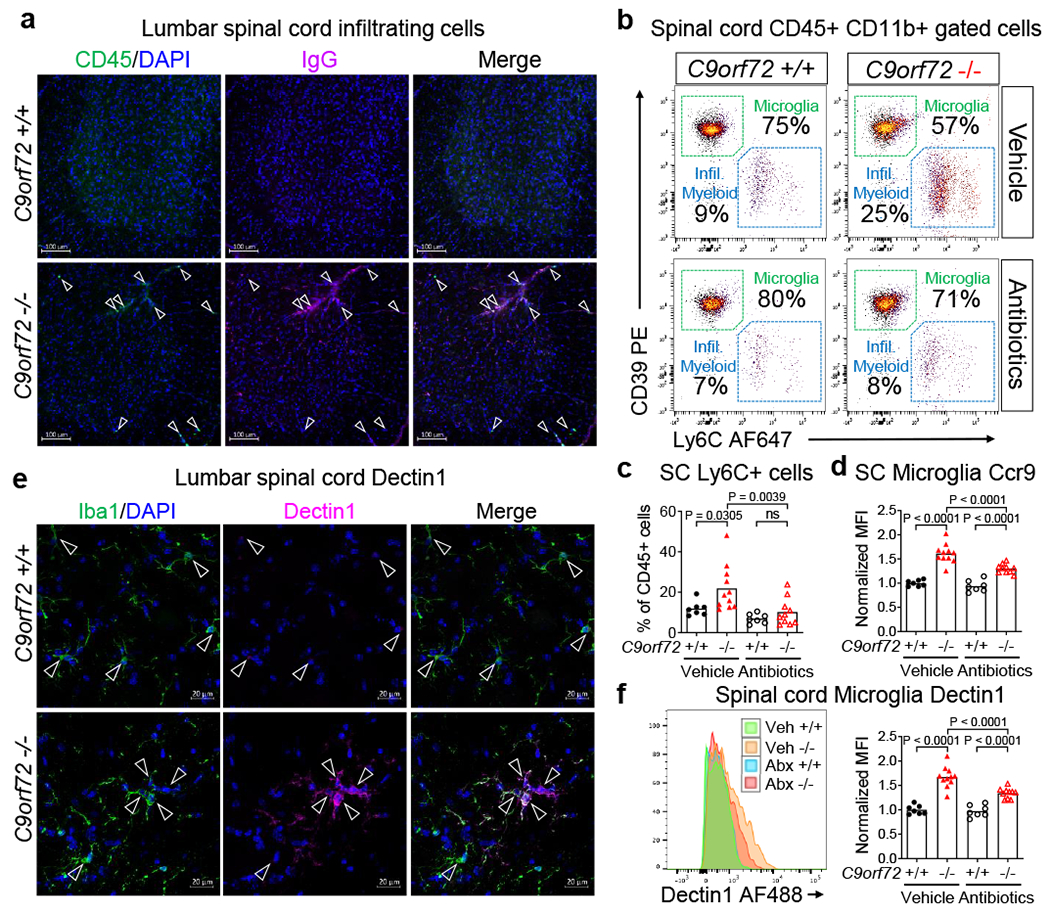
Gut microflora promotes myeloid cell infiltration and microgliosis in C9orf72 LOF spinal cord
a, Orthogonal projection of CD45 and mouse immunoglobulin G (IgG) in 55-week-old C9orf72Harvardneo deleted lumbar spinal cord (+/+ n=3; −/− n=3). b, Representative gating of CD45+ CD11b+ cells from spinal cord of C9orf72Harvard mice in Fig. 2. c, CD45hi CD11b+ Ly6C+ spinal cord infiltrating myeloid cells in b. c-d, f, One way ANOVA with Sidak multiple comparisons. Each dot represents one animal, d, Ccr9 expression on CD45mid CD11b+ CD39+ microglia from spinal cord of C9orf72Harvard mice in Fig. 2. e, Orthogonal projection of Dectin1 in Iba1+ microglia in 55-week-old C9orf72Harvardneo deleted lumbar spinal cord (+/+ n=3; −/− n=3 mice), f, Dectin1 in CD45mid CD11b+ CD39+ microglia from spinal cord of C9orf72Harvard mice in Fig. 2.
In addition to infiltrating peripheral immune cells, there are also substantial changes to resident microglia in the nervous system of ALS/FTD patients23. Studies from our group24 and others6,8,9 have implicated C9orf72 and its interactor Smcr8 in regulation of endo-lysosomal trafficking and autophagy, particularly in myeloid derivatives. We found that microglia from the spinal cord of C9orf72Harvard −/− animals expressed higher levels of the lysosome-associated proteins Lamp16 (Extended data Fig 9a and Supplementary Videos 3–4) and Cathepsin B (Extended data Fig 9b and Supplementary Videos 5–6). Lifelong suppression of gut microbiota did not significantly decrease Lamp1 or Cathepsin B levels in C9orf72Harvard −/− microglia (Extended data Fig 9c–d) suggesting that C9orf72 function regulates lysosomal constituents independently from microbial signals.
To examine the activation status of resident microglia in C9orf72Harvard mutant animals and to also ask whether it might be altered by signals from the microbiota, we measured levels of the pattern recognition receptor Dectin1, the chemokine receptor Ccr9, and the lipoprotein lipase Lpl which have previously been associated with pro-inflammatory microglial states25–27. Consistent with the notion that microglia become activated when C9orf72 levels decline, we found that Dectin1 and Ccr9 were enriched on microglia from C9orf72Harvard −/− mice (Fig 4d–f and Extended data Fig 9e–g and Supplementary Videos 7–8). Importantly, Dectin1 and Ccr9 expression were significantly reduced in microglia from C9orf72Harvard mutant animals whose gut microflora was chronically suppressed with antibiotics (Fig 4d–f). Together these results demonstrate that when C9orf72 function is reduced, peripheral immune cells can infiltrate the spinal cord where they associate with sites of neuroinflammation and that treatment with antibiotics, which suppresses the microbiota, modulates both infiltration and microglial activation.
Discussion
Our results indicate that when C9orf72 function declines, the environment generally and the gut microbiota specifically become potent modifiers of whether autoimmunity, neural inflammation, motor deficits and premature mortality occur. In fact, the effect of environment and accompanying changes of microbial microflora are so strong in this mouse model that in one environment, inflammatory disease and death were highly penetrant phenotypes, while in another they were essentially absent. We therefore provide the likely explanation for the considerable phenotypic variation that has been observed across groups studying this C9orf72 LOF allele in mice6,7,12,13. Our conclusions are important because they re-emphasize that the 50% reduction in the levels of C90RF72 found in C90RF72 ALS/FTD patients are a credible cause for the neural inflammation that are characteristic in their condition. Most provocatively, our findings also suggest that variance in microbiota could explain why some carriers of the C90RF72 mutation develop ALS/FTD or overt inflammatory conditions like lupus15,16, while others do not.
It should be re-emphasized that the microbes present in the environments we studied here are not considered mouse pathogens per se, and that their abundances were within the scope found in comparable institutions28. Importantly, the environmental conditions that triggered severe phenotypes in our C9orf72Harvard animals were reproducible elsewhere. Others12 reported a relationship between reduction in C9orf72 function and an increased rate of premature mortality comparable to that we described previously7 and replicated here. It is notable that these two environments were most similar in their microbial constituents and also shared many microbes that were not present in the two pro-survival locations we surveyed. Given the large number of species we found to significantly differ in their abundance between pro-inflammatory and pro-survival environments, future studies will be needed to elucidate the relative contribution of individual bacterial species to variation in the inflammatory and autoimmune phenotypes we reported herein. However, microbe by microbe analysis of varying environments and our transplant animals would seem to rule out reported protective effects of Akkermansia muciniphila29,30 (Extended data Fig 6f) and potential inflammatory influences of Tritrichomonas muris (Extended data Fig 5e).
It is increasingly appreciated that gut microbes alter the maturation and function of microglia31, can influence the activity of neurons in the central nervous system32 and contribute to neuroinflammation and neuropathology in models of Alzheimer’s33 and Parkinson’s disease34. However, only initial surveys of the gut microbiota have been reported in patients with neurological conditions35 and thus far results from initial studies in ALS patients have been mixed36,37. One study reported significant differences between the microbial constituencies of ALS/FTD patients and controls36 while another found no clear distinctions37. Given the genetic heterogeneity exhibited within ALS patients, it is perhaps not surprising that early studies have failed to reach consensus.
Consistent with the idea there are complex interactions between a patient’s germline genotype and their gut microflora in ALS, it was recently reported that SOD1 transgenic mice displayed a more rapid decline when bacterial load was reduced, which was linked to reduced bacterial production of nicotinamide29. While we cannot rule out the presence of protective microbes in some environments, our studies suggest that lowering bacterial load in C9orf72 mutants was in aggregate protective, likely by reducing exposure of their genetically sensitized innate immune response to microbially-derived inflammatory factors. In sum, our studies suggests that the microbiome may be an important governor of onset and progression in patients with C90RF72 mutations, including those experiencing autoimmune and inflammatory conditions prior to an ALS/FTD diagnosis15,16. To test this idea, a key future experiment will be to identify C90RF72 repeat expansion carriers within known families and to determine whether the gut microbiota differs between individuals that remain healthy and those acquiring ALS/FTD.
Extended Data
Extended Data Fig. 1 |.
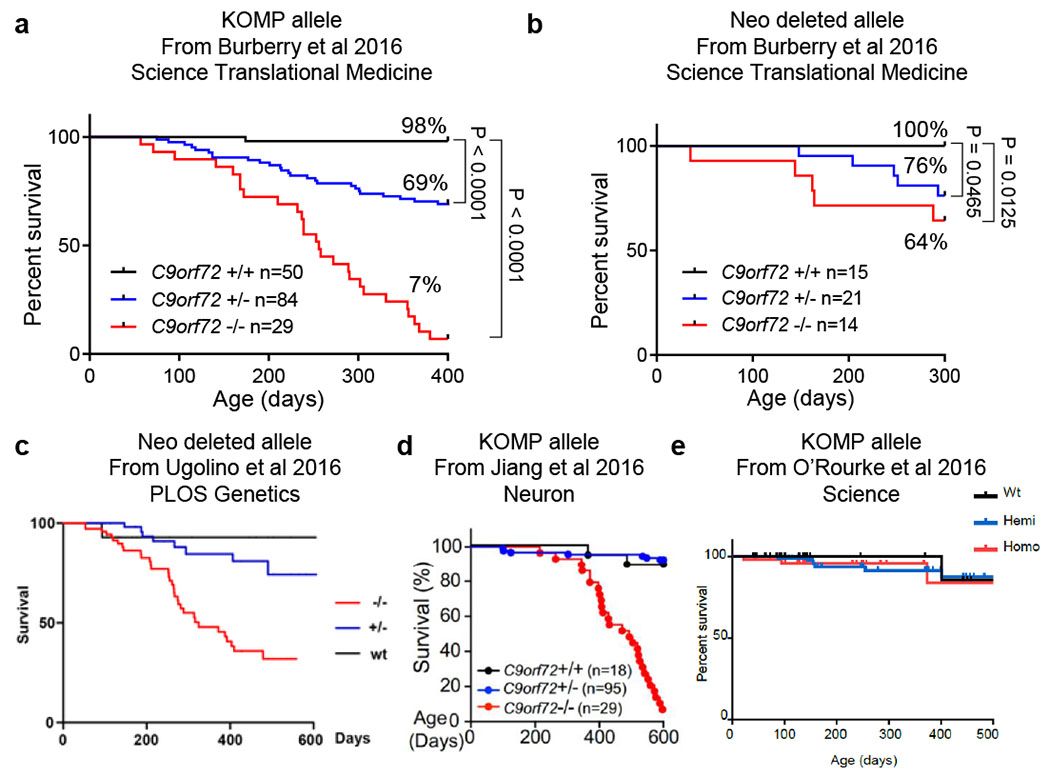
Previously published C9orf72 LOF survival studies
a, From Burberry et al (2016) Science Translational Medicine, originally Figure 2A. b, From Burberry et al (2016) Science Translational Medicine, originally Figure 2B. c, From Ugolino et al (2016) PLOS Genetics, originally Figure 1c. d, From Jiang et al (2016) Neuron, originally Figure 1D. e, From O’Rourke et al (2016) Science, originally Figure S1G. Permissions to reproduce these figures were obtained from each publishing group. Permission for c is licensed under “CC BY 4.0”.
Extended Data Fig. 2 |.
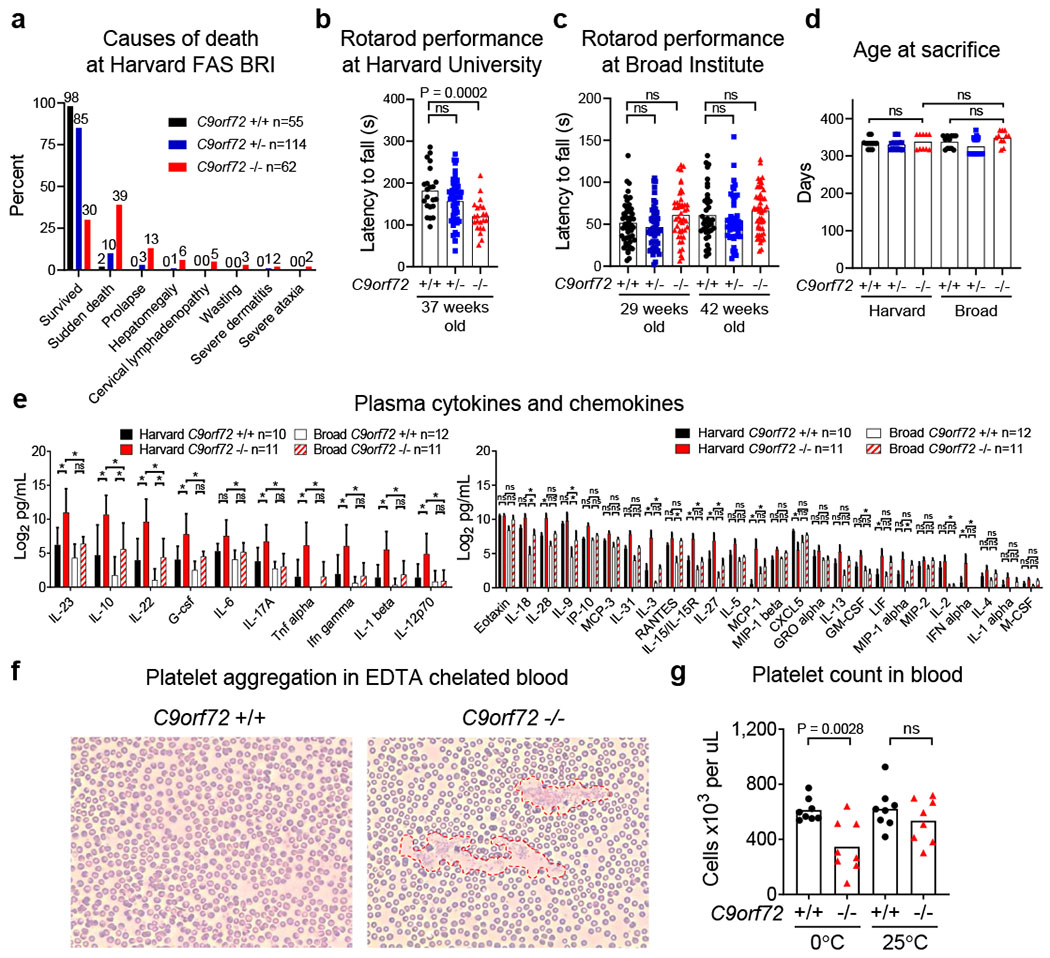
Causes of death, motor performance, plasma cytokines and identification of pseudothrombocytopenia in C9orf72 LOF mice
a, Causes of death or premature mortality of C9orf72Harvard mice in Fig. 1b. b, Accelerating rotarod performance of 37-week-old C9orf72Harvard neo deleted mice (+/+ n=22; +/− n=50; −/− n=22). C9orf72Broad mice . b-c, One way ANOVA with Dunnett multiple comparison. Each point represents the average of three trials per animal. c, Accelerating rotarod performance of C9orf72Broad neo deleted mice at 29-weeks of age (+/+ n=53; +/− n=52; −/− n=48) or 42-weeks of age (+/+ n=38; +/− n=48; −/− n=48). (One way ANOVA with Sidak multiple comparisons). d, Age at sacrifice of animals in Fig. 1d–g (One way ANOVA with Sidak multiple comparison). e, Plasma cytokines and chemokines at sacrifice from mice in Fig. 1d–g. Mean ± s.d. Two way ANOVA with Tukey multiple comparisons. f, Peripheral blood smear of 18-week-old C9orf72Harvard neo deleted mice. Platelets from C9orf72Harvard −/− mice were prone to aggregate (outlined by red dashed lines) in the presence of EDTA at 0ºC. g, This pseudothrombocytopenia could be reversed by warming the blood to room temperature. Reduced platelet count in this model therefore represents an indirect measure of anti-platelet autoantibodies, rather than a reduction in platelet abundance. Two way ANOVA with Tukey multiple comparison. Each dot represents one animal.
Extended Data Fig. 3 |.
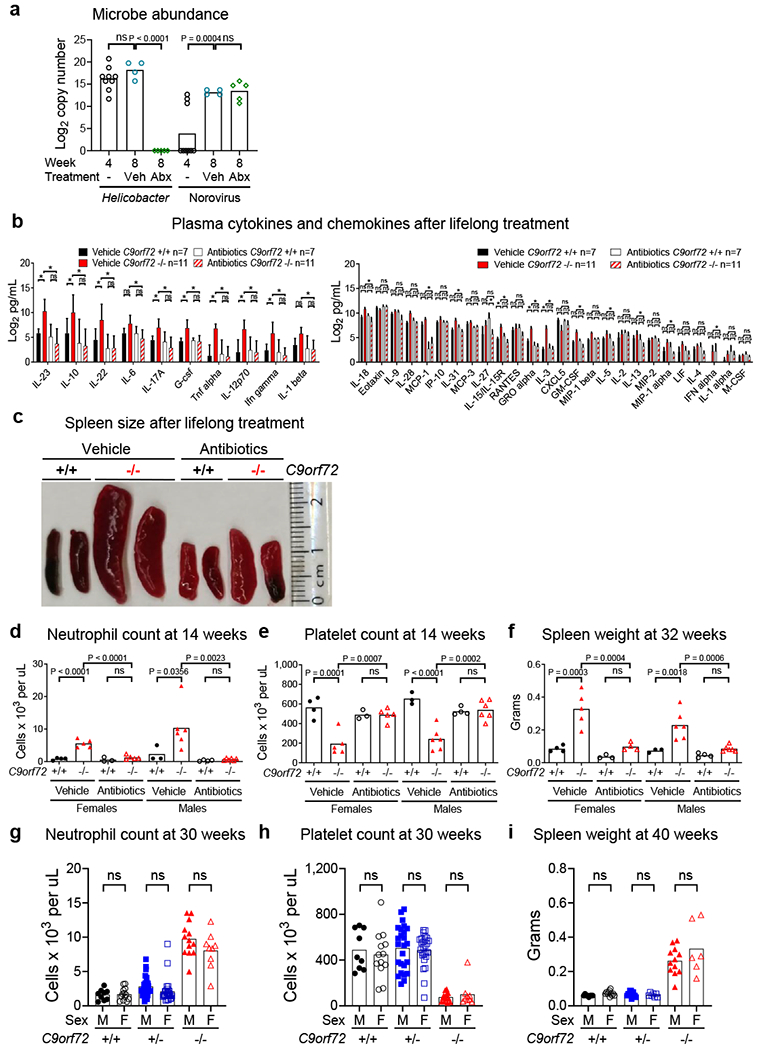
Cytokines and chemokines in lifelong antibiotics treated C9orf72 LOF mice and sex stratification of inflammatory phenotypes
a, PCR analysis of Helicobacter spp. and Norovirus DNA in fecal pellets. Each dot represents feces from one cage. One way ANOVA with Dunnett multiple comparisons. b, Plasma cytokines and chemokines of mice in Fig 2. Mean ± s.d. Two way ANOVA with Tukey multiple comparisons. c, Representative spleen size of mice in Fig 2. d-f, Total blood d, neutrophil count e, platelet count and f, spleen weight from mice in Fig 2 stratified by sex. d-i, Each dot represents one animal. One way ANOVA with Sidak multiple comparisons. Total blood g, neutrophil count and h, platelet count in 30-week-old C9orf72Harvard neo deleted mice stratified by sex (+/+ n=9M, n=13F; +/− n=25M n=27F; −/− n=13M n=9F). i, Spleen weight in 40-week-old C9orf72Harvard neo deleted mice stratified by sex (+/+ n=8M, n=11F; +/− n=13M n=7F; −/− n=12M n=6F).
Extended Data Fig. 4 |.
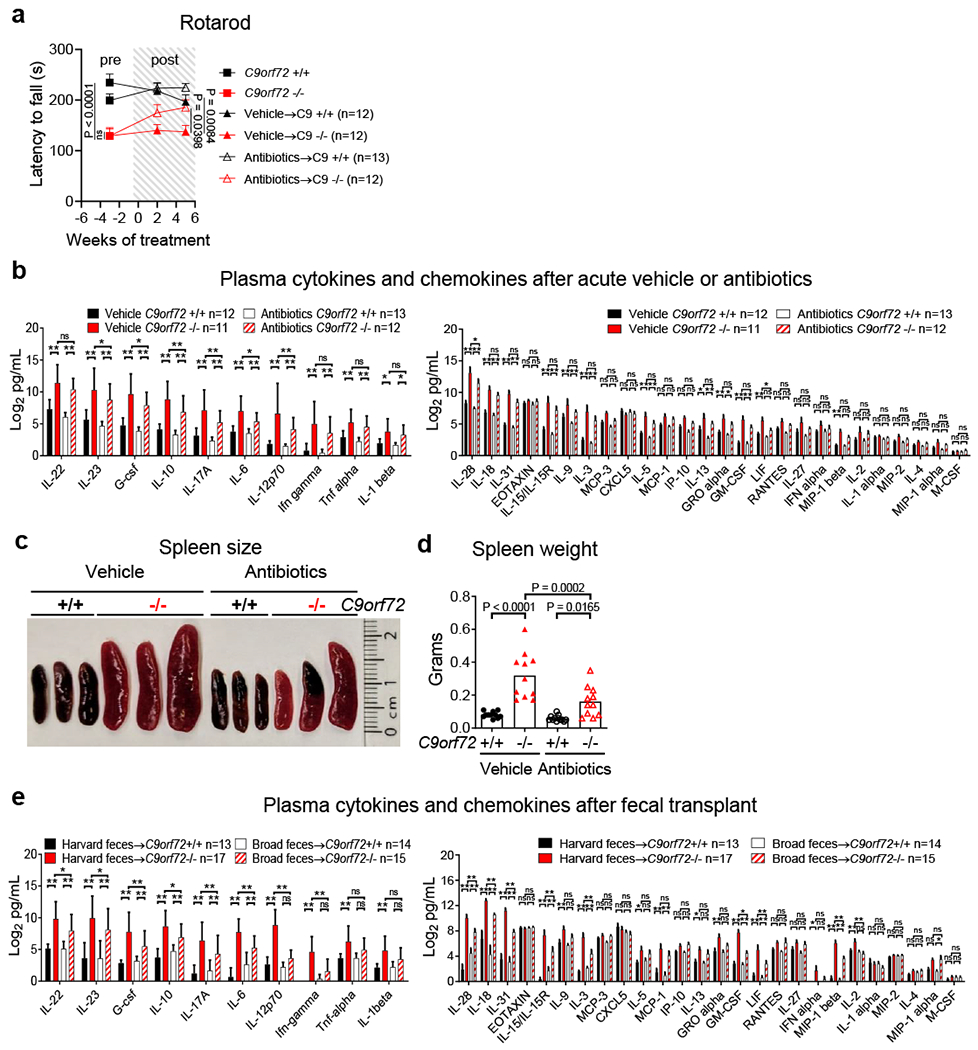
Acute antibiotics treatment improves motor function and mitigates splenomegaly and cytokine burden in C9orf72 LOF mice
a, Accelerating rotarod performance of mice in Fig 3a. Each point represents the average of three trials per animal. Two way ANOVA with Dunnett multiple comparisons. b, Plasma cytokines and chemokines of mice in Fig 3a–d after 7 weeks of treatment. b,e, Mean ± s.d. Two way ANOVA with Tukey multiple comparisons. c, Representative spleen size and d, spleen weight of mice in Fig 3a after 8 weeks of treatment. Each dot represents one animal. One way ANOVA with Sidak multiple comparisons. e, Plasma cytokines and chemokines of mice in Fig 3e–h 10 weeks after fecal transplant.
Extended Data Fig. 5 |.
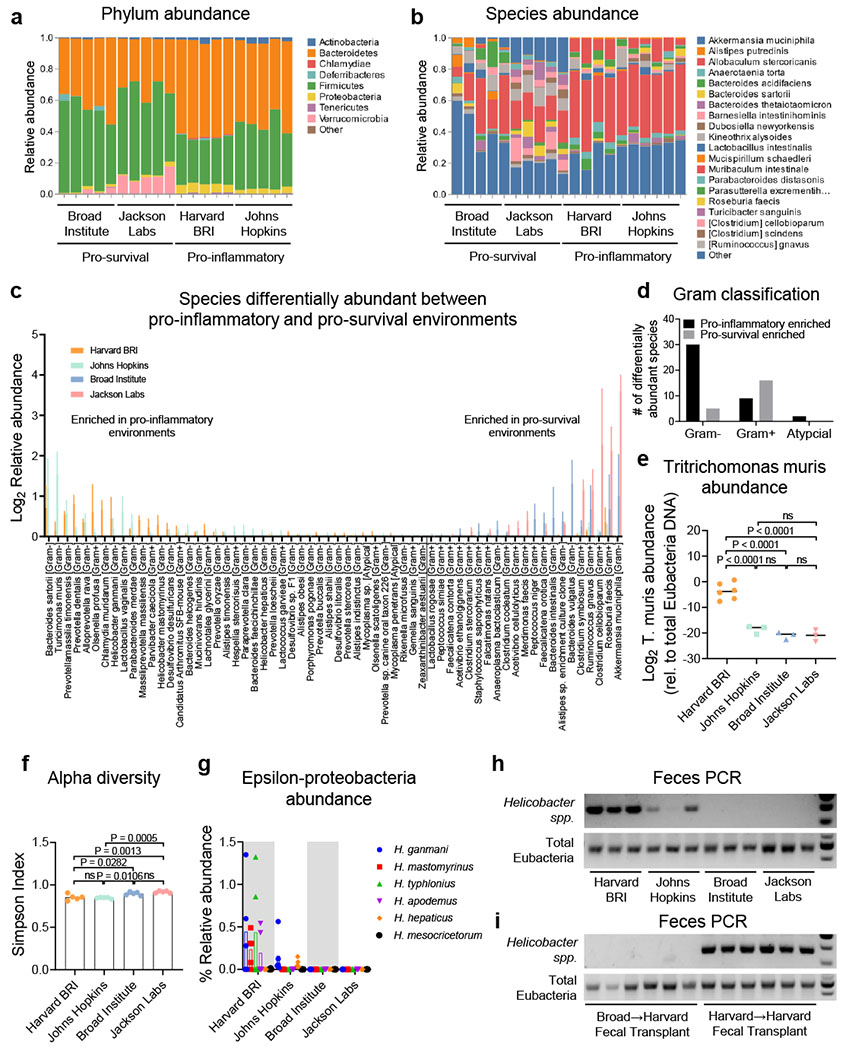
Bacteria and protozoa diversity across environments
a, Phylum level and b, species level relative abundance of bacteria from 16S rDNA sequencing in Fig. 3i. Each bar represents sequencing from one pellet per cage. c, Relative abundance and d, Gram stain classification of bacterial species whose abundance was significantly different between pro-inflammatory environments (Harvard BRI/JHU) and pro-survival environments (Broad Institute and Jackson Labs). (T test with Bonferroni multiple comparisons) 62/301 detected species had significance P < 0.0002. n=5 fecal pellets per environment. Mean ± s.d. e, Quantitative RT-PCR analysis of Tritrichomonas muris 28S rDNA relative to total Eubacteria 16S rDNA in feces, e-g Each dot represents a fecal pellet from one cage. One way ANOVA with Tukey multiple comparisons, f, Simpson index of fecal alpha diversity, g, Relative abundance of epsilon proteobacteria [Helicobacter], h, PCR analysis of Helicobacter spp. 16S rDNA and total Eubacteria 16S rDNA in feces, i, PCR analysis of Helicobacter spp. 16S rDNA and total Eubacteria 16S rDNA in feces (6 weeks-post-transplant) from Fig 3e.
Extended Data Fig. 6 |.
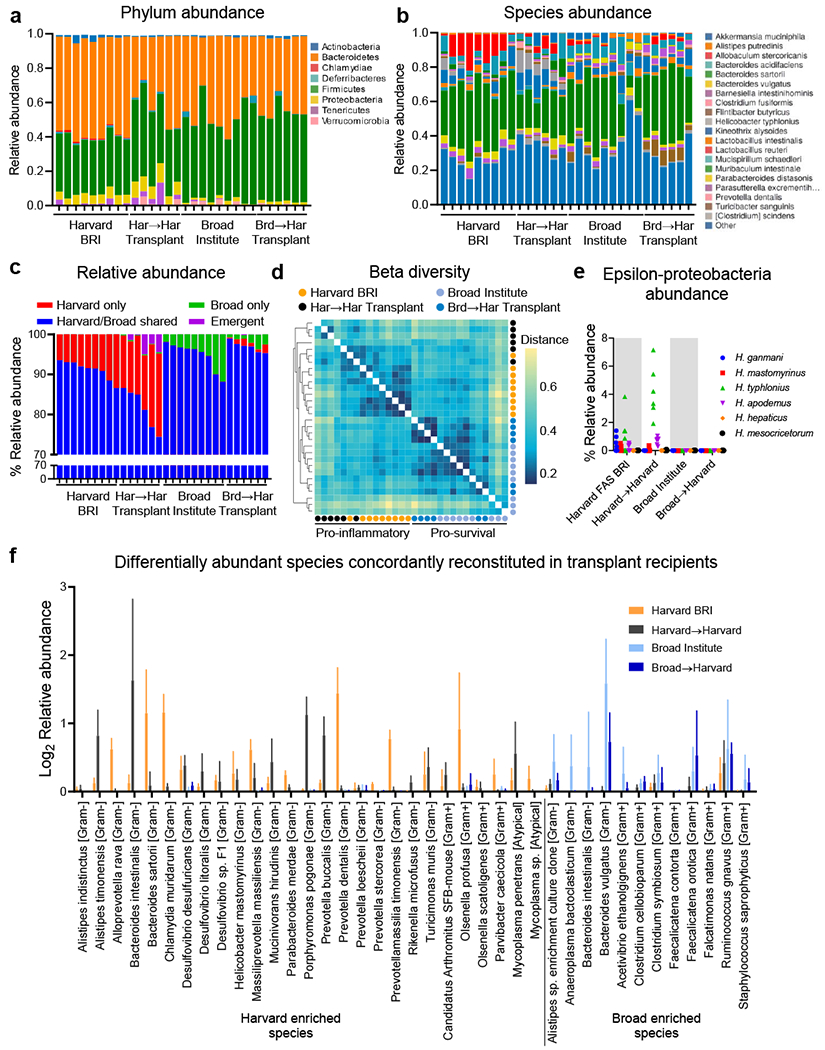
Environment enriched bacteria engraft fecal transplant recipients
a-f, Analysis of bacteria in feces 10-weeks-post-transplant from mice in Fig 3e by 16S rDNA sequencing. Each bar represents a fecal sample from an individual cage, a, Phylum level and b, species level relative abundance, c, Relative abundance of bacterial species grouped as those only observed in cages from Harvard BRI (Harvard-only), those only observed in cages from the Broad Institute (Broad-only), those observed in cages from Harvard BRI and the Broad Institute (Harvard/Broad-shared) or those not observed in Harvard BRI or Broad Institute cages but detectable in transplant recipient cages (Emergent), d, Bray-Curtis dissimilarity matrix of feces beta diversity, e, Relative abundance of epsilon proteobacteria [Helicobacter], (f) Putative pro-inflammatory species (n=27) enriched in pro-inflammatory environments (Harvard BRI/JHU) that were also enriched in Harvard->Harvard recipients and putative pro-survival species (n=12) enriched in pro-survival environments (Broad/Jackson Labs) and enriched in Broad->Harvard recipients.
Extended data Fig. 7 |.
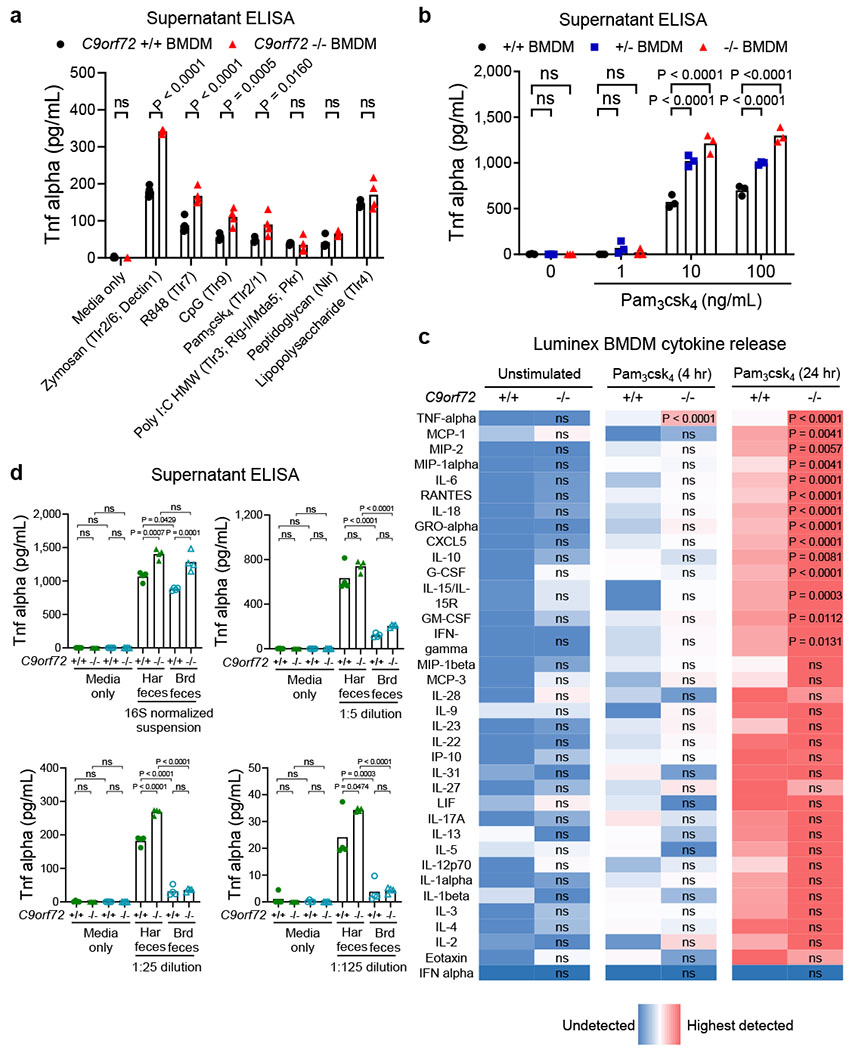
C9orf72 restricts myeloid cytokine release in response to foreign stimuli.
a-d, Analysis of cytokines and chemokines in supernatant 24 hours after stimulation of bone marrow derived macrophages (BMDM) with a-c, activators of Toll-like receptor (Tlr) or NOD-like receptor (Nlr) agonists or d, filtered Eubacteria-normalized fecal preparations. c, The abundance of cytokine and chemokine in supernatant was normalized and color coded (Blue low; Red high) relative to the average level of each molecule in unstimulated +/+ BMDM wells. Levels of each analyte were measured by Luminex in multiplex. d, The abundance of total Eubacteria in each fecal sample was measured by qPCR for 16S rDNA and this value was used to normalize fecal Eubacteria bacteria concentration prior to generation of the dilution curve. Each dot represents one well. Panels are representative of a n=2 replicate experiments; b n=5 replicate experiments; c, One representative experiment with average of n=3 technical replicates per condition; d n=2 replicate experiments). a Two way ANOVA with Sidak multiple comparison. b, Two way ANOVA with Dunnett multiple comparison. c, Two way ANOVA with Sidak multiple comparison for each analyte tested. d, One way ANOVA with Sidak multiple comparison.
Extended Data Fig. 8 |.
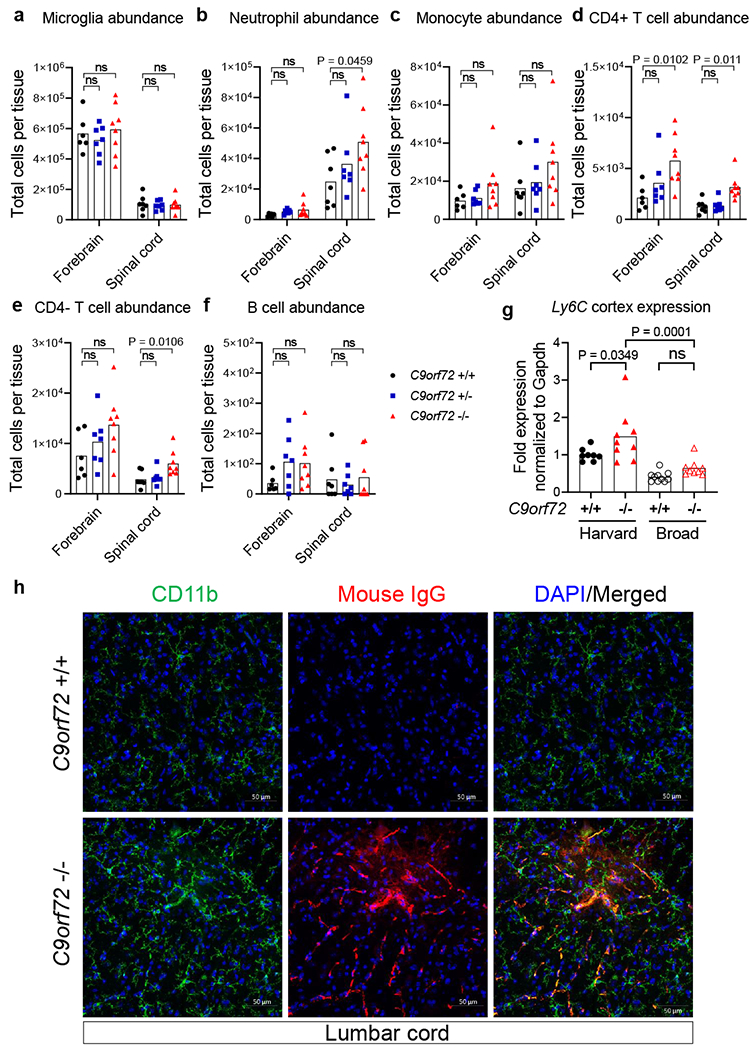
Neutrophils and T cells infiltrate C9orf72 LOF spinal cord.
a-f, Mass cytometry interrogation of single cell dissociated forebrain or spinal cord from 36-week-old C9orf72Harvard neo deleted male and female mice (+/+ n=7; +/− n=7; −/− n=8). One C9orf72 +/+ forebrain sample failed and was excluded from analysis. Representative gating scheme can be found in Supplementary Info. Populations were defined as a, CD45mid CX3CR1+ CD39+ Microglia, b, CD45hi Ly6C+ Ly6Ghi Neutrophils, c, CD45hi Ly6C+ Ly6Glo Monocytes, d, CD45hi CD3e+ CD4+ T cells, e, CD45hi CD3e+ CD4− T cells and f, CD45hi CD 19+ B cells. Quantitation of total cells per tissue was obtained by multiplying the percentage of each gated population by the total cells recovered from that mouse’s tissue. Each dot represents one mouse. Two Way ANOVA with Dunnett multiple comparison, g, Quantitative RT-PCR of Ly6C expression in 47-week-old C9orf72Harvard neo deleted (+/+ n=8; −/− n=9) or C9orf72Broad neo deleted (+/+ n=10; −/− n=9) total cortex tissue. Each dot represents one animal. One Way ANOVA with Sidak multiple comparison, h, Orthogonal projection of confocal imaging of CD11b and mouse immunoglobulin IgG in 43-week-old C9orf72Harvard lumbar spinal cord .
Extended data Fig. 9 |.
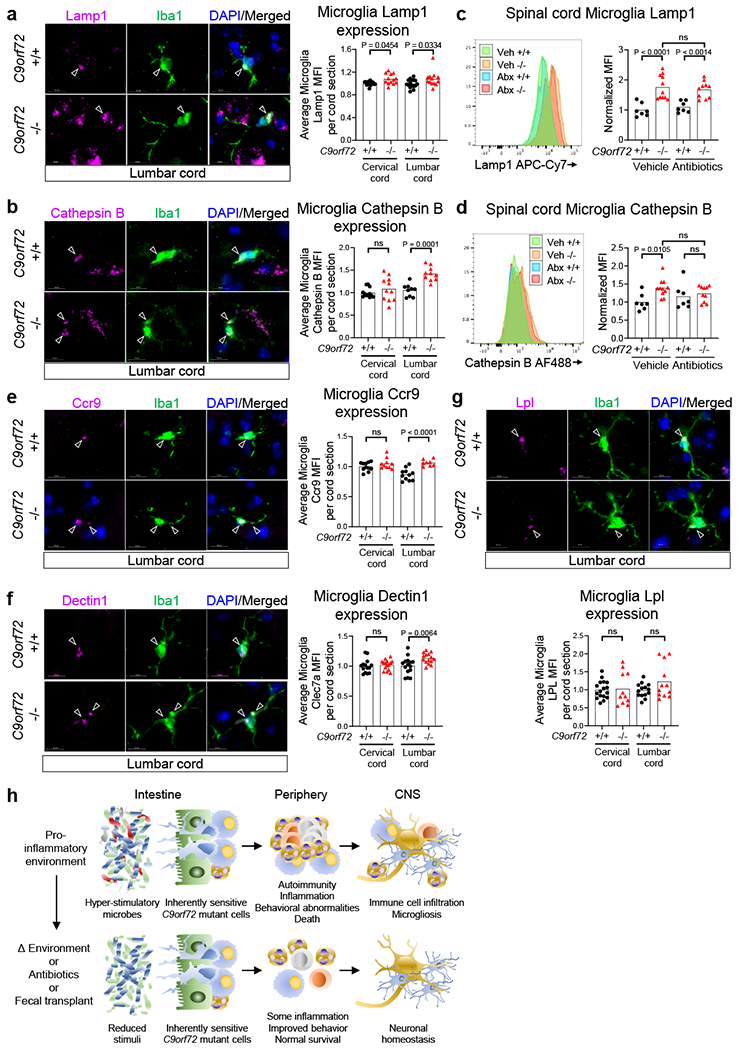
Elevated lysosomal proteins and microgliosis in C9orf72 LOF spinal cord.
a-b,e-g, Orthogonal projection and quantification of confocal imaging of a, Lamp1, b, Cathepsin B, e, Ccr9, f, Dectin1/Clec7a, and g, Lpl in Iba1+ microglia in 55-week-old C9orf72Harvardspinal cord (One way ANOVA with Sidak multiple comparisons). Each dot represents the average mean fluorescent intensity (MFI) of the antigen within microglia on a given spinal cord section. >100 microglia surveyed per section. Sections from n=3 C9orf72 +/+ and n=3 C9orf72 −/− mice surveyed, c-d, Flow cytometry quantification of c, Lamp1 or d, Cathepsin B in CD45mid CD11b+ CD39+ microglia from spinal cord of C9orf72Harvard neo deleted mice in Fig. 2. One way ANOVA with Sidak multiple comparisons, h, Graphical illustration of C9orf72 functioning within the hematopoietic system to restrict the development of inflammation, autoimmunity, peripheral immune infiltration into the central nervous system (CNS) and microgliosis in response to hyper-stimulatory communities of gut microflora. The microglia image was modified from Servier Medical Art (https://smart.servier.com/smartimage/microglia-2/) under license “CC BY 3.0”.
Supplementary Material
Acknowledgements
A.B., M.F.W., J.M., and K.E. conceived the study. Experiments performed by A.B. (All Figures), F.L.(Fig 1 and Extended data Fig 2), M.F.W. (Fig 1 and Extended data Fig 2), K.S.S. (Fig 4 and Extended data Fig 8-9), A.C. (Fig 2-4 and related Extended data), J.M. (Fig 1-3 and related Extended Data), N.V.G. (Extended Data Fig 2), J-Y.W. (Fig 4 and Extended data Fig 8), J.K. & G.G. (Extended Data Fig 8), M.Q. & P.E. & C.C. (Fig 2-3 and related Extended Data), A.B., F.L., K.S.S., J.K., G.G, N.V.G., J-Y.W., O.P., I.K., D.T.S., K.E. interpreted results. A.B. and K.E. wrote manuscript. Support to KE provided by The Merkin Fund at Broad Institute, Target ALS, NIH 5R01NS089742, the Harvard Stem Cell Institute and UCB. A.B. was supported by NIH 5K99AG057808-02, M.F.W. was supported by NIH 1K99MH119327-01. We thank Jiou Wang for providing JHU feces.
Footnotes
Conflicts of interest
KE is a co-founder of Q-State Biosciences, Quralis and Endear Therapies.
Data availability statement
The 16S rDNA sequencing datasets generated and analyzed herein are available through the GEO repository at GSE147325. All other data generated or analyzed herein are included in this published article (and supplementary information files).
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
References
- 1.DeJesus-Hernandez M et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C90RF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Majounie E et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 11, 323–330 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mori K et al. The C9orf72 GGGGCC Repeat Is Translated into Aggregating Dipeptide-Repeat Proteins in FTLD/ALS. Science 339, 1335–1338 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Ash PEA et al. Unconventional translation of C90RF72 GGGGCC expansion generates insoluble polypeptides specific to C9FTD/ALS. Neuron 77, 639–646 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donnelly CJ et al. RNA Toxicity from the ALS/FTD C90RF72 Expansion Is Mitigated by Antisense Intervention. Neuron 80, 415–428 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Rourke JG et al. C9orf72 is required for proper macrophage and microglial function in mice. Science 351, 1324–1329 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burberry A et al. Loss-of-function mutations in the C90RF72 mouse ortholog cause fatal autoiimnune disease. Sci. Transl. Med 8, 347ra93 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nassif M„ Woehlbier U & Manque PA. The Enigmatic Role of C90RF72 in Autophagy. Front. Neurosci 11, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi Y et al. Haploinsufficiency leads to neurodegeneration in C90RF72 ALS/FTD human induced motor neurons. Nat. Med 24, 313–325 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whary ΜT & Fox JG Natural and Experimental Helicobacter Infections. https://www.ingentaconnect.corn/content/aalas/cm/2004/00000054/00000002/art00002%3bjsessionidMhkpa2iwdtskb.x-ic-live-02# (2004). [PubMed] [Google Scholar]
- 11.Flannigan KL & Denning TL Segmented filamentous bacteria-induced iimnune responses: a balancing act between host protection and autoimmunity. Immunology 154, 537–546 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ugolino J et al. Loss of C9orf72 Enhances Autophagic Activity via Deregulated mTOR and TFEB Signaling. PLOS Genet. 12,el006443 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang J et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C90RF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron 90, 535–550 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Atanasio A et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci. Rep 6, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller ZA et al. Increased prevalence of autoiimnune disease within C9 and FTD/MND cohorts. Neurol. Neuroimmunol. Neuroinflammation 3, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fredi M et al. C9orf72 Intennediate Alleles in Patients with Amyotrophic Lateral Sclerosis, Systemic Lupus Erythematosus, and Rheumatoid Arthritis. NeuroMolecular Med. (2019) doi: 10.1007/sl2017-019-08528-8. [DOI] [PubMed] [Google Scholar]
- 17.Stine JG & Lewis JH Hepatotoxicity of antibiotics: a review and update for the clinician. Clin. Liver Dis 17, 609–642, ix (2013). [DOI] [PubMed] [Google Scholar]
- 18.Ransohoff RM How neuroinflammation contributes to neurodegeneration. Science 353, 777–783 (2016). [DOI] [PubMed] [Google Scholar]
- 19.McCauley ΜE & Baloh RH Inflammation in ALS/FTD pathogenesis. Acta Neuropathol. (Berl.) 137, 715–730 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao W, Beers DR & Appel SH Immune-mediated Mechanisms in the Pathoprogression of Amyotrophic Lateral Sclerosis. J. Neuroimmune Pharmacol. Off. J. Soc. Neurolmmune Pharmacol 8, 888–899 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zondler L et al. Peripheral monocytes are functionally altered and invade the CNS in ALS patients. Acta Neuropathol. (Berl.) 132, 391–411 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Zhang GX, Li J, Ventura E & Rostami A Parenchymal microglia of naive adult C57BL/6J mice express high levels of B7.1, B7.2, and MHC class II. Exp. Mol. Pathol 73, 35–45 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Lall D & Baloh RH Microglia and C9orf72 in nemoinflammation and ALS and frontotemporal dementia. J. Clin. Invest 127, 3250–3258 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y et al. The C9orf72-interacting protein Smcr8 is a negative regulator of autoimmunity and lysosomal exocytosis. Genes Dev. 32, 929–943 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H et al. Different Neurotropic Pathogens Elicit Neurotoxic CCR9- or Neurosupportive CXCR3-Expressing Microglia. J. Immunol 177, 3644–3656 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Krasemann S et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keren-Shaul H el al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290.el7 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Nilsson H-O et al. High Prevalence of Helicobacter Species Detected in Laboratory Mouse Strains by Multiplex PCR-Denaturing Gradient Gel Electrophoresis and Pyrosequencing. J. Clin. Microbiol 42, 3781–3788 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blacher E et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature 572, 474–480 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Zliai R et al. Strain-Specific Anti-inflammatory Properties of Two Akkennansia muciniphila Strains on Chronic Colitis in Mice. Front. Cell. Infect. Microbiol 9, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emy D et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. 18, 965–977 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olson CA el al. The Gut Microbiota Mediates the Anti-Seizure Effects of the Ketogenic Diet. Cell 173, 1728–1741.el3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harach T et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep 7, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sampson TR et al. Gut Microbiota Regulate Motor Deficits and Neuroinflaimnation in a Model of Parkinson’s Disease. Cell 167, 1469–1480.el2 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tremlett H, Bauer KC, Appel-Cresswell S, Finlay BB & Waubant E The gut microbiome in human neurological disease: A review. Ann. Neurol 81, 369–382 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Fang X et al. Evaluation of the Microbial Diversity in Amyotrophic Lateral Sclerosis Using Fhgh-Throughput Sequencing. Front. Microbiol 7, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brenner D et al. The fecal microbiome of ALS patients. Neurobiol. Aging 61, 132–137 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.