

Abstract
Most cancer cells exacerbate the pentose phosphate pathway (PPP) to enhance biosynthetic precursors and antioxidant defenses. Metformin, which is used as a first-line oral drug for the treatment of type 2 diabetes, has been proposed to inhibit the malignant progression of different types of cancers. However, metformin has shown poor efficacy as single agent in several clinical trials. Thus, the aim of the present work was to investigate whether the pharmacological inhibition of G6PDH, the first and rate-limiting enzyme of the PPP, by 6-amino nicotinamide (6-AN) potentiates the antitumoral activity of metformin on different human melanoma cell lines. Our results showed that 6-AN has sensitizing properties to metformin cytotoxicity. The combination of metformin and 6-AN decreased glucose consumption and lactate production, altered the mitochondrial potential and redox balance, and thereby blocked melanoma cell progression, directing cells to apoptosis and necrosis. To our knowledge, this is the first study describing the effect of this combination. Future preclinical studies should be performed to reveal the biological relevance of this finding.
Keywords: Metformin, Pentose phosphate pathway (PPP), NADPH, 6-Aminonicotinamide (6-AN)
Highlights
-
•
G6PDH is upregulated in many tumors and correlates with poor prognosis.
-
•
Metformin has been proposed to inhibit malignant progression.
-
•
Pentose phosphate pathway inhibition increases NADPH producing enzymes.
-
•
Inhibition of G6PDH by 6-AN potentiates the antitumoral activity of metformin.
Introduction
Despite the efforts of new therapeutics development, including BRAF inhibitors and immunomodulators (as anti-PDL1 and anti-CTLA-4), metastatic melanoma remains to be the principal life-threating skin cancer [1,2]. Cancer cell metabolism is particularly characterized by the Warburg effect [3,4] which includes an exacerbation of glucose consumption accompanied by an increase in lactate production. Thus, cancer cell metabolism appears as an attractive field for the development of clinical and pre-clinical therapy [5,6]. Beyond glycolysis, the second most important fate of intracellular glucose is the pentose phosphate pathway (PPP). This pathway enables cancer cells to adapt to anabolic demands that require rapid DNA, RNA, and lipid synthesis and to oxidative cellular stress [7,8]. The inhibition of key enzymes of the PPP, including glucose-6-phosphate dehydrogenase (G6PDH), strongly affects the malignant proliferation and metastases of cancer cells both in vitro and in vivo [9]. In many human cancers, G6PDH is upregulated and correlates with poor prognosis [10]. Interestingly, the inhibition of G6PDH restores the sensitivity of cancer cells to chemotherapy [11]. Therefore, the inhibition of the PPP has been proposed as an attractive therapeutic strategy against cancer.
Metformin is a biguanide anti-diabetic drug, which is clinically known as orally well tolerated that has been approved by the Food and Drug Administration (FDA). Retrospective epidemiological studies have revealed a decrease in the incidence of cancer in diabetic patients treated with metformin [12,13]. Metformin modulates cell metabolism at different cell levels by increasing glycolysis, inhibiting respiratory chain complex I and ultimately inhibiting mTOR pathway. This leads to growth arrest and apoptosis [14,15]. Interestingly, metformin has been shown to decrease cancer cell viability and tumor growth in different preclinical models [[16], [17], [18]], inhibit the malignant progression of oral premalignant lesions in chemically-induced experimental models [19] and diminish tumor growth in human head and neck squamous cell carcinoma xenografts [19]. However, metformin seems to have low efficacy as monotherapy against a number of different tumors, including melanoma [20]. Thus, the potential adjuvant role of metformin is currently being investigated in several clinical trials [5,16,[21], [22], [23], [24]] and, high efforts are being made to improve metformin performance.
Despite the fact that metformin cytotoxicity may be in part mediated by ROS increase [25,26], the role of the pentose phosphate pathway during metformin treatment remains to be investigated. In this context, the aim of this study was to investigate whether metformin and the G6PDH inhibitor 6-amino nicotinamide (6-AN) synergize to kill malignant melanoma cells and determine the mechanisms underlying this combinatory approach and its significance regarding the antitumor response against melanoma.
Materials and methods
Cell culture
Cells hM1, hM2, hM4, hM9 and hM10 were established from melanoma patients of Instituto de Oncología Ángel H. Roffo, Facultad de Medicina, Universidad de Buenos Aires, as it was previously described [27]. Also, we used other human melanoma cell lines as A375 (ATCC® CRL-1619™), SB2 [28] and M8 [29]. Cells were cultured at 37 °C in a humidified atmosphere of 95% air and 5% CO2 with DMEM/F12 medium (Invitrogen, Carlsbad, CA, USA) containing 10% FBS (Internegocios, Córdoba, Argentina), 10 mM HEPES (pH 7.4) and antibiotics (60 mg/L Penicillin G, 50 mg/L Streptomycin and 50 mg/L Gentamicin). 3D culture. Multicellular spheroids were obtained following the procedure of hanging drop culture [30] from trypsinized monolayers (0.8–1.4 × 104 cell/spheroid). Viability. Cells were seeded onto 96-well plates at 4–7 × 103 cells/well 24 h before treatments. After 5 days of treatments, cell viability was measured by acidic phosphatase assay [31] and crystal violet staining [32]. Combination studies. Cells were treated with a medium containing a combination of different concentrations of MET (0.1–10 nM) and a fixed concentration of 6-AN (50 μM) or a combination of different concentrations of 6-AN (0.01–100 μM) and a fixed concentration of MET (5 nM). To evaluate the possible effect between the combination of 6-AN and MET was determined using both CompuSyn and Combenefit software [[33], [34], [35]]. The three possibilities: CI < 1, CI = 1, and CI > 1, indicated synergy, additive effect, and antagonism, respectively.
Glucose and lactate content in cell culture media
After 48 h of treatments, 5 μL of each supernatant was transferred to a new 96-well plate. Then, the concentration of glucose and lactate was determined colorimetrically by specific commercial kits (Weiner Lab. and Cobas Roche, respectively).
Western blotting
(i) Whole-cell extracts were obtained using a lysis and extraction buffer (50 mM tris-HCl (pH 8); 100 mM NaCl; 1% Triton; 10 mM EDTA; protease inhibitor 1:10,000). The lysates were centrifuged at 10,000 rpm for 10 min at 4 °C, and the supernatant was stored at −20 °C until immunoblotting was performed. Protein content was determined by the Bradford method. (ii) Immunoblot. Proteins (70–100 μg) from whole-cell extracts were electrophoresed on SDS-PAGE and transferred to PVDF membranes. The membrane was blocked with 5% nonfat milk for 1 h, incubated with the primary antibody overnight at 4 °C and exposed to corresponding secondary antibody (1:5000) for 1 h at room temperature. The primary antibodies used were GLUT-1 (Abcam 115,730), PCNA (PC10, Cell-Signaling 2586) and PARP (46D11, Cell-Signaling 9532). Densitometry units were referred to β-actin (8H10D10, Cell-Signaling 3700). The secondary antibodies used were goat anti-rabbit IgG-HRP (Sigma A9169) and goat anti-mouse IgG-HRP (Santa Cruz sc-2031). (iii) Detection. The chemiluminescence was detected using the Image Quant LAS 500 (GE Healthcare Life Sciences).
Immunofluorescence assay
Immunofluorescence imaging was performed to determine the expression of the proliferative marker Ki67 of treated melanoma cells for 48 h and controls. Cells were fixed with fixation solution (2% formaldehyde - 0,2% glutaraldehyde) for 10 min. For immunostaining, the cells were permeabilized for 30 min in 0.1% Triton X–100 and incubated for 1 h with a blocking solution containing 1% BSA, 0.1% Triton X–100 and 0.3 M glycine in PBS. After overnight incubation with primary antibody solution (Ki67, Abcam 15580; γH2AX, 11174 Abcam) at 4 °C, the secondary antibody (1:1000, Abcam 150077) was added for 1 h at room temperature. To counterstain the nuclei, the coverslips were mounted with Fluoroshield Mounting Medium with DAPI (Abcam 104139). The images were acquired with a Nikon Eclipse™ TE2000-5 inverted fluorescence microscope and photographed with a Nikon DS-Fi1 digital camera at 400 magnification. The objective used was Nikon Plan Fluor ELWD 40× Ph2 DM.
Measurement of cellular production of reactive oxygen species (ROS) (i.e. intracellular oxidants) and mitochondrial membrane potential
After treatments, cells were harvested and incubated with 0.5 μM 2′,7′-dichlorodihydrofluorescein diacetate (DCF, Invitrogen) and 50 nM tetramethylrhodamine, methyl ester (TMRM, Invitrogen) in DMEM-F12 medium without FBS for 30 min. Then, cells were subjected to multiple-channel flow cytometry on a PARTEC PASS III at the Flow Cytometry Facility of the IDEHU (Instituto de Estudios de la Inmunidad Humoral, Buenos Aires, Argentina). The final data were analyzed using the Flowing software (2.5.1, Finland) and the medium intensity of fluorescence was calculated (Geometric Mean: Gm).
Determination of granularity or internal complexity of melanoma cells
The cell complexity of control untreated cells was analyzed by physical parameter by flow cytometry. Side scatter is a measure of the cell refractive index that depends on the cell granularity or internal complexity.
Bromodeoxyuridine labeling
Cells were grown and labeled with a thymidine analog BrdU for 48 h together with treatments. The complete medium of monolayer was replaced with medium containing 40 μM 5-Bromo-2′-deoxyuridine (Sigma), 5 μM uridine (Sigma) and 0.4 μM 5-fluoro-2′-deoxyuridine (Sigma). After treatments, the cells were fixed with fixation solution (2% formaldehyde - 0,2% glutaraldehyde) for 10 min. Then, samples were digested by Hind lll on React 2 buffer (Gibco) and Eco Rl on SH buffer (Sigma) for 1 h to create single-stranded regions in the DNA and to expose the incorporation of BrdU to the monoclonal mouse antibody [36]. After this, the samples were finished as previously described in a conventional immunofluorescence assay. Results are expressed as an index of BrdU positive nuclei relative to total number of nuclei.
Annexin V/propidium iodide labeling
The binding of annexin V to externalized phosphatidylserine was used as a measurement of the number of apoptotic cells with an FITC Annexin V Apoptosis Detection Kit II (BD Biosciences) according to the manufacturer's instructions. The samples were analyzed by flow cytometry as previously described and the results are presented as the percentage of cells that were viable (Annexin V and PI negative), early apoptotic (Annexin V positive but PI negative), late apoptotic (Annexin V and PI positive) or necrotic cells (Annexin V negative and PI positive).
Acridine orange/ethidium bromide staining
Melanoma cells treated for 72 h were incubated with acridine orange (10 μg/mL, green) and ethidium bromide (10 μg/mL, red) for 1 min and staining was evaluated by epifluorescent microscopy (Nikon Eclipse™ E400 fluorescence microscope and photographed with a Nikon Coolpix® 995 digital camera at 400 magnification, objective: Plan Fluor 40× DIC M). The co-localization of both stains indicates late apoptotic or necrotic events (orange/red).
Determination of enzyme activity
Cells were suspended in culture medium and the suspension was frozen at −20 °C until use (maximum two months). The frozen suspensions were sonicated at 100 W in 50% cycle at 4 °C using a VibraCell sonicator model 600 W (Sonics & Materials Inc., Newton, USA) for 4 min. After centrifugation of the homogenate (10,000 ×g, 20 min, 4 °C), the supernatants were collected and maintained at 4 °C during enzyme assays. Enzyme activity was measured in a Shimadzu spectrophotometer model UV-160 (Shimadzu Corporation, Tokyo, Japan). The protocols described by Kitto (1969) and Alp (1976) were used for the determination of malate dehydrogenase activity and NADP dependent isocitrate dehydrogenase activity, respectively. NADP dependent malate dehydrogenase activity was measured according to Brook's (1978) protocol [37,38]. Glucose 6-P dehydrogenase activity was determined with Kornberg and Horecker assays (1955).
Statistics
All experiments were performed at least in triplicate and data are expressed as the mean ± SEM. Differences between groups were analyzed with one- or two-way ANOVA followed by multiple comparisons Tukey's test. p < .05 was established as significant. Analyses were made using INFOSTAT free edition and GraphPad Prism 6 software (GraphPad Software Inc., USA).
Results
6-AN potentiated the cytotoxic effects of metformin
The inhibition of glycolysis, the main glucose metabolism pathway, is known to potentiate the effect of metformin (MET) [25,39,40]. Since glucose is also highly metabolized by the PPP, we investigated whether the inhibition of G6PDH, the key enzyme of the PPP, could sensitize melanoma cells to MET. Eight human melanoma cell lines were treated with 5 mM of MET, 50 μM of 6-AN, or a combination of both (MET/6-AN). After five days of treatment, the effect was visualized by crystal violet (CV) staining. The viability of all the melanoma cell lines evaluated was highly affected by the combination of MET/6-AN that showed almost negative CV staining (Fig. 1A). This effect was even observed in the M8 cell line, whose viability was not affected by the individual treatments. Since spheroids better resemble the tumor architecture, we further evaluated the effect of MET/6-AN on melanoma cells growing as multicellular spheroids. After 14 days of treatment, MET/6-AN combination was also effective in decreasing spheroids size and disaggregating them (Fig. 1B).
Fig. 1.

Cytotoxic effect of the combination of metformin and 6-aminonicotinamide on eight malignant melanoma cell lines. (A) Melanoma cells were treated with metformin (MET, 5 mM), 6-aminonicotinamide (6-AN, 50 μM) or a combination of both MET/6-AN (5 mM/50 μM). After 5 days of treatment, control and treated cells were subjected to crystal violet staining. (B) Multicellular melanoma spheroids after 14 days of treatment (40 magnification). (C) Melanoma cells were treated with increasing concentrations of metformin (1; 2.5; 5; 10 mM) with or without the addition of 6-AN 50 μM for 5 days. After that, cell viability was determined by APH. Results are expressed as a percentage of cell viability from control cells as means ± SEM of three independent experiments. Statistical analysis was performed with two-way ANOVA using Tukey's multiple comparisons test to calculate significance (*p < .05, **p < .01 and ***p < .001). (D) The half maximal inhibitory concentration (IC50) of each curve were calculated. Statistical analysis was performed with the extra sum-of-squares F test to compare if IC50 was different for each data set (***p < .001). (E) Mapped-surface of Loewe from Combenefit software analysis of MET and 6-AN combinations. The concentrations of each drug are plotted along the horizontal axes, while the percentage of cells viability is plotted on the vertical axis. A heat map represents the level of synergy (blue color) at each concentration.
To further evaluate this combinatory strategy, we incubated melanoma cells with a fixed concentration of 6-AN (50 μM) and variable concentrations of MET (0.1–10 mM). After five days of treatment, 1 mM MET was enough to significantly potentiate the effect of 6-AN in five out of the eight melanoma cell lines evaluated (62.5%, p < .05; hM1, hM2, hM9, hM10 and SB2), whereas 2.5 mM MET was necessary for the other three cell lines (hM4, M8 and A375) (Fig. 1C). The significant changes in the IC50s values (p < .05, Fig. 1D) demonstrated that 6-AN improved the in vitro potency of MET.
Next, we used two different software applications to analyze the extent of the combinatory effect of MET/6-AN. The Combination Index (CI) described by Chou-Talalay was determined by using CompuSyn software. We found that MET and 6-AN combination resulted in a CI < 1 thus indicating a synergist effect for all the evaluated cell lines (A375, hM1 and hM4). This result was also depicted by Loewe curves from Combenefit software, where blue areas show synergistic combinations as it is shown in Fig. 1E.
MET/6-AN treatment decreased the proliferation index of melanoma cells
Aggressive melanomas are characterized by a high proliferation index. Thus, we next evaluated Ki67 nuclei by labeling A375, hM1 and hM4 melanoma cells after 48 h of treatments. We found no significant differences between controls and any of the MET-treated cell lines evaluated (Fig. 2A). In addition, inhibition of G6PDH by 6-AN only decreased the Ki67 rate of the highly sensitive hM1 melanoma cell line from 0.73 to 0.65 (Fig. 2A, p < .05), without affecting the other cell lines. Notably, the proliferation index was affected by MET/6-AN combination in all cell lines as shown by the decrease in the number of positive nuclei (white arrows) (Fig. 2A). The quantification of Ki67-positive nuclei indicated that hM1 presented a highly antiproliferative response towards MET/6-AN combination, with an almost complete decrease in the Ki67 rate from 0.73 to 0.1 (~90%, Fig. 2B, p < .01). Consistently, the western blotting assay showed a mildly decrease in the proliferating cell nuclear antigen (PCNA) content caused by the MET/6-AN treatment, in hM1 (Fig. 2B, p < .05). Then, we further investigated the proliferation capacity of hM1 cells by the bromodeoxyuridine (BrdU) incorporation rate. In agreement with the results of Ki67 and PCNA, the proliferation capacity of hM1 was affected both by 6-AN and MET/6-AN combination, to a higher extent (Fig. 2C).
Fig. 2.

Decrease in the proliferative index, cells in S phase and PCNA expression after MET/6-AN treatment. Melanoma cells were treated with metformin (MET, 5 mM), 6-aminonicotinamide (6-AN, 50 μM) or a combination of both (MET/6-AN). After 48 h of treatment, nuclei were stained with DAPI and immuno-stained with (A) a monoclonal antiKi67 antibody to evaluate the proliferation index or (B) a monoclonal antiBrdU antibody to determine the number of cells in S phase. Images were obtained at 400 magnification. Results are expressed as means ± SEM of three independent experiments. Statistical analysis (A and B) was performed with one-way ANOVA using Tukey's multiple comparisons test to calculate significance (*p < .05, **p < .01 and ***p < .001 respect to control treated with PBS, ○○○p < .001 respect to MET 5 mM and ●●●p < .001 respect to 6-AN 50 μM). (C) Representative immunoblot analysis showing the level of PCNA after 48 h treatment of two independent experiments.
MET/6-AN increased late apoptotic/necrotic events
Once we had demonstrated that the MET/6-AN combinatory approach decreased the proliferation index, we moved one step forward and used the double staining acridine orange/ethidium bromide (AO/EB) to allow the observation of apoptotic/necrotic events. The cell membrane is permeable to acridine orange (green) whereas ethidium bromide (red) enters only in late apoptotic or necrotic cells. After 72 h of treatments, A375, hM1 and hM4 control cells or treated with 6-AN mostly appeared as green-stained healthy cells (Fig. 3A). In contrast, MET and MET/6-AN treatments increased the number of late apoptotic/necrotic cells as evidenced by the increase in the number of red-stained nuclei and apoptotic bodies (white and dashed arrows respectively). Interestingly, whereas red nuclei staining after MET treatment as single agent appeared among apparently green healthy cells, after the MET/6-AN combination, red nuclei appeared among notoriously affected (shrinkage and blebbing) remaining cells. To quantify this process, melanoma cells were analyzed by Flow cytometry for binding of annexin V to exteriorized phosphatidylserine. MET/6-AN increased the number of double-stained positive cells either highly (3- to 4-fold in A375 and hM1, respectively) or mildly (hM4), thus denoting an increase in the number of late apoptotic events (Fig. 3B). We found no significant increase in annexin V exposure after MET or 6-AN as monotherapies in any of the cell lines evaluated.
Fig. 3.

Apoptosis/necrosis induced by MET/6-AN treatment. Melanoma cells were treated with metformin (MET, 5 mM), 6-aminonicotinamide (6-AN, 50 μM) or a combination of both (MET/6-AN). (A) After 72 h of treatments, non-fixed cells were incubated with acridine orange/ethidium bromide (AO/EB) to be evaluated by epifluorescence microscopy. The arrows on the representative images of AO/EB staining indicate the co-localization of both stains showing late apoptotic or necrotic events (orange/red). (B) After 48 h treatment, Annexin V antibody and propidium iodide (PI, 5 μg/mL) label was detected by flow cytometry as described in M&M. Results are expressed as dot plot graph and stacked bar chart with means of three independent experiments. (C) After 48 h treatment, the SubG0 population was determined. Results are expressed as means ± SEM of two independent experiments. Statistical analysis was performed with one-way ANOVA using Tukey's multiple comparisons test to calculate significance (*p < .05 and ***p < .001 respect to control treated with PBS, ○○○p < .001 respect to MET 5 mM and ●●●p < .001 respect to 6-AN 50 μM). (D) Representative image of Poly (ADP-ribose) polymerase-1 (PARP1) expression after 48 h of treatments. Values refer to the relative density of cleaved PARP with the respective β-actin. Results are expressed as means ± SEM of two independent experiments.
Next, necrosis was also evaluated by propidium iodide (PI) uptake. hM1 and A375 cell lines exhibited a loss of membrane integrity denoted by an increase in PI uptake after 48 h of treatment with MET and MET/6-AN. These results were accompanied by an increase in the number of subG0 events after MET/6-AN treatment in A375 and hM1 but not in hM4 (Fig. 3C).
Finally, we also found an increase in cleaved Poly-(ADP-ribose) polymerase, a proapoptotic protein, after MET and MET/6-AN treatment compared to control, especially in hM1 melanoma cells (Fig. 3D).
Effects of MET/6-AN on metabolic parameters
To understand which mechanisms were underlying the antiproliferative and cytotoxic effects of MET/6-AN, we further investigate whether this combination was affecting glucose consumption and lactate production, both of which are particularly exacerbated in cancer cells.
Independently of cell sensitiveness, the color of the cell culture medium suggested that 6-AN and MET/6-AN decreased extracellular acidification, whereas MET increased (Fig. 4A). In agreement, we found that 6-AN significantly decreased the presence of lactate in the supernatant of all cell lines (Fig. 4B, gray bars), whereas MET significantly increased in hM4 supernatant (Fig. 4B, black bars). The effect of 6-AN was abolished by the presence of MET in A375 and hM4 but not in hM1, where MET/6-AN combination also decreased the concentration of lactate in the supernatant (Fig. 4B, white bars). In addition, the concentration of glucose in the extracellular medium was higher, thus indicating less consumption, than control cells after 6-AN and MET/6-AN treatment in the supernatant of hM1 and A375, but not in that of hM4 (Fig. 4C). We found comparable results at extracellular glucose concentration when referring glucose values to the number of viable cells (Supplementary Fig. S1). In addition, we found also comparable results by referring lactate values to the number of viable cells except for MET/6-AN treatment. This effect may be explained by the fact that we measured the accumulative lactate. As this metabolite was not present in the fresh medium, we hypothesize that during MET/6-AN treatment lactate production was almost zero. As Met/6-AN decreased cell viability, by referring this to viable cells, we increased the value of lactate even when its production was not augmented (Supplementary Fig. S1).
Fig. 4.

Modulation of glycolytic parameters by MET, 6-AN or the combination of both. Melanoma cells were treated with metformin (MET, 5 mM), 6 aminonicotinamide (6-AN, 50 μM) or a combination of both MET/6-AN (5 mM/50 μM). After 48 h of treatment, control and treated cells were subjected to determination of extracellular: (A) pH, (B) lactate and (C) glucose. Results are expressed relative to control cells as means ± SEM of three independent experiments. Statistical analysis (A, B and C) was performed with one-way ANOVA using Tukey's multiple comparisons test to calculate significance (*p < .05, **p < .01 and ***p < .001 respect to control treated with PBS, ○○○p < .001 respect to MET 5 mM and ●●●p < .001 respect to 6-AN 50 μM). (D) Representative image of the expression of glucose transporter 1 (Glut1). Values refer to the relative density of Glut1 with the respective β-actin of three independent experiments.
Supplementary Fig. S1.
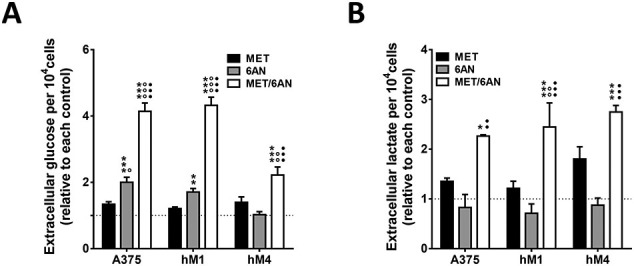
Modulation of glycolytic parameters by MET, 6-AN or the combination of both. Melanoma cells were treated with metformin (MET, 5 mM), 6 aminonicotinamide (6-AN, 50 μM) or a combination of both MET/6-AN (5 mM/50 μM). After 48 h of treatment, control and treated cells were subjected to determination of extracellular: (A) glucose and (B) lactate. Results are expressed relative to control cells as means ± SEM of three independent experiments. Statistical analysis (A and B) was performed with one-way ANOVA using Tukey's multiple comparisons test to calculate significance (*p < .05, **p < .01 and ***p < .001 respect to control treated with PBS, ○○○p < .001 respect to MET 5 mM and ●●●p < .001 respect to 6-AN 50 μM).
We further investigated whether the glucose transporter GLUT1 was being modulated by MET and/or 6-AN treatment. We found that the treatment with 6-AN alone (A375 and hM1) or in combination with MET (hM1) produced an increase in GLUT1 content. Notably, hM4 presented considerably less GLUT1 content than A375 and hM1 cells, without considerable changes after the treatments (Fig. 4D).
MET/6-AN increased intracellular oxidants and depolarized mitochondria
One of the most important roles of G6PDH (as the main enzyme of PPP) is to restitute the concentration of glutathione (the most abundant soluble antioxidant) from glutathione disulfide by the activity of the enzyme glutathione reductase and NADPH as a cofactor. Thus, after the inhibition of G6PDH, we expected an increase in the levels of intracellular oxidants. However, 6-AN as single agent was not enough to significantly increase the levels of intracellular oxidants (Fig. 5A, gray bars). A similar situation was observed after MET treatment in all cell lines. In contrast, the MET/6-AN combination increased the levels of intracellular oxidants at 48 h in A375 and hM1, with a maximum increase after 72 h as observed by the shift to the right of the green fluorescent abscissa (FL1) and by the quantification of DCF cell content (by Flow cytometry) from at least four independent experiments (Fig. 5A). Conversely, hM4 cells did not show an increase in the levels of intracellular oxidants after the treatments (Fig. 5A).
Fig. 5.

Intracellular oxidants, mitochondrial potential and cell complexity after treatment with MET, 6-AN or the combination of both.
Melanoma cells were treated with metformin (MET, 5 mM), 6-aminonicotinamide (6-AN, 50 μM) or a combination of both (MET/6-AN). After 24, 48, or 72 h of treatment, cells were incubated with (A) DCF (2′,7′-dichlorodihydrofluorescein diacetate, intracellular oxidants) for 20 min, (B) TMRM (tetramethylrhodamine, methyl ester mitochondrial membrane potential) for 30 min and evaluated by flow cytometry as described in M&M. (C) Cell complexity was determined from the physical parameter SSC (Side scatter) on a dot plot graph. From each label or SSC, a representative overlay histogram is shown at 48 h of treatment. Results are expressed as means ± SEM of three independent experiments. Statistical analysis was performed with one-way ANOVA using Tukey's multiple comparisons test to calculate significance (*p < .05, **p < .01 and ***p < .001 respect to control treated with PBS, ○○○p < .001 respect to MET 5 mM and ●●●p < .001 respect to 6-AN 50 μM).
Since mitochondria are one of the main sources of intracellular oxidants and mitochondria depolarization could be involved in melanoma cell death, we next studied the integrity of the mitochondrial potential after the treatments. We found that MET did not affect the mitochondrial potential of melanoma cells as monotherapy at any time. Interestingly, the mitochondrial potential of A375 remaining cells was significantly increased 72 h after the 6-AN and MET/6-AN treatment (Fig. 5B). On the other hand, hM1 cells presented an early depolarization at 24 h after the MET/6-AN treatment, which was not overcome at any of the times evaluated (Fig. 5B). Finally, the mitochondrial potential of hM4 was not significantly affected by the treatments (Fig. 5B). Nevertheless, MET/6-AN treatment tended to cause early mitochondrial depolarization in all cell lines.
During the flow cytometry assays, we were also able to observe changes in cell complexity. Interestingly, we found that not only MET/6-AN but also 6-AN as single agent significantly increased cell complexity at 24 h, with a maximum of effect at 72 h, being the magnitude of the effect of the combination higher than that of 6-AN as a single agent (Fig. 5C). In addition, hM4 did not increase its intracellular complexity at any time with any treatment, in concordance with the results regarding the levels of its mitochondrial and intracellular oxidants.
Metformin and 6-AN inhibitor increased the activity of NADPH producing enzymes
As cell reductive power should be accurately maintained to normal cell function, we hypothesized that other NADPH enzymatic sources may be increasing their activities in order to compensate G6PDH inhibition by 6-AN. Effectively, 6-AN treatment for 48 h increased the activity of NADP-dependent Malic Enzyme (ME(NADP), 4-fold, p < .001) and Isocitrate Dehydrogenase IDH enzyme (20-fold, p < .001) (Fig. 6A).Most interestingly, MET and MET/6-AN treatment also increased (NADP-dependent) IDH activity (10-fold and 5-fold, p < .01 and p < .05, respectively). Furthermore, MET, 6-AN or the combination of MET/6-AN drastically increased NAD dependent ME (ME(NAD), ~100-fold, p < .001).
Fig. 6.

Increased activity of NADPH producing enzymes by MET and 6-AN treatments.
Melanoma cells were treated with metformin (MET, 5 mM), 6-aminonicotinamide (6-AN, 50 μM) or a combination of both (MET/6-AN). After 48 h of treatment, cells were lysed, and enzyme activity was measured as described in M&M. (A) Results are expressed as mean activity ± SEM of two independent experiments. Statistical analysis was performed with one-way ANOVA using Tukey's multiple comparisons test to calculate significance (*p < .05, **p < .01 and ***p < .001 respect to control treated with PBS, ○○○p < .001 respect to MET 5 mM, ■■■p < .001 respect to MET/6-AN). (B) Representative image of the effect of MET and 6-AN effect. (C) Melanoma cells were treated with metformin (MET, 5 mM), 6-aminonicotinamide (6-AN, 50 μM) and a combination of both (MET/6-AN) with or without the addition of Nicotinamide Adenine Dinucleotide Phosphate Hydrate (NADPH, 50 μM). After 5 days of treatment, control and treated cells were subjected to the acid phosphatase assay as described in M&M. Results are expressed as means ± SEM of four independent experiments. Statistical analysis was performed with two-way ANOVA using Tukey's multiple comparisons test to calculate significance (***p < .001).
Since MET and 6-AN were probably affecting NADPH content, we explored whether NADPH supply could reverse or at least diminish the cytotoxic effect of its combination. Interestingly, we found that 50 μM NADPH was able to significantly reverse MET/6-AN effect on hM4 (Fig. 6C) but not in A375 nor hM1 cells. Additionally, 50 μM NADH or the combination of both, NADPH/NADH (50 μM/50 μM) was also able to produce this inhibitory effect only in hM4. Since transportation of NADPH and NADH across cell membrane remains controversial [41], we investigated the addition of nicotinamide (NAM), a soluble precursor of its synthesis by the savage pathway. Irrespectively of the cell line, NAM (5 mM) reversed MET/6-AN (p < .001). Interestingly, the effect of MET/6-AN was reduced up to the effect of MET alone. Moreover, 6-AN but not MET cytotoxic effect was completely reversed by NAM in hM1 (Fig. 6C).
Discussion
In agreement with previous studies [5,9,42,43], our results support the idea of bioenergetic modulation as an emerging strategy to treat cancer. In this aspect, Arbe et al., 2017, and other groups have published exciting results about the synergistic combination of MET and 2-deoxyglucose (2DG), a glucose analogous, on feline mammary carcinoma cells and a wide variety of different tumor cells, respectively [25,39,40]. However, only two (hM4 and hM9) out of eight human melanoma cells (25%) have shown a significant potentiation after MET and 2DG combination (data not shown, manuscript in progress). Remarkably, those cells were considered highly sensitive to glycolysis inhibition by 2DG. In contrast, we did find a synergistic effect on eight out of eight melanoma cell lines (100% effectiveness, Fig. 1) by combining MET with 6-AN, an inhibitor of G6PDH [[44], [45], [46]], the first and limiting enzyme of the PPP, irrespectively of the response of the monotherapies. These results propose a common or an interrelated mechanism between MET signaling and the PPP, suggesting NADPH as one of the molecules involved. Consistent with our results, down-regulation of ME, another NADPH producing enzyme, increased MET response in HNSCC [47]. The PPP is the second most important fate of intracellular glucose after glycolysis, and supplies the requirement of anabolic precursors [7,8]. Another aspect to take into consideration should be the fact that G6PDH inhibition induces an increase in its substrate G6P, which in turn, inhibits hexokinase, a key enzyme of glycolysis. This effect could explain the decrease observed in lactate production and glucose consumption after 6-AN treatment of melanoma cells (Fig. 4B). In contrast, MET increased lactate production in hM4 and this effect was not prevented by the addition of 6-AN. We hypothesized that these different behaviors among melanoma cell lines may be related to their differences in glycolysis dependence, being hM4 less glycolytic than hM1 and A375 as suggested by our compelling results (unpublished data, manuscript in progress).
During MET/6-AN treatment but not MET alone, A375 and hM1 cells increased the production of reactive oxygen species (ROS) within the first 48 h. Accordingly with Choi et al., 2014 metformin as monotherapy significantly increases ROS and decreases GSH levels during glucose deprivation [26]. We hypothesized that 6-AN not only may be mimicking a glucose-free-like condition but also decreasing NADPH which in turn affects GSH levels and the antioxidant capacity even at high glucose condition (all the experiments have been performed under high glucose condition). In accordance, we found that MET and 6-AN increased IDH activity probably to abrogate oxidative stress. Moreover, the inhibition of G6PDH, the main source of NADPH, promoted both IDH (NADP-dependent) and ME (NADP-dependent) activity increase (Fig. 6A and B). Probably, by means of these compensatory activities, 6-AN-treated melanoma cells did not present ROS increase (Fig. 4B). In contrast, the increase in the activities of IDH(NADP) and ME(NADP) was not enough for reversing the reductive power impairment displayed by MET, thus promoting almost 50% of A375 cells death. This disability was even worst during MET/6-AN combination, thus promoting over 80% of A375 cells death (Fig. 1, Fig. 6A and B). Oxidative stress was accompanied by a hyperpolarization of the mitochondria in remaining A375 cells probably in an attempt to resist cell death, as it has been previously reported to other antitumor therapies [49]. In contrast with A375, the mitochondrial potential of hM1 significantly decreased and did not hyperpolarize during the time evaluated. Simultaneously, this effect was accompanied by an increase in intracellular complexity in both cell lines, probably due to an autophagy process as previously described during the combination of MET and 2DG by Arbe et al., 2017 [25] and others [40]. Indeed, our preliminary results in canine and feline melanoma cells and human glioblastoma cells also support this hypothesis (data not shown).
Ki67 immunostaining and BrdUr assay allowed observing that MET/6-AN decreased the proliferation index in all cell lines, especially in hM1, where 6-AN had also a low but significant effect as a monotherapy (Fig. 2). We also found that hM1 displayed a higher γH2AX immunostaining than A375 (Supplementary Fig. S2). Among its different activities, PARP may be displaying a protective role (as part of the DNA repair response) during the cytotoxic effect of MET/6-AN, especially in hM1 cells, where not only cleaved PARP but also its full-length protein was higher than in the other tested cells (Fig. 3D). Furthermore, hM1 presented the highest Annexin V positivity in agreement with the increase in cleaved PARP levels (Fig. 3). On the other hand, MET/6-AN was also a cytotoxic treatment that promoted both apoptotic and necrotic events (Fig. 2, Fig. 3). However, flow cytometry analysis of cell death denoted a notorious increase in late apoptotic but not in necrotic events. This indicates that cells from AO/BET staining and those shown as SubG0 population (Fig. 5B red and C respectively) were probably secondary to apoptotic processes. Taking into account the differences between hM4 and the other two melanoma cell lines after MET/6-AN treatment in terms of ROS production, mitochondrial potential and cell complexity, our results suggest that the particular behavior of hM4 could be due to a less glycolytic phenotype as it has been suggested above. Although this hypothesis remains to be fully investigated, we found that hM4 presented less glucose consumption and GLUT1 content, which were not altered by the 6-AN or MET/6-AN treatment (Fig. 4C and D).
Supplementary Fig. S2.
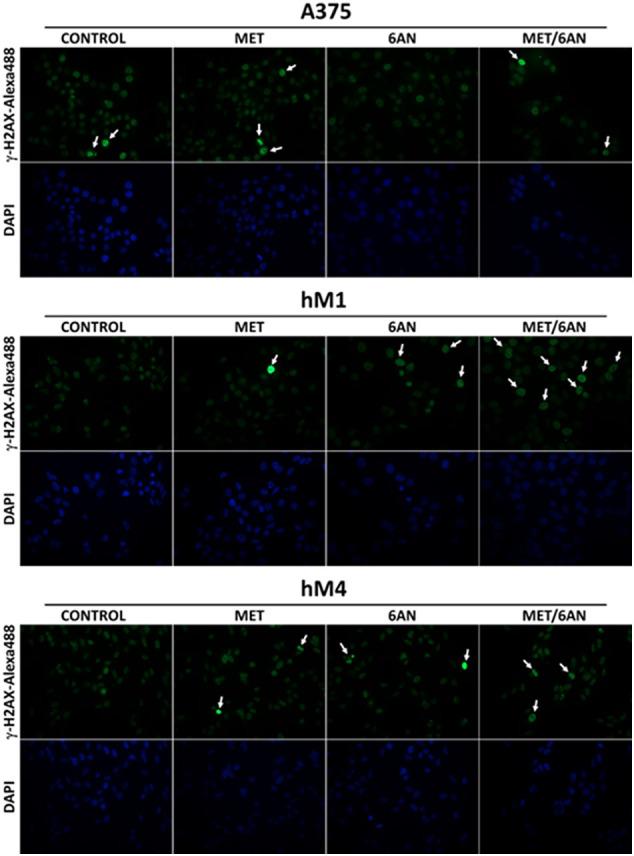
Induction of DNA damage after MET, 6-AN or the combination of both. Melanoma cells were treated with metformin (MET, 5 mM), 6 aminonicotinamide (6-AN, 50 μM) or a combination of both MET/6-AN (5 mM/50 μM). After 48 h of treatment, control and treated cells were subjected to γH2AX immunostaining.
As it has been recently reviewed by Jaune and Rocchi [50], MET antitumor activity is being extensively evaluated (more than 300 clinical trials, ClinicalTrials.gov) in different types of cancer. Less is known about the therapeutic opportunities of inhibiting the PPP [9].
Here, we demonstrate, for the first time, that G6PDH inhibition potentiates MET cytotoxicity leading to increased apoptotic/necrotic events, decreased growth and survival of melanoma cells, accompanied by a redox imbalance probably due to a large reductive power impairment (Fig. 7). Our work suggests that G6PDH inhibition represents a potential therapeutic strategy to potentiate MET antitumoral effects in melanoma.
Fig. 7.

Suggested metabolic changes displayed by MET in combination with 6-AN. Inhibition of G6PDH enhances MET cytotoxicity probably through decreasing cell reductive capacity. While MET alone increases glucose consumption and lactate production, MET/6-AN combination blocks this compensatory mechanism. In addition, during MET treatment, the induction of IDH(NADP) activity favors to decrease oxidative stress. In contrast, MET/6-AN treated cells may not overcome redox imbalance driving melanoma cells to increased apoptosis and necrosis.
The following are the supplementary data related to this article.
Author contributions statement
Arbe MF: Methodology, Conceptualization, Investigation, Formal analysis Visualization and Writing - Review & Editing.
Agnetti L: Methodology, Visualization.
Breininger E: Methodology, Formal analysis.
Finocchiaro LME: Writing - Review & Editing and Funding acquisition.
Glikin GC: Writing - Review & Editing and Funding acquisition.
Villaverde MS: Conceptualization, Investigation, Formal analysis, Visualization, Writing - Original Draft, Writing - Review & Editing, Supervision and Funding acquisition.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We thank Graciela B. Zenobi for technical advice and assistance. This work was supported by Agencia Nacional de Promoción Científica y Tecnológica (PICT 2014-1247 and PICT 2012-1738, Préstamo BID) and by Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET, D3646/14 and PIP 112 201101 00627). GCG, LMEF, and MSV are investigators and MFA, and LA are fellows of CONICET, Argentina.
References
- 1.Eggermont A.M.M., Spatz A., Robert C. Lancet. Lancet Publishing Group; 2014. Cutaneous melanoma; pp. 816–827. [DOI] [PubMed] [Google Scholar]
- 2.Weber J.S., O'Day S., Urba W., Powderly J., Nichol G., Yellin M., Snively J., Hersh E. Phase I/II study of ipilimumab for patients with metastatic melanoma. J. Clin. Oncol. 2008;26:5950–5956. doi: 10.1200/JCO.2008.16.1927. [DOI] [PubMed] [Google Scholar]
- 3.O. Warburg Berlin-Dahlem, The Metabolism of Carcinoma Cells, n.d.
- 4.Otto A.M. Warburg effect(s)—a biographical sketch of Otto Warburg and His impacts on tumor metabolism. Cancer Metab. 2016 doi: 10.1186/s40170-016-0145-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.S. Granja, C. Pinheiro, R.M. Reis, O. Martinho, F. Baltazar, Send Orders for Reprints to reprints@benthamscience.ae Glucose Addiction in Cancer Therapy: Advances and Drawbacks, 2015. [DOI] [PubMed]
- 6.Kroemer G., Pouyssegur J. Tumor cell metabolism: cancer's Achilles' heel. Cancer Cell. 2008;13:472–482. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 7.Stincone A., Prigione A., Cramer T., Wamelink M.M.C., Campbell K., Cheung E., Olin-Sandoval V., Grüning N.-M., Krüger A., Alam M.T., Keller M.A., Breitenbach M., Brindle K.M., Rabinowitz J.D., Ralser M. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway HHS public access. Biol. Rev. Camb. Philos. Soc. 2015;90:927–963. doi: 10.1111/brv.12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patra K.C., Hay N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014 doi: 10.1016/j.tibs.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mele L., Paino F., Papaccio F., Regad T., Boocock D., Stiuso P., Lombardi A., Liccardo D., Aquino G., Barbieri A., Arra C., Coveney C., La Noce M., Papaccio G., Caraglia M., Tirino V., Desiderio V. A new inhibitor of glucose-6-phosphate dehydrogenase blocks pentose phosphate pathway and suppresses malignant proliferation and metastasis in vivo article. Cell Death Dis. 2018 doi: 10.1038/s41419-018-0635-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang P., Du W., Wu M. Regulation of the pentose phosphate pathway in cancer. Protein Cell. 2014 doi: 10.1007/s13238-014-0082-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Catanzaro D., Gaude E., Orso G., Giordano C., Guzzo G., Rasola A., Ragazzi E., Caparrotta L., Frezza C., Montopoli M. Inhibition of glucose-6-phosphate dehydrogenase sensitizes cisplatin-resistant cells to death. Oncotarget. 2015;6:30102–30114. doi: 10.18632/oncotarget.4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Del Barco S., Vazquez-Martin A., Cufí S., Oliveras-Ferraros C., Bosch-Barrera J., Joven J., Martin-Castillo B., Menendez J.A. Metformin: multi-faceted protection against cancer. Oncotarget. 2011;2:896–917. doi: 10.18632/oncotarget.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chlebowski R.T., McTiernan A., Wactawski-Wende J., Manson J.A.E., Aragaki A.K., Rohan T., Ipp E., Kaklamani V.G., Vitolins M., Wallace R., Gunter M., Phillips L.S., Strickler H., Margolis K., Euhus D.M. Diabetes, metformin, and breast cancer in postmenopausal women. J. Clin. Oncol. 2012;30:2844–2852. doi: 10.1200/JCO.2011.39.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ochoa-Gonzalez F., Cervantes-Villagrana A.R., Fernandez-Ruiz J.C., Nava-Ramirez H.S., Hernandez-Correa A.C., Enciso-Moreno J.A., Castañeda-Delgado J.E. Metformin induces cell cycle arrest, reduced proliferation, wound healing impairment in vivo and is associated to clinical outcomes in diabetic foot ulcer patients. PLoS One. 2016;11 doi: 10.1371/journal.pone.0150900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo Q., Liu Z., Jiang L., Liu M., Ma J., Yang C., Han L., Nan K., Liang X. Metformin inhibits growth of human non-small cell lung cancer cells via liver kinase B-1-independent activation of adenosine monophosphate-activated protein kinase. Mol. Med. Rep. 2016;13:2590–2596. doi: 10.3892/mmr.2016.4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu X., Yeerna H., Goto Y., Ando T., Wu V.H., Zhang X., Wang Z., Amornphimoltham P., Murphy A.N., Tamayo P., Chen Q., Lippman S.M., Gutkind J.S. Metformin inhibits progression of head and neck squamous cell carcinoma by acting directly on carcinoma initiating cells. Cancer Res. 2019 doi: 10.1158/0008-5472.CAN-18-3525. canres.3525.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim K.H., Jeong Y.T., Kim S.H., Jung H.S., Park K.S., Lee H.Y., Lee M.S. Metformin-induced inhibition of the mitochondrial respiratory chain increases FGF21 expression via ATF4 activation. Biochem. Biophys. Res. Commun. 2013;440:76–81. doi: 10.1016/j.bbrc.2013.09.026. [DOI] [PubMed] [Google Scholar]
- 18.X. Wang, K. Chen, Y. Yu, Y. Xiang, J.H. Kim, W. Gong, J. Huang, G. Shi, Q. Li, M. Zhou, T. Sayers, P. Tewary, B. Gao, J.M. Wang, Metformin Sensitizes Lung Cancer Cells to Treatment by the Tyrosine Kinase Inhibitor Erlotinib, n.d. www.impactjournals.com/oncotarget (accessed November 27, 2019). [DOI] [PMC free article] [PubMed]
- 19.Martin D., Abba M.C., Molinolo A.A., Vitale-Cross L., Wang Z., Zaida M., Delic N.C., Samuels Y., Lyons G.J., Gutkind J.S. The head and neck cancer cell oncogenome: a platform for the development of precision molecular therapies. Oncotarget. 2014;5:1–18. doi: 10.18632/oncotarget.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Montaudié H., Cerezo M., Bahadoran P., Roger C., Passeron T., Machet L., Arnault J.-P., Verneuil L., Maubec E., Aubin F., Granel F., Giacchero D., Hofman V., Lacour J.-P., Maryline A., Ballotti R., Rocchi S. Metformin monotherapy in melanoma: a pilot, open-label, prospective, and multicentric study indicates no benefit. Pigment Cell Melanoma Res. 2017;30:378–380. doi: 10.1111/pcmr.12576. [DOI] [PubMed] [Google Scholar]
- 21.Pierotti M.A., Berrino F., Gariboldi M., Melani C., Mogavero A., Negri T., Pasanisi P., Pilotti S. Targeting metabolism for cancer treatment and prevention: metformin, an old drug with multi-faceted effects. Oncogene. 2013;32:1475–1487. doi: 10.1038/onc.2012.181. [DOI] [PubMed] [Google Scholar]
- 22.Schoors S., Bruning U., Missiaen R., Queiroz K.C.S., Europe PMC Funders Group Fatty acid carbon is essential for dNTP synthesis in endothelial cells. 2015;520:192–197. doi: 10.1038/nature14362.Fatty. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Queiroz E.A.I.F., Puukila S., Eichler R., Sampaio S.C., Forsyth H.L., Lees S.J., Barbosa A.M., Dekker R.F.H., Fortes Z.B., Khaper N. Metformin induces apoptosis and cell cycle arrest mediated by oxidative stress, AMPK and FOXO3a in MCF-7 breast cancer cells. PLoS One. 2014;9 doi: 10.1371/journal.pone.0098207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Samuel S.M., Varghese E., Kubatka P., Triggle C.R., Büsselberg D. Metformin: the answer to cancer in a flower? Current knowledge and future prospects of metformin as an anti-cancer agent in breast cancer. Biomolecules. 2019;9:846. doi: 10.3390/biom9120846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arbe M.F., Fondello C., Agnetti L., Álvarez G.M., Tellado M.N., Glikin G.C., Finocchiaro L.M.E., Villaverde M.S. Inhibition of bioenergetic metabolism by the combination of metformin and 2-deoxyglucose highly decreases viability of feline mammary carcinoma cells. Res. Vet. Sci. 2017;114 doi: 10.1016/j.rvsc.2017.07.035. [DOI] [PubMed] [Google Scholar]
- 26.Choi Y.W., Lim I.K. Sensitization of metformin-cytotoxicity by dichloroacetate via reprogramming glucose metabolism in cancer cells. Cancer Lett. 2014;346:300–308. doi: 10.1016/j.canlet.2014.01.015. [DOI] [PubMed] [Google Scholar]
- 27.Fondello C., Agnetti L., Villaverde M.S., Simian M., Glikin G.C., Finocchiaro L.M.E. The combination of bleomycin with suicide or interferon-β gene transfer is able to efficiently eliminate human melanoma tumor initiating cells. Biomed. Pharmacother. 2016;83 doi: 10.1016/j.biopha.2016.06.038. [DOI] [PubMed] [Google Scholar]
- 28.Lopez M.V., Viale D.L., Cafferata E.G.A., Bravo A.I., Carbone C., Gould D., Chernajovsky Y., Podhajcer O.L. Tumor associated stromal cells play a critical role on the outcome of the oncolytic efficacy of conditionally replicative adenoviruses. PLoS One. 2009;4 doi: 10.1371/journal.pone.0005119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gnjatic S., Cai Z., Viguier M., Chouaib S., Guillet J.G., Choppin J. Accumulation of the p53 protein allows recognition by human CTL of a wild-type p53 epitope presented by breast carcinomas and melanomas. J. Immunol. 1998;160:328–333. [PubMed] [Google Scholar]
- 30.Kelm J.M., Timmins N.E., Brown C.J., Fussenegger M., Nielsen L.K. Method for generation of homogeneous multicellular tumor spheroids applicable to a wide variety of cell types. Biotechnol. Bioeng. 2003;83:173–180. doi: 10.1002/bit.10655. [DOI] [PubMed] [Google Scholar]
- 31.Arbe M.F., Fondello C., Agnetti L., Álvarez G.M., Tellado M.N., Glikin G.C., Finocchiaro L.M.E., Villaverde M.S. Inhibition of bioenergetic metabolism by the combination of metformin and 2-deoxyglucose highly decreases viability of feline mammary carcinoma cells. Res. Vet. Sci. 2017;114:461–468. doi: 10.1016/j.rvsc.2017.07.035. [DOI] [PubMed] [Google Scholar]
- 32.Saotome K., Morita H., Umeda M. Cytotoxicity test with simplified crystal violet staining method using microtitre plates and its application to injection drugs. Toxicol. in Vitro. 1989;3:317–321. doi: 10.1016/0887-2333(89)90039-8. [DOI] [PubMed] [Google Scholar]
- 33.Bijnsdorp I.V., Giovannetti E., Peters G.J. Humana Press; 2011. Analysis of Drug Interactions; pp. 421–434. [DOI] [PubMed] [Google Scholar]
- 34.Chou T.C., Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 35.G.Y. Di Veroli, C. Fornari, D. Wang, S. Verine Mollard, J.L. Bramhall, F.M. Richards, D.I. Jodrell, Combenefit: An Interactive Platform for the Analysis and Visualization of Drug Combinations, (n.d.). doi: 10.1093/bioinformatics/btw230. [DOI] [PMC free article] [PubMed]
- 36.Finocchiaro L.M.E., Glikin G.C. Intracellular melatonin distribution in cultured cell lines. J. Pineal Res. 1998;24:22–34. doi: 10.1111/j.1600-079X.1998.tb00362.x. [DOI] [PubMed] [Google Scholar]
- 37.Brooks D.E. Activity and androgenic control of enzymes associated with the tricarboxylic acid cycle, lipid oxidation and mitochondrial shuttles in the epididymis and epididymal spermatozoa of the rat. Biochem. J. 1978;174:741–752. doi: 10.1042/bj1740741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Breininger E., Dubois D., Pereyra V.E., Rodriguez P.C., Satorre M.M., Cetica P.D. Participation of phosphofructokinase, malate dehydrogenase and isocitrate dehydrogenase in capacitation and acrosome reaction of boar spermatozoa. Reprod. Domest. Anim. 2017;52:731–740. doi: 10.1111/rda.12973. [DOI] [PubMed] [Google Scholar]
- 39.Cheong J.-H., Park E.S., Liang J., Dennison J.B., Tsavachidou D., Nguyen-Charles C., Wa Cheng K., Hall H., Zhang D., Lu Y., Ravoori M., Kundra V., Ajani J., Lee J.-S., Ki Hong W., Mills G.B. Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinical cancer models. Mol. Cancer Ther. 2011 doi: 10.1158/1535-7163.MCT-11-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ben Sahra I., Laurent K., Giuliano S., Larbret F., Ponzio G., Gounon P., Le Marchand-Brustel Y., Giorgetti-Peraldi S., Cormont M., Bertolotto C., Deckert M., Auberger P., Tanti J.F., Bost F. Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010 doi: 10.1158/0008-5472.CAN-09-2782. [DOI] [PubMed] [Google Scholar]
- 41.Xiao W., Wang R.-S., Handy D.E., Loscalzo J. NAD(H) and NADP(H) redox couples and cellular energy metabolism. Antioxid. Redox Signal. 2018;28:251–272. doi: 10.1089/ars.2017.7216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun R.C., Board P.G., Blackburn A.C. Targeting metabolism with arsenic trioxide and dichloroacetate in breast cancer cells. Mol. Cancer. 2011 doi: 10.1186/1476-4598-10-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Santo C., Booth S., Vardon A., Cousins A., Tubb V., Perry T., Noyvert B., Beggs A., Ng M., Halsey C., Kearns P., Cheng P., Mussai F. The arginine metabolome in acute lymphoblastic leukemia can be targeted by the pegylated-recombinant arginase I BCT-100. Int. J. Cancer. 2018;142:1490–1502. doi: 10.1002/ijc.31170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poulain L., Sujobert P., Zylbersztejn F., Barreau S., Stuani L., Lambert M., Palama T.L., Chesnais V. 2017. High mTORC1 Activity Drives Glycolysis Addiction and Sensitivity to G6PD Inhibition in Acute Myeloid Leukemia Cells; pp. 2326–2335. [DOI] [PubMed] [Google Scholar]
- 45.Sun Y., Gu X., Zhang E., Park M.A., Pereira A.M., Wang S., Morrison T., Li C., Blenis J., Gerbaudo V.H., Henske E.P., Yu J.J. Estradiol promotes pentose phosphate pathway addiction and cell survival via reactivation of Akt in mTORC1 hyperactive cells. Cell Death Dis. 2014;5 doi: 10.1038/cddis.2014.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharma P.K., Varshney R. 2-Deoxy-D-glucose and 6-aminonicotinamide-mediated Nrf2 down regulation leads to radiosensitization of malignant cells via abrogation of GSH-mediated defense. Free Radic. Res. 2012;46:1446–1457. doi: 10.3109/10715762.2012.724771. [DOI] [PubMed] [Google Scholar]
- 47.Woo S.H., Yang L.P., Chuang H.-C., Fitzgerald A., Lee H.-Y., Pickering C., Myers J.N., Skinner H.D. 2015. Down-regulation of malic enzyme 1 and 2: sensitizing head and neck squamous cell carcinoma cells to therapy-induced senescence. [DOI] [PubMed] [Google Scholar]
- 49.Villaverde M.S., Gil-Cardeza M.L., Glikin G.C., Finocchiaro L.M.E. Interferon-Β lipofection II. Mechanisms involved in cell death and bystander effect induced by cationic lipid-mediated interferon-Β gene transfer to human tumor cells. Cancer Gene Ther. 2012;19 doi: 10.1038/cgt.2012.19. [DOI] [PubMed] [Google Scholar]
- 50.Jaune E., Rocchi S. Metformin: focus on melanoma. Front. Endocrinol. (Lausanne) 2018;9 doi: 10.3389/fendo.2018.00472. [DOI] [PMC free article] [PubMed] [Google Scholar]