

Abstract
The therapeutic efficacy of adoptive transfer of T cells transduced with chimeric antigen receptors (CARs) has been limited in the treatment of solid cancers, partly due to tumor antigen heterogeneity. Overcoming lack of universal tumor antigen expression would be achieved if CAR T cells could induce bystander effects. To study this process, we developed a system where CAR T cells targeting mesothelin could cure tumors containing 100% antigen-positive cells in immunocompetent mice. Using this model, we found that the CAR T cells were unable to cure tumors, even when only 10% of the tumor cells were mesothelin negative. A bystander effect was not induced by co-administration of anti-PD-1, anti-CTLA-4, or anti-TGF-β (transforming growth factor β) antibodies; agonistic CD40 antibodies; or an IDO (indoleamine 2,3-dioxygenase) inhibitor. However, pretreatment with a non-lymphodepleting dose of cyclophosphamide (CTX) prior to CAR T cells resulted in cures of tumors with up to 25% mesothelin-negative cells. The mechanism was dependent on endogenous CD8 T cells but not on basic leucine zipper transcription factor ATF-like 3 (BATF3)-dependent dendritic cells. These data suggest that CAR T cell therapy of solid tumors, in which the targeted antigen is not expressed by the vast majority of tumor cells, will not likely be successful unless combination strategies to enhance bystander effects are used.
Graphical Abstract
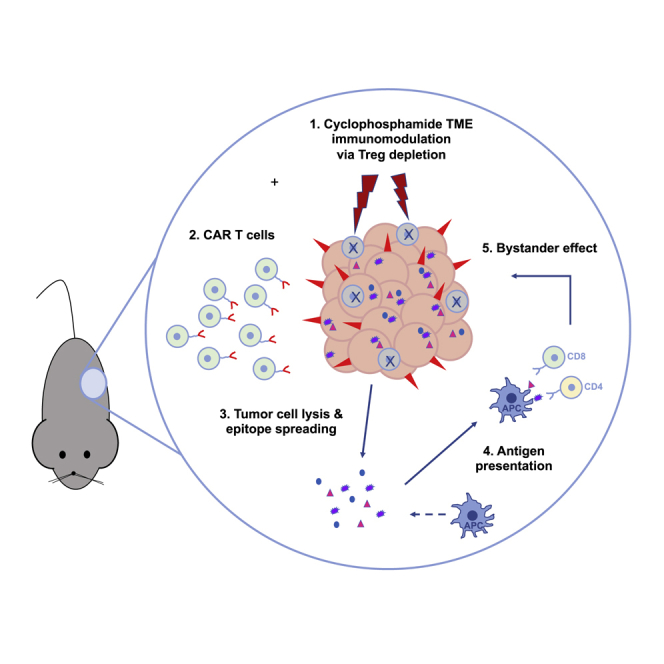
CAR T cell therapy has limited efficacy in solid tumors, partly due to tumor antigen heterogeneity. Klampatsa and colleagues demonstrated, using a single, non-lymphodepleting dose of cyclophosphamide prior to CAR T cells, that eradication of tumors <100% targeted antigen positive is possible, due to the induction of a bystander, CD8-dependent effect.
Introduction
Although use of the adoptive transfer of T cells transduced with chimeric antigen receptors (CARs) targeting CD19 has revolutionized the treatment of B cell malignancies,1 limited therapeutic efficiency of CAR T cells has been observed in solid tumors.2 Several barriers, not present in hematologic malignancies, likely prevent CAR T cells from being efficacious in solid tumors, including poor trafficking to the tumor, an immunosuppressive tumor microenvironment (TME), overexpression of checkpoint inhibitors, and suppression due to intrinsic inhibitory T cell programs.3, 4, 5, 6
In addition, no matter how active any antigen-specific CAR T cell might be, successful therapy will still need to overcome the critical challenge of tumor antigen heterogeneity.7 Unlike B cell malignancies, which uniformly express high levels of their CD19 CAR T target antigen, solid tumor cells express antigens heterogeneously and at different levels. Furthermore, therapy-induced immune editing can lead to the selection of CAR target antigen-negative tumor cells, allowing for tumor antigen “escape.”8,9
It has been hypothesized that tumor heterogeneity can be overcome by the induction of “bystander effects”; that is, the ability of the CART cells to also induce killing of tumor cells that are not expressing the CAR targeted antigen(s).10,11 This is an important issue, as the extent of bystander killing is critical in specifying a “cutoff value” for the percentage of tumor antigen positivity needed for eligibility in a clinical trial. However, this hypothesis has not be adequately tested and forms the focus of this work.
Although it is relatively straightforward to evaluate agents that augment the efficacy of CAR T cells by measuring tumor size,3,5 it is much more challenging to answer the specific questions of whether bystander effects are present and whether they can be enhanced. The majority of preclinical CAR T cell studies have been performed with human lymphocytes that have been injected into immunodeficient mice bearing human tumors. However, to assess immunologic bystander effects, mouse models with intact immune systems and the use of murine-derived CAR T cells are required. It would then be possible to define bystander effects by determining how well CAR T cells could treat defined mixtures of target antigen-positive and target antigen-negative tumor cells.
Although the tumor-mixing approach seems straightforward, it requires a system where 100% antigen-positive tumors can be eradicated by mouse CAR T cells in immune-competent animals. Unfortunately, mouse CAR T cell efficacy is usually not high, and augmentation strategies traditionally involve whole-body irradiation and/or lymphodepletion—interventions that make bystander interpretations difficult.12, 13, 14, 15
We were able to develop such a mixing model, using potent murine CAR T cells that react against a human mesothelin-expressing murine tumor cell line that grows in immunocompetent mice, allowing us to directly test the bystander hypothesis. Using this model, we show that these CAR T cells could cure 100% mesothelin-positive tumors but were unable to cure tumors that did not universally express mesothelin, demonstrating the lack of a bystander effect.
We next tested the hypothesis that specific immune modulatory agents that are directly or indirectly related to impaired T cell function could augment bystander effects in our model. These included anti-PD-1, anti-CTLA-4, or anti-TGF-β antibodies; an agonistic CD40 antibody; and an indoleamine 2,3-dioxygenase (IDO) inhibitor. However, none of these therapies helped induce bystander effects. In contrast, pre-treatment of the mice with low-dose cyclophosphamide (CTX) induced a bystander effect that resulted in cure of tumor mixtures. This effect was dependent on endogenous CD8 T cells but not on basic leucine zipper transcription factor ATF-like 3 (BATF3)-dependent type 1 dendritic cells (DCs).
Results
Development of an Immunocompetent Model in which Murine CAR T Cells Can Eliminate Established Malignant Mesothelioma (MM) Tumors
To test the hypothesis that CAR T cells induce significant bystander effects, we needed to create an immunocompetent mouse model in which we could cure established tumors that uniformly expressed the CAR target antigen. We previously developed a retroviral version of a CAR targeting human mesothelin (based on the SS1 anti-mesothelin antibody), which effectively transduced mouse T cells and had some anti-tumor activity against murine tumor cell lines expressing human mesothelin, including the MM AE17om cell line.16,17 We were unable, however, to obtain tumor cures, even when we used two injections of the SS1 CAR T cells to treat relatively small tumors (data not shown). Instead, we were able to cure tumors with two doses of 10 million “M11” CAR T cells administered on days 4 and 6 after tumor inoculation (Figures 1A and 1B). In contrast, this treatment regimen had no effect on AE17o cells not expressing human mesothelin (Figure 1C).
Figure 1.

M11 CAR T Cells Exert Minimal Bystander Effects in a Syngeneic Malignant Mesothelioma (MM) Mouse Model
A mouse model of MM was developed, using murine AE17 MM cells, transduced with chicken ovalbumin and human mesothelin (AE17om). 2 million AE17om cells were injected into the right flanks of B6 mice, which, by day 3 post-injection, developed into established tumors ∼50 mm3 in size. (A) The schema used to cure these tumors involved two doses of 107 transduced M11 CAR T cells each, given intravenously on day 4 and day 6 post-tumor inoculation. All experiments were repeated at least twice; n = 8 mice per group. (B) Tumor growth measurements over time show tumor cure. (C) M11 CAR T cells did not have an effect on tumors negative for mesothelin. AE17om cells were mixed with varying percentages of AE17o cells, i.e., cells that expressed ovalbumin but not human mesothelin, to assess whether our treatment schema could eradicate tumors that did not universally express the target antigen. (D) M11 CAR T cells could not cure tumors that were 50% mesothelin positive. (E) M11 CAR T cells could not cure tumors that were 75% mesothelin positive. (F) M11 CAR T cells could not cure tumors that were 90% mesothelin positive.
This new anti-mesothelin CAR, called M11, was developed in collaboration with Novartis (using a phage display library) that was much more effective in vitro and in vivo when expressed in human T cells. Accordingly, an M11 CAR (containing the mouse CD3 zeta and 41BB cytoplasmic domains; Figure S1A) was subcloned into a retroviral vector that was then used to efficiently transduce mouse T cells (Figure S1B). When co-cultured with the C57/B6-derived murine MM cell line AE17 that stably expressed both human mesothelin (in >98% of cells; Figure S1C) and the xeno-antigen chicken ovalbumin (AE17om cells), the M11 CAR T cells killed significantly more tumor cells than the SS1 CAR T cells and released significantly more interferon gamma (IFNγ) at the same effector:target (E:T) ratios (Figure S1D).
Mouse T cells were produced that expressed the M11 CAR in >70% of T cells (Figure S2A) and were highly effective in vitro (Figure S2B). A single dose of 10 million transduced M11 CAR T cells significantly decreased the size of established 100% AE17om tumors, but it did not result in cures (Figures S2C–S2E).
We attempted to replicate this model in three other tumor cell lines that were transduced to stably express human mesothelin in 100% of their cells: a lung cancer cell line growing in C57B6 mice (TC-1 m) and two MM cell lines growing in BALB/c mice (AB-1 meso [AB-1m] and AB12 meso [AB12m]). All these human mesothelin-expressing tumors grew well in flank models; however, unlike the AE17om model, they were unable to be cured using the regimen in Figure 1A. I, we observed temporary slowing in tumor growth in two models (TC-1 m and AB12m) and no effect of treatment in the AB-1m model (Figure S3).
M11 CAR T Cells Exert Minimal Bystander Effects
To assess bystander effects, we used the AE17om model by mixing the AE17om cells (in which >98% of the cells expressed mesothelin; Figure S1C) with non-mesothelin-expressing AE17o cells at various ratios. These cell mixtures were injected subcutaneously into the flanks of C57/B6 mice, allowed to grow to a size of ∼50 mm3, and then treated using the schema shown in Figure 1A. In contrast to the cures seen in 100% mesothelin-positive tumors (Figure 1B), we were unable to cure tumors that contained 50% mesothelin-expressing cells (Figure 1D), 75% mesothelin-expressing cells (Figure 1E), or even 90% mesothelin-expressing cells (Figure 1F). These data demonstrate that our M11 CAR T cells induce very little bystander effects on their own.
Failure of Immune Stimulants to Induce Bystander Effects
We next hypothesized that that the lack of bystander effect was due to activation of T cell-inhibitory receptors, inhibition of T cell activity by transforming growth factor β (TGF-β), poor DC activation, or inhibition by regulatory T cells (Tregs).
Accordingly, we attempted to induce a bystander effect by treating tumors generated from 90% mesothelin-positive AE17om tumors with two doses of the M11 CAR T cells combined with established immune-activating agents that modulate these pathways. Administration of either anti-PD-1 or anti-CTLA-4 antibodies (to block inhibitory receptors PD-1 and CTLA-4, respectively) alone to tumor-bearing mice slowed tumor growth, but combinations of these agents with M11 CARs did not result in cures (Figures 2A and 2B). The use of an anti-TGF-β antibody or an agonistic CD40 antibody (to activate DCs) alone had no effect and did not include cures in combination with M11 CAR T cells (Figures 2C and 2D). Administration of an oral IDO inhibitor (to help activate DC and T cells) alone significantly slowed tumor growth, but when combined with M11 CARs, it did not result in cures (Figure 2E).
Figure 2.

M11 CAR T Cells Combined with Immunomodulatory Therapies Are Unable to Induce Bystander Effects in 90% Mesothelin-Positive Tumors
We used the treatment schedule that cured the 100% mesothelin-positive AE17om tumors with the addition of several immunomodulatory therapies in order to augment the ability of M11 CAR T cells to cure 90% mesothelin-positive AE17om tumors. All experiments were repeated at least twice; n = 8 mice per group. (A) The use of anti-PD-1 antibody (5mg/kg, given i.p. two times per week) resulted in some tumor regressions both as a single agent and in combination with M11 CAR T cells; however, no cures were observed. (B) Anti-CTLA-4 antibody (200 μg per mouse, given i.p. two times per week) did not have an effect as a single agent and only minimally increased cytotoxicity of M11 CAR T cells when given in combination. (C) Administration of an anti-TGF-β antibody (150 μg per mouse, given i.p. two times per week) had a synergistic effect with M11 CAR T cells and stopped tumor growth; however, only temporarily. (D) Agonistic CD40 antibody (40 μg per mouse, given i.p. as a single dose on day 7; 1 day after the 2nd dose of M11 CAR T cells) had no effect, either as a single agent or in combination with M11 CAR T cells. (E) The use of an IDO inhibitor (300 mg/kg, given by oral gavage daily for 2 weeks, starting on day 3 after tumor inoculation) did not enhance efficiency of M11 CAR T cells.
Low-Dose CTX Induces Bystander Effects that Result in Cures of MM Tumors
Administration of CTX in combination with adoptive T cell transfer has been shown to augment anti-tumor efficacy through multiple mechanisms (including increasing T cell expansion, activating DCs, and reducing Tregs), but, to our knowledge, its ability to induce bystander effects has not been studied. Accordingly, we tested the effects of one dose of CTX pre-treatment alone or in combination with two doses of M11 CAR T cells given to mice with tumors generated from 90% mesothelin-positive AE17om cells. Figures 3A and 3B show the results from two independent experiments that demonstrate the ability of CTX to induce bystander effects. In both cases, CTX alone or two doses of the M11-CAR T cells slowed tumor growth temporarily but did not cause tumor regression or a cure. In contrast, the combination of one dose of CTX followed by two doses of the M11-CAR T cells consistently resulted in tumor cures. To test the “strength” of the CTX-induced bystander effect, we performed the same experiments using established tumors from 75% mesothelin-positive AE17om cells and observed that the combination of CTX plus two doses of M11 CARs was able to induce cures as well (Figure 3C).
Figure 3.

CTX Can Induce Cures in <100% Mesothelin-Positive AE17om Tumors
(A and B) In two separate experiments, we show that administration of CTX (100 mg/kg, given as a single i.p. dose on day 3 post-tumor inoculation, 1 day before the first dose of M11 CAR T cells) induces cures in 90% mesothelin-positive tumors. (C) We were able to cure mice with 75% mesothelin-positive AE17om tumors.
To quantify the effects of CTX by itself and choose the appropriate dose for our experiments, we first carried out a dose-response experiment using a single dose of 50, 100, or 150 mg/kg CTX on day 3 post-tumor inoculation (Figure S4A). Tumor measurements post-treatment showed some anti-tumor activity that was dose dependent, but there was subsequent tumor outgrowth in all groups (Figure S4B). We selected 100 mg/kg CTX as a treatment dose for follow-up experiments, because it was the lowest dose that had some anti-tumor effects but did not eradicate the tumors. This was not a lymphodepleting dose; Figures S4C and S4D shows only slight decreases in lymphocytes on days 1 and 5 post-CTX administration.
CTX Induces Increased Persistence of M11 CAR T Cells
The persistence of adoptively transferred mouse T cells is quite short, peaking at around 1 week post-transfer, with the cells detected at very low levels from around day 10 onward (data not shown). Since one previously reported consequence of the administration of CTX is increased persistence of adoptively transferred T cells,18 we analyzed tumor-draining lymph nodes (TDLNs) and spleens on day 17 after tumor inoculation (2 weeks post-CTX) for the detection of M11 CAR T cells. As expected, low numbers of M11 CAR T cells were present in TDLNs and spleens (∼0.2% of cells), which were only slightly increased in the mice treated with CTX and M11 cells (Figure 4A).
Figure 4.

Effect of CTX Treatment on M11 CAR T Cells, Endogenous T Cells, and Regulatory T Cells (Tregs)
TDLNs, spleens, and tumors were harvested from mice treated as described in Figures 3A and 3B. After digestion, they were subjected to flow cytometry. (A) M11 CAR T cells were tracked by flow cytometry through their GFP label. CTX treatment increased the persistence of M11 CAR T cells in TDLNs (left graph) and spleens (right graph) (numbers are in percent total live cells). Samples: n = 3 per group. Statistics by 2-way ANOVA: ∗∗p < 0.01. (B) The frequency of T cells in AE17om tumors post-treatment was assessed. When used alone, CTX decreased the percentage of CD3+ and CD4+ T cells of live tumor cells. Tumors treated with the combination of CTX plus M11 CAR T cells showed significantly increased infiltration of CD8+ T cells compared to the untreated group, the group treated with M11 CAR T cells only, or the CTX-only group. Samples: n = 3 per group. Statistics by 2-way ANOVA: ∗p < 0.05; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. (C) Treg (CD25+CD3+CD4+FoxP3+) frequency was seen to be significantly decreased in TDLNs of mice treated with CTX alone. This effect was not replicated in the spleens of these mice, where a slight decrease in Tregs was seen in CTX-treated samples, but it was not significant. Tumors treated with CTX and with CTX plus M11 CAR T cells had a significantly smaller percentage of Tregs compared to the Tregs in tumors of untreated mice or mice that were treated with M11 CAR T cells. Samples: n = 3 per group. Statistics by 2-way ANOVA: ∗p < 0.05; ∗∗∗∗p < 0.0001.
Low-Dose CTX Reduces the Frequency of CD4+ Tregs in TDLNs and Tumors
We first hypothesized that the immunosuppressive CD4+ Tregs might be important in augmenting endogenous T cell responses. Therefore, we assessed the frequency of these cells in tumor-bearing mice 5 days after injection of CTX and following treatment with M11 CAR T cells. At this time point, we saw a significant decrease in the percentage of CD4 T cells and Tregs, both within TDLNs and within the tumors (Figures 4B and 4C). Specifically, the frequency of Tregs in the TDLNs of tumored animals (untreated or treated with M11 CAR T cells) was around 2% of the total live cells; CTX treatment resulted in a decrease of this T cell subset to around 1%–1.4%. In tumors, 0.5% of the total live digest was CD4+ Treg, which was almost completely depleted following CTX treatment.
Endogenous CD8 T Cells Are a Requirement for CTX-Induced Bystander Effects
Since the direct effect of the dose of CTX used was limited, and we observed only a slight increase in the number of persisting M11 CAR T cells, we wanted to test the hypothesis that our observed bystander effect required the presence of endogenous CD8 T cells. Therefore, we repeated our curative regime on 90% mesothelin-positive AE17om tumors that were established in wild-type (WT) and CD8 T cell-deficient mice. Similar to the data shown earlier, one dose of CTX followed by two doses of the M11 CAR T cells resulted in tumor cures in WT mice (Figure 5A, left panel); however, the tumors were not cured by the same treatment when endogenous CD8 T cells were not present (Figure 5A, right panel).
Figure 5.

Bystander Effects Require CD8+ T Cells but Not BATF3+ DCs
(A) CD8 WT and CD8 KO C57BL/6J mice were inoculated with 2 × 106 90% mesothelin-positive AE17om tumor cells (day 0), treated with a single dose of 100 mg/kg CTX (day 3), and then given two doses of 107 M11 CAR T cells (days 4 and 6, respectively). Tumor growth measurements over time showed a curative effect of the CTX/M11 CAR T cell combination treatment in the CD8 WT mice but not in the CD8 KO mice. (B) Flow tracings from harvested TDLNs from the WT mice at the end of the CD8 WT/CD8 KO in vivo study that were stained for the presence of OVA-positive CD8 T cells. Cells were gated on live CD3+CD8+ T cells. Non-tumor-bearing (naive) mice were compared to tumor-bearing mice that received no treatment (NT), M11 CAR T cells, cells treated with CTX alone, or cells treated with a combination of CTX and M11 CAR T cells. (C) Summary of the flow cytometry data (expressed as percentage of CD8+ T cells or the percentage of total live cells) shows that CTX treatment significantly increased the ova-specific CD8+ response in TDLNs of the CTX/M11 CAR T cell combination treatment group compared to all others. (D) BATF3 WT and BATF3 KO C57BL/6J mice were treated as described above. Tumor growth measurements over time showed a curative effect of the CTX/M11 CAR T cell combination treatment in both the WT and BATF3 KO mice. Samples: n = 3 per group. Statistics by 2-way ANOVA: ∗∗p < 0.01; ∗∗∗p < 0.001.
Given that the bystander effect required endogenous CD8 T cells, we first analyzed CD8 T cell frequencies in the percentage of total live cells in tumors on day 21, when the mice were euthanized. We observed increased numbers of CD3 and CD8 lymphocytes in the groups treated with CTX, especially in the CTX plus M11 CAR combination group (Figure 4B). More importantly, we asked whether we could detect enhanced T cell responses to non-CAR T cell targeted antigens. Since the tumor cells expressed the xenoantigen ovalbumin, we used ova-specific tetramers to monitor the induction of endogenous ova-specific CD8 T cells in TDLNs (Figures 5B and 5C). The percentage of ova-tetramer+ CD8 T cells was very low in naive mice (0.14%) and slightly increased in the tumor-bearing non-treated mice (0.45%). Treatment with the M11 CAR T cells increased tetramer positivity to ∼4%. A similar increase was seen after CTX treatment alone (∼4%). However, the mice treated with CTX plus the M11 CAR T cells showed a marked and significant increase in the ova-tetramer+ cells (∼32%).
CTX-Induced Bystander Effects Are independent of the Role of BATF3+ DCs
Given that the bystander effect appeared dependent on endogenous T cells and was accompanied by increased reactivity against a model tumor antigen (and, thus, likely other tumor neoantigens), we hypothesized that this effect was due to the known ability of CTX to activate DCs.19,20 We first conducted flow cytometry of TDLNs and tumors on days 1 and 5 after CTX administration in order to assess frequencies of DC subsets, especially the cross-presenting CD103+ conventional DC 1 (cDC1) cells.21 In our model, we detected only very low numbers of both CD8a+ and CD103+ cDC1 cells in the TDLNs, regardless of treatment, and virtually no cDC1 cells in tumors (data not shown). The very low frequencies of these cells did not allow further analysis of their activation status.
To directly assess the functional importance of cDC1 cells, we utilized commercially available mice in which the BATF3 transcription factor has been deleted, leading to the absence of cDC1 cells.22 We treated AE17om tumors with 90% mesothelin-expressing cells in WT versus BATF3-deficient mice. As described earlier, one dose of CTX followed by two doses of the M11-CAR T cells resulted in tumor cures (Figure 5D, left panel), however, the tumors in the BATF3-deficient mice were also cured by the CTX/M11 combination (Figure 5D, right panel), suggesting that the increase in endogenous anti-tumor T cells was not cDC1 cell dependent.
Discussion
The hypothesis that CAR T cells induce cross-priming and epitope spreading has been proposed as a possibility;1 however, the data showing that these events actually occur in vivo are, so far, limited. Some supportive evidence has been provided from studies in immunocompetent mice, showing that animals that had been cured of their tumors by CAR T cells were resistant to re-challenge with tumor cells that were not expressing the CAR-targeted antigens.15,23,24 Although these data demonstrate the generation of host immunity against non-CAR-targeted tumor antigens, these studies are not definitive or direct evidence of CAR-induced epitope spreading, since many tumors induce endogenous anti-tumor T cell responses capable of preventing tumor re-challenge when the primary tumor has been controlled by any means. For example, we and others have found that even surgical removal of an immunogenic tumor will result in resistance to subsequent challenge with small numbers of tumor cells. In other studies, Kuhn et al.15 showed that mice treated with the CD19 CAR T cells had only a small increase in the number of tumor-reactive non-CAR T cells compared to untreated tumor-bearing mice, suggesting low levels of epitope spreading, similar to our tetramer data. Interestingly, an enzyme-linked immunospot (ELISPOT) assay was enhanced in mice treated with a CD19-CD40L CAR, suggesting some stimulation of endogenous T cells.15 In our model, systemic administration of an agonistic anti-CD40 antibody did not enhance bystander killing. Although not the specific goal of the study, recent data from Lai et al.25 showed minimal epitope spreading in their murine tumor model (similar to what we observed), but the authors observed an increase after attraction (with FLT3L) and activation (with poly(I:C) and agonistic 41BB antibodies) of DCs. However, to our knowledge, no direct tumor bystander studies have been described.
Even less data about bystander effects are available from clinical trials. Our group observed the appearance of new antibody reactivities against mesothelioma tumor cell lines and against the tumor antigen mesothelin in serum obtained from patients after treatment with mesothelin-CAR T cells (generated by electroporation of CAR mRNA), suggesting the engagement of CD4 and B cells.26 Ramos et al.27 looked for antitumor immune responses in a CD19 CAR trial in lymphoma patients by analyzing for newly emergent T cell-mediated immunity to known tumor-associated antigens, including NY-ESO, survivin, PRAME, or MAGE in peripheral blood collected before and after CD19 CAR T cell infusion. They observed no differences in the frequency of precursor T cells responding to these antigens in peripheral blood collected before and after CD19 CAR T cell infusion; thus, no evidence for T cell antigen spreading was obtained.
Given this knowledge gap, we developed a model system in which we could specifically quantify bystander effects using murine anti-mesothelin CAR T cells that could cure established tumors when 100% of the tumor cells expressed human mesothelin. In this model, we found that CAR T cell treatment could slightly enhance the number of endogenous T cells directed against a non-targeted antigen (chicken ovalbumin); however, the CAR T cells were unable to cure tumors when only 10% of the tumor cells were mesothelin negative, demonstrating that any bystander effects were very limited.
One hypothesis to explain these findings is the highly immunosuppressive microenvironment of solid tumors, which impedes endogenous T cell function,28, 29, 30 prevents efficient DC activation,31 and promotes the expansion of immunosuppressive Tregs.29,32 To assess whether these factors might account for the lack of bystander effects we were seeing in our model, we first blocked the inhibitory T cell receptors PD-1 and CTLA-4, given their known ability to inhibit T cell activation, leading to reduced proliferation, cytokine production, and T cell death.33,34 We hypothesized that an anti-PD-1 or anti-CTLA-4 antibody therapy in combination with CAR T cells might neutralize the inhibitory signals of PD-1 on T cells (endogenous or transferred cells) and induce bystander effects. However, although each agent had some anti-tumor effect by itself, no tumor cures were induced.
We next assessed the role of TGF-β in our system. In tumors, TGF-β functions primarily as an immunosuppressive cytokine due to its ability to interfere with the generation, expansion, and function of anti-tumor immune cells as well as its association with the suppression of growth and/or activity of T cells, natural killer (NK) cells, and DCs.35,36 Our hypothesis was that the CAR T cells and, hopefully, any anti-tumor endogenous T cells would function more efficiently and that bystander effects would be induced. However, using a bioactive TGF-β neutralizing antibody with our CAR T cells did not significantly enhance bystander effects.
DCs play a significant role in regulating the balance between CD8 T cell immunity and tolerance to tumor antigens.37 T cell cross-priming—a process in which DCs cross-present exogenous antigens to CD8 T cells, resulting in CD8 T cell activation—is critical in generating endogenous anti-tumor CD8 T cell immunity.31 Despite the presence of DCs in the TME, it appears that tumor-associated DCs are often defective in their differentiation and activation and are poor stimulators of immune responses.37 We hypothesized that agents capable of activating DC might thus augment CAR-induced antigen cross-presentation and stimulate endogenous anti-tumor T cells and bystander effects. One such agent is an agonistic CD40 antibody that has been shown to license DCs to promote antitumor T cell activation and re-educate macrophages to destroy tumor stroma.38 Another is an IDO inhibitor.39 IDO participates in a metabolic pathway implicated in a peripheral tolerance and also participates in the functional tolerance of the immune system toward tumors.39,40 However, in our study, neither the use of an agonistic CD40 antibody nor the use of an IDO inhibitor in combination with CAR T cells induced significant bystander effects. It should be noted that there is now some support for this DC activation hypothesis from the recent study of Lai et al.,25 who showed evidence of enhanced epitope spreading when CAR T cell therapy was combined with agents that both attracted DCs (FLT3L) and strongly activated the DCs (poly(I:C) in combination with an agonist 41BB antibody was required).
CTX is an alkylating agent that has been in clinical use for more than 40 years and has been extensively studied in mice as an immuno-modulating agent.13,41, 42, 43 Administration of CTX induces lymphopenia, and it has thus been used extensively as a way to precondition animals and patients before the adoptive transfer of lymphocytes, including CAR T cells.42,44, 45, 46 The lymphopenia is thought to both “make room in the bone marrow” and induce the secretion of homeostatic cytokines such as interleukin (IL)-7 and IL-15 that enhance the proliferation and persistence of the transferred lymphocytes. However, a number of other immunomodulatory activites (usually seen with low doses) have been reported,18 including a somewhat selective depletion of CD4 Tregs,41,47 activation of DCs,20,47,48 a TH2-to-TH1 polarization in CD4 cells,18,49 and induction of type 1 IFNs.50 Of note, these previous studies have focused primarily on the beneficial effects of CTX on anti-tumor activity due to enhanced efficacy and persistence of the adoptively transferred T cells, without specifically examining the potential ability of CTX to induce endogenous T cell bystander effects. In contrast to all the aforementioned treatments, pre-treatment of the mice with one low dose of CTX induced a bystander effect that resulted in the cures of tumors that contained 10% and up to 25% of mesothelin-negative tumor cells.
We wanted to explore the mechanisms of this observed bystander effect. There are a number of possible pathways by which a bystander effect could occur.1,5,51 Release of cytokines from the activated CAR T cells (i.e., IFNγ and tumor necrosis factor alpha [TNF-α]) could directly kill tumor cells,52 or these cytokines could activate innate immune cells (e.g., macrophages, neutrophils, or NK cells) that could then induce tumor destruction via phagocytosis or by other means (e.g., secretion of additional TNF-α or nitric oxide). CAR T cells could also directly kill non-target-expressing cells by engagement of death receptors, such as FAS or DR5 (the ligand of TRAIL), on the the non-targeted tumor cells, leading to cell death. Alternatively, CAR T cells could induce epitope (or antigen) spreading, resulting in tumor cell killing by stimulated endogenous T cells.11 When tumor cells die and release tumor antigens in an immunostimulatory environment (i.e., in the presence of TNF-α, IFNγ, and granulocyte-macrophage colony-stimulating factor [GM-CSF]), antigen-presenting cells (with type 1 DCs thought to be the most important) have the ability to take up and cross-present tumor antigens generating endogenous B cell and T cell (both CD4+ and CD8+) immunity against tumor antigens not originally targeted by the CAR T cells.10
To elucidate the possible mechansims of the CTX-induced bystander effect in our model, we first assessed the role endogenous CD8 T cells in the process. In contrast to WT mice (where tumors with 10% antigen-negative cells were cured by CTX plus CAR T cells), when we conducted the same study with mice genetically lacking only CD8 T cells, we lost the ability to cure the tumors. These results demonstrate that endogenous CD8 T cells were critical to our observed bystander effect. Consistent with this finding, we observed an increase in the number of endogenous CD8 T cells in the tumors and an increase in CD8 T cells in TDLNs that could react with ovalbumin. Thus, the observed bystander effect was not primarily due to cytokine-mediated activation of TME cells or death-receptor-mediated killing of tumor cells. Although we did see (as expected) a small increase in the number of the infused CAR T cells in the CTX-treated mice (Figure 4A), the bystander effect was not due to enhanced persistence of the infused CAR T cells, since endogenous CD8 T cells were required. However, it should be mentioned that it is possible that other endogenous cell types, such as helper CD4 T cells, also play a role. Sorting out the contributions of CD4 T helper cells versus CD4 Tregs (discussed later) is a subject of ongoing investigation.
Given that CTX enhanced the endogenous CD8 T cell anti-tumor response, we hypothesized that one mechanism might be through the activation of DCs that could then take up released tumor antigens and cross-present to CD8 T cells. BATF3 DCs (otherwise known as cDC1 cells or CD103+ DCs) are thought to be the most important DC subtype responsible for cross-presentation in tumors.10,22 However, when we administered CTX and M11 CAR T cells to BATF3-deficient mice bearing tumors that had 10% antigen-negative cells, we were still able to eradicate the tumors (Figure 5D), showing that BATF3-dependent cDC1 cells were not required for the bystander effect in our model. This finding does not agree with the prevailing dogma that cDC1 cells are the sole DC subset sufficient for optimal anti-tumor cytotoxic T cell generation. However, a number of studies have previously shown that BATF3/CD103+ DCs are not required for the generation of cytotoxic T cells.53, 54, 55, 56 For example, Desch et al.56 showed that depending on the type of DC activating agent present (i.e., Toll-like receptor [TLR]7 versus TLR3), cDC2 cells could also effectively cross-present. This may be the case in our model.
As an additional or alternative mechanism, we hypothesized that the known ability of CTX to reduce Tregs is important. The ability of Tregs to suppress anti-tumor endogenous CD8 T cell responses is well known.41,47 Since CTX significantly reduced the number of Tregs in tumors and TDLNs in our model (Figure 4C), we postulate that loss of these suppressive Tregs combined with the immunostimulatory effects of the CAR T cells allowed the expansion/emergence of endogenous anti-tumor CD8 T cells (including those reactive with neoantigens and the xenoantigen, ovalbumin) that could then exert more potent anti-tumor effects. Experiments to test this hypothesis by selectively depleting Tregs in our model are ongoing.
The conclusions of our paper should be tempered by the fact that we have presented data from only one cell line and one CAR. Our mixing model required an experimental system where we could cure established tumors in which 100% of the tumor cells expressed the targeted antigen—in our case, mesothelin—using mouse CAR T cells in an immune-competent mouse without added perturbations (i.e., radiation, vaccination, or lymphodepletion) that could affect the immune system. This proved to be surprisingly difficult. Despite evaluating multiple tumor cell lines expressing mesothelin, we were only able to achieve cures in the AE17om cell line. It is possible that the bystander effect might be stronger with other cell lines. We also acknowledge that our model is somewhat simplistic in that we used a “binary” system where tumor cells either expressed high levels of antigen or none at all. This does not precisely mimic the situation seen clinically, where tumor cells can express a range of levels of target antigen. Our data showing very limited epitope spreading after CAR T cell therapy are similar to the recent findings of Lai et al.,25 using a different CAR and a different tumor model.
Future studies aimed at detecting bystander effects in human CAR T cell clinical trials are thus needed. However, until convincing data are obtained in human studies, our study suggests that the hypothesis that CAR T cells induce strong bystander effects should be considered an open question and that treatment of solid tumors without expression of the target antigen on the vast majority of tumor cells using a CAR targeted to a single antigen may not be successful.
If CAR-T cell-induced bystander effects are, in fact, quite limited, a second implication of our study is that, since most solid tumors do not express target antigen on >90% of tumor cells, other approaches will likely be needed for tumor eradication. A number of strategies are being explored that include: injection of a mixture of CAR T cells that target multiple tumor antigens; transducing T cells with multiple CARs targeting different antigens;9,57,58 and designing CARs that target multiple antigens,7 such as tandem CARs that express two linked single-chain variable fragments (scFvs) to recognize different antigens.59 Another approach is to create CARs that also secrete payloads or cargos that are designed to kill tumor cells that are not expressing tumor antigens. Examples of these cargos include cytokines or TLR ligands (i.e., IL-12, IL-18, IL-21, and CD40L) that can activate endogenous TME cells (e.g., macrophages or NK cells)15,60, 61, 62 or bispecific T cell engagers (BiTEs) that could redirect endogenous T cells.63 Another way to enhance bystander effects would be to augment CAR-induced cross-presentation of tumor antigens to endogenous T cells using approaches like the one described here (i.e., CTX) or other strategies aimed at attracting and activating DCs (i.e., CD40L or FLT3L plus DC activating agents) or reducing the immunosuppressive TME.15,25
Materials and Methods
Animals
Female C57BL/6 and BALB/c mice (6–8 weeks old, 18–20 g) were purchased from Charles River Laboratories (Wilmington, MA, USA). BATF3-deficient (BATF3 knockout [KO]) and CD8-deficient (CD8 KO) mice on the C57BL/6 background were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). The animal use committees of the University of Pennsylvania approved all animal study protocols, and experiments were conducted in compliance with the Guide for the Care and Use of Laboratory Animals. See Supplemental Materials and Methods for more details.
Cell Cultures
Murine AE17o mesothelioma cells expressing chicken ovalbumin (which grow in C57/B6 mice) were provided by Dr. Delia Nelson, University of Western Australia.64 These cells were lentivirally transduced to stably express human mesothelin (AE17om cell line). The murine mesothelioma cell lines AB12 and AB1 (which grow in BALB/c mice) were obtained from Dr. Bruce Robinson, University of Western Australia.65 These cells were also transduced to express human mesothelin (AB12m and AB1m, respectively). Murine lung cancer cell line TC-1 was a gift from Dr. Yvonne Paterson, University of Pennsylvania, and were transduced to express human mesothelin as described earlier (TC-1 m).
Generation of Anti-mouse M11 CAR Constructs
The anti-mesothelin M11 scFv was generated from a human phage display library and selected for its ability to bind to purified human mesothelin. The VH and VL variable domains of the M11 scFv were fused with a mouse CD8a hinge, CD8a transmembrane domain, and two mouse cytoplasmic domains derived from 4-1BB and CD3z. This CAR was subcloned into the MIGR1 retroviral vector, which also expresses GFP, using an internal ribosomal entry site as we have previously described.17 Infective particles were generated from the supernatants of Phoenix cells transfected with the retroviral vector plasmid and helper plasmids using Lipofectamine 2000 (Invitrogen).17
Isolation, Transduction, and Expansion of Primary Mouse T Lymphocytes
Primary murine splenic CD3+ T cells were isolated from naive mice, activated, and transduced following a protocol that is detailed in Supplemental Materials and Methods.
In Vitro Cytotoxicity Assays and IFNγ ELISAs
Triplicate wells of 5,000 luciferase-expressing AE17om cells were co-cultured with M11 CAR T cells at various E:T ratios. Cytotoxicity of T cells was measured using the luciferase assay system (Promega), as previously described.66 IFNγ detection in the supernatants of the co-cultures was done using a mouse IFNγ ELISA kit (Biolegend).66
Xenograft Models and In Vivo Therapies
2 × 106 tumor cells (AE17om in various mixtures with AE17o; AB12m mixed with AB12; TC-1 m mixed with TC-1; and AB-1 m mixed with AB-1) were injected into the flanks of syngeneic mice. When tumors reached ∼50–70 mm3 (4 days after tumor cell inoculation for both cell lines), 107 transduced M11 CAR T cells were injected intravenously via tail vein in 2 doses, 2 days apart, and the tumors were measured by calipers every 3–4 days for up to 2 weeks. Non-treated tumor-bearing animals were used as controls. For combination treatments, mice were treated in one of four groups: (1) untreated controls, (2) M11 CAR T cells alone, (3) immune-stimulatory agent alone, or (4) a combination of M11 CAR T cell therapy and immune-stimulatory agent. The agents include anti-PD-1 antibody (BioXcell, RMPI-14) at 5 mg/kg, given via intraperitoneal (i.p.) injection biweekly; anti-CTLA-4 antibody (BioXcell, 9H10) at 200 μg per mouse, given via i.p. injection biweekly; anti-TGF-β antibody (BioXcell, ID11.16.8) at 150 μg per mouse, given via i.p. injection biweekly; agonistic CD40 (BioXcell, FGK4.5/FGK45) at 40 μg per mouse, given via i.p. injection as a single dose 1 day following CAR T cell treatment; IDO inhibitor (INCB023843) at 300 mg/kg, given via oral gavage daily for 2 weeks, starting on day 3 post-tumor inoculation; and CTX (MP Biomedicals, 150749) at 100 mg/kg, given via i.p. injection as a single dose on day 3 post-tumor inoculation, 1 day prior to CAR T cell injections.
The dosages of each agent were based on previous experiments or pilot experiments in our lab (unpublished data), where each agent was tested for its anti-tumor activity by itself. We used a standard 3- to 4-day injection of antibodies. In order to be able to detect their effects on the ability of CAR T cells to cure tumors, our goal in these studies was to choose a dose that had clear bioactivity but one that did not have anti-tumor activity so strong that it would eliminate or strongly suppress tumor growth by itself.
Flow Cytometry
Spleens, tumor-draining lymph nodes (TDLNs), tumors, and blood were isolated from mice at the end of in vivo experiments, and all assays were performed immediately after isolation and processing. Single-cell suspensions from these samples were then stained with Live Dead Blue (Invitrogen, 1:400 in PBS) and mouse Fc block (BD Biosciences, 1:200 in PBS) for 10 min at 4°C, followed by cell-surface marker antibodies for 45 min at 4°C. For FoxP3 intracellular staining, cells were washed, fixed with eBioscience Fix/Perm (1:3 Fixation/Permealization Concentrate: Perm Diluent) for 1 h at 4°C, washed twice, and stained in the presence of Perm/Wash (1X Permealization Buffer) for 45 min at 4°C. All marker antibodies used are summarized in Table S1. Flow-cytometric analysis was performed on a BD LSR Fortessa with BD FACSDiva software (BD Biosciences) and analyzed using FlowJo v.10.4.
Statistical Analysis
Graphical and statistical analyses were performed using Prism v.7.0c (GraphPad Software). Descriptive statistics were computed for all variables. p values were performed either by non-parametric Mann-Whitney test or by one-way or two-way ANOVA, where appropriate. p values < 0.05 were considered significant. In each figure, variability in the data is shown as standard error of the mean (SEM).
Author Contributions
Conceptualization, A.K. and S.M.A.; Methodology, A.K. and S.M.A.; Investigation, A.K., M.L., E.A., J.S., and M.S.L.; Writing – Original Draft, A.K. and S.M.A.; Writing – Review & Editing, A.K., M.S.L., and S.M.A.; Funding Acquisition, S.M.A.; Resources, A.K., M.L., and J.S.; Supervision, A.K. and S.M.A.
Conflicts of Interest
S.M.A. received sponsored research agreements from Novartis and Incyte. The other authors declare no competing interests.
Acknowledgments
Support for this work was provided by grants from the National Cancer Institute (P01-CA217805 to S.M.A., A.K., M.S.L.), the Saint Baldrick’s Foundation (to M.S.L.), Alex’s Lemonade Stand Foundation (to M.S.L.), and the Rotz Family Foundation (to J.S. and S.M.A.).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.07.005.
Supplemental Information
References
- 1.June C.H., Sadelain M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018;379:64–73. doi: 10.1056/NEJMra1706169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Majzner R.G., Mackall C.L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019;25:1341–1355. doi: 10.1038/s41591-019-0564-6. [DOI] [PubMed] [Google Scholar]
- 3.Newick K., Moon E., Albelda S.M. Chimeric antigen receptor T-cell therapy for solid tumors. Mol. Ther. Oncolytics. 2016;3:16006. doi: 10.1038/mto.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klebanoff C.A., Rosenberg S.A., Restifo N.P. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat. Med. 2016;22:26–36. doi: 10.1038/nm.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knochelmann H.M., Smith A.S., Dwyer C.J., Wyatt M.M., Mehrotra S., Paulos C.M. CAR T Cells in Solid Tumors: Blueprints for Building Effective Therapies. Front. Immunol. 2018;9:1740. doi: 10.3389/fimmu.2018.01740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmidts A., Maus M.V. Making CAR T Cells a Solid Option for Solid Tumors. Front. Immunol. 2018;9:2593. doi: 10.3389/fimmu.2018.02593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen K.H., Wada M., Pinz K.G., Liu H., Shuai X., Chen X., Yan L.E., Petrov J.C., Salman H., Senzel L. A compound chimeric antigen receptor strategy for targeting multiple myeloma. Leukemia. 2018;32:402–412. doi: 10.1038/leu.2017.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sotillo E., Barrett D.M., Black K.L., Bagashev A., Oldridge D., Wu G., Sussman R., Lanauze C., Ruella M., Gazzara M.R. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015;5:1282–1295. doi: 10.1158/2159-8290.CD-15-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruella M., Barrett D.M., Kenderian S.S., Shestova O., Hofmann T.J., Perazzelli J., Klichinsky M., Aikawa V., Nazimuddin F., Kozlowski M. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Invest. 2016;126:3814–3826. doi: 10.1172/JCI87366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sánchez-Paulete A.R., Teijeira A., Cueto F.J., Garasa S., Pérez-Gracia J.L., Sánchez-Arráez A., Sancho D., Melero I. Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann. Oncol. 2017;28(Suppl_12):xii74. doi: 10.1093/annonc/mdx237. [DOI] [PubMed] [Google Scholar]
- 11.Gulley J.L., Madan R.A., Pachynski R., Mulders P., Sheikh N.A., Trager J., Drake C.G. Role of Antigen Spread and Distinctive Characteristics of Immunotherapy in Cancer Treatment. J. Natl. Cancer Inst. 2017;109 doi: 10.1093/jnci/djw261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beavis P.A., Henderson M.A., Giuffrida L., Mills J.K., Sek K., Cross R.S., Davenport A.J., John L.B., Mardiana S., Slaney C.Y. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J. Clin. Invest. 2017;127:929–941. doi: 10.1172/JCI89455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahlmann M., Hempel G. The effect of cyclophosphamide on the immune system: implications for clinical cancer therapy. Cancer Chemother. Pharmacol. 2016;78:661–671. doi: 10.1007/s00280-016-3152-1. [DOI] [PubMed] [Google Scholar]
- 14.Di S., Zhou M., Pan Z., Sun R., Chen M., Jiang H., Shi B., Luo H., Li Z. Combined Adjuvant of Poly I:C Improves Antitumor Effects of CAR-T Cells. Front. Oncol. 2019;9:241. doi: 10.3389/fonc.2019.00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuhn N.F., Purdon T.J., van Leeuwen D.G., Lopez A.V., Curran K.J., Daniyan A.F., Brentjens R.J. CD40 Ligand-Modified Chimeric Antigen Receptor T Cells Enhance Antitumor Function by Eliciting an Endogenous Antitumor Response. Cancer Cell. 2019;35:473–488.e6. doi: 10.1016/j.ccell.2019.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moon E.K., Wang L.S., Bekdache K., Lynn R.C., Lo A., Thorne S.H., Albelda S.M. Intra-tumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines. OncoImmunology. 2018;7:e1395997. doi: 10.1080/2162402X.2017.1395997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Riese M.J., Wang L.C., Moon E.K., Joshi R.P., Ranganathan A., June C.H., Koretzky G.A., Albelda S.M. Enhanced effector responses in activated CD8+ T cells deficient in diacylglycerol kinases. Cancer Res. 2013;73:3566–3577. doi: 10.1158/0008-5472.CAN-12-3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bracci L., Moschella F., Sestili P., La Sorsa V., Valentini M., Canini I., Baccarini S., Maccari S., Ramoni C., Belardelli F., Proietti E. Cyclophosphamide enhances the antitumor efficacy of adoptively transferred immune cells through the induction of cytokine expression, B-cell and T-cell homeostatic proliferation, and specific tumor infiltration. Clin. Cancer Res. 2007;13:644–653. doi: 10.1158/1078-0432.CCR-06-1209. [DOI] [PubMed] [Google Scholar]
- 19.Radojcic V., Bezak K.B., Skarica M., Pletneva M.A., Yoshimura K., Schulick R.D., Luznik L. Cyclophosphamide resets dendritic cell homeostasis and enhances antitumor immunity through effects that extend beyond regulatory T cell elimination. Cancer Immunol. Immunother. 2010;59:137–148. doi: 10.1007/s00262-009-0734-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salem M.L., Al-Khami A.A., El-Naggar S.A., Díaz-Montero C.M., Chen Y., Cole D.J. Cyclophosphamide induces dynamic alterations in the host microenvironments resulting in a Flt3 ligand-dependent expansion of dendritic cells. J. Immunol. 2010;184:1737–1747. doi: 10.4049/jimmunol.0902309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Theisen D., Murphy K. The role of cDC1s in vivo: CD8 T cell priming through cross-presentation. F1000Res. 2017;6:98. doi: 10.12688/f1000research.9997.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hildner K., Edelson B.T., Purtha W.E., Diamond M., Matsushita H., Kohyama M., Calderon B., Schraml B.U., Unanue E.R., Diamond M.S. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kueberuwa G., Kalaitsidou M., Cheadle E., Hawkins R.E., Gilham D.E. CD19 CAR T Cells Expressing IL-12 Eradicate Lymphoma in Fully Lymphoreplete Mice through Induction of Host Immunity. Mol. Ther. Oncolytics. 2017;8:41–51. doi: 10.1016/j.omto.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuhn N.F., Purdon T.J., van Leeuwen D.G., Lopez A.V., Curran K.J., Daniyan A.F., Brentjens R.J. CD40 Ligand-Modified Chimeric Antigen Receptor T Cells Enhance Antitumor Function by Eliciting an Endogenous Antitumor Response. Cancer Cell. 2019;35:473–488.e6. doi: 10.1016/j.ccell.2019.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lai J., Mardiana S., House I.G., Sek K., Henderson M.A., Giuffrida L., Chen A.X.Y., Todd K.L., Petley E.V., Chan J.D. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat. Immunol. 2020;21:914–926. doi: 10.1038/s41590-020-0676-7. [DOI] [PubMed] [Google Scholar]
- 26.Beatty G.L., Haas A.R., Maus M.V., Torigian D.A., Soulen M.C., Plesa G., Chew A., Zhao Y., Levine B.L., Albelda S.M. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res. 2014;2:112–120. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramos C.A., Rouce R., Robertson C.S., Reyna A., Narala N., Vyas G., Mehta B., Zhang H., Dakhova O., Carrum G. In Vivo Fate and Activity of Second- versus Third-Generation CD19-Specific CAR-T Cells in B Cell Non-Hodgkin’s Lymphomas. Mol. Ther. 2018;26:2727–2737. doi: 10.1016/j.ymthe.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moon E.K., Wang L.C., Dolfi D.V., Wilson C.B., Ranganathan R., Sun J., Kapoor V., Scholler J., Puré E., Milone M.C. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin. Cancer Res. 2014;20:4262–4273. doi: 10.1158/1078-0432.CCR-13-2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klampatsa A., O’Brien S.M., Thompson J.C., Rao A.S., Stadanlick J.E., Martinez M.C., Liousia M., Cantu E., Cengel K., Moon E.K. Phenotypic and functional analysis of malignant mesothelioma tumor-infiltrating lymphocytes. OncoImmunology. 2019;8:e1638211. doi: 10.1080/2162402X.2019.1638211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Brien S.M., Klampatsa A., Thompson J.C., Martinez M.C., Hwang W.T., Rao A.S., Standalick J.E., Kim S., Cantu E., Litzky L.A. Function of Human Tumor-Infiltrating Lymphocytes in Early-Stage Non-Small Cell Lung Cancer. Cancer Immunol. Res. 2019;7:896–909. doi: 10.1158/2326-6066.CIR-18-0713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hargadon K.M. Tumor microenvironmental influences on dendritic cell and T cell function: A focus on clinically relevant immunologic and metabolic checkpoints. Clin. Transl. Med. 2020;10:374–411. doi: 10.1002/ctm2.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee G.R. Phenotypic and Functional Properties of Tumor-Infiltrating Regulatory T Cells. Mediators Inflamm. 2017;2017:5458178. doi: 10.1155/2017/5458178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu X., Gao R., Li Y., Zeng C. Regulation of PD-1 in T cells for cancer immunotherapy. Eur. J. Pharmacol. 2020;881:173240. doi: 10.1016/j.ejphar.2020.173240. [DOI] [PubMed] [Google Scholar]
- 34.Sharpe A.H., Pauken K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018;18:153–167. doi: 10.1038/nri.2017.108. [DOI] [PubMed] [Google Scholar]
- 35.Syed V. TGF-β Signaling in Cancer. J. Cell. Biochem. 2016;117:1279–1287. doi: 10.1002/jcb.25496. [DOI] [PubMed] [Google Scholar]
- 36.Thomas D.A., Massagué J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8:369–380. doi: 10.1016/j.ccr.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 37.Giovanelli P., Sandoval T.A., Cubillos-Ruiz J.R. Dendritic Cell Metabolism and Function in Tumors. Trends Immunol. 2019;40:699–718. doi: 10.1016/j.it.2019.06.004. [DOI] [PubMed] [Google Scholar]
- 38.Vonderheide R.H. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu. Rev. Med. 2020;71:47–58. doi: 10.1146/annurev-med-062518-045435. [DOI] [PubMed] [Google Scholar]
- 39.Hornyák L., Dobos N., Koncz G., Karányi Z., Páll D., Szabó Z., Halmos G., Székvölgyi L. The Role of Indoleamine-2,3-Dioxygenase in Cancer Development, Diagnostics, and Therapy. Front. Immunol. 2018;9:151. doi: 10.3389/fimmu.2018.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Munn D.H., Mellor A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016;37:193–207. doi: 10.1016/j.it.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.North R.J. Cyclophosphamide-facilitated adoptive immunotherapy of an established tumor depends on elimination of tumor-induced suppressor T cells. J. Exp. Med. 1982;155:1063–1074. doi: 10.1084/jem.155.4.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hughes E., Scurr M., Campbell E., Jones E., Godkin A., Gallimore A. T-cell modulation by cyclophosphamide for tumour therapy. Immunology. 2018;154:62–68. doi: 10.1111/imm.12913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sistigu A., Viaud S., Chaput N., Bracci L., Proietti E., Zitvogel L. Immunomodulatory effects of cyclophosphamide and implementations for vaccine design. Semin. Immunopathol. 2011;33:369–383. doi: 10.1007/s00281-011-0245-0. [DOI] [PubMed] [Google Scholar]
- 44.Proietti E., Greco G., Garrone B., Baccarini S., Mauri C., Venditti M., Carlei D., Belardelli F. Importance of cyclophosphamide-induced bystander effect on T cells for a successful tumor eradication in response to adoptive immunotherapy in mice. J. Clin. Invest. 1998;101:429–441. doi: 10.1172/JCI1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dudley M.E., Wunderlich J.R., Yang J.C., Sherry R.M., Topalian S.L., Restifo N.P., Royal R.E., Kammula U., White D.E., Mavroukakis S.A. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J. Clin. Oncol. 2005;23:2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Viaud S., Ma J.S.Y., Hardy I.R., Hampton E.N., Benish B., Sherwood L., Nunez V., Ackerman C.J., Khialeeva E., Weglarz M. Switchable control over in vivo CAR T expansion, B cell depletion, and induction of memory. Proc. Natl. Acad. Sci. USA. 2018;115:E10898–E10906. doi: 10.1073/pnas.1810060115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wada S., Yoshimura K., Hipkiss E.L., Harris T.J., Yen H.R., Goldberg M.V., Grosso J.F., Getnet D., Demarzo A.M., Netto G.J. Cyclophosphamide augments antitumor immunity: studies in an autochthonous prostate cancer model. Cancer Res. 2009;69:4309–4318. doi: 10.1158/0008-5472.CAN-08-4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nakahara T., Uchi H., Lesokhin A.M., Avogadri F., Rizzuto G.A., Hirschhorn-Cymerman D., Panageas K.S., Merghoub T., Wolchok J.D., Houghton A.N. Cyclophosphamide enhances immunity by modulating the balance of dendritic cell subsets in lymphoid organs. Blood. 2010;115:4384–4392. doi: 10.1182/blood-2009-11-251231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brode S., Cooke A. Immune-potentiating effects of the chemotherapeutic drug cyclophosphamide. Crit. Rev. Immunol. 2008;28:109–126. doi: 10.1615/critrevimmunol.v28.i2.20. [DOI] [PubMed] [Google Scholar]
- 50.Schiavoni G., Mattei F., Di Pucchio T., Santini S.M., Bracci L., Belardelli F., Proietti E. Cyclophosphamide induces type I interferon and augments the number of CD44(hi) T lymphocytes in mice: implications for strategies of chemoimmunotherapy of cancer. Blood. 2000;95:2024–2030. [PubMed] [Google Scholar]
- 51.Ross S.L., Sherman M., McElroy P.L., Lofgren J.A., Moody G., Baeuerle P.A., Coxon A., Arvedson T. Bispecific T cell engager (BiTE®) antibody constructs can mediate bystander tumor cell killing. PLoS ONE. 2017;12:e0183390. doi: 10.1371/journal.pone.0183390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kearney C.J., Lalaoui N., Freeman A.J., Ramsbottom K.M., Silke J., Oliaro J. PD-L1 and IAPs co-operate to protect tumors from cytotoxic lymphocyte-derived TNF. Cell Death Differ. 2017;24:1705–1716. doi: 10.1038/cdd.2017.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seillet C., Jackson J.T., Markey K.A., Brady H.J., Hill G.R., Macdonald K.P., Nutt S.L., Belz G.T. CD8α+ DCs can be induced in the absence of transcription factors Id2, Nfil3, and Batf3. Blood. 2013;121:1574–1583. doi: 10.1182/blood-2012-07-445650. [DOI] [PubMed] [Google Scholar]
- 54.Mott K.R., Maazi H., Allen S.J., Zandian M., Matundan H., Ghiasi Y.N., Sharifi B.G., Underhill D., Akbari O., Ghiasi H. Batf3 deficiency is not critical for the generation of CD8α+ dendritic cells. Immunobiology. 2015;220:518–524. doi: 10.1016/j.imbio.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gilfillan C.B., Kuhn S., Baey C., Hyde E.J., Yang J., Ruedl C., Ronchese F. Clec9A+ Dendritic Cells Are Not Essential for Antitumor CD8+ T Cell Responses Induced by Poly I:C Immunotherapy. J. Immunol. 2018;200:2978–2986. doi: 10.4049/jimmunol.1701593. [DOI] [PubMed] [Google Scholar]
- 56.Desch A.N., Gibbings S.L., Clambey E.T., Janssen W.J., Slansky J.E., Kedl R.M., Henson P.M., Jakubzick C. Dendritic cell subsets require cis-activation for cytotoxic CD8 T-cell induction. Nat. Commun. 2014;5:4674. doi: 10.1038/ncomms5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wilkie S., van Schalkwyk M.C., Hobbs S., Davies D.M., van der Stegen S.J., Pereira A.C., Burbridge S.E., Box C., Eccles S.A., Maher J. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J. Clin. Immunol. 2012;32:1059–1070. doi: 10.1007/s10875-012-9689-9. [DOI] [PubMed] [Google Scholar]
- 58.Anurathapan U., Leen A.M., Brenner M.K., Vera J.F. Engineered T cells for cancer treatment. Cytotherapy. 2014;16:713–733. doi: 10.1016/j.jcyt.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hegde M., Mukherjee M., Grada Z., Pignata A., Landi D., Navai S.A., Wakefield A., Fousek K., Bielamowicz K., Chow K.K. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Invest. 2016;126:3036–3052. doi: 10.1172/JCI83416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yeku O.O., Purdon T.J., Koneru M., Spriggs D., Brentjens R.J. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci. Rep. 2017;7:10541. doi: 10.1038/s41598-017-10940-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu Y., Di S., Shi B., Zhang H., Wang Y., Wu X., Luo H., Wang H., Li Z., Jiang H. Armored Inducible Expression of IL-12 Enhances Antitumor Activity of Glypican-3-Targeted Chimeric Antigen Receptor-Engineered T Cells in Hepatocellular Carcinoma. J. Immunol. 2019;203:198–207. doi: 10.4049/jimmunol.1800033. [DOI] [PubMed] [Google Scholar]
- 62.Hu B., Ren J., Luo Y., Keith B., Young R.M., Scholler J., Zhao Y., June C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017;20:3025–3033. doi: 10.1016/j.celrep.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsai A.K., Davila E. Producer T cells: Using genetically engineered T cells as vehicles to generate and deliver therapeutics to tumors. OncoImmunology. 2016;5:e1122158. doi: 10.1080/2162402X.2015.1122158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jackaman C., Bundell C.S., Kinnear B.F., Smith A.M., Filion P., van Hagen D., Robinson B.W., Nelson D.J. IL-2 intratumoral immunotherapy enhances CD8+ T cells that mediate destruction of tumor cells and tumor-associated vasculature: a novel mechanism for IL-2. J. Immunol. 2003;171:5051–5063. doi: 10.4049/jimmunol.171.10.5051. [DOI] [PubMed] [Google Scholar]
- 65.Davis M.R., Manning L.S., Whitaker D., Garlepp M.J., Robinson B.W. Establishment of a murine model of malignant mesothelioma. Int. J. Cancer. 1992;52:881–886. doi: 10.1002/ijc.2910520609. [DOI] [PubMed] [Google Scholar]
- 66.Newick K., O’Brien S., Sun J., Kapoor V., Maceyko S., Lo A., Puré E., Moon E., Albelda S.M. Augmentation of CAR T-cell Trafficking and Antitumor Efficacy by Blocking Protein Kinase A Localization. Cancer Immunol. Res. 2016;4:541–551. doi: 10.1158/2326-6066.CIR-15-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.