

SUMMARY
Cell proliferation changes concomitantly with fate transitions during reprogramming, differentiation, regeneration, and oncogenesis. Methods to resolve cell cycle length heterogeneity in real time are currently lacking. Here, we describe a genetically encoded fluorescent reporter that captures live-cell cycle speed using a single measurement. This reporter is based on the color-changing fluorescent timer (FT) protein, which emits blue fluorescence when newly synthesized before maturing into a red fluorescent protein. We generated a mouse strain expressing an H2B-FT fusion reporter from a universally active locus and demonstrate that faster cycling cells can be distinguished from slower cycling ones on the basis of the intracellular fluorescence ratio between the FT’s blue and red states. Using this reporter, we reveal the native cell cycle speed distributions of fresh hematopoietic cells and demonstrate its utility in analyzing cell proliferation in solid tissues. This system is broadly applicable for dissecting functional heterogeneity associated with cell cycle dynamics in complex tissues.
In Brief
Cell cycle speed greatly influences cell state but remains challenging to measure, particularly in dynamic or complex tissues. Here, Eastman et al. describe H2B-FT, a two-color reporter that resolves cell cycle speed ratiometrically in a single-snapshot measurement, enabling the identification and prospective isolation of live cells with distinct cycling rates.
Graphical Abstract
INTRODUCTION
Cell cycle speed varies widely and undergoes dynamic changes during development and tissue homeostasis, linking characteristic cycling behavior with fate-specifying events (Chen et al., 2015; Soufi and Dalton, 2016). The cleavage divisions initiating embryogenesis follow well-defined rapid and synchronous mitotic cycles (O’Farrell et al., 2004), with the onset of gastrulation coinciding with cell cycle lengthening and diversification (Deneke et al., 2016; Newport and Kirschner, 1982). In mammals, a characteristically fast cell cycle is seen in embryonic stem cells (ESCs), and pluripotency exit is coupled with dramatic restructuring and lengthening of the cell cycle (Calder et al., 2013; White and Dalton, 2005). Post-development, highly regulated cell cycles are seen across many tissues, including blood (Orford and Scadden, 2008; Pietras et al., 2011), brain (Yoshikawa, 2000), intestine (van der Flier and Clevers, 2009), and others (Liu et al., 2005; Tumbar et al., 2004). In tissues with low cellular turnover such as the heart, cells’ inability to re-enter the cell cycle appears to underlie poor regenerative capacity (Tzahor and Poss, 2017). In high-turnover tissues such as blood, lifelong hematopoiesis is sustained by hematopoietic stem cells (HSCs), which divide rarely (Wilson et al., 2008), and their ability to maintain quiescence is essential for function (Pietras et al., 2011). Contrastingly, committed myeloid progenitors proliferate rapidly under homeostasis (Passegué et al., 2005). Granulocyte-macrophage progenitors (GMPs) in particular appear to be one of the most proliferative cell types (Passegué et al., 2005) and are known to possess unique cell fate plasticity beyond the hematopoietic fate (Guo et al., 2014; Ye et al., 2015).
Cell cycle abnormalities characterize certain disease states, such as cancer. Many oncogenes and tumor suppressor genes, such as Rb, p53, and c-Myc (Chen, 2016; Gabay et al., 2014; Knudsen and Wang, 2010), converge on the (dys)regulation of the cell cycle. Conventional chemotherapies often attempt to blunt cancer growth by targeting the cell cycle (Hamilton and Infante, 2016; Schwartz and Shah, 2005), but the efficacy can be compromised by proliferative heterogeneity among cancer cells (Fisher et al., 2013). Relapse due to development of chemo-resistance is thought to be related to the presence of quiescent cancer cells at the time of treatment (Chen et al., 2016). Recently, cyclin D-CDK4 has been shown to destabilize PD-L1 to induce tumor immune surveillance escape (Zhang et al., 2018).
Overall, understanding the consequences of diverse cycling behaviors in development, regeneration, and disease is fundamentally important. However, convenient assessment of cell cycle speed, especially in live cells of complex tissues, remains technically challenging.
Existing strategies for cell cycle analysis have several limitations. First, they mostly convey cell cycle phase (Sakaue-Sawano et al., 2008), not duration. Although fast dividing populations tend to contain more S/G2/M cells at any given time, high S/G2/M frequency could also indicate cell-cycle arrest at these phases. Second, although image tracking is direct and accurate for determining cell cycle length, many cells in vivo are not amenable to microscopy, because of their deep location, their migratory behavior, and the prohibitively long duration to observe at least two consecutive mitoses. Microscopy-based analysis does not enable physical separation of fast versus slow cycling cells for downstream assays. Third, label retention assays (Lyons et al., 2001) reflect divisional history but give little information about the current cycling state. Although such techniques have yielded tremendous knowledge on stem cell quiescence in vivo (Falkowska-Hansen et al., 2010; Tumbar et al., 2004; Wilson et al., 2008), cycling kinetics become difficult to resolve after the label is chased beyond the detection limit. The resolution of such methods is also constrained by how similar the cells are to each other (Guo et al., 2014). Some of the labels are cytotoxic or require fixation to visualize (Wilson et al., 2008). To overcome these limitations, we developed a genetically encoded color-changing fluorescent protein that reports the cell cycle speed of live cells in vitro and in vivo in a ratiometric manner.
RESULTS
Mathematical Modeling Predicts Long- and Short-Lived Molecules to Be Differentially Susceptible to Reduction by Cell Division
The steady-state level of a given molecule is determined by the sum of its biogenesis and turnover. Turnover occurs by active catalysis and by passive dilution when cells divide (Eden et al., 2011). The extent to which cell division influences a molecule’s levels depends on its half-life (Data S1). When the timescale of degradation is significantly shorter than the length of the cell cycle, the contribution from active turnover dominates. An example of this is the inhibitor of NF-κB, or IκBa, which undergoes enhanced degradation upon phosphorylation within minutes of signaling, leading to NF-κB activation (Häcker and Karin, 2006). However, when molecular lifespan approaches the length of the cell cycle, dilution by cell division becomes the main mode of removal. This phenomenon is seen during myeloid fate commitment from multipotent hematopoietic progenitors, where the stable PU.1 protein accumulates as the cell cycle lengthens (Kueh et al., 2013). Therefore, the intracellular concentration of a stable species varies more than that of an unstable species in response to cell division rate. The different behaviors of short- versus long-lived molecules in relation to cell cycle length are illustrated in Figure 1A, suggesting a potential strategy for ranking cell cycle length by measuring two molecules (e.g., fluorescent proteins) that are distinct in their half-lives.
Figure 1. Design Principles of a Fluorescent Reporter of Cell Cycle Speed.

(A) On the basis of the equation in Data S1, intracellular levels of a molecule (M) depend on its half-life and cell cycle length. The ratio of M’s concentration in theoretical cells of different cycling speeds is plotted. When M is short lived (circle, far left), its concentration shows little difference between slow and fast cycling cells. With long-lived M (circle, far right), the concentration difference increases in proportion to the difference in cell cycle lengths.
(B) The fluorescent timer (FT) is short lived as a blue protein and long lived as a red protein.
(C) The average blue/red ratio (BR) of cells expressing FT drops as cell cycle lengthens (see Data S2). Because BR fluctuates within a cell cycle (see Figure S1), the relationship between BR and cell cycle length is best described by a probability distribution. For modeling, cells are assumed to maintain a constant cycling rate. Solid and dashed error bars denote 1 and 2 SDs, respectively.
(D) Anticipated positions of slow and fast asymmetrically cycling cells on a hypothetical plot of blue versus red fluorescence.
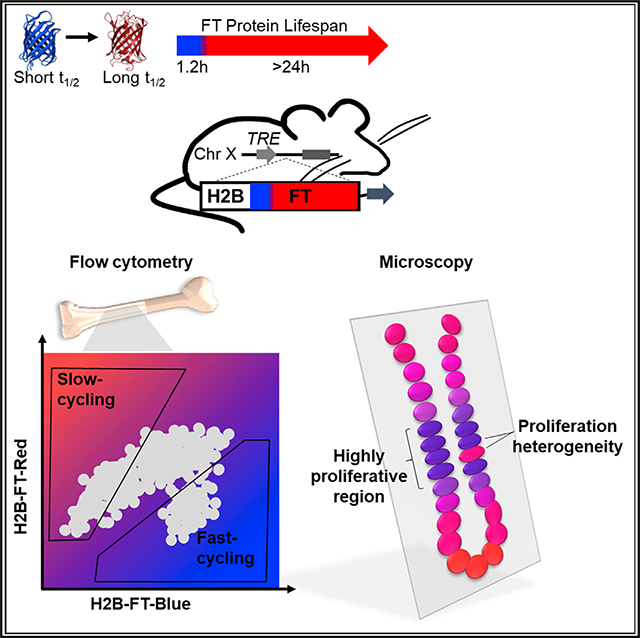
To exploit the differential susceptibility of labile and stable molecular species to cell division-mediated halving, we turned to the fluorescent timer (FT): a color-changing protein that emits blue fluorescence when newly synthesized, irreversibly turning red after a characteristic time delay (Subach et al., 2009) (Figure 1B; Figure S1A). The blue and red proteins are synthesized orthogonally in a stoichiometric 1:1 manner, because the blue species is a folding intermediate of the red, whose abundance can be measured separately by fluorescence microscopy or flow cytometry. Importantly, the blue form of the FT is lost soon after synthesis, when it converts to the red form, which has a much longer half-life (Figure 1B; Figure S1A). Thus, the blue form models the short-lived molecule, whose concentration is less variable across different cell cycle rates compared with the stable red form (Figures S1B–S1D). From a mathematical model describing the kinetics of this fluorescence conversion and degradation, we find that the steady-state blue/red fluorescence ratio (BR) scales inversely with cell cycle length (Data S2), with a shorter (longer) cell cycle length producing a higher (lower) BR. Thus, BR decreases as cell cycle lengthens or slows (Figure 1C; Figures S1B–S1D), providing a strategy to distinguish cycling rates (Figure 1D).
H2B-FT Fusion Proteins Undergo Blue-to-Red Conversion and Are Suitable for Proliferative Cells
The FT was fused to the C terminus of histone H2B (Figure 2A), a widely used strategy with minimal adverse consequences (Hadjantonakis and Papaioannou, 2004). This localized FT signal to the chromatin (Figure 2B), facilitating nuclear segmentation and image processing. To assess whether tagging the FT onto a histone interferes with its color-changing properties, we imaged HeLa cells expressing H2B-FT under the control of a tetracycline-inducible (TetO) promoter (Figure 2A). HeLa cells transduced with the “medium” FT variant (Subach et al., 2009) (Figure S1A) produced detectible blue signal ~2 h after adding doxycycline (Dox) (Figures 2C and 2D; Video S1), a time delay presumably required for Dox-induced transcription and translation. Approximately 1.5 h after the appearance of blue fluorescence, red signal emerged as the first wave of FT molecules underwent maturation (Figure 2C; Video S1). The average cellular level of red fluorescence rose for at least the next 26 h (Figure 2E). In contrast, the average blue level plateaued much sooner (after ~7 h) (Figure 2D), as expected for a molecule that is lost (in this case converted to red) shortly after synthesis. To confirm the divergent turnover times for the blue and red FT, we treated cells with Dox briefly (“Dox pulse”; Figure 2F). Upon Dox washout, blue signal dropped below detection within 7 h (Figures 2D and 2F), while red signal persisted for at least 24 h (Figures 2E and 2F), confirming that the red H2B-FT is more stable than the blue form. Mathematical simulation predicts that the FT reports cell cycle speed only after its expression reaches steady-state (Figures S1B–S1D). When newly expressed, the blue form should be over-represented independent of cell cycle effects because insufficient time has elapsed for red molecules to accumulate. In agreement with the model, cells had a higher BR within the first 24 h of Dox induction compared with cells maintained continuously in Dox (Figure 2G; Videos S1 and S2). As such, all subsequent in vitro experiments were conducted after at least 48 h on Dox.
Figure 2. Characterization of the H2B-FT Reporter in Cultured Cells.

(A) The inducible H2B-FT expression construct.
(B) HeLa cells expressing H2B-FT.
(C) Time series of H2B-FT fluorescence in HeLa cells after Dox induction.
(D and E) Quantification of average nuclear blue (D) and red (E) FT intensity over time following Dox treatment.
(F) Time series of HeLa cells after Dox washout at T = 4.5 h.
(G) BR over time following Dox treatment. Error bars represent SD across n = 3 culture wells. Scale bars, 50 μm.
We next assessed the reduction in H2B-FT-red signal contributed by mitosis-dependent and mitosis-independent processes. Single cells (primary mouse embryonic fibroblasts [MEFs] or HeLa cells) were tracked over 20 h after Dox washout (Figures S2A and S2B). Red fluorescence decreased by 15% in non-dividing MEFs (irradiated or naturally non-dividing by chance) (Figure S2C), while mitosis led to a drop of ~50% (Figures S2A and S2C). Similar to MEFs, mitosis caused a drop of ~50% in red fluorescence of dividing HeLa cells, as long as all progeny were accounted for (Figures S2B and S2D). These analyses confirmed that the predominant turnover mode of H2B-FT-red is mediated by cell division. In these analyses, the half-life of H2B-FT-red (~84 h in MEFs and ~37 h in HeLa cells) appeared longer than the 24 h half-life reported for the free FT-red (Subach et al., 2009). This gain in stability could result from fusion to a core histone, as histones are among the most stable proteins (Toyama et al., 2013). These results indicate that the resolution of the H2B-FT as a cell cycle speed reporter could become limited in slow dividing cells, when non-division-mediated decay becomes too substantial to be ignored.
BR Responds to Cell Cycle Manipulation in Cultured Cells
To test whether cell cycle acceleration causes BR to increase as predicted by the model (Figures 1C and 1D; Figures S1C and S1D), we expressed c-Myc in primary MEFs carrying allelic expression of H2B-FT-medium (details below) (Figure S1A). After transduction with c-Myc or an empty vector (EV) control virus, H2B-FT fluorescence was quantified using time-lapse microscopy (Figures S3A and S3B). As expected, cells overexpressing c-Myc proliferated more rapidly (Figure 3A). In agreement with the model, c-Myc-transduced MEFs had a higher BR than control MEFs after 70 h but not at 25 h post-transduction (Figure 3B), which was presumably before the exogenously expressed c-Myc had significantly altered the cell cycle. Notably, BR was heterogeneous in both control and c-Myc cultures (Figure 3C). To determine whether this reflected cell cycle speed heterogeneity, we classified cells on the basis of their relative BR using a flow cytometry-style gating strategy (Figure 3C) and systematically determined individual cell cycle lengths by tracking the time interval between consecutive mitoses. In c-Myc cultures, there were more cells in the bluer group (“blue cells”) and fewer red-gated cells (“red cells”) (Figures 3C and 3D). Importantly, the blue cells divided faster than the red ones in both conditions (Figure 3E). Whereas most red cells had a long cell cycle (>30 h), the blue cell cycle length centered around 10–15 h in control MEFs and 5–10 h in c-Myc MEFs (Figure 3E, blue cells). A small population (~1.5%) of c-Myc cells had cell cycle lengths shorter than 5 h (Figure 3E). Most cells with intermediate BR (cells between the gates in Figure 3C) centered around 25 h/cycle, likely representing the gap between the fast cycling blue and the slow cycling red cells (Figure 3E). These results concord with previous reports of MEF cell cycle length (White and Dalton, 2005). Importantly, BR reflected cell cycle length regardless of whether cells had been transduced with c-Myc (Figures 3E and 3F).
Figure 3. H2B-FT Color Profile Reflects Cell Proliferation Rate In Vitro.

(A) Proliferation of H2B-FT MEFs after transduction with c-Myc or empty vector control (EV). Error bars: SD across n = 4 culture wells.
(B) Cellular BR at 25 and 70 h post-transduction. Box plots represent median and interquartile range (IQR); whiskers, 5th–95th percentiles. p = 0.295 (25 h) and p < 0.0001 (70 h) on the basis of Mann-Whitney test with 99% confidence interval (CI).
(C) Representative blue versus red intensity of individual cells at 70 h post transduction. Flow cytometry-style gates define “red” and “blue” populations (see also Figures S3A and S3B).
(D) Percentage of cells within gates defined in (C). Percentages do not total 100%, as many cells lie between red and blue gates. Error bars show SD across n = 4 culture wells. p = 0.0035 (red cells) and p = 0.0498 (blue cells) on the basis of Student’s t test with Welch’s correction, 95% CI. dF = 4.057 (red cells) and dF = 3.221 (blue cells).
(E) Cell cycle length distributions in each gate as measured by image-based tracking. Cell cycle length refers to the time interval between consecutive mitoses.
(F) Correlation between cell cycle length and BR. All trackable cells (n = 220) from c-Myc and EV conditions are plotted. Spearman correlation coefficient = 0.7648; p < 0.0001 with 95% CI.
n values in (B) and (E) refer to the numbers of single cells analyzed.
The correspondence between short cell cycle and high BR supported the validity of H2B-FT as a cell cycle speed reporter and suggested that snapshot measurements by fluorescence microscopy could be used to estimate cell cycle length, which usually requires sophisticated long-term imaging and tracking with considerable phototoxicity (Skylaki et al., 2016). However, the fluorescence detection sensitivity by microscopy appeared limited, yielding a much narrower dynamic range compared with flow cytometry (Figure S3C). Therefore, quantitative estimation of cell cycle length was reserved for data acquired by flow cytometry, discussed below.
To test whether BR decreases upon cell cycle lengthening, we expressed H2B-FT in BaF3 cells, whose proliferation depends on IL-3 (Ajjappala et al., 2009). BaF3 cells cultured in full IL-3 (270 pg/mL) proliferated rapidly, slowed proliferation within 48 h when IL-3 was reduced to 14 pg/mL, and completely ceased proliferation in IL-3-free conditions (Figure 4A). Flow cytometry analysis of BR in these different growth conditions revealed that BaF3 cells grown in high IL-3 largely emitted blue fluorescence, which shifted toward the red axis at low IL-3 and became entirely red with IL-3 withdrawal (Figure 4B; Figures S3D and S3E).
Figure 4. H2B-FT Enables Fluorescence-Activated Cell Sorting of Live Cells with Different Proliferative Rates.

(A) BaF3 proliferation in varying IL-3 concentrations. Error bars: SD across n = 6 culture wells.
(B) Representative flow cytometry plots of BaF3 cells expressing H2B-FT-medium, grown as in (A).
(C) Density plots of BR derived from flow cytometry data in (B).
(D) H2B-FT knockin mESCs. Scale bar, 80 μm. Full-size image shown in Data S3A.
(E) DAPI/EdU cell cycle profile after 48 h RA treatment.
(F) Representative flow cytometry plots of pluripotent and RA-treated mESCs (top). Cells not exposed to Dox served as colorless control to demarcate H2B-FT+ gate. Data were rendered into BR density plots (bottom).
(G and H) Pluripotent and RA-treated mESCs stably expressing H2B-FT via Sleeping Beauty transposon were sorted on the basis of BR using the gating strategy shown. Non-transfected mESCs (gray) served as colorless control. (G) Representative FACS plots and gating. (H) Quantification of BR.
(I) DAPI/EdU cell cycle profiles of sub-populations sorted as in (G).
(J) Percentage of cells in G1 versus G2/M from the sorted populations in (G).
(K) Number of AP+ colonies recovered per 8,000 cells sorted in (G) after 6 day culture. Error bars: SD across n = 3 or 4 culture wells. p < 0.001, one-way ANOVA, dF = 3.
See also Figures S3D, S3E, and S4.
To facilitate direct comparison of flow cytometry data across diverse samples, we use a normalized BR, which is given by dividing a cell’s blue signal intensity by its total blue and red intensity:
This normalization eliminates differences in the amount of H2B-FT protein expressed by individual cells and places all on a scale of 0–1, with 0 being all red and 1 being all blue (Figures S4A–S4E). All BR data were normalized in this manner. As expected, BaF3 BR decreased in response to cell cycle lengthening (Figure 4C; Figure S3E). IL-3-induced differences in proliferation were captured by all three kinetic variants of the FT protein: H2B-FT-fast, H2B-FT-medium, and H2B-FT-slow (Figures 4B and 4C; Figures S3D and S3E). Furthermore, as predicted by the model (Data S2), the range of BR was lowest with FT-fast and highest with FT-slow (Figures S3D and S3E), which have the shortest and longest blue-to-red conversion times, respectively (Figure S1A). For simplicity, all data presented from here onward were obtained with H2B-FT-medium.
To examine the reporter’s response to changes in cell cycle speed accompanying a cell fate transition, we expressed H2B-FT in mouse ESCs (mESCs) from a targeted allele (Iacovino et al., 2011) (details below and Figure 4D) or by transposon (Hudecek et al., 2017). The characteristic mESC cell cycle of 8–10 h immediately lengthens upon differentiation (White and Dalton, 2005). mESCs expressing H2B-FT were induced to differentiate with retinoic acid (RA) for 48 h (Wichterle et al., 2002) (Figures S4F and S4G), which slowed the cell cycle as confirmed by EdU/DAPI staining (Figure 4E). As expected, RA-treated mESCs displayed a profound, population-wide shift toward the red axis (Figure 4F). This brief RA treatment is expected to induce only partial differentiation, yielding cell state heterogeneity, which was captured by the prominently red-shifted but overlapping population following treatment (Figure 4F; Figures S4H and S4I). Taken together, the two-color H2B-FT ratio responds to diverse modes of cell cycle manipulation in multiple cell types.
H2B-FT Enables Fluorescence-Activated Cell Sorting of Live Cells with Different Cycling Rates
To test the feasibility of physically separating live cells of distinct cell cycle rates, mESCs maintained in pluripotency conditions or treated with RA were sorted on the basis of BR (Figures 4G and 4H). Cell cycle profiles of the sorted populations were confirmed by EdU/DAPI staining (Figure 4I). As cells with longer cell cycles tend to increase their dwell time in G1 relative to other phases, slow dividing cells are expected to be enriched for G1 cells. Consistently, the reddest population (Figures 4G–4J, red) was enriched for G1 cells, while the bluest (Figures 4G–4J, blue) contained more G2/M cells. Cells from the middle FT gate (Figures 4G–4J, middle) had an intermediate cell cycle profile. These results are consistent with observations that mESCs do not respond homogeneously or synchronously to differentiation cues (Drukker et al., 2012; Semrau et al., 2017). Cell cycle rate heterogeneity was seen not only in the RA-treated samples but also in mESCs (Figures 4G–4J), corroborating reports that cellular heterogeneity exists under pluripotency maintenance conditions (Stewart et al., 2006) and is often associated with the cell cycle (Furusawa et al., 2006; Gonzales et al., 2015).
To evaluate functional heterogeneity associated with the H2B-FT color profile, cells sorted from different BR gates were re-plated in pluripotency conditions to evaluate their ability to give rise to new colonies expressing the pluripotency marker alkaline phosphatase (AP) (Kalkan et al., 2017; Sela et al., 2012). We expected the slow cycling (red) cells to be more differentiated and have reduced colony-forming ability, and any cells remaining pluripotent during the RA treatment to be among the fastest cycling (blue) group. In concordance with the DAPI/EdU results, RA treatment greatly reduced overall colony-forming activity (Figure 4K). Importantly, the residual colony-forming potential was most enriched in the blue cells and depleted in the red ones (Figure 4K, right). Our results also demonstrated that even among cells never induced to differentiate, colony-forming potential was highest in the bluest cells, although this difference was less pronounced (Figure 4K, left). These results confirmed the presence of cell cycle heterogeneity in both culture conditions, which could be prospectively separated by fluorescence activated cell sorting with the H2B-FT reporter.
The Proliferative Landscape of Live Hematopoietic Cells as Captured by H2B-FT
We harnessed the well-known proliferative differences among hematopoietic cells to test whether H2B-FT can report live-cell cycle speed in vivo (Passegué et al., 2005). As a first approach, H2B-FT was virally introduced into donor hematopoietic stem and progenitor cells (HSPCs), which were transplanted into recipient mice. Following engraftment and reconstitution, we analyzed HSPCs defined as Lin−Kit+Sca1+ (LKS), GMPs, and whole bone marrow (WBM) (Figures 5A and 5B; Figures S5A and S5B, gating strategy). The heterogeneity as reflected by the BR profile was most exaggerated in the WBM (Figure 5B), consistent with the fact that it contains the entire dynamic spectrum from rapidly cycling progenitors to post-mitotic differentiated cells. In contrast, the GMP and LKS distributions were narrower (Figure 5B). Among HSPCs positive for H2B-FT, LKS cells were red-shifted compared with GMPs (Figures 5A and 5B), in agreement with the LKS compartment containing quiescent and slow cycling hematopoietic progenitors, while GMPs are highly proliferative (Passegué et al., 2005; Wilson et al., 2008). The reconstituted LKS cells were heterogeneous, containing a bluer subset (Figures 5A and 5B) likely reflecting the more proliferative multipotent progenitors (Passegué et al., 2005; Wilson et al., 2008). Although the GMPs were collectively bluer than LKS and many other cells, they were not the bluest within the WBM (Figure 5B). Further investigation revealed the bluest cells to be Ter119+ (Figures 5C and 5D), indicating erythroid identity (Kina et al., 2000). Contrastingly, Mac1+ myeloid cells displayed a wide BR distribution, with a severely red-shifted subpopulation (Figures 5C and 5D). Therefore, the proliferative behavior of hematopoietic cells, as established by BrdU uptake in fixed cells from earlier studies (Passegué et al., 2005; Pop et al., 2010), can be conveniently recapitulated in live cells by the H2B-FT expressed from a viral vector.
Figure 5. The Proliferative Landscape of Live Hematopoietic Cells as Captured by the H2B-FT Reporter.

(A) Representative flow cytometry plots of LKS and GMP cells from mouse BM reconstituted by HSPCs virally expressing H2B-FT.
(B) Data from (A) plotted as BR distribution, with WBM BR added.
(C) Flow cytometry plots of myeloid (Mac1+) and erythroid (Ter119+) cells from the mouse in (A) and (B).
(D) BR distribution of data in (C). Gates in (A) and (C) excluded the non-transduced (H2B-FT−) cells.
(E) Targeting strategy for the iH2B-FT mouse allele.
(F) DAPI/EdU profiles of GMPs versus early erythroid cells after 2 h EdU labeling in vivo.
(G) BR distributions in LKS, GMPs, and early erythroid cells.
(H) Percentage of myeloid (Mac1+), B cell (B220+), and T cell (CD3+) cells in peripheral blood of healthy iH2B-FT mice versus those crossed with MLL-ENL (n = 3 mice per group). All mice were treated with Dox ≥8 days prior to analysis. Error bars represent SD; p = 0.0090 (Mac1+), p = 0.0072 (B220+), and p = 0.7629 (CD3+) on the basis of Student’s t test with 95% CI, dF = 4.
(I) Representative BR profiles of the PBMN subsets in (H).
To use the reporter in other tissues, we generated a knockin mouse by targeting the H2B-FT coding sequence into the universally active HPRT locus (Iacovino et al., 2011), under a Dox-inducible promoter (iH2B-FT) (Figure 5E). When crossed with specific rtTA or tTA, H2B-FT can be induced in desired cell types. To test this mouse model and confirm the results from the viral system, we crossed the iH2B-FT allele with Rosa26::rtTA (Hochedlinger et al., 2005), which enables inducible H2B-FT expression in most tissues, including the hematopoietic system. The compound mice were healthy and fertile. To directly compare the proliferative behavior of erythroid progenitors (ProE) and GMPs, we pulsed the iH2B-FT mice with EdU for 2 h and analyzed the cell cycle profile of CD71+/Ter119+ cells and GMPs (Figures S5A–S5C, gating strategy). Approximately 40% of the erythroid cells, compared with 27% of GMPs, were EdU labeled (Figure 5F). Accordingly, the CD71+/Ter119+ cells were further blue-shifted compared with GMPs (Figure 5G), corroborating results obtained with the viral approach. The LKS compartment was again robustly red-shifted and heterogeneous (Figure 5G), in agreement with virally expressed H2B-FT (Figures 5A and 5B). Taken together, the known cycling rates of bone marrow cells are faithfully and reproducibly captured by the H2B-FT irrespective of expression strategy.
Peripheral blood mononucleocytes (PBMNs) primarily consist of B cells (B220+), T cells (CD3+), and neutrophils/monocytes (Mac1+) (Figure 5H), with the myeloid cells turning over more rapidly than the lymphocytes (Passegué et al., 2005). This difference was readily reflected by the H2B-FT: Mac1+ cells were bluer than B220+ or CD3+ cells, whose BR was highly heterogeneous (Figure 5I, top). Although many circulating B and T cells did not express detectible H2B-FT, most myeloid cells (>95%) were H2B-FT+ (Figure 5I, top), on par with H2B-FT+ frequency in the WBM. Importantly, the H2B-FT+ B and T cells contained a severely red-shifted subpopulation, which could represent long-lived memory cells (Tough and Sprent, 1995). Although the exact identity and function of these various cellular subsets remain to be studied, cell sorting with H2B-FT should provide a convenient methodology.
Last, we explored the iH2B-FT BR profile in a disease state characterized by overt proliferation, such as acute myeloid leukemia (AML). To induce AML, iH2B-FT mice were crossed with Dox-inducible MLL-ENL (iMLL-ENL) allele (Ugale et al., 2014) and treated with Dox. As expected, the myeloid compartment expanded within 14 days, when the marrow contained a preponderance of L-GMPs (Figure S5D) and the blood became dominated by Mac1+ cells (Figures 5H and 5I; Figure S5E, bottom). The expanded myeloid compartment was immediately visible by the increased number of H2B-FT+ cells in the unstained whole blood (Figure 5I, bottom). Furthermore, the bluest shifted subpopulation became ~3 times larger than that in healthy control mice. Thus, the aberrant proliferative behaviors of malignant hematopoietic cells can be readily detected.
H2B-FT Unveils the Geography of Cell Proliferation In Situ
Because proliferation is compartmentally organized in many solid tissues, we evaluated the H2B-FT’s ability to resolve such borders in frozen tissue sections. In mice pulsed with EdU for 35 min, we first examined the kidney, representing low homeostatic proliferation (Kusaba et al., 2014), and the pylorus, which contains rapidly cycling cells. The pylorus is a gastric glandular tube lined with epithelium supported by non-dividing nerve, blood vessel, and muscle cells (Treuting et al., 2012). Gastric epithelial glands are organized into base, isthmus, and pit regions (Kim and Shivdasani, 2016), with the most rapidly dividing progenitors located in the isthmus. As expected, the BR of kidney cells was much lower overall than that of the gastric epithelium (Figures 6A and 6B; Figure S6, top and middle). In contrast, the pylorus was heterogeneous, with a subset of cells appearing as red as the kidney (Figure 6B, top and middle). When binned into five populations (P1–P5; Figure 6B), BR was rendered into a pseudo-colored heatmap and applied back onto the original images (Figures 6C and 6D). Notably, cells at the base of gastric glands were very red, juxtaposed abruptly with a region of bluer cells (Figures 6A and 6C, middle). The co-localization of this color boundary with a dense band of EdU+ nuclei confirmed that the BR identified the border between the mostly quiescent base compartment and the transit-amplifying isthmus region. Glands that were fully captured longitudinally from base to pit further showed a declining BR past the most proliferative zone (Figures 6C and 6D, “pylorus I”), as expected. Gastric glands captured in cross-section, representing assorted positions along the base-pit axis, showed the same co-localization between regions of high BR and high density of EdU+ cells (Figures 6C and 6D, “pylorus II”).
Figure 6. H2B-FT Profile Is Consistent with Relative Turnover Rates in Solid Tissue Sections.

(A) H2B-FT appearance in representative frozen sections from adult and embryonic iH2B-FT mice. Top: kidney. Middle: gastric glands in two orientations, longitudinal (pylorus I) and oblique/cross-sectional (pylorus II). L, lumen; S, submucosa. Bottom: neocortex of an E17.5 embryo. Dotted line indicates lateral ventricle (LV). See Figure S6 for single-channel images.
(B) BR histogram for each image in (A) was subdivided into five populations of increasing BR (P1–P5).
(C) Left, a heatmap corresponding to P1–P5 in (B) projected onto images from (A). Right: the same regions after DAPI/EdU labeling.
(D) Zoomed-in view of pyloric regions boxed in (C). Arrow: base of a gastric gland.
(E) Tiled images of an E17.5 embryonic brain hemisphere. Left: H2B-FT. Middle: heatmap of BR binned as in (B). Right: same region after DAPI/EdU labeling. Dotted line indicates LV. IZ, intermediate zone; CP, cortical plate; MZ, marginal zone; Th, thalamus.
(F) BR distributions from all tissues in (A). “Pylorus” includes combined data from both orientations (pylorus I and II, A).
Scale bars, 200 μm (A and C), 80 μm (D), and 500 μm (E). Full-size images are shown in Data S3B–S3H.
To test H2B-FT in embryogenesis, we examined the developing cortex, another tissue with well-recognized zones of proliferation and differentiation (Sun and Hevner, 2014). Ventricular radial glia divide rapidly during corticogenesis, while their progeny undergo a limited number of fate-specifying cell divisions as they migrate away from the ventricular zone (Uzquiano et al., 2018). In embryonic day (E) 17.5 iH2B-FT cortices, ventricular cells had the highest BR, followed by cells of the subventricular zone (Figures 6A–6C, bottom). Cells farther from the ventricle were redder, especially toward the interior of the brain. When tiled images are stitched together spanning a large tissue area, the H2B-FT color profile forms distinct layers in the embryonic cortex (Figure 6E). The proliferative behavior revealed by H2B-FT confirms known developmental timelines (Chen et al., 2017) and again is supported by the location of EdU+ cells (Figure 6C, bottom). For cells that exit cell cycle upon differentiation, the gradually decreasing BR should, up to a point, report relative time elapsed since their last division. Cells eventually plateau in their ability to become redder, reaching the resolution limit of the H2B-FT reporter. This is exemplified in the homogeneously red-shifted thalamus (Figure 6E), in which new neuron production peaks at E14 but trails off by E17 (Chen et al., 2017). Therefore, BR reveals the distribution of relative cell cycle lengths in situ (Figure 6F).
H2B-FT BR Provides an Estimate of Cell Cycle Length In Vivo
Because the two-color H2B-FT ratio agreed with known proliferative behaviors across all biological settings tested, we sought to establish a workflow to quantify cell cycle length from BR obtained in single-time point measurements. The equation in Data S2 describes a mathematical relationship between the steady-state BR,
, and the cell cycle length, τD:
and are quantified by fluorescence assays, preferably by flow cytometry given its high sensitivity (Figure S3C). The equation suggests that cell cycle length τD can be calculated as long as the constant C is known, as the color conversion time τC remains the same for a given FT variant (Figure S1A). The equation also states that C τC can be derived if τD can be experimentally determined. Therefore, we measured both cell cycle length distribution (Figure 7A) and BR distribution (Figure 7B) of the same cells using a combined live imaging/flow cytometry workflow. The details of the workflow to solve for C⋅ τC are described in the Mathematical Appendix in STAR Methods and in Table S1.
Figure 7. Estimating In Vivo Cell Cycle Length from H2B-FT BR.

(A and B) Cell cycle length distribution of n = 215 GMPs determined by time-lapse microscopy and BR distribution as analyzed by flow cytometry. Dotted lines show indicated percentiles.
(C) Calculated cell cycle length distributions of BM populations from a representative iH2B-FT mouse.
(D) Cell cycle length distribution of L-GMPs driven by MLL-ENL versus normal GMPs.
(E) Median cell cycle length variation among n = 3 iH2B-FT mice, shown by percentage coefficient of variation (CV%).
(F) Cell cycle length distributions of LKS and GMP from the individual mice in (E).
Boxes in (C), (D), and (F) represent median and IQR of each group; whiskers, 5th–95th percentiles. See also Figure S7.
Having determined the value of C τC (Figures 7A and 7B), we entered this constant into the equation above to obtain cell cycle lengths for freshly isolated bone marrow cell types. Specifically, we assessed BR in mature monocytes and granulocytes (Gran) (Lagasse and Weissman, 1996) as well as immunophenotypically defined stages of erythropoiesis (Koulnis et al., 2011) (Figures S5F and S5G, gating strategy). Gran were estimated to have a median cell cycle length of >30 h. The ProE appeared to have the shortest median cell cycle, ~4.3 h (Figure 7C). This result was particularly compelling, as it agrees with fetal liver ProE cell cycle length previously measured using sequential thymidine labeling (Hwang et al., 2017). L-GMPs from iMLL-ENL mice had a wider range of cell cycle speeds than normal GMPs, with the median shifted toward a faster cell cycle of ~9 h/cycle (Figure 7D). For most examined populations, cell cycle length distributions were similar across animals, and the variability generally decreased as the population became more immunophenotypically refined (Figure 7E). High variability was seen in WBM, as expected for a heterogeneous tissue (Figure 7E). However, the LKS compartment displayed striking variability across different animals (Figure 7E), contrasting the uniformity of GMPs from the same cohort. Specifically, while GMPs had a median cell cycle length of ~11.6 ± 1.2 h across three mice, the median cell cycle length of the corresponding LKS compartment was 22.7, 28.3, and 36.1 h (Figure 7F). We note that this median is most influenced by the predominant population, the more proliferative multipotent progenitors (Passegué et al., 2005). The LKS compartment is known to be heterogeneous (Copley et al., 2012; McKenzie et al., 2006), fluctuates diurnally (Méndez-Ferrer et al., 2009), remodels in response to injury (Haas et al., 2018), and evolves with aging (Beerman et al., 2010; Kowalczyk et al., 2015). Our reporter reveals an unexpected level of heterogeneity across individuals even under homeostatic conditions, offering a unique tool for unveiling the role of HSPC heterogeneity in regeneration, aging, and disease.
DISCUSSION
Harnessing the distinct lifespans of the FT’s two fluorescent states, we describe a novel, ratiometric method for resolving live-cell cycle length in vitro, in vivo, and in situ. Relative cell cycle speed can be determined from snapshot measurements by fluorescence imaging or flow cytometry: cellular BR faithfully reflects the proliferative state of all cell types tested, even in deep tissues inaccessible to imaging, making this a first-of-its-kind tool.
Measuring cell cycle length in vivo has previously been challenging and indirect. Detecting cell cycle dynamics in deep tissues relied largely on markers of specific cell cycle phases, such as the presence of EdU+ cells following a pulse label. Regions of EdU+ cells co-localize with areas of high BR (Figures 6C–6E), although cells marked by high BR are far less sparse. This is likely because EdU only marks cells that chanced to be in S phase during the pulse, leaving many blue (fast cycling) cells EdU−. Along with superior detection of fast cycling cells, BR also unveils hidden gradations in cell cycle length beyond the EdU+ zones. In general, interpreting pulse-label data assumes that S-phase duration varies minimally. In cases in which cell cycle accelerates by S-phase shortening (Hwang et al., 2017), a fast cell cycle could (counterintuitively) present with decreased frequency of labeled cells. Conversely, a slow cycling population arrested at S phase could show increased labeling frequency. These shortcomings are partly remedied by sequential labeling with two thymidine analogs (Bokhari and Raza, 1992). However, the accuracy of the dual-labeling method requires a largely homogeneous population, as it calculates the mean cell cycle length of all cells analyzed. As BR is determined for each cell individually free of such assumptions, the H2B-FT reporter begins to unveil hidden cell cycle heterogeneity.
The H2B-FT reporter resolves cell cycle speed in a single snapshot, a distinguishing ability from cell cycle phase reporters (Sakaue-Sawano et al., 2008). Besides revealing relative proliferation rate, BR can produce quantitative estimates of cell cycle length after a calibration step. Cell cycle length of ProE derived in this manner agree with results from dual thymine labeling (Hwang et al., 2017). We note that the relationship between BR and cell cycle length is nonlinear (Figure 1C). Experimental data from MEFs and briefly cultured GMPs, whose cell cycle length and BR can both be measured, support the model and show resolution plateauing beyond ~30 h/cycle (Figure 3F; Figure S7J). Although flow cytometry can help extend this dynamic range owning to its superior sensitivity (Basiji et al., 2007) (Figure S3C), this H2B-FT reporter is expected to better resolve faster cycling cells. For improved resolution at longer cell cycle lengths, future versions of the reporter could instead use the slow FT variant (Figure S1A; Figures S3D and S3E) or correct for BR “breathing” across the cell cycle (Figure S1) by simultaneous use of a phase reporter.
We anticipate wide use for H2B-FT as single-cell genomics underscore the importance of cellular heterogeneity, with cell cycle being a major driver (Battich et al., 2015; Olsson et al., 2016). Expressed from a knockin allele, even rare cell types such as HSCs can be reliably analyzed (Figures 5 and 7). As the H2B-FT wavelengths are compatible with additional colors, this allele can be crossed with GFP-expressing reporters to further refine cells of interest. Unlike assays based on label dilution (Tumbar et al., 2004), H2B-FT’s readout does not decay over cellular generations. We have also demonstrated that H2B-FT can be conveniently expressed, for example, by viral or transposon-based vectors, to enable cell cycle rate analysis in other models, including human cells. The ability to sort live cells free of additional labels or perturbation yields new opportunities to parse the interplay between cell fate regulation and cell cycle dynamics. For example, our previous work demonstrated the extraordinary response of naturally fast cycling GMPs (~8 h/cell cycle) to pluripotency induction (Guo et al., 2014) and to malignancy initiation (Chen et al., 2019). Sorting the fastest cycling GMPs would allow an unprecedented look into a unique state of cellular plasticity. Further purification of HSPC subsets is under way given the remarkable heterogeneity seen in LKS cells (Figure 7F). H2B-FT should also be helpful in the study of cell fate control during development. For example, it could pinpoint imbalances between proliferation and differentiation in the neocortex that may underlie neurodevelopmental disorders (Ernst, 2016). Finally, the method described here should be applicable to questions of immediate clinical relevance, such as the functional differences between fast- and slow dividing cancer cells in terms of metastatic potential, therapy response, and immune evasion. When expressed specifically in the host, the H2B-FT could also dissect proliferation heterogeneity in the complex tumor niche (Madar et al., 2013; Solito et al., 2014).
Beyond its technical utility, the mathematical principles underlying the H2B-FT reporter have important biological implications. The experimental data depict a generalized relationship between molecular half-life and intracellular concentration. These findings constitute direct evidence for a mechanism by which cells can selectively alter their molecular contents by modulating cell cycle dynamics. Simplistically, intracellular molecules are placed in two categories: those existing long enough such that their intracellular concentration depends on cell cycle length and shorter lived molecules that are less sensitive to dilution by cell division. As these principles do not discriminate against any specific class of molecule, they suggest a fundamental mechanism by which cell cycle speed might be tuned to optimize the concentration of key molecular regulators, including proteins, their various post-translationally modified derivatives, RNA species, and others. This mode of regulation could echo direct regulation on the half-life of proteins and RNA, as their effective concentration is determined not only by their turnover rate but also by cell cycle length.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Requests for resources and further information should be directed to and will be fulfilled by the Lead Contact, Shangqin Guo (shangqin.guo@yale.edu).
Materials Availability
Submission of H2B-FT plasmids to Addgene is in process. iH2B-FT-Medium mice have been deposited to the Mutant Mouse Resource & Research Centers (MMRRC). Other unique materials generated in this study, including cell lines, are available from the authors upon request.
Data and Code Availability
No unique code was generated in this study. Raw data used to support the conclusions of this study are available from the authors on request. Custom CellProfiler (Carpenter et al., 2006) image analysis pipelines along with sample images are available on request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cloning and reporter cell lines
pFast-FT-N1, pMedium-FT-N1, and pSlow-FT-N1 (Subach et al., 2009) were obtained from Addgene. Each FT coding sequence (~711 bp each) was cloned into a Dox-inducible lentiviral backbone and a constitutive retroviral backbone. The inducible lentiviral plasmid (pFU-TetO-Gateway-PGK-Puro) was constructed previously (Guo et al., 2012) by inserting a Gateway cassette (Invitrogen), a PGK promoter, and a puromycin resistance gene into the pFU-tetO-Klf4 vector (Stadtfeld et al., 2008) through blunted EcoRI sites. Each FT insert was then cloned into this destination vector through Gateway recombination. For constitutive expression, the FT sequences were inserted into a pSCMV retroviral backbone (Guo et al., 2012) using HindIII and XhoI restriction sites. The H2B-FT fusion transgenes were constructed by overlap extension PCR using the three FT plasmids as well as the human H2B.J coding sequence from PGK-H2B-mCherry (Kita-Matsuo et al., 2009, obtained from Addgene) as templates, and cloned into the pFU-TetO-Gateway-PGK-Puro and pSCMV expression plasmids. H2B-mCherry and H2B-BFP (BFP template obtained from Addgene) were similarly cloned to serve as single-color controls for the color-changing H2B-FT.
The MSCV-IRES-GFP (“empty vector”) retroviral expression plasmid was previously described (Lu et al., 2008). The LZRS-c-Myc-IRES-GFP retroviral expression plasmid was a gift from Sebastian Nijman.
Viral vectors were transfected in 293T cells using Fugene® 6 transfection reagent (Promega). Viruses harvested from the supernatant were used to transduce BaF3 and HeLa cells. Successfully transduced cells were selected by fluorescence activated cell sorting.
HPRT::iH2B-FT knock-in mouse embryonic stem cells (mESCs) were generated using inducible cassette exchange (ICE) to target the TetO-CMV-H2B-FT transgene to the HPRT locus by cre-recombination in the A2lox.cre mESC cell line (Iacovino et al., 2011). Briefly, H2B-FT inserts (all three kinetic variants, Subach et al., 2009 and Figure S1A), as well as H2B-BFP and H2B-mCherry were cloned into P2lox targeting plasmids (Iacovino et al., 2011) using HindIII and NotI restriction sites. The targeting plasmids were electroporated into Dox-activated A2lox.cre mESCs. After one day of recovery on neomycin-resistant feeder MEFs (Millipore Sigma), successfully recombined clones were selected by supplementing the mESC culture medium with 300μg/mL geneticin (G418, GIBCO). After 6 days of selection, healthy surviving colonies were hand-picked under an inverted microscope and replated onto C57BL/6J irradiated feeder MEFs. These “Passage 0” cultures were subsequently split for cryopreservation as well as continued culture and characterization of reporter activity by fluorescence microscopy and flow cytometry.
mESCs were made to constitutively express H2B-FT under an EF-1α promoter using the Sleeping Beauty transposon system (Hudecek et al., 2017). H2B-FT inserts (all three variants) were cloned into a modified empty transposon (pT3) backbone. A GFP sequence in the original backbone, pT3-Neo-EF1a-GFP (Chicaybam et al., 2017, obtained from Addgene), was replaced with a multiple cloning site (MCS) synthesized by IDT using MluI and NotI restriction cloning. The H2B-FT sequence was then inserted into the MCS using PmeI and NotI restriction sites. pT3-EF1a-H2B-FT vectors were transfected into C57BL/6J mESCs using Lipofectamine™ 2000 reagent (Invitrogen) along with a separate vector, pCMV(CAT)T7-SB100 (Mátés et al., 2009, obtained from Addgene), encoding the Sleeping Beauty transposase. Successfully transfected mESCs were enriched by fluorescence activated cell sorting.
Primer sequences used for cloning are provided in Table S2. Plasmid maps are available upon request.
Cell culture and mESC differentiation
MEFs, HeLa, and 293T cells were cultured in a standard growth medium (“MEF medium”) consisting of DMEM basal medium (GIBCO) with 10% heat-inactivated fetal bovine serum (FBS) (GIBCO) and 1% penicillin/streptomycin/L-Glutamine supplement (Thermo Fisher Scientific). For MEF derivation from E13.5 embryos, this medium was additionally supplemented with 1% Non-essential amino acid (NEAA) mixture (Thermo Fisher Scientific) for the first passage in vitro. 293T cells used for viral transfection were cultured in the standard growth medium additionally supplemented with 1% sodium pyruvate (Thermo Fisher Scientific). Phenol-red-free Medium 199 (Thermo Fisher Scientific) was used in place of DMEM for live fluorescence microscopy experiments with H2B-FT HeLa cells (Figure 2). Mouse embryonic stem cells (mESCs) were cultured either on γ-irradiated primary feeder MEFs or on plates coated with 0.1% gelatin in DMEM supplemented with 15% ESC-qualified FBS (Millipore Sigma), 1% penicillin/streptomycin/L-Glutamine, 1% NEAA, 1000U/ml LIF (Millipore Sigma), and 0.8μl/100mL β-mercaptoethanol. BaF3 cells were cultured in an RPMI-based growth medium consisting of 10% heat-inactivated FBS, 1% penicillin/streptomycin/L-Glutamine, and 270pg/mL IL-3 (Peprotech). Primary GMPs were cultured in x-vivo15 medium (Lonza) supplemented with 10% BSA (STEMCELL Technologies), 1% penicillin/streptomycin/L-Glutamine, 0.14μl/mL β-mercaptoethanol, and 100 ng/ml mSCF, 50 ng/ml mIL3, 50 ng/ml Flt3L, and 50 ng/ml mTPO (all from PeproTech).
In vitro, doxycycline (Sigma) was added to cell culture medium at 2μg/mL for inducible promoter activation. To induce iH2B-FT and/or iMLL-ENL expression in vivo, mice were fed drinking water containing 1g/L Dox supplemented with 10 g/L sucrose.
For the differentiation assay, mESCs maintained on feeder MEFs were transferred to feeder-free conditions (0.1% gelatin) for 2 passages to potentiate exit from pluripotency. The pluripotency of control cells was maintained with mESC culture medium, while the differentiation condition entailed switching the cells into standard MEF medium supplemented with 2μM retinoic acid (RA, Sigma).
For the colony recovery assay, sorted cells were plated onto feeder MEFs (n = 4 replicate wells) at a standardized seeding density and fed with mESC medium. After 6 days, the cultures were fixed and stained for alkaline phosphatase (AP) activity using the Stemgent AP II kit, in accordance with the protocol provided by the manufacturer. Culture wells were scanned under brightfield microscopy and colonies staining positive for AP activity were counted manually (RA treated group) or using MetaXpress® software automated image processing (mESCs).
Total mRNA was extracted with TRIzol® Reagent (Ambion) and reverse transcribed into cDNA using SuperScriptIII First-Strand Synthesis SuperMix (Invitrogen) according to the product manual. For quantitative real-time PCR, cDNA and gene-specific primers were mixed with iQ SYBR®Green Supermix (Bio-Rad) and carried out using a Bio-Rad CFX384 Real-Time PCR System. Gene expression levels were normalized to GAPDH level in the same sample. qPCR primer sequences are provided in Table S2.
Mice
All mouse work was approved by the Institutional Animal Care and Use Committee (IACUC) of Yale University. All research animals were housed and maintained in facilities of Yale Animal Resource Center (YARC).
HPRT::iH2B-FT-Medium chimeric mice were generated from passage 0 A2lox.cre H2B-FT targeted mouse embryonic stem cells by the Yale Genome Editing Center via blastocyst injection and implantation into C57BL/6J females. High-degree chimeric male offspring were selected by coat color and the iH2B-FT allele was subsequently crossed with mice carrying the Rosa26::rtTA allele (Hochedlinger et al., 2005), and backcrossed onto a C57BL/6J background. For all H2B-FT blue/red analysis of primary-harvested cells, H2B-FT expression was induced in vivo for at least one week by feeding Dox drinking water. Tissues from iH2B-FT mice were harvested and analyzed when the mice were 8–12 weeks of age. For experiments involving AML induction, iH2B-FT mice were crossed with iMLL-ENL mice (Ugale et al., 2014), and offspring were harvested at 5 weeks old. All cohorts reported here involved male mice. Genotyping primer sequences are provided in Table S2.
Primary feeder MEFs for pluripotent stem cell culture were derived from C57BL/6J (Jackson Laboratory) E13.5 mouse embryos and mitotically inactivated by 80 Gy γ-irradiation. H2B-FT-Medium MEFs were derived from HPRT::iH2B-FT-Medium E13.5 mouse embryos.
METHOD DETAILS
HSPC transplantation
Bone-marrow-derived LKS (Lineage-/Kit+/Sca-1+) cells from homozygous Rosa26::rtTA male donor mice were isolated by fluorescence activated cell sorting and transduced overnight in vitro with TetO-H2B-FT lentivirus and injected through the tail vein at > 10,000 cells/mouse into 9-week-old female C57BL/6J mice (Jackson Laboratory) γ-irradiated recipient mice (9 Gy) along with 500,000 WBM support cells. Alternatively, donor mice were injected intraperitoneally with 150μg/g body weight 5-fluorouracil (5-FU, APP Pharmaceuticals, LLC) 4 days before harvesting, and the HSPC-enriched WBM was virally transduced and then transplanted at a ratio of 1 donor per 2 recipients. Transplanted mice were given a one-time intraperitoneal injection of 200μg Dox and thereafter continuously maintained on Dox drinking water. After four weeks, peripheral blood was collected and analyzed by flow cytometry to evaluate H2B-FT expression coming from the engrafted virally transduced cells. Bone marrow HSPCs were harvested between 8–16 weeks post-transplantation and stained with fluorescent antibodies marking Lineage, Kit, Sca-1, CD34, and CD16/32. “Lineage” refers to a cocktail of antibodies included biotin-conjugated Ter119, CD3e, CD4, CD8, CD11b, Gr1, and B220. Cellular fluorescence was recorded on an LSRII flow cytometer (BD) or a FACSAria cell sorter (BD) using FACSDiva software (BD), and the data were subsequently analyzed using FlowJo software (FlowJo, LLC).
All antibodies used in this study are listed in the Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Biotin rat anti-mouse Ly-6G/6C Clone RB6-8C5 | BD Biosciences | Cat#553125; RRID:AB_394641 |
| Biotin hamster anti-mouse CD3e Clone 145-2C11 | BD Biosciences | Cat#553060; RRID:AB_394593 |
| Biotin rat anti-mouse CD45R/B220 Clone RA3-6B2 | BD Biosciences | Cat#553086; RRID:AB_394616 |
| Biotin rat anti-mouse CD11b Clone M1/70 | BD Biosciences | Cat#553309; RRID:AB_394773 |
| Biotin rat anti-mouse Ter119 | BD Biosciences | Cat#553672; RRID:AB_394985 |
| Biotin rat anti-mouse CD8a Clone 53-67 | BD Biosciences | Cat#553029; RRID:AB_394567 |
| Biotin rat anti-mouse CD4 Clone GK1.5 | BD Biosciences | Cat#553728; RRID:AB_395012 |
| APC rat anti-mouse CD117 Clone 2B8 | BD Biosciences | Cat#553356; RRID:AB_398536 |
| FITC rat anti-mouse Ly6A/E (Sca-1) Clone E13-161.7 | BD Biosciences | Cat#553335; RRID:AB_394791 |
| PerCP Streptavidin | BD Biosciences | Cat#554064; RRID:AB_2336918 |
| PE-Cy7 rat anti-mouse CD16/32 Clone 93 | Thermo Fisher Scientific | Cat#25-0161-82; RRID:AB_469598 |
| Alexa Fluor rat 700 anti-mouse CD34 Clone RAM34 | BD Biosciences | Cat#560518; RRID:AB_1727471 |
| APC rat anti-mouse CD11b Clone M1/70 | Thermo Fisher Scientific | Cat#17-0112-83; RRID:AB_469344 |
| FITC rat anti-mouse CD45R/B220 Clone RA3-6B2 | BD Biosciences | Cat#553087; RRID:AB_394617 |
| PE-Cy7 rat anti-mouse CD3 Clone 17A2 | BioLegend | Cat#100219; RRID:AB_1732068 |
| FITC rat anti-mouse CD71 Clone C2 | BD Biosciences | Cat#553266; RRID:AB_394743 |
| PE-Cy7 rat anti-mouse Ter119 | Thermo Fisher Scientific | Cat#25-5921-81; RRID:AB_469660 |
| FITC rat anti-mouse Ly6C Clone AL-21 | BD Biosciences | Cat#553104; RRID:AB_394628 |
| PE-Cy7 rat anti-mouse Ly6G Clone 1A8 | BD Biosciences | Cat#560601; RRID:AB_1727562 |
| Streptavidin MicroBeads | Miltenyi Biotec | Cat#130-048-101 |
| Bacterial and Virus Strains | ||
| pFast-FT-N1 | Dr. Vladislav Verkhusha (Subach et al., 2009) | RRID:Addgene_31910 |
| pMedium-FT-N1 | Dr. Vladislav Verkhusha (Subach et al., 2009) | RRID:Addgene_31911 |
| pSlow-FT-N1 | Dr. Vladislav Verkhusha (Subach et al., 2009) | RRID:Addgene_31912 |
| pFU-TetO-Gateway-PGK-Puro | Dr. Jun Lu (Guo et al., 2012) | N/A |
| pSCMV | Dr. Jun Lu (Guo et al., 2012) | N/A |
| PGK-H2B-mCherry | Dr. Mark Mercola (Kita-Matsuo et al., 2009) | RRID:Addgene_21217 |
| pMSCV-IRES-Blue FP | Dr. Dario Vignali | RRID:Addgene_52115 |
| MSCV-IRES-GFP | Dr. Jun Lu (Lu et al., 2008) | N/A |
| LZRS-c-Myc-IRES-GFP | Dr. Sebastian Nijman | N/A |
| pFU-TetO-H2B-FT-Fast-PGK-Puro | This Publication | N/A |
| pFU-TetO-H2B-FT-Medium-PGK-Puro | This Publication | N/A |
| pFU-TetO-H2B-FT-Slow-PGK-Puro | This Publication | N/A |
| pSCMV-H2B-FT-Fast | This Publication | N/A |
| pSCMV-H2B-FT-Medium | This Publication | N/A |
| pSCMV-H2B-FT-Slow | This Publication | N/A |
| p2Lox | Dr. Michael Kyba (Iacovino et al., 2011) | RRID:Addgene_34635 |
| pT3-Neo-EF1a-GFP | Dr. Martin Bonamino (Chicaybam et al., 2017) | RRID:Addgene_69134 |
| pCMV(CAT)T7-SB100 | Dr. Zsuzsanna Izsvak (Mátés et al., 2009) | RRID:Addgene_34879 |
| pT3-EF1a-H2B-FT-Fast | This Publication | N/A |
| pT3-EF1a-H2B-FT-Medium | This Publication | N/A |
| pT3-EF1a-H2B-FT-Slow | This Publication | N/A |
| Biological Samples | ||
| Primary neomycin resistant feeder MEFs, Mitomycin C treated | Millipore Sigma | Cat#PMEF-N |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ESGRO mLif Medium Supplement | Millipore Sigma | Cat#ESG1107 |
| Recombinant murine IL-3 | PeproTech | Cat#213-13 |
| Recombinant murine SCF | PeproTech | Cat#250-03 |
| Recombinant murine TPO | PeproTech | Cat#315-14 |
| Recombinant human Flt3 Ligand | PeproTech | Cat#300-19 |
| Doxycycline hyclate | Sigma Aldrich | Cat#D9891; CAS ID 24390-14-5 |
| Retinoic acid | Sigma Aldrich | Cat#R2625; CAS ID 302-79-4 |
| DAPI (4’,6-Diamidino-2-Phenylindole, Dihydrochloride) | Thermo Fisher Scientific | Cat#D1306; RRID:AB_2629482 |
| EdU (5-Ethynyl-2′-deoxyuridine) | Cayman Chemical | Cat#20518; CAS ID 61135-33-9 |
| 5-FU (5-Fluorouracil) | APP Pharmaceuticals, LLC | NDC 63323-117-10 |
| Critical Commercial Assays | ||
| Molecular Probes Click-iT Plus EdU Alexa Fluor 488 Imaging Kit Cat# C10637 | Thermo Fisher Scientific | Cat#C10637 |
| Stemgent AP Staining Kit II | REPROCELL | Cat#00-0055 |
| SuperScript III First-Strand Synthesis SuperMix | Thermo Fisher Scientific | Cat#18080400 |
| iQ SYBR Green Supermix | Bio-Rad | Cat#170-8882 |
| MACS LD Columns | Miltenyi Biotec | Cat#130-042-901 |
| QuadroMACS Separator | Miltenyi Biotec | Cat#130-090-976 |
| MACS MultiStand | Miltenyi Biotec | Cat#130-042-303 |
| Experimental Models: Cell Lines | ||
| HEK293T Cell Line | ATCC | Cat#CRL-3216; RRID:CVCL_0063 |
| HeLa Cell Line | ATCC | Cat#CRM-CCL-2; RRID:CVCL_0030 |
| BaF3 Cell Line | RCB | Cat#RCB0805; RRID:CVCL_0161 |
| A2lox.cre mESC Cell Line | Dr. Michael Kyba (Iacovino et al., 2011) | N/A |
| A2lox.cre H2B-FT-Fast mESC Cell Line | This Publication | N/A |
| A2lox.cre H2B-FT-Medium mESC Cell Line | This Publication | N/A |
| A2lox.cre H2B-FT-Slow mESC Cell Line | This Publication | N/A |
| A2lox.cre H2B-BFP mESC Cell Line | This Publication | N/A |
| A2lox.cre H2B-mCherry mESC Cell Line | This Publication | N/A |
| Experimental Models: Organisms/Strains | ||
| Mus musculus: HPRT::iH2B-FT-Medium: B6.Cg- Gt(ROSA)26Sortm1(rtTA*M2)Jae Hprttm2(tetO-H2B/mediumFT)Sguo | This Publication | MMRRC Cat#66959 |
| Mus musculus: iMLL-ENL: Col1a1tm1(tetO-KMT2A/ELL)Dbry | Dr. David Bryder (Ugale et al., 2014) | MGI ID 5825211 |
| Mus musculus: Rosa26::rtTA: Gt(ROSA) 26Sortm1(rtTA*M2)Jae | Jackson Laboratory | Cat#006965; RRID:IMSR_JAX:006965 |
| Mus musculus: C57BL/6J | Jackson Laboratory | Cat#000664; RRID:IMSR_JAX:000664 |
| Oligonucleotides | ||
| Primers for cloning | See Table S2 | N/A |
| Primers for genotyping | See Table S2 | N/A |
| Primers for qPCR | See Table S2 | N/A |
| Software and Algorithms | ||
| ImageJ | ImageJ (Schneider et al., 2012) | RRID:SCR_003070 |
| MetaXpress Software | Molecular Devices | RRID:SCR_016654 |
| CellProfiler Image Analysis Software | CellProfiler (Carpenter et al., 2006) | RRID:SCR_007358 |
| FCS Express Image Cytometry | De Novo Software | RRID:SCR_016431 |
| FACSDiva Software | BD Biosciences | RRID:SCR_001456 |
| FlowJo Flow Cytometry Analysis | FlowJo | RRID:SCR_008520 |
| Prism Software | GraphPad | RRID:SCR_002798 |
EdU/DAPI labeling
Cultured cells were treated with growth medium containing 10μM EdU (5-ethynyl-2′-deoxyuridine) for 15 minutes. EdU was rinsed away with PBS and cells were trypsinized into a single-cell suspension for immediate fixation or H2B-FT blue/red fluorescence activated cell sorting followed by fixation. For in vivo labeling of adult tissues, mice were pulsed with EdU at 50μg/g body weight via intraperitoneal injection either 2 hours (Figure 5F) or 35 minutes (Figure 6) before harvesting. Pregnant dams were given EdU intravenously at 50μg/g body weight 35 minutes before harvesting embryos (Figure 6, E17.5 cortex).
H2B-FT-blue and red signal was captured (by flow cytometry or microscopy) in unfixed cells/tissue prior to downstream steps, as exposure to fixative prematurely converts blue molecules into red.
Labeling in suspension: Bone-marrow-derived HSPCs plus Ter119+ cells were enriched via streptavidin-conjugated magnetic microbead separation (Miltenyi Biotec) of Lineage+ cells (this Lineage cocktail included biotin-conjugated CD3e, CD4, CD8, CD11b, Gr1, and B220); stained with GMP markers or CD71/Ter119; and sorted on a FACSAria (BD) directly into 70% ethanol. These fixed single-cell suspensions of EdU-treated cells were stored in 70% ethanol at −20C for > 24h, and then rinsed in PBS. Cells were permeablized in 0.2% Triton X-100 in PBS at room temperature for 15 minutes and then fluorescently labeled by Click chemistry using a Click-IT EdU-488 kit (Thermo Fisher Scientific) for 30 minutes at room temperature in an AF488-azide EdU labeling cocktail prepared according to the product manual. Cells were rinsed in PBS and incubated for 10 minutes at room temperature in 1μg/mL DAPI (Thermo Fisher Scientific) diluted in PBS. Cells were rinsed once more with PBS and resuspended in a PBS buffer containing 1% BSA, then analyzed by flow cytometry on a BD LSRII.
Labeling on slides: Intact organs (kidney, stomach) and whole embryos were embedded in cryomolds (Sakura) filled with OCT compound (Sakura), and flash-frozen in a 2-methylbutane/liquid nitrogen bath. 5μm sections were cut at −25C using a CM3050 S cryostat (Leica) and mounted onto SuperFrost® slides (Fisher Scientific). Frozen sections were scanned by fluorescence microscopy (details below) at 20x to capture blue and red H2B-FT signal, and then slides were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) for 12 minutes at room temperature. Samples were washed, permeablized, and stained as described above.
Microscopy
Live- and fixed-cell microscopy was performed using a Molecular Devices ImageXpress® Micro 4 high-throughput compound inverted epifluorescence microscope equipped with a live imaging environment. For time-lapse imaging, cells were plated into a Greiner Bio-One CellStar® 96well plate, sealed with a Breathe-Easy® gas-permeable membrane, and maintained at 37C/5% CO2 for the duration of the experiment. MetaXpress® 6 software was used for all image acquisition and for some image processing and analysis. Most of the image segmentation and image-based measurements were carried out using CellProfiler (Carpenter et al., 2006) and exported into FCS Express 6 Image Cytometry software (De Novo Software) for data analysis. ImageJ software (Schneider et al., 2012) was used for some image processing.
QUANTIFICATION AND STATISTICAL ANALYSIS
Mathematical Appendix
This appendix describes the details of cell cycle length quantification from the BR readout of the H2B-FT reporter. A comprehensive list of experimental considerations for acquiring BR data is provided in Table S1.
Determining the equation constant
Data S2B describes a mathematical relationship between the steady-state BR, , and the cell cycle length, τD:
where τC is the time required for the molecular conversion of blue to red, andCis a normalization constant. Because and are readily quantified in a fluorescence assay, the equation suggests that the cell cycle length τD can be calculated if the constant C can be experimentally determined, as the color conversion time τC remains the same for a given FT variant (Figure S1A). With the value normalized to the total cellular FT content, , the modified equation then becomes:
We found that C depended on the fluorescence detection parameters: exposure time in the case of fluorescence microscopy, and laser voltage for flow cytometry (Figures S7A–S7E). To determine the value of C, we directly measured the cell cycle lengths of iH2B-FT GMPs during a 24h culture period by live-cell imaging and single-cell tracking (Figure 7A). After 24h, the imaged iH2B-FT GMPs were analyzed by flow cytometry to determine their BR profile (Figure 7B). Thus, the BR distribution for these 24h cultured cells could be matched to their directly observed cell cycle speed distribution. This provided paired coordinate values for and cell cycle length τD such that the value of the combined constant C τC could be calculated (Figures S7F and S7G). Separate calculations were performed using coordinates representing the 10th, 25th, 50th and 80th percentile of cell cycle length, assumed to correspond to the 90th, 75th, 50th and 20th percentile of the bluest cells, respectively (Figure S7G). Within the examined cell cycle speed range, these calculated cell cycle values differed maximally from observed values by 2.06h at the 10th percentile and 6.25h at the 80th percentile (Figure S7G). Predictions 2 and 3 were closest to observed cell cycle lengths, differing at most by 1.17h and 3.43h at the 10th and 80th percentiles, respectively (Figure S7G). The resulting C ∙ τC values were then plugged back into the equation (Figure S7F) to produce curves which we evaluated for their similarity to each other, and to the experimental microscopy data (Figure S7H). Based on the plot in Figure S7H, a C τC value representing the average of predictions 2 and 3 was selected for estimating in vivo cell cycle lengths.
Considering mitosis-independent red decay
As H2B-FT-red does exhibit decay independent of cell division in MEFs and HeLa cells (Figure S2), we considered a modified version of the equation shown in Data S2 in which a rate constant for active degradation, τR, was incorporated into the red removal rate, δ (Figure S7I):
However, as the exact red degradation parameter in hematopoietic cells in vivo is unknown, we examined the relationship between BR and cell cycle length by modeling a wide range of red decay rates (Figures S7J and S7K). These exercises showed that for cell cycle lengths shorter than 30h, the red degradation rate was irrelevant; there was little difference in cell cycle lengths whether H2B-FT-red was assumed to be shorter (35h) or longer (135h), or whether it was simply ignored ((1 /τR)→0), when δ becomes simply inversely proportional to τD as in Data S2B. At longer cell cycle lengths (> 30h), ignoring red decay resulted in the shortest possible prediction of cell cycle length for a given BR value, while incorporating red decay adjusted these predictions upward in a manner inversely correlated with half-life (Figures S7J and S7K). To visualize these different scenarios in our actual bone marrow data, we proceeded to generate two estimates of cell cycle length with different red decay parameters: either by ignoring cell division-independent decay or by setting the red fluorescence half-life to 85h (Figure S7L). Although absolute cell cycle length for slow-dividing cells remains more uncertain than in fast-cycling cells, the relative differences between cell types are still maintained in any version of the equation. Therefore, in order to compare cell cycle length distributions across many hematopoietic cell types and different individual mice (Figures 7C–7F), we chose the simplest version of the equation (red decay ignored) for reporting results, while acknowledging that slow cell cycle lengths (> 30h) are likely to be underestimated using this assumption.
With these considerations and having determined the value of C τC, we proceeded to estimate the cell cycle length distributions for various hematopoietic cell types (Figure 7C).
Statistical Analysis
Details pertaining to each statistical analysis are provided in the figure legend accompanying the relevant data figure. All statistical tests used were two-tailed and were carried out using Prism software (GraphPad). All measurements were taken from distinct samples.
Supplementary Material
Highlights.
Stable molecule levels are more sensitive to cell cycle length than labile ones
Stable/labile species can be modeled by the two states of a fluorescent timer (FT)
H2B-FT resolves live-cell cycle speed heterogeneity in cultured cells and in mice
Fast proliferating cells appear bluer, while slow dividing cells are redder
ACKNOWLEDGMENTS
This work was funded by a National Institutes of Health (NIH) award (DP2GM123507), the Charles H. Hood Foundation, and Gilead Sciences. A.E.E. was supported in part by the NIH/National Institute of General Medical Sciences (NIGMS) Cellular and Molecular Biology Training Program under award T32GM007223 and Yale Cooperative Center for Excellence in Hematology NIH/U54DK106857. We thank the Yale Stem Cell Center, Dr. Diane Krause, the Yale Flow Cytometry Core, and the Yale Genome Editing Core.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107804.
REFERENCES
- Ajjappala BS, Kim YS, Kim MS, Lee MY, Lee KY, Ki HY, Cha DH, and Baek KH (2009). 14–3-3 gamma is stimulated by IL-3 and promotes cell proliferation. J. Immunol 182, 1050–1060. [DOI] [PubMed] [Google Scholar]
- Basiji DA, Ortyn WE, Liang L, Venkatachalam V, and Morrissey P (2007). Cellular image analysis and imaging by flow cytometry. Clin. Lab. Med 27, 653–670, viii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battich N, Stoeger T, and Pelkmans L (2015). Control of transcript variability in single mammalian cells. Cell 163, 1596–1610. [DOI] [PubMed] [Google Scholar]
- Beerman I, Bhattacharya D, Zandi S, Sigvardsson M, Weissman IL, Bryder D, and Rossi DJ (2010). Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc. Natl. Acad. Sci. U S A 107, 5465–5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokhari SA, and Raza A (1992). Novel derivation of total cell cycle time in malignant cells using two DNA-specific labels. Cytometry 13, 144–148. [DOI] [PubMed] [Google Scholar]
- Calder A, Roth-Albin I, Bhatia S, Pilquil C, Lee JH, Bhatia M, Levadoux-Martin M, McNicol J, Russell J, Collins T, and Draper JS (2013). Lengthened G1 phase indicates differentiation status in human embryonic stem cells. Stem Cells Dev. 22, 279–295. [DOI] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, et al. (2006). CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J (2016). The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb. Perspect. Med 6, a026104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Hartman A, and Guo S (2015). Choosing cell fate through a dynamic cell cycle. Curr. Stem Cell Rep. 1, 129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Dong J, Haiech J, Kilhoffer MC, and Zeniou M (2016). Cancer stem cell quiescence and plasticity as major challenges in cancer therapy. Stem Cells Int. 2016, 1740936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VS, Morrison JP, Southwell MF, Foley JF, Bolon B, and Elmore SA (2017). Histology atlas of the developing prenatal and postnatal mouse central nervous system, with emphasis on prenatal days E7.5 to E18.5. Toxicol. Pathol 45, 705–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Burkhardt DB, Hartman AA, Hu X, Eastman AE, Sun C, Wang X, Zhong M, Krishnaswamy S, and Guo S (2019). MLL-AF9 initiates transformation from fast-proliferating myeloid progenitors. Nat. Commun 10, 5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicaybam L, Barcelos C, Peixoto B, Carneiro M, Limia CG, Redondo P, Lira C, Paraguassú-Braga F, Vasconcelos ZF, Barros L, and Bonamino MH (2017). An efficient electroporation protocol for the genetic modification of mammalian cells. Front. Bioeng. Biotechnol 4, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copley MR, Beer PA, and Eaves CJ (2012). Hematopoietic stem cell heterogeneity takes center stage. Cell Stem Cell 10, 690–697. [DOI] [PubMed] [Google Scholar]
- Deneke VE, Melbinger A, Vergassola M, and Di Talia S (2016). Waves of Cdk1 activity in S-phase synchronize the cell cycle in Drosophila embryos. Dev. Cell 38, 399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drukker M, Tang C, Ardehali R, Rinkevich Y, Seita J, Lee AS, Mosley AR, Weissman IL, and Soen Y (2012). Isolation of primitive endoderm, mesoderm, vascular endothelial and trophoblast progenitors from human pluripotent stem cells. Nat. Biotechnol 30, 531–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Geva-Zatorsky N, Issaeva I, Cohen A, Dekel E, Danon T, Cohen L, Mayo A, and Alon U (2011). Proteome half-life dynamics in living human cells. Science 331, 764–768. [DOI] [PubMed] [Google Scholar]
- Ernst C (2016). Proliferation and differentiation deficits are a major convergence point for neurodevelopmental disorders. Trends Neurosci. 39, 290–299. [DOI] [PubMed] [Google Scholar]
- Falkowska-Hansen B, Kollar J, Grüner BM, Schanz M, Boukamp P, Siveke J, Rethwilm A, and Kirschner M (2010). An inducible Tet-Off-H2B-GFP lentiviral reporter vector for detection and in vivo isolation of label-retaining cells. Exp. Cell Res 316, 1885–1895. [DOI] [PubMed] [Google Scholar]
- Fisher R, Pusztai L, and Swanton C (2013). Cancer heterogeneity: implications for targeted therapeutics. Br. J. Cancer 108, 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusawa T, Ikeda M, Inoue F, Ohkoshi K, Hamano T, and Tokunaga T (2006). Gene expression profiling of mouse embryonic stem cell subpopulations. Biol. Reprod 75, 555–561. [DOI] [PubMed] [Google Scholar]
- Gabay M, Li Y, and Felsher DW (2014). MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med 4, a014241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales KA, Liang H, Lim YS, Chan YS, Yeo JC, Tan CP, Gao B, Le B, Tan ZY, Low KY, et al. (2015). Deterministic restriction on pluripotent state dissolution by cell-cycle pathways. Cell 162, 564–579. [DOI] [PubMed] [Google Scholar]
- Guo S, Bai H, Megyola CM, Halene S, Krause DS, Scadden DT, and Lu J (2012). Complex oncogene dependence in microRNA-125a-induced myeloproliferative neoplasms. Proc. Natl. Acad. Sci. U S A 109, 16636–16641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Zi X, Schulz VP, Cheng J, Zhong M, Koochaki SH, Megyola CM, Pan X, Heydari K, Weissman SM, et al. (2014). Nonstochastic reprogramming from a privileged somatic cell state. Cell 156, 649–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas S, Trumpp A, and Milsom MD (2018). Causes and consequences of hematopoietic stem cell heterogeneity. Cell Stem Cell 22, 627–638. [DOI] [PubMed] [Google Scholar]
- Häcker H, and Karin M (2006). Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006, re13. [DOI] [PubMed] [Google Scholar]
- Hadjantonakis A-K, and Papaioannou VE (2004). Dynamic in vivo imaging and cell tracking using a histone fluorescent protein fusion in mice. BMC Bio-technol. 4, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton E, and Infante JR (2016). Targeting CDK4/6 in patients with cancer. Cancer Treat. Rev 45, 129–138. [DOI] [PubMed] [Google Scholar]
- Hochedlinger K, Yamada Y, Beard C, and Jaenisch R (2005). Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell 121, 465–477. [DOI] [PubMed] [Google Scholar]
- Hudecek M, Izsvák Z, Johnen S, Renner M, Thumann G, and Ivics Z (2017). Going non-viral: the Sleeping Beauty transposon system breaks on through to the clinical side. Crit. Rev. Biochem. Mol. Biol 52, 355–380. [DOI] [PubMed] [Google Scholar]
- Hwang Y, Futran M, Hidalgo D, Pop R, Iyer DR, Scully R, Rhind N, and Socolovsky M (2017). Global increase in replication fork speed during a p57KIP2-regulated erythroid cell fate switch. Sci. Adv 3, e1700298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacovino M, Bosnakovski D, Fey H, Rux D, Bajwa G, Mahen E, Mitanoska A, Xu Z, and Kyba M (2011). Inducible cassette exchange: a rapid and efficient system enabling conditional gene expression in embryonic stem and primary cells. Stem Cells 29, 1580–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkan T, Olova N, Roode M, Mulas C, Lee HJ, Nett I, Marks H, Walker R, Stunnenberg HG, Lilley KS, et al. (2017). Tracking the embryonic stem cell transition from ground state pluripotency. Development 144, 1221–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T-H, and Shivdasani RA (2016). Stomach development, stem cells and disease. Development 143, 554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kina T, Ikuta K, Takayama E, Wada K, Majumdar AS, Weissman IL, and Katsura Y (2000). The monoclonal antibody TER-119 recognizes a molecule associated with glycophorin A and specifically marks the late stages of murine erythroid lineage. Br. J. Haematol 109, 280–287. [DOI] [PubMed] [Google Scholar]
- Kita-Matsuo H, Barcova M, Prigozhina N, Salomonis N, Wei K, Jacot JG, Nelson B, Spiering S, Haverslag R, Kim C, et al. (2009). Lentiviral vectors and protocols for creation of stable hESC lines for fluorescent tracking and drug resistance selection of cardiomyocytes. PLoS ONE 4, e5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen ES, and Wang JY (2010). Targeting the RB-pathway in cancer therapy. Clin. Cancer Res 16, 1094–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koulnis M, Pop R, Porpiglia E, Shearstone JR, Hidalgo D, and Socolovsky M (2011). Identification and analysis of mouse erythroid progenitors using the CD71/TER119 flow-cytometric assay. J. Vis. Exp (5), 2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk MS, Tirosh I, Heckl D, Rao TN, Dixit A, Haas BJ, Schneider RK, Wagers AJ, Ebert BL, and Regev A (2015). Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res. 25, 1860–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kueh HY, Champhekar A, Nutt SL, Elowitz MB, and Rothenberg EV (2013). Positive feedback between PU.1 and the cell cycle controls myeloid differentiation. Science 341, 670–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusaba T, Lalli M, Kramann R, Kobayashi A, and Humphreys BD (2014). Differentiated kidney epithelial cells repair injured proximal tubule. Proc. Natl. Acad. Sci. U S A 111, 1527–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagasse E, and Weissman IL (1996). Flow cytometric identification of murine neutrophils and monocytes. J. Immunol. Methods 197, 139–150. [DOI] [PubMed] [Google Scholar]
- Liu S, Dontu G, and Wicha MS (2005). Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast Cancer Res. 7, 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Guo S, Ebert BL, Zhang H, Peng X, Bosco J, Pretz J, Schlanger R, Wang JY, Mak RH, et al. (2008). MicroRNA-mediated control of cell fate in megakaryocyte-erythrocyte progenitors. Dev. Cell 14, 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons AB, Hasbold J, and Hodgkin PD (2001). Flow cytometric analysis of cell division history using dilution of carboxyfluorescein diacetate succinimidyl ester, a stably integrated fluorescent probe. Methods Cell Biol. 63, 375–398. [DOI] [PubMed] [Google Scholar]
- Madar S, Goldstein I, and Rotter V (2013). “Cancer associated fibroblasts”—more than meets the eye. Trends Mol. Med 19, 447–453. [DOI] [PubMed] [Google Scholar]
- Mátés L, Chuah MK, Belay E, Jerchow B, Manoj N, Acosta-Sanchez A, Grzela DP, Schmitt A, Becker K, Matrai J, et al. (2009). Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat. Genet 41, 753–761. [DOI] [PubMed] [Google Scholar]
- McKenzie JL, Gan OI, Doedens M, Wang JCY, and Dick JE (2006). Individual stem cells with highly variable proliferation and self-renewal properties comprise the human hematopoietic stem cell compartment. Nat. Immunol 7, 1225–1233. [DOI] [PubMed] [Google Scholar]
- Méndez-Ferrer S, Chow A, Merad M, and Frenette PS (2009). Circadian rhythms influence hematopoietic stem cells. Curr. Opin. Hematol 16, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newport J, and Kirschner M (1982). A major developmental transition in early Xenopus embryos: I. characterization and timing of cellular changes at the midblastula stage. Cell 30, 675–686. [DOI] [PubMed] [Google Scholar]
- O’Farrell PH, Stumpff J, and Su TT (2004). Embryonic cleavage cycles: how is a mouse like a fly? Curr. Biol 14, R35–R45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson A, Venkatasubramanian M, Chaudhri VK, Aronow BJ, Salomonis N, Singh H, and Grimes HL (2016). Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature 537, 698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orford KW, and Scadden DT (2008). Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat. Rev. Genet 9, 115–128. [DOI] [PubMed] [Google Scholar]
- Passegué E, Wagers AJ, Giuriato S, Anderson WC, and Weissman IL (2005). Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J. Exp. Med 202, 1599–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras EM, Warr MR, and Passegué E (2011). Cell cycle regulation in hematopoietic stem cells. J. Cell Biol 195, 709–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop R, Shearstone JR, Shen Q, Liu Y, Hallstrom K, Koulnis M, Gribnau J, and Socolovsky M (2010). A key commitment step in erythropoiesis is synchronized with the cell cycle clock through mutual inhibition between PU.1 and S-phase progression. PLoS Biol. 8, e1000484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. (2008). Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132, 487–498. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz GK, and Shah MA (2005). Targeting the cell cycle: a new approach to cancer therapy. J. Clin. Oncol 23, 9408–9421. [DOI] [PubMed] [Google Scholar]
- Sela Y, Molotski N, Golan S, Itskovitz-Eldor J, and Soen Y (2012). Human embryonic stem cells exhibit increased propensity to differentiate during the G1 phase prior to phosphorylation of retinoblastoma protein. Stem Cells 30, 1097–1108. [DOI] [PubMed] [Google Scholar]
- Semrau S, Goldmann JE, Soumillon M, Mikkelsen TS, Jaenisch R, and van Oudenaarden A (2017). Dynamics of lineage commitment revealed by single-cell transcriptomics of differentiating embryonic stem cells. Nat. Commun 8, 1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skylaki S, Hilsenbeck O, and Schroeder T (2016). Challenges in long-term imaging and quantification of single-cell dynamics. Nat. Biotechnol 34, 1137–1144. [DOI] [PubMed] [Google Scholar]
- Solito S, Marigo I, Pinton L, Damuzzo V, Mandruzzato S, and Bronte V (2014). Myeloid-derived suppressor cell heterogeneity in human cancers. Ann. N Y Acad. Sci 1319, 47–65. [DOI] [PubMed] [Google Scholar]
- Soufi A, and Dalton S (2016). Cycling through developmental decisions: how cell cycle dynamics control pluripotency, differentiation and reprogramming. Development 143, 4301–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Maherali N, Breault DT, and Hochedlinger K (2008). Defining molecular cornerstones during fibroblast to iPS cell reprogramming in mouse. Cell Stem Cell 2, 230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart MH, Bossé M, Chadwick K, Menendez P, Bendall SC, and Bhatia M (2006). Clonal isolation of hESCs reveals heterogeneity within the pluripotent stem cell compartment. Nat. Methods 3, 807–815. [DOI] [PubMed] [Google Scholar]
- Subach FV, Subach OM, Gundorov IS, Morozova KS, Piatkevich KD, Cuervo AM, and Verkhusha VV (2009). Monomeric fluorescent timers that change color from blue to red report on cellular trafficking. Nat. Chem. Biol 5, 118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T, and Hevner RF (2014). Growth and folding of the mammalian cerebral cortex: from molecules to malformations. Nat. Rev. Neurosci 15, 217–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tough DF, and Sprent J (1995). Life span of naive and memory T cells. Stem Cells 13, 242–249. [DOI] [PubMed] [Google Scholar]
- Toyama BH, Savas JN, Park SK, Harris MS, Ingolia NT, Yates JR 3rd, and Hetzer MW (2013). Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell 154, 971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treuting PM, Valasek MA, and Dintzis SM (2012). Upper Gastrointestinal Tract In Comparative Anatomy and Histology: A Mouse and Human Atlas, Treuting PM and Dintzis SM, eds. (San Diego: Academic Press; ), pp. 155–175. [Google Scholar]
- Tumbar T, Guasch G, Greco V, Blanpain C, Lowry WE, Rendl M, and Fuchs E (2004). Defining the epithelial stem cell niche in skin. Science 303, 359–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzahor E, and Poss KD (2017). Cardiac regeneration strategies: staying young at heart. Science 356, 1035–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugale A, Norddahl GL, Wahlestedt M, Säwén P, Jaako P, Pronk CJ, Soneji S, Cammenga J, and Bryder D (2014). Hematopoietic stem cells are intrinsically protected against MLL-ENL-mediated transformation. Cell Rep. 9, 1246–1255. [DOI] [PubMed] [Google Scholar]
- Uzquiano A, Gladwyn-Ng I, Nguyen L, Reiner O, Götz M, Matsuzaki F, and Francis F (2018). Cortical progenitor biology: key features mediating proliferation versus differentiation. J. Neurochem 146, 500–525. [DOI] [PubMed] [Google Scholar]
- van der Flier LG, and Clevers H (2009). Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu. Rev. Physiol 71, 241–260. [DOI] [PubMed] [Google Scholar]
- White J, and Dalton S (2005). Cell cycle control of embryonic stem cells. Stem Cell Rev. 1, 131–138. [DOI] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter JA, and Jessell TM (2002). Directed differentiation of embryonic stem cells into motor neurons. Cell 110, 385–397. [DOI] [PubMed] [Google Scholar]
- Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, et al. (2008). Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 135, 1118–1129. [DOI] [PubMed] [Google Scholar]
- Ye M, Zhang H, Yang H, Koche R, Staber PB, Cusan M, Levantini E, Welner RS, Bach CS, Zhang J, et al. (2015). Hematopoietic differentiation is required for initiation of acute myeloid leukemia. Cell Stem Cell 17, 611–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa K (2000). Cell cycle regulators in neural stem cells and postmitotic neurons. Neurosci. Res 37, 1–14. [DOI] [PubMed] [Google Scholar]
- Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, Tan Y, Ci Y, Wu F, Dai X, et al. (2018). Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 553, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No unique code was generated in this study. Raw data used to support the conclusions of this study are available from the authors on request. Custom CellProfiler (Carpenter et al., 2006) image analysis pipelines along with sample images are available on request.