

Abstract
Immune‐related adverse events induced by immune checkpoint inhibitor (ICI) therapy may affect diverse organ systems, including skeletal and cardiac muscle. ICI‐associated myositis may result in substantial morbidity and occasional mortality. We present a case of a patient with advanced non‐small cell lung cancer who developed grade 4 myositis with concurrent myocarditis early after initiation of anti‐programmed death ligand 1 therapy (durvalumab). Autoantibody analysis revealed marked increases in anti‐3‐hydroxy‐3‐methylglutaryl‐coenzyme A reductase antibody levels that preceded clinical toxicity, and further increased during toxicity. Notably, the patient had a history of intolerable statin myopathy, which had resolved clinically after statin discontinuation and prior to ICI initiation. This case demonstrates a potential association between statin exposure, autoantibodies, and ICI‐associated myositis.
Short abstract
This article presents the case of a patient with advanced non‐small cell lung cancer treated with PD‐L1 checkpoint blockade who developed grade 4 myositis. This case report describes the first known case of ICI‐induced myositis associated with anti‐HMGCR antibodies, a key enzyme in cholesterol biosynthesis.
Introduction
Immune checkpoint inhibitors (ICI) are now widely used for the treatment of multiple solid tumors and hematological malignancies. ICI therapy may result in immune‐related adverse events (irAEs), which may affect diverse organ systems and result in substantial morbidity and occasional mortality. Myositis is a rare but increasingly recognized irAE that is fatal in up to 20% of reported cases 1. ICI‐associated myositis may occur as an isolated entity or may overlap with myocarditis and myasthenia.
Anti‐3‐hydroxy‐3‐methylglutaryl‐coenzyme A reductase (HMGCR) myopathy is a rare autoimmune myopathy characterized by subacute proximal muscle weakness and discomfort with substantial creatine phosphokinase (CPK) elevation. A history of statin exposure is often present; however, most cases of statin‐induced myopathy do not result in anti‐HMGCR antibodies. Most cases of anti‐HMGRC myopathy have longstanding elevations in HMGCR antibodies, but it is unclear if antibodies are typically present before symptom onset 2.
We present a case of a patient with advanced non‐small cell lung cancer (NSCLC) treated with anti‐programmed death ligand 1 (PD‐L1) checkpoint blockade (durvalumab) who developed grade 4 myositis and presumptive myocarditis after 1 month of ICI therapy. Notably, he had a history of statin‐associated myalgia causing mild functional impairment, which had resolved after statin discontinuation and prior to ICI initiation. We performed longitudinal autoantibody analysis to provide insight into the humoral immune landscape accompanying these toxicities.
Case Presentation
A 65‐year‐old man with locally advanced, PD‐L1‐positive (tumor proportion score 90%) squamous NSCLC received concurrent thoracic radiation therapy with cisplatin and etoposide and was then initiated on the anti–PD‐L1 monoclonal antibody durvalumab 10 mg/kg every 2 weeks as consolidation therapy. Notably, he had a history of hyperlipidemia previously treated with atorvastatin and then rosuvastatin. These agents were both discontinued because of intolerable myalgias, the rosuvastatin being discontinued 2 weeks prior to ICI initiation. The myalgias resolved shortly after discontinuation of the statins and prior to ICI initiation.
Two weeks after the second durvalumab dose, the patient presented to the emergency department with symptoms of progressive fatigue, lower extremity weakness, myalgias, diplopia, and chest tightness. There was no fever or upper respiratory viral prodrome present. His physical examination was notable for normal strength throughout, with mild tenderness to palpation of bilateral thighs. Creatine phosphokinase level was 5,144 U/L (reference range, 20–200 U/L). High‐sensitivity cardiac troponin was 296 ng/L (reference range, <18 ng/L). Additional laboratory evaluation is shown in Table 1. Electrocardiogram revealed a previously documented right bundle branch block. Echocardiogram demonstrated no wall motion abnormalities and a left ventricular ejection fraction of 60% (reference range, 55%–70%). Cardiac magnetic resonance imaging was performed without contrast, so enhancement could not be evaluated.
Table 1.
Selected laboratory values over the hospital course
| Laboratory value | Reference range | Day 1 | Day 2 | Day 4 | Day 7 | Day 10 |
|---|---|---|---|---|---|---|
| Erythrocyte sedimentation rate | 0–9 mm/hr | 12 | 140 | x | x | x |
| Creatine phospho‐kinase | 20–200 units/L | 5,144 | 7,104 | 9,205 | 1,985 | 520 |
| Aldolase | <7.7 units/L | 71.3 | x | x | x | x |
| Myoglobin | <90 μg/L | 1,249 | x | x | x | x |
| Urine myoglobin | <21 μg/L | 1,139 | x | x | x | x |
| High‐sensitivity cardiac troponin | <18 ng/L | 296 | 397 | 455 | 251 | 272 |
| Lactate dehydrogenase | 135–225 units/L | 681 | x | x | x | x |
| C‐reactive protein | <10 mg/L | 3.13 | x | x | x | x |
Abbreviation: x, not measured.
The patient was diagnosed with ICI‐associated grade 4 myositis and presumptive myocarditis. Given the absence of fever, cardiac conduction abnormalities, or clinical features of myocardial infarction, prior radiation therapy, prior chemotherapy, or viral infection were considered less likely etiologies 3, 4. He initially received intravenous (IV) immunoglobulin (Ig) 0.5 g/kg daily for 4 days but developed worsening dyspnea and ptosis, accompanied by further increases in erythrocyte sedimentation rate, CPK, and troponin (Table 1). He was then treated with methylprednisolone 1 mg/kg IV twice daily, with some improvement in ptosis and weakness and CPK stabilization. By hospital day 14, CPK and troponin levels were decreasing, and he was transitioned to prednisone 1 mg/kg orally twice daily. After 37 days, he was discharged on a prednisone taper.
After discharge, myalgias, weakness, and diplopia persisted, but with physical therapy and cardiac rehabilitation, he was able to return to his baseline level of physical activity after 3 months. Eventually, the patient resumed his baseline level of physical activity. Fluorodeoxyglucose positron emission tomography‐computed tomography after discharge demonstrated complete radiographic and metabolic tumor response. Approximately 9 months after stopping durvalumab, he developed biopsy‐proven recurrent right upper lobe NSCLC.
Autoantibody Analysis
Prior to therapy initiation, the patient was enrolled in a prospective biospecimen collection protocol (institutional review board no. STU 082015‐053). Peripheral blood samples were collected from the patient at pretreatment baseline, 2 weeks after ICI initiation, and at time of toxicity. As previously described 5, we measured serum autoantibody levels against a panel of 124 autoantigens using an autoantigen microarray platform developed by the UT Southwestern Human Genomics Core. Briefly, diluted serum samples were incubated with autoantigen arrays, and autoantibodies were detected using a Genepix 4200A scanner. Median signal intensity was calculated and subtracted from local background intensity. Intensity was normalized to the average intensity of mouse IgG and IgM internal controls. Signal‐to‐noise ratio values ≥3 were considered significantly higher than background.
Results
Hierarchical clustering of 124 autoantibodies demonstrated marked changes in serum levels of multiple autoantibodies 2 weeks after ICI initiation. At time of toxicity, 69 autoantibodies were upregulated twofold or more (Fig. 1A), including those associated with autoimmune myocarditis (e.g., anti‐SRP) and myositis (anti‐synthetase, anti–Jo‐1). In particular, we noted substantial increases in anti‐HMGCR and anti‐cytosolic 5′‐nucleotidase 1A (cN‐1A) antibody levels at 2 weeks, with further elevation at time of toxicity (Fig. 1B).
Figure 1.
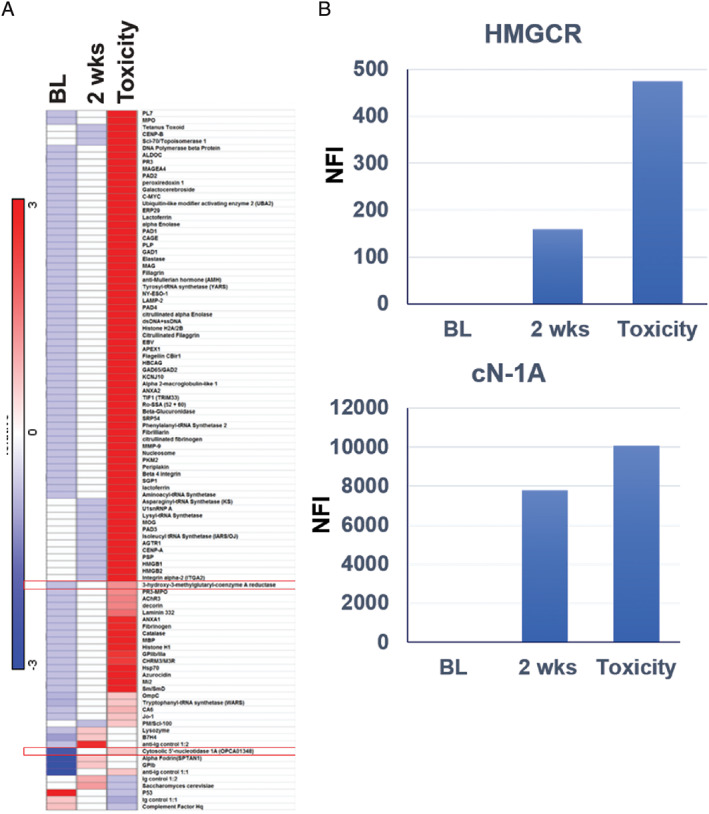
Relative antibody concentrations. (A): Heatmap of serum autoantibody levels at baseline, at 2‐weeks postimmunotherapy and at the time of toxicity. (B): Serum levels of anti‐HMGCR and anti‐cN‐1A autoantibodies over time. Values on the y‐axis represent NFI as described in the Methods and Materials.
Abbreviations: BL, baseline; cn‐1A, cytosolic 5′‐nucleotidase 1A; HMGCR, hydroxy‐3‐methylglutaryl‐coenzyme A reductase; NFI, new fluorescence intensity.
Discussion
Clinicians and researchers face challenges in diagnosing, managing, and understanding the pathophysiology of irAEs 6. One proposed mechanism posits that these autoimmune toxicities require sequential steps that progressively weaken immune regulation. Initially, environmental or genetic factors result in breakdown of tolerance, subsequently leading to autoantibody development 7.
In this case, it is not clear if prior statin exposure served as an inciting environmental factor, subsequently amplified by ICI exposure. An earlier report described heightened risk of immune‐related myopathy with concurrent ICI and statin administration, even if prior statin use did not cause musculoskeletal symptoms 8. History of prior statin myopathy was also reported in another case of ICI‐associated myositis 9. Other scenarios potentially supporting a “two‐hit” hypothesis of irAE include markedly increased rates of pulmonary toxicity with combination anti–PD‐L1 and epidermal growth factor receptor inhibition (up to 64% incidence, compared with ≤5% for either agent alone 10) and heightened incidence of hepatic toxicity with combination anti‐cytotoxic T lymphocyte antigen 4 and BRAF inhibition 11.
To our knowledge, this is the first described case of ICI‐induced myositis associated with anti‐HMGCR antibodies. HMGCR is a key enzyme in cholesterol biosynthesis. Anti‐HMGCR antibodies are considered relatively specific for necrotizing myositis and are usually associated with statin exposure 12, 13. As a single case report, the presented clinical and laboratory findings can only be considered hypothesis generating. It is unclear whether anti‐HMGCR antibodies mediated the myositis or were simply associations. Nor is it certain that prior statin exposure predisposed to the irAE. One possibility is that statin‐induced myopathy caused increased exposure of muscle antigens (HMGCR; Hit 1), and then ICI administration caused immune recognition of this newly exposed antigen and subsequent development of anti‐HMGCR antibodies (Hit 2). That is, statin exposure served as an environmental trigger, leading to breakdown of self‐tolerance and subsequent autoimmunity 14. Alternatively, patients with statin intolerance may be predisposed to develop myositis.
Although the clinical presentation in this case was not strongly suggestive of inclusion body myositis (IBM; which typically presents with insidious distal upper and lower extremity weakness), the increased cN‐1A antibody levels observed in this case have been associated with IBM 15. The significance of this finding is unclear. Unfortunately, blood samples at the time of clinical improvement were not available for autoantibody analysis, which we recognize as a limitation of this study. Although further research into humoral and cellular biomarkers of ICI‐associated myositis and myocarditis seems warranted given this potential high‐risk population (individuals with prior statin‐related myopathy or exposure), there is currently no evidence to suggest that statin therapy should be withheld in individuals receiving ICI.
Disclosures
Jason Y. Park: GLUT1 Foundation (RF), Miraca Holdings, including subsidiaries Baylor Genetics and Fujirebio Inc. (SAB), Guanylyl Cyclase C biomarkers licensed to Targeted Diagnostics and Therapeutics and to Takeda Oncology (IP, OI). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Acknowledgements
The authors thank Ms. Dru Gray for assistance with manuscript preparation.
Funding: Supported in part by a National Cancer Institute (NCI) Midcareer Investigator Award in Patient‐Oriented Research (K24 CA201543‐01, to D.E.G.), the David M. Crowley Foundation, the Peter Bradley Carlson Trust, an American Cancer Society‐Melanoma Research Alliance Team Award (MRAT‐18‐114‐01‐LIB; to D.E.G.), a V Foundation Robin Roberts Cancer Survivorship Award (DT2019‐007; to D.E.G.), the University of Texas Lung Cancer Specialized Program in Research Excellence (SPORE, P50‐CA‐070907‐08S1, to D.E.G.), a Melanoma Research Alliance‐Society for Immunotherapy of Cancer Young Investigator Award in Immune‐related Adverse Events (Award number‐ 619351; to S.K.), the Cancer Prevention and Research Institute of Texas (RP15096), the Human Genomics/Microarray Core, and the Biomarker Research Core and Biostatistics Shared Resources at the Harold C. Simmons Comprehensive Cancer Center, University of Texas Southwestern Medical Center, Dallas, Texas, which is supported in part by NCI Cancer Center Support Grant 1P30 CA142543‐01.
Ethics approval and consent to participate: The patient was enrolled in a prospective biospecimen collection protocol, which was approved by the UT Southwestern Institutional Review Board (no. STU 082015‐053).
Written informed consent for publication was obtained using our institutional consent form (included with manuscript submission).
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
Disclosures of potential conflicts of interest may be found at the end of this article.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact Commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
References
- 1. Anquetil C, Salem JE, Lebrun‐Vignes B et al. Immune checkpoint inhibitor‐associated myositis. Circulation 2018;138:743–745. [DOI] [PubMed] [Google Scholar]
- 2. Alshehri A, Choksi R, Bucelli R et al. Myopathy with anti‐HMGCR antibodies: Perimysium and myofiber pathology. Neurol Neuroimmunol Neuroinflamm 2015;2:e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yusuf SW, Sami S, Daher IN. Radiation‐induced heart disease: A clinical update. Cardiol Res Pract 2011;2011:317659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Florescu M, Cinteza M, Vinereanu D. Chemotherapy‐induced cardiotoxicity. Maedica (Buchar) 2013;8:59–67. [PMC free article] [PubMed] [Google Scholar]
- 5. Li QZ, Zhou J, Wandstrat AE et al. Protein array autoantibody profiles for insights into systemic lupus erythematosus and incomplete lupus syndromes. Clin Exp Immunol 2007;147:60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Khan S, Khan SA, Luo X et al. Immune dysregulation in cancer patients developing immune‐related adverse events. Br J Cancer 2019;120:63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Herbelet S, De Bleecker JL. Immune checkpoint failures in inflammatory myopathies: An overview. Autoimmun Rev 2018;17:746–54. [DOI] [PubMed] [Google Scholar]
- 8. Yoshioka M, Kambe N, Yamamoto Y et al. Case of respiratory discomfort due to myositis after administration of nivolumab. J Dermatol 2015;42:1008–1009. [DOI] [PubMed] [Google Scholar]
- 9. Fox E, Dabrow M, Ochsner G. A case of nivolumab‐induced myositis. The Oncologist 2016;21:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ahn MJ, Yang J, Yu J et al. 136O: Osimertinib combined with durvalumab in EGFR‐mutant non‐small cell lung cancer: Results from the TATTON phase Ib trial. J Thorac Oncol 2016;11(suppl): S115. [Google Scholar]
- 11. Ribas A, Hodi FS, Callahan M et al. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med 2013;368:1365–1366. [DOI] [PubMed] [Google Scholar]
- 12. Liewluck T, Kao JC, Mauermann ML. PD‐1 Inhibitor‐associated Myopathies: Emerging Immune‐mediated Myopathies. J Immunother 2018;41:208–211. [DOI] [PubMed] [Google Scholar]
- 13. Mohassel P, Mammen AL. Anti‐HMGCR myopathy. J Neuromuscul Dis 2018;5:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vojdani A, Pollard KM, Campbell AW. Environmental triggers and autoimmunity. Autoimmune Dis 2014;2014:798029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Felice KJ, Whitaker CH, Wu Q et al. Sensitivity and clinical utility of the anti‐cytosolic 5'‐nucleotidase 1A (cN1A) antibody test in sporadic inclusion body myositis: Report of 40 patients from a single neuromuscular center. Neuromuscul Disord 2018;28:660–664. [DOI] [PubMed] [Google Scholar]