

Abstract
Down syndrome (DS) is characterized by the occurrence of three copies of human chromosome 21 (HSA21). HSA21 contains a cluster of four interferon receptor (IFN-R) genes: IFNAR1, IFNAR2, IFNGR2 and IL10RB. DS patients often develop mucocutaneous infections and autoimmune diseases, mimicking patients with heterozygous gain-of-function (GOF) STAT1 mutations, which enhance cellular responses to three types of interferon (IFN). A gene dosage effect at these four loci may contribute to the infectious and autoimmune manifestations observed in individuals with DS. We report high levels of IFN-αR1, IFN-αR2 and IFN-γR2 expression on the surface of monocytes and EBV-transformed-B (EBV-B) cells from studying 45 DS patients. Total and phosphorylated STAT1 (STAT1 and pSTAT1) levels were constitutively high in unstimulated and IFN-α- and IFN-γ-stimulated monocytes from DS patients, but lower than those in patients with GOF STAT1 mutations. Following stimulation with IFN-α or -γ, but not with IL-6 or IL-21, pSTAT1 and IFN-γ activation factor (GAF) DNA binding activities were significantly higher in the EBV-B cells of DS patients than in controls. These responses resemble the dysregulated responses observed in patients with STAT1 GOF mutations. Concentrations of plasma type I IFNs were high in 12% of the DS patients tested (1.8% in the healthy controls). Levels of type I IFNs, IFN-Rs and STAT1 were similar in DS patients with and without recurrent skin infections. We performed a genome-wide transcriptomic analysis based on principal component analysis and interferon modules on circulating monocytes. We found that DS monocytes had levels of both IFN-α- and IFN-γ- inducible ISGs intermediate to those of monocytes from healthy controls and from patients with GOF STAT1 mutations. Unlike patients with GOF STAT1 mutations, patients with DS had normal circulating Th17 counts and a high proportion of terminally differentiated CD8+ T cells with low levels of STAT1 expression. We conclude a mild interferonopathy in Down syndrome lead to an incomplete penetrance at both cellular and clinical level, which is not correlate with recurrent skin bacterial or fungal infections. The constitutive upregulation of type I and type II IFN-R, at least in monocytes of DS patients, may contribute to the autoimmune diseases observed in these individuals.
Introduction
Down syndrome (DS), or trisomy 21, was the first human disease attributed to a chromosomal abnormality, and was even the first human genetic disease to be deciphered at the molecular level1. In the absence of medical intervention, it is the most common genetic cause of intellectual disability, with an incidence of about 1/700 newborns, and there were about 206,000 people with DS living in the USA in 20102. Infectious diseases have been reported to be one of the major causes of death in DS patients3,4. The prevalence of chronic mucocutaneous candidiasis (CMC) is high in DS patients (56%-76%)5,6, with clinical manifestations of folliculitis (50%), lip fissures7, angular stomatitis (up to 80%)6, periodontal disease (40%)8–11 and onychomycosis (2% to 76.6%) commonly reported12. Staphylococcus aureus infection (11%)13,14 and hidradenitis suppurativa (15%)15 are frequent in DS patients, and invasive fungal infections have also been reported16,17. Thyroid dysfunction is common, often related to Hashimoto’s thyroiditis or Grave’s disease, and typically manifests as hypothyroidism in −22% of DS patients18. Other autoimmune disorders, including type I diabetes mellitus (DM), alopecia areata19, vitiligo and celiac disease20, are more frequent in DS patients than in the general population21,22. Despite the occurrence of these infectious and autoimmune phenotypes, together with the underlying chromosomal abnormality and its complete penetrance for at least several cardinal features, DS has not traditionally be seen as a primary immunodeficiency, probably because the defect involves multiple genes, and the biological, immunological and clinical immunological phenotypes display incomplete penetrance23.
Intriguingly, DS patients have a number of key infectious and autoimmune phenotypes, including CMC and hypothyroidism24, in common with patients heterozygous for STAT1 GOF mutations, a bona fide inborn error of immunity. Hypothyroidism occurs in similar proportions (~20%) of patients with the two conditions24. Monoallelic GOF STAT1 mutations enhance cellular responses to the three types of IFN, IFN-α/β, γ, and λ, thereby promoting autoimmunity through a type I interferonopathy mechanism, whilst impairing IL-17 T cell immunity, thereby triggering CMC25–27. For activation of the JAK-STAT pathway, type I interferon (IFN-α/β) requires IFN-αR1 and IFN-αR2, whereas type II interferon (IFN-γ) requires IFN-γR1 and IFN-γR2, and type III interferon (IFN-λ) requires IFN-λR1 and IL-10RB28. Interestingly, four of the six IFN-R genes are located on chromosome 21 (Figure 1A)21. We therefore hypothesized that DS patients may suffer from CMC and thyroiditis due to hyperresponsiveness to IFNs. Abnormally strong IFN responses were first documented in the cells of individuals with DS in 1974, before the cloning of interferon receptors, with the demonstration of enhanced antiviral activities in DS fibroblasts29,30. In another study, quantitative PCR analyses showed mRNA levels for IFNAR1, IFNAR2, IL10RB, and IFNGR2 to be significantly higher than normal in both EBV-transformed B cells (n=36) and fibroblasts (n=33) from DS patients31. A third study showed that IFNAR1, IFNAR2, IFNGR2 and IL-10RB mRNA levels were high, with constitutive IFN activation, as measured by RNA-Seq, in fibroblasts (n=6), EBV-B cells (n=3), monocytes (n=10) and T cells (n=10) from individuals with DS32. As previously reported31,32, the mRNA levels of IFNAR1, IFNGR2 and IFNI OR, but not IFNAR2 were markedly higher in monocytes from DS patients than those of control (Supplementary Figure 1). However, no previous attempt has ever been made to connect clinical and cellular phenotypes in individuals with DS. We characterized in detail the impact of trisomy 21 at the protein level and explored the potential effects of hyperresponsiveness to IFN in T cells, focusing particularly on the development of Th17 cells in the pathogenesis of infectious and autoimmune diseases in DS patients.
Figure 1: Surface expression of IFN-Rs on EBV-B cells from DS patients.
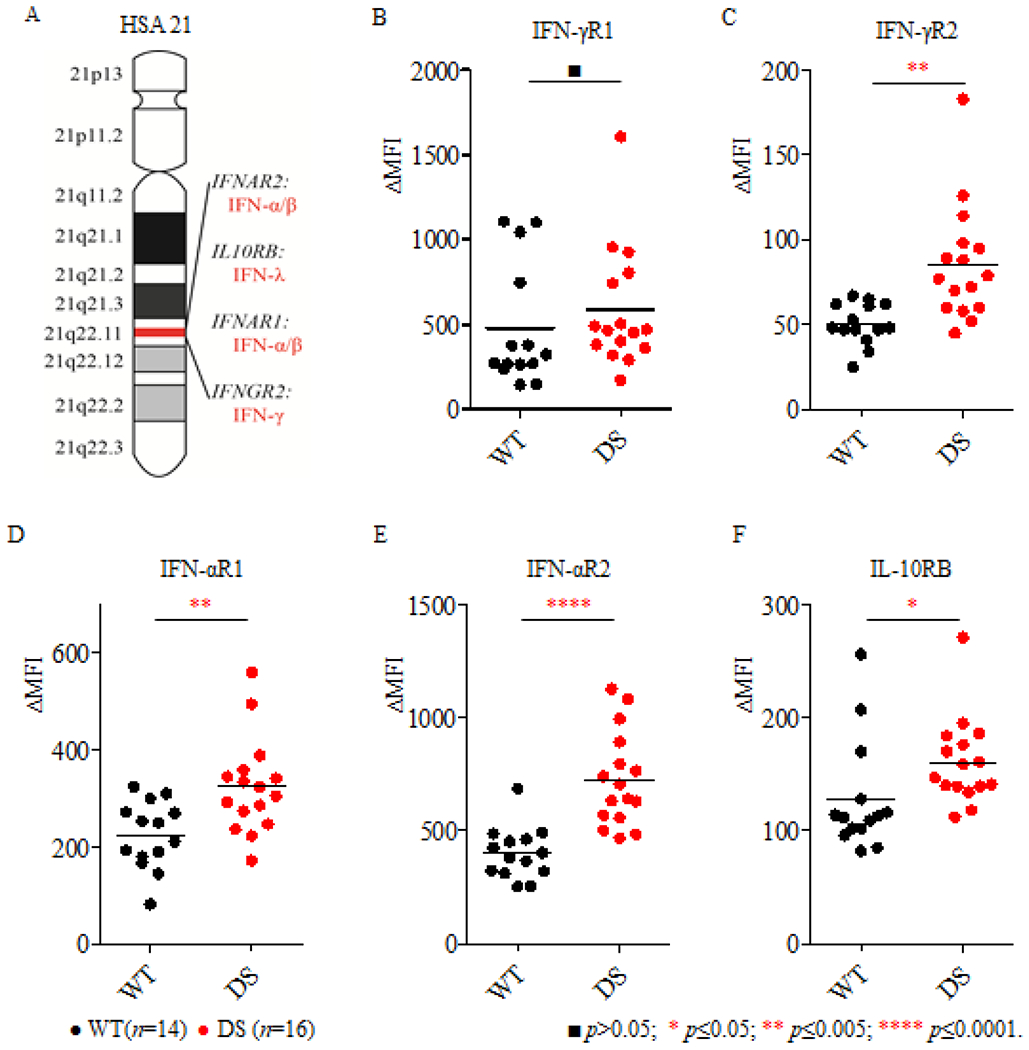
(A). Schematic diagram of the structure of HSA21, with the IFN-R locus including IFNAR2, IL-10RB, IFNAR1 and IFNGR2 as well as genes for the corresponding agonists; (B-F). IFN-R expression was analyzed in EBV-B cells from healthy controls (WT, n=14) and DS patients (DS, n=16) with the corresponding specific antibodies, against IFN-γR1 (B), IFN-γR2 (C), IFN-αR1 (D), IFN-αR2 (E) and IL-10RB (F). ΔMFI was calculated by subtracting the MFI for the isotype control from that for the specific anti-IFN-R antibody. Significance was assessed by calculating p values in unpaired t tests. ■p>0.05; * p≤ 0.05; * p≤0.005; *** p≤ 0.0005; **** p≤ 0.0001.
Results
1. IFN-R protein levels in EBV-B cells from DS patients
We extended these findings by using flow cytometry to quantify protein levels for the six interferon receptors. We were able to quantify surface levels of IFN-αR1, IFN-αR2, IFN-γR1, IFN-γR2 and IL-10RB, whereas IFN-λR1 levels were below the limit of detection. EBV-B cells from 14 healthy controls and 16 DS patients were assessed on at least three independent occasions to evaluate IFN-αR1, IFN-αR2, IFN-γR1, IFN-γR2 and IL-10RB levels, together with the B-cell surface marker CD20, as a control. The four IFN-Rs encoded by the genes located on chromosome 21, were expressed at significantly higher levels on the surface of EBV-B cells from DS patients than on those from healthy donors (Figure 1C, 1D, 1E, 1F). By contrast, levels of IFN-γR1 (chromosome 6) and CD20 (chromosome 11) were similar in EBV-B cells from DS patients and healthy controls (Figure 1B and Supplementary Figure 2). In conclusion, these experiments demonstrate that the levels of both chains of the type I interferon receptor (IFN-αR1 and IFN-αR2), one of the two chains of the type II interferon receptor (IFN-γR2), and one of the two chains of the type III interferon receptor (IL-10RB) are abnormally high on the surface of EBV-B cells from DS patients.
2. EBV-B cells from DS patients have enhanced responses to IFN-γ and IFN-α but not to IL-6 or IL-21
We then studied IFN responses by measuring STAT1 Tyr 701 phosphorylation (pSTAT1) and GAF DNA binding levels in EBV-B cells from healthy controls, two DS patients, one CMC patient with a GOF STAT1 mutation25, a patient with Mendelian susceptibility to mycobacterial disease (MSMD) and IFNGR2 haploinsufficiency33 and an MSMD patient with complete IFNGR2 deficiency33. IFN-α or IFN-γ induced significantly higher levels of GAF DNA binding in EBV-B cells from DS patients than in those of healthy controls (Figure 2A). However, these levels remained lower than those observed for patients with GOF STAT1 mutations (Figure 2A). GAF DNA-binding activities increased with IFN-γ concentrations in controls, and to an even greater extent in cells from DS patients (Figure 2B). By contrast, GOF STAT1 EBV-B cells systematically displayed high levels of GAF DNA binding regardless of the IFN-γ dose (Figure 2B). By contrast to the findings for patients with GOF STAT1 mutation, GAF DNA binding activity upon IL-6 or IL-21 stimulation was similar in cells from DS and cells from healthy controls (Figure 2C). We then assessed the levels of total and phosphorylated STAT1 by FACS. Total STAT1 and basal pSTAT1 levels in EBV-B cells from DS patients were similar to those in cells from healthy controls (Figure 3A, 3B). However, IFN-α induced significantly higher pSTAT1 levels in DS EBV-B cells than in the corresponding cells from healthy donors (Figure 3C). In summary, EBV-B cells from DS patients displayed enhanced responses to IFN-α and IFN-γ, but not to IL-6 or IL-21.
Figure 2: IFN responses in EBV-B cells from DS patients.
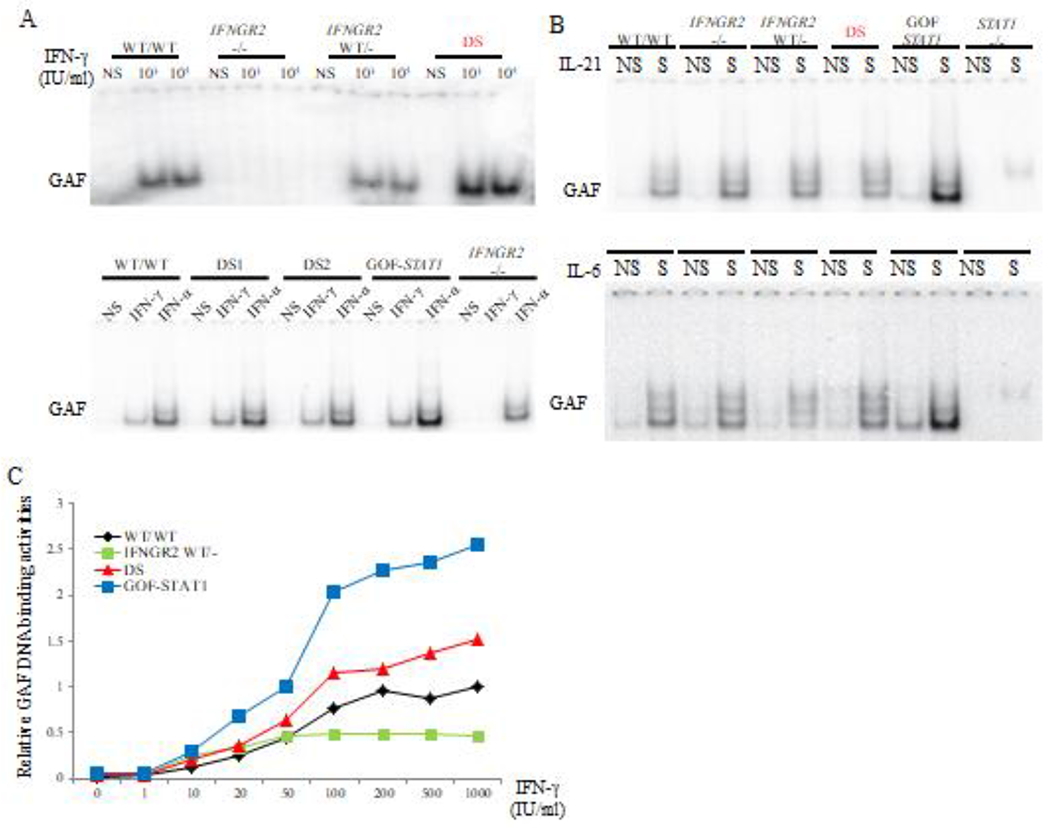
(A). EMSA analysis of GAF DNA-binding activity in the EBV-B cells from a healthy control, a patient with complete IFNGR2 deficiency, a patient with a heterozygous IFNGR2 mutation, a patient with GOF STAT1 mutation and two DS patients, with and without IFN stimulation. (B). EMSA analysis of GAF DNA-binding activity in the EBV-B cells as described above, in response to stimulation with IL-21 (upper panel) or IL-6 (lower panel). All the experiments were performed at least three times. (C). IFN-γ dose-dependent GAF DNA-binding activity in EBV-B cells, as indicated.
Figure 3: FACS analysis of total STAT1 and pSTAT1 in EBV-B cells from DS patients.
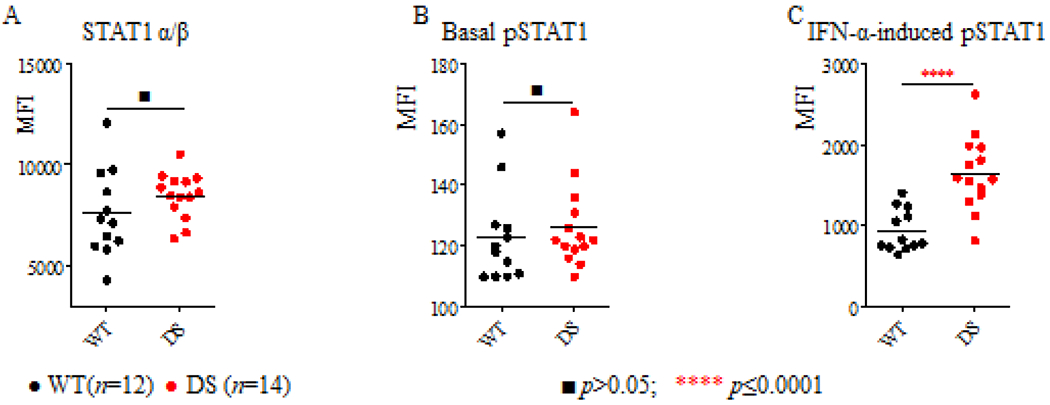
The levels of total STAT1 protein (A), basal pSTAT1(B) and IFN-α-induced pSTAT1(C) in the EBV-B cells of healthy controls (n=14) and DS patients (n=16) were compared. Thep values were obtained in unpaired t tests. ■p>0.05; **** p≤ 0.0001.
3. Enhanced surface expression of IFN-αR1, IFN-αR2 and IFN-γR2 on monocytes from DS patients
EBV-B cells are powerful tools for studying the human genetics of interferon responses33,35, but peripheral blood mononuclear cells are more physiologically relevant for elucidating disease pathogenesis. We investigated the potential effects of gene dosage effects on immune cells in two steps. We first explore the potential differences between several cell types in DS patients (n=5) and healthy controls (n=5) (Supplementary Figure 3). IFN-Rs have a cell type-specific expression pattern, with monocytes displaying the highest levels of IFN-αR2, IFN-γR1, IFN-γR2, and IL-10RB expression, and T and NK cells the lowest levels of IFN-γR2 expression33. We then focused on monocytes. Due to the variability of fluorescence intensity between independent FACS assays, the values for IFN-αR2, IFN-γR1, IFN-γR2, IL-10RB were expressed relative to those obtained from one healthy control as the reference in each independent experiment (Figure 4A). For the 15 DS patients and 17 healthy controls studied, IFN-αR2 and IFN-γR2 expression were 1.47 (p=0.0001) and 1.32 (p=0.0064) times more strongly expressed on the monocytes of DS patients than on those of healthy donors (Figure 4B, Supplementary Table 1). However, interindividual variation was greater for IL-10RB expression on monocytes, with no significant difference between healthy controls and DS patients. In summary, cell surface IFN-αR2 and IFN-γR2 levels were significantly higher on the monocytes of DS patients than on control monocytes, suggesting a role for enhanced type I and type II interferon responses in the pathogenesis of infectious and autoimmune manifestations of DS.
Figure 4: Surface expression of IFN-Rs on monocytes from DS patients.
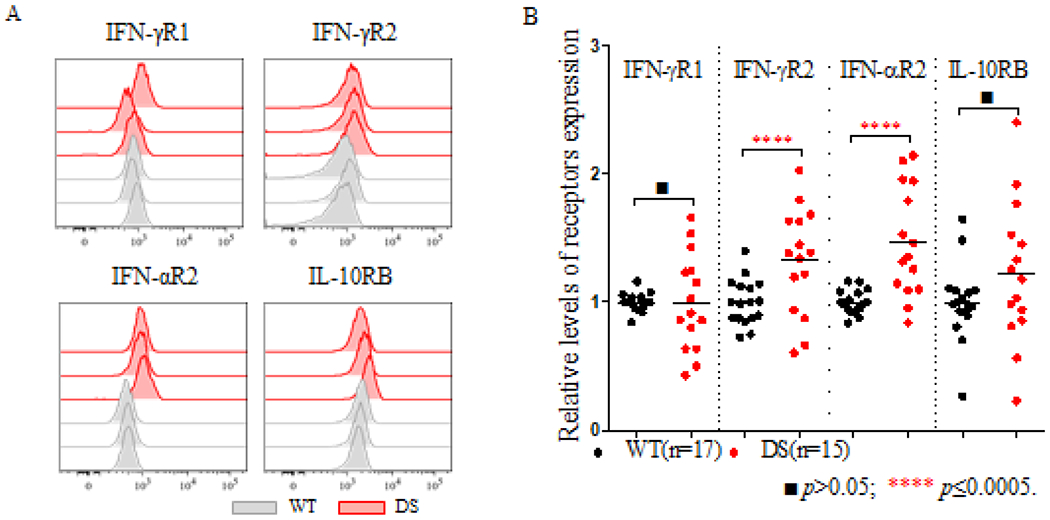
(A). Representative histogram of the levels of IFN-γR1, IFN-γR2, IFN-αR2 and IL-10RB expressed on monocytes from three healthy controls (WT) and three DS patients (DS) in one representative experiment; (B). The relative levels of IFN-Rs on monocytes were compared between healthy controls (n=17) and DS patients (n=15). The p values were obtained in unpaired t tests. ■ p>0.05; **** p≤ 0.0001.
4. Constitutively high of STAT1 and pSTAT1 levels in monocytes and high levels of type I IFN proteins in the plasma of DS patients
Monocytes are the cells with the highest known levels of human IFNAR and IFNGR2 expression. We therefore isolated monocytes for the quantification of total STAT1 and pSTAT1 levels (Figure 5A and Supplementary Figure 4). Ex vivo analyses of monocytes from 13 DS patients showed total STAT1 and pSTAT1 levels to be constitutively higher than those in 13 healthy controls (p=0.003, Figure 5B and 5C). We also treated monocytes ex vivo with 1,000 IU/ml either IFN-γ or IFN-α. Under these conditions, monocytes from DS patients had significantly higher pSTAT1 levels than monocytes from healthy controls (Figure 5C). Basal STAT and pSTAT1 levels were even higher in patients with GOF STAT1 mutations (p<0.0001, Figure 5B). Given the constitutive activation of STAT1 in DS patients, we then measured serum IFN-α levels with the high-affinity antibodies obtained from APECED patients and a single molecular array (Sigmoa) platform36. As shown in a previous study36, serum IFN-α concentration was < 10 fg/ml in healthy controls (Figure 5D). Most of the DS patients studied here had serum IFN-α concentrations within the normal range. However, five of the 43 DS patients tested (11.6%) had significantly higher levels of IFN-α (Figure 5D). We also found no significant differences in circulating IFN-α and monocyte IFN-Rs and STAT1 levels from DS patients with clinical manifestations of chronic mucocutaneous infections (Table 1), due to either Candida or Staphylococcus to those without infection (Supplementary Table 1). Thus, monocytes from DS patients had constitutively high levels of both STAT1 and its phosphorylated form, probably due to high levels of type I interferon and IFN receptor expression, however, we found no common clinical features characteristics of DS patients with high basal STAT1 levels, either for skin infection or hypothyroidism.
Figure 5: Total STAT1 and pSTAT1 levels in monocytes and plasma type I interferon levels in DS patients.
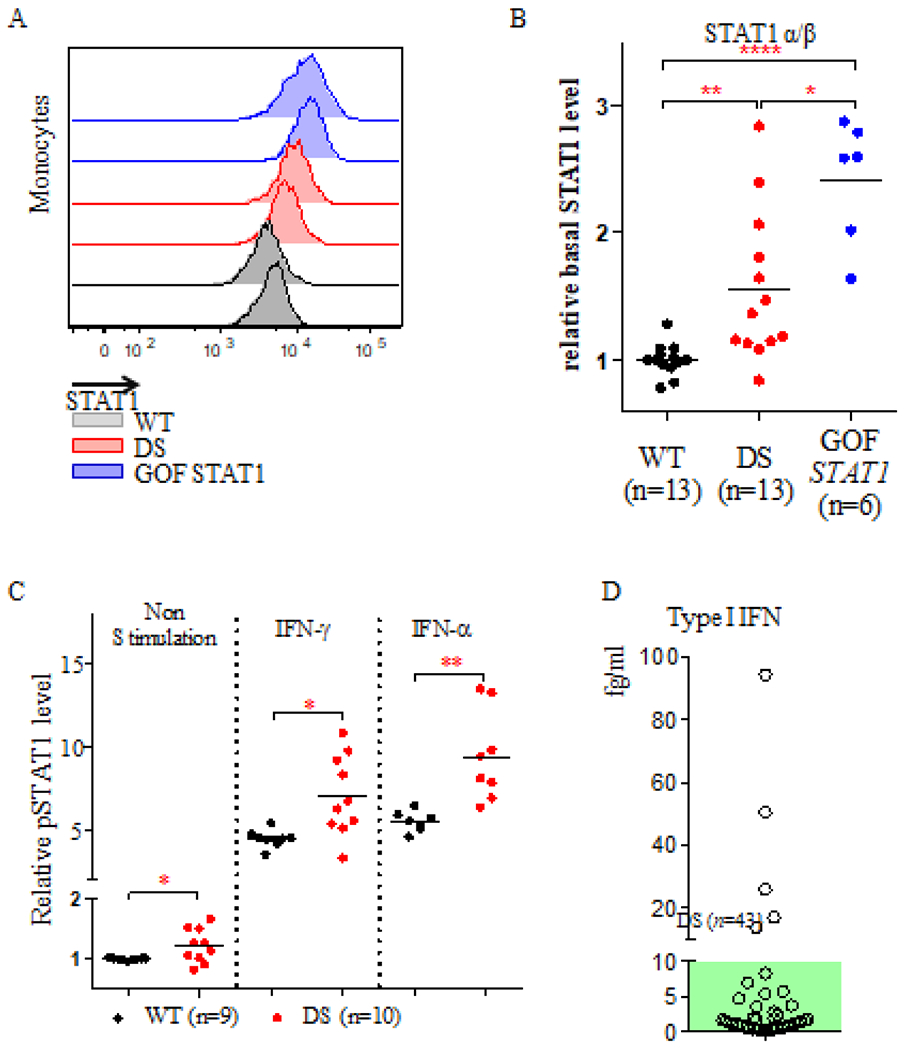
(A). Representative histogram of total STAT1 α/β protein levels in monocytes from two healthy controls (gray), two DS patients (red) and two patients with GOF STAT1 mutations (blue). (B). Relative levels of total STAT1 α/β protein in 13 healthy controls, 13 DS patients, and 6 patients with GOF STAT1 mutations. (C). Relative pSTAT1 levels in monocytes from 9 healthy controls and 10 DS patients, with and without IFN stimulation. Difference between the two groups were assessed in t tests. * p≤ 0.05; ** p≤0.005; **** p≤ 0.0001. (D). Plasma IFN-α protein levels were quantified in 43 DS patients with a single molecular array (Sigmoa). The green area indicates the normal range of IFN-α levels (≤ 10 fg/ml).
Table 1:
Clinical summary of the 45 Down Syndrome patients studied
| Category | Sub-category | Number (n=45) |
|---|---|---|
| Sex (n, %) | Male | 29 (64.4%) |
| Female | 16 (35.6%) | |
| Age (years) | Mean±SD | 23.5±12.1 |
| Range | 3-53 | |
| Age groups (n, %) | 3-10 | 7 (15.6%) |
| 11-20 | 14 (31.1%) | |
| 21-30 | 12 (26.7%) | |
| 31-40 | 8 (17.8%) | |
| Above 40 | 4 (8.9%) | |
| Ethnicity (n, %) | White or European | 34 (75.6%) |
| Black or African American | 10 (22.2%) | |
| Asian | 1 (2.2%) | |
| Mucocutaneous infection or inflammation (n, %) | Recurrent skin infection | 12 (26.7%) |
| Otitis media | 8 (17.8%) | |
| Onychomycosis | 7 (15.6%) | |
| Candidiasis | 3 (6.7%) | |
| Pustules | 3 (6.7%) | |
| Tonsillitis | 3 (6.7%) | |
| Conjunctivitis | 2 (4.4%) | |
| Intertriginous dermatitis | 1 (2.2%) | |
| Other infection, unspecified (n, %) | Varicella | 16 (35.6%) |
| Pneumonia | 4 (8.9%) | |
| Systemic Candida infection | 1 (2.2%) | |
| Urinary tract infection | 1 (2.2%) | |
| Hypothyroidism (n, %) | 7 (15.6%) | |
| Cardiac abnormality (n, %) | 7 (15.6%) | |
| Positive ANA (n, %) | 20 (44.4%) | |
5. ISGs are upregulated in monocytes from DS patients on transcriptome analysis
As monocytes have unique patterns of interferon receptor expression, we then isolated monocytes and extracted mRNA for transcriptomic analysis. We studied cells from 14 DS patients, eight patients with GOF STAT1 mutations, and 10 age- and sex-matched healthy controls. We performed principal component analysis (PCA) with all the genes passing our filters and found large overlaps between the GOF STAT1, DS, and healthy control groups. By contrast, PCA with the list of genes induced by IFN-α, IFN-β, or IFN-γ revealed a separation of DS patients from both the healthy control and GOF STAT1 groups (Supplementary Figure 5). Five of the six common ISGs used in screening assays for the diagnosis of a type I interferonpathy37 were significantly upregulated in DS patients (IFI44L, IFIT1, ISG15, RSAD2, SIGLEC1) (Supplementary Figure 6). We investigated the patterns of ISG expression by performing modular analysis and clustering six interferon modules for each individual38,39. Both DS patients and patients with GOF STAT1 mutations had upregulated IFN signatures, particularly for M10.1 and M8.3 (Figure 6A and 6B). For the three main clusters, the first cluster with normal ISG expression, contained nine of healthy controls and three DS patients, whereas the second cluster, with intermediate levels of ISG expression, contained nine DS patients, four patients with GOF STAT1 mutations and one healthy control. The third cluster, with high levels of ISG expression, contained four patients with GOF STAT1 mutations and two DS patients (Figure 6A). High circulating type I IFN levels tended to be correlated with enhanced interferon signatures (Figure 6A). Overall, the upregulation of ISGs was milder in DS patients than in patients with GOF STAT1 mutations.
Figure 6: Modular interferon signature in monocytes from DS patients and patients with STAT1 GOF.
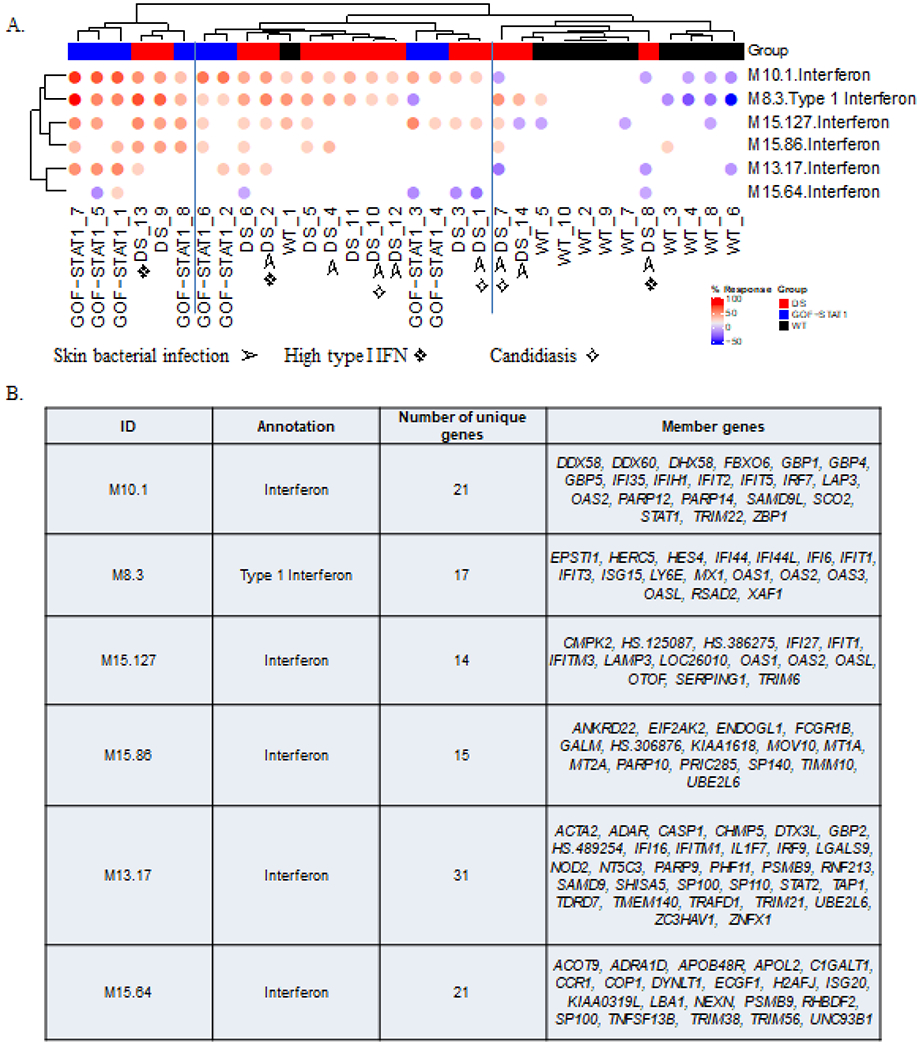
(A). Differences in the levels of transcripts from six “interferon modules” are shown on the heatmap above. Each column corresponds to a different subject (healthy controls are shown in black, DS in red and STAT1 GOF in blue). Each row corresponds to a different module. Both modules and subjects are arranged by hierarchical clustering. The colored spots on the map represent the percentage of the transcripts in a given module for which abundance was higher (in red) or lower (in blue) in DS patients than in controls (the cutoff was a 1.5-fold difference and 100-fold difference relative to the mean for the controls). ➢ : DS patient with history of recurrent skin bacterial infections; ✧: DS patient with history chronic mucocutaneous candidiasis; ❖: DS patient with high type I IFN level (≥10fg/ml). (B). The table below lists the genes in each of the transcriptional modules, with their associated functional annotations.
6. Normal circulating Th17 counts in DS patients
We tested the hypothesis that impaired Th17 immunity underlies CMC in DS patients, as it does in patients with GOF STAT1 mutations. We comprehensively tested six DS patients and 16 age-matched controls. DS patients had lower proportions of CD4+ T cells and higher proportions of CD8+ T cells than healthy controls (Supplementary Figure 7A). In analyses based on the expression of CD45RA and CCR7 expression, DS patients were found to have lower frequencies of naïve, and higher frequencies of central memory and Tfh CD4+ T cells (Supplementary Figure 7B and C), consistent with a recent report40. In assessments of subsets of memory CD4+ T cells (CD45RA−CXCR5−) based on their differential expression of the chemokine receptors CXCR3 and CCR6, DS patients were found to have high proportions of CXCR3+CCR6− Th1 cells, low proportions of CXCR3−CCR6− Th2 cells, and normal proportions of CXCR3−CCR6+ Th17 T cells (Supplementary figure 7D). We sorted naïve and memory CD4+ T cells from DS patients and healthy donors and cultured these cells under Th0, Th1, Th2, Th9 or Th17-polarizing conditions in vitro and assessed CD4+ T cell differentiation. No significant differences in the secretion of IL-2, IL-6, IL-10, IL-13, IL-17A or IL-17F were observed for naïve CD4+ T cells from DS patients and healthy controls under appropriate culture conditions (Supplementary Figure 8). However, naïve DS CD4+ T cells produced more IFN-γ under Th1-polarizing conditions than naïve CD4+ T cells from healthy donors (Supplementary Figure 8). For memory CD4+ T cells, IL-10, IFN-γ and TNF-α levels were normal under Th0 and Th1 conditions; levels of IL-17A and IL-17F secretion levels were low, but not significantly so, under Th0 conditions, but normal under Th17 polarization (data not shown). Ex-vivo Th17 quantification also demonstrated an absence of Th17 defects in 12 additional DS patients, including three with CMC (Supplementary Figure 9A and 9B). Finally, we assessed the inhibitory effect of IFN-β on the differentiation of naïve CD4+ T cells into IL-17A-producing T cells. IFN-β can inhibit Th17 differentiation, but no difference in the transition of naïve CD4+ cells to IL-17 T cells following exposure to IFN-β was observed between 11 healthy control and 5 DS patients (Supplementary Figure 9C). Overall, we detected no impairment of Th17 immunity in the peripheral blood of DS patients.
7. Low naïve CD4+ T cell levels and high terminal effector T cell levels in DS patients
IFNs exert various effects on T-cell differentiation, memory maintenance and effector T-cell activation and may determine T-cell fates in DS patients under physiological and pathological conditions. DS patients generally had much smaller proportions of naïve cells in the CD8+ T-cell compartment, with disproportionately higher levels of TEMRA cells (Supplementary Figure 10). Interestingly, CD57 and CX3CR1, two markers of T cell activation and immune senescence, were more strongly expressed on the naïve, TEM, TERMA CD8+ T-cell subset (Supplementary Figure 10), whereas CD28 was downregulated in a larger proportion of TCM and TEMRA CD8+ T cells than in healthy donors (Supplementary Figure 10). Our evaluation of interferon responses in T cells from DS patients surprisingly showed that fewer CD8+ T cells (25%) from DS patients than of CD8+ T cells from healthy controls (65%) could respond to IFN-α by STAT1 phosphorylation (Figure 7A). The levels of pSTAT1 generated in response to IFN-α were much lower in CD8+ T cells from DS patients than in CD8+ T cells from controls (Figure 7B). We found that total STAT1 levels were much lower in one subset of CD8+ T cells than in the others: Temra CD8+ T cells (Supplementary Figure 11). Collectively, these observations suggest an overactivation of T cells and the premature senescence of CD8+T cells in DS patients, consistent with a recent report of T cell lineages dysregulation toward an auto-immunity-prone state by interferon hyperactivity41.
Figure 7: IFN-α responses and STAT1 levels in the CD8+ T cells of DS patients.

(A). Representative histogram, (B). Summary of the relative levels of pSTAT1 in the CD8+ T cells of healthy controls, DS patients, and patients with STAT1 GOF with and without IFN-α stimulation. (C). Representative FACS histogram of STAT1 levels in the four subsets of CD8+ T cells from one DS patient. The p values were obtained in unpaired t tests. ■ p>0.05; * p≤ 0.05;.
Discussion
The infectious and autoimmune disorders seen in DS patients have remained unexplained despite improvements in our understanding of the pathogenesis of DS obtained through studies of gene dosage effects in various animal models42,43. Our hypothesis was based on both the phenotypic (similar infectious and autoimmune manifestations) and genotypic (gene dosage effect of four IFN receptors signaling via STAT1 in DS patients and a gain of STAT1 function in patients with GOF STAT1 mutations) similarities between DS patients and patients with GOF STAT1 mutations. We therefore tested whether the immunological features of DS constitute a phenocopy of STAT1 GOF. We found that levels of the IFN receptors IFN-αR1, IFN-αR2 and IFN-γR2 in DS were high in DS patients, resulting in constitutive IFN activation in monocytes, similar to that observed in patients with GOF STAT1 mutations. The cellular phenotype appears to be milder in DS, consistent with the milder clinical phenotypes of DS patients. Unlike patients with GOF STAT1 mutation, DS patients have cells with normal GAF DNA binding activity upon stimulation with IL-6 and IL-21, and normal proportions and functions of peripheral Th17 cells. Significant, but mild, IFN overactivation may directly contribute to the autoimmune disorders, such as thyroiditis, frequently observed in DS patients. However, we were unable to establish a causal relationship for the susceptibilities of DS patients to CMC. Indeed, circulating Th17 cell counts were normal ex vivo and IFN-β inhibited Th17 differentiation normally in vitro. CMC may be related to a dysregulation of mucosal Th17 immunity in situ, in conditions in which T cells interact closely with the pathogens, epithelium and connective tissues. Moreover, we were unable to study the cellular response to IFN-λ, which is crucial for epithelial and tissue innate immunity44. Future studies should focus on testing the immune cells interacting with organoid systems in DS45.
Our findings of an enhanced IFN response raise the intriguing possibility that treatments targeting the JAK-STAT pathway might be beneficial in patients with DS. One JAK inhibitor, tofacitinib, has been used successfully for the treatment of alopecia areata in DS patients19. This approach might also alleviate CMC and autoimmunity in these patients. This and other studies suggest that IFN activation is a striking cellular phenotype in DS, and that DS may therefore be considered to belong to the expanding group of type I interferonopathies. This group includes monogenic Aicardi-Goutieres Syndrome and a constellation of disorders, characterized by enhanced type I IFN activity. Type I interferonopathies are caused by type I interferon overproduction or hyperresponsiveness to type I interferon46. An IFN signature has also been shown to act as a biomarker in more common conditions, including SLE, and chronic viral infections, and in the identification of immune checkpoint inhibitor non-responders46,47. It remains unclear why having too much interferon is pathogenic and why type I interferonopathies differ so markedly clinically. Future studies of immunological abnormalities in DS patients will shed light on their pathogenesis and open up new possibilities for managing DS patients.
Patients, Materials and Methods
Patients
We enrolled 45 Down syndrome patients (29 males and 16 females patients, details provided in the Table 1) from 2012 onwards, between the age of three and 53 years (mean ± standard deviation: 24.6±12.5) in this study. Our inclusion criteria were: DS patients, aged from 3 to 70 years old. Our exclusion criteria were: Mosaic DS, Robertsonian translocation DS, a diagnosis of cancer or leukemia in the past six months, treatment with immunomodulators, steroids, chemotherapy or immunotherapy in the past three months, acute infection or sepsis in the past month. One of the patients was of Asian origin, 10 were of African origin and 34 were Caucasian. Three patients had a history of chronic mucocutaneous candidiasis (6.7%), seven patients had onychomycosis (15.6%), eight had recurrent otitis media (17.8%), 12 had recurrent bacterial infections of the skin (17.8%), 16 developed a clinical presentation of varicella infection (35.6%), one developed systemic Candida infection (2.2%), seven patients were on treatment for hypothyroidism (15.6%), and seven had cardiac abnormalities (15.5%). Various immunological characterizations including flowcytometry, EMSA, Simoa and microarray analysis, were summarized in Supplementary Table 2. This study was approved by two institutional IRB at Rockefeller University, New York (Protocol No.XFK-0815) and Necker-Enfants Malades / IHU Imagine, Paris (Protocol C10-14). This study was performed in accordance with the Declaration of Helsinki and the institutional regulations.
Flow cytometry analysis and cell sorting
We used FACS to assess IFN-R expression on the surface of EBV-B cells and monocytes. Briefly, 0.1 to 1 million monocytes or EBV-B cells were first blocked by incubation with 1 μl of FcR blocking reagent in 1% SVF in PBS for 10 minutes. We then added 125 ng/ml anti-IFN-γR2-APC (FAB773A, R&D Systems), anti-IFN-γR1-PE (558934, BD), anti-IFN-αR1 (AA-3, from Dr. Sandra Pellegreni), anti-IFN-αR2-PE (21385-3, PBL) or anti-IL-10RB-FITC (FAB245F, R&D systems) antibody and incubated the cells for 30 minutes on ice before washing them twice with 1% FBS in PBS. The corresponding fluorochrome-conjugated IgG was used as the isotype control.
We purchased antibodies against the following molecules: CD4 (mouse RPA-T4, APC, BD Pharmingen555349), CD8(V450, BD Horizon560347), CD19 (PerCP-Cy5.5, BD 340951), CD45RA (APC-H7BD Pharmingen560674), CD27 (Alexa-Fluor 700 BioLegend356416), STAT1 (PE, BD Phosflow 558537), pSTAT1 (pY705, Alexa-Fluor 647, BD Phosflow 557815), CD14 (PE-Cy7 BD Pharmingen 557742), CD20 (PE, BD Pharmingen 555623), IL-17A (Alexa-Fluor 700, BD Pharmingen 560613), IFN-γ (PE,BD559356). For the PBMC experiment, we first isolated CD14+ monocytes with a positive selection kit from MiltenyiBiotec. The remaining lymphocytes were stained with antibodies recognizing CD4, CD8, CD 19, CD45RA and CD27, conjugated to different fluorochromes to distinguish between the various cell populations in PBMCs. Dead cells were excluded by Aqua dead cell staining (L34965, Thermo Fisher Scientific). Cells were sorted in an Aria flow cytometer. STAT1 and pSTAT1 levels were assessed with the BD Pharmingen Phospho Buffer kit. Briefly, 0.1-1 million cells, at a concentration of 1-10 million cells/ml in 10% FBS RPMI 1640, were left unstimulated or stimulated with IFNs for 30 minutes. Cells were then washed with cold IX PBS, fixed with Fix Buffer I (BD557870) and permeabilized with Perm Buffer III (BD558050). Cells were then incubated with anti-STAT1-PE (BD558537) or anti-pSTAT1-Alexa Fluor 647 (BD557815) antibodies in IX Perm/Wash buffer I (BD557885). Fluorescence signals were measured with a BD LSRII and analyzed with FlowJo software (Tree Star, Inc).
Transcriptome analysis
CD14+ monocytes were isolated with the microbeads from MiltenyiBiotec (130-050-201). We extracted mRNAs with the ZR RNA MicroPrep™ kit (R1061, ZYMO RESEARCH) according to the manufacturer’s instructions. We subjected about 100ng total mRNA to transcriptomic analysis on GeneChip® Human Transcriptome Array 2.0 (Affymetrix), according to the manufacturer’s instruction. Data were analyzed with R Studio (version 0.98.1103). After RMA normalization, expression levels (CHF file) were log2-transformed. The expression files were then matched by probe ID with the annotation file: HTA-2_0.na35.1. hg 19.transcript. Modules were constructed via co-expression analysis conducted on a collection of 16 blood transcriptome datasets, encompassing nearly one thousand individual subjects. Each dataset corresponds to a different pathological or physiological “state” (including autoimmune and infectious diseases, pregnancy, transplantation, cancer). This collection of datasets and the module construction process are described in detail in a preprint48 and are based on published work38,49.
Simoa quantification of IFN-α protein levels
Interferon alpha (IFN-α) protein levels were quantified with a digital-ELISA assay (Simoa, Quanterix) developed by Rodero et al.36, in accordance with the instructions provided by the manufacturer of the Homebrew Simoa assay kit, with two autoantibodies specific for IFN-α isolated and cloned from two patients with autoimmune polyendocrinopathy syndrome type 1 (APSl)/autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED)50. The 8H1 antibody clone was used to coat paramagnetic beads (0.3 mg/ml), as a capture antibody, and the 12H5 antibody was biotinylated (biotin-to-antibody ratio 30: 1) and used as the detector. SBG was used at concentrations of 0.3 μg/ml and 150 pM as a detector and revelation enzyme, respectively. Recombinant IFN-α17/-αI (PBL Assay Science) was used to generate a standard curve, after we had tested for cross-reactivity. Each sample was analyzed in duplicate and at a 1:3 dilution. The limit of detection (LOD) was calculated as the mean value +3SD (positivity with 99% confidence) of reactivity for all blank runs. The LOD, taking the dilution factor into account, was 0.19 fg/ml.
Electrophoretic mobility shift assay (EMSA)
EMSA was carried out as previously described34. Briefly, cells were stimulated by incubation for 20 minutes with various cytokines, including IFN-γ, IFN-α, IL-6 and IL-21 at the concentrations indicated. We incubated 10pg of nuclear extract with a 32P-labeled (a-dATP) GAS (from the FCGR1 promoter) probe and subjected the mixture to electrophoresis in a polyacrylamide gel. GAF DNA-binding activity was assessed with ImageQuant (Amersham Bioscience).
Statistics
Student’s t-tests were used to assess the significance of differences in quantitative variables between two groups.
Supplementary Material
Acknowledgments
We thank Yanick Crow, Barry Coller, Charlie Rice, James Krueger and Timothy Wang for helpful discussions and critical reading. We thank Y. Nemirovskaya, T. Kochetkov, M. Romanick, L. Amar, C. Patissier, C. Desvallees, M. Woollett, A. Gall and J. Gonzalez for technical and secretarial assistance and all members of the Laboratory of Human Genetics of Infectious Diseases for helpful discussions. X-F.K was supported by the Jerome Lejeune Foundation and Alexander’s Angels Inc. X-F.K is currently a gastroenterology fellow in CUIMC supported by NIH grant T32 DK083256. The Laboratory of Human Genetics of Infectious Diseases is supported by grants from the St. Giles Foundation, the Jeffrey Modell Foundation, The Rockefeller University Center for Clinical and Translational Science grant number UL1TR001866 from the National Center for Research Resources and the National Center for Advancing Sciences (NCATS), the National Institutes of Health, the National Institute of Allergy and Infectious Diseases (grants 5R01AI127564 and 5R37AI095983), grants from the Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID) and the French National Research Agency (ANR) under the “Investments for the future” program (ANR-10-IAHU-01), ANR-IFN GPHOX (ANR-13-ISV3-0001-01) and ANR-GENMSMD (ANR-16-CE17-0005-01), LTh-MSMD-CMCD (ANR-18-CE93-0008-01), HGDIFD (ANR-14-CE15-0006-01), Institut National de la Santé et de la Recherche Médicate (INSERM), University of Paris, and The Rockefeller University and the Helmut Horten Foundation.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of Interest:
Dr. Casanova reports personal fees from Celgene, personal fees from ADMA, personal fees from Nimbus, personal fees from Vitae Pharmaceuticals, Inc, personal fees from KymeraTX, personal fees from Sanofi, personal fees from Asahi Kasei, personal fees from Pfizer, personal fees from Elixiron Immunotherapeutics, outside the submitted work. None of the other authors have no conflict of interest to declare.
References
- 1.Lejeune J, Gautier M & Turpin R A study of somatic chromosomes in nine infants with mongolism. C.R. Acad. Sci 248, 1721–1722 (1959). [PubMed] [Google Scholar]
- 2.de Graaf G, Buckley F & Skotko BG Estimation of the number of people with Down syndrome in the United States. Genet. Med 19, 439–447 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Oster J, Mikkelsen M & Nielsen A Mortality and life-table in Down’s syndrome. Acta Paediatr. Scand 64, 322–6 (1975). [DOI] [PubMed] [Google Scholar]
- 4.Bell JA, Pearn JH & Firman D Childhood deaths in Down’s syndrome. Survival curves and causes of death from a total population study in Queensland, Australia, 1976 to 1985. J. Med. Genet 26, 764–768 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nagarajan A et al. Oral Candidal and Streptococcal carriage in Down syndrome patients. J. Nat. Sci. Biol. Med 6, 300 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scully C et al. Down syndrome: lip lesions (angular stomatitis and fissures) and Candida albicans. Br. J. Dermatol 147, 37–40 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Donnelly LF, Shott SR, LaRose CR, Chini BA & Amin RS Causes of Persistent Obstructive Sleep Apnea Despite Previous Tonsillectomy and Adenoidectomy in Children with Down Syndrome as Depicted on Static and Dynamic Cine MRI. Am. J. Roentgenol 183, 175–181 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Madan V, Williams J & Lear JT Dermatological manifestations of Down’s syndrome. Clinical and Experimental Dermatology 31, 623–629 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Pikora TJ et al. Health Conditions and Their Impact among Adolescents and Young Adults with Down Syndrome. PLoS One 9, e96868 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ram G & Chinen J Infections and immunodeficiency in Down syndrome. Clinical and Experimental Immunology 164, 9–16 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abanto J et al. Medical problems and oral care of patients with Down syndrome: a literature review. Spec. Care Dentist 31, 197–203 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Svejgaard EL & Nilsson J Onychomycosis in Denmark: Prevalence of fungal nail infection in general practice. Mycoses 47, 131–135 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Johnston JN, Kaplan SL, Mason EO & Hulten KG Characterization of Staphylococcus aureus infections in children with Down syndrome. J. Infect. Chemother 21, 790–794 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Eldars W, Eldegla H, Yahia S, Ela MA & Hawas S Prevalence of community acquired infections in down syndrome children: A single center study. Brazilian Journal of Infectious Diseases 17, 624–625 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poizeau F et al. Prevalence and description of hidradenitis suppurativa in down syndrome: A cross-sectional study of 783 subjects. Acta Derm. Venereol 99, 351–352 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Romanio MR et al. FUNGEMIA POR SACCHAROMYCES CEREVISIAE EM PACIENTE PEDIÁTRICO APÓS TRATAMENTO COM PROBIÓTICO. Rev. Paul. Pediatr 35, 361–364 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shetty S, Kini U & Joy R Isolated lingual mucormycosis in an infant with Down syndrome. Ear. Nose. Throat J 87, 34–5, 43 (2008). [PubMed] [Google Scholar]
- 18.Lavigne J et al. Thyroid dysfunction in patients with Down syndrome: Results from a multi-institutional registry study. Am. J. Med. Genet. Part A (2017). doi: 10.1002/ajmg.a.38219 [DOI] [PubMed] [Google Scholar]
- 19.Rachubinski AL et al. Janus kinase inhibition in Down syndrome: 2 cases of therapeutic benefit for alopecia areata. JAAD Case Reports 5, 365–367 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ludvigsson JF, Lebwohl B, Green PHR, Chung WK & Mårild K Celiac disease and Down syndrome mortality: a nationwide cohort study. BMC Pediatr. 17, 41 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Antonarakis SE, Lyle R, Dermitzakis ET, Reymond A & Deutsch S Chromosome 21 and down syndrome: from genomics to pathophysiology. Nat. Rev. Genet 5, 725–38 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Föister-Holst R, Rohrer T & Jung AM Dermatological aspects of the S2k guidelines on Down syndrome in childhood and adolescence. JDDG - J. Ger. Soc. Dermatology 16, 1289–1295 (2018). [DOI] [PubMed] [Google Scholar]
- 23.De Hingh YCM et al. Intrinsic abnormalities of lymphocyte counts in children with Down syndrome. J. Pediatr 147, 744–747 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Toubiana J et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood 127, 3154–3164 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu L et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J. Exp. Med 208, 1635–1648 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Puel A et al. Inborn errors of human IL-17 immunity underlie chronic mucocutaneous candidiasis. Carr. Opin. Allergy Clin. Immunol 12, 616–22 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van de Veerdonk FL et al. STAT1 Mutations in Autosomal Dominant Chronic Mucocutaneous Candidiasis. N. Engl. J. Med 365, 54–61 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Borden EC et al. Interferons at age 50: past, current and future impact on biomedicine. Nat. Rev. Drug Discov 6, 975–990 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan YH, Schneider EL, Tischfield J, Epstein CJ & Ruddle FH Human chromosome 21 dosage: effect on the expression of the interferon induced antiviral state. Science 186, 61–63 (1974). [DOI] [PubMed] [Google Scholar]
- 30.Cupples CG & Tan YH Effect of human interferon preparations on lymphoblastogenesis in Down’s syndrome. Nature 267, 165–7 (1977). [DOI] [PubMed] [Google Scholar]
- 31.Prandini P et al. Natural gene-expression variation in Down syndrome modulates the outcome of gene-dosage imbalance. Am. J. Hum. Genet 81, 252–63 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sullivan KD et al. Trisomy 21 consistently activates the interferon response. Elife 5, el6220 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kong XF et al. Haploinsufficiency at the human IFNGR2 locus contributes to mycobacterial disease. Hum. Mol. Genet 22, 769–781 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kong XF et al. A novel form of human STAT1 deficiency impairing early but not late responses to interferons. Blood 116, 5896–5906 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kong XF et al. A novel form of cell type-specific partial IFN-gammaRl deficiency caused by a germ line mutation of the IFNGR1 initiation codon. Ham Mol Genet 19, 434–444 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodero ΜP et al. Detection of interferon alpha protein reveals differential levels and cellular sources in disease. J. Exp. Med 214, 1547–1555 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rice GI et al. Assessment of interferon-related biomarkers in Aicardi-Gouti??res syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: A case-control study. Lancet Neurol. 12, 1159–1169 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Obermoser G et al. Systems Scale Interactive Exploration Reveals Quantitative and Qualitative Differences in Response to Influenza and Pneumococcal Vaccines. Immunity 38, 831–844 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chaussabel D et al. A Modular Analysis Framework for Blood Genomics Studies: Application to Systemic Lupus Erythematosus. Immunity 29, 150–164 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ottaviano G et al. A Prevalent CXCR3+ Phenotype of Circulating Follicular Helper T Cells Indicates Humoral Dysregulation in Children with Down Syndrome. J. Clin. Immunol 1–9(2020). doi: 10.1007/sl0875-020-00755-0 [DOI] [PubMed] [Google Scholar]
- 41.Araya P et al. Trisomy 21 dysregulates T cell lineages toward an autoimmunity-prone state associated with interferon hyperactivity. Proc. Natl. Acad. Sci. U. S. A 116, 24231–24241 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Letourneau A et al. Domains of genome-wide gene expression dysregulation in Down’s syndrome. Nature 508, 345–50 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Reeves RH et al. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat. Genet 11, 177–184(1995). [DOI] [PubMed] [Google Scholar]
- 44.Syedbasha M & Egli A Interferon Lambda: Modulating Immunity in Infectious Diseases. Front. Immunol 8, 119 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fatehullah A, Tan SH & Barker N Organoids as an in vitro model of human development and disease. Nat. Cell Biol 18, 246–254 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Rodero ΜP & Crow YJ Type I interferon-mediated monogenic autoinflammation: The type I interferonopathies, a conceptual overview. J. Exp. Med 213, 2527–2538 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bennett L et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med 197, 711–723 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Altman MC et al. A Novel Repertoire of Blood Transcriptome Modules Based on Co-expression Patterns Across Sixteen Disease and Physiological States. bioRxiv 525709 (2019). doi: 10.1101/525709 [DOI] [Google Scholar]
- 49.Chaussabel D et al. A modular analysis framework for blood genomics studies: application to systemic lupus erythematosus. Immunity 29, 150–64 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meyer S et al. AIRE-Deficient Patients Harbor Unique High-Affinity Disease-Ameliorating Autoantibodies. Cell 166, 582–595 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.