

Abstract
Macrovascular complications develop in over a half of the diabetic individuals, resulting in high morbidity and mortality. This poses a severe threat to public health and a heavy burden to social economy. It is therefore important to develop effective approaches to prevent or slow down the pathogenesis and progression of macrovascular complications of diabetes (MCD). Oxidative stress is a major contributor to MCD. Nuclear factor (erythroid‐derived 2)‐like 2 (NRF2) governs cellular antioxidant defence system by activating the transcription of various antioxidant genes, combating diabetes‐induced oxidative stress. Accumulating experimental evidence has demonstrated that NRF2 activation protects against MCD. Structural inhibition of Kelch‐like ECH‐associated protein 1 (KEAP1) is a canonical way to activate NRF2. More recently, novel approaches, such as activation of the Nfe2l2 gene transcription, decreasing KEAP1 protein level by microRNA‐induced degradation of Keap1 mRNA, prevention of proteasomal degradation of NRF2 protein and modulation of other upstream regulators of NRF2, have emerged in prevention of MCD. This review provides a brief introduction of the pathophysiology of MCD and the role of oxidative stress in the pathogenesis of MCD. By reviewing previous work on the activation of NRF2 in MCD, we summarize strategies to activate NRF2, providing clues for future intervention of MCD. Controversies over NRF2 activation and future perspectives are also provided in this review.
Keywords: complications, diabetes, macrovascular, NRF2, oxidative stress
1. INTRODUCTION
Diabetes is predicted to be the seventh leading cause of death in the world in 2030. 1 , 2 Macrovascular complications of diabetes (MCD)—basically ischaemic heart disease, cerebrovascular disease and peripheral vascular disease—develop in over a half of the diabetic population, resulting in high morbidity and mortality. 3 This poses a severe threat to the world's public health. Despite the successful control of hyperglycaemia, hypertension and hyperlipidaemia, the diabetic patients are still at risk of developing MCD. 4 Therefore, it is crucial to identify more viable drug targets and develop more effective approaches, in order to prevent or slow down the pathogenesis and progression of MCD.
Oxidative stress is a key mechanism by which diabetes induces its complications. 5 Under diabetic condition, excessive reactive oxygen species (ROS) are produced, causing detrimental cellular events, such as formation of advanced glycation end products (AGEs) and overexpression of receptor for AGEs (RAGE), along with activation of polyol pathway, hexosamine pathway and protein kinase C (PKC). 5 , 6 These contribute to the pathogenesis and progression of MCD.
The transcription factor nuclear factor (erythroid‐derived 2)‐like 2 (NRF2) plays a critical role in cellular defence against oxidative stress. 7 NRF2 turns on the transcription of various antioxidant genes, producing cellular antioxidants 7 that act as scavengers of free radicals and prevent the oxidative stress‐driven pathogenesis of MCD. NRF2 is negatively regulated by Kelch‐like ECH‐associated protein 1 (KEAP1) in the cytoplasm. KEAP1 restricts NRF2 from nuclear translocation on one hand and facilitates proteasomal degradation of NRF2 on the other. 8 Therefore, small molecule‐induced structural inhibition of KEAP1 protein—a canonical way to activate NRF2—has become a research hotspot in the past two decades, with the protective outcome verified in animal models of MCD.
In addition to structural inhibition of KEAP1 protein, activation of NRF2 in MCD has been achieved through other approaches. These include activation of Nfe2l2 gene transcription, decrease in KEAP1 protein level by microRNA‐induced degradation of Keap1 mRNA, prevention of proteasomal degradation of NRF2 protein and modulation of other upstream regulators of NRF2. Taken together, these investigations have provided alternative strategies to activate NRF2, and shed light upon novel targets upstream of NRF2 for the intervention of MCD as well.
In this review, we summarize and discuss the various strategies arisen to activate NRF2 and their outcome in MCD, with the aim of providing insights into future management of MCD.
2. PATHOPHYSIOLOGY OF MCD
The diabetes‐driven atherosclerosis is the main cause of MCD. The typical pathological features of atherosclerosis in arteries are characterized by generation of fibrosis, proliferation and structural derangement of smooth muscle cells, thickening of tunica media, accumulation of lipid, formation of plaque, calcification and thrombosis. 9 Under microscope, adhesion of blood cells to the endothelium and extension of interendothelial connection may be observed, indicating the diabetes‐enhanced adhesion and permeability of the endothelium. 10 The arterial pathophysiological events induced by diabetes (Figure 1) eventually lead to vascular dysfunction and ischaemic complications.
FIGURE 1.
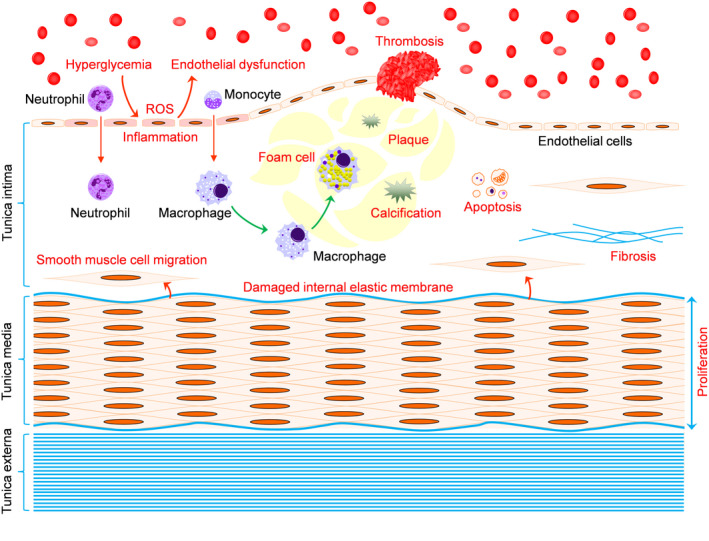
Pathophysiology of macrovascular complications of diabetes (MCD). Hyperglycaemia causes formation of reactive oxygen species (ROS) and inflammation in the endothelium, leading to endothelial dysfunction as a critical first step towards MCD. Under diabetic condition, the permeability of the inflamed endothelium is increased, allowing recruitment of neutrophils and monocytes into the tunica intima, where the macrophages—differentiated from monocytes—engulf lipids and become foam cells that gradually form a plaque. Calcium can be deposited in the plaque. Thrombus forms at the location where the plaque breaks. Apoptosis is induced, and fibrosis is accumulated. Smooth muscle cells proliferate, thereby thickening the tunica media. Smooth muscle cells can migrate into tunica intima through damaged internal elastic membrane, contributing to atherogenesis. Red characters, detrimental processes resulting in atherosclerosis
3. ROLE OF OXIDATIVE STRESS IN THE PATHOGENESIS OF MCD
Oxidative stress reflects an imbalance between the status of ROS and the antioxidant ability of a biological system. Diabetes induces generation of ROS through formation of AGEs, overexpression of RAGE and activation of polyol pathway, hexosamine pathway and PKC. 5 Upon diabetes, the excessive ROS exceed the scavenging capacity of the cellular antioxidant system, resulting in damage to proteins, lipids and DNAs. 11 Moreover, the diabetes‐induced ROS provokes inflammation, which in turn exacerbates oxidative stress. This vicious circle formed by ROS and inflammation 12 contributes to fibrosis and calcification in the plaque, 13 , 14 during a later stage of atherosclerosis.
Endothelial dysfunction is a critical pathophysiological event prior to MCD, with oxidative stress and inflammation as major contributors (Figure 1). 15 Once formed, typical pathological features of atherosclerosis—such as fibrosis, tunica media thickening, plaque and calcification—are impossible to be reversed. It is therefore crucial to improve diabetes‐induced endothelial dysfunction, the effect of which may efficiently prevent or slow down atherogenesis. In this regard, targeting oxidative stress is a viable strategy.
4. PROTECTIVE EFFECTS OF NRF2 ON MCD
As supplementation of antioxidants, such as vitamin E, vitamin C, coenzyme Q10, alpha‐lipoic acid, L‐carnitine and ruboxistaurin, has proven non‐beneficial to diabetic complications, 16 attention has been transferred to activation of the inner cellular antioxidant capacity. NRF2—the governor of the cellular antioxidant defence system—activates the transcription of downstream antioxidant genes by binding antioxidant response element in the promoter regions of the antioxidant genes, such as haem oxygenase 1 (Hmox1), NAD(P)H dehydrogenase (quinone 1) (Nqo1), glutathione (Gsh), superoxide dismutase (Sod), gamma‐glutamylcysteine synthetase and catalase (Cat). 17 , 18 , 19
NRF2 is negatively regulated by KEAP1. 7 , 8 , 20 , 21 Oxidative stress disrupts critical cysteine residues in KEAP1, resulting in the release of NRF2 from the KEAP1‐NRF2 complex. 22 Hence, a short‐term oxidative stress generated upon diabetes activates NRF2 as a compensatory protective mechanism through which the cells protect against hyperglycaemia‐induced injuries. 23 However, accumulating evidence has demonstrated that cardiovascular NRF2 antioxidant signalling is impaired after a long‐term exposure to hyperglycaemia. 24 , 25 , 26 , 27 , 28 , 29 This decompensatory effect exposes the cells to more severe injuries. Thus, activation of NRF2 is beneficial to the vasculature under both short‐term and long‐term diabetic conditions prior to atherosclerosis, especially the latter.
To date, many studies have reported the protective effects of NRF2 activation on diabetic complications, such as diabetic cardiomyopathy, nephropathy and retinopathy. However, less is known for the impact of NRF2 activation on MCD, as summarized below.
4.1. Canonical structural inhibition of KEAP1 in MCD
4.1.1. Sulforaphane (SFN)
SFN derives from broccoli sprouts 30 and modifies specific cysteine residues in KEAP1 protein, thereby changing its conformation. This enables NRF2 to dissociate from KEAP1, promoting NRF2 nuclear translocation and preventing proteasomal degradation of NRF2. 31
The beneficial effects of SFN on MCD have been reported. 32 In a mouse model of type 2 diabetes, SFN activated NRF2 antioxidant signalling in the aorta and attenuated the diabetes‐induced oxidative stress, inflammation, apoptosis, cell proliferation, thickening of the tunica media and accumulation of collagen in the aorta. 32 In an animal model of non‐obese type 2 diabetes (Goto‐Kakizaki rats), SFN reversed the diabetes‐repressed expression of aortic NRF2, attenuated the production of aortic O2 and AGEs and improved nitric oxide (NO)‐dependent and nitric oxide nitric oxide‐independent vasorelaxation. 29
Moreover, SFN inhibits nuclear factor‐kappa B (NF‐κB) 33 —a key pro‐inflammatory factor in diabetes‐induced vascular inflammation. Crosstalk may exist between NRF2 and NF‐κB. HO‐1, a potent antioxidant downstream of NRF2, has been shown to repress NF‐κB activity by reducing cellular labile iron content. 34 NF‐κB, in turn, suppresses NRF2‐induced HO1 production. 35 The predominant activity of NF‐κB under diabetes might explain the impaired NRF2 antioxidant activity in the vasculature under a long‐term diabetic condition. Given that NF‐κB is a transcription factor, it would be interesting to explore the effect of NF‐κB on the expression of Keap1 in further studies.
The finding that SFN improves insulin resistance in patients with type 2 diabetes 36 suggests that SFN may prevent MCD at much earlier stages including diabetes and obesity.
4.1.2. Dh404
Dh404 is a derivative of bardoxolone methyl. The latter was tested in clinical trials for treatment of diabetic nephropathy (DN). 37 Dh404 activates NRF2 via modification of KEAP1, 38 a mechanism similar to SFN. Tan et al showed that Dh404 lessened diabetes‐induced atherosclerosis with reduction in oxidative stress and inflammatory factors at lower (3 and 10 mg/kg/d) doses in streptozotocin (STZ)‐induced diabetic apolipoprotein E knockout mice. 38 However, at a higher dose (20 mg/kg/d), Dh404 increased the expression of pro‐inflammatory mediators monocyte chemoattractant protein‐1 and NF‐κB in the kidney. This side effect of the higher dose needs to be further investigated. Because the expression of the NRF2 downstream genes was not further enhanced in the higher‐dose group compared with the lower‐dose group, it is insufficient to conclude that a drastic activation of NRF2 has detrimental effects. Instead, other mechanisms might account for Dh404's toxicity. Further studies are needed to determine the pharmacological activities of dh404.
4.1.3. Dimethyl fumarate (DMF, BG‐12)
DMF is a known NRF2 activator and has been used for clinical treatment of multiple sclerosis. 39 Ha et al researched the effect of DMF on vascular calcification in vascular smooth muscle cells (VSMCs) and aortas of C57BL/6J mice. 40 DMF activated NRF2, attenuating calcification in both the VSMCs and aortas. Moreover, this effect of DMF was abolished when NRF2 was knocked down in VSMCs. 40 This study may indicate a potential protective effect of DMF on MCD. The approval of DMF in clinical use has granted DMF a unique advantage in its potential application in MCD.
4.1.4. Tert‐butyl hydroquinone (tBHQ)
tBHQ activates NRF2 by targeting Cys‐151 within KEAP1 protein. 41 tBHQ was reported to ameliorate diabetes‐driven atherosclerosis in apolipoprotein E‐deficient mice. 42 In this study, tBHQ was found to enhance NRF2 activity in macrophages and VSMCs within atherosclerotic lesions, promoting autophagic activity. This led to decrease in size, extension and lipid content of the atheroma plaques, as well as reduction of lesional macrophages, foam cell size and chemokine expression. 42
4.2. Non‐canonical ways to activate NRF2 in MCD
In recent years, strategies towards NRF2 activation in MCD have evolved from structural inhibition of KEAP1 to other regulatory mechanisms, shedding light upon novel targets and approaches for the intervention of MCD.
4.2.1. Targeting Keap1 mRNA
MicroRNA‐200a (miR‐200a)/Keap1 mRNA
We and others have reported that miR‐200a targets Keap1 mRNA, leading to degradation of Keap1 mRNA and activation of NRF2. 24 , 43 , 44 Recently, our group found that the miR‐200a/Keap1/NRF2 axis played an essential role in protecting against diabetes‐induced endothelial dysfunction. 45 In aortic ECs isolated from C57BL/6 wild‐type mice, miR‐200a mimic or inhibitor modulated KEAP1/NRF2 antioxidant signalling and manipulated oxidative stress and inflammation under high glucose (HG) condition. These effects were completely abrogated by knockdown of Keap1, indicating that Keap1 mRNA is a major target of miR‐200a. Moreover, the protective effect of miR‐200a mimic on HG‐induced endothelial oxidative stress and inflammation was completely abolished in aortic ECs isolated from C57BL/6 Nfe2l2 knockout mice, suggesting that NRF2 is required for miR‐200a's actions. Further, supplementation of miR‐200a inhibited aortic Keap1 expression, activated NRF2 signalling and attenuated hyperglycaemia‐induced oxidative stress, inflammation and endothelial dysfunction in the wild‐type, but not Nfe2l2 knockout, mice. 45 These findings have verified that domestic miR‐200a/KEAP1/NRF2 directly controls endothelial antioxidant activity, providing miR‐200a and Keap1 mRNA as viable targets for the intervention of MCD.
MiR‐200a/KEAP1/NRF2 not only provides direct protection to the endothelium, but also benefits the endothelium through a remote route by its regulation of fibroblast growth factor 21 (FGF21). FGF21 regulates glucose and lipid metabolism and has potential for the treatment of diabetes. 46 FGF21 was shown to prevent aortic pathologies in STZ‐induced diabetic mice. 46 In OVE26 type 1 diabetic mice, Fgf21 mRNA and protein levels were found to be increased in the liver and plasma, leading to protection against the diabetes‐induced aortic fibrosis and inflammation. 47 Further investigation showed that inhibition of HDAC3 with RGFP‐966 enhanced hepatic miR‐200a expression, the effect of which decreased Keap1 mRNA and protein levels, thereby promoting NRF2‐mediated Fgf21 gene expression. FGF21, produced in the liver, was then released to the plasma and protected the diabetic aorta. 47
MicroRNA‐24 (MiR‐24)/Keap1 mRNA
Endothelial repair after stent implantation is delayed in diabetic patients compared with that in non‐diabetic patients. 48 MiR‐24, which targets Keap1 mRNA, 44 was found to activate NRF2/HO1 signalling, restore SOD and GSH, and suppress ROS and malondialdehyde production in HG‐stimulated VSMCs. 48 In vivo, adenovirus‐induced overexpression of miR‐24 promoted re‐endothelialization in balloon‐injured diabetic rats. 48
4.2.2. Modulation of c‐Jun N‐terminal kinase (JNK)
JNK has been shown to regulate vascular NRF2 expression and function upon diabetes. However, the role of JNK in control of NRF2 is controversial. The pioneer work by He et al showed that AGEs induced rapid phosphorylation of JNK in bovine aortic endothelial cells. Inhibition of JNK by SP600125 abrogated the expression of the NRF2 downstream antioxidant HO1, 49 suggesting that JNK positively regulates NRF2 signalling. However, this study did not look into the impact of SP600125 on the expression of NRF2 and its various downstream antioxidant genes other than Hmox1. Additionally, the impact of HG on the activity of JNK and NRF2 was not studied. Therefore, the impact of JNK on vascular NRF2 expression and function under diabetes/HG conditions was still unclear. More recently, naringenin—a flavanone found in various plants—elevated NRF2/HO1 and inhibited HG‐ or free fatty acid–induced apoptosis of human umbilical vein endothelial cells (HUVECs). 50 Naringenin phosphorylated AKT serine/threonine kinase (AKT) and JNK in HUVECs under the normal glucose condition and failed to increase HO1 protein level in the presence of SP600125 or the phosphoinositide 3‐kinase (PI3K) inhibitor Y294002, indicating that naringenin might activate NRF2/HO1 through activation of JNK and AKT. 50
On the contrary, JNK was found by other groups to negatively regulate NRF2. C66—a novel curcumin analogue—has shown NRF2‐activating efficacy in both the aortas 51 and kidneys 24 of STZ‐induced diabetic mice. C66 was primarily identified to be an inhibitor of JNK. 51 Inhibition of JNK by either C66 or SP600125 activated NRF2 expression and function and attenuated the diabetes‐induced aortic pathological injuries to a similar extent, 51 suggesting that JNK negatively regulates NRF2 in the aorta. Moreover, a recent study showed that C66 predominantly targeted JNK2, as both Jnk2 gene deletion and C66 treatment could similarly activate NRF2 and alleviate diabetes‐induced aortic oxidative stress, inflammation and fibrosis. 43 Supporting these findings, our group found that SP600125 prevented DN through activation of NRF2, with enhanced expression of Hmox1 and Nqo1. 52 SP600125 hampered the HG‐induced phosphorylation of JNK and c‐Jun, repressing Keap1 gene expression. 52 This inhibitory impact of SP600125 on KEAP1 was viable because SP600125 induced remarkable nuclear translocation of NRF2. 52 Supporting the inhibitory effect of SP600125 on Keap1 gene expression, 36 c‐Jun‐binding sites were found between −3000 bp and −1 bp within the promoter region of the mouse Keap1 gene. 52 Luciferase reporter assay should be helpful to further confirm the regulatory effect of JNK on Keap1 gene transcription.
As reviewed above, controversies exist in defining the role of JNK in NRF2 activity in the vasculature. These controversies may be owing to the differences between cell types (BACEs, HUVECs, whole aortas and mouse mesangial cells) and (or) disease models (stimulation with AGEs vs HG/hyperglycaemia). Further studies are needed to elucidate the regulatory effect of JNK on NRF2 in MCD.
4.2.3. Transcriptional activation of Nfe2l2
Recently, NRF2 activation has been achieved in diabetes‐induced endothelial dysfunction at the transcription level. Sodium butyrate (NaB), a polyunsaturated fatty acid found in food, was primarily reported by our group to activate renal Nfe2l2 gene expression in an STZ‐induced type 1 diabetic mice. 53 We further found that NaB improved the diabetes‐induced aortic endothelial dysfunction in the wild‐type, but not Nfe2l2 gene knockout, mice. 28 Mechanistically, in HG‐treated aortic endothelial cells, NaB elevated Nfe2l2 mRNA and protein levels without facilitating NRF2 nuclear translocation, an effect distinct from that of SFN. Further, NaB inhibited the activity of histone deacetylase (HDAC) and increased the occupancy of the transcription factor aryl hydrocarbon receptor and the co‐activator P300 at the Nfe2l2 gene promoter. Moreover, the P300 inhibitor C646 completely abolished these efficacies of NaB. 28 These results suggest that NaB prevented diabetes‐induced aortic endothelial dysfunction through HDAC/P300‐mediated transcriptional activation of Nfe2l2.
4.2.4. Preservation of NRF2 protein
Preservation of NRF2 protein from degradation is another efficacious way to maintain cellular NRF2 protein level, facilitating NRF2 activation. MG132—a proteasome inhibitor—preserved NRF2 protein and attenuated aortic pathological injuries in OVE26 diabetic mice. 54 MG132, administered 3 months starting from the age of 3 months old, was shown in this study to lower the diabetes‐provoked aortic levels of tumour necrosis factor‐alpha (TNF‐α), plasminogen activator inhibitor‐1, 3‐nitrotyrosine and 4‐hydroxynonenal. Moreover, MG132 ameliorated the diabetes‐induced thickening and structural derangement of the aortic wall. 54 It is noted that the pathological aortic changes at the initiation of the treatment (3‐month‐old OVE diabetic mice) were not evident. MG132 should have yielded greater beneficial effects if administered at earlier stages of diabetes, since once formed, it is impossible to reverse the diabetes‐induced typical pathological features. 55
4.2.5. Inhibition of nuclear export of NRF2
Zinc (Zn)
Zn is an essential trace element that has antioxidant activity. Zn deficiency in endothelial cells enhanced inflammatory response and oxidative stress. 56 , 57 On the contrary, Zn supplementation benefitted the aortas of 3‐month‐old OVE26 diabetic mice, 58 including the attenuation of aortic fibrosis, oxidative stress, inflammation, apoptosis and proliferation. 58 Moreover, Zn elevated both mRNA and protein levels of Nfe2l2, 58 suggesting that Zn might regulate Nfe2l2 gene expression at the transcription or post‐transcription levels.
The same group performed a further study to investigate the effect of Zn supplementation on DN. In OVE26 type 1 diabetic mice, Zn induced renal AKT and glycogen synthase kinase‐3 beta (GSK‐3β) phosphorylation with a decrease in proto‐oncogene tyrosine‐protein kinase Fyn (Fyn), a nuclear exporter of NRF2. 59 Although this mechanism still cannot explain the former Zn‐increased Nfe2l2 mRNA level, this study showed AKT/ GSK‐3β/Fyn as a mechanism, at least in part, by which Zn activated NRF2. Given that Zn deficiency is prevalent in patients with both type 1 diabetes and type 2 diabetes, 60 Zn supplementation has a potential in the management of MCD.
Hydrogen sulphide (H2S)
H2S, together with NO and carbon monoxide, is an important member of the family of gasotransmitters. 61 Liu et al observed a decreased H2S production in the aortas of 28‐week‐old db/db mice and in HG‐treated endothelial cells. 62 Exogenous administration of sodium hydrosulphide decreased the levels of adhesive and apoptotic factors, increased nuclear accumulation of NRF2 and the expression of Sod and Cat, and suppressed the excessive autophagy induced by oxidative stress. 62 More recently, the same group showed an inhibitory effect of H2S on VSMC proliferation under hyperglycaemic condition via inhibiting mitochondrial fragmentation. 63 It was indicated in another study that PI3K/AKT was involved in H2S activation of NRF2 in cerebral ischaemia/reperfusion injury, 64 suggesting a possible way through which H2S induced nuclear accumulation of NRF2—a mechanism similar to that of Zn. 59 However, another study showed that sodium hydrosulphide S‐sulfhydrated KEAP1 at cysteine‐151, and facilitated NRF2 nuclear translocation, protecting against senescence in mouse embryonic fibroblasts. 65 It would be interesting to investigate whether H2S structurally inhibits KEAP1 in MCD.
Baicalin
Baicalin is the major component found in Scutellaria baicalensis root, exhibiting anti‐inflammatory activity. 66 Baicalin was reported to restore the activity of hyperglycaemia‐impaired aortic NRF2 signalling in STZ‐induced diabetic mice. 66 Mechanistically, baicalin reversed the HG‐induced dephosphorylation of AKT and GSK‐3β in HUVECs, leading to the inactivation of Fyn. This promoted nuclear localization of NRF2 and transcription of HMOX1, NQO1, NQO2, CAT and SOD2, ameliorating HG‐induced oxidative stress. 66
4.3. Other mechanisms to activate vascular NRF2 antioxidant signalling
Several other mechanisms have been studied.
4.3.1. Insulin‐like growth factor 1 (IGF‐1)
In a mouse model of vascular ageing, liver‐specific knockdown of IGF‐1 decreased vascular oxidative stress resistance by impairing NRF2 signalling. 67 In the aortas of IGF‐1‐deficient mice, the expression of Nfe2l2 and downstream glutamate‐cysteine ligase catalytic subunit, Nqo1 and Hmox1 was decreased. 67 When challenged with HG, the expression of these NRF2 downstream genes was activated in the wild‐type, but not IGF‐1‐deficient, aortas. 67 This study indicates that IGF‐1 positively regulates NRF2. IGF‐1 activation or overexpression in animal or cell models of MCD could be helpful to confirm IGF‐1 as an activator of NRF2.
4.3.2. HO1
HO1 is a known downstream antioxidant of NRF2 and has shown beneficial effects on endothelial cells and animal models of vascular disease. 68 Inhibition of HO1 resulted in HG‐mediated endothelial cell damage. 69 Supplementation of bilirubin, a HO1‐derived metabolite, 68 restored endothelial cell viability under HG condition. 69 Given the robust protective effect of HO1, two points of view are indicated based on this study. Firstly, HO1 might also be controlled by other regulators in addition to NRF2. Secondly, although NRF2 targets multiple downstream antioxidant genes, HO1 might exert the most robust antioxidant efficacy in MCD. This view is supported by the finding that metallothionein, another NRF2 downstream antioxidant, provided approximately 50% protection against DN upon SFN‐induced NRF2 activation. 70
In summary, a number of NRF2‐activating strategies, either KEAP1‐inhibiting or not, exhibited beneficial effects on MCD (Table 1), providing versatile approaches for the management of MCD.
TABLE 1.
Effect of NRF2 activators on MCD
| Activator | Target | NRF2 dependence | Dose and period | Model | Effect | Reference |
|---|---|---|---|---|---|---|
| SFN | KEAP1 protein | Not verified | 0.5 mg/kg, 5 d/wk, 16 wk | High‐fat diet + STZ‐induced type 2 diabetic mice | Aortic NRF2, HO1 and SOD1 proteins↑; 3‐NT↓, 4‐HNE↓, TNF‐α↓, VCAM‐1↓, apoptosis↓, proliferation↓, tunica media thickness↓, collagen accumulation↓ | 32 |
| Dh404 | KEAP1 protein | Not verified | 3, 10 or 20 mg/kg/d, 18 wk | STZ‐induced type 1 diabetic mice | Oxidative stress↓, TNF‐α↓, ICAM‐1↓, MCP‐1↓, atherosclerosis↓, at lower (3 or 10 mg/kg/d), but not higher (20 mg/kg/d), doses | 38 |
| DMF | KEAP1 protein | Yes (Nfe2l2 knockdown by siRNA) | 25 or 50 mg/kg/d in mice; 5‐50 µmol/L in VSMCs | Vitamin D3‐induced aortic calcification mouse model; calcification media‐cultured VSMCs | Calcification↓, expression of bone marker genes Runx2, Oc and Alp↓ | 40 |
| tBHQ | KEAP1 protein | Not verified | 50 mg/kg, every other day, for 6 wk; 5‐24 µmol/L, 1‐3 h in VSMCs | STZ‐induced diabetes in ApoE ‐/‐ mice; VSMCs and macrophages exposed to IL‐6 and IFN‐γ | Atherosclerosis↓, lesional macrophages↓, foam cell size↓, chemokine expression↓ | 42 |
| MiR‐200a mimic | Keap1 mRNA | Yes (aortas and ECs isolated from Nfe2l2 gene knockout mice) | 1 mg/kg, 3 times weekly 30 nmol/L, 48 h | STZ‐induced type 1 diabetic mice HG‐treated mouse aortic ECs |
Aorta: expression of Keap1↓, NRF2 protein↑; mRNA levels of Nqo1↑, Homx1↑, iNos↓, Vcam‐1↓ and Mcp‐1↓; 4‐HNE↓; endothelial dysfunction↓ ECs: expression of Keap1↓, t‐NRF2↑, n‐NRF2↑; mRNA levels of Nqo1↑, Homx1↑, Vcam‐1↓ and Mcp‐1↓; levels of ROS↓ and MDA↓; NO production↑ |
45 |
| RGFP‐966 | HDAC3/miR‐200a/Keap1 mRNA | Not verified | 200 mg/kg, every other day, for 3 mo | OVE26 type 1 diabetic mice | Aortic fibrosis and inflammation↓; hepatic HDAC3↓, miR‐200a↑, Keap1 expression↓, NRF2/FGF21↑ | 47 |
| Adenovirus‐induced overexpression of miR‐24 | MiR‐24/Keap1 mRNA | Not verified | Balloon‐injured diabetic rats; HG‐stimulated VSMCs | Re‐endothelialization↑; NRF2, HO1, SOD, GSH expression↑, ROS and MDA↓ | 48 | |
| SP600125 | JNK | Not verified | 20 µmol/L, 24 h | AGE‐stimulated BAECs | Hmox1 expression↓ | 49 |
| C66 or SP600125 | JNK | Not verified | 5 mg/kg, every other day, 12 wk | STZ‐induced type 1 diabetic mice | Aortic JNK phosphorylation↓, 3‐NT↓, TNF‐α↓, PAI‐1↓, apoptosis↓, proliferation↓, tunica media thickness↓, collagen accumulation↓ | 51 |
| Naringenin | JNK and AKT | Not verified | 50 µmol/L, 72 h | HG‐cultured HUVECs | Phosphorylation of JNK and AKT↑, NRF2 and HO1 protein↑ | 50 |
| MG132 | Proteasome | Not verified | 10 µg/kg, 12 wk | OVE26 type 1 diabetic mice | Aortic 3‐NT↓, 4‐HNE↓, TNF‐α↓, PAI‐1↓, TGF‐β↓, CTGF↓, tunica media thickness↓, collagen accumulation↓ | 54 |
| NaB | HDAC | Yes (aortas isolated from Nfe2l2 gene knockout mice) |
5 g/kg/d, 20 wk 10 µmol/L, 48 h |
STZ‐induced type 1 diabetic mice HG‐treated mouse aortic ECs |
Aortic expression of Nfe2l2/Hmox1↑, oxidative stress and inflammation↓, endothelial dysfunction↓ HDAC activity↓, AHR, P300 and H3K9ac occupancy at Nfe2l2 gene promoter↑, endothelial expression of Nfe2l2, Hmox1 and Nqo1↑, oxidative stress and inflammation↓ |
28 |
| Zn (ZnSO4) | Possibly AKT/Fyn | Not verified | 5 mg/kg, every other day, 12 wk | OVE26 type 1 diabetic mice | Nfe2l2 mRNA and protein↑, MT↑, 3‐NT↓, 4‐HNE↓, VCAM‐1↓, TNF‐α↓, TGF‐β1↓, CTGF↓, PAI‐1↓, COL4↓, tunica media thickness↓ | 58 |
| H2S (NaHS) |
Possibly PI3K/AKT Not indicated |
Not verified Not verified |
100 µg/kg, every other day, 12 wk in mice; 100 µmol/L, 48 h, in cells 100 µmol/L, 24 h, in cells |
db/db type 2 diabetic mice; HG + palmitate‐cultured rat aortic ECs HG‐cultured VSMCs |
ATP↑, respiratory complex activity↑, AMPK phosphorylation↓, autophagy↓, apoptosis↓, adhesive molecules↓ Proliferation and migration of VSMCs↓, mitochondrial fragmentation in VSMCs↓ |
|
| Baicalin | AKT/GSK‐3β/Fyn | Yes (Nfe2l2 knockdown by shRNA) |
50 mg/kg/d for 4 wk 50 µmol/L |
STZ‐induced type 1 diabetic mice HG‐treated HUVECs |
Phosphorylation of AKT and GSK‐3β↑, Fyn‐mediated nuclear export of NRF2↓, CAT, HO1 and NQO1↑, oxidative damage↓, endothelial impairment↓ | 66 |
| Bilirubin | HO1 | Not verified | 0.5 µmol/L | HG‐cultured bovine aortic ECs | Cell viability↑, 4‐HNE↓ | 69 |
| IGF‐1 | Not indicated | Not verified | None | HG‐cultured aorta segments of liver IGF‐1‐deficient mice | Serum IGF‐1↓; NRF2 signalling in HG‐cultured aorta segments↓, oxidative stress↑, apoptosis↑, endothelial dysfunction↑ | 67 |
Although DMF was tested in experimental models of vascular calcification, but not diabetes, it is listed in this table as calcification is a pathological feature of MCD. Abbreviations: 3‐NT, 3‐nitrotyrosine; 4‐HNE, 4‐hydroxynonenal; AHR, aryl hydrocarbon receptor; AKT, AKT serine/threonine kinase; Alp, alkaline phosphatase; AMPK, AMP‐activated protein kinase; ApoE−/−, atherosclerosis‐prone apolipoprotein E‐deficient; ATP, adenosine triphosphate; BEACs, bovine aortic endothelial cells; CAT, catalase; COL4, collagen IV; CTGF, connective tissue growth factor; Dh404, dihydro‐CDDO‐trifluoroethyl amide DMF, dimethyl fumarate; EC, endothelial cell; FGF21, fibroblast growth factor 21; GSH, glutathione; GSK‐3β, glycogen synthase kinase 3 beta; H2S, hydrogen sulphide; H3K9ac, acetylated histone H3 lysine 9; HDAC, histone deacetylase; HG, high glucose; HO1, haem oxygenase 1; HUVEC, human umbilical vein endothelial cells; ICAM‐1, intercellular adhesion molecule 1; IFN‐γ, interferon‐gamma; IGF‐1, insulin‐like growth factor 1; IL‐6, interleukin‐6; JNK, c‐Jun N‐terminal kinase; KEAP1, Kelch‐like ECH‐associated protein 1; MCP‐1, monocyte chemoattractant protein‐1; MDA, malondialdehyde; miR‐200a, microRNA‐200a; miR‐24, microRNA‐24; MT, metallothionein; n‐NRF2, nuclear NRF2; NaB, sodium butyrate; Nqo1, NAD(P)H dehydrogenase (quinone 1); NRF2, nuclear factor (erythroid‐derived 2)‐like 2; Oc, osteocalcin; P300, E1A binding protein P300; PAI‐1, plasminogen activator inhibitor‐1; PI3K, phosphoinositide 3‐kinase; ROS, reactive oxygen species; Runx2, runt‐related transcription factor 2; SFN, sulforaphane; shRNA, short hairpin RNA; SOD1, superoxide dismutase 1; STZ, streptozotocin; tBHQ, tert‐butyl hydroquinone; t‐NRF2, total cellular NRF2; TGF‐β, transforming growth factor beta; TNF‐α, tumour necrosis factor alpha; VCAM‐1, vascular cell adhesion molecule‐1; VSMC, vascular smooth muscle cell. Symbols: ↑, upregulation or activation; ↓, downregulation or inhibition.
5. NRF2‐ACTIVATING STRATEGIES BORROWED FROM DIABETES AND CARDIOVASCULAR COMPLICATIONS OTHER THAN MCD
NRF2 activation in diabetes and cardiovascular complications other than MCD, such as DN, cardiomyopathy and retinopathy, has implications on MCD.
5.1. Sirtuin 1 (SIRT1)
SIRT1 is an HDAC and benefits diabetes and complications. 71 Antioxidant efficacy contributes to SIRT1’s protective effects. 71 , 72 In a mouse model of STZ‐induced diabetes, SIRT1 and NRF2 antioxidant signalling were less expressed in the kidneys of the diabetic mice compared with non‐diabetic control mice. 73 Paeonol (PA), a single phenolic compound extracted from the root bark of Cortex Moutan, increased renal SIRT1 protein level and activated NRF2 antioxidant signalling in the diabetic mice. 73 In HG and PA cotreated rat mesangial cells, knockdown of Sirt1 abrogated the PA‐induced activation of NRF2 signalling and reversed the inhibitory effect of PA on the expression of pro‐inflammatory and pro‐fibrotic genes. 73 These results indicate that SIRT1 positively regulates NRF2 antioxidant signalling in protection against DN. Activation of SIRT1 may thus have potential in the intervention of MCD. Confirming this speculation, our group has demonstrated the beneficial effect of SIRT1 activation in diabetes‐induced endothelial dysfunction. 74 , 75 We found that SRT2104—a potent activator of SIRT1—attenuated diabetes‐induced aortic oxidative stress, inflammation and endothelial dysfunction in STZ‐induced diabetic mice through inhibition of P53. 74 Because P53 inhibits Nfe2l2 expression and function, 26 , 76 SIRT1 may activate NRF2 via inhibition of P53.
P53 is a transcription factor that activates the expression of miR‐34a which targets Sirt1 mRNA. 75 By inhibiting SIRT1 in the presence of either the P53 inhibitor pifithrin‐α or the miR‐34a inhibitor, our other work found that inhibition of P53/miR‐34a improved diabetes‐induced endothelial dysfunction through activation of SIRT1. 75 Taken together, these studies have found a SIRT1/P53/miR‐34a circuit that acts upstream of NRF2 antioxidant signalling and controls endothelial function under diabetes.
5.2. Mouse double minute 2 (MDM2)/P53
MDM2 is a suppressor of P53. 77 As P53 is primarily known as a guardian of the genome coordinating cellular responses to genotoxic stress, 78 MDM2 is considered as an oncogene in tumours. 77 However, under diabetic conditions, MDM2/P53 shows a different profile from that in carcinogenesis. We and others have found that P53 is elevated and activated upon diabetes, contributing to endothelial dysfunction, 75 , 79 nephropathy 26 , 80 and cardiomyopathy. 81 Moreover, renal Mdm2 expression is inhibited in STZ‐induced diabetic mice. 26 Interestingly, inhibition of MDM2 by nutlin3a activated P53 and generated DN‐like pathologies in the non‐diabetic healthy mice. 26 Notably, inhibition of P53 by pifithrin‐α activated renal NRF2 signalling and attenuated renal injuries in the wild‐type, but not Nfe2l2 gene knockout, diabetic mice. 26 In HG‐treated mouse mesangial cells, P53 gene silencing completely abolished nutlin3a's inhibitory effect on NRF2 signalling. Together, these findings demonstrate that MDM2 controls NRF2 antioxidant signalling via inhibition of P53 in DN, providing MDM2 as a novel target.
5.3. Protein kinase C delta (PKC‐δ)
Oxidative stress contributes to diabetes‐induced pancreatic beta‐cell damage. 82 , 83 Glucagon‐like peptide‐1 receptor (GLP‐1) is a peptide released by the gastrointestinal tract in response to nutrients such as carbohydrates, proteins and lipids, 84 stimulating insulin secretion in the presence of elevated blood glucose concentrations.
Kim et al investigated whether NRF2 is involved in the protective effect of GLP‐1 on oxidative stress‐induced beta‐cell apoptosis. 85 In this study, NRF2 antioxidant signalling was activated by exendin‐4 (EX4)—an agonist of GLP‐1—in palmitic acid‐ or hydrogen peroxide–stimulated beta cells. 85 NRF2 was required for the protective effects of EX4 on ROS production and insulin secretion, as these effects were abolished when Nfe2l2 was knocked down. 85 Mechanistic study revealed that EX4 phosphorylated PKC‐δ. By using PKC‐δ siRNA in the presence of EX4, PKC‐δ was found to mediate the EX4‐induced activation of NRF2. Hence, PKC‐δ might be a positive regulator upstream of NRF2 in beta cells under oxidative stress.
5.4. PKR‐like ER kinase (PERK) and thrombospondin 1
Endoplasmic reticulum (ER) stress and oxidative stress are important mediators of beta‐cell failure in diabetes. 82 ER stress develops when the protein load in the ER exceeds the capacity of the organelle to handle proper protein folding. In response, the cell activated an adaptive mechanism namely unfolded protein response which is controlled by the ER transmembrane protein PERK. 86 NRF2 was reported to be a direct substrate of PERK. 87 PERK phosphorylates NRF2, leading to dissociation of NRF2 from KEAP1. 87 Cunha et al showed that thrombospondin 1—a multimeric Ca2+‐binding glycoprotein—was able to activate PERK and NRF2 in the endoplasmic reticulum, protecting against palmitate‐induced death, 86 providing PERK and thrombospondin 1 as potential candidates upstream of NRF2 in the management of MCD.
5.5. DJ‐1 (Parkinson disease protein 7, PARK7)
DJ‐1, also known as PARK7, stabilizes NRF2 by preventing association of NRF2 with KEAP1. 88 In support of the positive regulatory effect of DJ‐1 on NRF2, a recent study found that the protein levels of DJ‐1 and NRF2/HO1 were increased in the kidneys of STZ‐induced Sprague Dawley diabetic (4, 8 and 12 weeks of diabetes) rats. 89 The role of DJ‐1 in the regulation of NRF2 antioxidant signalling in MCD warrants further investigation.
In summary, several targets identified in diabetes and its vascular complications other than MCD might shed light on future studies for MCD, as summarized in Table 2.
TABLE 2.
Drawing lessons from diabetes and cardiovascular complications other than MCD
| Target | Approach | Dose and period | Model | Effect | Reference |
|---|---|---|---|---|---|
| SIRT1/NRF2 | Paeonol |
150 mg/kg, 6 times/wk 5, 10 and 20 μg/mL |
STZ‐induced diabetic mice HG‐treated rat mesangial cells |
Renal SIRT1↑; NRF2↑; SOD activity↑; MDA↓; ICAM‐1↓; FN↓; mesangial matrix index↓ SIRT1↑; NRF2, HO1, SOD1↑; FN and ICAM‐1↓ |
73 |
| MDM2/P53/NRF2 |
Nutlin3a PFT‐α |
10 mg/kg, or 4 wk 1.1 mg/kg, 3 times weekly, for 24 wk |
Non‐diabetic healthy mice DN (STZ‐induced diabetic mice) |
DN‐like renal pathologies↑; UACR↑; renal oxidative stress, inflammation and fibrosis↑; P53↑; expression of Nfe2l2 and downstream Hmox1 and Nqo1↓ UACR↓; renal pathologies↓; renal oxidative stress, inflammation and fibrosis↓; P53↓; Nfe2l2 and Hmox1 expression↑ |
26 |
| GLP‐1/PKC‐δ/NRF2 | Exendin‐4 | 10 nmol/L | Palmitic acid‐ or hydrogen peroxide–stimulated beta cells | NRF2 protein level↑; GCLC and HO‐1 mRNA levels↑; beta‐cell dysfunction↓ | 85 |
| PERK/NRF2 | Lyophilized thrombospondin 1 or overexpression of thrombospondin 1 | 2 μg/mL | Palmitate‐induced beta‐cell death | PERK↑; NRF2‐dependent ARE activity↑; GSTM1↑; CAT↑; beta‐cell death↓ | 86 |
| DJ‐1/NRF2 | Not available | Not available | DN (STZ‐induced diabetic Sprague Dawley rats) | Renal DJ‐1↑; NRF2/HO1↑ | 89 |
Abbreviations: ↓, downregulation or inhibition; DJ‐1, Parkinson disease protein 7 (PARK7); DN, diabetic nephropathy; FN, fibronectin; GCLC, glutamate‐cysteine ligase catalytic subunit; GLP‐1, glucagon‐like peptide‐1 receptor; GSTM1, glutathione s‐transferase mu 1; MDM2, mouse double minute 2; PERK, PKR‐like ER kinase; PFT‐α, pifithrin‐α; PKC‐δ, protein kinase C delta; SIRT1, sirtuin 1; UACR, urinary albumin‐to‐creatinine ratio. Other abbreviations are the same as in Table 1. Symbols: ↑, upregulation or activation.
6. NRF2 ACTIVATORS IN CLINICAL TRIALS FOR VASCULAR COMPLICATIONS OF DIABETES
Although no NRF2 activator has been applied in clinical trials for MCD, the clinical trials of NRF2 activators used in diabetic complications other than MCD may provide clues for the future trials of MCD.
6.1. Bardoxolone methyl in treatment of DN
The most well‐known NRF2 activator tested in clinical trials for the treatment of diabetic complications is bardoxolone methyl. The initial application of bardoxolone methyl in treatment of type 2 diabetic patients with 3b‐4 chronic kidney disease (CKD) yielded promising beneficial outcome. 90 The 20 patients with moderate‐to‐severe DN received bardoxolone methyl at 25 mg/d for 28 days, followed by a dose of 75 mg/d for additional 28 days. 90 By the end of the study, bardoxolone methyl dose‐ and time‐dependently elevated estimated glomerular filtration rate and significantly reduced serum creatinine and blood urea nitrogen, accompanied by an increase in creatinine clearance. 90 Additionally, bardoxolone methyl blunted the biomarkers of vascular injury and inflammation. 90 It is noted that no severe side effect was observed in all the patients. 90
The success in the initial clinical trial of bardoxolone methyl brought light to the management of DN. Bardoxolone methyl was applied in a much larger phase 3 clinical trial in type 2 diabetic patients with stage 4 CKD thereafter. 91 However, the trial was terminated because of severe heart complications. 92 It is speculated that bardoxolone methyl may pharmacologically induce acute sodium and fluid retention, and therefore increase blood pressure and result in heart failure in patients with advanced CKD, despite its improvement of kidney injury in some of the patients. 92 , 93 Thus, attention should be paid to the off‐target effects of a therapeutic in addition to its dose. Moreover, criticism has been raised regarding the use of the NRF2‐activating drug at an inappropriate stage of the disease. 55 , 94 Application of the antioxidant approach should be more beneficial at a much earlier stage of DN, as typical pathological features of DN are impossible to be reversed, 55 , 94 whereas prevention of DN by NRF2 activators starting from the initiation of diabetes has proven successful in numerous experimental studies. 24 , 53 , 70 , 94 , 95 , 96
The benefits earned and lessons learned from the use of bardoxolone methyl in the clinical trials of DN have a positive impact on the potential application of NRF2‐activating approaches in future clinical trials of MCD, in which special attention should be paid to off‐target effects and occasion for intervention.
6.2. Curcumin in treatment of diabetic microangiopathy
Curcumin is a natural compound that derives from turmeric. 97 Meriva, a lecithinized formulation of curcumin, was used in a pilot study on the management of diabetic microangiopathy. 97 Meriva was given to 25 patients with diabetic microangiopathy at 1 g/d, for 4 weeks, improving diabetic microangiopathy and reducing oedema in the skin of the patients. 97 Larger clinical trials with more detailed design are needed to further test the effect of Meriva on diabetic microangiopathy.
Despite the beneficial effects of Meriva, it is still unknown whether Meriva functioned through activating NRF2, as curcumin targets multiple factors. It is needed to elucidate the molecular mechanism of Meriva, which may provide evidence for potential off‐target effects.
The poor bioavailability restricts curcumin from clinical application. 24 C66, a novel analogue of curcumin with much lower effective dose demonstrated in animals, has anti‐inflammatory, antifibrotic and antioxidative effects, and protects against diabetic cardiomyopathy, 98 DN 24 and MCD. 51 C66's protection was shown to be partially mediated by NRF2. 24 Therefore, C66 has a potential for clinical use in the management of MCD, based on its advantage in bioavailability and the solid preliminary work.
7. CONCLUSIONS AND PERSPECTIVES
In recent years, the strategies for NRF2 activation have evolved from the canonical structural inhibition of KEAP1 protein to others such as microRNA‐induced inhibition of KEPA1 production, inhibition of proteasomal degradation of NRF2, inhibition of AKT/GSK‐3β/Fyn‐mediated nuclear export of NRF2 and modulation of HDAC/P300‐controlled Nfe2l2 gene transcription (Figure 2, red characters). In these novel strategies for NRF2 activation, epigenetic mechanisms such as microRNAs 45 , 48 and histone modifications 28 , 47 have recently emerged as efficient approaches that show good potentials in the intervention of MCD. Other epigenetic mechanisms including circular RNAs, long non‐coding RNAs and DNA methylation also play important roles in the regulation of NRF2 antioxidant signalling. 99 , 100 These epigenetic mechanisms should provide new clues for future studies on NRF2 activation in MCD.
FIGURE 2.

Schematic diagram for NRF2 activation in diabetes and its cardiovascular complications. Red characters, targets identified in macrovascular complications of diabetes (MCD); blue characters, targets identified in diabetes and cardiovascular complication other than MCD; symbols: ↓, activation; ┴, inhibition. AHR, aryl hydrocarbon receptor; CAT, catalase; Dh404, dihydro‐CDDO‐trifluoroethyl amide; DMF, dimethyl fumarate; DJ‐1, Parkinson disease protein 7 (PARK7); GCLC, glutamate‐cysteine ligase catalytic subunit; GLP‐1, glucagon‐like peptide‐1 receptor; GSH, glutathione; GSK‐3β, glycogen synthase kinase 3 beta; GSTM1, glutathione s‐transferase mu 1; H2S, hydrogen sulphide; HDAC, histone deacetylase; HO1, haem oxygenase 1; IGF‐1, insulin‐like growth factor 1; JNK, c‐Jun N‐terminal kinase; KEAP1, Kelch‐like ECH‐associated protein 1; MDM2, mouse double minute 2; miR‐200a, microRNA‐200a; miR‐24, microRNA‐24; MT, metallothionein; NaB, sodium butyrate; NQO1, NAD(P)H dehydrogenase (quinone 1); NRF2, nuclear factor (erythroid‐derived 2)‐like 2; P300, E1A binding protein P300; p‐AKT, phosphorylated AKT serine/threonine kinase 1; PERK, PKR‐like ER kinase; PFT‐α, pifithrin‐α; p‐Fyn, phosphorylated proto‐oncogene tyrosine‐protein kinase Fyn; PKC‐δ, protein kinase C delta; p‐NRF2, phosphorylated NRF2; ROS, reactive oxygen species; SFN, sulforaphane; SIRT1, sirtuin 1; SOD1, superoxide dismutase 1; tBHQ, tert‐butyl hydroquinone; Ub, ubiquitination; Zn, zinc
Moreover, regulators of NRF2 that were found in diabetes and cardiovascular complications other than MCD, including SIRT1, MDM2/P53, GLP‐1/PKC‐δ, PERK and DJ‐1, warrant further investigation for their roles in MCD (Figure 2, blue characters).
Taken together, NRF2 activation has a good potential in future clinical intervention of MCD. However, to date, the molecular mechanisms of NRF2 activation have not been fully understood. Further mechanistic studies are warranted. In addition, more novel and effective approaches should be developed as these will benefit the population with diabetes and MCD.
CONFLICTS OF INTEREST
None.
AUTHOR CONTRIBUTION
Junduo Wu: Data curation (lead); Investigation (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Xiaodan Sun: Data curation (lead); Investigation (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Ziping Jiang: Data curation (equal); Funding acquisition (equal); Investigation (equal); Writing‐review & editing (equal). Jun Jiang: Funding acquisition (equal); Investigation (supporting); Writing‐review & editing (equal). Linlin Xu: Funding acquisition (equal); Investigation (supporting); Supervision (equal); Writing‐review & editing (equal). Ao Tian: Investigation (supporting); Writing‐review & editing (equal). Xuechun Sun: Investigation (supporting); Writing‐review & editing (equal). Huali Meng: Investigation (supporting); Writing‐review & editing (equal). Ying Li: Funding acquisition (equal); Investigation (supporting); Supervision (equal); Writing‐review & editing (equal). Wenlin Huang: Investigation (supporting); Supervision (equal); Writing‐review & editing (equal). Ye Jia: Investigation (supporting); Supervision (equal); Writing‐review & editing (equal). Hao Wu: Conceptualization (lead); Data curation (lead); Funding acquisition (lead); Investigation (lead); Project administration (lead); Supervision (lead); Writing‐original draft (lead); Writing‐review & editing (lead).
ACKNOWLEDGEMENTS
This work was supported in part by National Natural Science Foundation of China (81973031) and Cheeloo Young Scholar Program of Shandong University (21320089963054) to Hao Wu; Natural Science Foundation of Jilin Province (2018SCZWSZX‐045) to Ziping Jiang; Key Technology Research and Development Program of Shandong Province (2019GSF108147) to Jun Jiang; National Natural Science Foundation of China (81901106) and Youth Talent Fund of The Second Hospital of Shandong University (2018YT09) to Linlin Xu; and Natural Science Foundation of Jilin Province (JJKH20180362KJ) to Ying Li.
Wu J, Sun X, Jiang Z, et al. Protective role of NRF2 in macrovascular complications of diabetes. J Cell Mol Med. 2020;24:8903–8917. 10.1111/jcmm.15583
Junduo Wu and Xiaodan Sun authors contributed equally to this work.
REFERENCES
- 1. Giovannini P, Howes MJ, Edwards SE. Medicinal plants used in the traditional management of diabetes and its sequelae in Central America: a review. J Ethnopharmacol. 2016;184:58‐71. [DOI] [PubMed] [Google Scholar]
- 2. Lopes G, Andrade PB, Valentao P. Phlorotannins: towards new pharmacological interventions for diabetes mellitus type 2. Molecules. 2017;22(1):56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Forbes JM, Yee LT, Thallas V, et al. Advanced glycation end product interventions reduce diabetes‐accelerated atherosclerosis. Diabetes. 2004;53:1813‐1823. [DOI] [PubMed] [Google Scholar]
- 4. Tan SM, de Haan JB. Combating oxidative stress in diabetic complications with Nrf2 activators: how much is too much? Redox Rep. 2014;19:107‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058‐1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aronson D. Hyperglycemia and the pathobiology of diabetic complications. Adv Cardiol. 2008;45:1‐16. [DOI] [PubMed] [Google Scholar]
- 7. Kaspar JW, Niture SK, Jaiswal AK. Nrf 2:INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol Med. 2009;47:1304‐1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2‐Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal. 2005;7:385‐394. [DOI] [PubMed] [Google Scholar]
- 9. Woolf N. Pathology of atherosclerosis. Br Med Bull. 1990;46:960‐985. [DOI] [PubMed] [Google Scholar]
- 10. Dahlfors G, Chen Y, Gustafsson B, et al. Inhibitory effect of diabetes on proliferation of vascular smooth muscle after balloon injury in rat aorta. Int J Exp Diab Res. 2000;1:101‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ceriello A. New insights on oxidative stress and diabetic complications may lead to a "causal" antioxidant therapy. Diab Care. 2003;26:1589‐1596. [DOI] [PubMed] [Google Scholar]
- 12. Hulsmans M, Holvoet P. The vicious circle between oxidative stress and inflammation in atherosclerosis. J Cell Mol Med. 2010;14:70‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Suga T, Iso T, Shimizu T, et al. Activation of receptor for advanced glycation end products induces osteogenic differentiation of vascular smooth muscle cells. J Atheroscler Thromb. 2011;18:670‐683. [DOI] [PubMed] [Google Scholar]
- 14. Demer LL, Tintut Y. Inflammatory, metabolic, and genetic mechanisms of vascular calcification. Arterioscler Thromb Vasc Biol. 2014;34:715‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mittal M, Siddiqui MR, Tran K, et al. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. 2014;20:1126‐1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Golbidi S, Ebadi SA, Laher I. Antioxidants in the treatment of diabetes. Curr Diab Rev. 2011;7:106‐125. [DOI] [PubMed] [Google Scholar]
- 17. Itoh K, Ishii T, Wakabayashi N, et al. Regulatory mechanisms of cellular response to oxidative stress. Free Radical Res. 1999;31:319‐324. [DOI] [PubMed] [Google Scholar]
- 18. Jaiswal AK. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med. 2004;36:1199‐1207. [DOI] [PubMed] [Google Scholar]
- 19. Kobayashi A, Ohta T, Yamamoto M. Unique function of the Nrf2‐Keap1 pathway in the inducible expression of antioxidant and detoxifying enzymes. Methods Enzymol. 2004;378:273‐286. [DOI] [PubMed] [Google Scholar]
- 20. Itoh K, Mimura J, Yamamoto M. Discovery of the negative regulator of Nrf2, Keap1: a historical overview. Antioxid Redox Signal. 2010;13:1665‐1678. [DOI] [PubMed] [Google Scholar]
- 21. Zhang DD. Mechanistic studies of the Nrf2‐Keap1 signaling pathway. Drug Metab Rev. 2006;38:769‐789. [DOI] [PubMed] [Google Scholar]
- 22. Yamamoto T, Suzuki T, Kobayashi A, et al. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol Cell Biol. 2008;28:2758‐2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Long M, Rojo de la Vega M, Wen Q, et al. An essential role of NRF2 in diabetic wound healing. Diabetes. 2016;65:780‐793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu H, Kong L, Tan Y, et al. C66 ameliorates diabetic nephropathy in mice by both upregulating NRF2 function via increase in miR‐200a and inhibiting miR‐21. Diabetologia. 2016;59:1558‐1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gu J, Cheng Y, Wu H, et al. Metallothionein is downstream of Nrf2 and partially mediates sulforaphane prevention of diabetic cardiomyopathy. Diabetes. 2017;66:529‐542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guo W, Tian D, Jia Y, et al. MDM2 controls NRF2 antioxidant activity in prevention of diabetic kidney disease. Biochim Biophys Acta. 2018;1865:1034‐1045. [DOI] [PubMed] [Google Scholar]
- 27. Sun W, Liu X, Zhang H, et al. Epigallocatechin gallate upregulates NRF2 to prevent diabetic nephropathy via disabling KEAP1. Free Radic Biol Med. 2017;108:840‐857. [DOI] [PubMed] [Google Scholar]
- 28. Wu J, Jiang Z, Zhang H, et al. Sodium butyrate attenuates diabetes‐induced aortic endothelial dysfunction via P300‐mediated transcriptional activation of Nrf2. Free Radic Biol Med. 2018;124:454‐465. [DOI] [PubMed] [Google Scholar]
- 29. Pereira A, Fernandes R, Crisostomo J, et al. The Sulforaphane and pyridoxamine supplementation normalize endothelial dysfunction associated with type 2 diabetes. Sci Rep. 2017;7:14357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang Y, Talalay P, Cho CG, et al. A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proc Natl Acad Sci USA. 1992;89:2399‐2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1‐dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137‐8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang Y, Zhang Z, Sun W, et al. Sulforaphane attenuation of type 2 diabetes‐induced aortic damage was associated with the upregulation of Nrf2 expression and function. Oxid Med Cell Longev. 2014;2014:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Patel B, Mann GE, Chapple SJ. Concerted redox modulation by sulforaphane alleviates diabetes and cardiometabolic syndrome. Free Radic Biol Med. 2018;122:150‐160. [DOI] [PubMed] [Google Scholar]
- 34. Seldon MP, Silva G, Pejanovic N, et al. Heme oxygenase‐1 inhibits the expression of adhesion molecules associated with endothelial cell activation via inhibition of NF‐kappa B RelA phosphorylation at serine 276. J Immunol. 1950;2007(179):7840‐7851. [DOI] [PubMed] [Google Scholar]
- 35. Liu GH, Qu J, Shen X. NF‐kappaB/p65 antagonizes Nrf2‐ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochem Biophys Acta. 2008;1783:713‐727. [DOI] [PubMed] [Google Scholar]
- 36. Bahadoran Z, Tohidi M, Nazeri P, et al. Effect of broccoli sprouts on insulin resistance in type 2 diabetic patients: a randomized double‐blind clinical trial. Int J Food Sci Nutr. 2012;63:767‐771. [DOI] [PubMed] [Google Scholar]
- 37. Ellison DH. Bardoxolone methyl in type 2 diabetes and advanced chronic kidney disease. N Eng J Med. 2014;370:1768. [DOI] [PubMed] [Google Scholar]
- 38. Tan SM, Sharma A, Stefanovic N, et al. Derivative of bardoxolone methyl, dh404, in an inverse dose‐dependent manner lessens diabetes‐associated atherosclerosis and improves diabetic kidney disease. Diabetes. 2014;63:3091‐3103. [DOI] [PubMed] [Google Scholar]
- 39. Gold R, Kappos L, Arnold DL, et al. Placebo‐controlled phase 3 study of oral BG‐12 for relapsing multiple sclerosis. N Engl J Med. 2012;367:1098‐1107. [DOI] [PubMed] [Google Scholar]
- 40. Ha CM, Park S, Choi YK, et al. Activation of Nrf2 by dimethyl fumarate improves vascular calcification. Vascul Pharmacol. 2014;63:29‐36. [DOI] [PubMed] [Google Scholar]
- 41. Tebay LE, Robertson H, Durant ST, et al. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic Biol Med. 2015;88:108‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lazaro I, Lopez‐Sanz L, Bernal S, et al. Nrf2 activation provides atheroprotection in diabetic mice through concerted upregulation of antioxidant, anti‐inflammatory, and autophagy mechanisms. Front Pharmacol. 2018;9:819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Eades G, Yang M, Yao Y, et al. miR‐200a regulates Nrf2 activation by targeting Keap1 mRNA in breast cancer cells. J Biol Chem. 2011;286:40725‐40733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhao XJ, Yu HW, Yang YZ, et al. Polydatin prevents fructose‐induced liver inflammation and lipid deposition through increasing miR‐200a to regulate Keap1/Nrf2 pathway. Redox Biol. 2018;18:124‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jiang Z, Wu J, Ma F, et al. MicroRNA‐200a improves diabetic endothelial dysfunction by targeting KEAP1/NRF2. J Endocrinol. 2020;245(1):129‐140. [DOI] [PubMed] [Google Scholar]
- 46. Yan X, Chen J, Zhang C, et al. Fibroblast growth factor 21 deletion aggravates diabetes‐induced pathogenic changes in the aorta in type 1 diabetic mice. Cardiovasc Diabetol. 2015;14:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang J, Xu Z, Gu J, et al. HDAC3 inhibition in diabetic mice may activate Nrf2 preventing diabetes‐induced liver damage and FGF21 synthesis and secretion leading to aortic protection. Am J Physiol Endocrinol Metab. 2018;315:E150‐E162. [DOI] [PubMed] [Google Scholar]
- 48. Zhang J, Cai W, Fan Z, et al. MicroRNA‐24 inhibits the oxidative stress induced by vascular injury by activating the Nrf2/Ho‐1 signaling pathway. Atherosclerosis. 2019;290:9‐18. [DOI] [PubMed] [Google Scholar]
- 49. He M, Siow RC, Sugden D, et al. Induction of HO‐1 and redox signaling in endothelial cells by advanced glycation end products: a role for Nrf2 in vascular protection in diabetes. Nutr Metab Cardiovasc Dis. 2011;21:277‐285. [DOI] [PubMed] [Google Scholar]
- 50. Feng J, Luo J, Deng L, et al. Naringenin‐induced HO‐1 ameliorates high glucose or free fatty acids‐associated apoptosis via PI3K and JNK/Nrf2 pathways in human umbilical vein endothelial cells. Int Immunopharmacol. 2019;75:105769. [DOI] [PubMed] [Google Scholar]
- 51. Liu Y, Wang Y, Miao X, et al. Inhibition of JNK by compound C66 prevents pathological changes of the aorta in STZ‐induced diabetes. J Cell Mol Med. 2014;18:1203‐1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang H, Liu X, Zhou S, et al. SP600125 suppresses Keap1 expression and results in NRF2‐mediated prevention of diabetic nephropathy. J Mol Endocrinol. 2018;60:145‐157. [DOI] [PubMed] [Google Scholar]
- 53. Dong W, Jia Y, Liu X, et al. Sodium butyrate activates NRF2 to ameliorate diabetic nephropathy possibly via inhibition of HDAC. J Endocrinol. 2017;232:71‐83. [DOI] [PubMed] [Google Scholar]
- 54. Miao X, Cui W, Sun W, et al. Therapeutic effect of MG132 on the aortic oxidative damage and inflammatory response in OVE26 type 1 diabetic mice. Oxid Med Cell Longev. 2013;2013:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang DD. Bardoxolone brings Nrf2‐based therapies to light. Antioxid Redox Signal. 2013;19:517‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hennig B, Meerarani P, Toborek M, et al. Antioxidant‐like properties of zinc in activated endothelial cells. J Am Coll Nutr. 1999;18:152‐158. [DOI] [PubMed] [Google Scholar]
- 57. Meerarani P, Ramadass P, Toborek M, et al. Zinc protects against apoptosis of endothelial cells induced by linoleic acid and tumor necrosis factor alpha. Am J Clin Nutr. 2000;71:81‐87. [DOI] [PubMed] [Google Scholar]
- 58. Miao X, Wang Y, Sun J, et al. Zinc protects against diabetes‐induced pathogenic changes in the aorta: roles of metallothionein and nuclear factor (erythroid‐derived 2)‐like 2. Cardiovasc Diabetol. 2013;12:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Li B, Cui W, Tan Y, et al. Zinc is essential for the transcription function of Nrf2 in human renal tubule cells in vitro and mouse kidney in vivo under the diabetic condition. J Cell Mol Med. 2014;18:895‐906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jansen J, Rosenkranz E, Overbeck S, et al. Disturbed zinc homeostasis in diabetic patients by in vitro and in vivo analysis of insulinomimetic activity of zinc. J Nutr Biochem. 2012;23:1458‐1466. [DOI] [PubMed] [Google Scholar]
- 61. Yang G, An SS, Ji Y, et al. Hydrogen sulfide signaling in oxidative stress and aging development. Oxid Med Cell Longev. 2015;2015:1‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu J, Wu J, Sun A, et al. Hydrogen sulfide decreases high glucose/palmitate‐induced autophagy in endothelial cells by the Nrf2‐ROS‐AMPK signaling pathway. Cell Biosci. 2016;6:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sun A, Wang Y, Liu J, et al. Exogenous H2S modulates mitochondrial fusion‐fission to inhibit vascular smooth muscle cell proliferation in a hyperglycemic state. Cell Biosci. 2016;6:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ji K, Xue L, Cheng J, et al. Preconditioning of H2S inhalation protects against cerebral ischemia/reperfusion injury by induction of HSP70 through PI3K/Akt/Nrf2 pathway. Brain Res Bull. 2016;121:68‐74. [DOI] [PubMed] [Google Scholar]
- 65. Yang G, Zhao K, Ju Y, et al. Hydrogen sulfide protects against cellular senescence via S‐sulfhydration of Keap1 and activation of Nrf2. Antioxid Redox Signal. 2013;18:1906‐1919. [DOI] [PubMed] [Google Scholar]
- 66. Chen G, Chen X, Niu C, et al. Baicalin alleviates hyperglycemia‐induced endothelial impairment 1 via Nrf2. J Endocrinol. 2018;240(1):81‐98. [DOI] [PubMed] [Google Scholar]
- 67. Bailey‐Downs LC, Mitschelen M, Sosnowska D, et al. Liver‐specific knockdown of IGF‐1 decreases vascular oxidative stress resistance by impairing the Nrf2‐dependent antioxidant response: a novel model of vascular aging. J Gerontol A Biol Sci Med Sci. 2012;67:313‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Calay D, Mason JC. The multifunctional role and therapeutic potential of HO‐1 in the vascular endothelium. Antioxid Redox Signal. 2014;20:1789‐1809. [DOI] [PubMed] [Google Scholar]
- 69. He M, Nitti M, Piras S, et al. Heme oxygenase‐1‐derived bilirubin protects endothelial cells against high glucose‐induced damage. Free Radic Biol Med. 2015;89:91‐98. [DOI] [PubMed] [Google Scholar]
- 70. Wu H, Kong L, Cheng Y, et al. Metallothionein plays a prominent role in the prevention of diabetic nephropathy by sulforaphane via up‐regulation of Nrf2. Free Radic Biol Med. 2015;89:431‐442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Strycharz J, Rygielska Z, Swiderska E, et al. SIRT1 as a therapeutic target in diabetic complications. Curr Med Chem. 2018;25:1002‐1035. [DOI] [PubMed] [Google Scholar]
- 72. Kitada M, Ogura Y, Koya D. The protective role of Sirt1 in vascular tissue: its relationship to vascular aging and atherosclerosis. Aging. 2016;8:2290‐2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang L, Chen Z, Gong W, et al. Paeonol ameliorates diabetic renal fibrosis through promoting the activation of the Nrf2/ARE pathway via up‐regulating Sirt1. Front Pharmacol. 2018;9:512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wu H, Wu J, Zhou S, et al. SRT2104 attenuates diabetes‐induced aortic endothelial dysfunction via inhibition of P53. J Endocrinol. 2018;237:1‐14. [DOI] [PubMed] [Google Scholar]
- 75. Wu J, Liang W, Tian Y, et al. Inhibition of P53/miR‐34a improves diabetic endothelial dysfunction via activation of SIRT1. J Cell Mol Med. 2019;23(5):3538‐3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Faraonio R, Vergara P, Di Marzo D, et al. p53 suppresses the Nrf2‐dependent transcription of antioxidant response genes. J Biol Chem. 2006;281:39776‐39784. [DOI] [PubMed] [Google Scholar]
- 77. Oliner JD, Pietenpol JA, Thiagalingam S, et al. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature. 1993;362:857‐860. [DOI] [PubMed] [Google Scholar]
- 78. Cancer LDP. p53, guardian of the genome. Nature. 1992;358:15‐16. [DOI] [PubMed] [Google Scholar]
- 79. Yokoyama M, Shimizu I, Nagasawa A, et al. p53 plays a crucial role in endothelial dysfunction associated with hyperglycemia and ischemia. J Mol Cell Cardiol. 2019;129:105‐117. [DOI] [PubMed] [Google Scholar]
- 80. Saito R, Rocanin‐Arjo A, You YH, et al. Systems biology analysis reveals role of MDM2 in diabetic nephropathy. JCI Insight. 2016;1:e87877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gu J, Wang S, Guo H, et al. Inhibition of p53 prevents diabetic cardiomyopathy by preventing early‐stage apoptosis and cell senescence, reduced glycolysis, and impaired angiogenesis. Cell Death Dis. 2018;9:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ihara Y, Toyokuni S, Uchida K, et al. Hyperglycemia causes oxidative stress in pancreatic beta‐cells of GK rats, a model of type 2 diabetes. Diabetes. 1999;48:927‐932. [DOI] [PubMed] [Google Scholar]
- 83. Gorogawa S, Kajimoto Y, Umayahara Y, et al. Probucol preserves pancreatic beta‐cell function through reduction of oxidative stress in type 2 diabetes. Diabetes Res Clin Pract. 2002;57:1‐10. [DOI] [PubMed] [Google Scholar]
- 84. Layer P, Holst JJ, Grandt D, et al. Ileal release of glucagon‐like peptide‐1 (GLP‐1). Association with inhibition of gastric acid secretion in humans. Digest Dis Sci. 1995;40:1074‐1082. [DOI] [PubMed] [Google Scholar]
- 85. Kim MH, Kim EH, Jung HS, et al. EX4 stabilizes and activates Nrf2 via PKCdelta, contributing to the prevention of oxidative stress‐induced pancreatic beta cell damage. Toxicol Appl Pharmacol. 2017;315:60‐69. [DOI] [PubMed] [Google Scholar]
- 86. Cunha DA, Cito M, Carlsson PO, et al. Thrombospondin 1 protects pancreatic beta‐cells from lipotoxicity via the PERK‐NRF2 pathway. Cell Death Differ. 2016;23:1995‐2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Cullinan SB, Zhang D, Hannink M, et al. Nrf2 is a direct PERK substrate and effector of PERK‐dependent cell survival. Mol Cell Biol. 2003;23:7198‐7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Clements CM, McNally RS, Conti BJ, et al. DJ‐1, a cancer‐ and Parkinson's disease‐associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc Nat Acad Sci. 2006;103(41):15091‐15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Sun Q, Shen ZY, Meng QT, et al. The role of DJ‐1/Nrf2 pathway in the pathogenesis of diabetic nephropathy in rats. Ren Fail. 2016;38:294‐304. [DOI] [PubMed] [Google Scholar]
- 90. Pergola PE, Krauth M, Huff JW, et al. Effect of bardoxolone methyl on kidney function in patients with T2D and Stage 3b–4 CKD. Am J Nephrol. 2011;33:469‐476. [DOI] [PubMed] [Google Scholar]
- 91. de Zeeuw D, Akizawa T, Agarwal R, et al. Rationale and trial design of bardoxolone methyl evaluation in patients with chronic kidney disease and type 2 diabetes: the occurrence of renal events (BEACON). Am J Nephrol. 2013;37:212‐222. [DOI] [PubMed] [Google Scholar]
- 92. Chin MP, Reisman SA, Bakris GL, et al. Mechanisms contributing to adverse cardiovascular events in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. Am J Nephrol. 2014;39:499‐508. [DOI] [PubMed] [Google Scholar]
- 93. Chin MP, Wrolstad D, Bakris GL, et al. Risk factors for heart failure in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. J Cardiac Fail. 2014;20:953‐958. [DOI] [PubMed] [Google Scholar]
- 94. Sun W, Liu X, Zhang H, et al. Epigallocatechin gallate upregulates NRF2 to prevent diabetic nephropathy via disabling KEAP1. Free Radic Biol Med. 2017;108:840‐857. [DOI] [PubMed] [Google Scholar]
- 95. Wei J, Zhang Y, Luo Y, et al. Aldose reductase regulates miR‐200a‐3p/141‐3p to coordinate Keap1‐Nrf2, Tgfbeta1/2, and Zeb1/2 signaling in renal mesangial cells and the renal cortex of diabetic mice. Free Radic Biol Med. 2014;67:91‐102. [DOI] [PubMed] [Google Scholar]
- 96. Zheng H, Whitman SA, Wu W, et al. Therapeutic potential of Nrf2 activators in streptozotocin‐induced diabetic nephropathy. Diabetes. 2011;60:3055‐3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Appendino G, Belcaro G, Cornelli U, et al. Potential role of curcumin phytosome (Meriva) in controlling the evolution of diabetic microangiopathy. A pilot study. Panminerva Med. 2011;53:43‐49. [PubMed] [Google Scholar]
- 98. Pan Y, Wang Y, Zhao Y, et al. Inhibition of JNK phosphorylation by a novel curcumin analog prevents high glucose‐induced inflammation and apoptosis in cardiomyocytes and the development of diabetic cardiomyopathy. Diabetes. 2014;63:3497‐3511. [DOI] [PubMed] [Google Scholar]
- 99. Guo Y, Yu S, Zhang C, et al. Epigenetic regulation of Keap1‐Nrf2 signaling. Free Radic Biol Med. 2015;88:337‐349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Fabrizio FP, Sparaneo A, Trombetta D, et al. Epigenetic versus genetic deregulation of the KEAP1/NRF2 axis in solid tumors: focus on methylation and noncoding RNAs. Oxid Med Cell Longev. 2018;2018:1‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]