

Key Points
Question
Can metabolic brain changes be detected in presymptomatic individuals who are carriers of a hexanucleotide repeat expansion in the C9orf72 gene (preSxC9) using time-of-flight fluorine 18–labeled fluorodeoxyglucose positron emission tomographic imaging and magnetic resonance imaging, and what is the association between the mutation and clinical and fluid biomarkers of amyotrophic lateral sclerosis and frontotemporal dementia?
Findings
In a case-control study including 17 preSxC9 participants and 25 healthy controls, fluorine 18–labeled fluorodeoxyglucose positron emission tomographic imaging noted significant clusters of relative hypometabolism in frontotemporal regions, the insular cortices, basal ganglia, and thalami in the preSxC9 participants. Use of this strategy allowed detection of changes at an individual level.
Meaning
Glucose metabolic changes appear to occur early in the sequence of events leading to manifest amyotrophic lateral sclerosis and frontotemporal dementia. Fluorine 18–labeled fluorodeoxyglucose positron emission tomographic imaging may provide a sensitive biomarker of a presymptomatic phase of disease.
Abstract
Importance
During a time with the potential for novel treatment strategies, early detection of disease manifestations at an individual level in presymptomatic carriers of a hexanucleotide repeat expansion in the C9orf72 gene (preSxC9) is becoming increasingly relevant.
Objectives
To evaluate changes in glucose metabolism before symptom onset of amyotrophic lateral sclerosis or frontotemporal dementia in preSxC9 using simultaneous fluorine 18–labeled fluorodeoxyglucose ([18F]FDG positron emission tomographic (PET) and magnetic resonance imaging as well as the mutation’s association with clinical and fluid biomarkers.
Design, Setting, and Participants
A prospective, case-control study enrolled 46 participants from November 30, 2015, until December 11, 2018. The study was conducted at the neuromuscular reference center of the University Hospitals Leuven, Leuven, Belgium.
Main Outcomes and Measures
Neuroimaging data were spatially normalized and analyzed at the voxel level at a height threshold of P < .001, cluster-level familywise error–corrected threshold of P < .05, and statistical significance was set at P < .05 for the volume-of-interest level analysis, using Benjamini-Hochberg correction for multiple correction. W-score maps were computed using the individuals serving as controls as a reference to quantify the degree of [18F]FDG PET abnormality. The threshold for abnormality on the W-score maps was designated as an absolute W-score greater than or equal to 1.96. Neurofilament levels and performance on cognitive and neurologic examinations were determined. All hypothesis tests were 1-sided.
Results
Of the 42 included participants, there were 17 with the preSxC9 mutation (12 women [71%]; mean [SD] age, 51 [9] years) and 25 healthy controls (12 women [48%]; mean [SD] age, 47 [10] years). Compared with control participants, significant clusters of relative hypometabolism were found in frontotemporal regions, basal ganglia, and thalami of preSxC9 participants and relative hypermetabolism in the peri-Rolandic region, superior frontal gyrus, and precuneus cortex. W-score frequency maps revealed reduced glucose metabolism with local maxima in the insular cortices, central opercular cortex, and thalami in up to 82% of preSxC9 participants and increased glucose metabolism in the precentral gyrus and precuneus cortex in up to 71% of preSxC9 participants. Other findings in the preSxC9 group were upper motor neuron involvement in 10 participants (59%), cognitive abnormalities in 5 participants (29%), and elevated neurofilament levels in 3 of 16 individuals (19%) who underwent lumbar puncture.
Conclusions and Relevance
The results suggest that [18F]FDG PET can identify glucose metabolic changes in preSxC9 at an individual level, preceding significantly elevated neurofilament levels and onset of symptoms.
This case-control study examines the use of fluorine 18–labeled fluorodeoxyglucose positron emission tomographic imaging to detect glucose metabolic changes in individuals who are presymptomatic carriers of a mutation in the C9orf72 gene that leads to amyotrophic lateral sclerosis and frontotemporal dementia.
Introduction
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are related neurodegenerative disorders. Amyotrophic lateral sclerosis primarily affects the motor system with upper and lower motor neuron involvement, but extramotor manifestations may occur.1,2,3 Frontotemporal dementia is the second most common form of presenile dementia, caused by degeneration of frontal and anterior temporal cortices. It affects brain regions implicated in executive control, language, behavior, and personality.4 The disease course of both ALS and FTD is progressive and invariably fatal. The molecular link between ALS and FTD has been confirmed by the discovery of the hexanucleotide repeat expansions in the 3′ untranslated region of the chromosome 9 open reading frame 72 gene (C9orf72, OMIM 614260), the most common known monogenetic cause of both ALS and FTD.5,6,7
During this time of antisense oligonucleotides and other interventional gene therapies, research in the presymptomatic stage may contribute to the development of novel treatment strategies8 and detection of individuals at risk of developing ALS and/or FTD, and ultimately lay the foundation for future clinical studies to slow or even prevent clinical disease manifestation.9 Presymptomatic carriers of disease-causing mutations permit in vivo research of the brain at a unique time to gain a better understanding of the early mechanisms that precede the onset of symptoms.
Over the past 10 years, study findings have suggested that several neurodegenerative diseases are preceded by an intermediate presymptomatic phase.10,11 Research in presymptomatic carriers of a hexanucleotide repeat expansion in the C9orf72 gene (preSxC9) reported the occurrence of cognitive and behavioral changes, neuropsychiatric symptoms, and degeneration of gray matter (GM) and white matter (WM).12,13,14,15,16,17,18,19,20,21
Neurofilaments (Nfs), such as neurofilament light chain (NfL) and phosphorylated neurofilament heavy chain (pNfH), have been studied extensively in ALS and FTD. Elevated levels of NfL and pNfH, both markers of neuronal injury and neurodegeneration, demonstrated high diagnostic performance.22 Previous research has shown that NfL is increased in symptomatic, but not presymptomatic, preSxC9 at the group level.23 Recent studies suggested that a slow increase in Nf levels can be observed in presymptomatic individuals who carry the mutation as far as 3.5 years before diagnosable illness,24,25,26 while another study described an association between higher NfL levels and GM atrophy.27
It has often been suggested that assessing glucose metabolism using positron emission tomographic (PET) imaging with fluorine 18–labeled fluorodeoxyglucose ([18F]FDG) is a useful diagnostic marker in the earliest stage of ALS and FTD.28,29,30,31 Moreover, [18F]FDG PET imaging serves as a relevant biomarker for disease staging, cognitive impairment, and survival prediction.29,32
However, little is known about the glucose metabolic changes that may occur before clinical disease manifestation in preSxC9. The goal of our study was to evaluate changes in glucose metabolism that occur before diagnosable illness, ie, the presymptomatic disease stage,33 in preSxC9. In addition, we wanted to explore the association between cerebral glucose metabolism and other known indicators of disease, such as Nf levels in cerebrospinal fluid (CSF), neuropsychological capacities, and clinical neurologic examination.
Methods
Participants
A total of 29 healthy individuals serving as controls were included in this study, of whom 25 were considered in the analysis. None of the volunteers had a first-degree relative with dementia or a history of neurologic illness, psychiatric illness, or substance use. Participants with brain lesions noted on structural magnetic resonance imaging (MRI) were excluded. Demographic characteristics are detailed in Table 1.
Table 1. Demographics and Clinical Data of PreSxC9 and Control Group.
| Characteristic | PreSxC9 (n=17) | Healthy controls (n=25) | Statistical test for group difference | P value |
|---|---|---|---|---|
| Age, mean (SD), y | 51 (9) | 47 (10) | Mann-Whitney, 154 | .13 |
| Sex, No. (%) | ||||
| Women | 12 (71) | 12 (48) | χ21 = 2.11 | .13 |
| Men | 5 (29) | 13 (52) | ||
| Educational level, ISCED, No. (%)a | ||||
| 0-4 | 7 (41) | 8 (32) | χ21 = 0.37 | .39 |
| 5-6 | 10 (59) | 17 (68) | NA | NA |
| MMSE score, median (range)b | 29 (26-30) | 30 (28-30) | Mann-Whitney, 156.5 | .12 |
| BDI, median (range)c | NA | 2 (0-5) | NA | NA |
| Psychiatric drugs, No. (%) | 1 (6) | NA | NA | NA |
| Antidepressants, No. (%) | 1 (6) | NA | NA | NA |
Abbreviations: BDI, Beck Depression Inventory; ISCED, International Standard Classification of Education Scale; MMSE, Mini-Mental-State Examination; NA, not applicable; preSxC9, presymptomatic carrier of a hexanucleotide repeat expansion in the C9orf72 gene.
Categorized according to the ISCED 1997 definitions. Scale numbers represent nontertiary education, 0-4, and tertiary education, 5-6.
Total score ranges, 0 to 30; lower scores indicate worse cognitive function.
Total score ranges, 0 to 63; higher scores indicate more severe depressive symptoms.
The study was conducted from November 30, 2015, to December 11, 2018, at the neuromuscular reference center of the University Hospitals Leuven, Leuven, Belgium. All participants provided written informed consent, and this study was approved by the ethics committee of the University Hospitals Leuven, Leuven, Belgium. This study followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline for case-control studies.
We compared the Nf levels in the preSxC9 group with those of a control group (n = 10; mean [SD] age, 49 [14] years) previously reported.34 A consecutive series of 17 preSxC9 participants was included in this study. A pathogenic expansion of C9orf72 was considered as having more than 30 repeats. All preSxC9 participants were native Flemish speakers, and their educational levels were between 3 ([upper] secondary education) and 6 (second stage of tertiary education) on the International Standard Classification of Education scale.35 None of the preSxC9 participants met the clinical diagnostic criteria for ALS or FTD.36,37 Exclusion criteria were the presence of clinically apparent ALS or FTD, severe and chronic illness, substance use, and traumatic brain injury.
All participants with preSxC9 were evaluated with the Dutch version of the Edinburgh Cognitive and Behavioral ALS Screen (ECAS) by an experienced neuropsychologist (J.D.V.).35 The ECAS is a brief, multidomain screening battery that assesses cognitive functions typically affected in patients with ALS (language, verbal fluency, and executive functioning), as well as cognitive functions not typically affected in patients with ALS (memory, visuospatial functioning).38 Dutch normative data were used, with the fifth percentile as a threshold for abnormality.35 Results of the ECAS are presented in Table 2.
Table 2. Clinical Characteristics in PreSxC9 Participants.
| Characteristic | Median (range) | Percentile rank scores ≤5%, No. (%) |
|---|---|---|
| ECAS performancea | ||
| Total score | 117 (93-124) | 1 (6) |
| ALS specificb | ||
| Total score | 86 (69-92) | 1 (6) |
| Language | 28 (25-28) | 0 |
| Verbal fluency | 20 (14-20) | 1 (6) |
| Executive functions | 39 (26-44) | 3 (18) |
| ALS nonspecificc | ||
| Total score (of 36) | 30 (23-33) | 0 |
| Memory (of 24) | 18 (11-21) | 1 (6) |
| Visuospatial functions (of 12) | 12 (11-12) | 0 |
Abbreviations: ALS, amyotrophic lateral sclerosis; ECAS, Edinburgh Cognitive and Behavioral ALS Screen; preSxC9, presymptomatic carrier of a hexanucleotide repeat expansion in the C9orf72 gene.
Possible score range, 0 to 136; lower scores indicate more severe cognitive dysfunction.
Evaluates functions typically affected in ALS. Total score ranges from 0 to 100; language, 0 to 28; verbal fluency, 0 to 20; and executive functions, 0 to 42. Lower scores indicate more severe cognitive dysfunction.
Evaluates cognitive functions not typically affected in ALS. Total score ranges from 0 to 36; memory, 0 to 24; and visuospatial functions, 0 to 12. Lower scores indicate more severe cognitive dysfunction.
All of preSxC9 participants underwent a standard clinical neurologic examination by a neurologist experienced in neuromuscular disorders (P.V.D.).
Sixteen of 17 preSxC9 participants agreed to undergo a lumbar puncture according to standardized protocol at the University Hospitals Leuven to determine the Nf levels in the CSF within 48 hours following the [18F]FDG PET MRI scan. Neurofilament levels in CSF were measured using commercially available kits for NfL (UD51001, with an intraassay variability of 1.6% and interassay variability of 8.7%; UmanDiagnostics AB) and pNfH (with an intraassay variability of 5.2% and interassay variability of 8.7%; Euroimmun AG). Assessment of Nf levels was done using predefined diagnostic cutoff values for NfL (1227 pg/mL)34 and pNfH (750 pg/mL).39
All participants underwent simultaneous [18F]FDG PET and MR imaging on a hybrid scanner (Signa PET/MR, release MP26; GE Healthcare) with integrated time-of-flight PET scan acquisition and 3-T MRI. All participants fasted for at least 6 hours before [18F]FDG administration. [18F]FDG was injected intravenously as a bolus (mean [SD], controls: 153 [11] MBq; preSxC9: 149 [6] MBq).
The [18F]FDG PET images were acquired in list mode for 25 minutes (30 minutes postinjection). The PET images were reconstructed with ordered subset maximum likelihood expectation maximization with 4 iterations and 28 subsets followed by postfiltering with 4.5-mm gaussian postsmoothing in the transaxial direction and standard smoothing along the Z direction. Images had an initial voxel size of 1.56 × 1.56 × 2.78 mm3. A vendor-provided, atlas-based method was used for attenuation correction.40
Simultaneous to the PET acquisition, a 3-dimensional volumetric sagittal T1-weighted image (3D BRAVO, repetition time/echo time [TR/TE] = 8.5/3.2 milliseconds, 0.6 × 1 × 1 mm3 voxel size, dimensions: 312 × 256 × 256 voxels) and T2-weighted fluid-attenuated inversion recovery image (3D CUBE, TR/TE = 8500/130 milliseconds, 0.7 × 1 × 1 mm3 voxel size, dimensions: 268 × 256 × 256 voxels) were acquired.
Statistical Analysis
Statistical analyses of clinical data were performed using SPSS software, version 25 (IBM Software) and GraphPad Prism, version 8.0 (GraphPad Software). Demographic characteristics and clinical test results were compared between groups using a χ2 test for dichotomous and categorical variables or Mann-Whitney test for numeric variables. All hypothesis tests were 1-sided, and statistical significance was set at P < .05.
Image Analysis
ANTS, version 2.1.0., and SPM, version 12 (Wellcome Trust Centre for Neuroimaging) software, combined with in-house scripts implemented in Matlab (R2018b; The MathWorks Inc), were used to process the T1-weighted and fluid-attenuated inversion recovery images. After visual inspection of the raw T1 images, the T1 images were processed in native space using the antsCorticalThickness pipeline in ANTS,39 which performs a brain extraction and segments the image of the individual’s brain by means of 5 specific tissue priors: CSF, cortical GM, WM, subcortical GM, and the brainstem. After visual inspection of the segmentations, 3 control scans were excluded because of poor image quality and subsequent suboptimal brain data extraction and segmentation. Gray matter tissue probability maps were warped to the Nathan Kline Institute template (Rockland Sample, dimensions = 182 × 218 × 182 voxels), which was warped to Montreal Neurological Institute space (voxel size = 1 × 1 × 1 mm3, matrix = 182 × 218 × 182) and modulated with the jacobian warp parameters, all using nonlinear symmetric diffeomorphic registration.
All [18F]FDG PET images were first quality checked for complete acquisition and motion, then dynamically reconstructed and corrected for potential head motion. The frames, which were reconstructed over a series of 5 minutes, were then averaged. After visual inspection, PET images were coregistered to their respective native MRI and spatially normalized to Montreal Neurological Institute space using ANTS, applying the normalization parameters described above. After visual inspection, 1 control scan was considered an outlier (>3 SD from the mean) and subsequently excluded. The [18F]FDG PET images were corrected for partial volume effects with the Müller-Gartner method (PMOD, version 3.9), which considers both GM spill-out and WM spill-in based on the MRI-based GM and WM tissue probability maps. Partial volume correction (PVC) was done using a point-spread function with a 5.5-mm isotropic full width at half maximum to mimic the PET image resolution, while a regression approach was applied for all voxels with a WM probability greater than 0.95 to determine the [18F]FDG uptake in WM. The GM probability threshold was set at 0.3 to correct uptake values of GM voxels for WM activity. Partial volume effect–corrected [18F]FDG PET images were spatially normalized to Montreal Neurological Institute space using ANTS, applying the normalization parameters described above.
For the group comparison, both MR and [18F]FDG PET images were smoothed with an isotropic gaussian smoothing kernel of 8-mm full width at half maximum to blur individual variations. Owing to a difference in ambient conditions (ie, visual input) following tracer injection, the occipital lobe was excluded from all neuroimaging analyses. All PET images were proportionally normalized to the average activity in a GM mask generated from the voxel-based morphometry comparative analysis applying an absolute threshold of 0.1 (excluding the occipital lobe).
The spatially normalized and smoothed images were then entered into a generalized linear model. All hypothesis tests were 1-sided, with a height threshold of P < .001 and a cluster-level familywise error (FWE)-corrected threshold of P < .05, applying a minimum extent threshold of 150 voxels for the [18F]FDG PET analyses. Age was included as a nuisance covariate in [18F]FDG PET analyses as well as in the voxel-based morphometry analyses, where total intracranial volume was also considered a nuisance variable. The Talairach atlas41 was used to define Brodmann areas and the Harvard-Oxford Atlas42,43,44,45 was used for the anatomic localization of significant clusters.
A volume-of-interest–based analysis after region-based voxelwise correction for GM atrophy was conducted using the Hammers N30R83 maximum probability atlas to confirm our findings at the voxel level in PMOD, version 3.9 (PMOD Inc) and SPSS, version 25 (IBM Software). We applied the Benjamini-Hochberg method to correct for multiple testing.
W-Score Maps
W-score maps ([raw value for each patient − value expected in the control group for the patient’s age] / SD of the residuals in the control group) were computed for preSxC9 using the control group as a reference to quantify the degree of [18F]FDG PET imaging abnormality at the voxel level. W-score maps are analogous to z-score maps, adjusted for covariates of interest. For our study, we considered age as a covariate of interest.46 The threshold for abnormality was defined as an absolute W-score greater than or equal to 1.96, which corresponds to 95% of the area under the curve in a normal distribution. Hypometabolic maps, binarized at a W-score less than or equal to −1.96, and hypermetabolic maps, binarized at a W-score greater than or equal to 1.96, were summed across participants to generate W-score frequency maps to illustrate the fraction of preSxC9 participants surpassing the threshold for abnormality at the voxel level.
Results
A total of 46 participants (17 preSxC9 and 29 healthy controls) were included in this study. After data inspection, all preSxC9 participants (mean [SD] age, 51 [9] years; 12 women [71%], 5 men [29%]) and 25 (4 of 29 excluded owing to poor image quality) healthy controls (mean [SD] age, 47 [10] years; 12 women [48%], 13 men [25%]) were considered for the analyses. The demographics of the study population are given in Table 1. The preSxC9 and control groups did not differ significantly in sex distribution (χ21 = 2.11; P = .13), educational level (χ21 = 0.37; P = .39), or age (Mann-Whitney, 1.54; P = .13).
Neuroimaging
Relative glucose metabolism was compared between the preSxC9 and control cohorts. This analysis revealed significant clusters of relative hypometabolism in the preSxC9 group compared with the control group (range, 27%-36%) situated in the basal ganglia, thalamus, and frontotemporal and insular cortices. All analyses were thresholded at a height of P < .001 and FWE-corrected level of P < .05 at the cluster level (Figure 1; eTable 1 in the Supplement). At the group level, we observed no significant clusters of relative hypermetabolism.
Figure 1. Relative Glucose Metabolism and Gray Matter Volume in Presymptomatic Carriers of a Hexanucleotide Repeat Expansion in the C9orf72 Gene (PreSxC9) Corrected for Age on Axial, Coronal, and Sagittal Sections .
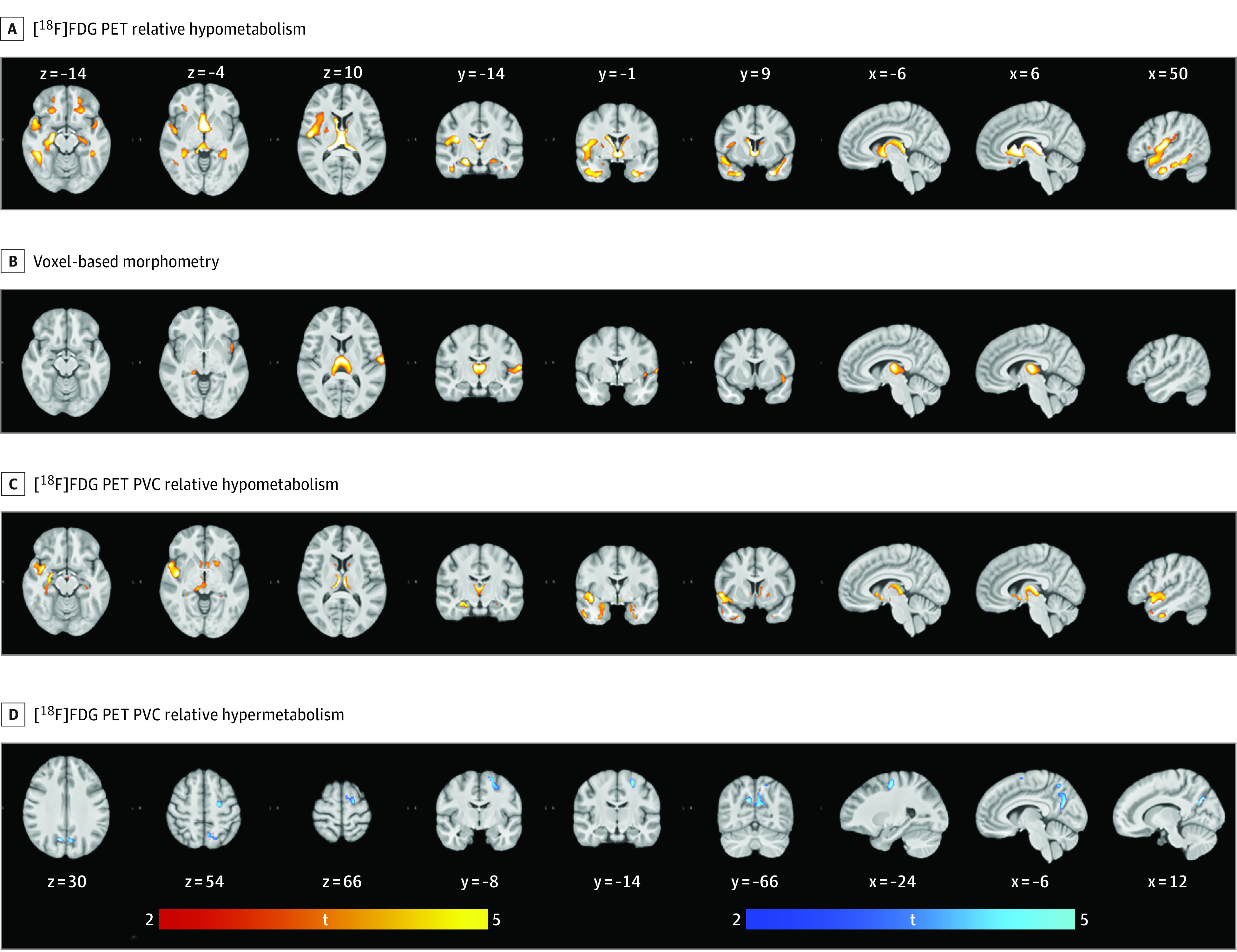
Reduced glucose metabolism and gray matter volume depicted in red-yellow, and increased glucose metabolism depicted in blue-white. Data were analyzed at a height threshold of P < .001 and were cluster level corrected for familywise error at P < .05. A, Projections of areas with relative hypometabolism in preSxC9 participants vs healthy controls. B, Volume decline in preSxC9 participants vs healthy controls. C, Relative hypometabolism in preSxC9 participants and healthy controls following voxel-based PVC. D, Relative hypermetabolism in preSxC9 and healthy controls following voxel-based PVC. [18F]FDG indicates fluorine 18–labeled fluorodeoxyglucose; PET, positron emission tomography; PVC, partial volume correction; and t, t value. Section numbers refer to Montreal Neurological Institute coordinates.
The comparative voxel-based volumetric analysis (voxel-based morphometry) revealed significant clusters of reduced GM volume (range, 19%-25%) located in the frontotemporal regions, including the peri-Rolandic region, insular cortices, basal ganglia, and thalami. All analyses were thresholded at a height of P < .001 and FWE-corrected level of P < .05 at the cluster level (Figure 1; eTable 2 in the Supplement). A voxel-based regression analysis of the association between age and GM volume failed to show a significant difference in the slopes of the preSxC9 and control participants.
The [18F]FDG PET imaging data were also analyzed with partial volume effect correction to account for GM atrophy. Significant clusters of relative hypometabolism (range, 16%-22%) persisted in frontotemporal regions, including the insular cortices, as well as the basal ganglia and thalami. All analyses were thresholded at a height of P < .001 and FWE-corrected level of P < .05 at the cluster level (Figure 1; eTable 3 in the Supplement; Video). A voxel-based regression analysis of the association between age and cerebral metabolism failed to show a significant difference in the slopes of the preSxC9 and healthy control participants.
Video. Relative Glucose Metabolism on Fluorine 18–Labeled Fluorodeoxyglucose Positron Emission Tomographic (PET) Imaging in PreSxC9 Participants vs Controls.
Relative hypometabolism and relative hypermetabolism following partial volume correction (PVC) in presymptomatic carriers of a hexanucleotide repeat expansion in the C9orf72 gene (preSxC9) in relation to healthy controls in red-yellow and blue-white, respectively, while correcting for age (height-corrected threshold of P < .001 at a cluster-level familywise error–corrected threshold of P < .05).
These findings were supported in a volume-of-interest–based analysis applying region-based voxelwise correction for GM atrophy (eFigure 1 and eTable 5 in the Supplement). Significant clusters of relative hypermetabolism (range, 6%-7%) emerged in the peri-Rolandic region, the superior frontal gyrus, and the precuneus cortex following PVC. All analyses were thresholded at a height of P < .001 and FWE-corrected level of P < .05 at the cluster level (Figure 1; eTable 4 in the Supplement; Video). To confirm the presence of relative hypermetabolic clusters in preSxC9 participants, the analysis was repeated using standardized uptake value ratio images; cortical uptake was scaled to the average uptake in cerebellar structures not reported as being affected by a mutation in the C9orf72 gene, supporting our findings (eResults, eFigure 4, eTable 6 in the Supplement).47
Clinical Parameters
Neurologic examination revealed mild signs of upper motor neuron (UMN) abnormalities in 12 of the 17 preSxC9 participants (71%). As the presence of a Hoffman sign or ankle clonus is not necessarily abnormal in young people, we only considered the presence of a jaw jerk, a Babinski sign, hyperreflexia, and increased muscle tone for further analyses. This was apparent overall in 10 preSxC9 participants (59%). In 5 participants (29%) increased muscle tone was observed in the lower extremities; 1 (6%) also presented with increased muscle tone in upper extremities.
Five participants (29%) of the preSxC9 cohort presented with abnormal neuropsychological performance. Executive functioning was affected in 3 preSxC9 participants (18%), 1 participant presented with isolated abnormal performance on verbal fluency, and another individual showed isolated impairment on the memory subdomain (Table 2).
Examination of the CSF showed median NfL levels of 652 pg/mL (range, 276-1510) and pNfH levels of 195 pg/mL (range, 123-490). The NfL and pNfH levels did not differ significantly between the preSxC9 and healthy controls at the group level. However, elevated NfL levels, ie, surpassing the diagnostic cutoff, were observed at the individual level in the CSF of 19% of the preSxC9 group (Figure 2). All 3 of the 16 who underwent lumbar puncture displayed signs of UMN involvement on clinical neurologic examination, and 1 of the 3 individuals with elevated Nf levels displayed an abnormal score on the memory domain using the ECAS. The pNfH levels in the CSF remained within the reference range in all preSxC9 participants (Figure 2).
Figure 2. Neurofilament Levels in the Cerebrospinal Fluid (CSF) of Presymptomatic Carriers of a Hexanucleotide Repeat Expansion in the C9orf72 Gene (PreSxC9).

A, Neurofilament light chain in the CSF of healthy controls and preSxC9 participants. B, Phosphorylated neurofilament heavy chain in the CSF of healthy controls and preSxC9 participants. The dotted line indicates diagnostic cutoff level. The upward arrowheads indicate the controls, and the downward arrowheads indicate the preSxC9 participants. Error bars indicate 95% CIs.
We were unable to identify a significant association between relative tracer uptake and age, UMN involvement, ECAS performance, or Nf levels in CSF at the group level in preSxC9 participants using regression analyses at a height-corrected threshold of P < .001 and with a cluster-level FWE-corrected threshold of P < .05. Similarly, no significant association was identified between GM volume and age, UMN involvement, ECAS performance, or Nf levels in CSF, applying the same threshold for significance.
We generated voxel-level W-score maps to evaluate how many preSxC9 participants presented with suprathreshold voxels in key regions. A frequency image of the W-score maps, generated from the [18F]FDG PET images without correcting for partial volume effect, showed that 14 preSxC9 participants (82%) had significantly reduced tracer uptake in the insular cortices, central opercular cortex, and thalami (eFigure 2A in the Supplement). In addition, a frequency image of the W-score maps, generated from the [18F]FDG PET images corrected for partial volume effect, showed that up to 71% of preSxC9 patients had significantly increased tracer uptake, surpassing the predefined threshold of an absolute W-score of 1.96, which corresponds to the 2.5th percentile on both sides in the peri-Rolandic region (eFigure 2B in the Supplement). A mean image of the W-score maps in the preSxC9 cohort reflected the consistency of the changes observed at the group level (eFigure 3 in the Supplement). Individual W-score maps of relative hypometabolism supported the pattern observed at the group level in up to 82% of preSxC9 participants (Figure 3). A W-score frequency map of GM volume reduction revealed suprathreshold voxels in the thalami and central opercular cortex in 11 preSxC9 participants (65%) (eFigure 2C in the Supplement). In addition, using the W-score maps, we were unable to identify a clear association between the extent of abnormality and UMN involvement, ECAS performance, and Nf levels in CSF.
Figure 3. Individual W-Score Maps of Relative Hypometabolism Observed in PreSxC9 Participants.
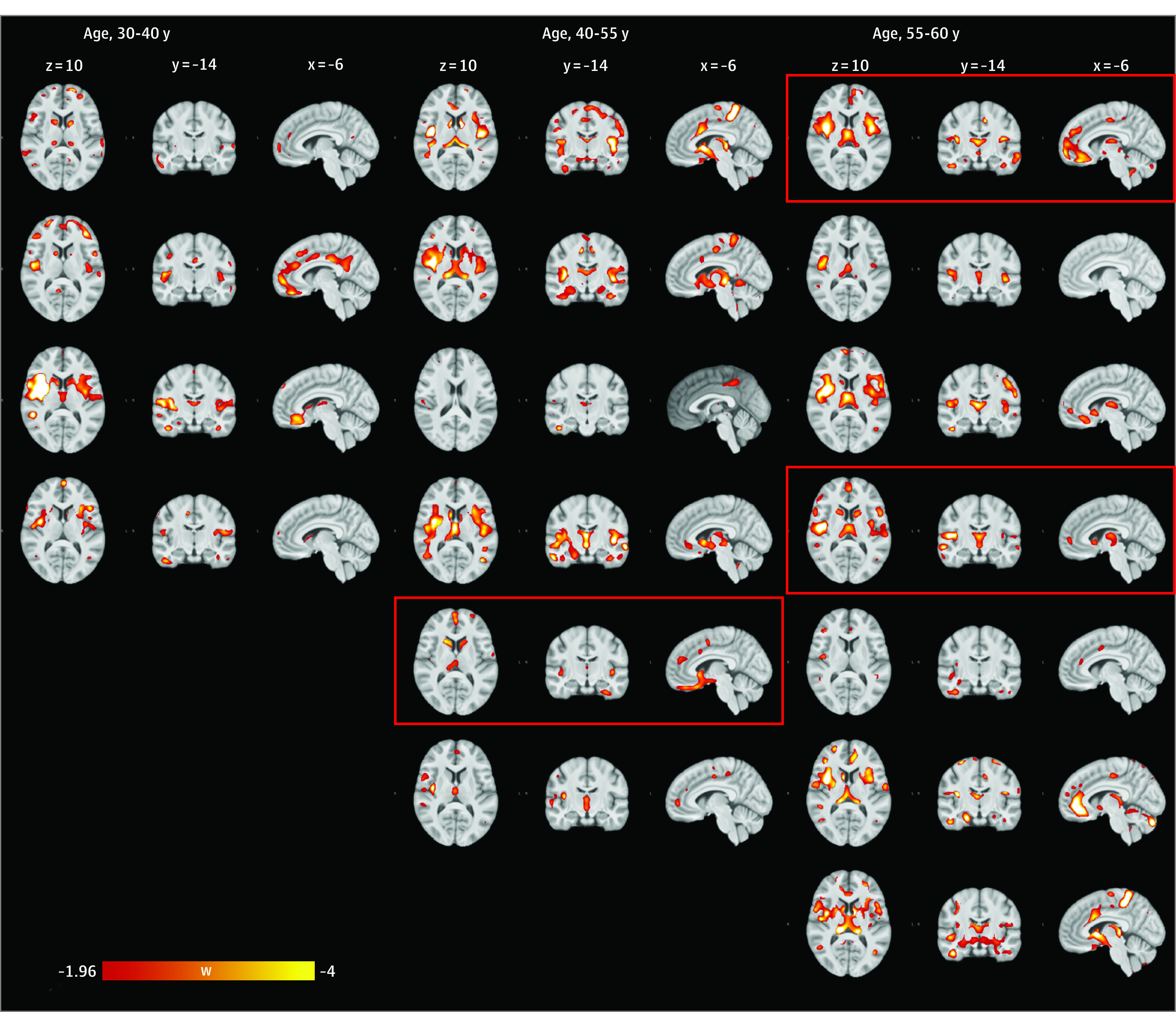
Thresholds for W-score maps were set at a W score less than or equal to −1.96 and sorted by age, displayed on axial (z = 10), coronal (y = −14), and sagittal (x = −6) sections. Section numbers refer to Montreal Neurological Institute coordinates. Images from participants with elevated neurofilament levels are within red boxes. w indicates W score.
Discussion
A voxelwise comparison of glucose metabolic patterns revealed clusters of relative glucose hypometabolism situated in frontotemporal and insular cortices, the basal ganglia, and thalami. Moreover, GM volume reductions revealed a widespread neuroanatomic signature in the frontotemporal and insular cortices, basal ganglia, and thalami. The observed volumetric differences are consistent with structural changes reported in previous studies of preSxC9.13 Even though regional hypometabolism in subcortical and extramotor regions may be explained in part by neuronal loss, the functional disruption identified by [18F]FDG PET imaging was supported, as clusters of reduced glucose metabolism in aforementioned regions withstood PVC, and thus correction for GM atrophy.
Significant clusters of relative hypermetabolism were observed in the precentral and superior frontal gyrus and the precuneus cortex following PVC. This finding may be interpreted as compensatory neuronal activity or a possible abnormal function of cortico-striatal-thalamic-cortical circuits resulting in UMN abnormalities. In addition, we can speculate that the observed clusters of relative hypermetabolism reflect neuroinflammation associated with activated astrocytes or microglia.29
The observed structural and metabolic changes in the preSxC9 participants suggest that brain regions corresponding to cognitive and motor processes are impaired in the presymptomatic stage of ALS and FTD. These findings are in line with previous [18F]FDG PET imaging studies in symptomatic carriers of a C9orf72 hexanucleotide repeat expansion, demonstrating relative hypometabolism in frontotemporal and subcortical regions.48,49 Moreover, our findings support the role of the thalamus in C9-related disease.49,50
The role of the cerebellum in C9-related disease remains unclear. A recent voxel-based morphometry study from the multicenter Genetic Frontotemporal Dementia Initiative consortium described GM volume reductions in the superior-posterior cerebellum.14 We, however, did not observe significant GM volume reductions in the cerebellum, supporting the findings of another study.15 To our knowledge, there are no consistent findings on volumetric changes in the cerebellum of preSxC9 individuals.
For this study, W-score maps were generated to observe individual effects, as individual differences may have been washed out in a group-level voxel-based analysis. W-score frequency maps reflected the consistency of the pattern observed at the group level in individual W-score maps of a number of preSxC9 participants. These maps demonstrated that the highest frequencies (up to 82%) of reduced glucose metabolic uptake, below the threshold for abnormality, were found in the insular cortices, central opercular cortex, and thalami of preSxC9 participants. The highest frequencies (up to 71%) of increased glucose metabolism, above the threshold for abnormality, following PVC were found in the peri-Rolandic region and superior frontal gyrus of the preSxC9 participants. Given that only part of the preSxC9 cohort had cognitive, pyramidal, or Nf changes, we suggest that the metabolic changes may occur early in the sequence of events leading to manifest ALS and FTD.
Because the age at disease onset is variable in C9orf72 repeat expansion carriers, the preSxC9 cohort in the present study most likely consists of a mixture of individuals who are relatively close to or far from disease onset. In addition, a hexanucleotide repeat expansion is known to be associated with a clinically heterogeneous disease spectrum.2 The conceivable high degree of clinical variability within the preSxC9 group could potentially blur correlations with clinical parameters. As we did not observe an association with deviation from the norm and increasing age, longitudinal studies are needed to establish how the patterns of hypometabolism evolve and their predictive value for clinical disease onset.
We did not identify significant differences in CSF Nf levels at the group level between healthy controls and preSxC9 participants. Other studies were also unable to identify significant differences for this marker for axonal injury between healthy controls and individuals who are preSxC9.24,51 At the individual level, NfL appears to be more sensitive than pNfH in the phase preceding diagnosable illness: no preSxC9 participant displayed a pathologic increase in pNfH, but 3 preSxC9 participants displayed abnormally high NfL levels.
We did not identify a significant association between Nf levels, ECAS performance, clinical neurologic screening, and findings on neuroimaging. This finding may, at least in part, be explained by sample size, as few preSxC9 participants presented with cognitive changes, elevated Nf levels, and UMN signs. Changes in cerebral metabolism may also precede clinical signs, which is in line with a recent study describing functional reorganization and network resilience in individuals who are preSxC9.18
Longitudinal, multimodal PET and MRI studies are needed to gain a better understanding of the sequence of events that precede diagnosable illness. In addition, no significant association was observed between GM volume and increasing age in the preSxC9 cohort. We could therefore speculate that the observed volumetric differences between the preSxC9 and healthy control participants may represent not only GM atrophy but may, at least in part, indicate neurodevelopmental differences. Adolescent neuroimaging studies could assist in gaining more insight into the natural history of brain development in preSxC9.
Limitations
This study has limitations. First, the sample size was relatively small, which may have been a factor in the power of group comparisons for signs of upper motor neuron involvement and the association between neuroimaging data and clinical indicators of disease. Second, the absence of convertors in our cohort prevented us from exploring the predictive values of these markers for diagnosis. Third, we did not perform neurologic examinations in the control cohort. We also did not use cognitive screening with the ECAS in the healthy controls; however, we administered a Mini-Mental-State Examination in all participants, which did not reflect any cognitive abnormalities. Fourth, the difference in ambient conditions (visual input) between the preSxC9 and control cohorts necessitated masking the occipital lobe from our comparative analyses between preSxC9 and healthy controls, therefore preventing us from performing a whole-brain, voxel-based comparative analysis. However, to ensure the robustness of the patterns of relative hypometabolism, we performed a second whole-brain analysis in the preSxC9 group. This second analysis revealed the same clusters that we observed previously as well as a cluster of relative hypermetabolism in the occipital lobe, supporting our findings.
Conclusions
This study showed regional glucose metabolic alterations in presymptomatic carriers of a C9orf72 hexanucleotide repeat expansion before diagnosable illness that remained after correction for volume differences. Within the preSxC9 cohort on W-score maps of [18F]FDG PET images, up to 82% (n = 14) presented with voxels surpassing the threshold of abnormality in key regions, Nf levels were elevated in only 19% (n = 3), deviation from the norm according to ECAS performance was observed in 29% (n = 5), 59% (n = 10) presented with subtle UMN signs, and abnormalities were noted on W-score maps on MR images in 65% (n = 11). The individual W-score image suggests that [18F]FDG PET might be able to detect neuronal injury in an earlier stage than motor or cognitive changes or Nf levels.
To our knowledge, this is the first study that closely examines cerebral glucose metabolism in preSxC9 carriers and its association with GM volume and indicators of disease. Our findings suggest that [18F]FDG PET imaging could provide a sensitive biomarker of a presymptomatic phase of disease, which can be of relevance for future therapeutic strategies. Multimodal and longitudinal imaging studies with an augmented sample size are needed to gain more insight into the sequence of events in the presymptomatic stage of C9orf72-related disease.
eResults. Supplementary Analysis
eReferences
eFigure 1. Violin Plots of 18F FDG Uptake in Volumes of Interest, After Correcting for GM Atrophy With PVC, in Healthy Controls and PreSxC9
eFigure 2. W-Score Frequency Maps Detailing the Fraction of PreSxC9 With Suprathreshold W-Scores at Voxel Level
eFigure 3. Mean W-Score Images Detailing Tracer Uptake in PreSxC9, Thresholded From 0.5 Standard Deviations From the Norm
eFigure 4. Relative Hypermetabolism in PreSxC9 in relation to healthy controls (pheight <0.001, at Cluster Level pFWE<0.05) Following Voxel-Based Partial Volume Corrections, Using SUVr Images
eTable 1. Significant Areas of Decreased Cortical Glucose Metabolism in PreSxC9 Compared to HC, Identified Using Voxel-Wise Testing
eTable 2. Significant Areas of Volume Decline in PreSxC9 Compared to HC, Identified Using Voxel-Wise Testing
eTable 3. Significant Areas of Decreased Metabolism in PreSxC9 Compared to HC, Identified Using Voxel-Wise Testing Following Partial Volume Correction
eTable 4. Significant Areas of Increased Cortical Glucose Metabolism in PreSxC9 Compared to HC, Identified Using Voxel-Wise Testing Following Partial Volume Correction
eTable 5. A Volume-of-Interest (VOI)-Based Analysis After Region-Based Voxel-Wise (RBV) Correction for GM Atrophy, Using the Hammers N30R83 Maximum Probability Atlas
eTable 6. Significant Areas of Increased Cortical Glucose Metabolism in PreSxC9 Compared to HC, Identified Using Voxel-Wise Testing Following Partial Volume Correction
References
- 1.Hardiman O, Al-Chalabi A, Chio A, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3:17085. Published online October 20, 2017. doi: 10.1038/nrdp.2017.71 [DOI] [PubMed] [Google Scholar]
- 2.van Es MA, Hardiman O, Chio A, et al. Amyotrophic lateral sclerosis. Lancet. 2017;390(10107):2084-2098. doi: 10.1016/S0140-6736(17)31287-4 [DOI] [PubMed] [Google Scholar]
- 3.Brown RH, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377(2):162-172. doi: 10.1056/NEJMra1603471 [DOI] [PubMed] [Google Scholar]
- 4.Olney NT, Spina S, Miller BL. Frontotemporal dementia. Neurol Clin. 2017;35(2):339-374. doi: 10.1016/j.ncl.2017.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Renton AE, Majounie E, Waite A, et al. ; ITALSGEN Consortium . A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21–linked ALS-FTD. Neuron. 2011;72(2):257-268. doi: 10.1016/j.neuron.2011.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p–linked FTD and ALS. Neuron. 2011;72(2):245-256. doi: 10.1016/j.neuron.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gijselinck I, Van Langenhove T, van der Zee J, et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 2012;11(1):54-65. doi: 10.1016/S1474-4422(11)70261-7 [DOI] [PubMed] [Google Scholar]
- 8.Mestre TA, Guttman M. The dawn of a new era for neurodegenerative disorders: Huntington’s disease leading the way. Mov Disord. 2019;34(9):1301-1302. doi: 10.1002/mds.27826 [DOI] [PubMed] [Google Scholar]
- 9.Benatar M, Wuu J. Presymptomatic studies in ALS: rationale, challenges, and approach. Neurology. 2012;79(16):1732-1739. doi: 10.1212/WNL.0b013e31826e9b1d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rohrer JD, Nicholas JM, Cash DM, et al. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal dementia Initiative (GENFI) study: a cross-sectional analysis. Lancet Neurol. 2015;14(3):253-262. doi: 10.1016/S1474-4422(14)70324-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tentolouris-Piperas V, Ryan NS, Thomas DL, Kinnunen KM. Brain imaging evidence of early involvement of subcortical regions in familial and sporadic Alzheimer’s disease. Brain Res. 2017;1655:23-32. doi: 10.1016/j.brainres.2016.11.011 [DOI] [PubMed] [Google Scholar]
- 12.Mutsaerts HJMM, Mirza SS, Petr J, et al. ; GENetic Frontotemporal dementia Initiative (GENFI) . Cerebral perfusion changes in presymptomatic genetic frontotemporal dementia: a GENFI study. Brain. 2019;142(4):1108-1120. doi: 10.1093/brain/awz039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greaves CV, Rohrer JD. An update on genetic frontotemporal dementia. J Neurol. 2019;266(8):2075-2086. doi: 10.1007/s00415-019-09363-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cash DM, Bocchetta M, Thomas DL, et al. ; Genetic FTD Initiative, GENFI . Patterns of gray matter atrophy in genetic frontotemporal dementia: results from the GENFI study. Neurobiol Aging. 2018;62:191-196. doi: 10.1016/j.neurobiolaging.2017.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee SE, Sias AC, Mandelli ML, et al. Network degeneration and dysfunction in presymptomatic C9ORF72 expansion carriers. Neuroimage Clin. 2016;14:286-297. doi: 10.1016/j.nicl.2016.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caverzasi E, Battistella G, Chu SA, et al. Gyrification abnormalities in presymptomatic c9orf72 expansion carriers. J Neurol Neurosurg Psychiatry. 2019;90(9):1005-1010. doi: 10.1136/jnnp-2018-320265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiskoot LC, Rombouts SARB, Panman JL, et al. Neuropsychological and gray matter volume decline in presymptomatic C9Orf72 mutation carriers. Alzheimers Dement. 2016;12(7):62. doi: 10.1016/j.jalz.2016.06.108 [DOI] [Google Scholar]
- 18.Rittman T, Borchert R, Jones S, et al. ; Genetic Frontotemporal Dementia Initiative (GENFI) . Functional network resilience to pathology in presymptomatic genetic frontotemporal dementia. Neurobiol Aging. 2019;77:169-177. doi: 10.1016/j.neurobiolaging.2018.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Papma JM, Jiskoot LC, Panman JL, et al. Cognition and gray and white matter characteristics of presymptomatic C9orf72 repeat expansion. Neurology. 2017;89(12):1256-1264. doi: 10.1212/WNL.0000000000004393 [DOI] [PubMed] [Google Scholar]
- 20.Popuri K, Dowds E, Beg MF, et al. Gray matter changes in asymptomatic C9orf72 and GRN mutation carriers. Neuroimage Clin. 2018;18(January):591-598. doi: 10.1016/j.nicl.2018.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sellami L, Bocchetta M, Masellis M, et al. ; Genetic FTD Initiative, GENFI . Distinct neuroanatomical correlates of neuropsychiatric symptoms in the three main forms of genetic frontotemporal dementia in the GENFI Cohort. J Alzheimers Dis. 2018;65(1):147-163. doi: 10.3233/JAD-180053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poesen K, Van Damme P. Diagnostic and prognostic performance of neurofilaments in ALS. Front Neurol. 2019;9:1167. doi: 10.3389/fneur.2018.01167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meeter LH, Dopper EG, Jiskoot LC, et al. Neurofilament light chain: a biomarker for genetic frontotemporal dementia. Ann Clin Transl Neurol. 2016;3(8):623-636. doi: 10.1002/acn3.325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benatar M, Wuu J, Andersen PM, Lombardi V, Malaspina A. Neurofilament light: a candidate biomarker of presymptomatic amyotrophic lateral sclerosis and phenoconversion. Ann Neurol. 2018;84(1):130-139. doi: 10.1002/ana.25276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benatar M, Wuu J, Lombardi V, et al. Neurofilaments in pre-symptomatic ALS and the impact of genotype. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20(7-8):538-548. doi: 10.1080/21678421.2019.1646769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van der Ende EL, Meeter LH, Poos JM, et al. ; Genetic Frontotemporal dementia Initiative (GENFI) . Serum neurofilament light chain in genetic frontotemporal dementia: a longitudinal, multicentre cohort study. Lancet Neurol. 2019;18(12):1103-1111. doi: 10.1016/S1474-4422(19)30354-0 [DOI] [PubMed] [Google Scholar]
- 27.Meeter LHH, Gendron TF, Sias AC, et al. Poly(GP), neurofilament and grey matter deficits in C9orf72 expansion carriers. Ann Clin Transl Neurol. 2018;5(5):583-597. doi: 10.1002/acn3.559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Weehaeghe D, Ceccarini J, Delva A, Robberecht W, Van Damme P, Van Laere K. Prospective validation of 18F-FDG brain PET discriminant analysis methods in the diagnosis of amyotrophic lateral sclerosis. J Nucl Med. 2016;57(8):1238-1243. doi: 10.2967/jnumed.115.166272 [DOI] [PubMed] [Google Scholar]
- 29.Chew S, Atassi N. Positron emission tomography molecular imaging biomarkers for amyotrophic lateral sclerosis. Front Neurol. 2019;10:135. doi: 10.3389/fneur.2019.00135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katisko K, Cajanus A, Korhonen T, Remes AM, Haapasalo A, Solje E. Prodromal and early bvFTD: evaluating clinical features and current biomarkers. Front Neurosci. 2019;13:658. doi: 10.3389/fnins.2019.00658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Laere K, Vanhee A, Verschueren J, et al. Value of 18fluorodeoxyglucose–positron-emission tomography in amyotrophic lateral sclerosis: a prospective study. JAMA Neurol. 2014;71(5):553-561. doi: 10.1001/jamaneurol.2014.62 [DOI] [PubMed] [Google Scholar]
- 32.van Weehaeghe D, Ceccarini J, Willekens SM, de Vocht J, van Damme P, van Laere K. Is there a glucose metabolic signature of spreading TDP-43 pathology in amyotrophic lateral sclerosis? Q J Nucl Med Mol Imaging. 2020;64(1):96-104. doi: 10.23736/S1824-4785.17.03009-6 [DOI] [PubMed] [Google Scholar]
- 33.Benatar M, Turner MR, Wuu J. Defining pre-symptomatic amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20(5-6):303-309. doi: 10.1080/21678421.2019.1587634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Poesen K, De Schaepdryver M, Stubendorff B, et al. Neurofilament markers for ALS correlate with extent of upper and lower motor neuron disease. Neurology. 2017;88(24):2302-2309. doi: 10.1212/WNL.0000000000004029 [DOI] [PubMed] [Google Scholar]
- 35.Bakker LA, Schröder CD, Spreij LA, et al. Derivation of norms for the Dutch version of the Edinburgh Cognitive and Behavioral ALS Screen. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20(1-2):19-27. doi: 10.1080/21678421.2018.1522352 [DOI] [PubMed] [Google Scholar]
- 36.Strong MJ, Abrahams S, Goldstein LH, et al. Amyotrophic lateral sclerosis—frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(3-4):153-174. doi: 10.1080/21678421.2016.1267768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schrooten M, Smetcoren C, Robberecht W, Van Damme P. Benefit of the Awaji diagnostic algorithm for amyotrophic lateral sclerosis: a prospective study. Ann Neurol. 2011;70(1):79-83. doi: 10.1002/ana.22380 [DOI] [PubMed] [Google Scholar]
- 38.Abrahams S, Newton J, Niven E, Foley J, Bak TH. Screening for cognition and behaviour changes in ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(1-2):9-14. doi: 10.3109/21678421.2013.805784 [DOI] [PubMed] [Google Scholar]
- 39.Das SR, Avants BB, Grossman M, Gee JC. Registration based cortical thickness measurement. Neuroimage. 2009;45(3):867-879. doi: 10.1016/j.neuroimage.2008.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wollenweber SD, Ambwani S, Delso G, et al. Evaluation of an atlas-based PET head attenuation correction using PET/CT & MR patient data. IEEE Trans Nucl Sci. 2013;60(5):3383-3390. doi: 10.1109/TNS.2013.2273417 [DOI] [Google Scholar]
- 41.Talairach J, Tournoux P. Co-Planar Stereotactic Atlas of the Human Brain. Thieme Medical Publishers; 1988. [Google Scholar]
- 42.Makris N, Goldstein JM, Kennedy D, et al. Decreased volume of left and total anterior insular lobule in schizophrenia. Schizophr Res. 2006;83(2-3):155-171. doi: 10.1016/j.schres.2005.11.020 [DOI] [PubMed] [Google Scholar]
- 43.Frazier JA, Chiu S, Breeze JL, et al. Structural brain magnetic resonance imaging of limbic and thalamic volumes in pediatric bipolar disorder. Am J Psychiatry. 2005;162(7):1256-1265. doi: 10.1176/appi.ajp.162.7.1256 [DOI] [PubMed] [Google Scholar]
- 44.Desikan RS, Ségonne F, Fischl B, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31(3):968-980. doi: 10.1016/j.neuroimage.2006.01.021 [DOI] [PubMed] [Google Scholar]
- 45.Goldstein JM, Seidman LJ, Makris N, et al. Hypothalamic abnormalities in schizophrenia: sex effects and genetic vulnerability. Biol Psychiatry. 2007;61(8):935-945. doi: 10.1016/j.biopsych.2006.06.027 [DOI] [PubMed] [Google Scholar]
- 46.La Joie R, Perrotin A, Barré L, et al. Region-specific hierarchy between atrophy, hypometabolism, and β-amyloid (Aβ) load in Alzheimer’s disease dementia. J Neurosci. 2012;32(46):16265-16273. doi: 10.1523/JNEUROSCI.2170-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bocchetta M, Cardoso MJ, Cash DM, Ourselin S, Warren JD, Rohrer JD. Patterns of regional cerebellar atrophy in genetic frontotemporal dementia. Neuroimage Clin. 2016;11:287-290. doi: 10.1016/j.nicl.2016.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Castelnovo V, Caminiti SP, Riva N, Magnani G, Silani V, Perani D. Heterogeneous brain FDG-PET metabolic patterns in patients with C9orf72 mutation. Neurol Sci. 2019;40(3):515-521. doi: 10.1007/s10072-018-3685-7 [DOI] [PubMed] [Google Scholar]
- 49.Diehl-Schmid J, Licata A, Goldhardt O, et al. ; FTLDc Study Group . FDG-PET underscores the key role of the thalamus in frontotemporal lobar degeneration caused by C9ORF72 mutations. Transl Psychiatry. 2019;9(1):54. doi: 10.1038/s41398-019-0381-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schönecker S, Neuhofer C, Otto M, et al. ; Deutsches FTLD-Konsortium . Atrophy in the thalamus but not cerebellum is specific for C9orf72 FTD and ALS patients—an atlas-based volumetric MRI study. Front Aging Neurosci. 2018;10(FEB):45. doi: 10.3389/fnagi.2018.00045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meeter LHH, Gendron TF, Sias AC, et al. Poly-GP dipeptide repeats and neurofilament light chain as biomarkers in presymptomatic and symptomatic frontotemporal dementia caused by C9ORF72 repeat expansions. Alzheimers Dement. 2017;13(7):1228-1229. doi: 10.1016/j.jalz.2017.07.425 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eResults. Supplementary Analysis
eReferences
eFigure 1. Violin Plots of 18F FDG Uptake in Volumes of Interest, After Correcting for GM Atrophy With PVC, in Healthy Controls and PreSxC9
eFigure 2. W-Score Frequency Maps Detailing the Fraction of PreSxC9 With Suprathreshold W-Scores at Voxel Level
eFigure 3. Mean W-Score Images Detailing Tracer Uptake in PreSxC9, Thresholded From 0.5 Standard Deviations From the Norm
eFigure 4. Relative Hypermetabolism in PreSxC9 in relation to healthy controls (pheight <0.001, at Cluster Level pFWE<0.05) Following Voxel-Based Partial Volume Corrections, Using SUVr Images
eTable 1. Significant Areas of Decreased Cortical Glucose Metabolism in PreSxC9 Compared to HC, Identified Using Voxel-Wise Testing
eTable 2. Significant Areas of Volume Decline in PreSxC9 Compared to HC, Identified Using Voxel-Wise Testing
eTable 3. Significant Areas of Decreased Metabolism in PreSxC9 Compared to HC, Identified Using Voxel-Wise Testing Following Partial Volume Correction
eTable 4. Significant Areas of Increased Cortical Glucose Metabolism in PreSxC9 Compared to HC, Identified Using Voxel-Wise Testing Following Partial Volume Correction
eTable 5. A Volume-of-Interest (VOI)-Based Analysis After Region-Based Voxel-Wise (RBV) Correction for GM Atrophy, Using the Hammers N30R83 Maximum Probability Atlas
eTable 6. Significant Areas of Increased Cortical Glucose Metabolism in PreSxC9 Compared to HC, Identified Using Voxel-Wise Testing Following Partial Volume Correction