

Abstract

3,3-Bis(2,4,6-triisopropylphenyl)-1,1-binaphthyl-2,2-diyl hydrogenphosphate (TRIP) catalyzes the asymmetric allylation of aldehydes with organozinc compounds, leading to highly valuable structural motifs, like precursors to lignan natural products. Our previously reported mechanistic proposal relies on two reaction intermediates and requires further investigation to really understand the mode of action and the origins of stereoselectivity. Detailed ab initio calculations, supported by experimental data, render a substantially different mode of action to the allyl boronate congener. Instead of a Brønsted acid-based catalytic activation, the chiral phosphate acts as a counterion for the Lewis acidic zinc ion, which provides the activation of the aldehyde.
Introduction
Computational efforts have become a frequent tool in the elucidation of mechanistic scenarios and understanding the course of chemical reactions.1−3 They allow the evaluation of different events by the comparison of their energetic profiles and deliver useful information to understand and improve the chemistry behind the process of interest.1−4 Especially in combination with asymmetric synthesis, density functional theory (DFT) calculations have facilitated the finding of asymmetric induction and have delivered or confirmed the accepted mechanistic concept of activation and stereopreference. Regarding asymmetric synthesis, allylation reactions have a pronounced foundation and are an indispensable methodology when it comes to the creation of chiral molecules.5−10 One of these examples is the asymmetric preparation of dibenzylbutyrolactones, which proceeds via the asymmetric allylation of an aldehyde precursor with an organozinc reagent.11,12 The catalyst, namely, 3,3-bis(2,4,6-triisopropylphenyl)-1,1-binaphthyl-2,2-diyl hydrogenphosphate (TRIP, 2)13shows high stereoinduction on the process and delivers a natural product precursor with an enantiomeric excess as high as 98%. Important to note is the fact that the reaction has a boron congener, using allyl boronate reagents and the same catalyst.14 Some efforts have been undertaken to explain the stereoselectivity in case of these reagents, and yielded a mechanistic mode of action, as depicted in Figure 1.15−17 The decrease of the activation barrier is explained by a hydrogen bond to one pinacol oxygen, activating the reagent, whereas the required rigidity for the alignment is reasoned by a formyl-H interaction with the P=O oxygen Lewis base.
Figure 1.

Asymmetric allylation of aldehydes with bromolactone 3 and previously reported rationales for the reactivity and the stereoselectivity of the catalyst 2 for the zinc- and boron-based reagents.
For the zinc-based reaction, the absence of the pinacol system renders this activation impossible, and our initial mechanistic proposal was based on the energetical assessment of two reaction intermediates. Therefore, additional investigations are required to understand the differences of these allylation reactions.
Results and Discussion
To investigate the reactivity of the TRIP acid (2) under reaction conditions, we probed the acid in the presence of compound 3 and zinc dust (see Scheme 1). The NMR spectra show strong peak broadening due to the unfavorable properties of the NMR-active zinc. The resolution of the recorded spectra slightly improves when the allylic bromide was changed to compound 4 (see Scheme 1a). The reaction with this open-chain, allylic bromide (4) yields the allylated product 6 under TRIP catalysis too, albeit with significantly reduced stereoselectivity [99% conv., 60% isolated yield for product 7 (over two steps), 28% ee, see Scheme 1a]. Alternatively, the initially envisaged complex 8b was prepared via the preformed zinc salt 10 and its subjection to the in-situ formed reagent 9 (see Scheme 1d). The observed shift in the 31P NMR spectrum is in almost perfect agreement with the one observed for complex 8a – a reasonable observation due to their highly similar environment for the zinc core [4.63 ppm (8a) and 4.54 ppm (8b)]. The formation of a zinc phosphate salt under reaction conditions finds additional support in the detection of the protodemetalated reagent 9 via headspace gas chromatography–mass spectrometry (GC–MS) analysis. Whereas, only little amounts of the quenched reagent 9 form in the absence of TRIP (presumably via a quench by the ammonium salt), significantly higher amounts were detected in the presence of the chiral phosphoric acid reflecting the 20 mol % used in the experiment (for details see Experimental Section and Supporting Information, Chapter 2.5).
Scheme 1. (a) Asymmetric Allylation with Reagent 4 and (b) 31P NMR Studies of the Catalyst under Reaction Conditions in the Absence of Benzaldehyde (5), (c) Rendered Structures of the Most Stable Conformers B1 and B1h,18 (d) Preparation of Zinc Salt 10 and Complex 8b, (e) Calculations of the Quench and Coordination of TRIP (2) and Reagent 9 at the DLPNO–CCSD(T)/cc-pVTZ ΔG + COSMO Level [compared to ΔG(edfav.) = −88.3 kJ/mol].

This observed quench of 2 by zinc organyls has precedence in literature and has been documented even by a crystal structure of a homolog phosphate.19 Important to note is that within the homolog zinc complex, both oxygen atoms of the phosphate interact with the zinc species forming a dimeric zinc salt – a fact that our calculations suggest for the outlined transition states herein too (vide infra).19 Finally, the question remained, which complexes of the observed ones are catalytically active: under resubmission to the reaction conditions, all complexes (namely, acid 2, zinc salt 10 and complex 8b) proved to be active and provided the homoallylic alcohol product with 86–88% conversion and close to perfect selectivity (96% ee).
These findings, in combination with the fact that educt complexes involving a mono-zinc phosphate complex are energetically significantly higher (see Supporting Information, Chapter 3.9.2), render a dinuclear catalytic species with respect to zinc the most likely scenario. All experimental data as well as the theoretical results, point to complex 8 to be the product of the treatment of 2 with reagent 4/9 (see Scheme 1b). It results from a quench of the first reagent equivalent by the acidic proton of TRIP (2) and the subsequent coordination of a second equivalent to the chiral zinc phosphate salt.
Our computational efforts support this hypothesis of a coordinated allylzinc species and the involvement of two zinc atoms in the active species, as such complexes are thermodynamically and kinetically highly favored (see Scheme 1d). Note that the subscripts of ΔG and ΔΔG values give the point of reference (“ed” refers to educts; “ISR” stands for “infinitely separated reactants”).
Phosphoric acid 2 is quenched with zinc reagent 9 (formed by the zinc insertion into allylic bromide 3) with a negligible barrier [ΔGISReduct = −3.6 kJ/mol; ΔΔGed = +6.7 kJ/mol; ΔGISRZn-salt = −85.3 kJ/mol; obtained from the resolution of identity-approximation-Perdew–Burke–Ernzerhof (RI-PBE)20,21 -D3/def222 -TZVPPD22,23 +COSMO24,25 single-point calculations]. Subsequently, the formed zinc salt can coordinate another equivalent of reagent 9, leading to structures B1 and B1h. Whereas, B1 represents the local minimum, the formation of the final educt complex edfav. requires severe geometrical rearrangements and thus B1 is regarded as an off-cycle intermediate, which leads to a nonproductive pathway (see Supporting Information, Chapter 3.9.1.; pathways leading to the favored and disfavored enantiomers are labeled with the subfixes “fav.” and “dis.”, respectively). B1h however, which resembles the assumed structure 8 (except the double coordination of the lactone carbonyl), demonstrates a perfect starting point for the formation of edfav.: Following the reaction trajectory, the most likely subsequent event is the coordination of the aldehyde oxygen to the Lewis acidic zinc salt of B1h (see Figure 2: mechanistic cycle, B1h to edfav.). This weakens and finally breaks the coordination of the lactone-carbonyl group.
Figure 2.

Left side: energy diagrams for the catalyzed and the uncatalyzed allylation step (blue = catalyzed reaction leading to the observed enantiomer; red = catalyzed reaction leading to the nonobserved enantiomer; gray = uncatalyzed reaction); right side: reactants, products, and transition state structures of the corresponding pathways (newly formed bond in the transition states is marked in dark blue).18 Only the energetically lowest pathways are shown, for all others, see the Supporting Information. All presented data are given on the DLPNO–CCSD(T)/cc-pVTZ ΔG + COSMO level; the reference point (0 kJ/mol) represents the indefinitely separated reactants [the zinc salt of TRIP, reagent 9 and benzaldehyde (5)].
The obtained monocoordination of the allylating reagent provides the possibility of an interaction of the aldehyde with both zinc atoms, increasing the aldehyde’s polarization and translating to a more reactive electrophile [see edfav. and eddis. in Figures 2 and 3; note that all calculated transition states with reasonable energies require this double coordination (for additional transition states see Supporting Information, Chapter 3)].
Figure 3.

Mechanistic proposal of the catalytic cycle.
The latter structures (edfav. and eddis.) were originally computed from the coordination of the educt complex of the uncatalyzed reaction edunc. to the zinc salt of the catalyst. Thus, they represent perfect starting points for the asymmetric catalysis.
Complex edfav. readily transforms into the product complex pfav. via an energy barrier of ΔΔGedTS = +39.0 kJ/mol (see Figure 2, left side). This compares to the lowest energy barrier for the formation of the experimentally disfavored enantiomer of ΔΔGed = +51.0 kJ/mol, giving a difference of 12.0 kJ/mol in favor of the experimentally observed results. This difference very well reflects the observed enantiomeric excess, which has been determined to be 94% at 4 °C.11
The rationale for the difference in the transition states may be found in the minimization of the dipole moment, especially with consideration of the apolar reaction medium (toluene). Whereas, the favored TSfav. exhibits a dipole moment of 4.13 D, the disfavored TSdis. yields 6.85 D. This is reasoned by a different alignment of the lactone moiety and its dipole, which is positioned along the overall dipole moment in TSdis.. In contrast to structure TSdis., the dipole moment of the lactone subunit is almost orthogonal to the overall dipole in TSfav. (see Figure 4). Furthermore, the dipole moment of edfav. was calculated to be 5.10 D, while the one of eddis. was determined to be 4.74 D. Hence, for the favored pathway, the polarity of the complex decreases along the reaction coordinate toward the transition state, while for the disfavored pathway, it increases significantly, reflected by the complementary educt energies (eddis. is slightly favored over edfav., see Figure 2). This adds to the energetic barriers and further favors the pathway of the observed stereoselectivity.
Figure 4.
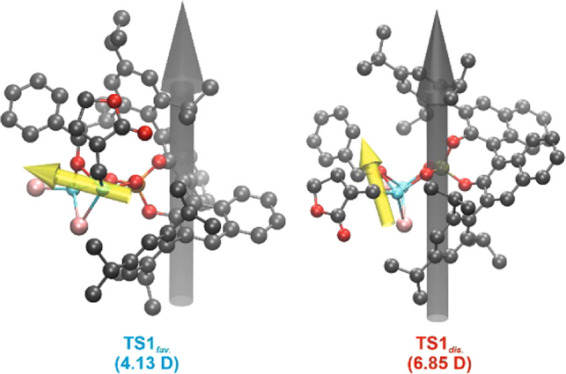
TS structures,18 overall dipole moment (gray arrow), and dipole moment of the lactone (yellow arrow); the overall dipole moment is given in brackets. All presented data are given on the DLPNO–CCSD(T)/cc-pVTZ level.
In addition, the experimental observation that allyl bromide leads to no detected stereoinduction may be rationalized by this hypothesis, as allyl bromide exhibits a significantly lower dipole moment.
Moreover, the alignment of the aldehyde carbon and the β-carbon of the lactone forming the future C–C bond is much more favorable in structure edfav. than in eddis., which can be attributed to unfavorable steric interactions with the isopropyl groups of the catalyst in the latter educt complex. Attempts to improve the alignment in eddis. reverted to the original geometry when optimized. Hence, we conclude that the pathway leading to the experimentally disfavored enantiomer requires significantly more reorganization, contributing to the higher energy barrier of TSdis. additionally.
The uncatalyzed reaction is disfavored by 36.5 kJ/mol (ΔΔGedTS = +75.5 kJ/mol). Other coordination patterns of the educt complexes to the zinc salt of the TRIP catalyst yielded unfavorable pathways (see Supporting Information, Chapter 3.9.1). Important to note is that the catalysis provides the stereoinduction, but the experimental data suggests that the preceding in situ formation of the allyl-zinc reagent demonstrates the rate-determining step of the overall process, as at no time of the reaction, the level of an active zinc reagent exceeds the catalyst loading (see Supporting Information, Chapter 2.3–2.4, for details).
Finally, the energies of the formed product complexes deserve some attention, as all reactions are endergonic (for energies of products see Supporting Information, Chapter 3.8). This fact may reveal the role of the NH4Cl additive, which has been either believed to activate the zinc dust or quench the obtained zinc alcoholate product, yielding the free protonated alcohol and ZnX2 (please note that without the ammonium salt, no reaction is observed regarding the catalyzed and the uncatalyzed reaction). With the reaction(s) being endergonic, the latter role of the additive seems to be supported by our calculations, as it provides the final pull the equilibrium to the product side. An alternative for this shift of the equilibrium would be a cluster formation of the zinc alcoholate product molecules, as described for Noyori’s dimethylaminoisoborneol (DAIB)-catalyzed, asymmetric alkylation of aldehydes with dialkylzinc reagents.26 Nevertheless, the presence of the NH4+ proton and the significant bulk around the zinc–oxygen cluster favor the equilibrium push by the quenching scenario.
Conclusions
We have shown that the TRIP-catalyzed asymmetric allylation via zinc reagents proceeds via the zinc salt of the chiral phosphoric acid. Addition of a second equivalent of the in-situ formed reagent yields complex B1h, which, upon an approaching aldehyde, gives educt complex edfav., an ideal precursor for the final C–C bond formation. The catalysis is rationalized by the double coordination of the aldehyde oxygen by both Lewis acidic zinc atoms (see the structure of TSfav.), and the required rigidity of the catalytic system stems from the Lewis basic interaction of the P=O oxygen with the allylzinc reagent. Perfectly aligned in edfav., the favorable enantiomer is formed via an energy barrier of 39.0 kJ/mol (ΔΔGedTS). Thus, the formation of the experimentally nonobserved enantiomer and the uncatalyzed reaction are outrun by 12.0 and 36.5 kJ/mol, respectively. The energy difference in the transition states can be rationalized by the dipole moment of the corresponding complexes, which is minimized for the favored enantiomer. Important to note is the endergonic character of the final reaction step, which requires an external pull to the reaction system. This pull can be found in the protonic quench of the zinc alcoholate by the ammonium chloride additive. All of these findings render a rather clear picture of the asymmetric catalysis in the case of this allylation process and provide another example for a counterion-directed asymmetric catalytic process. Additionally, they may lead to a better understanding of the Lewis acidic/basic interactions of this outstanding catalyst and stimulate new catalytic procedures of a similar type.
Experimental Section
General
All chemicals were purchased from Sigma-Aldrich or Acros Organics and were used as received unless otherwise noted. All solvents were purchased from Roth, except Dioxane (Alfa Aasar) and dry toluene (Sigma-Aldrich). Moisture sensitive reactions were performed using standard Schlenk techniques with argon 5.0. Analytical thin-layer chromatography (TLC) was carried out on Merck TLC silica gel aluminum sheets (silica gel 60, F254, 20 cm × 20 cm) and spots were visualized by UV light (λ = 254 nm), by staining with cerium ammonium molybdate solution [50 g of (NH4)6Mo7O24 was dissolved in 400 mL of H2O and 50 mL of conc. H2SO4 was added followed by 2.0 g of Ce(SO4)2] or KMnO4 solution (1 g of KMnO4 and 2 g of Na2CO3 were dissolved in 100 mL of H2O), and developed by heating with a heat gun. Column chromatography was performed on silica gel 60 from Merck with particle sizes 40–63 μm. A 30- to 100-fold excess of silica gel was used with respect to the mass of the dry crude product, depending on the separation problem. For sticky crude products, the crude material was dissolved in MeOH and subsequently adsorbed on the 2.5-fold excess of silica gel. Afterward, the solvent was removed in vacuum, and the adsorbed crude material was dried in an oil pump vacuum. The dimension of the column was adjusted to the required amount of silica gel and formed a pad between 20 and 40 cm of height. In general, the silica gel was mixed with the eluent and charged into the column before equilibration. Subsequently, the dissolved or adsorbed crude material was loaded onto the top of the silica gel, and the mobile phase was forced through the column by pressure exerted by a rubber bulb pump.
Instrumentation
1H, 13C, and 31P NMR spectra were recorded on a Bruker AVANCE III 300 spectrometer (1H: 300.13 MHz; 13C: 75.47 MHz; 31P: 121.49 MHz) with an autosampler. Chemical shifts are referenced to the residual proton and carbon signal of the deuterated solvent [CDCl3: δ = 7.26 ppm (1H), 77.16 ppm (13C)]. Chemical shifts δ are given in ppm (parts per million) and coupling constants J in Hz (Hertz). Deuterated solvents for nuclear resonance spectroscopy were purchased from Roth. Melting points were determined on a Gallenkamp MPD350.BM2.5 apparatus with an integrated microscopical support. They were measured in open capillary tubes with a mercury-in-glass thermometer and were not corrected. Infrared (IR) spectra were recorded neatly on a Bruker Alpha-P (ATR) instrument. The specific optical rotation was determined on a PerkinElmer Polarimeter 341 with an integrated sodium vapor lamp. All samples were measured in CHCl3 and CH2Cl2 (both were purchased from Sigma-Aldrich, ACS spectrophotometric grade, ≥99.8%) at the D-line of the sodium light (λ = 589 nm) under nontempered conditions between 22 and 27 °C. High-resolution mass spectra were recorded on an Agilent 6230 time-of-flight (TOF) liquid chromatography–MS (LC/MS) using electrospray ionization (ESI) (positive mode, capillary voltage 3.5 kV) or APCI (negative mode, 5.0 kV) methods. Chiral high-performance liquid chromatography (HPLC) analysis was performed on a Shimadzu HPLC system [DGU-20A (degasser), LC-20A (pump), SIL-20A (autosampler), CTO-20AC (column oven), SPD-M20A (detector), and CBM-20AC (controller)] with n-heptane/2-PrOH as an eluent using Daicel columns [dimension: 4.6 mm × 250 mm, 5 μm particle size, except Chiralpak AD (10 μm) and Chiralcel OJ (10 μm)] and conditions as specified below. All GC–MS measurements were carried out on an Agilent 7890A GC system, equipped with an Agilent 5975C mass-selective detector (electron impact, 70 eV), a HP-5-MS column (30 m × 0.25 mm × 0.25 μm film), and an Agilent 7697A headspace autosampler using He as a carrier gas at a flow of 0.7 mL/min. The following temperature program was used in all GC–MS headspace measurements: initial temperature 40 °C, hold for 5 min, 10 °C/min, to 200 °C. Headspace parameters: vial pressurization gas: He; loop size: 1 mL; transfer line: DB-ProSteel (0.53 mm diameter); oven temperature: 50 °C; loop temperature: 55 °C; transfer line: 60 °C; vial equilibration time: 4 min; Injection duration: 0.5 min; vial size: 20 mL; vial shaking: level 5, 71 shakes per min with an acceleration of 260 cm/s2; fill pressure: 15 psi.
3-Methylene-5-phenyldihydrofuran-2(3H)-one (7)
A 5 mL screw cap vial with a magnetic stirring bar was charged with zinc (33.0 mg, 500 μmol, 5 equiv), ammonium chloride (43.0 mg, 800 μmol, 8 equiv), and (S)-3,3′-bis(2,4,6-triisopropylphenyl)-1,1′-binaphthyl-2,2′-diyl hydrogenphosphate (TRIP, 7.5 mg, 10.0 μmol, 0.1 equiv), followed by toluene (1.0 mL), benzaldehyde (10.6 mg, 100 μmol), and allyl bromide 4 (29.0 mg, 150 μmol, 1.5 equiv). The mixture was stirred (720 rpm) at room temperature for 16 h and was consecutively quenched by the addition of a saturated aqueous NH4Cl solution (5 mL). The mixture was extracted with EtOAc (3 × 10 mL) and the combined organic phase was dried over Na2SO4, filtered, and concentrated under reduced pressure. The obtained crude product [mixture of compound 6 (free alcohol) and 7 (lactone)] was dissolved in CH2Cl2 and para-toluenesulfonic acid hydrate (4 mg, 20 μmol, 20 mol %) was added. The reaction mixture was stirred for 16 h, quenched with a NaHCO3 aqueous solution (saturated), and extracted with CH2Cl2 (3 × 10 mL). The combined organic phase was dried over Na2SO4, filtered, and concentrated. The crude product was purified via flash chromatography (SiO2, hexanes/EtOAc 5/1) to give product 7 (20 mg, 60 μmol, 60%) as a colorless oil.
[α]D20 = +4.4 (c = 2.0, CHCl3); 1H NMR (300.13 MHz, CDCl3): 7.46–7.28 (m, 5H), 6.31 (t, J = 2.8 Hz, 1H), 5.69 (t, J = 2.5 Hz, 1H), 5.53 (dd, J1 = 7.9, J2 = 6.6 Hz, 1H), 3.41 (ddt, J1 = 17.1, J2 = 8.1, J3 = 2.5 Hz, 1H), 2.91 (ddt, J1 = 17.1, J2 = 6.4, J3 = 2.9 Hz, 1H); 13C{1H} NMR (75.47 MHz, CDCl3): 170.3, 139.9, 134.3, 129.0, 128.7, 125.5, 122.6, 78.1, 36.4; IR (film) υ̃ = 3093, 3066, 3050, 3037, 2974, 2919, 2853, 1752, 1602, 1551, 1496, 1459, 1437, 1402, 1375, 1319, 1277, 1240, 1215, 1126, 1080, 1020, 985, 962, 938, 818, 761, 701, 639, 562 cm–1; HPLC analysis on a chiral stationary phase {Daicel Chiralpak OD-H, n-heptane/2-propanol 90/10, 1.0 mL/min, 30 °C, UV 215 nm, tret(enantiomer 1) = 8.7 min, tret(enantiomer 2) = 9.8 min}: tret(major isomer) = 8.7 min, 28% ee; HRMS(ESI): m/z: calcd for C11H11O2+: 175.0754 [M + H]+, found, 175.0755.
General Procedure for the Preparation of Racemic Reference Material
An HPLC vial with a magnetic stirring bar was charged with a ketone or aldehyde (25.0 μmol, 1 equiv), zinc (8.0 mg, 125 μmol, 5 equiv), NH4Cl (11.0 mg, 200 μmol, 8 equiv), and diphenyl phosphate (2.0 mg, 8.0 μmol, 0.3 equiv). Allylic bromide 4 (36.0 μmol, 1.5 equiv), dissolved in toluene (200 μL) was added and the reaction mixture was stirred at room temperature for 16 h. The suspension was filtered through a plug of silica gel (∼1 g), the plug was rinsed with additional EtOAc (ca. 1 mL), and the combined filtrates were concentrated. The residue was dissolved in a small amount of EtOAc (ca. 100 μL) and half of the solution was adsorbed on the starting line of a silica gel TLC plate (∼8 cm wide). The plate was developed in the solvent indicated for flash chromatography for the specific compound (indicated below), and the product band was scratched off. The obtained silica gel with the adsorbed product was transferred into an HPLC vial with a magnetic stirring bar and was extracted by stirring with 2-propanol (800 μL) for 30 min at 22 °C. The suspension was filtered through a syringe filter (Nylon, 0.2 μm) and subjected to HPLC-MS and HPLC-UV analyses on a chiral stationary phase.
Mechanistic Studies
General
All reactions were performed in dry and degassed CDCl3. All samples were prepared under a dry argon atmosphere in flame-dried glass ware. 31P NMR spectra were referenced to dimethyl methylphosphonate (δ = +33.8 ppm) unless otherwise noted. The reference was filled into a small capillary (50 μL volume), which was sealed by flame and added to the corresponding sample to avoid interaction of the standard with the reaction mixture. 1H and 13C spectra were referenced to the residual solvent peak of CDCl3 [δ = 7.26 ppm (1H) and 77.16 ppm (13C)].
TRIP (2) Spectra in the Presence of Allylic Zinc Reagent Derived from 4
(S)-TRIP [(S)-2, 22.5 mg, 30.0 μmol], zinc (98.1 mg, 1.50 mmol, 50 equiv), and ammonium chloride (128 mg, 2.40 mmol, 80 equiv) were combined in a Schlenk tube with a magnetic stirring bar and suspended in CDCl3 (600 μL). Compound 4 (29.0 mg, 150 μmol, 21.0 μL, 5 equiv) was added and the reaction mixture was stirred for 1 h. The reaction mixture was filtered through a syringe filter [poly(vinylidene difluoride) (PVDF), 0.45 μm, 33 mm diameter], and the filtrate was filled into an NMR tube and analyzed. After 1H, 13C, and a NOESY spectra were recorded, the capillary with dimethyl methylphosphonate was added to reference the 31P NMR, which was recorded subsequently.
Time Study of the Zinc Insertion Reaction Forming Reagent 9
Zinc dust (10.1 mg, 155 μmol, 5 equiv) and ammonium chloride (13.3 mg, 248 μmol, 8 equiv) were combined in a 1 mL HPLC vial. A stock solution (200 μL toluene and 50 μL 2-iPr2O) containing the bromolactone 3 (6.5 mg, 37 μmol, 1.2 equiv) was added and the reaction mixture was stirred at room temperature. Samples of 50 μL were withdrawn after the times given below, applied to a syringe with filter, diluted with 500 μL of toluene, and filtered. The filtrate was quenched with aqueous HCl (1 M, 100 μL), the phases were separated, the organic phase was dried over Na2SO4, and subjected to GC-flame ionization detection (FID) analysis [for conditions and instrumentation see general part; tret(quenched 9) = 6.65 min, tret(3) = 11.20 min].
Time Study of the Catalytic Reaction with Regard to the Accumulation of 9 over Time
Zinc dust (10.1 mg, 155 μmol, 5 equiv) and ammonium chloride (13.3 mg, 248 μmol, 8 equiv) were combined in a 1 mL HPLC vial. A stock solution (200 μL toluene and 50 μL 2-iPr2O) containing the bromolactone 3 (6.5 mg, 37 μmol, 1.2 equiv), benzaldehyde (5, 3.3 mg, 31 μmol, 1 equiv), and (R)-TRIP (2, 2.3 mg, 3.1 μmol, 0.1 equiv, 10 mol %) was added and the reaction mixture was stirred at room temperature. Samples of 50 μL were withdrawn after the times given below, applied to a syringe with filter, diluted with 500 μL of toluene, and filtered. The filtrate was quenched with aqueous HCl (1 M, 100 μL), the phases were separated, the organic phase was dried over Na2SO4, and subjected to GC-FID analysis [for conditions and instrumentation see general part; tret(quenched 9) = 6.65 min, tret(3) = 11.20 min, tret(homoallylic alcohol product) = 18.41 min]. The final sample (after 23 h reaction time) was dried by a positive stream of air, redissolved in 2-PrOH (500 μL), and subjected to HPLC-UV analysis on a chiral stationary phase {Daicel Chiralpak AD, n-heptane/2-propanol 85/15, 0.7 mL/min, 18 °C, UV 215 nm, tret(enantiomer 1) = 13.0 min, tret(enantiomer 2) = 14.1 min}: tret(major isomer) = 13.7 min, 92% ee.
GC–MS Headspace Measurements for the Indirect Detection of the Zinc Phosphate Salt of 2
Zinc dust (33 mg, 500 μmol), ammonium chloride (43 mg, 800 μmol), TRIP (2, 22.5 mg, 30 μmol), and a magnetic stirring bar were placed into an agilent headspace vial. The vial was capped and crimped, evacuated, and refilled with dry N2. A solution of 4 (29 mg, 150 μmol, 21 μL) in dry dichloromethane (200 μL) was added. The reaction mixture was stirred at room temperature for 3 h and subjected directly to the GC–MS headspace sampling under the conditions outlined in the general section. t(ret)(quenched reagent 9) = 6.03 min.
Preparation of Zinc Phosphate 10
TRIP (2, 26 mg, 34 μmol) and [ZnCO3]2·[Zn(OH)2]3 (26 mg, >58% zinc basis, 230 μmol zinc) were combined in a Schlenk tube, and dry, degassed CDCl3 (700 μL) was added. The slurry was stirred for 16 h at room temperature, and transferred via a syringe, cannula, and syringe filter (PVDF, 0.45 μm, 33 mm diameter) into an NMR tube under an argon protective gas atmosphere. A capillary with dimethyl methylphosphonate was added to reference the 31P NMR and the tube was capped and subjected to NMR analysis. After the analysis, 70 μL was withdrawn from the sample to test its catalytic activity. The remaining 630 μL was directly subjected to the next reaction step. 1H NMR (CDCl3, 300 MHz): δ = 7.87 (d, J = 8.2, 2H), 7.46 (ddd, J1 = 8.1, J2 = 6.3, J3 1.5 Hz, 2H), 7.33–7.20 (m, 4H), 7.01 (d, J = 1.7 Hz, 2H), 6.94 (d, J = 1.7 Hz, 2H), 2.88 (p, J = 6.9 Hz, 2H), 2.66 (tt, J1 = 13.5, J2 = 6.8 Hz, 4H), 1.26 (d, J = 4.5 Hz, 6H), 1.23 (d, J = 4.4 Hz, 6H), 1.07 (d, J = 6.8 Hz, 6H), 0.97 (d, J = 6.7 Hz, 6H), 0.91 (d, J = 6.8 Hz, 6H), 0.79 (d, J = 6.7 Hz, 6H); 13C{1H} NMR (CDCl3, 75 MHz): δ = 148.6, 148.5, 147.9, 146.8, 146.6, 132.7, 132.5, 132.3, 132.2, 130.8, 128.2, 127.3, 126.2, 125.4, 122.3, 121.0, 120.3, 34.3, 31.0, 30.8, 26.3, 25.1, 24.3, 24.1, 23.4, 23.3; 31P{1H} NMR (CDCl3, 121.5 MHz): δ = 6.67.
Preparation of Complex 8b from Zinc Phosphate 10
Zinc dust (10 mg, 153 μmol, 5 equiv) and ammonium chloride (13 mg, 243 μmol, 8 equiv) were combined in a flame-dried Schlenk tube. The solution containing zinc phosphate 10 in CDCl3 (ca. 630 μL) was added, followed by bromolactone 3 (6.8 μL, 11.6 mg, 66 μmol), and the reaction mixture was stirred for 16 h at room temperature. Subsequently, it was transferred via a syringe, cannula, and syringe filter (PVDF, 0.45 μm, 33 mm diameter) into an NMR tube under an argon protective gas atmosphere. A capillary with dimethyl methylphosphonate was added to reference the 31P NMR, and the tube was capped and subjected to NMR analysis. As the solubility of the complex proved low, 100 μL of DMSO-d6 was added and the NMR analysis was repeated. Subsequently, 70 μL of this reaction mixture was used to test the catalytic potential of complex 8b, as described below.
Testing the Catalytic Activity of Salt 10 and Complex 8b
Zinc dust (10 mg, 153 μmol, 5 equiv) and ammonium chloride (13 mg, 243 μmol, 8 equiv) were combined in a flame-dried Schlenk tube. Dry toluene (1 mL), the catalyst preparation (3.4 μmol in 70 μL CDCl3, 11 mol %), benzaldehyde (5, 3.0 μL, 2.9 mg, 30 μmol, 1 equiv) and bromolactone 3 (3.5 μL, 5.8 mg, 33 μmol, 1.1 equiv) were added. The remaining slurry was stirred for 16 h at room temperature, filtered through a pad of silica, and concentrated to dryness. The residue was dissolved in 2-PrOH and subjected to HPLC-UV analysis on a chiral stationary phase {Daicel Chiralpak IA, n-heptane/2-PrOH 95/5, 1.0 mL/min, 30 °C, UV 215 nm, t(ret)(enantiomer 1) = 26.4 min, t(ret)(enantiomer 2) = 28.3}: t(ret)(major isomer) = 28.7 min (catalyst 2), 28.7 min (catalyst 10), 28.7 min (catalyst 8b); ee = 96% for all catalysts. All other physical data have been reported previously.11
Computational Methods
Conformational searches of the catalyst and the substrates were performed with the COSMO-conf program27 at the BP8628,29/def-SVP22,23 level, and single points were calculated at the BP86/def-TZVP level. All calculations were run with the TURBOMOLE program (version 6.6),30 while the DLPNO–CCSD(T)/cc-pVTZ calculations were performed with ORCA 4.0.1.2.31 The geometries were optimized using the PBE functional,20,21 the def2-SVP basis set, and D3-dispersion correction. To reduce the computational cost, the RI-approximation was utilized. To model solvent effects, the geometries were reoptimized at the RI-PBE-D3/def2-SVP level employing the COSMO-solvation model for toluene (εr = 2.438 at 0 °C).24,25 Transition states were located using the TURBOMOLE’s woelfling program,32 followed by subsequent geometry optimization. Analytical normal modes were determined using the TURBOMOLE’s aoforce program for confirmation of the stationary points and transition state search. After scaling of the frequencies,33 the rigid-rotor harmonic oscillator (RRHO) approximation was used to calculate zero-point vibrational energies and thermal properties (at 4 °C). Single points of the transition states and minima were calculated with RI-PBE-D3/def2-TZVPPD (with and without computing solvation effects), B3LYP-D3/def2-TZVPPD, and DLPNO–CCSD(T)/cc-pVTZ (without solvation effects). Zero-point energies and thermal corrections for the COSMO-reoptimized structures and the single-point calculations were taken from the RI-PBE-D3/def2-SVP gas-phase calculations. COSMO corrected energies for the B3LYP28,34,35-D3/def2-TZVPPD and DLPNO–CCSD(T)36/cc-pVTZ37,38 were obtained from the RI-PBE-D3/def2-TZVPPD single-point calculations. All presented data are given on the DLPNO–CCSD(T)/cc-pVTZ ΔG + COSMO level unless otherwise noted.
Acknowledgments
M.L. and M.F. are grateful for a grant of the Austrian Science Fund (FWF, P 31276-B21). K. Faber, K. Zangger, B. Werner, E. Schwarz, J. Schachner, and R. Fischer are acknowledged for fruitful discussion. J. Goodman, J. Antilla, K. N. Houk, and M. Grayson are acknowledged for discussion of the content prior to submission.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.0c00992.
The authors declare no competing financial interest.
Supplementary Material
References
- Cheong P. H.-Y.; Legault C. Y.; Um J. M.; Çelebi-Ölçüm N.; Houk K. N. Quantum Mechanical Investigations of Organocatalysis: Mechanisms, Reactivities, and Selectivities. Chem. Rev. 2011, 111, 5042–5137. 10.1021/cr100212h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunoj R. B. Proline-derived organocatalysis and synergism between theory and experiments. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2011, 1, 920–931. 10.1002/wcms.37. [DOI] [Google Scholar]
- Sunoj R. B. Transition State Models for Understanding the Origin of Chiral Induction in Asymmetric Catalysis. Acc. Chem. Res. 2016, 49, 1019–1028. 10.1021/acs.accounts.6b00053. [DOI] [PubMed] [Google Scholar]
- Allemann C.; Gordillo R.; Clemente F. R.; Cheong P. H.-Y.; Houk K. N. Theory of Asymmetric Organocatalysis of Aldol and Related Reactions: Rationalizations and Predictions. Acc. Chem. Res. 2004, 37, 558–569. 10.1021/ar0300524. [DOI] [PubMed] [Google Scholar]
- Ramachandran P. V.; Gagare P. D.; Nicponski D. R.. 2.01 Allylborons. In Comprehensive Organic Synthesis II, 2nd ed. Knochel P., Ed.; Elsevier: Amsterdam, 2014; pp 1–71. [Google Scholar]
- Kim S. W.; Zhang W.; Krische M. J. Catalytic Enantioselective Carbonyl Allylation and Propargylation via Alcohol-Mediated Hydrogen Transfer: Merging the Chemistry of Grignard and Sabatier. Acc. Chem. Res. 2017, 50, 2371–2380. 10.1021/acs.accounts.7b00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denmark S. E.; Fu J. Catalytic Enantioselective Addition of Allylic Organometallic Reagents to Aldehydes and Ketones. Chem. Rev. 2003, 103, 2763–2794. 10.1021/cr020050h. [DOI] [PubMed] [Google Scholar]
- Lachance H.; Hall D. G. Allylboration of Carbonyl Compounds. Org. React. 2009, 73, 1–574. 10.1002/0471264180.or073.01. [DOI] [Google Scholar]
- Yamamoto H.; Wadamoto M. Silver-Catalyzed Asymmetric Allylation: Allyltrimethoxysilane as a Remarkable Reagent. Chem. - Asian J. 2007, 2, 692–698. 10.1002/asia.200600413. [DOI] [PubMed] [Google Scholar]
- Yus M.; González-Gómez J. C.; Foubelo F. Catalytic Enantioselective Allylation of Carbonyl Compounds and Imines. Chem. Rev. 2011, 111, 7774–7854. 10.1021/cr1004474. [DOI] [PubMed] [Google Scholar]
- Fuchs M.; Schober M.; Orthaber A.; Faber K. Asymmetric Synthesis of β-Substituted α-Methylenebutyrolactones via TRIP-Catalyzed Allylation: Mechanistic Studies and Application to the Synthesis of (S)-(−)-Hydroxymatairesinol. Adv. Synth. Catal. 2013, 355, 2499–2505. 10.1002/adsc.201300392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann P.; Lazzarotto M.; Steiner L.; Cigan E.; Poschenrieder S.; Sagmeister P.; Fuchs M. TRIP-Catalyzed Asymmetric Synthesis of (+)-Yatein, (−)-α-Conidendrin, (+)-Isostegane, and (+)-Neoisostegane. J. Org. Chem. 2019, 84, 5831–5837. 10.1021/acs.joc.9b00065. [DOI] [PubMed] [Google Scholar]
- Hoffmann S.; Seayad A. M.; List B. A Powerful Brønsted Acid Catalyst for the Organocatalytic Asymmetric Transfer Hydrogenation of Imines. Angew. Chem. 2005, 117, 7590–7593. 10.1002/ange.200503062. [DOI] [PubMed] [Google Scholar]
- Jain P.; Antilla J. C. Chiral Brønsted Acid-Catalyzed Allylboration of Aldehydes. J. Am. Chem. Soc. 2010, 132, 11884–11886. 10.1021/ja104956s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson M. N.; Pellegrinet S. C.; Goodman J. M. Mechanistic Insights into the BINOL-Derived Phosphoric Acid-Catalyzed Asymmetric Allylboration of Aldehydes. J. Am. Chem. Soc. 2012, 134, 2716–2722. 10.1021/ja210200d. [DOI] [PubMed] [Google Scholar]
- Rodríguez E.; Grayson M. N.; Asensio A.; Barrio P.; Houk K. N.; Fustero S. Chiral Brønsted Acid-Catalyzed Asymmetric Allyl(propargyl)boration Reaction of ortho-Alkynyl Benzaldehydes: Synthetic Applications and Factors Governing the Enantioselectivity. ACS Catal. 2016, 6, 2506–2514. 10.1021/acscatal.6b00209. [DOI] [Google Scholar]
- Wang H.; Jain P.; Antilla J. C.; Houk K. N. Origins of Stereoselectivities in Chiral Phosphoric Acid Catalyzed Allylborations and Propargylations of Aldehydes. J. Org. Chem. 2013, 78, 1208–1215. 10.1021/jo302787m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Lacasse M.-C.; Poulard C.; Charette A. B. Iodomethylzinc Phosphates: Powerful Reagents for the Cyclopropanation of Alkenes. J. Am. Chem. Soc. 2005, 127, 12440–12441. 10.1021/ja0529687. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple [Phys. Rev. Lett. 77, 3865 (1996)]. Phys. Rev. Lett. 1997, 78, 1396. 10.1103/PhysRevLett.78.1396. [DOI] [PubMed] [Google Scholar]
- Weigend F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. 10.1039/b515623h. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Klamt A.; Moya C.; Palomar J. A Comprehensive Comparison of the IEFPCM and SS(V)PE Continuum Solvation Methods with the COSMO Approach. J. Chem. Theory Comput. 2015, 11, 4220–4225. 10.1021/acs.jctc.5b00601. [DOI] [PubMed] [Google Scholar]
- Klamt A.; Schüürmann G. COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc., Perkin Trans. 2 1993, 2, 799–805. 10.1039/P29930000799. [DOI] [Google Scholar]
- Noyori R.; Kitamura M. Enantioselective Addition of Organometallic Reagents to Carbonyl Compounds: Chirality Transfer, Multiplication, and Amplification. Angew. Chem., Int. Ed. 1991, 30, 49–69. 10.1002/anie.199100491. [DOI] [Google Scholar]
- COSMOconf 4.1, COSMOlogic; GmbH & Co KG: http://www.cosmologic.de.
- Becke A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- Perdew J. P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. 10.1103/PhysRevB.33.8822. [DOI] [PubMed] [Google Scholar]
- TURBOMOLE 6.6. A Development of the University Ofkarlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007,TURBOMOLE GmbH, since 2007. 2015, http://www.turbomole.com.
- Neese F. Software update: the ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327 10.1002/wcms.1327. [DOI] [Google Scholar]
- Plessow P. Reaction Path Optimization without NEB Springs or Interpolation Algorithms. J. Chem. Theory Comput. 2013, 9, 1305–1310. 10.1021/ct300951j. [DOI] [PubMed] [Google Scholar]
- Kesharwani M. K.; Brauer B.; Martin J. M. L. Frequency and Zero-Point Vibrational Energy Scale Factors for Double-Hybrid Density Functionals (and Other Selected Methods): Can Anharmonic Force Fields Be Avoided?. J. Phys. Chem. A 2015, 119, 1701–1714. 10.1021/jp508422u. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Perspective on “Density functional thermochemistry. III. The role of exact exchange”. J. Chem. Phys. 1993, 98, 5648–5652. 10.1007/s002149900065. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Neese F.; Hansen A.; Liakos D. G. Efficient and accurate approximations to the local coupled cluster singles doubles method using a truncated pair natural orbital basis. J. Chem. Phys. 2009, 131, 064103 10.1063/1.3173827. [DOI] [PubMed] [Google Scholar]
- Dunning T. H. A Road Map for the Calculation of Molecular Binding Energies. J. Phys. Chem. A 2000, 104, 9062–9080. 10.1021/jp001507z. [DOI] [Google Scholar]
- Kendall R. A.; Dunning T. H. D. Jr.; Harrison R. J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. 10.1063/1.462569. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.