

Summary
BRCA1 promotes the DNA end-resection and RAD51 loading steps of homologous recombination (HR). Whether these functions can be uncoupled, or if mutant proteins retaining partial activity can complement one another, is unclear, and could impact the severity of BRCA1-associated Fanconi anemia (FA). Here, we generated a Brca1CC mouse with a coiled-coil (CC)-domain deletion. Brca1CC/CC mice are born at low frequencies and post-natal mice have FA-like abnormalities, including bone marrow failure. Intercrossing with Brca1Δ11, which is homozygous lethal, generated Brca1CC/Δ11 mice at Mendelian frequencies that were indistinguishable from Brca1+/+ mice. Brca1CC and Brca1Δ11 proteins were individually responsible for counteracting 53BP1-RIF1-Shieldin activity and promoting RAD51 loading, respectively. Thus, Brca1CC and Brca1Δ11 alleles represent separation-of-function mutations, which combine to provide a level of HR that is sufficient for normal development and hematopoiesis. Because BRCA1 activities can be genetically separated, compound heterozygosity for functional complementary mutations may protect individuals from FA.
In Brief
Nacson et al. show that compound heterozygosity for distinct Brca1 mutant alleles, which generate proteins capable of functional complementation, restores development and protects against Fanconi anemia (FA) in mice. Their observations suggest that BRCA1’s DNA end resection and RAD51 loading activities are genetically separable.
Graphical Abstract
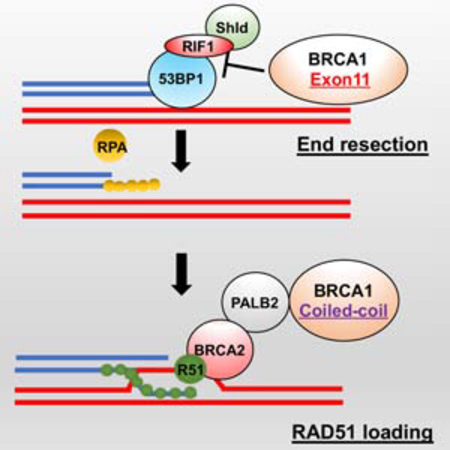
Introduction
Individuals that inherit a single BRCA1 mutation-containing allele in their germline are predisposed to breast and ovarian cancer, but develop normally. In contrast, biallelic BRCA1 germline mutations have been reported in patients with FA-like abnormalities, with the BRCA1 gene also designated FANCS (Domchek et al., 2013; Freire et al., 2018; Keupp et al., 2019; Sawyer et al., 2015; Seo et al., 2018). Developmental problems arise from defective DNA repair (Moynahan et al., 1999; Scully et al., 1997), where BRCA1 contributes to distinct steps of HR. BRCA1 counteracts 53BP1-RIF1-Shieldin activity, thereby activating DNA end resection. Second, as double stranded DNA breaks (DSBs) are resected to form single stranded DNA (ssDNA), BRCA1 promotes RAD51 filament formation (Roy et al., 2012). BRCA1 and PALB2 directly interact with one another via their respective CC domains, facilitating the formation of a larger BRCA1-PALB2-BRCA2-RAD51 (B1-P2-B2-R1) complex that directs RAD51 loading onto ssDNA (Sy et al., 2009; Zhang et al., 2009a; Zhang et al., 2009b). In this study, we generated and characterized a Brca1 CC domain deletion mouse model, which specifically blocks the Brca1-Palb2 interaction.
Results
Brca1CC/CC mice have FA-like developmental defects
To investigate BRCA1’s RAD51 loading activity, we used CRISPR/Cas9 to generate a Brca1 CC domain deletion allele, referred to as Brca1CC. Mice were established harboring an in-frame deletion of amino acids 1361–1363 (IKL), equivalent to human 1405–1407 (Fig. 1A). Brca1CC/CC cells generated Brca1 protein (Brca1CC) at levels comparable to wild-type cells (Brca1WT) (Fig. 1B). In line with the known function of the Brca1 CC domain in promoting RAD51 loading, Brca1CC/CC cells had undetectable RAD51 γ-irradiation-induced foci (IRIF) (Fig. 1C).
Fig 1. Brca1CC/CC mice have FA.

(A) The Brca1CC allele was generated using the indicated sgRNA sequence that targeted exon 13. DNA and amino acids deleted are shown (red). Below, electropherogram showing DNA sequences of Brca1+/+ and Brca1CC/CC mice, deleted base pairs (box) and deletion location (arrow) are indicated. (B) MEFs with the indicated genotypes were assessed for Brca1 protein expression by Western blotting. (C) MEFs with the indicated genotypes were assessed for RAD51 IRIF by immunofluorescence (IF). Nuclei with >5 foci were considered positive. Mean and S.E.M. are shown, n=3. Representative images, scale bar is 10 μm. (D) Summary of live pups born as well as live embryos at E16.5 generated from Brca1+/CC x Brca1+/CC intercross. (E) Representative photographs of E16.5 embryos, scale bar 0.5 cm; and 3-week old littermates, scale bar 2 cm. Hypopigmented fur area is indicated with an arrow. (F) Weights of individual mice at 3–4 weeks of age. (G) White blood cell (WBC), red blood cell (RBC) numbers from peripheral blood, and (H) mononuclear bone marrow (BM) cell numbers were measured in 3–4-week-old littermates with the indicated genotypes. Inset, representative H&E staining of BM, scale bar 100 μm. (I) Total number of long-term hematopoietic stem cells (LT-HSC) (Linlow; cKit+; Sca+; CD150+; CD48−; CD34−) analyzed by FACS in 3–4-week-old mice. See Fig. S1 for more information. (J) Representative H&E staining of testes and ovaries, scale bar 100 μm. (K) Representative images of T-ALL from a Brca1CC/CC mouse. H&E staining of thymic tumor and CD3+ staining of tumor cell infiltrates from the same mouse, scale bar 100 μm. Summary of Brca1CC/CC lifespans (days) and cause of death when known (inset). *p < 0.05 **p < 0.01, *** p < 0.001 (unpaired t-test).
Brca1+/CC mice from a mixed genetic background were interbred and the genotypes of offspring measured. Brca1CC/CC homozygous mice were born at sub-Mendelian ratios, with approximately 15% of the expected number of embryos reaching birth (Fig. 1D). Loss of viability occurred late in embryogenesis, as near Mendelian ratios could be detected at E16.5 (Fig. 1D). Brca1CC/CC embryos and mice were smaller in size and weight compared to littermates, and mice frequently showed distinctive hypopigmented fur spots on their underside, as well as kinked tails (Fig. 1E-F). Three-week-old Brca1CC/CC mice also presented with decreased erythrocytes and leukocytes in the peripheral blood (Fig. 1G), as well as bone marrow (BM) hypoplasia (Fig. 1H and S1A). Additionally, mice displayed a severe reduction in hematopoietic stem cells (HSCs) and committed progenitor cells (Fig. 1I and S1B-C). Atrophic changes were observed in the testes and ovaries accounting for male and female infertility (Fig. 1J). Mice subsequently developed disseminated T-cell acute lymphoblastic leukemia (T-ALL) and did not survive beyond 100 days of age (Fig. 1K). These collective phenotypes confirm that Brca1CC/CC induces late embryonic lethality in the majority of mice, with a portion reaching birth, but with developmental defects that parallel human FA (Parmar et al., 2009).
Brca1 compound heterozygosity rescues FA
BRCA1-associated FA can arise from various combinations of mutant alleles, and documented cases have shown variability in the range and severity of symptoms (Domchek et al., 2013; Freire et al., 2018; Keupp et al., 2019; Sawyer et al., 2015; Seo et al., 2018). We hypothesized that the specific combination of BRCA1 mutant alleles could impact FA phenotypes. To examine developmental abnormalities associated with Brca1 compound heterozygous mutations, we interbred mice harboring the Brca1CC allele with previously established Brca1Δ11 and Brca1ΔC alleles. Brca1ΔC has a stop codon in the CC domain that results in protein degradation and undetectable expression (Nacson et al., 2018); whereas, Brca1Δ11 is unable to produce full-length Brca1, but can generate the hypomorphic Brca1Δ11 isoform (Xu et al., 2001). Both Brca1Δ11 and Brca1ΔC alleles result in loss of HR and embryonic lethality in the homozygous setting. Similar to homozygous embryos, compound heterozygous Brca1ΔC/Δ11 and Brca1ΔC/CC genotypes were either embryonic lethal or born at sub-Mendelian ratios. Strikingly, intercrossing Brca1CC/+ and Brca1Δ11/+ mice produced Brca1CC/Δ11 pups at Mendelian frequencies (Fig. 2A and S2A).
Fig. 2. Compound heterozygosity can rescue FA.

(A) Cartoon showing compound heterozygous allele combinations and potential Brca1 protein products with their observed live birth frequency as a percentage of the expected Mendelian live births. See Fig. S2A for tables. (B) Representative photographs of 3.5-week-old littermates, scale bar 2 cm. Weights of individual mice with the indicated genotypes at 3–4 weeks of age. (C) WBC, RBC, BM, and LT-HSCs were measured as described in Fig. 1. (D) Summary of live pups generated from Brca1CC/Δ11 x Brca1CC/Δ11 intercross. (E) Kaplan-Meyer analyses of survival showing Brca1CC/CC (n=7) and Brca1CC/Δ11 (n=20) mice. (F) MEFs were treated with MMC and colony formation assays performed. Mean and S.E.M. IC50 values are shown, n=3. *** p < 0.001 (unpaired t-test). Non-significant (ns) p > 0.05 (unpaired t-test).
Brca1CC/Δ11 compound heterozygous mice were physically indistinguishable from Brca1+/+ mice (Fig. 2B), and demonstrated normal hematopoiesis (Fig. 2C and S2B-C). Male and female mice were fertile, generating Brca1CC/Δ11 offspring at the predicted frequency (Fig.2D). Brca1CC/Δ11 mice did not develop tumors and had lifespans similar to Brca1+/+ mice (Fig. 2E). Moreover, derived MEFs were significantly more resistant than either individual homozygous allele to mitomycin C treatment, a FA diagnostic test (Fig.2F). Taken together, these data show that, while both homozygous Brca1Δ11 and Brca1CC embryos fail to undergo normal development, compound heterozygosity resulted in synthetic viability, producing phenotypically normal mice that were protected from tumorigenesis and FA.
Brca1 mutations have distinct HR defects
We used MEFs to gain insights into the molecular mechanism by which the combination of Brca1CC and Brca1Δ11 alleles rescue development. Both Brca1CC and Brca1Δ11 proteins were detected in Brca1CC/Δ11 MEFs, while individual proteins were detected for homozygous genotypes (Fig. 3A). HR DNA repair is initiated when DSBs are resected by nucleases to generate ssDNA. We quantified the efficiency of resection indirectly by measuring phosphorylated RPA32 foci formation after PARPi-induced DSBs (Symington, 2016). Here, Brca1+/+, Brca1CC/CC, and Brca1CC/Δ11 were similarly proficient for resection, but Brca1Δ11/Δ11 MEFs demonstrated significantly reduced numbers of RPA32 foci positive cells (Fig. 3B). Downstream of resection, RAD51 loading requires B1-P2-B2-R1 complex formation. As expected, PARPi treated Brca1CC/CC MEFs entirely lacked RAD51 foci, but Brca1Δ11/Δ11 had detectable foci, and Brca1CC/Δ11 MEFs showed increased numbers of RAD51 foci positive cells compared to the latter genotypes (Fig. 3B).
Fig. 3. Brca1 alleles confer distinct functional deficiencies.

(A) MEFs were assessed for Brca1 protein expression by Western blotting. Predicted isoform migration patterns are indicated. (B) RPA32 and RAD51 foci formation was measured by IF after treatment with 1 μM rucaparib for 24 hours. For each protein, a minimum of 100 nuclei per cell line were counted per experiment. Mean and S.E.M. percentages of cells containing 5 or more foci are shown (n=3). Representative foci images are inset, scale bar 10 μm. (C) Heatmap of END-seq signals across individual AsiSI sites in the indicated MEFs measured 5 h after AsiSI induction. (Right), boxplots showing the quantification of resection endpoints in the top 10% resected breaks in the indicated MEFs at AsiSI-cleaved DSB sites. Welch’s t test was used to determine statistical significance. (D) MEFs were subject to CRISPR/Cas9 and sgRNA targeting gfp or 53bp1 (bp1). Cells were subsequently assessed for RAD51 IRIF as described in Fig.1C (left), or treated with the PARPi rucaparib and colony formation assays performed (right), mean and S.D. are shown (n=3), inset western blotting showing 53bp1 (bp1) protein levels. (E) Metaphase spreads were prepared from the indicated genotypes of MEFs treated with 500 nM rucaparib for 24 hours. The mean number of radial chromosomes per 10 metaphases are shown. The number of metaphases assessed were 30 for Brca1+/+, 26 for Brca1CC/CC; 25 for Brca1Δ11/Δ11, and 25 for Brca1CC/Δ11 MEFs. Scale bars, 10 μm. Red arrows point to aberrations. (F) MEFs were treated with 100 nM (left) or 1000 nM (right) rucaparib for 2 weeks and assessed for colony formation. Mean and S.D. colony formation is shown as a percentage of DMSO treated cells, n = 3. (G) Brca1CC/Δ11 mice 5–6 weeks old were treated with vehicle (V) or 200 mg/kg rucaparib (R) bi-daily for 5 days and peripheral blood collected 2 days after the last dose. Blood was assessed for lymphocyte cell numbers (left) and mice measured for body weight (right). (H) MEFs were sequentially pulsed with 50 μM CldU for 30 min, 250 μM IdU for 30 min, and 2 mM hydroxyurea for 3h. DNA fiber tract lengths and are presented as CldU/ IdU length ratio. Inset, representative fibers. *p < 0.05 **p < 0.01, *** p < 0.001, ns p > 0.05 (unpaired t-test).
To confirm differences in end resection proficiencies, we generated site-specific DSBs using inducible expression of the AsiSI restriction enzyme and performed END-seq, which maps the double-stranded DNA (dsDNA)/single stranded DNA (ssDNA) junctions at DSB sites (Canela et al., 2016). Similar to RPA32 foci trends, Brca1Δ11/Δ11 MEFs had significantly reduced resection lengths, while Brca1CC/CC and Brca1CC/Δ11 cells demonstrated a more modest reduction compared to Brca1+/+ MEFs (Fig. 3C). Given that Brca1CC/CC cells are highly defective for RAD51 loading, we measured the effects of 53bp1 KO, which promotes DNA end resection, on RAD51 IRIF and PARPi sensitivity. As expected, Brca1Δ11/Δ11 cells, which have defective end resection, showed dramatically increased RAD51 IRIF and PARPi resistance with 53bp1 KO. However, 53bp1 KO had little impact on RAD51 IRIF and PARPi sensitivity in Brca1CC/CC cells (Fig. 3D). These data provide further evidence that Brca1Δ11 and Brca1CC alleles have severe defects in either end resection or RAD51 loading, respectively, while retaining, although sub-optimally, the ability to carry out the opposing activity.
Brca1CC/Δ11 confers partial HR proficiency
HR deficient cells are highly sensitive to PARPi treatment, and the response to PARPi can be used as a readout for cellular HR-proficiency (Bryant et al., 2005; Farmer et al., 2005). In analyses of metaphase chromosomes from short-term PARPi treated cells, aberrations were abundant in Brca1Δ11/Δ11 and Brca1CC/CC MEFs, but few were detected in Brca1+/+ and Brca1CC/Δ11 cells (Fig. 3E). In long-term colony assays, Brca1CC/Δ11 cells formed more colonies in the presence of 100 nM rucaparib compared to Brca1Δ11/Δ11 and Brca1CC/CC MEFs. However, at 1000 nM PARPi, Brca1CC/Δ11 cells formed significantly fewer colonies compared with Brca1+/+ cells (Fig. 3F). Similar patterns were observed in response to cisplatin, but not taxol (Fig. S2D). Moreover, administration of rucaparib doses previously established to have no effect on HR-proficient mice (Nacson et al., 2018), resulted in a reduction in blood lymphocytes of Brca1CC/Δ11 mice, although body weight was unaffected and mice survived treatment (Fig. 3G). Thus, experiments assessing PARPi sensitivity suggest that the Brca1CC/Δ11 genotype confers a moderate level of HR proficiency that is greater than either homozygous allele, but less than Brca1+/+ cells.
Brca1CC protects replication forks from degradation
Brca1 has a well-documented role in protecting stalled DNA replication forks from nuclease degradation (Schlacher et al., 2012). In line with previous work (Chaudhuri et al., 2016; Daza-Martin et al., 2019), forks were subject to degradation upon replication stress in Brca1Δ11/Δ11 cells. In contrast, forks were protected from degradation in Brca1CC/CC and Brca1CC/Δ11 cells (Fig. 3H). Of note, differences in HR and fork protection patterns could not be attributed to differences in cell cycle profiles between cell lines (Fig. S2E). Thus, Brca1CC allele represents a separation-of-function mutation, and suggests BRCA1’s ability to protect stalled forks does not require the CC or efficient HR. Similarly, we previously found that the RING domain, though important for HR, is dispensable for fork protection (Zong et al., 2019).
BRCA1 exon 11 counteracts RIF1 to promote resection
To confirm that differences between the efficiency of DNA end resection and RAD51 loading were directly attributable to Brca1Δ11 and Brca1CC protein activity, we expressed the human homologs HA-BRCA1Δ11q and HA-BRCA1CC as well as BFP and HA-BRCA1WT controls in the BRCA1 null MDA-MB-436 cell line (Johnson et al., 2013). In immunoprecipitation experiments, HA-BRCA1CC was unable to interact with PALB2, and consequently BRCA2 and RAD51; however, the BRCT-mediated CtIP interaction was intact. In contrast, HA-BRCA1Δ11q was able to complex with all proteins tested; although interactions with P2-B2-R1 were less efficient compared to HA-BRCA1WT (Fig. S3A). HA-BRCA1WT, HA-BRCA1Δ11q and HA-BRCA1CC were equally efficient at forming BRCA1 IRIF (Fig. 4A and S3B), as well as accumulating as foci at stalled replication forks (Fig. S3C). All BRCA1 proteins also increased CtIP IRIF relative to BFP control cells. HA-BRCA1WT and HA-BRCA1CC expressing cells had increased RPA32 foci positive cells relative to BFP-expressing cells. Despite increasing CtIP foci, HA-BRCA1Δ11q cells did not demonstrate elevated RPA32 IRIF. In contrast, HA-BRCA1Δ11q did increase the number of RAD51 foci positive cells, although at lower levels than HA-BRCA1WT cells. HA-BRCA1CC expressing cells had undetectable RAD51 IRIF (Fig. 4A and S3B). These results parallel observations made with Brca1Δ11/Δ11 and Brca1CC/CC MEFs and confirm that differences in functions retained stem from BRCA1 protein expression.
Fig. 4. BRCA1 mutants have complimentary functions.

(A) MDA-MB-436 cells expressing BFP control, HA-BRCA1WT, HA-BRCA1Δ11q, and HA-BRCA1CC were used to assess BRCA1, CtIP, RPA32, and RAD51 IRIF by IF. Mean and S.E.M. are shown, n=3, grey bar indicates BFP control levels. See Fig. S3B for representative foci images. (B) Cells from A were assessed for 53BP1, p(T543)53BP1, and RIF1 IRIF by IF after treatment with 2 Gy IR. Cells were also co-stained with geminin to distinguish S/G2 phase cells. Foci positive cells that were also geminin positive were quantified. Mean and S.D. are shown, n=3, *** p < 0.001; ** p < 0.01; * p < 0.05; no marking p > 0.05, compared to WT (two-way ANOVA corrected for multiple comparisons). See Fig. S3D for representative foci images. (C) BRCA1 cDNA constructs from Fig. S4A, including full-length (1863aa), and exon 11 deletions of the central 800 (Δ8), 900 (Δ9), 1000 (Δ10), 11000 aa’s (Δ11) were expressed in MDA-MB-436 cells and assessed for BRCA1 and RIF1 IRIF. The mean and S.E.M. number of BRCA1 foci positive cells that also contained RIF1 foci were quantified. Representative images are shown, see Fig. S4C for images. ** p < 0.01; ns p > 0.05, unpaired t-test compared to FL. (D) MDA-MB-436 cells expressing a BFP control, BRCA1*WT, BRCA1*M1411T, BRCA1Δ11q, and BRCA1*M1411T+ BRCA1Δ11q were incubated with 100 nM PARPi for 2 weeks and colony formation assessed. Mean and S.E.M. colonies relative to BRCA1*WT cells are shown, n=3. Inset, Western blot showing HA, GFP and BRCA1 protein expression, upper and lower arrows indicate migration of BRCA1* and BRCA1Δ11q, respectively. (E) Cells from D were assessed for RPA32 and RAD51 IRIF as described in A. *** p < 0.001; ** p < 0.01; * p < 0.05; ns p > 0.05, unpaired t-test compared to BFP. (F) BRCA1*M1411T+BRCA1Δ11q cells were assessed for HA (M1411T) and GFP (Δ11q) IRIF. Representative image of GFP and HA foci positive cells, scale bar is 10 μm.
To further investigate why BRCA1Δ11q failed to promote RPA32 IRIF and DNA end resection, we assessed 53BP1 and RIF1 IRIF in BRCA1 add back MDA-MB-436 cells. 53BP1 IRIF followed similar patterns across cell lines, immediately increasing after IR, and remaining abundant up to 8 hours post-IR (Fig. 4B and S3D). Moreover, as previously shown (Isono et al., 2017), phospho-(p)53BP1 and RIF1 increased after IR, then steadily decreased over time in BRCA1WT and BRCA1CC expressing cells. By contrast, the reduction in p-53BP1 and RIF1 IRIF was delayed in BFP control and BRCA1Δ11q expressing cells (Fig. 4B and S3D), suggesting that BRCA1Δ11q fails to efficiently counteract 53BP1-RIF1-Shieldin.
Because exon 11 accounts for more than half of the full-length protein, we sought to determine the minimal peptide region that is required to activate resection. A series of increasingly truncated exon 11 deletions in BRCA1 full-length (FL) cDNA were generated and expressed in MDA-MB-436 cells (Fig. S4A). Similar to BRCA1-Δ11q (Δ1100aa), all deletion mutants formed foci and promoted CtIP IRIF (Fig. S4B). Surprisingly, deletions of up to Δ800aa’s remained effective at promoting RPA32 foci, but cells expressing Δ900-Δ1100aa deletions had decreased RPA32 IRIF, and consequently reduced RAD51 foci (Fig. S4B).
Despite their opposing effects on DSB resection, BRCA1 and 53BP1 foci are frequently found within the same nuclei and co-localizing foci can readily be detected (Chapman et al., 2012). However, BRCA1 and RIF1 have been shown to form foci in a mutually exclusive manner (Feng et al., 2013). To gain insight into why Δ900-Δ1100aa constructs were ineffective at promoting RPA32 IRIF, we measured the number of BRCA1 foci positive cells that also contained RIF1 foci. Here, few cells that were positive for BRCA1-FL and -Δ800aa foci were also positive for RIF1 foci. In contrast, more than half of all cells that were positive for BRCA1-Δ900, -Δ1000aa, or -Δ1100aa foci also contained, and often co-localized with, RIF1 foci (Fig. 4C and S4C). These data suggest that a large portion of the central region of BRCA1 is dispensable, but the terminal peptide regions of exon 11 contribute to counteracting RIF1 focal accumulation at DSB sites, consequently promoting DNA end resection.
Germline BRCA1 mutations retain hypomorphic activity
BRCA1 CC domain missense variants present in individuals with cancer also blocked the BRCA1-PALB2 interaction and produced proteins that behaved identically to BRCA1CC (Fig. S5A-C). Similarly, we confirmed our previous observations that BRCA1 exon 11 mutation-containing cells express the BRCA1Δ11q splice isoform (Wang et al., 2016; Wang et al., 2019) (Fig. S5D). To examine the possibility that BRCA1 proteins generated from alleles present in individuals with germline mutations could retain complementary activities, we expressed ectopic BRCA1M1411T and BRCA1Δ11q in the same cell line. To avoid viral packaging problems and achieve similar expression of both proteins, we used the BRCA1-Δ800 construct (herein BRCA1*), which had similar end resection and RAD51 loading activity as FL, but protein expression is similar to BRCA1Δ11q (Fig. S4A-B). Here, BRCA1*M1411T+BRCA1Δ11q cells showed elevated PARPi resistance (Fig. 4D), as well as RPA32 and RAD51 foci at levels comparable to individual protein expressing cells (Fig. 4E and S5E). Moreover, HA-tagged BRCA1*M1411T and GFP-tagged BRCA1Δ11q proteins were readily detected at co-localizing foci within the same nuclei (Fig. 4F), although proteins did not co-immunoprecipitate with one another (Fig. S5F). Therefore, mutations that recapitulate those found in the germline of patients are capable of expressing proteins that independently promote the DNA end resection or RAD51 loading steps of HR.
Discussion
BRCA1 CC missense mutations such as L1407P and M1411T have been reported in families with a history of breast and ovarian cancer (Anantha et al., 2017; Phelan et al., 2005; Sy et al., 2009). We found that disruption of the Brca1 CC domain is highly deleterious for mouse development and hematopoiesis. The Brca1CC protein is hypomorphic and can promote DNA end resection, but is entirely defective for RAD51 loading and consequently HR.
BRCA1 forms discrete biochemical protein complexes that perform distinct activities in DNA repair and checkpoint signaling (Greenberg et al., 2006). Our work underscores the requirement for BRCA1 in promoting RAD51 loading through the P2-B2-R1 axis. Cancer-associated PALB2CC missense mutations such as L35P have also been shown to abrogate RAD51 foci (Foo et al., 2017). Intriguingly, a Palb2CC domain substitution mutation produced normal mice, besides male infertility, and derived cells showed smaller but intact RAD51 foci (Simhadri et al., 2014). Differences in RAD51 foci and mouse development observed between Brca1 and Palb2 mutations could result from the severity of CC structural disruptions and possible residual Brca1-Palb2 association.
In addition to directing PALB2 to DSBs, BRCA1 has a central role in the initiation of DNA end resection. We discovered that a relatively small region of BRCA1 exon 11 is sufficient to counteract RIF1 accumulation at DSB sites. Although the precise mechanism by which exon 11 coding peptides counteract RIF1 requires further elucidating. Nevertheless, these results partly explain the BRCA1Δ11q isoforms inability to provide efficient HR, despite retaining conserved domains.
Brca1CC and Brca1Δ11 mutations elicit distinct functional defects; nevertheless, appear to be overall equally disruptive to HR, with homozygous cells demonstrating exquisite PARPi sensitivity. Although Brca1CC/Δ11 cells were able to form more colonies in the presence of PARPi than homozygous mutant cells, they remained significantly more sensitive to PARPi treatment compared to Brca1+/+ cells, suggesting that HR was only partially rescued. Similarly, Brca1S1598F/S1598F mice, which have impaired BRCT domain-protein interactions, develop normally apart from male sterility, yet are HR defective (Chen et al., 2017; Shakya et al., 2011). We propose that while severe HR deficiency induces embryonic lethality or FA, embryos and mice with less efficient HR can develop normally, but may demonstrate sensitivity to genotoxic agents or PARPi’s.
Brca1CC/CC mice have several features of FA, including short stature, pigmentation lesions, growth defects, BMF, and cancer predisposition. Intriguingly, BMF has not been reported in human patients with BRCA1-associated FA. Moreover, while adult patients have presented with breast and ovarian cancer (Domchek et al., 2013; Sawyer et al., 2015), childhood malignancies are less prevalent, with a single report of a 5-year old patient with BRCA1 homozygous exon 11 mutations that developed T-ALL (Seo et al., 2018). Interestingly, Brca1CC/CC mice also succumbed to T-ALL.
Hypomorphic BRCA1 protein activity likely contributes to phenotypes observed in patients. For example, the BRCA1 p.Arg1699Gln mutant has partial activity and prevented chromosome fragility (Keupp et al., 2019). Four patients with FA-like defects were also reported to carry homozygous exon 11 frameshift mutations and BRCA1-Δ11q expression was detected (Seo et al., 2018). Thus, particular BRCA1 mutant proteins may enable lifespans to extend beyond embryogenesis, but viability is coupled with FA. In contrast, our data raises the possibility that individuals with two distinct hypomorphic BRCA1 alleles that retain complementary functions could have a reduced risk of developmental disorders.
In summary, the precise or most critical function of BRCA1 within HR has been ambiguous and difficult to discern; the current study establishes that BRCA1 is equally important for DNA end resection and RAD51 loading. Future insights into mutant BRCA/FANC protein activity and compound heterozygous phenotypes might aid genetic counseling in determining FA risk and predicting the severity of associated diseases.
STAR Methods
Lead contact and materials availability
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact, Neil Johnson (neil.johnson@fccc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Cell lines were obtained from ATCC and were tested for mycoplasma. Cell line identities were confirmed by short tandem repeat (STR) profiling using IDEXX analysis. Except otherwise mentioned, cell lines were grown in media with 10% FBS and penicillin/streptomycin. MEFs were generated by dissection of embryos (12.5–13.5 days post-coitum). Dissociation was performed by aspiring embryos through a 16G needle followed by incubation with 0.25%Trypsin at 37°C. The solution was mixed again by trituration and incubated for an additional 5–10 minutes. After centrifugation cell pellet was resuspended in 75 cm2 flasks with media. MEFs were immortalized before passage 3 by electroporation of pBabe-SV40LTAg vector with the Amaxa Mouse/Rat Hepatocyte Nucleofector Kit (VAPL-1004 Lonza). MEF media (DMEM) was supplemented with non-essential amino acids, sodium pyruvate, and L-Glutamine. All cell lines were incubated at 37 degrees with 5% CO2 in a humidified incubator.
Mice
All experiments involving animals were approved by the Fox Chase Cancer Center (FCCC) Institutional Animal Care and Use Committee. The mouse Brca1CC allele was generated by the FCCC transgenic mouse facility (detailed below). Before initiating experiments, germline transmission of the allele was confirmed. Embryos harboring the Brca1Δ11 allele (Xu et al., 2001) were obtained from the NCI. The generation of mice harboring the Brca1ΔC allele was described previously (Nacson et al., 2018) and were produced by the FCCC transgenic mouse core facility during the same process as described for generating the Brca1CC allele using the same constructs and materials. For bone marrow analyses, 3–4-week-old males and females were used. For PARPi treatments, 6–8-week-old males and females were randomly selected for treatment.
Generation of Brca1CC mutant mice
Candidate guide RNAs targeting the coiled-coil region of Brca1 were selected using crispr.mit.edu and cleavage efficiency was tested in vitro using Cas9 nuclease (NEB). The selected guide (ctatttcgacgtcgtccttt) targeted close to the Leucine 1363 residue, had high cut efficiency and a low off-target score. Cas9 and sg-RNA (IDT) were resuspended (20 ng/μl) in injection buffer (1 mM Tris HCl, pH 7.5; 0.1 mM EDTA) and 1–2μl injected in single-cell 0.5-day embryos (pronuclei). The embryos were transferred to Swiss Webster recipient females (Taconic Tac:SW). Both Brca1CC and Brca1ΔC alleles were generated in B6C3F1/J background (offspring of a cross between C57BL/6J females (B6) and C3H/HeJ males (C3) acquired from the Jax Laboratory). The offspring were crossed with B6 mice. Brca1Δ11 embryos acquired from the NCI are described as being from a mixed background (STOCK). Compound heterozygotes offspring have a mixed background from the above strains.
METHOD DETAILS
Mouse Genotyping
DNA was extracted using 240 μl of 50 mM NaOH. To evaluate Brca1CC status a 2kb fragment was generated by PCR and fragments were subjected to restriction analysis with EcoNI restriction enzyme. WT animals showed 1 band, heterozygous 3 bands, and homozygous mutant 2 bands. Brca1Δ11 and Brca1ΔC mice were genotyped as previously described (Xu et al., 2001; Nacson et al. 2018).
Embryo dissection
Pregnant mice were euthanized 16.5 days postcoitum, uterus was removed and separated from the mesometrium. Individual embryos were dissected under a microscope vision (Nikon SMZ1500).
PARPi treatment and peripheral blood analysis
To evaluate tolerability to PARPi, 6 to 8 weeks old compound heterozygous mice were randomly selected to receive 200 mg/kg rucaparib orally bi-daily for 5 days. Mice were euthanized in accordance with institutional guidelines. Peripheral blood was collected from rucaparib treated animals and analyzed using an automated analyzer using mouse specific parameters (Abaxis VetScan5).
Peripheral blood analysis and staining of hematopoietic stem cells
After anesthesia, peripheral blood was collected from 3–4 weeks old mice via retro-orbital bleeding and analyzed in an automated analyzer using mouse-specific parameters (VectScan5 - Abaxis). After sacrifice, tibias and femurs were isolated and bone marrow flushed with 2% FBS/PBS (F-PBS). Red blood cell lysis was performed after centrifugation and cells were suspended in 0.16 M ammonium chloride solution for 5 minutes on ice. Cells were then resuspended in 0.5 ml of F-PBS, counted, and incubated on ice for 60 minutes with the following antibodies: CD3 (145–2C11 Biolegend), CD4 (RM4–5 eBiosience), CD8 (53–6.7 eBioscience), CD19 (6D5 Biolegend), B220 (RA3–6B2 Biolegend), Gr1 (RB6–8C5 Biolegend), Ter119 (Ter119 Biolegend), cKit (2B8 BD, Pharmingen), Sca-1 (D7, eBioscience), CD150 (TC15–12F12.2 Biolegend), CD34 ( RAM34 BD Pharmingen), FcgRII/III (93 Biolegend), CD48 (HM48–1 Biolegend). Cells were washed, resuspended in F-PBS and analyzed by Flow Cytometry (BD LSR II).
END-seq
MEFs with the indicated genotypes were stably infected with a retrovirus encoding AsiSI (pTRE3G-HA-ER-AsiSI) and clones with high expression were selected. Exponentially growing AsiSI-expressing MEFs were treated with 1 μM doxycycline (DOX) for 24 hours and then, for 5 hours with 4-hydroxytamoxifen, resulting in the nuclear translocation of the restriction enzyme. MEFs were harvested and 9 million cells were embedded in agarose plugs, after which END-seq was performed as described (Canela et al., 2017). END-seq reads were aligned to the mouse (GRCm38p2/mm10) genomes using Bowtie (version 1.1.2) (Langmead et al., 2009) with parameters (-n 3 -k 1 −1 50). Break intensity was measured by integrating the RPKM values within 100 bp of each AsiSI break site. To quantify the width of maximum resection endpoint (in bp), a sliding window containing twenty 50 bp bins was used, starting from the AsiSI site out to 15 kb until more than 20% of the bins within this sliding window had signals lower than the background, the last bin within the window with a detectable signal over background was regarded as the maximum resection endpoint. The background was determined by the maximum END-seq signal for 50 bp bins more than 15 kb (within 15 kb-20 kb) away from individual AsiSI sites.
BRCA1 mutant cDNA generation
HA-BRCA1 cDNA was cloned into the pENTR1A Gateway Entry vector (Invitrogen) and shuttled into a pDest-IRES Destination vector (Life Technologies) using Gateway™ LR Clonase™ II Enzyme Mix (11791 Invitrogen). The BRCA1-Δ11q construct was previously described (Wang et al., 2016). The L1407P, M1411T, R1443G and CC mutations were generated by site-directed mutagenesis (Agilent). BRCA1 exon 11 deletion constructs were generated from full-length BRCA1 construct using Q5 Site-Directed Mutagenesis Kit (NEB E0554S). (See key resources table for primer sequences).
Lentivirus cell infection
Cell lines were infected with lentivirus using polybrene (Boston Bioproducts) and infected cells were selected based on either fluorescent protein expression or antibiotic resistance. sgRNA targeting 53BP1 or GFP was cloned into the lentiCRISPR v2 vector (gift from Feng Zhang) using BsmBI sites. After puromycin selection protein expression was determined by Western Blotting.
Colony formation assay
MEFS were seeded in 6-well plates at 300 cells per well in the presence of increasing concentrations of mitomycin C or rucaparib. For taxol and cisplatin sensitivity experiments 5,000–10,000 cells were seeded in 24 well plates and exposed to the drugs for 24 hours. After, cells were reseeded in 6 well plates and cultured in drug-free media. Colony formation was assessed after 10 days with crystal violet staining. Lethal concentration (LC50) values (concentration required to reduce colony formation by 50%) were calculated using GraphPad Prism software. Clovis Oncology provided rucaparib. Taxol and cisplatin were obtained from the Fox Chase Cancer Center pharmacy. Mitomycin C was from Selleckchem.
PARPi resistance assays
MDA-MB-436 cells expressing different BRCA1 proteins were seeded in 6 well plates in the following seeding densities: 2×104, 1×104, 0.5×104, 0.25×104, 1.25×103 and 0.625×103 cells/ well. The cells were treated with 100 nM of rucaparib for 2–3 weeks. Media containing rucaparib was changed every 48–72 hr. After 3 weeks resistant colonies were fixed with 3:1 methanol: acid acetic solution and stained with crystal violet. The mean colony formation from 3 different experiments was expressed as the mean percentage of resistant colonies ± S.E.M. relative to BRCA1 full-length expressing PARPi treated cells.
DNA replication fork protection
To measure replication fork protection MEFs were labeled with 50 μM CldU for 30 min, washed and exposed to 250 μM IdU for 30 minutes. This was followed by exposure of 2 μM of hydroxyurea for 3 hours (Sigma). Cells were collected and resuspended in PBS. Cells were lysed (200 mM Tris-HCl, 50 mM EDTA, 0.5% SDS, pH 7.4), and DNA fibers stretched onto glass slides (Superfrost Microscope Slides precleaned, Fisher Scientific, 12–550-123) by tilting slides 30 degrees, followed by fixing in a methanol: acetic acid solution (3:1) for 10 minutes. The fibers were denatured with 2.5 M HCl for 2.5 h, washed with PBS and blocked with 2% BSA in PBST for 40 min. Labeled DNA with CldU and IdU were stained with anti-BrdU antibodies recognizing CldU and IdU (Novus 1:500 and BD Bioscience 1:100, respectively). Anti-mouse Alexa 594 (1:300) and anti-rat Alexa 488 (1:300) were used for secondary antibodies. Images were captured with Nikon NIU Upright Fluorescence Microscope. Fiber lengths were quantified using ImageJ software.
Immunofluorescence and microscopy
Cells were fixed with 4% paraformaldehyde (PFA) and permeabilized with 1% Triton-X in PBS. Primary antibodies used: RAD51 ([N1C2] GeneTex), RPA32 (19/NA18 CalBiochem), RPA32 (4E4 Cell Signaling), CtIP (271339 Santa Cruz), RIF1 (A300–569A Bethyl), 53BP1 (3802 Millipore), phospho53BP1 (3428 Cell Signaling), BRCA1 (6954 Santa Cruz) BRCA1 (07–434 Millipore), PCNA (sc-56 Santa Cruz) and, HA tag (Cell Signaling 1:500). Primary antibodies were incubated overnight at 4 degrees in 5% goat serum. To evaluate RPA32 and PCNA we pre-extracted cells prior to fixation. Briefly, we treated cells with ice-cold cytoskeleton buffer (10 mM Pipes pH 6.8, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA, 0.5% Triton X-100) for 5 min on ice followed by 5 min incubation with ice-cold cytoskeleton stripping buffer (10 mM TrisHCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 1% Tween 40 (v/v), 0.5% sodium deoxycholate) then fixed in 4% PFA and continued to be processed as described above. Alexa-Fluor or FITC conjugated antibodies (Jackson ImmunoResearch Labs or Thermo Fisher Scientific 1:1000) were incubated for 1 hour at room temperature. For PCNA and BRCA1 co-localization experiments cells were treated with 2 μM of HU for 3 hours before fixation. Slides were mounted with mounting media containing DAPI. Images were captured using a Nikon NIU Upright Fluorescence Microscope and generated images using Nikon NIS Elements software. Unless otherwise specified, we routinely fixed cells 24 hours after exposure to 1 μM of PARPi or 8 hours after 10 Gy of γ-irradiation. For analyses, we counted a minimum of 100 cells per condition per cell line. Foci positive cells were nuclei that contained at least 5 foci for RAD51 and BRCA1 or 10 foci for 53BP1, phospho53BP1, RIF1, RPA32, CtIP, and PCNA. Each experiment was performed at least 3 times with biological replicates. Confocal microscopy was used to evaluate co-localization of BRCA1Δ11q-GFP and HA-BRCA1*M1411T (Leica SP8 advanced).
Western Blotting and Immunoprecipitation
Nuclear extracts were derived using the NE-PER Nuclear and Cytoplasmic Extraction Kit following the manufacturer’s instructions (Thermo Scientific). Protease inhibitor and phosphatase inhibitors were added (Millipore, Cat#524625 and #539131). Membranes were blocked for 1 hour with 5% nonfat milk in PBS. Lysates were separated by SDS-PAGE and transferred to a PDVF membrane (ImmobilonP Millipore). Primary Antibodies were incubated in 5% non-fat milk overnight at 4 degrees directed against: BRCA1 (Millipore MS110, 1:500), BRCA2 (Bethyl 1:2000), CtIP (Bethyl 1:2000), PALB2 (Bethyl 1:2000), RAD51 (Genetex 1:1000), RPA32 (Santa Cruz 1:1000), Tubulin (Cell signaling 1:2000), HA tag (Cell Signaling 1:1000), GFP (Santa Cruz 1:1000). Brca1 recognizing mouse species was a gift from A. Nussenzweig. HRP conjugated secondary antibodies were used. Immunoprecipitation of BRCA1 complexes was done using the Pierce Classic IP Kit (Thermo Fischer). Briefly, 500 μg of nuclear extract was pre-cleared with Control Agarose Resin Slurry for 1 hour at 4 degrees. Each lysate was incubated with 10 μg of anti-HA antibody (Cell Signaling) or with GFP trap (Chromotek) and incubated at 4 degrees overnight. To retrieve the complex 20 μl of Protein A/G agarose beads was incubated with the antibody/lysate sample for 3 hours. The beads were washed three times and eluted with 50 μl of acidic Elution Buffer provided by the manufacturer. To neutralize the low pH of the buffer, 5μl of 1 M Tris pH 9.5 was added.
Cell cycle analyses
Cells were harvested and fixed with 50% ethanol. Cells were then washed with PBS and re-suspended in 0.5 mL FxCycle PI/RNase Staining Solution (Life Technologies, Thermo Fisher Scientific) and incubated at room temperature for 30 min. Data were acquired using a BD LSR II Flow Cytometer and analyzed using FlowJo software.
Metaphase Spreads
The assay was performed by the Fox Chase Cancer Center Research Cytogenetics Core Facility. Briefly, MEFs were treated with 500 nM of rucaparib for 24 hours, then were exposed to Colcemid (0.01 μg/ml) for 12 h, harvested with 0.075 M KCl and fixed with methanol-acetic acid (3:1).
Histologic and immunohistochemical staining
Slides were deparaffinized with xylene and hydrated with ethanol. Antigen retrieval was performed using Tris/EDTA buffer (DAKO Target Retrieval Solution) and endogenous peroxidase was quenched by immersing slides in 3% hydrogen peroxide solution (30% H2O2, Fisher BP2633–500) diluted in methanol. Primary antibody for CD3 (0452 DAKO) was diluted with DaVinci Green Diluent (Biocare) and incubated on slides overnight at 4°C in a humidified slide chamber. Slides were then washed and incubated with EnVision+ System HRP Labeled Polymer Anti-Rabbit or Anti-Mouse for 1 hour at RT. Specimens were washed, then developed with DAB solution (Dako) and counterstained in Meyer’s Hematoxylin (Sigma-Aldrich).
Quantification and Statistical Analysis
Statistical analysis was performed using GraphPad Prism software. Statistically tests, significant p-values and number of replicates are indicated in the figure legends. There were similar variances between statistical groups compared.
Data and code availability
Mouse END-seq data are available at the GEO repository under accession number GSE147468. All code used are listed in the “Software and algorithms” section of the Key Resources Table.
KEY RESOURCES TABLE
The table highlights the genetically modified organisms and strains, cell lines, reagents, software, and source data essential to reproduce results presented in the manuscript. Depending on the nature of the study, this may include standard laboratory materials (i.e., food chow for metabolism studies), but the Table is not meant to be comprehensive list of all materials and resources used (e.g., essential chemicals such as SDS, sucrose, or standard culture media don’t need to be listed in the Table). Items in the Table must also be reported in the Method Details section within the context of their use. The number of primers and RNA sequences that may be listed in the Table is restricted to no more than ten each. If there are more than ten primers or RNA sequences to report, please provide this information as a supplementary document and reference this file (e.g., See Table S1 for XX) in the Key Resources Table.
Please note that ALL references cited in the Key Resources Table must be included in the References list. Please report the information as follows:
REAGENT or RESOURCE: Provide full descriptive name of the item so that it can be identified and linked with its description in the manuscript (e.g., provide version number for software, host source for antibody, strain name). In the Experimental Models section, please include all models used in the paper and describe each line/strain as: model organism: name used for strain/line in paper: genotype. (i.e., Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtm1.1Wsy/J). In the Biological Samples section, please list all samples obtained from commercial sources or biological repositories. Please note that software mentioned in the Methods Details or Data and Software Availability section needs to be also included in the table. See the sample Table at the end of this document for examples of how to report reagents.
SOURCE: Report the company, manufacturer, or individual that provided the item or where the item can obtained (e.g., stock center or repository). For materials distributed by Addgene, please cite the article describing the plasmid and include “Addgene” as part of the identifier. If an item is from another lab, please include the name of the principal investigator and a citation if it has been previously published. If the material is being reported for the first time in the current paper, please indicate as “this paper.” For software, please provide the company name if it is commercially available or cite the paper in which it has been initially described.
IDENTIFIER: Include catalog numbers (entered in the column as “Cat#” followed by the number, e.g., Cat#3879S). Where available, please include unique entities such as RRIDs, Model Organism Database numbers, accession numbers, and PDB or CAS IDs. For antibodies, if applicable and available, please also include the lot number or clone identity. For software or data resources, please include the URL where the resource can be downloaded. Please ensure accuracy of the identifiers, as they are essential for generation of hyperlinks to external sources when available. Please see the Elsevier list of Data Repositories with automated bidirectional linking for details. When listing more than one identifier for the same item, use semicolons to separate them (e.g. Cat#3879S; RRID: AB_2255011). If an identifier is not available, please enter “N/A” in the column.
A NOTE ABOUT RRIDs: We highly recommend using RRIDs as the identifier (in particular for antibodies and organisms, but also for software tools and databases). For more details on how to obtain or generate an RRID for existing or newly generated resources, please visit the RII or search for RRIDs.
Please use the empty table that follows to organize the information in the sections defined by the subheading, skipping sections not relevant to your study. Please do not add subheadings. To add a row, place the cursor at the end of the row above where you would like to add the row, just outside the right border of the table. Then press the ENTER key to add the row. Please delete empty rows. Each entry must be on a separate row; do not list multiple items in a single table cell. Please see the sample table at the end of this document for examples of how reagents should be cited.
TABLE FOR AUTHOR TO COMPLETE
Please upload the completed table as a separate document. Please do not add subheadings to the Key Resources Table. If you wish to make an entry that does not fall into one of the subheadings below, please contact your handling editor. (NOTE: For authors publishing in Current Biology, please note that references within the KRT should be in numbered style, rather than Harvard.)
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse Anti-phospho53BP1 Polyclonal antibody | Cell Signaling | Cat#3428 RRID:AB_2206631 |
| Rabbit Anti-53BP1 Polyclonal antibody | Novus | Cat#NB100-305SS RRID: AB_920464 |
| Mouse Anti-53BP1 Monoclonal antibody | Millipore | Cat# MAB3802, RRID:AB_2206767 |
| Mouse Anti-BRCA1 Monoclonal antibody | Santa Cruz | Cat# sc-6954 RRID: AB_626761 |
| Rabbit Anti-BRCA1 Polyclonal antibody (C-terminal) | Millipore | Cat# 07-434, RRID:AB_2275035 |
| Mouse Anti-Brca1 (Mouse) | Gift from A. Nussenzweig | Custom made |
| Mouse Anti-BRCA1 Monoclonal antibody (N-terminal) | Millipore | Cat# OP92T-10UG, RRID:AB_564282 |
| Rabbit Anti-BRCA2 Polyclonal antibody | Bethyl | Cat# A303-434A, RRID:AB_10952240 |
| Rabbit Anti-CtIP Polyclonal antibody | Bethyl | Cat# A300-488A, RRID:AB_2175262 |
| Mouse Anti-Geminin Monoclonal antibody | Abnova | H00051053-M01, RRID:AB_606308 |
| Rabbit Anti-RIF1 Polyclonal antibody | Bethyl | Cat# A300-569A RRID: AB_669804 |
| Mouse Anti-HA Monoclonal antibody | Cell Signaling | Cat# 2367S, RRID:AB_10691311 |
| Rabbit Anti-PALB2 Polyclonal antibody | Bethyl | Cat# A301-247A, RRID:AB_890608 |
| Rabbit Anti-Rad51 Polyclonal antibody | GeneTex | Cat# GTX100469, RRID:AB_1951602 |
| Rabbit Anti-RPA 32 Polyclonal antibody | Santa Cruz | Cat# sc-28709, RRID:AB_2238546 |
| Rat Anti-RPA32 (4E4) Monoclonal antibody | Cell Signaling | Cat# 2208S RRID: AB_2238543 |
| Mouse anti-GFP Monoclonal antibody | Santa Cruz | Cat# Sc-9996 RRID: AB_627695 |
| Rabbit Anti-Tubulin Polyclonal antibody | Cell Signaling | Cat# 2148, RRID:AB_2288042 |
| Mouse Anti-PCNA Monoclonal antibody | Santa Cruz | Cat# sc-56 RRID: AB_628110 |
| Hamster Anti-CD3 Monoclonal antibody (mouse) Clone: 145-2C11 | Biolegend | Cat# 100310 RRID AB_312675 |
| Rat Anti-CD4 Monoclonal antibody (mouse) Clone: RM4-5 | eBioscience | Cat# 15-0041-81 RRID AB_468695 |
| Rat Anti-CD8 Monoclonal antibody (mouse) Clone: 53-6.7 | eBioscience | Cat# 15-0081-81 RRID AB_468706 |
| Rat Anti-CD19 Monoclonal antibody (mouse) Clone: 6D5 | Biolegend | Cat# 115510 RRID AB_313645 |
| Rat Anti-B220 Monoclonal antibody (mouse) Clone: RA3-6B2 | Biolegend | Cat# 103210 RRID AB_312995 |
| Rat Anti-GR1 Monoclonal antibody (mouse) Clone: RB6-8C5 | Biolegend | Cat# 108410 RRID AB_313375 |
| Rat Anti-Ter119 Monoclonal antibody (mouse) Clone: Ter19 | Biolegend | Cat# 116210 RRID AB_313711 |
| Rat Anti-Sca-1 Monoclonal antibody (mouse) Clone: D7 | eBioscience | Cat# 25-5981-81 RRID AB_469669 |
| Rat Anti CD150 Monoclonal antibody (mouse) Clone: TC15-12F12.2 | Biolegend | Cat# 115903 RRID AB_313682 |
| Rat Anti-CD34 Monoclonal antibody (mouse) Clone: RAM34 | BD Pharmingen | Cat# 553733 RRID |
| Rat Anti-FcgRII/III Monoclonal antibody (mouse) Clone: 93 | Biolegend | Cat# 101327 RRID AB_1967102 |
| Hamster Anti-CD48 Monoclonal antibody (mouse) Clone: HM-48 | Biolegend | Cat# 103418 RRID AB_756140 |
| Rat Anti-cKIT Monoclonal antibody (mouse) Clone: 2B8 | BD Pharmingen | Cat# 553356 RRID |
| Alexa Fluor 594-AffiniPure Donkey Anti-Mouse IgG | Jackson ImmunoResearch Labs | Cat# 715-585-150, RRID:AB_2340854 |
| Alexa Fluor 594- AffiniPure Donkey anti-rabbit antibody | Jackson ImmunoResearch Labs | Cat# 711-585-152, RRID:AB_2340621 |
| Fluorescein (FITC)-AffiniPure Donkey Anti Mouse | Jackson ImmunoResearch Labs | Cat# 715-095-150, RRID:AB_2340792 |
| Fluorescein (FITC)-AffiniPure Donkey Anti-Rabbit | Jackson ImmunoResearch Labs | Cat# 711-095-152, RRID:AB_2315776 |
| Donkey Anti-Rabbit IgG ECL Antibody, HRP Conjugated | GE Healthcare | Cat# NA9340-1ml, RRID:AB_772191 |
| Sheep Anti-Mouse IgG ECL Antibody, HRP Conjugated | GE Healthcare | Cat# NA9310-1ml, RRID:AB_772193 |
| Rabbit anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Cat# A-11062, RRID:AB_2534109 |
| Chicken anti-Rat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-21470, RRID:AB_2535873 |
| Mouse anti-BrdU antibody | BD Biosciences | Cat# 347580, RRID:AB_400326 |
| Rat anti-BrdU antibody | Bio-Rad | Cat# OBT0030, RRID:AB_609568 |
| Rabbit anti-CD3 Polyclonal antibody | DAKO | Cat# A0452 RRID: AB_2335677 |
| Dako EnVision+ System- HRP Labelled Polymer Anti-Mouse | DAKO | Cat#K4000 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Rucaparib | Clovis Oncology | N/A |
| Cisplatin | Fox Chase Cancer Center Pharmacy | N/A |
| Taxol | Fox Chase Cancer Center Pharmacy | N/A |
| Mitomycin | Selleckchem | Cat# S8146 |
| EcoNI | New England Biolabs | Cat# R0521 |
| Vectashield mounting medium with DAPI | Vector | Cat#H-1200 |
| 5-Iodo-2′-deoxyuridine | Millipore Sigma | Cat#I7125 |
| 5-Chloro-2′-deoxyuridine | Millipore Sigma | Cat#C6891 |
| Tween-20 | Millipore Sigma | Cat#1379 |
| Triton X-100 | Millipore Sigma | Cat#X100 |
| Paraformaldehyde | Millipore Sigma | Cat#158127 |
| OneTaq® Hot Start 2X Master Mix | New England Biolabs | Cat#M048S |
| Transit-LT1 | Mirus | Cat#MIR 2304 |
| Sodium hydroxide | Millipore Sigma | Cat#221465 |
| DAKO Target Retrieval Solution | DAKO | Cat#S1699 |
| Dako Liquid DAB+ Substrate Chromogen System | DAKO | Cat#K3467 |
| DA VINCI green diluent | BIOCARE | Cat#PD900L |
| Background Sniper | BIOCARE | Cat#BS966H |
| Meyer’s Hematoxylin | Sigma-Aldrich | Cat#MHS32 |
| Polybrene | Boston Bioproducts | Cat#BM-862W |
| Alt-R Cas9 | IDT | Cat#1074181 |
| Critical Commercial Assays | ||
| FxCycle PI/RNase Staining Solution | Thermo Fisher | Cat#F10797 |
| Q5 Site-Directed Mutagenesis Kit | NEB | Cat# E0554S |
| QuikChange Lightning Site-Directed Mutagenesis Kit | Agilent | Cat#210518 |
| Gateway™ LR Clonase™ II Enzyme Mix | Invitrogen | Cat#11791 |
| Pierce™ Classic IP Kit | Thermo Fisher | Cat#26146 |
| NE-PER Nuclear and Cytoplasmic Extraction Kit | Thermo Fisher | Cat#78833 |
| Experimental Models: Cell Lines | ||
| MDA-MB-436 | ATCC | Cat#HTB-130 RRID:CVCL_0623 |
| HEK293T | ATCC | Cat#CRL-3216 RRID:CVCL_0063 |
| MEFS Brca1+/+ | This paper | N/A |
| MEFS Brca1CC/CC | This paper | N/A |
| MEFS Brca1CC/Δ11 | This paper | N/A |
| MEFS Brca1Δ11/Δ11 | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| B6C3F1/J | The Jackson Laboratory | Cat#100010 |
| Mouse: Brca1+/+ | This paper | N/A |
| Mouse: Brca1CC/CC | This paper | N/A |
| Mouse: Brca1CC/Δ11 | This paper | N/A |
| STOCK Brca1tm2.1Cxd/Nci (Δ11) | NCI | Cat#01XC9 |
| Oligonucleotides | ||
| SEE SUPLEMENTARY TABLE 1 | ||
| Recombinant DNA | ||
| Gateway™ pENTR™ 1A vector | Invitrogen | Cat#A10462 |
| pDest-IRES Destination vector | Life Technologies | Custom modified |
| pLentiCRISPR V2 | Addgene | Cat#52961 |
| pTRE3G-HA-ER-AsiSI | Gift from A. Nussenzweig | Custom made |
| pBabe-SV40LTAg | Gift from from W. Hahn | Addgene#13970 |
| Software and Algorithms | ||
| Prism Software v6.0 | GraphPad Software | www.GraphPad.com |
| Adobe Photoshop CS v8.0 | Adobe | www.Adobe.com |
| NIS Elements | Nikon | www.Nikon.com |
| FlowJo v10.1 | FlowJo | www.FlowJo.com |
Highlights.
A Brca1 coiled-coil (CC) mutation results in FA in mice
Brca1 exon 11 coding region is required for DNA end resection
Brca1 CC domain is essential for efficient RAD51 loading
Combining complementary mutant alleles partially restores HR
Acknowledgments
This work was supported by US National Institutes of Health (NIH) Grants R01CA214799 and R01HL150190, as well as Susan Komen CCRCR17499048 and Department of Defense OC130212. Clovis Oncology supplied rucaparib. We are grateful to FCCC Genomics, Cell Culture and Cell Sorting facilities. We thank Dr. Dali Zong for critical reading of the manuscript. We thank Dr. Zemin Liu at the FCCC Cytogenetics facility for help analyzing metaphase spreads.
Footnotes
Declaration of interests
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anantha RW, Simhadri S, Foo TK, Miao S, Liu J, Shen Z, Ganesan S, and Xia B (2017). Functional and mutational landscapes of BRCA1 for homology-directed repair and therapy resistance. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, and Helleday T (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917. [DOI] [PubMed] [Google Scholar]
- Canela A, Sridharan S, Sciascia N, Tubbs A, Meltzer P, Sleckman BP, and Nussenzweig A (2016). DNA Breaks and End Resection Measured Genome-wide by End Sequencing. Mol Cell 63, 898–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JR, Sossick AJ, Boulton SJ, and Jackson SP (2012). BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. Journal of cell science 125, 3529–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri AR, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, et al. (2016). Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535, 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Kass EM, Yen WF, Ludwig T, Moynahan ME, Chaudhuri J, and Jasin M (2017). ATM loss leads to synthetic lethality in BRCA1 BRCT mutant mice associated with exacerbated defects in homology-directed repair. Proc Natl Acad Sci U S A 114, 7665–7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daza-Martin M, Starowicz K, Jamshad M, Tye S, Ronson GE, MacKay HL, Chauhan AS, Walker AK, Stone HR, Beesley JFJ, et al. (2019). Isomerization of BRCA1-BARD1 promotes replication fork protection. Nature 571, 521–527. [DOI] [PubMed] [Google Scholar]
- Domchek SM, Tang J, Stopfer J, Lilli DR, Hamel N, Tischkowitz M, Monteiro AN, Messick TE, Powers J, Yonker A, et al. (2013). Biallelic deleterious BRCA1 mutations in a woman with early-onset ovarian cancer. Cancer Discov 3, 399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921. [DOI] [PubMed] [Google Scholar]
- Feng L, Fong KW, Wang J, Wang W, and Chen J (2013). RIF1 counteracts BRCA1-mediated end resection during DNA repair. J Biol Chem 288, 11135–11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo TK, Tischkowitz M, Simhadri S, Boshari T, Zayed N, Burke KA, Berman SH, Blecua P, Riaz N, Huo Y, et al. (2017). Compromised BRCA1-PALB2 interaction is associated with breast cancer risk. Oncogene. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire BL, Homma TK, Funari MFA, Lerario AM, Leal AM, Velloso E, Malaquias AC, and Jorge AAL (2018). Homozygous loss of function BRCA1 variant causing a Fanconi-anemia-like phenotype, a clinical report and review of previous patients. Eur J Med Genet 61, 130–133. [DOI] [PubMed] [Google Scholar]
- Greenberg RA, Sobhian B, Pathania S, Cantor SB, Nakatani Y, and Livingston DM (2006). Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev 20, 34–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isono M, Niimi A, Oike T, Hagiwara Y, Sato H, Sekine R, Yoshida Y, Isobe SY, Obuse C, Nishi R, et al. (2017). BRCA1 Directs the Repair Pathway to Homologous Recombination by Promoting 53BP1 Dephosphorylation. Cell reports 18, 520–532. [DOI] [PubMed] [Google Scholar]
- Johnson N, Johnson SF, Yao W, Li YC, Choi YE, Bernhardy AJ, Wang Y, Capelletti M, Sarosiek KA, Moreau LA, et al. (2013). Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc Natl Acad Sci U S A 110, 17041–17046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keupp K, Hampp S, Hubbel A, Maringa M, Kostezka S, Rhiem K, Waha A, Wappenschmidt B, Pujol R, Surralles J, et al. (2019). Biallelic germline BRCA1 mutations in a patient with early onset breast cancer, mild Fanconi anemia-like phenotype, and no chromosome fragility. Mol Genet Genomic Med 7, e863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan ME, Chiu JW, Koller BH, and Jasin M (1999). Brca1 controls homology-directed DNA repair. Mol Cell 4, 511–518. [DOI] [PubMed] [Google Scholar]
- Nacson J, Krais JJ, Bernhardy AJ, Clausen E, Feng W, Wang Y, Nicolas E, Cai KQ, Tricarico R, Hua X, et al. (2018). BRCA1 Mutation-Specific Responses to 53BP1 Loss-Induced Homologous Recombination and PARP Inhibitor Resistance. Cell reports 24, 3513–3527 e3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmar K, D’Andrea A, and Niedernhofer LJ (2009). Mouse models of Fanconi anemia. Mutat Res 668, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan CM, Dapic V, Tice B, Favis R, Kwan E, Barany F, Manoukian S, Radice P, van der Luijt RB, van Nesselrooij BP, et al. (2005). Classification of BRCA1 missense variants of unknown clinical significance. J Med Genet 42, 138–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R, Chun J, and Powell SN (2012). BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer 12, 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer SL, Tian L, Kahkonen M, Schwartzentruber J, Kircher M, University of Washington Centre for Mendelian, G., Consortium, F. C., Majewski J, Dyment DA, Innes AM, et al. (2015). Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov 5, 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Wu H, and Jasin M (2012). A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22, 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, and Livingston DM (1997). Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 88, 265–275. [DOI] [PubMed] [Google Scholar]
- Seo A, Steinberg-Shemer O, Unal S, Casadei S, Walsh T, Gumruk F, Shalev S, Shimamura A, Akarsu NA, Tamary H, and King MC (2018). Mechanism for survival of homozygous nonsense mutations in the tumor suppressor gene BRCA1. Proc Natl Acad Sci U S A 115, 5241–5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakya R, Reid LJ, Reczek CR, Cole F, Egli D, Lin CS, deRooij DG, Hirsch S, Ravi K, Hicks JB, et al. (2011). BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science 334, 525–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simhadri S, Peterson S, Patel DS, Huo Y, Cai H, Bowman-Colin C, Miller S, Ludwig T, Ganesan S, Bhaumik M, et al. (2014). Male fertility defect associated with disrupted BRCA1-PALB2 interaction in mice. J Biol Chem 289, 24617–24629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sy SM, Huen MS, and Chen J (2009). PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci U S A 106, 7155–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS (2016). Mechanism and regulation of DNA end resection in eukaryotes. Crit Rev Biochem Mol Biol 51, 195–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Bernhardy AJ, Cruz C, Krais JJ, Nacson J, Nicolas E, Peri S, van der Gulden H, van der Heijden I, O’Brien SW, et al. (2016). The BRCA1-Delta11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin. Cancer Res 76, 2778–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Bernhardy AJ, Nacson J, Krais JJ, Tan YF, Nicolas E, Radke MR, Handorf E, Llop-Guevara A, Balmana J, et al. (2019). BRCA1 intronic Alu elements drive gene rearrangements and PARP inhibitor resistance. Nature communications 10, 5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Qiao W, Linke SP, Cao L, Li WM, Furth PA, Harris CC, and Deng CX (2001). Genetic interactions between tumor suppressors Brca1 and p53 in apoptosis, cell cycle and tumorigenesis. Nat Genet 28, 266–271. [DOI] [PubMed] [Google Scholar]
- Zhang F, Fan Q, Ren K, and Andreassen PR (2009a). PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Molecular cancer research : MCR 7, 1110–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, and Yu X (2009b). PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr Biol 19, 524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong D, Adam S, Wang Y, Sasanuma H, Callen E, Murga M, Day A, Kruhlak MJ, Wong N, Munro M, et al. (2019). BRCA1 Haploinsufficiency Is Masked by RNF168-Mediated Chromatin Ubiquitylation. Mol Cell 73, 1267–1281 e1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mouse END-seq data are available at the GEO repository under accession number GSE147468. All code used are listed in the “Software and algorithms” section of the Key Resources Table.