

Abstract
The diastereoselective synthesis of highly substituted β-lactams by intramolecular Tsuji–Trost allylation is reported. Judicious selection of the ligand on palladium allows selective access to either the trans isomer (in generally good to excellent yield with very high diastereomeric excess) or cis isomer (with yields and diastereoselectivity ranging from modest to excellent depending on the substrate). The reaction proceeds under exceedingly mild conditions (rt, no additives) with a broad range of substrates, which are readily accessible by the Ugi reaction.
Introduction
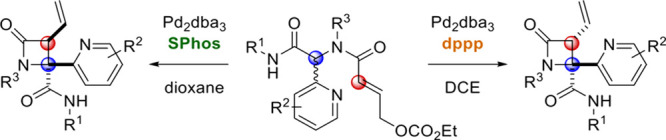
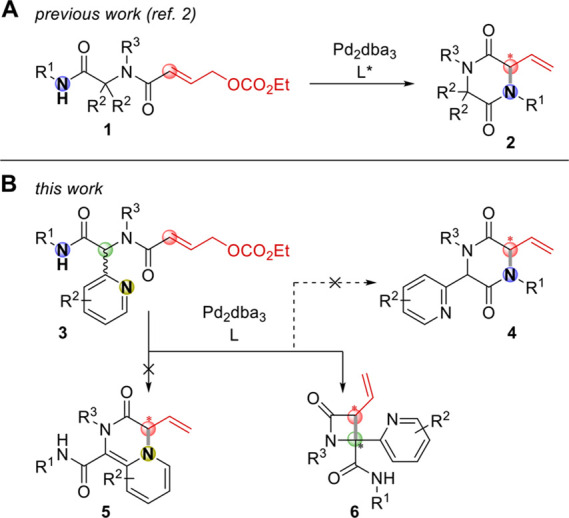
Transition metal-catalyzed formation of C–C, C–N, and C–O bonds has evolved into an essential tool for the construction of medicinally relevant heterocycles. In particular, the Tsuji–Trost reaction and related allylation reactions allow the efficient construction of C(sp3)–C(sp3) and C(sp3)–N bonds, resulting in highly substituted (hetero)cyclic frameworks when conducted in an intramolecular fashion.1 Recently, we demonstrated that diamides 1 functionalized with an allylic carbonate handle (readily produced by the Ugi four-component reaction [U4CR]) efficiently undergo catalytic asymmetric intramolecular allylation to give highly substituted diketopiperazines 2 (DKPs; Scheme 1A).2 In continuation of our work in this area, we realized that replacing symmetric (cyclic) ketones with heterocyclic aldehydes presented interesting opportunities for alternative cyclization modes (Scheme 1B). While formation of DKPs 4 may still occur, especially for small R1 substituents, the presence of the heterocyclic substituent significantly increases the α-acidity of the substrate, allowing cyclization of the π-allylpalladium intermediate either via the heterocyclic N atom (leading to 5, as reported by You et al. with iridium catalysis,3 albeit with a nonconjugated electrophile) or the Cα atom (giving 6). Preliminary experiments (see the Supporting Information) soon revealed that formation of 4 and 5 is outcompeted by the formation of β-lactams 6.
Scheme 1. Intramolecular Tsuji–Trost Reaction of Ugi Products for the Synthesis of Diverse Heterocycles.
It is hard to overstate the importance of β-lactams as antibiotics, with a prominent role for the penicillins and cephalosporins, as exemplified by penicillin G (I; Figure 1). The rise of antibiotic resistance against first-line antibiotics has led to the development of β-lactamase inhibitors such as aztreonam (II).4 In addition, the recently launched cholesterol absorption inhibitor ezetimibe (III) also features a β-lactam ring. Consequently, robust and efficient synthetic access to β-lactams with control over relative and absolute stereochemistry is in high demand.
Figure 1.
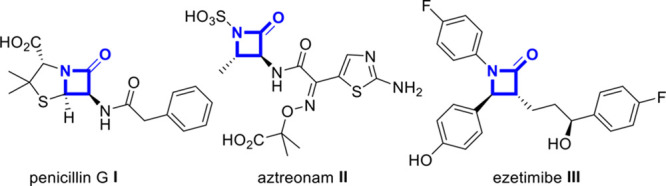
Pharmaceuticals containing β-lactam motifs.
Currently, the production of β-lactam drugs typically relies on semisynthesis starting from simplified penicillin and cephalosporin derivatives obtained by fermentation. In addition to the cyclization of β-amino acids,5 synthetic methods for the construction of β-lactams include the Staudinger reaction6 (i.e., the formal [2 + 2] cycloaddition of ketenes and imines), the Kinugasa reaction,7 intramolecular carbene insertion,8 and various other methods.9 In addition, several methods for the synthesis of β-lactams based on the U4CR have been reported,10−14 but these do not offer the same flexibility, substitution pattern, and stereocontrol. Given the mild conditions of our (likely kinetically controlled) reaction and opportunities to fine-tune the reaction outcome via the Pd ligand, we set about to investigate possibilities to control the relative stereochemistry of the process.
Results and Discussion
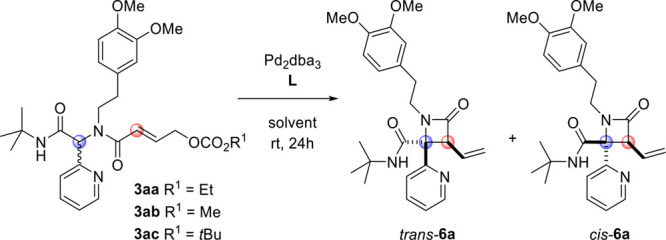
Fortuitously, we soon discovered that the ligand on Pd has a profound effect on the diastereoselectivity of our reaction (Table 1; see the Supporting Information for the full optimization study). Notably, we found that the use of monodentate phosphine ligands afforded predominantly the trans isomer, while the cis isomer was the major product when bidentate phosphines were used. Thus, while 6a was obtained as a 31:69 trans/cis diastereomeric mixture under the initial conditions (Pd2dba3, dppe, CH2Cl2, rt, 24 h, entry 1), simply replacing the dppe ligand with SPhos gave 6a in near-quantitative yield as a 91:9 mixture of diastereomers (entry 2). The stereoselectivity was lower in more polar solvents (DMF, MeCN; entries 3 and 4) but even higher in toluene and 1,4-dioxane (entries 5 and 6). Having selected the latter as the optimal solvent, we studied the influence of the carbonate leaving group. As in our previous work,2 we found that the ethyl carbonate is superior to the corresponding methyl and tert-butyl carbonates (3ab and 3ac, entries 7 and 8). Next, we focused on identifying conditions that allow selective access to the cis isomer of 6a. Testing several bidentate phosphine ligands, we noted a marked dependence on the bite angle, with dppp giving the highest selectivity (for details, see the Supporting Information). Thus, simply replacing dppe with dppp increased the diastereoselectivity from 31:69 to 11:89 (entry 9). Interestingly, in this case, solvents such as toluene and 1,4-dioxane gave lower selectivity (entries 10 and 11), while the selectivity was maintained in polar solvents (entries 12 and 13). Performing the reaction in 1,2-dichloroethane gave 6a with 9:91 dr, albeit still in only modest yield (entry 14). After observing various other reaction parameters not leading to either an improved yield or dr (see the Supporting Information), we next performed the reaction at different concentrations (entries 15–17) and found that the reaction performs optimally at 0.066 M. Again, the use of carbonates 3ab and 3ac offered no further improvement (entries 18 and 19).
Table 1. Optimization of Diastereoselectivitya.
| entry | substrate | ligand | solvent | yield (%)b | trans:cisc |
|---|---|---|---|---|---|
| 1 | 3aa | dppe | CH2Cl2 | 75 | 31:69 |
| 2 | 3aa | SPhos | CH2Cl2 | 99 | 91:9 |
| 3 | 3aa | SPhos | DMF | 91 | 62:38 |
| 4 | 3aa | SPhos | MeCN | 92 | 69:31 |
| 5 | 3aa | SPhos | PhMe | 91 | >95:5 |
| 6 | 3aa | SPhos | dioxane | 92 | >95:5 |
| 7 | 3ab | SPhos | dioxane | 87 | >95:5 |
| 8 | 3ac | SPhos | dioxane | 85 | 94:6 |
| 9 | 3aa | dppp | CH2Cl2 | 45 | 11:89 |
| 10 | 3aa | dppp | PhMe | 17 | 24:76 |
| 11 | 3aa | dppp | dioxane | n.d. | |
| 12 | 3aa | dppp | DMF | 30 | 11:89 |
| 13 | 3aa | dppp | MeCN | 47 | 11:89 |
| 14 | 3aa | dppp | DCE | 50 | 9:91 |
| 15d | 3aa | dppp | DCE | 42 | 10:90 |
| 16e | 3aa | dppp | DCE | 55 | 9:91 |
| 17f | 3aa | dppp | DCE | 70 | 9:91 |
| 18 | 3ab | dppp | DCE | 73 | 27:73 |
| 19 | 3ac | dppp | DCE | 70 | 10:90 |
Reagents and conditions: 3a (0.20 mmol), Pd2(dba)3 (0.01 mmol), monodentate (0.04 mmol), or bidentate (0.02 mmol) ligand in the indicated solvent (0.2 M).
Determined by 1H NMR analysis with an internal standard.
Determined by 1H NMR analysis of the crude product.
Performed at 0.4 M concentration.
Performed at 0.1 M concentration.
Performed at 0.066 M concentration. n.d., not detected. DCE, 1,2-dichloroethane.
Having identified conditions to selectively access either the trans isomer [conditions A: 5 mol % Pd2(dba)3, 20 mol % SPhos, 1,4-dioxane (0.2 M), rt, 24 h] or the cis isomer [conditions B: 5 mol % Pd2(dba)3, 10 mol % dppp, 1,2-dichloroethane (0.066 M), rt., 24 h], we set out to investigate the scope and limitations of the stereodivergent and chemoselective procedures with respect to R1 and R2 as well as the heterocyclic substituent. Thus, we studied the cyclization of diversely substituted Ugi products 3aa–3r under both conditions A and B (Scheme 2). To our delight, nearly all reactants were converted to the corresponding β-lactams 6a–6r under both sets of conditions, often with high selectivity and no trace of either diketopiperazines 4 or dearomatization products 5.
Scheme 2. Scope of the Reaction,,

Isolated yields.
Diastereomeric ratios are reported as trans/cis ratios as determined by 1H NMR analysis of the crude reaction product.
Yield of 3.0 mmol-scale experiment.
Reagents and conditions: A: 3 (0.20 mmol), Pd2(dba)3 (0.01 mmol), SPhos (0.04 mmol), 1,4-dioxane (1 mL), 24 h, rt. B: 3 (0.20 mmol), Pd2(dba)3 (0.01 mmol), dppp (0.02 mmol), 1,2-dichloroethane (3 mL), 24 h, rt.
Pyridine-substituted substrates 3aa–3d were converted to β-lactams 6a–6d in good to excellent yield with (nearly) complete selectivity for the trans isomer under conditions A.
Under conditions B, products 6a–6d were formed with moderate selectivity for the cis isomer in modest to reasonable yield. Notably, the cyclization of 3d (featuring a primary R1 substituent) was accompanied by the formation of the corresponding diketopiperazine 2d as a side product (15%, 67:33 dr) only under conditions B. Isoxazole-functionalized substrates 3e–3h were also converted to the corresponding β-lactams in modest to excellent yield, always with full diastereoselectivity under conditions A. Under conditions B, the yields were generally lower, and the selectivity for the cis isomer ranges from moderate (6e and 6f) to excellent (6g and 6h). The regioisomeric isoxazole substrate 3i was converted to 6i under both sets of conditions, in both cases with moderate selectivity for the expected diastereomer. Imidazole-substituted substrates 3j–3m were selectively converted to trans-β-lactams 6j–6m under conditions A, but no conversion took place under conditions B. The product 6l was an exception, being formed in good yield under conditions B, curiously with complete selectivity for the trans isomer. Isoquinolin-3-yl-substituted β-lactam 6n was also only formed under conditions A, still with excellent dr. Reactions of substrates with other heterocyclic substituents (3o–3q and 3s) or an ester (3r) all afforded the corresponding β-lactams in reasonable to excellent yield under both conditions A and B, but the selectivity was completely lost; nearly identical dr’s were observed regardless of the conditions. Apparently, the nature of the heterocyclic substituent plays a key role in the diastereoselection. X-ray crystallographic analysis of cis-6s15 confirmed the relative stereochemistry assigned by 1H NMR.16 With regard to the R1 and R2 substituents, the mild reaction conditions tolerate a wide variety of functional groups, including nitro groups, ethers, esters, amides, alkenes, alkynes, acetals, and aryl bromides. Intrigued by the stereodivergence of the reaction under conditions A and B, we sought to rationalize this remarkable difference in diastereoselectivity. We speculate that, in reactions employing monodentate ligands (i.e., conditions A), the heterocyclic N atom is actively involved in the mechanism, specifically by intramolecular coordination of the π-allylpalladium(II) complex (Figure 2), thus leading to the trans diastereoisomer (with a syn arrangement of the vinyl group and heterocycle). Indeed, pyridines and related N-heterocycles are common directing groups in Pd(II)-catalyzed C–H activation.17 However, we could not find any literature precedent of their use in directing reactions of π-allylpalladium(II) intermediates by intramolecular coordination. In contrast, with bidentate ligands (conditions B), the bidentate complex is proposed to remain intact throughout the catalytic cycle, with C–C bond formation occurring via an outer-sphere mechanism.
Figure 2.
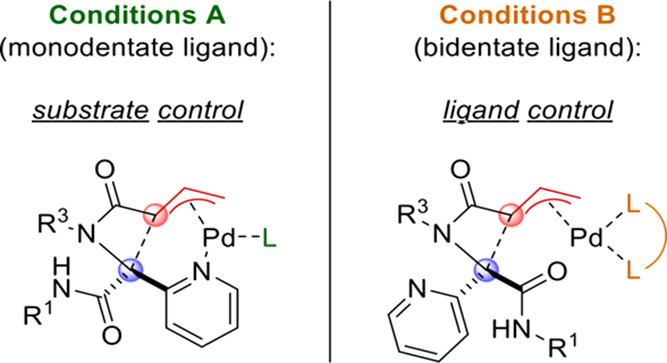
Proposed model for observed diastereodivergence.
This would also explain why substrates bearing heterocycles that are good potential Pd ligands (e.g., pyridines, isoxazoles, and imidazoles) react much more selectively under conditions A than those with heterocycles that are poor ligands for steric or electronic reasons (e.g., quinoline, quinoxaline, and pyrazine). To test this hypothesis, we reacted a series of substrates (3t–3zc) bearing electronically diverse 2-pyridyl moieties as well as substituents that cannot coordinate to the Pd center. Based on our mechanistic proposal (Figure 2), we expected that the introduction of electron-donating substituents on the pyridine ring would enhance its coordination capacity, thus leading to high trans selectivity under conditions A. Conversely, electron-withdrawing substituents would weaken the proposed interaction, leading to lower selectivity and/or yield. Under conditions B, we expected no particular influence of the substituents on the diastereoselectivity based on the proposed mechanism. On the other hand, electron-poor pyridines should increase the acidity of the α-proton, thereby increasing the reaction rate. The results (Scheme 3) are in line with our expectations. Substrates bearing relatively electron-rich pyridines were converted to the corresponding β-lactams (6t, 6w, and 6x) in excellent yield and selectivity under conditions A, while at best, poor conversion was observed under conditions B. Conversely, β-lactams 6u, 6v, 6y, and 6z bearing moderately electron-deficient pyridine rings were obtained in good yield under conditions B, while the selectivity (and in some cases the yield) was reduced under conditions A.
Scheme 3. Further Variation of the Pyridine Ring,
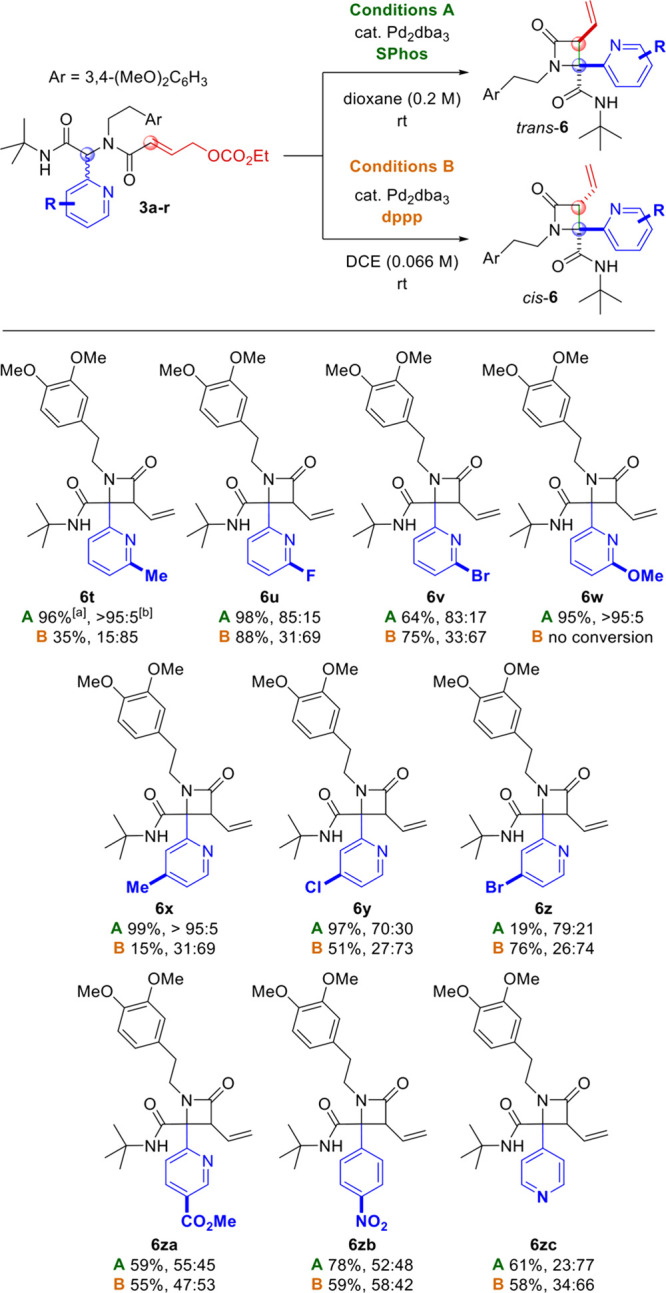
Isolated yields.
Diastereomeric ratios are reported as trans/cis ratios as determined by 1H NMR analysis of the crude reaction product.
Reagents and conditions: A: 3 (0.20 mmol), Pd2(dba)3 (0.01 mmol), SPhos (0.04 mmol), 1,4-dioxane (1 mL), 24 h, rt. B: 3 (0.20 mmol), Pd2(dba)3 (0.01 mmol), dppp (0.02 mmol), 1,2-dichloroethane (3 mL), 24 h, rt.
To gain further insight into the reaction mechanism, we next performed density functional theory (DFT) calculations at COSMO-ZORA-BLYP/TZ2P18 on the reaction of 3zd (Het = 2-pyridyl, R1 = R2 = Me) using ADF.19 Extensive benchmarking of ZORA-BLYP/TZ2P for oxidative addition to palladium shows that the reactivity trends compare very well with ab initio reference from hierarchical series up until CCSD(T).20 Furthermore, computed trends in reactivity for the studied reactions are the same across multiple level of theories, including when explicit dispersion corrections are applied (COSMO-ZORA-BLYP-D3(BJ)/TZ2P) and when energies are computed at the meta-hybrid level (COSMO-ZORA-M06/TZ2P//COSMO-ZORA-BLYP-D3(BJ)/TZ2P) (see Table S4 in the Supporting Information). An activation strain analysis21 was performed on the computed transition state structures using the PyFrag 2019 program (Figure S3).22
The potential energy surface (PES) in Figure 3 (left) reveals that the reactions involving the monodentate Pd catalyst (conditions A) favor the formation of the trans-β-lactam, which is the experimentally obtained diastereomer (kinetic control).23 The PES in Figure 3 (right) shows that the reactions involving the bidentate Pd catalyst (conditions B) favor the formation of the cis-β-lactam. This is also the experimentally obtained diastereomer (kinetically and thermodynamically favored). The mono-TS-trans is associated with a lower activation barrier than mono-TS-cis (ΔΔG‡ = 4.9 kcal mol–1), supporting the experimentally observed full selectivity for the trans isomer. The activation barrier for mono-TS-cis is higher due to a more destabilizing activation strain caused by a substantially later and more product-like transition state (Figure S3). The bi-TS-cis is associated with a lower barrier than bi-TS-trans (ΔΔG‡ = 3.7 kcal mol–1), again supporting the experimentally observed selectivity. The activation strain in both bi-TS-cis and bi-TS-trans is similar and only differs by 0.4 kcal mol–1. A close hydrogen bond contact (2.22 Å) between the carbonyl oxygen of the amide group and a phenyl group on the bidentate ligand was found in the bi-TS-cis, which was absent in bi-TS-trans (Figure 4).
Figure 3.

Reaction energy profiles and activation strain analyses for the (left) monodentate (conditions A) and (right) bidentate (conditions B) Pd-catalyzed reactions leading to the trans-β-lactam (black) and cis-β-lactam (gray) 3zd computed at COSMO-ZORA-BLYP/TZ2P.
Figure 4.
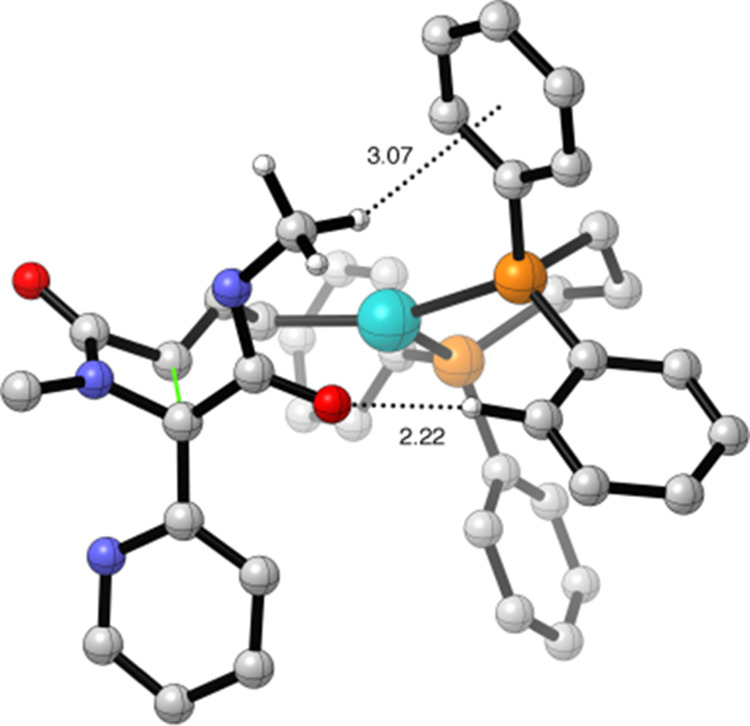
Stabilizing hydrogen bonding and C–H → π interactions in bi-TS-cis. All nonessential hydrogens are removed for clarity.
An additional stabilizing C–H → π interaction was identified between the methyl on the amide group and the bidentate ligand. These combined effects (likely even more so when the methyl group is replaced by a tert-butyl group) result in a more stabilizing interaction energy (ΔΔE‡int = –3.8 kcal mol–1) for bi-TS-cis compared to bi-TS-trans and thus a lower activation barrier of the former.
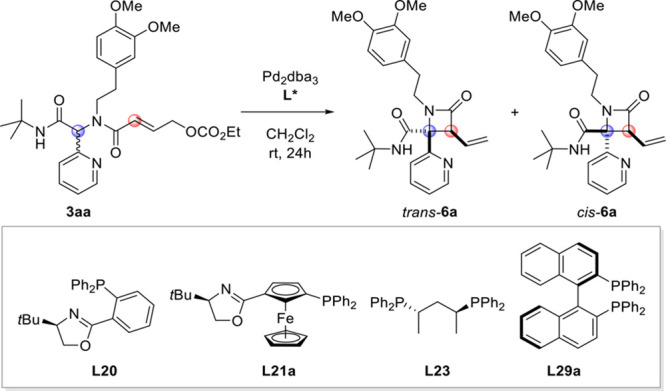
Encouraged by the possibilities to control the relative stereochemistry and to obtain additional insight into the mechanism, we tested a wide variety of chiral mono- and bidentate ligands in an attempt to also control the absolute stereochemistry of the product (see Table S3 for details). Unfortunately, our attempts met with limited success, and only very few ligands gave >50% ee (Table 2). For example, tBuPHOX (L20), which was highly efficient and selective in the intramolecular allylation of very similar substrates to give diketopiperazines,2 gave low ee for both the cis and trans isomers. In this class of ligands, the oxazoline nitrogen is proposed to act as a hemilabile ligand for palladium. In light of our mechanistic considerations, it is perhaps not surprising that L20 gives only low ee: the observed dr suggests it acts as a monodentate ligand, and the oxazoline is likely displaced by the pyridine of the substrate. In fact, our DFT calculations suggest that Pd already coordinates to the pyridine prior to generation of the π-allyl complex, which is the enantiodetermining step. Ligand L21a proved to be more efficient in controlling the absolute stereochemistry (72% ee for the trans isomer), albeit at the expense of the diastereoselectivity. Unfortunately, our attempts to further improve the ee remained fruitless. All chiral bisphosphine ligands appear to follow the bidentate scenario as described above, affording cis-6a as the main product. However, the conversion was very slow, resulting in modest yields even after 1 week of reaction time. (R)-BINAP (L29a) gave the highest ee for the cis isomer, but also in this case, any modifications made to the reaction conditions or the ligand structure proved to be counterproductive.
Table 2. Diastereoselectivity versus Enantioselectivitya.
| entry | ligand | yield (%)b | trans:cisc | eetransd | eecisd |
|---|---|---|---|---|---|
| 1 | L20 | 96 | 81:19 | 24% | –33% |
| 2 | L21a | 87 | 60:40 | –72% | –16% |
| 3 | L23 | 42 | 10:90 | n.d. | 20% |
| 4 | L29a | 22 | 14:86 | n.d. | –56% |
All reactions were performed with 3aa (0.20 mmol), Pd2(dba)3 (0.01 mmol), monodentate (0.04 mmol), or bidentate (0.02 mmol) ligand in CH2Cl2 (0.2 M).
Isolated yield.
Determined by 1H NMR analysis of the crude product.
Determined by chiral HPLC. Positive and negative signs refer to the earlier or later eluting enantiomer, respectively, being the major enantiomer. n.d., not determined.
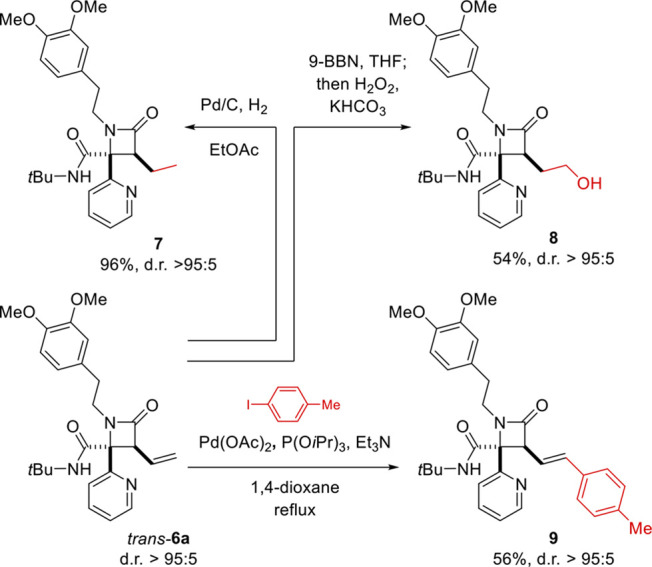
Finally, to further expand the range of accessible products, we performed some further transformations of the vinyl moiety of trans-6a (Scheme 4). Catalytic hydrogenation smoothly afforded 7 in excellent yield with full retention of the dr. Hydroboration/oxidation furnished 8 in moderate yield as a single diastereomer. Heck reaction with 4-iodotoluene afforded the cross-coupling product 9 without epimerization.
Scheme 4. Transformations of the Vinyl Moiety of trans-6a.
Conclusions
In conclusion, we developed a new ligand-directed stereodivergent synthesis of β-lactams by Pd-catalyzed intramolecular C(sp3)–C(sp3) bond formation. The divergent diastereoselective outcome of the reaction under different conditions is proposed to result from the presence or absence of intramolecular coordination of the Pd(II) π-allyl complex to the heterocyclic moiety. This hypothesis was supported by further studies and DFT calculations. The latter further indicated that the origin of diastereoselectivity in the monodentate scenario primarily results from a difference in activation strain between the two diastereomeric transition states, while a hydrogen bonding interaction in one of the diastereomeric transition states was found to be the origin of the diastereoselectivity in the bidentate scenario. The exceedingly mild reaction conditions tolerate a wide range of functional groups. The accessible product range of our method was further illustrated by various transformations of the vinyl moiety.
Experimental Section
General Information
Commercially available reagents were purchased from Sigma-Aldrich, Fischer, Strem Chemicals, or Fluorochem and were used as purchased, unless mentioned otherwise. Solvents were purchased from VWR Chemicals or Sigma-Aldrich and used without purification, unless stated otherwise. Anhydrous, air-free solvents were obtained from a PureSolv MD 5 solvent purification system. The infrared (IR) spectra were recorded neat using a Shimadzu FTIR-8400 s spectrophotometer and wavelengths are reported in cm–1. The nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance 600 (150.90 MHz for 13C), Bruker Avance 500 (125.78 MHz for 13C), or Bruker Avance 300 using the residual CHCl3 as the internal standard (1H: δ 7.26 ppm; 13C{1H}: δ 77.16 ppm). Chemical shifts (δ) are given in ppm and coupling constants (J) are quoted in Hertz (Hz). Resonances are described as s (singlet), d (doublet), t (triplet), q (quartet), br (broad singlet), and m (multiplet) or combinations thereof. Electrospray ionization (ESI) high-resolution mass spectrometry was carried out using a Bruker micrOTOF-Q instrument in positive ion mode (capillary potential of 4500 V). Flash chromatography was performed on Silicycle Silica-P Flash Silica Gel (particle size, 40–63 μm; pore diameter, 60 Å) using the indicated eluent. Thin-layer chromatography (TLC) was performed using TLC plates from Merck (SiO2, Kieselgel 60 F254 neutral, on aluminum with a fluorescence indicator) and compounds were visualized by UV detection (254 nm) and/or KMnO4 stain. SFC-MS analysis was conducted using a Shimadzu Nexera SFC-MS equipped with a Nexera X2 SIL-30 AC autosampler, Nexera UC LC-30 AD SF CO2 pump, Nexera X2 LC-30 AD liquid chromatograph, Nexera UC SFC-30A back pressure regulator, prominence SPD-M20A diode array detector, prominence CTO-20 AC column oven, and CBM-20A system controller. Enantiomeric excess was determined by SFC-MS analysis using Lux 3 μm Cellulose-1 column (cellulose tris(3,5-dimethylphenylcarbamate) (column 1) and Lux 3 μm Cellulose-3 column (cellulose tris(4-methylbenzoate), 150 × 4.6 mm) (column 3). A gradient of supercritical CO2 (A) and methanol (B) was used. Method 1 (column 1) was 2% B/98% A to 25% B/75% A over the course of 8 min and was maintained at 25% B/75% A for 1 min (flow: 1.5 mL/min). Method 2 (column 3) was 2% B/98% A to 7% B/93% A over the course of 6 min and then to 30% B/70% A over the course of 1 min and was maintained at 30% B/70% A for 1 min (flow: 1.5 mL/min). The sample injection volume was 5 μL. Mass spectrometry analyses were performed using a Shimadzu LCMS-2020 mass spectrometer. The data were acquired in full-scan APCI mode (MS) from m/z 100 to 800 in positive ionization mode. Data were processed using Shimadzu Labsolutions 5.82. Specific rotations were measured with an Automatic AA-10 polarimeter.
General Procedures
Procedure A: Synthesis of the Ugi Precursors (GP-A)
A solution of the corresponding aldehyde (3 mmol, 1 equiv) and amine (3 mmol, 1 equiv) in MeOH (1 M, 3 mL) was stirred for 30 min, then the carboxylic acid (3 mmol, 1 equiv) was added, and after 5 min, the corresponding isocyanide (3 mmol, 1 equiv) was added. The reaction mixture was stirred for 24 h or until full conversion of the starting material (monitored by TLC), concentrated, and purified by silica gel column chromatography, as described in the corresponding synthetic procedure.
Procedure B: Diastereoselective Tsuji–Trost Reaction Using Monodentate Ligand (SPhos) (GP-B)
A solution of Pd2(dba)3 (9 mg, 0.01 mmol, 0.05 equiv), SPhos (17 mg, 0.04 mmol, 0.2 equiv), and the corresponding linear precursor (0.2 mmol, 1 equiv) in dioxane (0.2 M, 1 mL) was stirred overnight or until full conversion of the starting material (monitored by TLC). Then, the reaction mixture was filtrated, concentrated, and purified by silica gel column chromatography, as described in the corresponding synthetic procedure.
Procedure C: Diastereoselective Tsuji–Trost Reaction Using Bidentate Ligand (dppp) (GP-C)
A solution of Pd2(dba)3 (9 mg, 0.01 mmol, 0.05 equiv), dppp (9 mg, 0.04 mmol, 0.2 equiv), and the corresponding linear precursor (0.2 mmol, 1 equiv) in DCE (0.066 M, 3 mL) was stirred overnight or until full conversion of the starting material (monitored by TLC). Then, the reaction mixture was filtrated, concentrated, and purified by silica gel column chromatography, as described in the corresponding synthetic procedure.
Ugi Product 3aa
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (40% EtOAc/cHex) afforded the title compound as a beige solid in 65% yield (1.020 g, 1.93 mmol). Rf = 0.36 (70% EtOAc/cHex). Two rotamers were present on NMR timescale (R1:R2 = 4:1), of which the signals of the major rotamer are reported. 1H NMR (600 MHz, CDCl3) δ 8.60 (d, J = 4.1 Hz, 1H), 7.70 (t, J = 7.4 Hz, 1H), 7.46 (d, J = 7.8 Hz, 1H), 7.24 (s, 1H), 7.08 (s, 1H), 6.91 (dt, J = 15.1, 4.4 Hz, 1H), 6.75 (d, J = 7.8 Hz, 1H), 6.66–6.60 (m, 2H), 6.52 (d, J = 15.1 Hz, 1H), 5.92 (s, 1H), 4.78 (d, J = 3.7 Hz, 2H), 4.25–4.18 (m, 2H), 3.84 (s, 3H), 3.83 (s, 3H), 3.75–3.66 (m, 2H), 2.82–2.73 (m, 1H), 2.55–2.47 (m, 1H), 1.38 (s, 9H), 1.31 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (150 MHz, CDCl3) δ 167.6, 166.5, 160.7, 156.4, 154.9, 149.1, 147.9, 138.9, 137.2, 130.9, 124.1, 123.0, 121.7, 120.8, 112.2, 111.5, 66.3, 64.8, 64.5, 56.0, 56.0, 51.7, 49.6, 36.2, 28.8 (3C), 14.4. IR (neat) νmax (cm–1): 3722, 3296, 2966, 2907, 1740, 1664, 1616, 1514, 1460, 1425, 1257, 1157, 1026, 995, 781, 770, 565, 426. m.p.: 129–134 °C. HRMS (ESI) m/z: [M + Na]+ calcd. for C28H37N3O7Na+, 550.2524; found, 550.2530.
Ugi Product 3ab
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (50% EtOAc/cHex) afforded the title compound as a colorless solid in 31% yield (0.471 g, 0.91 mmol). Rf = 0.31 (50% EtOAc/cHex). Two rotamers were present on NMR timescale (R1:R2 = 4:1), of which the signals of the major rotamer are reported. 1H NMR (600 MHz, CDCl3) δ 8.59 (d, J = 7.7 Hz, 1H), 7.70 (t, J = 7.6 Hz, 1H), 7.45 (d, J = 7.8 Hz, 1H), 7.24 (d, J = 7.3 Hz, 1H), 7.09 (s, 1H), 6.90 (dt, J = 15.1, 4.5 Hz, 1H), 6.74 (d, J = 8.0 Hz, 1H), 6.66–6.59 (m, 2H), 6.50 (d, J = 15.1 Hz, 1H), 5.93 (s, 1H), 4.78 (d, J = 4.5 Hz, 2H), 3.84 (s, 3H), 3.82 (s, 3H), 3.78 (s, 3H), 3.72–3.68 (m, 2H), 2.79–2.73 (m, 1H), 2.54–2.47 (m, 1H), 1.37 (s, 9H). 13C{1H}NMR (151 MHz, CDCl3) δ 167.5, 166.4, 156.3, 155.4, 149.1, 149.1, 147.8, 138.7, 137.2, 130.9, 124.1, 123.0, 121.6, 120.7, 112.2, 111.4, 66.5, 64.7, 56.0, 55.9, 55.1, 51.7, 49.6, 36.2, 28.8 (3C). IR (neat) νmax (cm–1): 3298, 2959, 1745, 1663, 1612, 1514, 1427, 1259, 1230, 1157, 1136, 1026, 953. HRMS (ESI) m/z: [M + H]+ calcd. for C27H36N3O7+, 514.2548; found. 514.2549.
Ugi Product 3ac
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (35% EtOAc/cHex) afforded the title compound as a light brown solid in 19% yield (0.320 g, 0.85 mmol). Rf = 0.57 (50% EtOAc/cHex). Two rotamers were present on NMR timescale (R1:R2 = 9:4), of which the signals of the major rotamer are reported. 1H NMR (600 MHz, CDCl3) δ 8.59 (d, J = 4.6 Hz, 1H), 7.70 (t, J = 7.2 Hz, 1H), 7.45 (d, J = 7.8 Hz, 1H), 7.24 (t, J = 6.5 Hz, 1H), 7.08 (s, 1H), 6.92 (dt, J = 15.1, 4.5 Hz, 1H), 6.74 (d, J = 8.0 Hz, 1H), 6.66–6.62 (m, 2H), 6.51 (d, J = 15.1 Hz, 1H), 5.91 (s, 1H), 4.71 (d, J = 4.5 Hz, 2H), 3.84 (s, 3H), 3.83 (s, 3H), 3.73–3.69 (m, 2H), 2.79 (t, J = 14.8 Hz, 1H), 2.56–2.50 (m, 1H), 1.47 (s, 9H), 1.38 (s, 9H). 13C{1H} NMR (151 MHz, CDCl3) δ 167.6, 166.5, 156.4, 153.1, 149.1, 149.0, 147.9, 139.4, 137.3, 130.9, 124.1, 123.0, 121.6, 120.8, 112.3, 111.5, 82.7, 65.6, 64.9, 56.0 (2C), 51.7, 49.6, 36.2, 28.8 (3C), 27.9 (3C). IR (neat) νmax (cm–1): 3298, 1732, 1666, 1618, 1516, 1421, 1281, 1252, 1236, 1159, 1134, 1030. HRMS (ESI) m/z: [M + H]+ calcd. for C30H42N3O7+, 556.3017; found, 556.3015.
Ugi Product 3b
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (75% EtOAc/cHex) afforded the title compound as a yellow solid in 59% yield (0.937 g, 1.75 mmol). Rf = 0.18 (70% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 8.52 (s, 1H), 8.49 (d, J = 4.5 Hz, 1H), 7.57 (td, J = 7.7, 1.7 Hz, 1H), 7.42 (d, J = 7.8 Hz, 1H), 7.38–7.28 (m, 2H), 7.21 (d, J = 8.0 Hz, 1H), 7.13 (dd, J = 7.1, 5.1 Hz, 1H), 7.04 (t, J = 7.5 Hz, 1H), 6.98 (t, J = 7.4 Hz, 1H), 6.84 (d, J = 1.7 Hz, 1H), 6.71 (dt, J = 15.1, 4.8 Hz, 1H), 6.24 (d, J = 15.1 Hz, 1H), 5.88 (s, 1H), 4.42 (d, J = 4.8 Hz, 2H), 4.07 (q, J = 7.1 Hz, 2H), 3.83–3.63 (m, 3H), 2.93–2.81 (m, 1H), 2.74–2.60 (m, 1H), 1.84 (s, 2H), 1.59–1.50 (m, 2H), 1.46 (dd, J = 8.9, 3.9 Hz, 1H), 1.30–1.20 (m, 2H), 1.18 (t, J = 7.1 Hz, 3H), 1.13–1.01 (m, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 167.6, 166.5, 156.2, 154.7, 149.0, 138.5, 137.3, 136.3, 127.2, 123.9, 123.00, 122.98, 121.9, 121.7, 119.4, 118.3, 111.8, 111.4, 66.2, 64.6, 64.4, 48.6, 48.5, 32.8, 32.7, 26.0, 25.6, 24.7 (2C), 14.3. IR (neat) νmax (cm–1): 2934, 1749, 1678, 1510, 1375, 1254, 1203, 1032. HRMS (ESI) m/z: [M + Na]+ calcd. for C30H36N4O5Na+, 555.2578; found, 555.2566.
Ugi Product 3c
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (60% EtOAc/cHex) afforded the title compound as a colorless solid in 51% yield (0.716 g, 1.53 mmol). Rf = 0.18 (70% EtOAc/cHex). Three rotamers were present on NMR timescale (R1:R2:R3 = 3:2:1), of which the signals of the major rotamer are reported. 1H NMR (500 MHz, CDCl3) δ 8.15 (d, J = 7.6 Hz, 1H), 7.55 (t, J = 8.0 Hz, 1H), 7.22 (d, J = 7.9 Hz, 1H), 7.09–7.04 (m, 1H), 6.92–6.83 (m, 1H), 6.68 (d, J = 15.2 Hz, 1H), 5.57 (s, 1H), 4.79 (d, J = 3.4 Hz, 2H), 4.21 (q, J = 7.1 Hz, 2H), 3.88–3.81 (m, 1H), 3.78–3.63 (m, 2H), 3.59–3.40 (m, 2H), 3.28 (s, 3H), 2.53 (s, 3H), 2.00–1.86 (m, 2H), 1.71–1.50 (m, 4H), 1.43–1.15 (m, 7H). 13C{1H} NMR (126 MHz, CDCl3) δ 167.2, 157.3, 155.7, 150.9, 143.9, 138.8, 137.9, 137.5, 122.5, 120.5, 71.0, 66.5, 64.9, 64.4, 59.0, 48.3, 48.1, 47.4, 32.9 (2C), 25.8 (2C), 24.5, 14.49. IR (neat) νmax (cm–1): 2932, 1746, 1665, 1622, 1452, 1248, 1115, 995, 7317. HRMS (ESI) m/z: [M + Na]+ calcd. for C24H35N3NaO6+, 484.2418; found, 484.2412.
Ugi Product 3d
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (75% EtOAc/cHex) afforded the title compound as a yellow oil in 60% yield (0.923 g, 1.80 mmol). Rf = 0.20 (70% EtOAc/cHex). Two rotamers were present on NMR timescale (R1:R2 = 7:1), of which the signals of the major rotamer are reported. 1H NMR (500 MHz, CDCl3) δ 8.45 (d, J = 4.1 Hz, 1H), 7.68–7.56 (m, 2H), 7.42–7.37 (m, 1H), 7.32–7.27 (m, 2H), 7.26–7.21 (m, 3H), 7.18–7.13 (m, 1H), 7.05 (d, J = 8.1 Hz, 2H), 6.98 (dt, J = 15.2, 4.8 Hz, 1H), 6.74 (d, J = 8.3 Hz, 2H), 6.52 (d, J = 15.1 Hz, 1H), 5.86 (s, 1H), 4.85–4.80 (m, 2H), 4.74 (d, J = 3.0 Hz, 2H), 4.56–4.42 (m, 2H), 4.16 (q, J = 7.2 Hz, 2H), 3.75 (s, 3H), 1.27 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 168.2, 167.1, 164.9, 159.0, 154.7, 148.5, 139.9, 138.3, 137.3, 128.9, 128.7 (2C), 128.2 (2C), 127.7 (2C), 127.4, 123.0, 121.9, 114.1 (2C), 114.0, 66.3, 64.5, 64.5, 55.4, 51.2, 43.8, 14.4. IR (neat) νmax (cm–1): 3300, 2957, 1744, 1664, 1612, 1512, 1433, 1244, 1202, 1175, 1028, 995. HRMS (ESI) m/z: [M + Na]+ calcd. for C29H31N3NaO6+, 540.2105; found, 540.2101.
Ugi Product 3e
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a yellow oil in 57% yield (1.005 g, 1.67 mmol). Rf = 0.71 (50% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 7.41 (d, J = 8.4 Hz, 2H), 7.07 (d, J = 8.3 Hz, 2H), 6.97 (dt, J = 15.1, 4.6 Hz, 1H), 6.31 (dd, J = 15.4, 1.8 Hz, 1H), 6.22 (s, 1H), 6.10 (s, 1H), 5.65 (s, 1H), 4.76–4.69 (m, 3H), 4.64 (d, J = 17.8 Hz, 1H), 4.14 (q, J = 7.1 Hz, 2H), 2.33 (s, 3H), 1.68 (d, J = 15.0 Hz, 1H), 1.62 (d, J = 17.2 Hz, 1H), 1.36 (s, 3H), 1.35 (s, 3H), 1.26 (t, J = 7.1 Hz, 3H), 0.92 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 170.2, 166.9, 165.2, 159.9, 154.6, 140.6, 135.9, 131.9 (2C), 128.5 (2C), 121.6, 121.3, 102.9, 66.1, 64.5, 57.3, 55.9, 52.2, 50.8, 31.7, 31.5 (3C), 28.9, 28.7, 14.3, 12.4. IR (neat) νmax (cm–1): 2953, 1747, 1666, 1624, 1398, 1366, 1252, 1227, 1203, 1011, 791. HRMS (ESI) m/z: [M + Na]+ calcd. for C28H38BrN3NaO6+, 614.1836; found, 614.1828.
Ugi Product 3f
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (70% EtOAc/cHex) afforded the title compound as an amber oil in 47% yield (0.894 g, 1.40 mmol). Rf = 0.13 (50% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 6.94 (dt, J = 15.2, 4.7 Hz, 1H), 6.67 (d, J = 7.9 Hz, 1H), 6.61 (dt, J = 15.0, 1.6 Hz, 1H), 6.13 (s, 1H), 5.33 (s, 1H), 4.80–4.77 (m, 2H), 4.52 (t, J = 5.1 Hz, 1H), 4.21 (q, J = 7.1 Hz, 2H), 4.03–3.89 (m, 3H), 3.63 (ddt, J = 14.2, 8.9, 7.1 Hz, 3H), 3.56–3.41 (m, 3H), 2.86 (s, 2H), 2.40 (s, 3H), 1.98–1.81 (m, 5H), 1.72 (s, 1H), 1.45–1.41 (m, 11H), 1.31 (d, J = 14.3 Hz, 3H), 1.19 (t, J = 7.1 Hz, 6H). 13C{1H} NMR (126 MHz, CDCl3) δ 170.2, 166.1, 166.0, 160.5, 154.7, 154.7, 139.8, 121.1, 102.6, 100.6, 79.6, 66.1, 64.4, 62.2, 61.9, 58.1, 47.2 (2C), 44.7, 33.6, 31.6 (4C), 28.4 (3C), 15.3, 15.3, 14.3, 12.4. IR (neat) νmax (cm–1): 2974, 1747, 1666, 1423, 1366, 1252, 1171, 1138, 1059, 1005, 7317. HRMS (ESI) m/z: [M + H]+ calcd. for C31H51N4O10+, 639.3600; found, 639.3605.
Ugi Product 3g
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a colorless waxy solid in 86% yield (1.094 mg, 2.58 mmol). Rf = 0.38 (50% EtOAc/cHex). 1H NMR (600 MHz, CDCl3) δ 6.94 (dt, J = 15.1, 4.6 Hz, 1H), 6.52–6.43 (m, 2H), 6.17 (s, 1H), 5.55 (s, 1H), 4.81 (d, J = 4.3 Hz, 2H), 4.22 (d, J = 7.2 Hz, 2H), 3.43 (t, J = 8.1 Hz, 2H), 2.42 (s, 3H), 1.67–1.57 (m, 1H), 1.47 (dq, J = 14.2, 6.9 Hz, 1H), 1.35–1.31 (m, 12H), 1.30–1.24 (m, 2H), 0.89 (t, J = 7.4 Hz, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ 170.0, 166.2, 166.1, 160.7, 154.8, 139.5, 121.5, 102.8, 66.2, 64.5, 58.0, 51.8, 48.5, 32.0, 28.7 (3C), 20.1, 14.4, 13.7, 12.5. IR (neat): νmax (cm–1): 3286, 2964, 2934, 2872, 1738, 1688, 1663, 1605, 1549, 1477, 1454, 1454, 1367, 1288, 1246, 1217, 1136, 995, 918, 851, 797, 662, 471, 401. HRMS (ESI) m/z: [M + Na]+ calcd. for C21H33N3NaO6+, 446.2262; found, 446.2268.
Ugi Product 3h
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (15% EtOAc/cHex) afforded the title compound as a light yellow solid in 56% yield (0.908 g, 1.69 mmol). Rf = 0.78 (50% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 7.65 (d, J = 8.0 Hz, 2H), 7.25 (d, J = 8.0 Hz, 2H), 7.01 (dt, J = 15.1, 4.4 Hz, 1H), 6.70 (s, 1H), 6.68 (d, J = 15.6 Hz, 1H), 6.46 (s, 1H), 6.07 (s, 1H), 4.84 (d, J = 3.2 Hz, 2H), 4.29 (s, 2H), 4.23 (q, J = 7.0 Hz, 2H), 2.39 (s, 3H), 2.30 (s, 1H), 1.76–1.66 (m, 2H), 1.44 (s, 3H), 1.43 (s, 3H), 1.33 (t, J = 7.1 Hz, 3H), 0.98 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 170.5, 166.5, 165.0, 160.2, 154.8, 140.8, 140.6, 129.8 (2C), 125.9 (2C), 124.5, 121.1, 100.2, 79.1, 73.8, 66.2, 64.6, 56.4, 56.1, 52.1, 37.3, 31.7, 31.5 (3C), 28.9, 28.9, 21.6, 14.4. IR (neat) νmax (cm–1): 2953, 1747, 1666, 1618, 1452, 1252, 1227, 1211, 1188, 822. HRMS (ESI) m/z: [M + Na]+ calcd. for C30H39N3NaO6+, 560.2731; found, 560.2721.
Ugi Product 3i
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a colorless solid in 59% yield (0.305 g, 0.59 mmol). Rf = 0.40 (50% EtOAc/cHex). 1H NMR (600 MHz, CDCl3) δ 7.72 (dd, J = 6.7, 3.0 Hz, 2H), 7.44–7.38 (m, 3H), 7.09 (d, J = 7.9 Hz, 2H), 7.00–6.93 (m, 3H), 6.71 (s, 1H), 6.51 (s, 1H), 6.23 (s, 1H), 5.87 (dt, J = 15.3, 1.7 Hz, 1H), 4.66 (dt, J = 5.0, 2.9, 1.7 Hz, 2H), 4.13 (q, J = 7.1 Hz, 2H), 2.30 (s, 3H), 1.40 (s, 9H), 1.25 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ 166.1, 165.9, 165.5, 162.5, 154.7, 139.5, 139.2, 136.1, 130.2 (2C), 130.2, 129.1 (2C), 129.0 (2C), 128.9, 126.9 (2C), 122.5, 104.8, 66.2, 64.4, 58.0, 52.0, 28.7 (3C), 21.3, 14.3. IR (neat) νmax (cm–1): 3313, 2972, 2935, 1740, 1693, 1668, 1605, 1541, 1512, 1464, 1447, 1371, 1269, 1246, 993, 764, 687, 590, 509. HRMS (ESI) m/z: [M + H]+ calcd. for C29H34N3O6+, 520.2442; found, 520.2463.
Ugi Product 3j
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (65% EtOAc/cHex) afforded the title compound as an off-white solid in 74% yield (0.991 g, 2.22 mmol). Rf = 0.11 (50% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 7.95–7.88 (m, 1H), 6.85–6.74 (m, 3H), 6.56 (s, 1H), 6.22 (d, J = 15.2 Hz, 1H), 4.66–4.61 (m, 2H), 4.52 (s, 1H), 4.32 (s, 1H), 4.06 (q, J = 7.1 Hz, 2H), 3.97 (d, J = 18.6 Hz, 1H), 3.81 (d, J = 18.6 Hz, 1H), 3.70–3.60 (m, 1H), 3.53 (s, 3H), 1.80 (d, J = 9.4 Hz, 1H), 1.70 (d, J = 11.4 Hz, 1H), 1.59–1.49 (m, 2H), 1.47–1.41 (m, 1H), 1.33 (s, 3H), 1.25–1.21 (m, 2H), 1.17 (t, J = 7.1 Hz, 3H), 1.08 (dq, J = 25.9, 11.9, 11.4 Hz, 3H).13C{1H} NMR (126 MHz, CDCl3) δ 166.7, 165.2, 154.5, 142.6, 141.0, 139.4, 127.5, 121.9, 121.6, 110.1, 66.0, 64.2, 51.8, 50.0, 48.5, 33.0, 32.5, 25.5 (2), 24.6 (2C), 19.8, 14.2. IR (neat) νmax (cm–1): 3259, 2932, 2853, 1745, 1651, 1618, 1562, 1377, 1249, 1202, 993. HRMS (ESI) m/z: [M + Na]+ calcd. for C23H34N4NaO5+, 469.2421; found, 469.2427.
Ugi Product 3k
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (50% EtOAc/cHex) afforded the title compound as a light yellow oil in 65% yield (0.935 g, 1.56 mmol). Rf = 0.38 (50% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 8.47 (s, 1H), 6.97–6.89 (m, 3H), 6.64 (s, 1H), 6.48 (d, J = 8.4 Hz, 1H), 6.34 (d, J = 2.0 Hz, 1H), 6.29 (d, J = 15.2 Hz, 1H), 6.25 (dd, J = 8.4, 1.9 Hz, 1H), 4.83–4.73 (m, 2H), 4.65 (d, J = 4.7 Hz, 2H), 4.42 (d, J = 18.0 Hz, 1H), 4.11 (q, J = 7.1 Hz, 2H), 3.78 (s, 3H), 3.73 (s, 3H), 1.76 (d, J = 14.9 Hz, 1H), 1.48 (d, J = 14.9 Hz, 1H), 1.38 (d, J = 6.6 Hz, 3H), 1.35 (d, J = 6.6 Hz, 3H), 1.29 (s, 3H), 1.27 (s, 3H), 1.24 (t, J = 7.1 Hz, 3H), 0.87 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 167.1, 164.8, 159.9, 157.2, 154.7, 142.2, 139.1, 128.04, 127.99, 122.4, 118.0, 116.3, 103.8, 98.1, 66.4, 64.3, 55.5, 55.4, 55.3, 53.2, 52.2, 47.4, 43.2, 31.6, 31.5 (3C), 28.5, 28.5, 24.7, 23.3, 14.3. IR (neat) νmax (cm–1): 2949, 1747, 1672, 1614, 1508, 1462, 1454, 1254, 1207, 1157, 1119, 1032, 7301. HRMS (ESI) m/z: [M + H]+ calcd. for C32H49N4O7+, 601.3596; found, 601.3588.
Ugi Product 3l
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (80% EtOAc/cHex) afforded the title compound as a brown oil in 65% yield (0.295 g, 0.65 mmol). Rf = 0.18 (100% EtOAc). 1H NMR (600 MHz, CDCl3) δ 8.43 (s, 1H), 7.01 (s, 1H), 6.95–6.89 (m, 2H), 6.78 (d, J = 15.3 Hz, 1H), 6.60 (s, 1H), 4.78 (d, J = 4.6 Hz, 2H), 4.20 (q, J = 7.1 Hz, 2H), 3.65–3.51 (m, 4H), 3.39–3.28 (m, 2H), 3.25 (s, 3H), 3.18 (s, 3H), 1.38 (s, 9H), 1.30 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ 167.7, 165.1, 154.9, 143.2, 138.8, 127.5, 123.1, 122.3, 103.5, 66.5, 64.4, 55.6, 55.4, 52.8, 51.8, 47.5, 33.2, 28.8 (3C), 14.4. IR (neat) νmax (cm–1): 3298, 2968, 2920, 1749, 1659, 1616, 1553, 1454, 1423, 1394, 1367, 1254, 1223, 1176, 1121, 1051, 989, 920, 795, 756, 602, 401. m.p.: 111–112 °C. HRMS (ESI) m/z: [M + H]+ calcd. for C21H35N4O7+, 455.2500; found, 455.2510.
Ugi Product 3m
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (60% EtOAc/cHex) afforded the title compound as a colorless solid in 37% yield (0.587 g, 1.11 mmol). Rf = 0.45 (70% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 8.46 (s, 1H), 7.23–7.11 (m, 3H), 7.05–6.99 (m, 3H), 6.96 (dt, J = 15.4, 4.4 Hz, 1H), 6.89 (d, J = 1.0 Hz, 1H), 6.55 (s, 1H), 6.52 (dt, J = 15.4, 1.8 Hz, 1H), 4.76 (dd, J = 4.4, 1.8 Hz, 2H), 4.17 (q, J = 7.1 Hz, 2H), 3.62 (ddd, J = 16.7, 11.6, 4.7 Hz, 1H), 3.58 (s, 3H), 3.42 (dt, J = 15.4, 5.8 Hz, 1H), 2.57 (td, J = 12.5, 5.3 Hz, 1H), 1.98 (td, J = 13.1, 12.5, 5.4 Hz, 1H), 1.80 (d, J = 14.8 Hz, 1H), 1.62 (d, J = 14.8 Hz, 1H), 1.42 (s, 3H), 1.40 (s, 3H), 1.27 (t, J = 7.1 Hz, 3H), 0.92 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 165.8, 164.8, 154.6, 142.9, 139.5, 137.9, 128.6 (2C), 128.5 (2C), 127.4, 126.5, 122.2, 120.7, 66.0, 64.3, 55.5, 52.4, 52.0, 46.9, 36.2, 33.0, 31.5, 31.4 (3C), 28.8, 28.6, 14.2. IR (neat) νmax (cm–1): 3306, 2949, 1738, 1657, 1610, 1547, 1421, 1281, 1250, 1178, 1032, 762, 746. HRMS (ESI) m/z: [M + H]+ calcd. for C29H43N4O5+, 527.3228; found, 527.3216.
Ugi Product 3n
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (40% EtOAc/cHex) afforded the title compound as a brown oil in 63% yield (0.318 mg, 0.63 mmol). Rf = 0.35 (70% EtOAc/cHex). 1H NMR (600 MHz, CDCl3) δ 9.15 (s, 1H), 7.92 (d, J = 8.2 Hz, 1H), 7.82 (s, 1H), 7.76 (d, J = 8.3 Hz, 1H), 7.65 (t, J = 7.5 Hz, 1H), 7.58 (t, J = 7.5 Hz, 1H), 7.38 (s, 1H), 7.17–7.10 (m, 2H), 7.04 (d, J = 8.0 Hz, 2H), 6.86 (dt, J = 15.3, 5.2 Hz, 1H), 6.10 (s, 1H), 5.89 (d, J = 15.3 Hz, 1H), 4.60 (d, J = 5.2 Hz, 2H), 4.09 (q, J = 7.1 Hz, 2H), 2.27 (s, 3H), 1.28 (s, 9H), 1.22 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ 167.3, 165.5, 154.6, 151.7, 148.8, 138.1, 137.93, 137.91, 136.3, 130.7, 129.7 (2C), 127.74, 127.73, 127.4 (2C), 127.2 (2C), 123.5, 121.5, 68.0, 66.3, 64.2, 51.4, 28.6 (3C), 21.1, 14.3. IR (neat) νmax (cm–1): 3325, 2970, 2928, 1670, 1628, 1543, 1510, 1450, 1366, 1248, 1005, 960, 789, 754, 604, 532, 472. M.p.: 136–138 °C. HRMS (ESI) m/z: [M + Na]+ calcd. for C29H33N3O5Na+, 526.2312; found, 526.2318.
Ugi Product 3o
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (40% EtOAc/cHex) afforded the title compound as an orange solid in 45% yield (0.406 g, 0.89 mmol). Rf = 0.23 (50% EtOAc/cHex). Two rotamers were present on NMR timescale (R1:R2 = 4:1), of which the signals of the major rotamer are reported. 1H NMR (600 MHz, CDCl3) δ 13.06 (s, 1H), 8.00 (d, J = 2.1 Hz, 1H), 7.60 (d, J = 7.9 Hz, 1H), 7.42–7.32 (m, 1H), 7.18–7.12 (m, 1H), 7.09 (dd, J = 8.1, 0.9 Hz, 1H), 6.98 (dt, J = 15.3, 5.0 Hz, 1H), 6.30 (dt, J = 15.3, 1.7 Hz, 1H), 6.04 (ddt, J = 17.1, 10.0, 7.1 Hz, 1H), 5.50 (s, 1H), 5.32–5.27 (m, 1H), 5.25–5.20 (m, 1H), 4.74–4.64 (m, 2H), 4.30 (dd, J = 13.8, 7.0 Hz, 1H), 4.09 (qd, J = 7.1, 1.9 Hz, 2H), 4.04 (dd, J = 13.7, 7.2 Hz, 1H), 1.37 (s, 9H), 1.22 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ 169.0, 168.0, 154.7, 146.9, 140.1, 139.8, 134.1, 132.1, 131.3, 130.1, 129.4, 123.3, 121.7, 121.1, 115.7, 66.1, 64.4, 62.8, 53.0, 51.6, 29.2 (3C), 14.3. IR (neat) νmax (cm–1): 3304, 2968, 2930, 1744, 1663, 1628, 1556, 1456, 1406, 1364, 1261, 1200, 1119, 987, 945, 793, 762, 677, 507. m.p.: 101–107 °C. HRMS (ESI) m/z: [M + H]+ calcd. for C24H31N4O5+, 455.2289; found, 455.2276.
Ugi Product 3p
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (20% EtOAc/cHex) afforded the title compound as an orange solid in 87% yield (1.550 mg, 2.61 mmol). Rf = 0.73 (70% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 13.70 (s, 1H), 7.44–7.38 (m, 3H), 7.33 (d, J = 7.6 Hz, 1H), 7.27 (d, J = 7.7 Hz, 2H), 7.24–7.18 (m, 2H), 7.06 (t, J = 7.5 Hz, 1H), 6.96 (dt, J = 15.3, 5.2 Hz, 1H), 6.39–6.33 (m, 1H), 6.10 (dd, J = 9.6, 1.6 Hz, 1H), 5.11 (d, J = 13.3 Hz, 1H), 4.94 (s, 1H), 4.66 (d, J = 5.0 Hz, 2H), 4.21 (d, J = 13.3 Hz, 1H), 4.13–4.04 (m, 2H), 1.67 (d, J = 14.8 Hz, 1H), 1.30 (s, 3H), 1.24–1.18 (m, 4H), 1.15 (s, 3H), 0.89 (s, 9H). 13C{1H} NMR (151 MHz, CDCl3) δ 169.0, 168.8, 154.7, 148.0, 139.6, 137.9, 136.2, 135.3, 134.2, 131.9 (2C), 131.1, 129.1 (2C), 127.7, 122.4, 122.1, 120.8, 117.0, 116.3, 96.9, 66.3, 64.3, 55.0, 53.0, 52.4, 31.6 (3C), 29.3, 28.4 (2C), 14.3. IR (neat) νmax (cm–1): 3350, 2959, 2943, 1742, 1622, 1585, 1506, 1443, 1373, 1250, 1213, 1148, 1094, 987, 847, 802, 748, 613. m.p.: 106–108 °C. HRMS (ESI) m/z: [M + H]+ calcd. for C33H41ClN3O5+, 594.2729; found, 594.2737.
Ugi Product 3q
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (60% EtOAc/cHex) afforded the title compound as an amber oil in 56% yield (0.880 g, 1.67 mmol). Rf = 0.22 (50% EtOAc/cHex). Two rotamers were present on NMR timescale (R1:R2 = 5:1), of which the signals of the major rotamer are reported. 1H NMR (500 MHz, CDCl3) δ 7.62 (d, J = 3.2 Hz, 1H), 7.45 (d, J = 8.1 Hz, 2H), 7.25–7.20 (m, 4H), 6.97 (dt, J = 15.1, 4.5 Hz, 1H) 6.28 (d, J = 15.2 Hz, 1H), 6.15 (s, 1H), 4.86 (d, J = 17.6 Hz, 1H), 4.78 (d, J = 17.6 Hz, 1H), 4.67 (dd, J = 4.3, 1.6 Hz, 2H), 4.05 (q, J = 7.1 Hz, 2H), 1.25 (s, 9H), 1.17 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 167.0, 165.6, 164.3, 154.5, 142.2, 140.95, 140.91, 129.6 (q, J = 32.4 Hz), 126.9 (2C), 125.4 (q, J = 3.6 Hz, 2H), 124.0 (q, J = 272.0 Hz), 121.1, 120.8, 65.9, 64.3, 59.9, 51.8, 50.2, 28.4 (3C), 14.1. IR (neat) νmax (cm–1): 1747, 1666, 1323, 1254, 1202, 1163, 1121, 1113, 1067, 1016, 733, 625, 590. HRMS (ESI) m/z: [M + Na]+ calcd. for C24H28F3N3NaO5S+, 550.1594; found, 550.1592.
Ugi Product 3r
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a clear oil in 37% yield (0.367 g, 0.73 mmol). Rf = 0.31 (50% EtOAc/cHex). 1H NMR (600 MHz, CDCl3) δ 7.50 (d, J = 7.8 Hz, 1H), 7.25 (d, J = 8.5 Hz, 2H), 6.94 (dd, J = 15.2, 4.5 Hz, 1H), 6.85 (d, J = 8.5 Hz, 2H), 6.49 (d, J = 15.2 Hz, 1H), 4.82 (d, J = 16.7 Hz, 1H), 4.75 (d, J = 4.5 Hz, 2H), 4.62 (d, J = 16.7 Hz, 1H), 4.46 (s, 1H), 4.25–4.07 (m, 4H), 3.77 (s, 3H), 3.74–3.68 (m, 1H), 1.81 (t, J = 13.9 Hz, 2H), 1.68–1.59 (m, 1H), 1.56–1.49 (m, 1H), 1.34–1.24 (m, 6H), 1.23–1.13 (m, 6H). 13C{1H} NMR (151 MHz, CDCl3) δ 167.6, 167.0, 164.8, 159.3, 154.6, 140.1, 128.7 (2C), 127.7, 120.9, 114.1 (2C), 66.0, 64.4, 63.8, 62.0, 55.3, 52.7, 48.4, 32.6, 32.4, 25.5 (2C), 24.5, 14.2, 13.9. IR (neat) νmax (cm–1): 3306, 2979, 2932, 2854, 1745, 1664, 1616, 1539, 1514, 1447, 1371, 1246, 1203, 1176, 1097, 1028, 966, 868, 818, 791, 555, 509, 459. HRMS (ESI) m/z: [M + H]+ calcd. for C26H37F3N2O8+, 505.2544; found, 505.2541.
Ugi Product 3s
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (40% EtOAc/cHex) afforded the title compound as a yellow solid in 78% yield (1.236 g, 2.09 mmol). Rf = 0.34 (60% EtOAc/cHex). 1H NMR (600 MHz, CDCl3) δ 8.96 (s, 1H), 8.60 (s, 1H), 8.50 (d, J = 5.3 Hz, 2H), 7.31–7.27 (m, 12H), 7.25–7.21 (m, 3H), 6.90 (dt, J = 15.6, 4.5 Hz, 1H), 6.50 (d, J = 15.6 Hz, 1H), 5.54 (s, 1H), 4.81 (d, J = 4.5 Hz, 2H), 4.23 (q, J = 7.1 Hz, 2H), 3.52–3.34 (m, 2H), 1.58–1.46 (m, 1H), 1.43–1.35 (m, 1H), 1.33 (t, J = 7.1 Hz, 3H), 0.80 (t, J = 7.3 Hz, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ 166.8, 166.3, 154.8, 152.0, 144.6, 144.5 (3C), 143.6, 143.1, 139.8, 128.8 (6C), 128.0 (6C), 127.0 (3C), 121.4, 70.9, 66.2, 65.7, 64.5, 51.5, 23.3, 14.4, 11.3. IR (neat) νmax (cm–1): 3722, 3263, 2974, 2303, 1745, 1688, 1672, 1599, 1529, 1433, 1366, 1248, 1219, 1030, 756, 698, 633, 590, 430. m.p.: 110–114 °C. HRMS (ESI) m/z: [M + H]+ calcd. for C35H37N4O5+, 593.2578; found, 593.2588.
Ugi Product 3t
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (40% EtOAc/cHex) afforded the title compound as a beige solid in 70% yield (1.134 g, 2.10 mmol). Rf = 0.30 (70% EtOAc/cHex). Two rotamers were present on NMR timescale (R1:R2 = 7:3), of which the signals of the major rotamer are reported. 1H NMR (500 MHz, CDCl3) δ 7.78 (s, 1H), 7.58 (t, J = 7.2 Hz, 1H), 7.23 (d, J = 7.5 Hz, 1H), 7.12–7.06 (m, 1H), 6.90 (dt, J = 15.1, 4.4 Hz, 1H), 6.79–6.70 (m, 1H), 6.66–6.61 (m, 2H), 6.50 (d, J = 15.1 Hz, 1H), 5.86 (s, 1H), 4.77 (d, J = 4.4 Hz, 2H), 4.20 (q, J = 7.1 Hz, 2H), 3.87–3.81 (m, 6H), 3.67 (dt, J = 14.0, 7.3 Hz, 2H), 2.83–2.74 (m, 1H), 2.60–2.53 (m, 4H), 1.38 (s, 9H), 1.30 (d, J = 7.1 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 167.2, 166.3, 157.5, 155.8, 154.8, 149.0, 147.8, 138.7, 137.6, 131.0, 123.2, 122.4, 121.7, 120.7, 112.1, 111.3, 66.3, 64.5, 64.2, 56.0, 56.0, 51.5, 49.6, 36.2, 28.8 (3C), 24.5, 14.4. IR (neat) νmax (cm–1): 3304, 2959, 2932, 1742, 1664, 1624, 1560, 1514, 1456, 1416, 1364, 1246, 1153, 1024, 959, 864, 787, 644, 557, 473. m.p.: 118–121 °C. HRMS (ESI) m/z: [M + Na]+ calcd. for C29H39N3O7Na+, 564.2680; found, 564.2701.
Ugi Product 3u
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (40% EtOAc/cHex) afforded the title compound as a colorless solid in 81% yield (1.330 g, 2.44 mmol). Rf = 0.23 (50% EtOAc/cHex). 1H NMR (600 MHz, CDCl3) δ 7.79 (q, J = 7.9 Hz, 1H), 7.33 (d, J = 7.1 Hz, 1H), 6.92–6.87 (m, 2H), 6.76 (d, J = 8.1 Hz, 2H), 6.71 (d, J = 1.8 Hz, 1H), 6.68 (d, J = 8.1 Hz, 1H), 6.50 (d, J = 15.1 Hz, 1H), 5.77 (s, 1H), 4.78 (s, 2H), 4.21 (q, J = 7.1 Hz, 2H), 3.86 (s, 3H), 3.84 (s, 3H), 3.73 (d, J = 8.5 Hz, 2H), 2.81 (t, J = 14.0, 8.5 Hz, 1H), 2.62 (dd, J = 14.0, 8.5 Hz, 1H), 1.38 (s, 9H), 1.31 (d, J = 7.1 Hz, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ 167.3, 166.6, 162.9 (d, J = 241.5 Hz), 154.9 (d, J = 9.8 Hz), 154.8, 149.2, 147.9, 142.0 (d, J = 7.3 Hz), 139.3, 130.7, 121.4, 121.0 (d, J = 3.8 Hz), 120.8, 112.2, 111.5, 108.9 (d, J = 36.6 Hz), 66.2, 65.1, 64.5, 56.03 55.98, 51.8, 50.1, 36.2, 28.7 (3C), 14.4. IR (neat) νmax (cm–1): 3290, 2966, 1742, 1664, 1618, 1574, 1555, 1514, 1450, 1425, 1362, 1254, 1240, 1157, 1024, 991, 933, 878, 787, 764, 656, 577, 555, 467, 432.03. m.p.: 122–128 °C. HRMS (ESI) m/z: [M + Na]+ calcd. for C28H36N3O7FNa+, 568.2429; found, 568.2446.
Ugi Product 3v
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (40% EtOAc/cHex) afforded the title compound as a colorless solid in 68% yield (1.241 g, 2.05 mmol). Rf = 0.42 (50% EtOAc/cHex). 1H NMR (600 MHz, CDCl3) δ 7.55 (t, J = 7.8 Hz, 1H), 7.41 (dd, J = 16.8, 7.7 Hz, 2H), 7.04 (s, 1H), 6.90 (dt, J = 15.1, 4.5 Hz, 1H), 6.77 (d, J = 8.2 Hz, 1H), 6.70 (d, J = 6.5 Hz, 2H), 6.50 (d, J = 15.1 Hz, 1H), 5.75 (s, 1H), 4.78 (d, J = 4.5 Hz 2H), 4.22 (q, J = 7.1 Hz, 2H), 3.86 (s, 3H), 3.84 (s, 3H), 3.71 (t, J = 7.9 Hz, 2H), 2.81 (dt, J = 15.1, 7.8 Hz, 1H), 2.65 (dt, J = 14.6, 7.8 Hz, 1H), 1.38 (s, 9H), 1.31 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ 166.8, 166.5, 157.6, 154.8, 149.2, 147.9, 141.2, 139.4, 139.2, 130.7, 127.3, 122.7, 121.5, 120.9, 112.2, 111.5, 66.2, 65.0, 64.5, 56.0, 51.8, 50.2, 36.2, 28.8 (3C), 14.4. IR (neat) νmax (cm–1): 3304, 2966, 2908, 1744, 1663, 1628, 1555, 1514, 1445, 1414, 1259, 1232, 1157, 1130, 1028, 997, 787, 741, 640, 567, 434. m.p.: 130–132 °C. HRMS (ESI) m/z: [M + Na]+ calcd. for C28H36O7N3BrNa+, 628.1629; found, 628.1633.
Ugi Product 3w
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (40% EtOAc/cHex) afforded the title compound as a beige solid in 87% yield (1.456 g, 2.61 mmol). Rf = 0.33 (50% EtOAc/cHex). Two rotamers were present on NMR timescale (R1:R2 = 4:1), of which the signals of the major rotamer are reported. 1H NMR (600 MHz, CDCl3) δ 7.58 (t, J = 7.8 Hz, 1H), 7.01 (d, J = 7.2 Hz, 1H), 6.93 (dt, J = 15.1, 4.4 Hz, 1H), 6.73 (dd, J = 16.9, 8.2 Hz, 2H), 6.59 (d, J = 8.2 Hz, 1H), 6.55 (d, J = 13.0 Hz, 2H), 6.40 (s, 1H), 5.92 (s, 1H), 4.80 (d, J = 4.5 ,2H), 4.21 (q, J = 7.1 Hz, 2H), 3.87 (s, 3H), 3.83 (s, 6H), 3.73 (t, J = 8.3 Hz, 2H), 2.77 (dt, J = 15.7, 8.3 Hz, 1H), 2.35 (dd, J = 15.7, 8.3 Hz, 1H), 1.37 (s, 9H), 1.30 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ 167.7, 166.3, 163.7, 154.8, 153.3, 149.1, 147.8, 139.6, 138.9, 130.9, 121.6, 120.6, 116.8, 112.0, 111.4, 110.5, 66.3, 64.45, 64.43, 56.0, 55.9, 53.5, 51.6, 49.3, 36.4, 28.8 (3C), 14.4. IR (neat) νmax (cm–1): 3306, 2968, 2935, 1747, 1686, 1663, 1595, 1553, 1514, 1460, 1420, 1254, 1157, 1026, 991, 816, 789, 733, 648, 567, 548. m.p.: 99–101 °C. HRMS (ESI) m/z: [M + Na]+ calcd. for C29H39N3O8Na+, 580.2629; found, 580.2655.
Ugi Product 3x
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (40% EtOAc/cHex) afforded the title compound as a beige solid in 66% yield (1.079 g, 2.00 mmol). Rf = 0.31 (70% EtOAc/cHex). Two rotamers were present on NMR timescale (R1:R2 = 3:1), of which the signals of the major rotamer are reported. 1H NMR (500 MHz, CDCl3) δ 8.44 (d, J = 4.5 Hz, 1H), 7.25 (s, 1H), 7.17 (s, 1H), 7.07 (d, J = 4.5 Hz, 1H), 6.92 (dd, J = 15.1, 3.9 Hz, 1H), 6.74 (d, J = 7.9 Hz, 1H), 6.66–6.60 (m, 2H), 6.52 (d, J = 15.1 Hz, 1H), 5.93 (s, 1H), 4.78 (d, J = 3.9 Hz, 2H), 4.20 (q, J = 7.1 Hz, 2H), 3.84 (s, 3H), 3.83 (s, 3H), 3.69 (tt, J = 15.4, 7.9 Hz, 2H), 2.77 (td, J = 13.7, 10.3, 6.1 Hz, 1H), 2.50 (ddd, J = 13.7, 10.3, 6.1 Hz, 1H), 2.34 (s, 3H), 1.37 (s, 9H), 1.30 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 167.6, 166.3, 156.3, 156.1, 154.8, 149.0, 148.7, 147.8, 138.8, 130.9, 125.1, 124.1, 121.6, 120.7, 112.2, 111.3, 66.3, 64.5, 64.3, 56.0, 55.9, 51.6, 49.4, 36.2, 28.8 (3C), 21.4, 14.4. IR (neat) νmax (cm–1): 3300, 2968, 2930, 1742, 1680, 1599, 1545, 1514, 1431, 1259, 1155, 1026, 820, 781, 646, 569, 467, 430. m.p.: 118–121 °C. HRMS (ESI) m/z: [M + Na]+ calcd. for C29H39N3O7Na+, 564.2680; found, 564.2683.
Ugi Product 3y
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (40% EtOAc/cHex) afforded the title compound as a colorless solid in 83% yield (1.401 g, 2.50 mmol). Rf = 0.34 (70% EtOAc/cHex). Two rotamers were present on NMR timescale (R1:R2 = 7:1), of which the signals of the major rotamer are reported. 1H NMR (500 MHz, CDCl3) δ 8.46 (d, J = 5.2 Hz, 1H), 7.43 (s, 1H), 7.24 (dd, J = 5.2, 1.6 Hz, 1H), 7.04 (s, 1H), 6.92 (dt, J = 15.2, 4.5 Hz, 1H), 6.75 (d, J = 8.6 Hz, 1H), 6.69–6.62 (m, 2H), 6.52 (d, J = 15.2 Hz, 1H), 5.82 (s, 1H), 4.80–4.76 (m, 2H), 4.21 (q, J = 7.1 Hz, 2H), 3.85 (s, 3H), 3.83 (s, 3H), 3.72 (d, J = 7.9 Hz, 2H), 2.81 (dt, J = 14.4, 8.0 Hz, 1H), 2.59 (dd, J = 14.4, 7.9 Hz, 1H), 1.37 (s, 9H), 1.30 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 167.0, 166.5, 157.9, 154.8, 149.8, 149.1, 147.8, 145.3, 139.4, 130.6, 124.1, 123.4, 121.2, 120.8, 112.03, 111.3, 66.2, 64.8, 64.5, 56.0, 55.9, 51.8, 49.9, 36.2, 28.7 (3C), 14.4. IR (neat) νmax (cm–1): 3294, 2964, 2939, 1745, 1688, 1664, 1595, 1570, 1545, 1514, 1470, 1448, 1423, 1389, 1360, 1254, 1230, 1190, 1159, 1103, 1028, 966, 868, 818, 787, 764, 706, 567, 467. m.p.: 118–122 °C. HRMS (ESI) m/z: [M + Na]+ calcd. for C28H36N3O7ClNa+, 584.2134; found, 584.2135.
Ugi Product 3z
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (40% EtOAc/cHex) afforded the title compound as a colorless solid in 50% yield (0.913 g, 1.50 mmol). Rf = 0.33 (70% EtOAc/cHex). Two rotamers were present on NMR timescale (R1:R2 = 3:1), of which the signals of the major rotamer are reported. 1H NMR (500 MHz, CDCl3) δ 8.39 (d, J = 5.3 Hz, 1H), 7.62 (s, 1H), 7.45–7.39 (m, 1H), 7.00 (s, 1H), 6.93 (dt, J = 15.1, 4.5 Hz, 1H), 6.76 (d, J = 8.6 Hz, 1H), 6.69–6.63 (m, 2H), 6.53 (d, J = 15.1 Hz, 1H), 5.84 (s, 1H), 4.83–4.77 (m, 2H), 4.22 (q, J = 7.1 Hz, 2H), 3.86 (s, 3H), 3.84 (s, 3H), 3.73 (d, J = 7.5 Hz, 2H), 2.81 (dd, J = 14.0, 7.5 Hz, 1H), 2.60 (dd, J = 14.0, 7.5 Hz, 1H), 1.38 (s, 9H), 1.31 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 167.0, 166.6, 157.7, 154.8, 149.5, 149.13, 147.90, 147.88, 139.5, 130.6, 127.2, 126.5, 121.2, 120.8, 112.1, 111.4, 66.2, 64.7, 64.5, 56.0 (2C), 51.8, 49.9, 36.2, 28.8 (3C), 14.4. IR (neat) νmax (cm–1): 3269, 2962, 2930, 1738, 1663, 1622, 1560, 1514, 1460, 1418, 1389, 1366, 1296, 1259, 1159, 1030, 993, 962, 870, 835, 793, 702, 679, 571, 546, 465, 436. m.p.: 106–109 °C. HRMS (ESI) m/z: [M + Na]+ calcd. for C28H36N3O7BrNa+, 628.1629; found, 628.1636.
Ugi Product 3za
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (60% EtOAc/cHex) afforded the title compound as an amber oil in 94% yield (1.691 g, 2.8 mmol). Rf = 0.22 (50% EtOAc/cHex). Two rotamers were present on NMR timescale (R1:R2 = 5:2), of which the signals of the major rotamer are reported. 1H NMR (500 MHz, CDCl3) δ 9.16 (s, 1H), 8.26 (d, J = 8.2 Hz, 1H), 7.49 (d, J = 8.2 Hz, 1H), 7.11 (s, 1H), 6.90 (dt, J = 15.1, 4.4 Hz, 1H), 6.75 (dd, J = 13.7, 6.2 Hz, 1H), 6.65 (s, 2H), 6.51 (d, J = 15.2 Hz, 1H), 5.83 (s, 1H), 4.78 (d, J = 4.5 Hz, 2H), 4.20 (q, J = 7.0 Hz, 2H), 3.94 (s, 3H), 3.84–3.80 (m, 6H), 3.73 (t, J = 7.9 Hz, 2H), 2.82 (dt, J = 13.5, 6.1 Hz, 1H), 2.61 (dt, J = 14.8, 7.9 Hz, 1H), 1.37 (s, 9H), 1.29 (t, J = 7.0 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 168.1, 167.1, 164.7, 160.5, 154.8, 150.2, 149.1, 147.7, 138.1, 134.8, 130.5, 125.1, 123.2, 121.2, 112.0 (2C), 111.4, 66.2, 65.5, 64.5, 56.0, 55.97, 55.9, 50.3, 36.1, 34.1, 28.7 (3C), 14.4. IR (neat) νmax (cm–1): 2959, 1728, 1514, 1256, 1236, 1190, 1157, 1140, 1117, 1025, 731. HRMS (ESI) m/z: [M + Na]+ calcd. for C30H39N3NaO9+, 608.2579; found, 608.2573.
Ugi Product 3zb
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (40% EtOAc/cHex) afforded the title compound as a yellow solid in 60% yield (1.031 g, 1.80 mmol). Rf = 0.42 (70% EtOAc/cHex). 1H NMR (600 MHz, CDCl3) δ 8.22 (d, J = 8.6 Hz, 2H), 7.59 (d, J = 8.6 Hz, 2H), 7.00 (dt, J = 15.0, 4.0 Hz, 1H), 6.73 (d, J = 8.1 Hz, 1H), 6.61–6.51 (m, 3H), 6.16 (s, 1H), 6.01 (s, 1H), 4.88–4.79 (m, 2H), 4.22 (q, J = 7.1 Hz, 2H), 3.83 (s, 3H), 3.82 (s, 3H), 3.61 (t, J = 8.1 Hz, 2H), 2.77 (dd, J = 15.5, 6.9 Hz, 1H), 2.42 (dd, J = 15.5, 8.1 Hz, 1H), 1.38 (s, 9H), 1.31 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ 167.9, 166.7, 154.8, 149.2, 148.1, 147.8, 143.2, 140.2, 130.2, 129.8 (2C), 123.9 (2C), 120.9, 120.7, 112.0, 111.5, 66.1, 64.6, 62.2, 56.05, 55.99, 52.1, 49.0, 36.5, 28.7 (3C), 14.4. IR (neat) νmax (cm–1): 3302, 2962, 1738, 1661, 1610, 1516, 1460, 1421, 1348, 1246, 1153, 1001, 953, 851, 793, 696, 596, 544, 473, 424. M.p.: 138–139 °C. HRMS (ESI) m/z: [M + Na]+ calcd. for C29H37N3O9Na+, 594.2422; found, 594.2438.
Ugi Product 3zc
Prepared according to GP-A. Purification of the crude material by silica gel column chromatography (60% EtOAc/cHex) afforded the title compound as an orange solid in 27% yield (0.424 g, 0.81 mmol). Rf = 0.21 (100% EtOAc). 1H NMR (500 MHz, CDCl3) δ 8.62 (d, J = 5.9 Hz, 1H), 7.31 (d, J = 5.7 Hz, 2H), 7.00 (dd, J = 15.1, 4.2 Hz, 2H), 6.73 (d, J = 8.1 Hz, 1H), 6.62–6.50 (m, 3H), 6.17 (s, 1H), 5.90 (s, 1H), 4.84 (dd, J = 4.2, 1.7 Hz, 2H), 4.22 (d, J = 7.1 Hz, 2H), 3.83 (s, 3H), 3.83 (s, 3H), 3.59 (t, J = 8.3 Hz, 2H), 2.82–2.72 (m, 1H), 2.51–2.35 (m, 1H), 1.38 (s, 9H), 1.31 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 167.7, 166.6, 154.8, 150.3 (2C), 149.1, 147.9, 144.8, 140.0, 130.3, 123.6 (2C), 120.9, 120.7, 111.9, 111.4, 66.1, 64.6, 62.0, 56.02, 55.98, 52.1, 49.0, 36.5, 28.7 (3C), 14.4. IR (neat) νmax (cm–1): 3302, 2972, 1740, 1661, 1620, 1551, 1514, 1456, 1420, 1369, 1254, 1159, 1024, 995, 947, 874, 783, 550, 432. m.p.: 110–112 °C. HRMS (ESI) m/z: [M + Na]+ calcd. for C28H37N3O7Na+, 550.2524; found, 550.2524.
β-Lactam Synthesis
trans-β-Lactam 6a
Prepared according to GP-B using 3a. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a brown oil in 92% yield (77 mg, 0.18 mmol).
Three Millimole-Scale Experiment (trans-6a)
Prepared according to GP-B. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a brown oil in 82% yield (1115.7 mg, 2.55 mmol).
Rf = 0.67 (60% EtOAc/cHex). 1H NMR (600 MHz, CDCl3) δ 8.79 (s, 1H), 8.56–8.54 (m, 1H), 7.58 (td, J = 7.8, 1.8 Hz, 1H), 7.21 (ddd, J = 7.8, 4.8, 1.2 Hz, 1H), 7.02 (d, J = 7.8 Hz, 1H), 6.84 (s, 1H), 6.82–6.77 (m, 2H), 5.22 (dd, J = 15.2, 1.7 Hz, 1H), 5.07–4.88 (m, 2H), 4.09 (d, J = 7.3 Hz, 1H), 3.85 (s, 3H), 3.83 (s, 3H), 3.72 (ddd, J = 13.9, 9.6, 4.8 Hz, 1H), 3.42 (ddd, J = 13.9, 9.6, 4.8 Hz, 1H), 3.38–3.32 (m, 1H), 3.16 (ddd, J = 13.9, 9.6, 4.8 Hz, 1H), 1.38 (s, 9H). 13C{1H} NMR (151 MHz, CDCl3) δ 168.5, 168.0, 156.4, 149.0, 148.5, 147.7, 137.4, 132.1, 128.5, 122.8, 122.6, 121.1, 120.9, 112.4, 111.4, 69.9, 65.7, 56.1, 55.9, 51.5, 46.6, 33.6, 28.7 (3C). IR (neat): νmax (cm–1): 2964, 2930, 1751, 1672, 1585, 1545, 1512, 1460, 1261, 1230, 1148, 1028, 995, 930, 808, 758, 650, 623, 463, 401. HRMS (ESI) m/z: [M + Na]+ calcd. for C25H31N3O4Na+, 460.2207; found, 460.2216.
cis-β-Lactam 6a
Prepared according to GP-C using 3a. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a brown oil in 64% yield (55 mg, 0.12 mmol). Rf = 0.61 (60% EtOAc/cHex). 1H NMR (600 MHz, CDCl3) δ 8.56 (d, J = 5.8 Hz, 1H), 7.96 (s, 1H), 7.63 (dd, J = 7.8, 1.8 Hz, 1H), 7.25 (t, J = 7.8, 5.8 Hz, 1H), 7.19 (d, J = 7.8 Hz, 1H), 6.79–6.76 (m, 3H), 5.91 (dt, J = 17.1, 10.3, 8.6 Hz, 1H), 5.37 (d, J = 17.1 Hz, 1H), 5.33 (d, J = 10.3 Hz, 1H), 3.96 (d, J = 8.6 Hz, 1H), 3.85 (s, 3H), 3.84 (s, 3H), 3.66 (dd, J = 14.0, 7.8 Hz, 1H), 3.44 (dd, J = 14.0, 7.8 Hz, 1H), 3.16 (t, J = 7.8 Hz, 2H), 1.33 (s, 9H). 13C{1H} NMR (151 MHz, CDCl3) δ 168.8, 166.5, 158.3, 149.1, 148.9, 147.8, 137.6, 131.9, 128.9, 123.0, 121.9, 121.2, 120.9, 112.3, 111.4, 70.6, 66.2, 56.1, 56.0, 51.7, 46.4, 33.9, 28.9 (3C). IR (neat) νmax (cm–1): 2964, 2926, 2339, 1751, 1672, 1583, 1545, 1512, 1458, 1261, 1230, 1148, 1028, 995, 922, 806, 762, 733, 640. HRMS (ESI) m/z: [M + Na]+ calcd. for C25H31N3O4Na+, 460.2207; found, 460.2216.
trans-β-Lactam 6b
Prepared according to GP-B using 3db. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a light yellow solid in 82% yield (0.073 g, 0.16 mmol). Rf = 0.62 (50% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 8.72 (d, J = 7.1 Hz, 1H), 8.56–8.53 (m, 1H), 8.48 (s, 1H), 7.61 (d, J = 7.9 Hz, 1H), 7.40–7.35 (m, 2H), 7.21–7.14 (m, 2H), 7.13 (d, J = 2.2 Hz, 1H), 7.12–7.06 (m, 2H), 5.24 (dt, J = 17.0, 1.3 Hz, 1H), 5.05 (ddd, J = 16.8, 10.3, 8.0 Hz, 1H), 4.97 (dd, J = 10.3, 1.5 Hz, 1H), 4.14 (d, J = 8.0 Hz, 1H), 3.88–3.75 (m, 2H), 3.65 (ddd, J = 13.6, 8.8, 7.2 Hz, 1H), 3.55 (ddd, J = 16.0, 8.8, 7.4 Hz, 1H), 3.42 (ddd, J = 14.4, 8.7, 5.5 Hz, 1H), 1.97–1.91 (m, 1H), 1.83–1.77 (m, 1H), 1.72–1.66 (m, 1H), 1.65–1.54 (m, 2H), 1.43–1.07 (m, 5H). 13C{1H} NMR (126 MHz, CDCl3) δ 168.6, 168.4, 155.9, 148.5, 137.2, 136.4, 128.5, 127.4, 122.8, 122.8, 122.7, 122.0, 121.3, 119.4, 118.9, 113.0, 111.4, 69.6, 65.9, 48.6, 45.1, 32.6, 25.6 (2C), 24.6, 23.7 (2C). IR (neat) νmax (cm–1): 3290, 2932, 2851, 1730, 1672, 1649, 1529, 1431, 1402, 1339, 924, 750, 729. HRMS (ESI) m/z: [M + Na]+ calcd. for C27H30N4NaO2+, 465.2261; found, 465.2260.
cis-β-Lactam 6b
Prepared according to GP-C using 3b. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a yellow solid in 9.9% trans diastereoisomer (0.009 g, 0.020 mmol) and 12.1% cis diastereoisomer (0.011 g, 0.024 mmol). Rf= 0.66 (50% EtOAc/cHex). Two diastereoisomers were present in NMR (trans: cis = 1: 4.5) of which the signals of the trans diastereoisomer are marked with ■ and cis diastereoisomer are marked with ●. 1H NMR (500 MHz, CDCl3) ■ δ 8.66 (d, J = 6.6 Hz, 1H), ●■ 8.61–8.56 (m, 1H), 8.55 ■ (dd, J = 4.1, 1.6 Hz, 1H), ● 8.23 (s, 1H), ■ 8.19 (s, 1H), ■ 7.62 (d, J = 8.0 Hz, 1H), ● 7.56 (d, J = 7.9 Hz, 1H), ● 7.51 (td, J = 7.8, 1.8 Hz, 1H), ●■ 7.40–7.33 (m, ● 2H, ■ 1H), ● 7.26 (d, J = 7.9 Hz, 1H), ●■ 7.23–7.16 (m, 3H), ●■ 7.12–7.08 (m, ● 1H, ■ 2H), ● 5.84 (ddd, J = 17.2, 10.3, 8.2 Hz, 1H), ● 5.39 (dt, J = 17.1, 1.2 Hz, 1H), ● 5.29 (d, J = 10.3 Hz, 1H), ■ 5.24 (dt, J = 17.0, 1.3 Hz, 1H), ■ 5.09–4.94 (m, 2H), ● 4.28 (d, J = 8.2 Hz, 1H), ■ 4.13 (d, J = 7.9 Hz, 1H), ■ 3.87–3.75 (m, 2H), ●■ 3.73–3.63 (m, ● 2H, ■ 1H), ■ 3.59–3.52 (m, 1H), ●■ 3.50–3.40 (m, 1H), ● 3.35 (dt, J = 13.4, 6.6 Hz, 1H), ● 3.26 (dt, J = 15.1, 7.6 Hz, 1H), ■ 1.96–1.90 (m, 1H), ■ 1.84–1.76 (m, 1H), ●■ 1.70 (d, J = 8.4 Hz, ● 2H, ■ 1H), ●■ 1.61–1.45 (m, ● 3H, ■ 2H), ●■ 1.28–1.15 (m, ● 2H, ■ 5H), ● 0.97–0.87 (m, 1H), ● 0.74–0.56 (m, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ ● 169.6, ■ 168.6, ■ 168.4, ● 166.9, ● 156.8, ■ 156.0, ● 149.2, ■ 148.5, ■ 137.2, ● 137.2, ● 136.5, ■ 136.4, ● 128.7, ■ 128.5, ■ 127.4, ● 127.4, ● 123.1, ■ 122.83, ■ 122.80, ■ 122.7, ● 122.5, ● 122.4, ● 122.3, ■ 122.2, ● 121.6, ■ 121.3, ● 119.7, ■ 119.5, ■ 119.0, ● 118.9, ■ 113.3, ● 112.9, ● 111.4, ■ 111.3, ● 70.7, ■ 69.7, ■ 65.9, ● 65.3, ■ 48.6, ● 48.2, ■ 45.1, ● 44.0, ● 32.7, ■ 32.5, ■ 25.7 (2C), ● 25.3 (2C), ● 24.83, ● 24.77, ■ 24.7, ●■ 23.8 (● 1C, ■ 2C). IR (neat) νmax (cm–1): 3290, 2932, 2851, 1730, 1672, 1649, 1529, 1431, 1402, 1339, 924, 750, 729. HRMS (ESI) m/z: [M + Na]+ calcd. for C27H30N4NaO2+, 465.2261; found, 465.2263.
trans-β-Lactam 6c
Prepared according to GP-B using 3c. Purification of the crude material by silica gel column chromatography (35% EtOAc/cHex) afforded the title compound as a yellow oil in 87% yield (0.070 g, 0.188 mmol). Rf = 0.50 (50% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 8.78 (d, J = 7.3 Hz, 1H), 7.61–7.51 (m, 2H), 7.09–6.99 (m, 1H), 5.30–5.16 (m, 2H), 5.06–4.97 (m, 1H), 4.10 (d, J = 7.2 Hz, 1H), 3.87–3.78 (m, 2H), 3.75–3.62 (m, 2H), 3.49–3.42 (m, 1H), 3.30 (s, 3H), 2.50 (s, 3H), 1.95–1.86 (m, 2H), 1.76–1.67 (m, 2H), 1.60 (dt, J = 12.5, 3.6 Hz, 1H), 1.41–1.31 (m, 2H), 1.26–1.12 (m, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 169.4, 168.9, 157.2, 154.6, 136.9, 128.6, 122.2, 121.4, 120.8, 70.0, 69.8, 65.7, 58.5, 48.5, 44.2, 32.9, 32.8, 25.7, 24.9, 24.8, 24.5. IR (neat) νmax (cm–1): 2930, 2853, 1757, 1663, 1649, 1535, 1456, 1340, 1107, 1094, 924, 750. HRMS (ESI) m/z: [M + Na]+ calcd. for C21H29N3NaO3+, 394.2101; found, 394.2109.
cis-β-Lactam 6c
Prepared according to GP-C using 3c. Purification of the crude material by silica gel column chromatography (35% EtOAc/cHex) afforded the title compound as a yellow oil in 9% trans diastereoisomer and 28% cis diastereoisomer (0.017 g, 0.045 mmol). Rf= 0.50 (50% EtOAc/cHex). Two diastereoisomers were present in NMR (trans:cis = 1:4), of which the signals of the trans diastereoisomer are marked with ■ and cis diastereoisomer are marked with ●. 1H NMR (500 MHz, CDCl3) δ ■ 8.78 (d, J = 6.8 Hz, 1H), ● 8.41 (d, J = 7.6 Hz, 1H), ●■ 7.63–7.53 (m, ● 1H, ■ 2H), ● 7.38 (d, J = 7.8 Hz, 1H), ●■ 7.07 (d, J = 7.7 Hz, 1H), ● 5.87 (ddd, J = 17.9, 10.3, 8.0 Hz, 1H), ● 5.39 (d, J = 17.2 Hz, 1H), ● 5.29 (d, J = 10.4 Hz, 1H), ■ 5.25–5.16 (m, 2H), ■ 5.02 (dd, J = 9.6, 2.1 Hz, 1H), ● 4.39 (d, J = 7.9 Hz, 1H), ■ 4.12 (d, J = 7.3 Hz, 1H), ●■ 3.96–3.81 (m, 2H), ■ 3.77–3.67 (m, 2H), ● 3.61–3.53 (m, 2H), ■ 3.51–3.46 (m, 1H), ● 3.43 (s, 3H), ■ 3.33 (s, 3H), ● 3.06 (ddd, J = 15.3, 9.6, 5.2 Hz, 1H), ■ 2.52 (s, 3H), ● 2.51 (s, 3H), ●■ 1.98–1.93 (m, 2H), ●■ 1.78–1.71 (m, 2H), ●■ 1.68–1.60 (m, 1H), ●■ 1.44–1.31 (m, 2H), ●■ 1.26–1.12 (m, 3H). 13C{1H} NMR (126 MHz, CDCl3 δ ● 170.3, ■ 169.5, ■ 168.9, ● 167.4, ● 158.1, ■ 157.2, ● 155.7, ■ 154.7, ● 137.0, ■ 136.9, ● 129.0, ■ 128.7, ● 122.7, ■ 122.3, ■ 121.5, ● 120.9, ●■ 120.2, ■ 70.9, ● 69.9, ●■ 69.7, ■ 65.8, ● 64.9, ● 58.7, ■ 58.6, ■ 48.6, ● 48.5, ■ 44.3, ● 43.6, ● 33.6, ● 33.3, ■ 33.0, ■ 32.9, ●■ 25.7, ● 25.2 (2C), ■ 24.95, ■ 24.92, ● 24.64, ■ 24.58. IR (neat) νmax (cm–1): 2928, 1757, 1663, 1533, 1454, 1342, 1103, 995, 625, 442. HRMS (ESI) m/z: [M + Na]+ calcd. for C21H29N3NaO3+, 394.2101; found, 394.2112.
trans-β-Lactam 6d
Prepared according to GP-B using 3d. Purification of the crude material by silica gel column chromatography (50% EtOAc/cHex) afforded the title compound as a yellow oil in 85% yield (0.078 g, 0.176 mmol). Rf = 0.38 (50% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 9.04 (t, J = 5.8 Hz, 1H), 8.53–8.48 (m, 1H), 7.50 (td, J = 7.8, 1.8 Hz, 1H), 7.35–7.31 (m, 2H), 7.28 (d, J = 6.7 Hz, 2H), 7.25 (s, 1H), 7.24–7.20 (m, 2H), 7.20–7.16 (m, 2H), 6.78 (d, J = 8.7 Hz, 2H), 5.26 (dt, J = 16.9, 1.3 Hz, 1H), 5.07 (ddd, J = 16.9, 10.3, 7.9 Hz, 1H), 4.98 (ddd, J = 10.2, 1.7, 0.7 Hz, 1H), 4.86 (d, J = 15.3 Hz, 1H), 4.49 (d, J = 2.8 Hz, 1H), 4.48 (d, J = 3.0 Hz, 1H), 4.41 (d, J = 15.2 Hz, 1H), 4.24 (d, J = 8.0 Hz, 1H), 3.76 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 169.4, 168.5, 159.2, 155.6, 148.3, 138.1, 137.0, 130.6 (2C), 128.7 (2C), 128.4, 128.2, 127.6 (2C), 127.5, 123.6, 122.8, 121.6, 114.1 (2C), 70.4, 66.3, 55.3, 46.7, 43.9. IR (neat) νmax (cm–1): 2930, 1751, 1666, 1512, 1433, 1244, 1176, 1030, 727. HRMS (ESI) m/z: [M + H]+ calcd. for C27H28N3O3+, 442.2125; found, 442.2130.
cis-β-Lactam 6d and Diketopiperazine 2d
Prepared according to GP-C using 3d. Purification of the crude material by silica gel column chromatography (50% EtOAc/cHex) afforded the title compound as a yellow oil in 37% 6d and 15% 2d (0.046 g, 0.10 mmol). Rf= 0.38 (50% EtOAc/cHex). Two products were present in NMR (18b:18c = 2:1), of which the signals of 2d are marked with ■ and 6d are marked with ●. 1H NMR (500 MHz, CDCl3) δ ●■ 8.62–8.55 (m, 1H), ●■ 7.77–7.69 (m, 1H), ● 7.60 (td, J = 7.8, 1.8 Hz, 1H), ●■ 7.35–7.27 (m, ● 3H, ■ 4H), ●■ 7.25–7.21 (m, ● 1H, ■ 3H), ● 7.18 (d, J = 8.6 Hz, 2H), ● 7.16–7.11 (m, 2H), ■ 7.04 (d, J = 8.6 Hz, 2H), ● 7.03–6.99 (m, 1H), ■ 6.83 (d, J = 8.7 Hz, 2H), ● 6.72 (d, J = 8.6 Hz, 2H), ●■ 5.93–5.77 (m, 1H), ■ 5.51 (d, J = 10.0 Hz, 1H), ●■ 5.42–5.35 (m, ● 1H, ■ 2H), ■ 5.33–5.29 (m, 1H), ● 5.29–5.24 (m, 1H), ■ 4.98 (s, 1H), ■ 4.74 (d, J = 8.4 Hz, 1H), ● 4.54 (d, J = 15.3 Hz, 1H), ● 4.38 (dd, J = 14.7, 5.9 Hz, 1H), ● 4.35–4.29 (m, 2H), ● 4.27 (d, J = 8.1 Hz, 1H), ■ 3.95 (d, J = 15.0 Hz, 1H), ■ 3.80 (s, 3H), ● 3.75 (s, 3H), ■ 3.39 (d, J = 14.7 Hz, 1H). 13C{1H} NMR (126 MHz, CDCl3) δ ● 168.0, ● 166.9, ■ 165.4, ■ 164.1, ■ 158.4, ● 158.2, ● 155.8, ■ 155.0, ■ 149.1, ● 148.1, ● 136.7, ● 136.2, ■ 136.2, ■ 134.4, ● 132.2, ■ 129.6, ● 129.3 (2C), ■ 129.1 (2C), ● 127.65 (2C), ■ 127.60 (2C), ● 127.55, ■ 127.02 (2C), ● 126.96 (2C), ■ 126.64, ● 126.57, ■ 126.2, ■ 123.1, ■ 122.8, ● 122.2, ● 121.7, ■ 121.0, ● 120.9, ●■ 113.3 (2C), ● 70.8, ● 65.3, ■ 62.9, ■ 61.2, ● 54.4, ■ 54.3, ■ 46.0, ■ 45.7, ● 44.9, ● 42.8. IR (neat) νmax (cm–1): 2932, 1753, 1663, 1512, 1452, 1433, 1302, 1244, 1176, 1030, 930, 731. HRMS (ESI) m/z: [M + H]+ calcd. for C27H28N3O3+, 442.2125; found, 442.2130.
trans-β-Lactam 6e
Prepared according to GP-B using 3e. Purification of the crude material by silica gel column chromatography (20% EtOAc/cHex) afforded the title compound as a yellow solid in 78% yield (0.079 g, 0.15 mmol). Rf = 0.34 (20% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 7.46 (d, J = 8.4 Hz, 2H), 7.24 (d, J = 8.4 Hz, 2H), 6.61 (s, 1H), 5.86 (s, 1H), 5.37–5.32 (m, 2H), 5.19–5.16 (m, 1H), 4.62 (d, J = 15.6 Hz, 1H), 4.58 (d, J = 15.6 Hz, 1H), 4.17 (d, J = 6.2 Hz, 1H), 2.37 (s, 3H), 1.59 (d, J = 14.9 Hz, 1H), 1.52 (d, J = 14.9 Hz, 1H), 1.21 (s, 3H), 1.20 (s, 3H), 0.90 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 169.9, 168.0, 166.4, 160.9, 135.4, 132.2 (2C), 131.0 (2C), 127.7, 122.5, 122.3, 103.3, 65.9, 65.7, 56.0, 51.5, 46.0, 31.7, 31.5 (3C), 28.8, 28.3, 12.3. IR (neat) νmax (cm–1): 3369, 2945, 1759, 1672, 1522, 1377, 1364, 1155, 1013, 908, 839, 806, 721, 573. HRMS (ESI) m/z: [M + Na]+ calcd. for C25H32BrN3NaO3+, 524.1519; found, 524.1516.
cis-β-Lactam 6e
Prepared according to GP-C using 3e. Purification of the crude material by silica gel column chromatography (20% EtOAc/cHex) afforded the title compound as a light yellow solid in 21% trans diastereoisomer and 44% cis diastereoisomer (0.065 g, 0.13 mmol). Rf = 0.29 (20% EtOAc/cHex). Two diastereoisomers were present in NMR (trans:cis = 1:2), of which the signals of the trans diastereoisomer are marked with ■ and cis diastereoisomer are marked with ●. 1H NMR (500 MHz, CDCl3) δ ■ 7.46 (d, J = 8.4 Hz, 2H), ● 7.43 (d, J = 8.4 Hz, 2H), ■ 7.24 (d, J = 8.4 Hz, 2H), ● 7.19 (d, J = 8.4 Hz, 2H), ■ 6.61 (s, 1H), ● 6.26 (s, 1H), ●■ 5.90–5.80 (m, 1H), ● 5.78–5.74 (m, 1H), ● 5.44 (dt, J = 17.1, 1.2 Hz, 1H), ●■ 5.37–5.32 (m, ● 1H, ■ 2H), ■ 5.18 (dd, J = 8.5, 3.2 Hz, 1H), ●■ 4.65–4.56 (m, ● 1H, ■ 2H), ● 4.50 (d, J = 15.6 Hz, 1H), ● 4.21 (d, J = 8.1 Hz, 1H), ■ 4.17 (d, J = 6.2 Hz, 1H), ■ 2.37 (s, 3H), ● 2.35 (s, 3H), ■ 1.59 (d, J = 14.9 Hz, 1H), ●■ 1.55–1.50 (m, 1H), ● 1.44 (d, J = 14.9 Hz, 1H), ● 1.27 (s, 3H), ● 1.25 (s, 3H), ■ 1.22 (s, 3H), ■ 1.20 (s, 3H), ■ 0.90 (s, 9H), ● 0.88 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ ● 170.5, ■ 169.9, ■ 168.0, ● 167.9, ■ 166.4, ● 165.0, ● 162.3, ■ 161.0, ● 135.4, ■ 135.4, ■ 132.2 (2C), ● 132.1 (2C), ■ 131.0 (2C), ● 130.8 (2C), ●■ 127.7, ● 122.6, ■ 122.5, ■ 122.3, ● 122.2, ■ 103.3, ● 101.8, ■ 65.9, ● 65.8, ● 65.73, ■ 65.71, ● 56.4, ■ 56.0, ● 52.6, ■ 51.5, ■ 46.0, ● 45.8, ■ 31.7, ● 31.6, ●■ 31.5 (3C), ■ 28.8, ● 28.8, ■ 28.3, ● 28.1, ■ 12.33, ● 12.31. IR (neat) νmax (cm–1): 2955, 1763, 1678, 1601, 1514, 1489, 1474, 1445, 1383, 1366, 1350, 1225, 1070, 1013, 914. HRMS (ESI) m/z: [M + Na]+ calcd. for C25H32BrN3NaO3+, 512.1519; found, 524.1513.
trans-β-Lactam 6f
Prepared according to GP-B using 3f. Purification of the crude material by silica gel column chromatography (35% EtOAc/cHex) afforded the title compound as an off-white solid in 94% yield (0.104 g, 0.189 mmol). Rf= 0.63 (50% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 7.88 (d, J = 7.1 Hz, 1H), 6.24 (s, 1H), 5.35–5.22 (m, 2H), 5.16–5.09 (m, 1H), 4.61 (dd, J = 6.0, 4.5 Hz, 1H), 4.13–3.97 (br, 3H), 3.97–3.89 (m, 1H), 3.72–3.64 (m, 1H), 3.64–3.56 (m, 2H), 3.56–3.45 (m, 2H), 3.34 (dt, J = 14.3, 6.3 Hz, 1H), 2.89–2.75(br, 2H), 2.41 (s, 3H), 2.08 (dt, J = 14.4, 5.4 Hz, 1H), 1.96 (tt, J = 14.3, 6.4 Hz, 2H), 1.92–1.83 (br, 2H), 1.50–1.32 (br, 12H), 1.17 (td, J = 7.0, 4.9 Hz, 6H). 13C{1H} NMR (126 MHz, CDCl3) δ 170.0, 167.9, 167.7, 160.8, 154.7, 127.7, 122.4, 103.3, 102.3, 79.7, 66.0 (2C), 64.8, 63.1 (2C), 62.0, 47.8 (2C), 39.3, 31.5 (2C), 28.4 (3C), 26.9, 15.5, 15.4, 12.4. IR (neat) νmax (cm-1): 3313, 2974, 1749, 1686, 1653, 1427, 1364, 1167, 1136, 1124, 1057. HRMS (ESI) m/z: [M + H]+ calcd. for C28H45N4O7+, 549.3283; found, 549.3284.
cis-β-Lactam 6f
Prepared according to GP-C using 3f. Purification of the crude material by silica gel column chromatography (50% EtOAc/cHex) afforded the title compound as a light yellow oil in 19% cis stereoisomer and 7% trans stereoisomer (0.028 g, 0.052 mmol). Rf= 0.31 (50% EtOAc/cHex). Two diastereoisomers were present in NMR (trans:cis = 2:5), of which the signals of the trans diastereoisomer are marked with ■ and cis diastereoisomer are marked with ●. 1H NMR (500 MHz, CDCl3) ■ δ 7.90 (d, J = 7.2 Hz, 1H), ● 7.68 (d, J = 7.7 Hz, 1H), ■ 6.26 (q, J = 0.8 Hz, 1H), ● 6.21 (q, J = 0.8 Hz, 1H), ● 5.77 (ddd, J = 17.9, 10.3, 7.7 Hz, 1H), ● 5.42 (dt, J = 17.2, 1.2 Hz, 1H), ●■ 5.35–5.28 (m, ● 1H, ■ 2H), ■ 5.20–5.09 (m, 1H), ●■ 4.67–4.60 (m, 1H), ●■ 4.25–3.89 (m, 4H), ●■ 3.77–3.42 (m, ● 5H, ■ 6H), ● 3.26 (dd, J = 13.3, 7.3 Hz, 1H), ●■ 2.88–2.78 (m, 2H), ●■ 2.44 (s, 3H), ● 2.42–2.38 (m, 1H), ●■ 2.13–1.81 (m, ● 4H, ■ 5H), ●■ 1.49–1.37 (m, 12H), ●■ 1.25–1.15 (m, 6H). 13C{1H} NMR (126 MHz, CDCl3) δ ● 170.6, ■ 170.0, ■ 168.0, ● 167.9, ■ 167.8, ● 166.5, ● 161.9, ■ 160.9, ●■ 154.8, ■ 127.7, ● 127.4, ■ 122.5, ● 122.4, ■ 103.4, ● 102.7, ■ 102.4, ● 102.0, ■ 79.8, ● 79.8, ■ 66.1, ● 65.3, ● 65.1, ■ 64.9, ● 63.6 (2C), ■ 63.1 (2C), ■ 62.14 (2C), ● 62.07 (2C), ■ 47.85 (2C), ● 47.80 (2C), ■ 39.3 (2C), ● 39.0 (2C), ● 31.7 (2C), ■ 31.6 (2C), ●■ 28.5 (3C), ● 15.6, ■ 15.6, ●■ 15.4, ● 12.5, ■ 12.4. IR (neat) νmax (cm–1): 2978, 1761, 1420, 1366, 1169, 1138, 1059, 629, 467. HRMS (ESI) m/z: [M + H]+ calcd. for C28H45N4O7+, 549.3283; found, 549.3282.
trans-β-Lactam 6g
Prepared according to GP-B using 3g. Purification of the crude material by silica gel column chromatography (20% EtOAc/cHex) afforded the title compound as a brown oil in 78% yield (52 mg, 0.16 mmol). Rf= 0.47 (50% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 7.12 (s, 1H), 6.08 (s, 1H), 5.35–5.31 (m, 2H), 5.15 (dd, J = 7.5, 4.3 Hz, 1H), 4.18–4.14 (m, 1H), 3.37 (ddd, J = 14.0, 9.8, 6.2 Hz, 1H), 3.26 (ddd, J = 14.0, 9.8, 6.2 Hz, 1H), 2.46 (s, 3H), 1.75–1.64 (m, 2H), 1.38 (s, 9H), 1.37–1.30 (m, 2H), 0.91 (t, J = 7.4 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 170.2, 167.8, 166.8, 161.6, 128.1, 122.2, 103.0, 65.1 (2C), 52.2, 43.3, 30.1, 28.6 (3C), 20.7, 13.8, 12.4. IR (neat) νmax (cm–1): 3366, 2959, 2930, 2878, 1753, 1672, 1601, 1518, 1450, 1360, 1223, 941, 816, 594, 465. HRMS (ESI) m/z: [M + Na]+ calcd. for C18H27N3O3Na+, 356.1945; found, 356.1958.
cis-β-Lactam 6g
Prepared according to GP-C using 3g. Purification of the crude material by silica gel column chromatography (20% EtOAc/cHex) afforded the title compound as a brown oil in 28% yield (26 mg, 0.08 mmol). Rf= 0.52 (50% EtOAc/cHex). 1H NMR (600 MHz, CDCl3) δ 6.56 (s, 1H), 6.11 (s, 1H), 5.83 (ddd, J = 17.1, 10.3, 8.2 Hz, 1H), 5.39 (dt, J = 17.1, 1.2 Hz, 1H), 5.31 (dd, J = 10.3, 1.2 Hz, 1H), 4.12 (d, J = 8.2 Hz, 1H), 3.28 (t, J = 7.9 Hz, 2H), 2.45 (s, 3H), 1.70–1.59 (m, 2H), 1.34 (s, 9H), 1.34–1.28 (m, 2H), 0.89 (t, J = 7.4 Hz, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ 170.7, 167.7, 165.5, 162.6, 128.01122.4, 101.4, 65.1 (2C), 52.3, 43.2, 30.4, 28.8 (3C), 20.7, 13.8, 12.4. IR (neat) νmax (cm–1): 3366, 2959, 2930, 2878, 1753, 1672, 1601, 1518, 1450, 1361, 1223, 941, 816, 594, 465. HRMS (ESI) m/z: [M + Na]+ calcd. for C18H27N3O3Na+, 356.1945; found, 356.1958.
trans-β-Lactam 6h
Prepared according to GP-B using 3h. Purification of the crude material by silica gel column chromatography (15% EtOAc/cHex) afforded the title compound as a light yellow oil in 27% yield (0.024 g, 0.05 mmol). Rf= 0.86 (50% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 7.67 (d, J = 8.2 Hz, 2H), 7.36 (s, 1H), 7.28 (d, J = 8.0 Hz, 2H), 6.88 (s, 1H), 5.40–5.35 (m, 2H), 5.22–5.17 (m, 1H), 4.44 (dd, J = 18.3, 2.6 Hz, 1H), 4.37 (dd, J = 18.3, 2.5 Hz, 1H), 4.19 (d, J = 7.1 Hz, 1H), 2.41 (s, 3H), 2.33 (t, J = 2.6 Hz, 1H), 1.88 (d, J = 14.9 Hz, 1H), 1.75 (d, J = 14.9 Hz, 1H), 1.48 (s, 3H), 1.47 (s, 3H), 1.00 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 170.5, 167.4, 166.4, 161.4, 141.2, 129.9 (2C), 127.3, 126.0 (2C), 124.2, 122.8, 100.8, 78.1, 74.2, 66.4, 66.0, 56.6, 51.5, 31.9, 31.6 (3C), 31.5, 29.4, 28.8, 21.7. IR (neat) νmax (cm–1): 2955, 1774, 1678, 1510, 1423, 1364, 932, 822, 824, 625. HRMS (ESI) m/z: [M + Na]+ calcd. for C27H33N3NaO3+, 470.2414; found, 470.2423.
cis-β-Lactam 6h
Prepared according to GP-C using 3h. Purification of the crude material by silica gel column chromatography (15% EtOAc/cHex) afforded the title compound as a light yellow oil in 7.5% trans diastereoisomer and 21.5% cis diastereoisomer (0.026 g, 0.05 mmol). Rf= 0.42 (20% EtOAc/cHex). Two diastereoisomers were present in NMR (trans:cis = 1:3), of which the signals of the trans diastereoisomer are marked. 1H NMR (500 MHz, CDCl3) δ ●■ 7.67 (d, J = 8.2 Hz, 2H), ■ 7.35 (s, 1H), ●■ 7.28 (d, J = 8.0 Hz, 2H), ● 7.09 (s, 1H), ■ 6.88 (s, 1H), ● 6.87 (s, 1H), ● 5.88 (ddd, J = 17.2, 10.3, 8.0 Hz, 1H), ● 5.46 (dt, J = 17.2, 1.1 Hz, 1H), ●■ 5.41–5.35 (m, ● 1H, ■ 2H), ■ 5.20–5.16 (m, 1H), ●■ 4.47–4.29 (m, 2H), ●■ 4.21–4.13 (m, 1H), ●■ 2.41 (s, 3H), ■ 2.33 (t, J = 2.6 Hz, 1H), ● 2.32 (t, J = 2.6 Hz, 1H), ■ 1.88 (d, J = 14.9 1H), ●■ 1.79–1.69 (m, ● 2H, ■ 1H), ■ 1.48 (s, 3H), ■ 1.47 (s, 3H), ● 1.44 (s, 3H), ● 1.42 (s, 3H), ■ 1.00 (s, 9H), ● 0.97 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ ● 170.9, ■ 170.5, ■ 167.3, ● 167.3, ■166.4, ● 164.8, ● 163.0, ■ 161.4, ● 141.3, ■ 141.2, ● 129.93 (2C), ■ 129.91 (2C), ●■ 127.3, ● 126.0 (2C), ■ 125.9 (2C), ■ 124.24, ● 124.19, ● 122.85, ■ 122.82, ■ 100.8, ● 99.4, ● 78.12, ■ 78.09, ■ 74.2, ● 74.0, ● 67.0, ■ 66.4, ■ 66.0, ● 65.6, ● 56.8, ■ 56.6, ● 52.5, ■ 51.5, ■ 31.9, ● 31.8, ●■ 31.6 (3C), ● 31.5, ■ 31.3, ■ 29.4, ● 29.2, ■ 28.8, ● 28.5, ●■ 21.7. IR (neat) νmax (cm–1): 2955, 1772, 1678, 1510, 1452, 1423, 1366, 1354, 1225, 932, 733. HRMS (ESI) m/z: [M + Na]+ calcd. for C27H33N3NaO3+, 470.2414; found, 470.2414.
trans/cis-β-Lactam 6i
Prepared according to GP-B using 3i. Purification of the crude material by silica gel column chromatography (20% EtOAc/cHex) afforded the title compound as a brown oil in 48% yield trans diastereoisomer and 21% yield cis diastereoisomer (63 mg, 0.15 mmol). Also prepared according to GP-C using 3i. Purification of the crude material by silica gel column chromatography (20% EtOAc/cHex) afforded the title compound as a brown oil in 10% yield cis diastereoisomer and 23% yield trans diastereoisomer (35 mg, 0.08 mmol). Rf= 0.55 (50% EtOAc/cHex). Two diastereoisomers were present in NMR (trans:cis = 5:2), of which the signals of the trans diastereoisomer are marked with ● and cis diastereoisomer are marked with ■. 1H NMR (500 MHz, CDCl3) δ ■ 7.82–7.77 (m, 5H), ● 7.47–7.43 (m, 5H), ●■ 7.26–7.21 (m, 2H), ●■ 7.13–7.07 (m, 2H), ■ 7.02 (s, 1H), ● 7.00 (s, 1H), ● 6.20 (s, 1H), ■ 6.12 (s, 1H), ■ 5.97–5.87 (m, 1H), ●■ 5.60–5.51 (m, 1H), ●■ 5.49–5.41 (d, J = 1.6 Hz, 1H), ● 5.30–5.25 (m, 1H), ■ 4.41 (d, J = 7.5 Hz, 1H), ● 4.32 (d, J = 7.7 Hz, 1H), ● 2.30 (s, 3H), ■ 2.28 (s, 3H), ● 1.36 (s, 9H), ■ 1.35 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ ■ 167.0, ● 165.6, ● 165.5, ■ 164.05, ● 164.04, ■ 163.8, ● 162.9, ■ 162.8, ■ 135.1, ● 135.0, ● 133.8, ■ 133.6, ● 130.55, ■ 130.50, ■ 130.1 (2C), ● 130.0 (2C), ● 129.1 (2C), ■ 129.0 (2C), ■ 128.4, ● 128.3, 127.1 (2C), ■ 127.0 (2C), ■ 126.6, ● 126.5, ● 123.8, ■ 123.2, ● 117.35 (2C), ■ 117.33 (2C), ● 104.6, ■ 103.9, ● 66.2, ■ 65.5, ● 65.0, ■ 64.8, ■ 52.7, ● 52.5, ■ 28.8 (3C), ● 28.6 (3C), ●■ 21.1. IR (neat) νmax (cm–1): 3354, 2966, 2926, 2311, 1757, 1680, 1516, 1367, 1221, 1192, 943, 916, 818, 766, 727,689, 592, 507, 401. m.p.: 170–172 °C. HRMS (ESI) m/z: [M + Na]+ calcd. for C26H27N3O3Na+, 452.1945; found, 452.1958.
trans-β-Lactam 6j
Prepared according to GP-B using 3j. Purification of the crude material by silica gel column chromatography (50% EtOAc/cHex) afforded the title compound as a yellow solid in 94% yield (0.067 g, 0.18 mmol). Rf= 0.31 (50% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 7.03 (d, J = 1.2 Hz, 1H), 6.91 (d, J = 1.1 Hz, 1H), 5.74 (d, J = 7.2 Hz, 1H), 5.40–5.33 (m, 1H), 5.24–5.12 (m, 2H), 4.86 (s, 1H), 4.81 (s, 1H), 4.79 (d, J = 9.3 Hz, 1H), 4.36 (d, J = 16.5 Hz, 1H), 4.17 (d, J = 16.4 Hz, 1H), 3.71 (dtd, J = 10.8, 7.4, 4.0 Hz, 1H), 3.40 (s, 3H), 1.84–1.76 (m, 1H), 1.73 (s, 3H), 1.64–1.50 (m, 3H), 1.35–1.21 (m, 3H), 1.13–0.93 (m, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 167.5, 166.1, 142.9, 140.4, 128.3, 128.1, 123.4, 122.7, 113.5, 66.1, 62.2, 49.4, 48.3, 33.9, 33.0, 32.2, 25.4, 24.73, 24.66, 20.5. IR (neat) νmax (cm–1): 2931, 2854, 1749, 1655, 1518, 1377, 1281, 914, 729. HRMS (ESI) m/z: [M + Na]+ calcd. for C20H28N4NaO2+, 379.2104; found, 379.2117.
trans-β-Lactam 6k
Prepared according to GP-B using 3k. Purification of the crude material by silica gel column chromatography (35% EtOAc/cHex) afforded the title compound as an off-white solid in 76% yield (0.078 g, 0.15 mmol). Rf= 0.57 (50% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 7.21 (d, J = 8.4 Hz, 1H), 7.09–7.06 (m, 1H), 7.00 (d, J = 1.2 Hz, 1H), 6.42 (dd, J = 8.4, 2.3 Hz, 1H), 6.37 (d, J = 2.3 Hz, 1H), 5.72 (s, 1H), 5.39 (dd, J = 16.8, 1.7 Hz, 1H), 5.33–5.24 (m, 1H), 5.21 (dd, J = 9.9, 1.8 Hz, 1H), 5.11 (d, J = 17.3 Hz, 1H), 4.87 (d, J = 17.4 Hz, 1H), 4.80 (d, J = 9.4 Hz, 1H), 4.10 (hept, J = 6.9 Hz, 1H), 3.73 (s, 6H), 1.32 (s, 3H), 1.30 (s, 3H), 1.20–1.15 (m, 4H), 1.10 (d, J = 14.9 Hz, 1H), 0.79 (s, 3H), 0.73 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 168.2, 165.9, 160.3, 157.5, 142.1, 129.4, 129.3, 128.8, 122.4, 117.5, 117.3, 104.3, 98.5, 67.7, 61.6, 56.3, 55.5, 55.3, 53.9, 48.1, 40.5, 31.4 (3C), 31.3, 27.1, 26.8, 24.5, 24.4. IR (neat) νmax (cm–1): 2955, 1751, 1678, 1508, 1375, 1256, 1203, 1124, 1032, 932. HRMS (ESI) m/z: [M + H]+ calcd. for C29H43N4O4+, 511.3279; found, 511.3270.
trans-β-Lactam 6l
Prepared according to GP-B using 3l. Purification of the crude material by silica gel column chromatography (50% EtOAc/cHex) afforded the title compound as a brown oil in 95% yield (69 mg, 0.19 mmol). Rf= 0.18 (70% EtOAc/cHex). 1H NMR (600 MHz, CDCl3) δ 7.46 (s, 1H), 6.97 (s, 1H), 6.87 (s, 1H), 5.38–5.31 (m, 1H), 5.27–5.18 (m, 1H), 5.15 (dd, J = 10.2, 1.3 Hz, 1H), 5.05 (t, J = 5.6 Hz, 1H), 4.39 (d, J = 9.0 Hz, 1H), 3.72 (dd, J = 14.8, 5.6 Hz, 1H), 3.66 (dd, J = 14.8, 5.6 Hz, 1H), 3.57 (s, 3H), 3.42 (s, 3H), 3.31 (s, 3H), 1.34 (s, 9H). 13C{1H} NMR (151 MHz, CDCl3) δ 167.8, 166.7, 143.3, 128.4, 127.2, 123.6, 122.4, 101.8, 67.5, 63.9, 55.1, 54.2, 52.0, 45.7, 35.1, 28.6 (3C). IR (neat) νmax (cm–1): 2966, 2934, 2340, 1759, 1676, 1528, 1367, 1121, 1067, 984, 760, 652. HRMS (ESI) m/z: [M + H]+ calcd. for C18H29N4O4+, 365.2183; found, 365.2190.
trans-β-Lactam 6m
Prepared according to GP-B using 3m. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a yellow solid in 81% yield (0.075 g, 0.162 mmol). Rf= 0.76 (50% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 7.40 (s, 1H), 7.30–7.24 (m, 2H), 7.20 (d, J = 7.5 Hz, 3H), 7.01 (d, J = 1.1 Hz, 1H), 6.88 (d, J = 1.1 Hz, 1H), 5.39–5.34 (m, 1H), 5.31–5.22 (m, 1H), 5.16–5.12 (m, 1H), 4.57 (d, J = 7.9 Hz, 1H), 3.75 (ddd, J = 13.9, 11.7, 5.5 Hz, 1H), 3.54 (ddd, J = 13.9, 11.6, 5.4 Hz, 1H), 3.49 (s, 3H), 3.25 (td, J = 12.5, 11.7, 5.5 Hz, 1H), 3.04 (ddd, J = 13.3, 11.9, 5.4 Hz, 1H), 1.65 (d, J = 15.0 Hz, 1H), 1.58 (d, J = 15.0 Hz, 1H), 1.47 (s, 3H), 1.45 (s, 3H), 0.90 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 167.6, 166.2, 142.7, 138.8, 128.7 (2C), 128.6 (2C), 128.4, 127.8, 126.5, 123.7, 122.0, 66.4, 62.7, 56.2, 53.5, 45.1, 34.5, 34.0, 31.7, 31.5 (3C), 28.4, 28.2. IR (neat) νmax (cm–1): 2955, 1753, 1670, 1510, 1454, 1364, 1281, 1223, 731, 700, 501, 457. HRMS (ESI) m/z: [M + Na]+ calcd. for C26H36N4NaO2+, 459.2730; found, 459.2733.
trans-β-Lactam 6n
Prepared according to GP-B using 3n. Purification of the crude material by silica gel column chromatography (20% EtOAc/cHex) afforded the title compound as a brown oil in 59% yield (49 mg, 0.12 mmol). Rf= 0.65 (60% EtOAc/cHex). 1H NMR (600 MHz, CDCl3) δ 9.37 (s, 1H), 9.29 (s, 1H), 8.03 (d, J = 8.1 Hz, 1H), 7.75 (s, 1H), 7.73–7.65 (m, 3H), 7.32 (d, J = 8.5 Hz, 2H), 7.09 (d, J = 8.2 Hz, 2H), 5.32 (td, J = 17.0, 1.4 Hz, 1H), 5.06 (ddd, J = 17.0, 10.3, 8.1 Hz, 1H), 4.96 (td, J = 10.3, 1.4 Hz, 1H), 4.32 (td, J = 8.1 Hz, 1H), 2.29 (s, 3H), 1.42 (s, 9H). 13C{1H} NMR (151 MHz, CDCl3) δ 166.8, 164.7, 151.6, 148.2, 136.3, 135.3, 133.6, 131.5, 129.6 (2C), 128.5, 128.1, 127.6, 127.5, 127.4, 121.8, 120.4, 117.7(2C), 69.0, 65.9, 51.7, 28.7 (3C), 21.1. IR (neat) νmax (cm–1): 2966, 2926, 2339, 2312, 1755, 1678, 1541, 1514, 1366, 1273, 1221, 1188, 933, 812, 752, 521, 471. HRMS (ESI) m/z: [M + Na]+ calcd. for C26H27N3O2Na+, 436.1995; found, 436.2003.
trans/cis-β-Lactam 6o
Prepared according to GP-B using 3o. Purification of the crude material by silica gel column chromatography (20% EtOAc/cHex) afforded the title compound as a brown oil in 66% yield cis diastereoisomer and 29% yield trans diastereoisomer (55 mg, 0.15 mmol). Also prepared according to GP-C using 3o. Purification of the crude material by silica gel column chromatography (20% EtOAc/cHex) afforded the title compound as a brown oil in 64% yield cis diastereoisomer and 26% yield trans diastereoisomer (60 mg, 0.16 mmol). Rf= 0.39 (50% EtOAc/cHex). Two diastereoisomers were present in NMR (cis:trans = 7:3), of which the signals of the trans diastereoisomer are marked with ■ and cis diastereoisomer are marked with ●. 1H NMR (600 MHz, CDCl3) δ ■ 9.10 (s, 1H), ● 9.08 (s, 1H), ■ 8.16–8.12 (m, 3H), ●■ 8.07–7.99 (m, 1H), ■ 7.98 (s, 1H), ● 7.84–7.80 (m, 3H), ● 7.32 (s, 1H), ■ 6.08 (ddq, J = 15.0, 10.1, 6.6, 4.0 Hz, 1H), ● 5.99–5.90 (m, 2H), ● 5.47 (d, J = 17.1 Hz, 1H), ● 5.39 (d, J = 10.1 Hz, 1H), ■ 5.36–5.29 (m, 2H), ● 5.24 (d, J = 16.9 Hz, 1H), ■ 5.22 (d, J = 9.0 Hz, 1H), ● 5.18 (d, J = 10.1 Hz, 1H), ■ 5.13 (dt, J = 17.5, 10.3, 8.0 Hz, 1H), ■ 5.02 (d, J = 10.3 Hz, 1H), ● 4.46 (d, J = 7.9 Hz, 1H), ■ 4.34 (dd, J = 15.4, 6.6 Hz, 1H), ■ 4.30 (d, J = 7.9 Hz, 1H), ■ 4.25 (dd, J = 15.4, 6.6 Hz, 1H), ● 4.16 (dd, J = 15.6, 6.5 Hz, 1H), ● 4.02 (dd, J = 15.6, 6.5 Hz, 1H), ■ 1.42 (s, 9H), ● 1.40 (s, 9H). 13C{1H} NMR (151 MHz, CDCl3) δ ● 168.3, ■ 167.9, ■ 167.5, ● 166.3, ● 152.7, ■ 151.7, ■ 145.4, ● 144.5, ● 141.9, ■ 141.5, ● 140.9, ■ 140.5, ● 132.3, ■ 132.2, ●■ 130.9, ●■ 130.8, ■ 129.6, ● 129.5, ● 129.2, ■ 128.9, ● 128.1, ■ 127.8, ■ 122.6, ● 122.2, ■ 120.2, ● 120.0, ● 69.9, ■ 69.2, ■ 66.3, ● 65.6, ●52.5, ■ 52.2, ■ 46.6, ● 45.9, ● 28.9 (3C), ■ 28.7 (3C). IR (neat) νmax (cm–1): 3340, 2976, 2922, 2339, 1753, 1668, 1520, 1385, 1215, 1124, 951, 837, 756, 619, 563, 411. m.p.: 101–106 °C. HRMS (ESI) m/z: [M + Na]+ calcd. for C21H24N4O2Na+, 387.1791; found, 387.1810.
trans/cis-β-Lactam 6p
Prepared according to GP-B using 3p. Purification of the crude material by silica gel column chromatography (20% EtOAc/cHex) afforded the title compound as a brown oil in 50% yield (cis diastereoisomer) and 36% yield (trans diastereoisomer) (93 mg, 0.18 mmol). Also prepared according to GP-C using 3p. Purification of the crude material by silica gel column chromatography (20% EtOAc/cHex) afforded the title compound as a brown oil in 41% yield cis diastereoisomer and 35% yield trans diastereoisomer (80 mg, 0.16 mmol). Rf= 0.56 (60% EtOAc/cHex). Two diastereoisomers were present in NMR (cis:trans = 5:4), of which the signals of the trans diastereoisomer are marked with ● and cis diastereoisomer are marked with ■. 1H NMR (500 MHz, CDCl3) δ ■ 9.25 (s, 1H), ● 8.29 (s, 1H), ●■ 8.03–7.97 (m, 2H), ●■ 7.80 (d, J = 8.2 Hz, 1H), ●■ 7.78–7.74 (m, 1H), ●■ 7.60–7.56 (m, 1H), ■ 7.35 (d, J = 8.4 Hz, 2H), ● 7.30 (d, J = 8.6 Hz, 1H), ●■ 7.28–7.25 (m, 2H), ■ 7.22 (d, J = 8.7 Hz, 1H), ● 7.20 (d, J = 8.4 Hz, 2H), ● 6.04–5.95 (m, 1H), ● 5.42 (dt, J = 17.1, 1.1 Hz, 1H), ● 5.37 (d, J = 10.3 Hz, 1H), ■ 5.25 (dt, J = 17.0, 1.2 Hz, 1H), ■ 5.06 (ddd, J = 17.0, 10.4, 7.9 Hz, 1H), ■ 5.00 (d, J = 15.5 Hz, 1H), ■ 4.91 (d, J = 10.4 Hz, 1H), ● 4.85 (d, J = 15.6 Hz, 1H), ● 4.47 (d, J = 7.6 Hz, 1H), ■ 4.44 (d, J = 7.9 Hz, 1H), ■ 4.30 (d, J = 7.9 Hz, 1H), ● 4.24 (d, J = 8.5 Hz, 1H), ■ 1.85 (d, J = 14.9 Hz, 1H), ● 1.81 (d, J = 14.9 Hz, 1H), ■ 1.73 (d, J = 14.9 Hz, 1H), ● 1.50 (d, J = 14.9 Hz, 1H), ■ 1.46 (s, 3H), ●■ 1.43 (s, 3H), ● 1.41 (s, 3H), ■ 0.97 (s, 9H), ● 0.88 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ ● 169.1, ■ 168.7, ■ 167.3, ● 166.0, ● 158.0, ■ 156.6, ■ 146.5, ● 146.3, ● 137.4, ■ 136.8, ● 135.1, ■ 135.0, ■ 133.64, ● 133.58, ■ 130.7 (2C), ● 130.42 (2C), ■ 130.39, ● 130.37, ■ 128.9 (2C), ●■ 128.8 (● 2C, ■ 1C), ● 128.7, ● 128.5, ■ 128.1, ■ 127.8, ● 127.7, ■ 127.32, ● 127.31, ● 126.9, ■ 126.7, ● 122.3, ■ 121.5, ■ 120.7, ● 119.3, ● 71.3, ■ 70.7, ● 66.8, ■ 66.2, ● 55.9, ■ 55.6, ● 52.5, ■ 51.6, ■ 46.9, ● 46.4, ■ 31.7, ● 31.6, ■ 31.52 (3C), ● 31.47 (3C), ■ 29.2, ■ 29.0, ● 28.8, ● 28.7. IR (neat) νmax (cm–1): 2955, 2907, 2341, 2309, 1759, 1672, 1499, 1371, 1225, 1143, 1094, 924, 825, 731, 642, 623, 505, 453, 401. HRMS (ESI) m/z: [M + Na]+ calcd. for C30H34N3O2ClNa+, 526.2232; found, 526.2249.
trans-β-Lactam 6q
Prepared according to GP-B using 3q. Purification of the crude material by silica gel column chromatography (15% EtOAc/cHex) afforded the title compound as a colorless solid in 40% yield (0.035 g, 0.079 mmol). Rf= 0.84 (50% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 7.82 (d, J = 3.3 Hz, 1H), 7.77 (s, 1H), 7.60 (d, J = 8.1 Hz, 2H), 7.51 (d, J = 8.1 Hz, 2H), 7.33 (d, J = 3.3 Hz, 1H), 5.33 (d, J = 17.0 Hz, 1H), 5.22 (ddd, J = 17.1, 10.2, 7.5 Hz, 1H), 5.10 (d, J = 10.1 Hz, 1H), 4.73 (d, J = 15.5 Hz, 1H), 4.59 (d, J = 15.5 Hz, 1H), 4.18 (d, J = 7.5 Hz, 1H), 1.25 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 168.6, 166.6, 166.0, 142.9, 140.3, 130.4 (q, J = 32.6 Hz), 129.7 (2C), 126.7, 125.9 (q, J = 3.7 Hz, 2C), 124.1 (d, J = 272.2 Hz), 122.8, 121.3, 69.0, 67.8, 51.9, 46.7, 28.4 (3C). IR (neat) νmax (cm–1): 2968, 1767, 1672, 1366, 1321, 1163, 1242, 1113, 1067, 1020. HRMS (ESI) m/z: [M + Na]+ calcd. for C21H22F3N3NaO2S+, 460.1277; found, 460.1282.
cis-β-Lactam 6q
Prepared according to GP-C using 3q. Purification of the crude material by silica gel column chromatography (15% EtOAc/cHex) afforded the title compound as a colorless solid in 27% yield (0.013 g, 0.027 mmol). Rf= 0.82 (50% EtOAc/cHex). 1H NMR (600 MHz, CDCl3) δ 7.80 (d, J = 3.3 Hz, 1H), 7.59 (d, J = 8.1 Hz, 2H), 7.47 (d, J = 8.0 Hz, 2H), 7.37 (s, 1H), 7.34 (d, J = 3.3 Hz, 1H), 5.87 (ddd, J = 17.2, 10.3, 8.1 Hz, 1H), 5.41 (dt, J = 17.2, 1.2 Hz, 1H), 5.35 (dd, J = 10.3, 0.9 Hz, 1H), 4.60 (d, J = 1.9 Hz, 2H), 4.04 (d, J = 8.0 Hz, 1H), 1.21 (s, 9H). 13C{1H} NMR (151 MHz, CDCl3) δ 168.7, 167.7, 165.1, 142.9, 140.3, 130.4 (q, J = 32.5 Hz), 129.6 (2C), 127.3, 126.0 (q, J = 3.7 Hz, 2C), 124.1 (d, J = 272.2 Hz), 122.9, 121.2, 69.4, 69.2, 52.1, 46.4, 28.5 (3C). IR (neat) νmax (cm–1): 3265, 2920, 1761, 1672, 1560, 1321, 1313, 1223, 1161, 1138, 1111, 1065, 1013, 943. HRMS (ESI) m/z: [M + Na]+ calcd. for C21H22F3N3NaO2S+, 460.1277; found 460.1290.
trans/cis-β-Lactam 6r
Prepared according to GP-B using 3r. Purification of the crude material by silica gel column chromatography (20% EtOAc/cHex) afforded the title compound as a clear oil in 63% yield as a mixture of diastereoisomers (ratio, 2:1) (47 mg, 0.11 mmol). Also prepared according to GP-C using 3r. Purification of the crude material by silica gel column chromatography (20% EtOAc/cHex) afforded the title compound as a clear oil in 76% yield as a mixture of diastereoisomers (ratio, 3:2) (61 mg, 0.15 mmol). Rf= 0.43 (50% EtOAc/cHex). Two diastereoisomers were present in NMR (ratio, 2: 1), of which the signals of the major diastereoisomer are marked with ● and minor diastereoisomer are marked with ■. 1H NMR (600 MHz, chloroform-d) δ ● 7.29 (d, J = 8.6 Hz, 2H), ■ 7.23 (d, J = 8.6 Hz, 2H), ■ 7.18 (d, J = 7.7 Hz, 2H), ● 6.86 (d, J = 8.6 Hz, 2H), ■ 6.82 (d, J = 8.6 Hz, 1H), ● 6.20 (d, J = 7.7 Hz, 1H), ■● 5.76–5.62 (m, 1H), ■● 5.41 (d, J = 17.1 Hz, 1H), ■● 5.30 (d, J = 12.0 Hz, 1H), ■ 4.58 (d, J = 14.7 Hz, 1H), ● 4.54 (d, J = 15.0 Hz, 1H), ● 4.42 (d, J = 15.0 Hz, 1H), ●■ 4.37 (d, J = 14.7 Hz, 1H), ● 4.25 (d, J = 7.9 Hz, 1H), ● 4.21–4.13 (m, 1H), ■● 4.13–4.08 (m, 1H), ■ 4.04 (d, J = 7.9 Hz, 1H), ■ 3.93 (dq, J = 10.8, 7.2 Hz, 1H), ● 3.79 (s, 3H), ■ 3.77 (s, 3H), ■ 3.75–3.69 (m, 1H), ● 3.64 (ddt, J = 14.7, 7.8, 4.1 Hz, 1H), ■● 1.89–1.51 (m, 5H), ■● 1.43–1.02 (m, 7H), ● 0.91 (qd, J = 11.9, 3.1 Hz, 1H), ■ 0.82–0.74 (m, 1H). 13C{1H} NMR (151 MHz, CDCl3) δ ■ 169.7, ● 169.0, ● 167.4, ■ 166.4, ■ 164.8, ● 163.9, ● 159.6, ■ 159.3, ■ 130.8 (2C), ● 130.5 (2C), ● 128.1, ● 127.4, ■ 127.2, ■ 127.0, ■ 123.1, ● 122.6, ● 114.4 (2C), ■ 113.9 (2C), ● 68.9, ■ 67.8, ■ 64.5, ● 62.7, ● 62.6, ■ 62.5, ● 55.5, ■ 55.4, ■ 48.8, ● 48.5, ■ 46.1, ● 45.6, ■ 32.8, ● 32.7, ● 32.62, ■ 32.58, ● 25.6, ■ 25.50, ■ 24.8, ● 24.7, ● 24.6, ■● 14.07, ■ 14.03. IR (neat) νmax (cm–1): 3339, 2930, 2854, 2338, 1757, 1668, 1612, 1516, 1450, 1381, 1246, 1175, 1103, 1034, 814, 623, 513. HRMS (ESI) m/z: [M + Na]+ calcd. for C23H30N2O5Na+, 437.2047; found, 437.2057.
trans-β-Lactam 6s
Prepared according to GP-B using 3s. Purification of the crude material by silica gel column chromatography (20% EtOAc/cHex) afforded the title compound as yellow crystals in 36% yield (36 mg, 0.07 mmol). Rf= 0.57 (60% EtOAc/cHex). 1H NMR (600 MHz, CDCl3) δ 10.07 (s, 1H), 8.84 (s, 1H), 8.63 (d, J = 2.1 Hz, 1H), 8.56–8.42 (m, 1H), 7.39–7.13 (m, 15H), 5.32–5.20 (m, 1H), 5.16–4.93 (m, 2H), 4.04 (d, J = 6.8 Hz, 1H), 3.37 (ddd, J = 13.8, 9.8, 6.5 Hz, 1H), 3.24 (dd, J = 10.0, 6.7 Hz, 1H), 1.80 (dq, J = 13.8, 3.9, 0.9 Hz, 2H), 0.90 (t, J = 7.4 Hz, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ 167.5, 166.9, 152.3, 144.9, 144.5 (3C), 144.3, 142.5, 128.6 (6C), 128.1 (6C), 127.8 (3C), 127.3, 122.6, 71.3, 68.5, 65.9, 46.2, 21.3, 12.1. IR (neat) νmax (cm–1): 3227, 3057, 2930, 2336, 1751, 1688, 1551, 1483, 1447, 1391, 1263, 1022, 930, 758, 700, 602, 401. m.p.: 193–198 °C. HRMS (ESI) m/z: [M + Na]+ calcd. for C32H30N4O2Na+, 525.2261; found, 525.2279.
cis-β-Lactam 6s
Prepared according to GP-C using 3s. Purification of the crude material by silica gel column chromatography (20% EtOAc/cHex) afforded the title compound as yellow crystals in 45% yield (40 mg, 0.08 mmol). Rf= 0.51 (60% EtOAc/cHex). 1H NMR (600 MHz, CDCl3) δ 9.10 (s, 1H), 8.74 (d, J = 1.4 Hz, 1H), 8.63 (d, J = 2.4 Hz, 1H), 8.57–8.46 (m, 1H), 7.36–7.19 (m, 15H), 5.73 (ddd, J = 17.1, 10.3, 8.6 Hz, 1H), 5.37 (d, J = 17.1 Hz, 1H), 5.26 (d, J = 10.3 Hz, 1H), 4.08 (d, J = 8.6 Hz, 1H), 3.33–3.25 (m, 1H), 3.22–3.12 (m, 1H), 1.64 (dddd, J = 24.8, 14.6, 8.1, 5.0 Hz, 2H), 0.83 (t, J = 7.4 Hz, 3H). 13C{1H} NMR (151 MHz, CDCl3) δ 168.1, 165.7, 153.9, 144.5, 144.2 (3C), 143.7, 143.0, 128.8 (6C), 128.6, 128.1 (6C), 127.3 (3C), 122.7, 71.5, 69.4, 67.1, 45.9, 21.7, 11.9. IR (neat) νmax (cm–1): 3225, 3053, 2925, 1783, 1664, 1651, 1485, 1448, 1391, 1263, 1022, 930, 757, 700, 602, 401. m.p.: 193–198 ° C. HRMS (ESI) m/z: [M + Na]+ calcd. for C32H30N4O2Na+, 525.2261; found, 525.2279.
trans-β-Lactam 6t
Prepared according to GP-B using 3t. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a brown oil in 93% yield (87 mg, 0.19 mmol). Rf= 0.37 (60% EtOAc/cHex). 1H NMR (600 MHz, CDCl3) δ 9.41 (s, 1H), 7.47 (t, J = 7.8 Hz, 1H), 7.06 (d, J = 7.8 Hz, 1H), 6.85 (d, J = 7.8 Hz, 1H), 6.83 (s, 1H), 6.79 (s, 2H), 5.22 (d, J = 16.7 Hz, 1H), 5.06–4.97 (m, 1H), 4.95 (d, J = 10.3 Hz, 1H), 4.05 (d, J = 7.6 Hz, 1H), 3.85 (s, 3H), 3.84 (s, 3H), 3.70 (ddd, J = 13.7, 9.9, 4.8 Hz, 1H), 3.45–3.38 (m, 1H), 3.34 (dt, J = 13.7, 9.9, 7.4 Hz, 1H), 3.17 (ddd, J = 13.7, 9.9, 4.8 Hz, 1H), 2.53 (s, 3H), 1.40 (s, 9H). 13C{1H} NMR (151 MHz, CDCl3) δ 168.4, 168.2, 157.3, 155.7, 149.0, 147.7, 137.6, 132.2, 128.6, 122.3, 120.93, 120.92, 119.7, 112.4, 111.4, 69.7, 65.8, 56.1, 56.0, 51.4, 46.5, 33.6, 28.8 (3C), 24.3. IR (neat) νmax (cm–1): 2962, 2934, 2341, 1753, 1674, 1585, 1547, 1512, 1456, 1360, 1261, 1230, 1149, 1028, 991, 922, 731, 646, 536, 501, 461. HRMS (ESI) m/z: [M + Na]+ calcd. for C26H33N3O4Na+, 474.2363; found, 474.2381.
cis-β-Lactam 6t
Prepared according to GP-C using 3t. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a brown oil in 30% yield (29 mg, 0.06 mmol). Rf= 0.33 (50% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 8.78 (s, 1H), 7.52 (t, J = 7.8 Hz, 1H), 7.10 (d, J = 7.8 Hz, 1H), 6.99 (d, J = 7.8 Hz, 1H), 6.81–6.77 (m, 3H), 5.92 (ddd, J = 17.1, 10.2, 8.8 Hz, 1H), 5.39–5.29 (m, 2H), 3.85 (s, 3H), 3.84 (s, 3H), 3.81 (d, J = 8.7 Hz, 1H), 3.68 (dt, J = 14.0, 9.3, 6.9 Hz, 1H), 3.44 (dt, J = 14.0, 9.3, 6.9 Hz, 1H), 3.27–3.13 (m, 2H), 2.55 (s, 3H), 1.36 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 168.7, 166.4, 157.8, 157.6, 149.0, 147.7, 138.0, 132.0, 128.9, 122.5, 121.9, 120.9, 117.8, 112.2, 111.3, 70.3, 66.8, 56.1, 56.0, 51.6, 46.4, 33.8, 28.9 (3C), 24.4. IR (neat) νmax (cm–1): 3381, 2959, 2926, 2858, 2297, 1745, 1664, 1583, 1514, 1448, 1358, 1259, 1232, 1026, 935, 798, 579, 462, 401. HRMS (ESI) m/z: [M + Na]+ calcd. for C26H33N3O4Na+, 474.2363; found, 474.2381.
trans-β-Lactam 6u
Prepared according to GP-B using 3u. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a brown oil in 83% yield (70 mg, 0.15 mmol). Rf= 0.37 (50% EtOAc/cHex). 1H NMR (600 MHz, CDCl3) δ 7.81 (s, 1H), 7.67 (q, J = 7.9 Hz, 1H), 6.90 (d, J = 7.5 Hz, 1H), 6.87–6.84 (m, 2H), 6.84–6.74 (m, 2H), 5.26 (d, J = 16.8 Hz, 1H), 5.13–5.04 (m, 1H), 5.02 (d, J = 10.3 Hz, 1H), 4.10 (d, J = 7.8 Hz, 1H), 3.86 (d, J = 2.5 Hz, 3H), 3.84 (d, J = 2.5 Hz, 3H), 3.78–3.67 (m, 1H), 3.49–3.42 (m, 1H), 3.39–3.28 (m, 1H), 3.21–3.09 (m, 1H), 1.38 (s, 9H). 13C{1H} NMR (151 MHz, CDCl3) δ 168.2, 167.7, 162.3 (d, J = 243.3 Hz), 154.9 (d, J = 11.8 Hz), 149.1, 147.8, 142.3 (d, J = 7.6 Hz), 132.0, 128.1, 121.7, 121.0, 120.1 (d, J = 4.0 Hz), 112.4, 111.4, 108.9 (d, J = 35.6 Hz), 69.9, 65.6, 56.1, 56.0, 51.9, 46.7, 33.7, 28.7 (3C). IR (neat) νmax (cm–1): 2964, 2934, 2301, 1751, 1674, 1591, 1514, 1450, 1261, 1229, 1148, 1028, 802, 731, 646, 461, 401. HRMS (ESI) m/z: [M + H]+ calcd. for C25H31N3O4F+, 456.2293; found, 456.2306.
cis-β-Lactam 6u
Prepared according to GP-C using 3u. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a brown oil in 61% yield (57 mg, 0.13 mmol). Rf= 0.32 (50% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 7.74 (d, J = 7.9 Hz, 1H), 7.10 (dd, J = 7.9, 2.4 Hz, 1H), 7.02 (s, 1H), 6.89 (dd, J = 7.9, 2.4 Hz, 1H), 6.78–6.73 (m, 3H), 5.87 (ddd, J = 17.1, 10.4, 8.5 Hz, 1H), 5.42 (dt, J = 17.1, 1.2 Hz, 1H), 5.35 (d, J = 10.4 Hz, 1H), 4.09 (d, J = 8.4 Hz, 1H), 3.85 (s, 3H), 3.84 (s, 3H), 3.62 (ddd, J = 13.8, 8.8, 5.8 Hz, 1H), 3.46 (dt, J = 13.8, 8.8, 7.5 Hz, 1H), 3.18–3.02 (m, 2H), 1.31 (s, 9H). 13C{1H} NMR (151 MHz, CDCl3) δ 168.4, 166.3, 162.5 (d, J = 243.0 Hz), 156.4 (d, J = 11.9 Hz), 149.1, 147.8, 142.3 (d, J = 7.7 Hz), 131.6, 128.5, 122.1, 120.9, 119.2 (d, J = 4.1 Hz), 112.2, 111.4, 109.3 (d, J = 36.0 Hz), 70.3, 65.2, 56.04, 55.96, 52.1, 46.1, 33.9, 28.8 (3C). IR (neat) νmax (cm–1): 3377, 2961, 2934, 2328, 1749, 1668, 1599, 1576, 1516, 1448, 1151, 1026, 941, 860, 802, 744, 584, 463, 401. HRMS (ESI) m/z: [M + H]+ calcd. for C25H31N3O4F+, 456.2293; found, 456.2306.
trans-β-Lactam 6v
Prepared according to GP-B using 3v. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a brown oil in 54% yield (53 mg, 0.10 mmol). Rf= 0.40 (50% EtOAc/cHex). 1H NMR (600 MHz, CDCl3) δ 8.19 (s, 1H), 7.43–7.35 (m, 2H), 6.92 (dd, J = 7.4, 1.1 Hz, 1H), 6.84 (d, J = 1.5 Hz, 1H), 6.82–6.79 (m, 2H), 5.29–5.23 (m, 1H), 5.11–5.00 (m, 2H), 4.08 (d, J = 6.9 Hz, 1H), 3.85 (s, 3H), 3.83 (s, 3H), 3.78–3.70 (m, 1H), 3.42–3.32 (m, 2H), 3.18–3.11 (m, 1H), 1.39 (s, 9H). 13C{1H} NMR (151 MHz, CDCl3) δ 168.1, 167.6, 157.5, 149.1, 147.8, 140.5, 139.4, 132.0, 128.1, 127.2, 121.69, 121.66, 121.0, 112.4, 111.4, 69.7, 65.7, 56.1, 56.0, 51.8, 46.7, 33.6, 28.7 (3C). IR (neat) νmax (cm–1): 3377, 2951, 2330, 1749, 1668, 1587, 1516, 1447, 1231, 1149, 1028, 939, 800, 744, 581, 463. m.p.: 115–125 °C. HRMS (ESI) m/z: [M + Na]+ calcd. for C25H30N3O4BrNa+, 538.1312; found, 538.1334.
cis-β-Lactam 6v
Prepared according to GP-C using 3v. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a brown oil in 50% yield (56 mg, 0.11 mmol). Rf= 0.32 (50% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 7.52 (s, 1H), 7.48 (t, J = 7.6 Hz, 1H), 7.43 (d, J = 7.6 Hz, 1H), 7.12 (d, J = 7.6 Hz, 1H), 6.83–6.74 (m, 3H), 5.88 (ddd, J = 17.1, 10.3, 8.4 Hz, 1H), 5.41 (d, J = 17.1 Hz, 1H), 5.35 (d, J = 10.3 Hz, 1H), 3.96 (d, J = 8.4 Hz, 1H), 3.86 (s, 3H), 3.85 (s, 3H), 3.68 (ddd, J = 13.9, 9.0, 6.0 Hz, 1H), 3.43 (ddd, J = 13.9, 9.0, 6.0 Hz, 1H), 3.24–3.09 (m, 2H), 1.34 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 168.3, 165.9, 159.2, 149.0, 147.7, 140.9, 139.6, 131.6, 128.3, 127.5, 122.2, 120.9, 120.4, 112.1, 111.3, 70.2, 66.1, 56.01, 55.97, 51.9, 46.3, 33.8, 28.8 (3C). IR (neat) νmax (cm–1): 3377, 2951, 2330, 1749, 1668, 1587, 1516, 1447, 1231, 1149, 1028, 939, 800, 744, 581, 463. HRMS (ESI) m/z: [M + Na]+ calcd. for C25H30N3O4BrNa+, 538.1312; found, 538.1331.
trans-β-Lactam 6w
Prepared according to GP-B using 3w. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a brown oil in 95% yield (88 mg, 0.19 mmol). Rf= 0.33 (50% EtOAc/cHex). 1H NMR (600 MHz, CDCl3) δ 7.77 (s, 1H), 7.50–7.47 (m, 1H), 6.83 (s, 1H), 6.80 (s, 2H), 6.68 (t, J = 7.9 Hz, 2H), 5.24 (d, J = 17.0 Hz, 1H), 5.10 (ddd, J = 17.0, 10.2, 8.1 Hz, 1H), 4.99 (d, J = 10.2 Hz, 1H), 4.11 (d, J = 8.1 Hz, 1H), 3.89 (s, 3H), 3.85 (s, 3H), 3.84 (s, 3H), 3.70–3.63 (m, 1H), 3.51 (ddd, J = 13.6, 10.1, 7.0 Hz, 1H), 3.32 (ddd, J = 13.6, 10.1, 7.0 Hz, 1H), 3.22–3.15 (m, 1H), 1.39 (s, 9H). 13C{1H} NMR (600 MHz, CDCl3) δ 168.4, 168.3, 163.4, 153.5, 149.1, 147.7, 139.7, 132.0, 128.7, 121.0, 120.9, 115.3, 112.4, 111.4, 110.6, 70.5, 65.4, 56.1, 56.0, 53.7, 51.7, 46.5, 33.8, 28.9 (3C). IR (neat) νmax (cm–1): 2964, 2934, 1751, 1674, 1580, 1514, 1464, 1414, 1327, 1261, 1232, 1153, 1026, 989, 924, 800, 733, 644, 617, 544, 503, 463. HRMS (ESI) m/z: [M + Na]+ calcd. for C26H33N3O5Na+, 490.2312; found, 490.2328.
trans-β-Lactam 6x
Prepared according to GP-B using 3x. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a brown oil in 99% yield (90 mg, 0.20 mmol). Rf= 0.40 (50% EtOAc/cHex). 1H NMR (600 MHz, CDCl3) δ 8.92 (s, 1H), 8.40 (d, J = 5.0 Hz, 1H), 7.03 (d, J = 5.0 Hz, 1H), 6.87 (s, 2H), 6.85–6.77 (m, 2H), 5.23 (d, J = 16.5 Hz, 1H), 5.06–4.90 (m, 2H), 4.09 (d, J = 7.5 Hz, 1H), 3.85 (s, 3H), 3.85 (s, 3H), 3.75–3.70 (m, 1H), 3.48–3.38 (m, 2H), 3.19–3.12 (m, 1H), 2.22 (s, 3H), 1.38 (s, 9H). 13C{1H} NMR (151 MHz, CDCl3) δ 168.7, 168.5, 156.2, 149.05, 149.01, 148.2, 147.7, 132.3, 128.6, 123.8, 123.3, 121.0, 120.9, 112.4, 111.4, 69.8, 65.8, 56.03, 55.97, 51.5, 46.7, 33.5, 28.8 (3C), 21.5. IR (neat) νmax (cm–1): 2962, 2930, 2309, 1753, 1674, 1601, 1545, 1514, 1456, 1261, 1230, 1148, 1028, 926, 810, 644, 451, 401. HRMS (ESI) m/z: [M + Na]+ calcd. for C26H33N3O4Na+, 474.2363; found, 474.2375.
cis-β-Lactam 6x
Prepared according to GP-C using 3x. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a brown oil in 10% yield (10 mg, 0.02 mmol). Rf = 0.33 (50% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 8.40 (d, J = 4.9 Hz, 1H), 8.30 (s, 1H), 7.06 (d, J = 4.9 Hz, 1H), 7.00 (s, 1H), 6.83–6.78 (m, 3H), 5.92 (ddd, J = 17.1, 10.2, 8.7 Hz, 1H), 5.41–5.30 (m, 2H), 3.88–3.84 (m, 7H), 3.71 (ddd, J = 13.8, 9.3, 5.3 Hz, 1H), 3.46 (dt, J = 13.8, 9.3, 7.3 Hz, 1H), 3.25 (dt, J = 13.8, 9.3, 7.3 Hz, 1H), 3.17 (ddd, J = 13.8, 9.3, 5.3 Hz, 1H), 2.27 (s, 3H), 1.34 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 168.8, 166.5, 158.2, 149.2, 149.0, 148.4, 147.7, 132.1, 128.9, 123.9, 122.0, 121.8, 120.8, 112.2, 111.3, 70.4, 66.5, 56.02, 55.99, 51.7, 46.5, 33.8, 28.9 (3C), 21.5. IR (neat) νmax (cm–1): 2962, 2930, 2309, 1753, 1674, 1601, 1545, 1514, 1456, 1261, 1230, 1148, 1028, 926, 810, 644, 451, 401. HRMS (ESI) m/z: [M + Na]+ calcd. for C26H33N3O4Na+, 474.2363; found, 474.2375.
trans-β-Lactam 6y
Prepared according to GP-B using 3y. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a brown oil in 68% yield (60 mg, 0.13 mmol). Rf= 0.46 (50% EtOAc/cHex). 1H NMR (600 MHz, CDCl3) δ 8.47–8.43 (m, 2H), 7.24 (dd, J = 5.3, 1.7 Hz, 1H), 7.08 (s, 1H), 6.84 (s, 1H), 6.82 (d, J = 7.6 Hz, 2H), 5.27–5.23 (m, 1H), 5.08–4.98 (m, 2H), 4.10 (d, J = 7.1 Hz, 1H), 3.85 (s, 6H), 3.79–3.72 (m, 1H), 3.40 (tt, J = 16.4, 8.1 Hz, 2H), 3.19–3.10 (m, 1H), 1.37 (s, 9H). 13C{1H} NMR (151 MHz, CDCl3) δ 168.4, 167.5, 158.2, 149.3, 149.1, 147.9, 146.0, 131.8, 128.2, 123.4, 123.2, 121.6, 120.8, 112.2, 111.5, 69.9, 65.9, 56.00, 55.96, 51.7, 46.8, 33.6, 28.7 (3C). IR (neat) νmax (cm–1): 2964, 2932, 1755, 1674, 1566, 1551, 1514, 1456, 1261, 1231, 1148, 1028, 920, 729, 644, 457, 401. HRMS (ESI) m/z: [M + Na]+ calcd. for C25H30N3O4ClNa+, 494.1817; found, 494.1827.
cis-β-Lactam 6y
Prepared according to GP-C using 3y. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a brown oil in 37% yield (35 mg, 0.07 mmol). Rf= 0.42 (50% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 8.46 (d, J = 5.3 Hz, 1H), 7.73 (s, 1H), 7.29–7.24 (m, 1H), 7.21 (d, J = 1.8 Hz, 1H), 6.82–6.76 (m, 3H), 5.89 (ddd, J = 17.1, 10.3, 8.5 Hz, 1H), 5.39 (d, J = 17.1 Hz, 1H), 5.34 (d, J = 10.3 Hz, 1H), 3.98 (d, J = 8.5 Hz, 1H), 3.88–3.83 (m, 6H), 3.75–3.64 (m, 1H), 3.48–3.38 (m, 1H), 3.17 (ttd, J = 16.9, 8.4, 5.3 Hz, 2H), 1.32 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 168.6, 166.0, 159.8, 149.7, 149.1, 147.8, 145.9, 131.6, 128.5, 123.5, 122.2, 122.0, 120.8, 112.0, 111.4, 70.3, 66.2, 56.00, 55.95, 51.91, 46.5, 33.8, 28.8 (3C). IR (neat) νmax (cm–1): 2964, 2932, 1755, 1674, 1566, 1551, 1514, 1456, 1261, 1230, 1148, 1028, 920, 729, 644, 457, 401. HRMS (ESI) m/z: [M + Na]+ calcd. for C25H30N3O4ClNa+, 494.1817; found, 494.1825.
trans-β-Lactam 6z
Prepared according to GP-B using 3z. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a brown oil in 15% yield (15 mg, 0.03 mmol). Rf= 0.38 (50% EtOAc/cHex). 1H NMR (600 MHz, CDCl3) δ 8.41 (s, 1H), 8.38 (d, J = 5.2 Hz, 1H), 7.42 (dd, J = 5.2, 1.7 Hz, 1H), 7.31 (d, J = 1.7 Hz, 1H), 6.83 (dd, J = 5.8, 1.8 Hz, 2H), 6.81 (d, J = 8.7 Hz, 1H), 5.31–5.23 (m, 1H), 5.09–5.00 (m, 2H), 4.11 (d, J = 7.4 Hz, 1H), 3.86 (s, 3H), 3.86 (s, 3H), 3.78–3.71 (m, 1H), 3.44 (ddd, J = 13.5, 9.4, 7.5 Hz, 1H), 3.38 (dt, J = 13.5, 9.4, 7.5 Hz, 1H), 3.17 (td, J = 8.8, 4.4 Hz, 1H), 1.38 (s, 9H). 13C{1H} NMR (151 MHz, CDCl3) δ 168.4, 167.5, 158.0, 149.2, 149.1, 147.9, 134.7, 131.8, 128.2, 126.4, 126.2, 121.7, 120.8, 112.2, 111.5, 69.9, 66.0, 56.03, 55.98, 51.7, 46.7, 33.6, 28.8 (3C). IR (neat) νmax (cm–1): 2962, 2932, 2339, 1753, 1674, 1553, 1514, 1456, 1387, 1261, 1230, 1148, 1028, 920, 729, 453. HRMS (ESI) m/z: [M + Na]+ calcd. for C25H30N3O4BrNa+, 538.1312; found, 538.1332.
cis-β-Lactam 6z
Prepared according to GP-C using 3z. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a brown oil in 56% yield (59 mg, 0.11 mmol). Rf= 0.33 (50% EtOAc/cHex). 1H NMR (600 MHz, CDCl3) δ 8.37 (d, J = 5.6 Hz, 1H), 7.66 (s, 1H), 7.43 (d, J = 7.1 Hz, 2H), 6.84–6.70 (m, 3H), 5.89 (ddd, J = 17.1, 10.3, 8.5 Hz, 1H), 5.38 (d, J = 17.1 Hz, 1H), 5.34 (d, J = 10.3 Hz, 1H), 3.99 (d, J = 8.5 Hz, 1H), 3.85 (s, 3H), 3.85 (s, 3H), 3.72–3.60 (m, 1H), 3.50–3.38 (m, 1H), 3.20–3.09 (m, 2H), 1.32 (s, 9H). 13C{1H} NMR (600 MHz, CDCl3) δ 168.6, 166.0, 159.6, 149.5, 149.1, 147.8, 134.5, 131.6, 128.6, 126.5, 125.0, 122.2, 120.8, 112.1, 111.5, 70.3, 66.1, 56.02, 55.96, 51.9, 46.4, 33.8, 28.8 (3C). IR (neat) νmax (cm–1): 2962, 2932, 2339, 1753, 1674, 1555, 1514, 1456, 1387, 1261, 1230, 1145, 1028, 920, 729, 453. HRMS (ESI) m/z: [M + Na]+ calcd. for C25H30N3O4BrNa+, 538.1312; found, 538.1331.
trans/cis-β-Lactam 6za
Prepared according to GP-B using 3za. Purification of the crude material by silica gel column chromatography (40% EtOAc/cHex) afforded the title compound as an amber oil in 27% trans diastereoisomer and 32% cis diastereoisomer yield (0.059 g, 0.11 mmol). Rf= 0.35 (50% EtOAc/cHex). Two diastereoisomers were present in NMR (trans: cis = 1: 2.8) of which the signals of the cis diastereoisomer are marked with ■ and trans diastereoisomer are marked with ●. 1H NMR (500 MHz, CDCl3) δ ●■ 9.14 (s, 1H), ■ 8.48 (s, 1H), ● 8.19 (dd, J = 8.3, 2.0 Hz, 1H), ■ 8.14 (dd, J = 8.3, 2.0 Hz, 1H), ● 7.68 (s, 1H), ● 7.26 (d, J = 4.9 Hz, 1H), ■ 7.08 (d, J = 8.3 Hz, 1H), ●■ 6.88–6.73 (m, 3H), ● 5.96–5.83 (m, 1H), ● 5.38 (d, J = 17.1 Hz, 1H), ● 5.35 (d, J = 10.3 Hz, 1H), ■ 5.27–5.19 (m, 1H), ■ 5.02–4.92 (m, 2H), ■ 4.12 (d, J = 6.5 Hz, 1H), ● 3.98 (d, J = 8.5 Hz, 1H), ● 3.96 (s, 3H), ■ 3.95 (s, 3H), ■ 3.87 (s, 3H), ● 3.85 (s, 3H), ■ 3.84 (s, 3H), ● 3.83 (s, 3H), ■ 3.79–3.74 (m, 1H), ● 3.67 (td, J = 15.1, 14.6, 7.0 Hz, 1H), ●■ 3.43 (dq, J = 14.3, 7.3, 6.6 Hz, 1H), ■ 3.37 (dd, J = 8.0, 4.7 Hz, 1H), ●■ 3.21–3.11 (m, ● 2H, ■ 1H), ■ 1.38 (s, 9H), ● 1.32 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ ● 168.5, ■ 168.2, ■ 167.4, ● 166.0, ● 165.14, ■ 165.13, ● 162.2, ■ 160.6, ● 150.0, ■ 149.6, ■ 149.03, ● 149.00, ●■ 147.8, ● 138.5, ■ 138.3, ■ 131.9, ● 131.6, ● 128.5, ■ 128.1, ● 125.3, ■ 125.1, ■ 122.33, ● 122.28, ■ 121.6, ● 121.0, ■ 120.9, ● 120.8, ■ 112.4, ● 112.1, ●■ 111.3, ● 70.7, ■ 70.6, ● 66.1, ■ 66.0, ■ 56.05, ● 56.01, ●■ 55.95, ●■ 52.7, ● 51.9, ■ 51.7, ■ 46.9, ● 46.5, ■ 33.8, ● 33.6, ● 28.8 (3C), ■ 28.7 (3C). IR (neat) νmax (cm–1): 2955, 1751, 1728, 1676, 1514, 1290, 1261, 1236, 1142. HRMS (ESI) m/z: [M + Na]+ calcd. for C27H32N3NaO6+, 518.2262; found, 518.2257.
trans-β-Lactam 6zb
Prepared according to GP-B using 3zb. Purification of the crude material by silica gel column chromatography (40% EtOAc/cHex) afforded the title compound as a brown oil in 41% yield (25 mg, 0.05 mmol). Rf= 0.38 (50% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 8.17 (d, J = 8.9 Hz, 2H), 7.21 (d, J = 8.9 Hz, 2H), 6.84 (d, J = 8.1 Hz, 1H), 6.81 (d, J = 1.9 Hz, 1H), 6.77 (dd, J = 8.1, 1.9 Hz, 1H), 5.30 (dt, J = 16.8, 1.2 Hz, 1H), 5.25 (s, 1H), 5.09–5.02 (m, 1H), 4.95 (ddd, J = 16.8, 10.3, 8.3 Hz, 1H), 4.66 (d, J = 8.3 Hz, 1H), 3.89 (s, 3H), 3.86 (s, 3H), 3.52 (ddd, J = 13.7, 9.1, 7.7 Hz, 1H), 3.44 (dd, J = 9.1, 4.9 Hz, 1H), 3.34–3.23 (m, 1H), 2.98 (ddd, J = 13.7, 9.1, 4.9 Hz, 1H), 1.38 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 168.7, 167.7, 149.2, 148.1, 147.9, 142.8, 130.9, 128.5 (2C), 128.4, 124.1 (2C), 122.3, 120.7, 112.2, 111.4, 71.1, 63.1, 56.1, 56.0, 52.9, 45.7, 33.8, 28.8 (3C). IR (neat) νmax (cm–1): 2966, 2932, 1747, 1676, 1599, 1514, 1454, 1346, 1261, 1230, 1148, 1026, 918, 851, 808, 729, 401. HRMS (ESI) m/z: [M + Na]+ calcd. for C26H31N3O6Na+, 504.2105; found, 504.2119.
cis-β-Lactam 6zb
Prepared according to GP-C using 3zb. Purification of the crude material by silica gel column chromatography (40% EtOAc/cHex) afforded the title compound as a brown oil in 31% yield (41 mg, 0.09 mmol). Rf = 0.40 (50% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 8.18 (d, J = 8.9 Hz, 2H), 7.49 (d, J = 8.9 Hz, 2H), 6.71 (d, J = 8.1 Hz, 1H), 6.65–6.57 (m, 2H), 5.89 (ddd, J = 17.2, 10.3, 8.1 Hz, 1H), 5.50–5.44 (m, 2H), 5.39 (d, J = 10.3 Hz, 1H), 4.18 (d, J = 8.1 Hz, 1H), 3.84 (s, 3H), 3.81 (s, 3H), 3.42 (ddd, J = 14.2, 8.1, 6.1 Hz, 1H), 3.33 (dq, J = 14.0, 7.8, 6.8 Hz, 1H), 3.00–2.86 (m, 2H), 1.32 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 168.0, 167.0, 149.0, 148.0, 147.8, 144.0, 130.6, 128.8, 128.3 (2C), 124.1 (2C), 122.3, 120.6, 111.9, 111.3, 69.8, 65.1, 56.01, 55.97, 52.7, 45.2, 33.7, 28.8 (3C). IR (neat) νmax (cm–1): 2966, 2932, 1747, 1676, 1599, 1514, 1454, 1346, 1261, 1230, 1148, 1026, 918, 851, 808, 729, 401. HRMS (ESI) m/z: [M + Na]+ calcd. for C26H31N3O6Na+, 504.2105; found, 504.2116.
trans-β-Lactam 6zc
Prepared according to GP-B using 3zc. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a brown oil in 16% yield (12 mg, 0.03 mmol). Rf= 0.24 (100% EtOAc). Two diastereoisomers were present in NMR (trans:cis = 5:2), of which the signals of the cis diastereoisomer are marked with ■ and trans diastereoisomer are marked with ●. 1H NMR (500 MHz, CDCl3) δ ■ 8.61 (d, J = 6.1 Hz, 2H), ● 8.58 (d, J = 6.0 Hz, 2H), ■ 7.24 (d, J = 6.2 Hz, 2H), ● 6.96 (d, J = 6.1 Hz, 2H), ● 6.83–6.75 (m, 3H), ■ 6.73 (d, J = 8.1 Hz, 1H), ■ 6.66–6.60 (m, 2H), ■ 5.87 (ddd, J = 17.2, 10.2, 8.2 Hz, 1H), ■ 5.47 (s, 1H), ■ 5.44 (d, J = 17.1 Hz, 1H), ■ 5.37 (d, J = 10.3 Hz, 1H), ● 5.32–5.26 (m, 2H), ● 5.07–5.03 (m, 1H), ● 4.97 (ddd, J = 16.9, 10.2, 8.3 Hz, 1H), ● 4.61 (d, J = 8.2 Hz, 1H), ■ 4.13 (d, J = 8.2 Hz, 1H), ● 3.86 (s, 3H), ● 3.85 (s, 3H), ■ 3.82 (s, 3H), ■ 3.82 (s, 3H), ● 3.52 (ddd, J = 13.7, 9.5, 7.4 Hz, 1H), ●■ 3.44–3.32 (m, ● 1H, ■ 2H), ● 3.24 (ddd, J = 13.6, 9.5, 7.4 Hz, 1H), ●■ 3.01–2.93 (m, 1H), ■ 2.92–2.85 (m, 1H), ● 1.37 (s, 9H), ■ 1.31 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ ● 168.7, ■ 168.1, ● 167.5, ■ 166.6, ■ 150.6 (2C), ● 150.5 (2C), ● 149.1, ■ 149.0, ● 148.0, ■ 147.9, ■ 145.9, ● 144.6, ● 130.9, ■ 130.7, ■ 128.9, ● 128.4, ● 122.2 (2C), ●■ 122.1, ■ 122.0 (2C), ● 120.7, ■ 120.6, ● 112.2, ■ 111.9, ● 111.40, ■ 111.38, ● 70.6, ■ 69.2, ■ 64.7, ● 62.8, ● 56.05, ■ 56.01, ● 56.00, ■ 55.96, ● 52.8, ■ 52.6, ● 45.5, ■ 45.1, ● 33.8, ■ 33.7, ■ 28.8 (3C), ● 28.7 (3C). IR (neat) νmax (cm–1): 2964, 2934, 1747, 1672, 1595, 1512, 1456, 1263, 1232, 1148, 1028, 916, 731, 644, 519. HRMS (ESI) m/z: [M + Na]+ calcd. for C25H31N3O4Na+, 460.2207; found, 460.2215.
cis-β-Lactam 6zc
Prepared according to GP-C using 3zc. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound as a brown oil in 35% yield (37 mg, 0.09 mmol). Rf= 0.12 (100% EtOAc). 1H NMR (500 MHz, CDCl3) δ 8.62 (d, J = 6.2 Hz, 2H), 7.32–7.14 (m, 2H), 6.73 (d, J = 8.1 Hz, 1H), 6.63 (dd, J = 8.1, 1.8 Hz, 2H), 5.88 (ddd, J = 17.2, 10.2, 8.2 Hz, 1H), 5.53–5.33 (m, 3H), 4.14 (d, J = 8.2 Hz, 1H), 3.85 (s, 3H), 3.83 (s, 3H), 3.36 (d, J = 7.5 Hz, 2H), 2.98 (dd, J = 14.3, 7.5 Hz, 1H), 2.89 (dd, J = 14.3, 7.5 Hz, 1H), 1.32 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 168.1, 166.7, 150.7 (2C), 149.1, 147.9, 145.9, 130.7, 128.9, 122.2, 122.0 (2C), 120.6, 111.9, 111.4, 69.3, 64.8, 56.05, 56.00, 52.6, 45.2, 33.7, 28.8 (3C). IR (neat) νmax (cm–1): 2964, 2934, 1747, 1672, 1595, 1512, 1456, 1263, 1232, 1148, 1028, 916, 731, 644, 519. HRMS (ESI) m/z: [M + Na]+ calcd. for C25H31N3O4Na+, 460.2207; found, 460.2212.
trans-N-(tert-Butyl)-1-(3,4-dimethoxyphenethyl)-3-ethyl-4-oxo-2-(pyridin-2-yl)azetidine-2-carboxamide (7)
To a stirred solution of 6a (44 mg, 0.1 mmol, 1 equiv) in EtOAc (2 mL, 0.05 M) was added Pd/C (11 mg; 25%, w/w). The suspension was filled with H2 and then degassed and backfilled with H2: the procedure was repeated three times and stirred overnight at room temperature. The crude was filtrated over a Celite pad with EtOAc, and the solvent was removed under vacuum, affording the title compound 7 as a yellow oil in 96% yield (0.044 g, 0.096 mmol). Rf = 0.43 (80% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 8.89 (s, 1H), 8.55 (d, J = 4.7 Hz, 1H), 7.58 (td, J = 7.8, 1.7 Hz, 1H), 7.23 (dd, J = 7.4, 4.9 Hz, 1H), 7.06 (d, J = 8.0 Hz, 1H), 6.86–6.76 (m, 3H), 3.85 (s, 3H), 3.83 (s, 3H), 3.66 (ddd, J = 17.9, 9.3, 5.6 Hz, 1H), 3.42–3.30 (m, 3H), 3.16 (td, J = 11.1, 9.6, 4.9 Hz, 1H), 1.37 (s, 9H), 1.05 (ddq, J = 28.5, 14.1, 7.3 Hz, 2H), 0.81 (t, J = 7.4 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 171.1, 168.7, 156.8, 148.9, 148.2, 147.6, 137.3, 132.2, 122.8, 122.6, 120.9, 112.3, 111.2, 69.6, 63.9, 56.0, 55.9, 51.3, 46.4, 33.7, 28.7 (3C), 19.6, 11.7. IR (neat) νmax (cm–1): 2964, 2933, 1747, 1672, 1514, 1261, 1234, 1155, 1140, 1028, 729, 646, 461, 401. HRMS (ESI) m/z: [M + Na]+ calcd. for C25H33N3NaO4+, 462.2363; found, 462.2362.
trans-N-(tert-Butyl)-1-(3,4-dimethoxyphenethyl)-3-(2-hydroxy-ethyl)-4-oxo-2-(pyridin-2-yl)azetidine-2-carboxamide (8)
To a solution of 6a (44 mg, 0.1 mmol, 1 equiv) in THF (1 mL, 0.1 M) cooled at 0 °C was added a solution of 9-BBN (0.5 M in THF, 0.6 mL, 3 equiv), and the mixture was stirred for 3 h at room temperature. Then, 3 M KHCO3 (150 μL) and 30% H2O2 (400 μL) were added to the mixture, which was stirred at room temperature for 1 h. EtOAc (5 mL) and a saturated solution of NH4Cl (2 mL) were added and the organic layer was washed with H2O (2 mL) and brine (2 mL) and dried over Na2SO4 and the solvent was removed under vacuum. Purification of the crude material by silica gel column chromatography (70% EtOAc/cHex) afforded the title compound 87 as a clear oil in 54% yield (0.032 g, 0.054 mmol). Rf= 0.13 (80% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 8.58 (d, J = 4.1 Hz, 1H), 8.45 (s, 1H), 7.62 (td, J = 7.8, 1.8 Hz, 1H), 7.27–7.23 (m, 1H), 7.09 (d, J = 8.0 Hz, 1H), 6.84–6.80 (m, 3H), 3.86 (s, 3H), 3.85 (s, 3H), 3.70 (ddd, J = 14.4, 10.0, 5.0 Hz, 1H), 3.64–3.50 (m, 4H), 3.30 (ddd, J = 13.4, 9.8, 7.2 Hz, 1H), 3.16 (ddd, J = 14.1, 9.7, 5.1 Hz, 1H), 2.16 (s, 1H), 1.44–1.39 (m, 1H), 1.37 (s, 9H), 1.30–1.21 (m, 1H). 13C{1H} NMR (126 MHz, CDCl3) δ 171.2, 168.7, 156.7, 149.1, 148.9, 147.8, 137.9, 132.0, 123.2, 122.7, 121.0, 112.4, 111.4, 69.6, 61.1, 60.1, 56.2, 56.1, 51.7, 46.8, 33.9, 28.8 (3C), 28.4. IR (neat) νmax (cm–1): 3302, 2957, 1744, 1663, 1514, 1431, 1259, 1230, 1157, 1138, 1026. HRMS (ESI) m/z: [M + Na]+ calcd. for C25H33N3NaO5+, 478.2312; found, 478.2314.
trans-N-(tert-Butyl)-1-(3,4-dimethoxyphenethyl)-3-((E)-4-methylstyryl)-4-oxo-2-(pyridine-2-yl)azetidine-2-carboxamide (9)
A solution of 6a (44 mg, 0.1 mmol, 1.0 equiv), Pd(OAc)2 (5 mg, 0.02 mmol, 0.2 equiv), P(OiPr)3 (5.5 μL, 0.02 mmol, 0.2 equiv), p-iodotoluene (44 mg, 0.2 mmol, 2.0 equiv), and TEA (27.9 μL, 0.25 mmol, 2.5 equiv) in dioxane (0.05 M, 2 mL) was refluxed overnight. The reaction mixture was diluted with DCM (5 mL), washed with 1 M HCl (5 mL) and brine (5 mL), and dried over Na2SO4 and the solvent was removed under vacuum. Purification of the crude material by silica gel column chromatography (30% EtOAc/cHex) afforded the title compound 9 as a clear oil in 56% yield (0.014 g, 0.027 mmol). Rf= 0.71 (50% EtOAc/cHex). 1H NMR (500 MHz, CDCl3) δ 8.85 (s, 1H), 8.55–8.53 (m, 1H), 7.58 (td, J = 7.8, 1.8 Hz, 1H), 7.22–7.18 (m, 1H), 7.09 (d, J = 8.0 Hz, 1H), 6.99 (d, J = 8.0 Hz, 2H), 6.89 (d, J = 8.1 Hz, 2H), 6.87–6.80 (m, 3H), 6.49 (d, J = 15.8 Hz, 1H), 5.27 (dd, J = 15.8, 8.3 Hz, 1H), 4.26 (d, J = 8.3 Hz, 1H), 3.87 (s, 3H), 3.85 (s, 3H), 3.76 (ddd, J = 14.1, 9.9, 4.8 Hz, 1H), 3.53–3.45 (m, 1H), 3.43–3.35 (m, 1H), 3.21 (ddd, J = 13.9, 9.6, 4.8 Hz, 1H), 2.26 (s, 3H), 1.41 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 168.8, 168.1, 156.6, 149.0, 148.5, 147.7, 137.8, 137.4, 135.5, 133.7, 132.1, 129.2 (2C), 126.3 (2C), 122.8, 122.6, 120.9, 118.8, 112.4, 111.3, 70.5, 65.6, 56.1, 56.0, 51.6, 46.7, 33.7, 28.8 (3C), 21.3. IR (neat) νmax (cm–1): 2930, 1753, 1672, 1514, 1261, 1234, 1157, 1142, 1028, 808, 731, 457, 401. HRMS (ESI) m/z: [M + Na]+ calcd. for C32H37N3NaO4+, 550.2676; found, 550.2676.
Acknowledgments
This work was financially supported by the Netherlands Organisation for Scientific Research (NWO). We thank Daniel Preschel for HRMS measurements and Elwin Janssen for the NMR support (both Vrije Universiteit Amsterdam). We thank SURFsara for the use of the Cartesius and Lisa supercomputer. Dr. Christophe Vande Velde (University of Antwerp) is gratefully acknowledged for X-ray crystallographic measurements and solving the structure of cis-6s. We also thank Prof. Kristof Van Hecke (Ghent University) for providing diffractometer time, and the Hercules Foundation (project AUGE/11/029 “3D-SPACE: 3D Structural Platform Aiming for Chemical Excellence”) for funding of the diffractometer.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.0c00575.
Author Contributions
The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- For selected reviews, see:; a Trost B. M.; Van Vranken D. L. Asymmetric Transition Metal-Catalyzed Allylic Alkylations. Chem. Rev. 1996, 96, 395–422. 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]; b Weaver J. D.; Recio A. III; Grenning A. J.; Tunge J. A. Transition Metal-Catalyzed Decarboxylative Allylation and Benzylation Reactions. Chem. Rev. 2011, 111, 1846–1913. 10.1021/cr1002744. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Cheng Q.; Tu H.-F.; Zheng C.; Qu J.-P.; Helmchen G.; You S.-L. Iridium-Catalyzed Asymmetric Allylic Substitution Reactions. Chem. Rev. 2019, 119, 1855–1969. 10.1021/acs.chemrev.8b00506. [DOI] [PubMed] [Google Scholar]
- Faltracco M.; Cotogno S.; Vande Velde C. M. L.; Ruijter E. Catalytic Asymmetric Synthesis of Diketopiperazines by Intramolecular Tsuji–Trost Allylation. J. Org. Chem. 2019, 84, 12058–12070. 10.1021/acs.joc.9b01994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yang Z.-P.; Wu Q.-F.; Shao W.; You S.-L. Iridium-Catalyzed Intramolecular Asymmetric Allylic Dearomatization Reaction of Pyridines, Pyrazines, Quinolines, and Isoquinolines. J. Am. Chem. Soc. 2015, 137, 15899–15906. 10.1021/jacs.5b10440. [DOI] [PubMed] [Google Scholar]; b Cheng Q.; Wang Y.; You S. L. Chemo-, Diastereo-, and Enantioselective Iridium-Catalyzed Allylic Intramolecular Dearomatization Reaction of Naphthol Derivatives. Angew. Chem., Int. Ed. 2016, 55, 3496–3499. 10.1002/anie.201511519. [DOI] [PubMed] [Google Scholar]; c Yang Z.-P.; Zheng C.; Huang L.; Qian C.; You S. L. Iridium-Catalyzed Intramolecular Asymmetric Allylic Dearomatization Reaction of Benzoxazoles, Benzothiazoles, and Benzimidazoles. Angew. Chem. Int. Ed. 2017, 56, 1530–1534. 10.1002/anie.201611056. [DOI] [PubMed] [Google Scholar]; d Yang Z.-P.; Wu Q.-F.; You S.-L. Direct Asymmetric Dearomatization of Pyridines and Pyrazines by Iridium-Catalyzed Allylic Amination Reactions. Angew. Chem., Int. Ed. 2014, 53, 6986–6989. 10.1002/anie.201404286. [DOI] [PubMed] [Google Scholar]; e Yang Z.-P.; Jiang R.; Zheng C.; You S.-L. Iridium-Catalyzed Intramolecular Asymmetric Allylic Alkylation of Hydroxyquinolines: Simultaneous Weakening of the Aromaticity of Two Consecutive Aromatic Rings. J. Am. Chem. Soc. 2018, 140, 3114–3119. 10.1021/jacs.8b00136. [DOI] [PubMed] [Google Scholar]
- a Worthington R. J.; Melander C. Overcoming Resistance to β-Lactam Antibiotics. J. Org. Chem. 2013, 78, 4207–4213. 10.1021/jo400236f. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Llarrull L. I.; Testero S. A.; Fisher J. F.; Mobashery S. The future of the β-lactams. Curr. Opin. Microbiol. 2010, 13, 551–557. 10.1016/j.mib.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Qin W.; Panunzio M.; Biondi S. β-Lactam Antibiotics Renaissance. Antibiotics 2014, 3, 193–215. 10.3390/antibiotics3020193. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Tehrani K. H. M. E.; Martin N. I. β-lactam/β-lactamase inhibitor combinations: an update. Med. Chem. Commun 2018, 1439–1456. 10.1039/C8MD00342D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See, for example:; a Weiner B.; Szymański W.; Janssen D. B.; Minnaard A. J.; Feringa B. L. Recent advances in the catalytic asymmetric synthesis of β-amino acids. Chem. Soc. Rev. 2010, 39, 1656–1691. 10.1039/b919599h. [DOI] [PubMed] [Google Scholar]; b Magriotis P. A. Recent Progress in the Enantioselective Synthesis of β-Lactams: Development of the First Catalytic Approaches. Angew. Chem., Int. Ed. 2001, 40, 4377–4379. . [DOI] [PubMed] [Google Scholar]
- Staudinger H. Zur Kenntniss der Ketene. Diphenylketen. Justus Liebigs Ann Chem. 1907, 356, 51–123. 10.1002/jlac.19073560106. [DOI] [Google Scholar]
- Shintani R.; Fu G. C. Catalytic enantioselective synthesis of beta-lactams: intramolecular Kinugasa reactions and interception of an intermediate in the reaction cascade. Angew. Chem., Int. Ed. 2003, 42, 4082–4085. 10.1002/anie.200352103. [DOI] [PubMed] [Google Scholar]
- a Pedroni J.; Boghi M.; Saget T.; Cramer N. Access to β-lactams by enantioselective palladium(0)-catalyzed C(sp3)-H alkylation. Angew. Chem., Int. Ed. 2014, 53, 9064–9067. 10.1002/anie.201405508. [DOI] [PubMed] [Google Scholar]; b Synofzik J.; Dar’in D.; Novikov M. S.; Kantin G.; Bakulina O.; Krasavin M. α-Acyl-α-diazoacetates in Transition-Metal-Free β-Lactam Synthesis. J. Org. Chem. 2019, 84, 12101–12110. 10.1021/acs.joc.9b02030. [DOI] [PubMed] [Google Scholar]
- For a review, see:Magriotis P. A. Progress in Asymmetric Organocatalytic Synthesis of β-Lactams. Eur. J. Org. Chem. 2014, 2647–2657. 10.1002/ejoc.201301720. [DOI] [Google Scholar]
- Ugi I. From Isocyanides via Four-Component Condensations to Antibiotic Syntheses. Angew. Chem., Int. Ed. Engl. 1982, 21, 810–819. 10.1002/anie.198208101. [DOI] [Google Scholar]
- Zidan A.; Garrec J.; Cordier M.; El-Naggar A. M.; Abd el-Sattar N. E.; Ali A. K.; Hassan M. A.; El Kaim L. β-Lactam Synthesis through Diodomethane Addition to Amide Dianions. Angew. Chem., Int. Ed. 2017, 56, 12179–12183. 10.1002/anie.201706315. [DOI] [PubMed] [Google Scholar]
- Cheibas C.; Cordier M.; Li Y.; El Kaïm L. A Ugi Straightforward Access to Bis-β-lactam Derivatives. Eur. J. Org. Chem. 2019, 4457–4463. 10.1002/ejoc.201900678. [DOI] [Google Scholar]
- Ghabraie E.; Balalaie S.; Mehrparvar S.; Rominger F. Synthesis of Functionalized β-Lactams and Pyrrolidine-2,5-diones through a Metal-Free Sequential Ugi-4CR/Cyclization Reaction. J. Org. Chem. 2014, 79, 7926–7934. 10.1021/jo5010422. [DOI] [PubMed] [Google Scholar]
- Gao X.; Shan C.; Chen Z.; Liu Y.; Zhao X.; Zhang A.; Yu P.; Galons H.; Lan Y.; Lu K. One-pot synthesis of β-lactams by the Ugi and Michael addition cascade reaction. Org. Biomol. Chem. 2018, 16, 6096–6105. 10.1039/C8OB01176A. [DOI] [PubMed] [Google Scholar]
- CCDC 1957914 contains the supplementary crystallographic data for this paper (cis-6s). These data can be obtained free of charge via www.ccdc.cam.ac.uk/structures/ by emailing data_request@ccdc.cam.ac.uk or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, U.K. (fax: +44 1223 336033). See also the Supporting Information.
- The trans diastereomers of 3 were identified by a characteristic upfield shift of the vinylic protons resulting from the spatial proximity to the aromatic ring. The cis diastereomers presented a regular pattern.
- a Xu Y.; Dong G. sp3 C–H activation via exo-type directing groups. Chem. Sci. 2018, 9, 1424–1432. 10.1039/C7SC04768A. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Colby D. A.; Bergman R. G.; Ellman J. A. Rhodium-Catalyzed C–C Bond Formation via Heteroatom-Directed C–H Bond Activation. Chem. Rev. 2010, 110, 624–655. 10.1021/cr900005n. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lyons T. W.; Sanford M. S. Palladium-catalyzed ligand-directed C-H functionalization reactions. Chem. Rev. 2010, 110, 1147–1169. 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Dutta P. K.; Sen S. (Benz)Imidazole-Directed Cobalt(III)-Catalyzed C–H Activation of Arenes: A Facile Strategy to Access Polyheteroarenes by Oxidative Annulation. Eur. J. Org. Chem. 2018, 5512–5519. 10.1002/ejoc.201801056. [DOI] [Google Scholar]; e Park Y.; Kim Y.; Chang S. Transition Metal-Catalyzed C-H Amination: Scope, Mechanism, and Applications. Chem. Rev. 2017, 117, 9247–9301. 10.1021/acs.chemrev.6b00644. [DOI] [PubMed] [Google Scholar]; f Kim D. S.; Park W. J.; Jun C. H. Metal-Organic Cooperative Catalysis in C-H and C-C Bond Activation. Chem. Rev. 2017, 117, 8977–9015. 10.1021/acs.chemrev.6b00554. [DOI] [PubMed] [Google Scholar]; g Hummel J. R.; Boerth J. A.; Ellman J. A. Transition-Metal-Catalyzed C-H Bond Addition to Carbonyls, Imines, and Related Polarized π Bonds. Chem. Rev. 2017, 117, 9163–9227. 10.1021/acs.chemrev.6b00661. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Gensch T.; Hopkinson M. N.; Glorius F.; Wencel-Delord J. Mild metal-catalyzed C–H activation: examples and concepts. Chem. Soc. Rev. 2016, 45, 2900–2936. 10.1039/C6CS00075D. [DOI] [PubMed] [Google Scholar]; i Yang L.; Huang H. Transition-Metal-Catalyzed Direct Addition of Unactivated C–H Bonds to Polar Unsaturated Bonds. Chem. Rev. 2015, 115, 3468–3517. 10.1021/cr500610p. [DOI] [PubMed] [Google Scholar]
- See the Supporting Information for computational details, along with energies and coordinates of all stationary points.
- a te Velde G.; Bickelhaupt F. M.; Baerends E. J.; Fonseca Guerra C.; van Gisbergen S. J. A.; Snijders J. G.; Ziegler T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. 10.1002/jcc.1056. [DOI] [Google Scholar]; b Fonseca Guerra C.; Snijders J. G.; te Velde G.; Baerends E. J. Towards an order- N DFT method. Theor. Chem. Acc. 1998, 99, 391–403. 10.1007/s002140050353. [DOI] [Google Scholar]; c ADF2017, SCM Theoretical Chemistry, Vrije Universiteit: Amsterdam (The Netherlands); 2017, http:// www.scm.com.
- de Jong G. T.; Bickelhaupt F. M. Oxidative Addition of the Chloromethane C–Cl Bond to Pd, An Ab Initio Benchmark and DFT Validation Study. J. Chem. Theory Comput. 2006, 2, 322–335. 10.1021/ct050254g. [DOI] [PubMed] [Google Scholar]
- a Vermeeren P.; van der Lubbe S. C. C.; Fonseca Guerra C.; Bickelhaupt F. M.; Hamlin T. A. Understanding chemical reactivity using the activation strain model. Nat. Protoc. 2020, 15, 649–667. 10.1038/s41596-019-0265-0. [DOI] [PubMed] [Google Scholar]; b Bickelhaupt F. M.; Houk K. N. Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem., Int. Ed. 2017, 56, 10070–10086. 10.1002/anie.201701486. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Fernández I.; Bickelhaupt F. M. The activation strain model and molecular orbital theory: understanding and designing chemical reactions. Chem. Soc. Rev. 2014, 43, 4953–4967. 10.1039/C4CS00055B. [DOI] [PubMed] [Google Scholar]; d Bickelhaupt F. M. Understanding reactivity with Kohn–Sham molecular orbital theory: E2–SN2 mechanistic spectrum and other concepts. J. Comput. Chem. 1999, 20, 114–128. . [DOI] [Google Scholar]
- Sun X.; Soini T. M.; Poater J.; Hamlin T. A.; Bickelhaupt F. M. PyFrag 2019-Automating the exploration and analysis of reaction mechanisms. J. Comput. Chem. 2019, 40, 2227–2233. 10.1002/jcc.25871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- In our mechanistic rationalization, we assume that only syn-π-allylpalladium species are involved. Additional DFT calculations do not support the involvement of other potential intermediates such as anti-π-allylpalladium species or O-coordinated palladium enolates (see the Supporting Information for details). For additional evidence that intramolecular Tsuji–Trost reactions can occur with stereospecific formation of a π-allylpalladium intermediate, which does not undergo syn/anti interconversion, see:; Braun J.; Ariëns M. I.; Matsuo B. T.; de Vries S.; van Wordragen E. D. H.; Ellenbroek B. D.; Vande Velde C. M. L.; Orru R. V. A.; Ruijter E. Stereoselective Synthesis of Fused Vinylcyclopropanes by Intramolecular Tsuji–Trost Cascade Cyclization. Org. Lett. 2018, 20, 6611–6615. 10.1021/acs.orglett.8b02232. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.