

Abstract
Background
Programmed death‐ligand 1 (PD‐L1) expression is associated with clinical outcomes of epidermal growth factor receptor (EGFR) mutant lung adenocarcinoma (ADC) treated with tyrosine kinase inhibitors (TKIs). However, whether PD‐L1 expression plays a role in anaplastic lymphoma kinase (ALK)‐positive lung ADC is unknown. We aimed to evaluate the impact of PD‐L1 in patients with ALK‐positive lung ADC receiving crizotinib.
Materials and Methods
PD‐L1 expression was identified by immunohistochemistry (IHC). Reverse transcriptase‐polymerase chain reaction was used for ALK variant detection, and immunofluorescence‐based multiplex staining was applied for exploring immune cells in tumor microenvironments.
Results
A total of 78 patients with ALK‐positive advanced ADC were enrolled in our study, of whom 52 received crizotinib. Compared with EGFR/ALK wild‐type tumors, PD‐L1 expression was lower in ALK‐positive ADC. ALK fusion variants were identified in 32 patients, and those with variant 3 and 5 (short variants) had higher PD‐L1 expression than those with other variants. The crizotinib objective response rate (ORR) and progression‐free survival (PFS) was better in tumors with negative PD‐L1 expression (ORR/PFS in PD‐L1 0% vs. 1%–49% vs. 50%–100%: 60.7%/11.8 months vs. 38.5%/6.5 months vs. 36.4%/4.0 months, p = .007/.022). The multivariate Cox proportional hazards model revealed that PD‐L1 0% (vs. ≥1%) was an independent factor for longer PFS (adjusted hazard ratio 0.322, 95% confidence interval 0.160–0.650, p = .002). Multiplex IHC in three cases showed a varied extent of immune cell infiltrations in tumors with different PD‐L1 expression.
Conclusion
Positive PD‐L1 expression was associated with unfavorable clinical outcomes in patients with ALK‐positive lung ADC receiving crizotinib.
Implications for Practice
Not all lung adenocarcinoma with sensitizing driver mutations experienced durable responses to small‐molecule tyrosine kinase inhibitors (TKIs). Similar to the negative impact of programmed death‐ligand 1 (PD‐L1) in epidermal growth factor receptor mutant tumors treated with TKIs, this study demonstrated that positive PD‐L1 expression was also associated with worse response rate and shorter progression‐free survival of anaplastic lymphoma kinase (ALK)‐positive adenocarcinoma treated with crizotinib. Among different ALK fusion partners, tumors with short variants (V3 and V5) had higher PD‐L1 compared with long variants (V1, V2, and V6). Testing PD‐L1 before initiating crizotinib for ALK‐positive lung cancer could be a simple method to provide important prognostic information.
Keywords: Anaplastic lymphoma kinase, Crizotinib, Fusion variant, Lung cancer, Programmed death‐ligand 1
Short abstract
This article focuses on the effect of PD‐L1 on ALK‐positive lung adenocarcinoma and examines the association of pretreatment tumor PD‐L1 and clinical outcomes of patients receiving the first approved ALK inhibitor, crizotinib.
Introduction
Lung cancer has been the leading cause of cancer‐related death worldwide for many years [1]. In addition to conventional chemotherapies, molecular targeted therapies and immune checkpoint inhibitors (ICIs) are the two mainstay treatments for advanced‐stage non‐small cell lung cancer (NSCLC). Tumors harboring specific driver mutations, such as epidermal growth factor receptor (EGFR) mutations [2], anaplastic lymphoma kinase (ALK) [3], and ROS1 rearrangements [4], have responded well to relevant targeted therapies; moreover, these oncogene‐driven tumors exhibit a poor response to ICIs [5]. In contrast, tumors without identified driver mutations are more likely to benefit from ICIs acting on the programmed death‐ligand 1 (PD‐L1)/programmed death 1 (PD‐1) axis [6], especially if the tumor cells exhibit higher PD‐L1 expression levels [7, 8] or higher tumor mutation burdens [9].
Recently, growing evidence suggests that PD‐L1 expression in EGFR mutant adenocarcinoma (ADC) is associated with the treatment outcomes of EGFR tyrosine kinase inhibitors (TKIs) [10, 11, 12, 13, 14, 15, 16, 17]. Although some inconsistencies existed, most of these studies showed that tumors with low or negative PD‐L1 expression tended to have better disease control from EGFR TKIs, suggesting that immune factors might influence the underlying mechanisms of TKI responses. Whether PD‐L1 expression also implicates effective targeted therapy in the second most common actionable driver mutation, ALK rearrangements, has not been well studied. ALK rearrangements are found in approximately 5% of patients with lung ADC [18, 19], in which an inversion of chromosome 2p leads to the fusion of echinoderm microtubule‐associated protein‐like 4 (EML4) and ALK, resulting in the constitutive activation of tyrosine kinase and subsequent tumor cell growth [3]. Similar to EGFR, many small‐molecule inhibitors are available for ALK‐rearranged ADC and have exhibited promising responses, including the first‐ (crizotinib [20]) and second‐generation (alectinib [21] and ceritinib [22]) ALK TKIs. Whereas first‐line crizotinib exhibited a better progression‐free survival (PFS) than chemotherapy regimens (10.9 vs. 7 months) in the PROFILE 1014 clinical trial [20], alectinib and ceritinib further extended the PFS to 34.8 and 16.6 months in the phase III ALEX [21] and ASCEND‐4 [22] clinical trials, respectively. Although the treatment response from targeted therapy is remarkable, several issues may still affect patient prognosis, including clinical factors (brain metastases and performance status) [23, 24] or genetic factors (fusion variants) [25]. Although immunotherapy has revolutionized the treatments of lung cancer, unraveling the immune factors within tumor microenvironments, which may implicate the efficacy of targeted therapy in ALK‐positive patients, has become increasingly important.
Herein, we aimed to study the impact of PD‐L1 on patients with ALK‐positive lung ADC. Our primary objective is to examine the association of pretreatment tumor PD‐L1 and clinical outcomes of patients receiving the first approved ALK inhibitor, crizotinib. We also analyzed the follow‐up results of post‐crizotinib treatment courses and overall survival (OS). Our secondary objective is to study the difference of PD‐L1 expression in ALK‐positive, EGFR mutant, and wild‐type EGFR/ALK tumors. The prevalence of PD‐L1 in ALK‐positive tumors with different fusion variants were investigated, too. Finally, we explored the immune microenvironments by multiplex immunohistochemical (IHC) staining in three index patients.
Materials and Methods
Patient Demographics and Outcome Measurements
The study CONSORT diagram is illustrated in Figure 1. From 2010 to 2018 in National Taiwan University Hospital (NTUH), we screened 129 patients with lung cancer with positive ALK IHC. After excluding 40 cases with early‐stage lung cancer undergoing surgical treatment only, 7 cases with focal IHC staining deemed false‐positive results of ALK IHC, and 4 cases with EGFR/ALK comutation, there were 78 patients with advanced‐stage ALK‐positive ADC who were analyzed for the PD‐L1 expression. The ALK IHC staining was performed using D5F3 antibody, which yielded a diagnostic value comparable to that of fluorescence in situ hybridization or next‐generation sequencing (NGS) for ALK rearrangements [26]. The prevalence of PD‐L1 expression in this cohort was compared with that of the other two NTUH cohorts [27] with EGFR mutations (EGFRm cohort, n = 153) and wild‐type EGFR/ALK (EGFR/ALK WT cohort, n = 148). Among the 78 patients, 52 received crizotinib as the first ALK TKI during their treatment courses and were assessed for the impact of PD‐L1 on crizotinib efficacy and clinical outcomes. The study was approved by the Research Ethics Committee of NTUH (201705049RIND).
Figure 1.
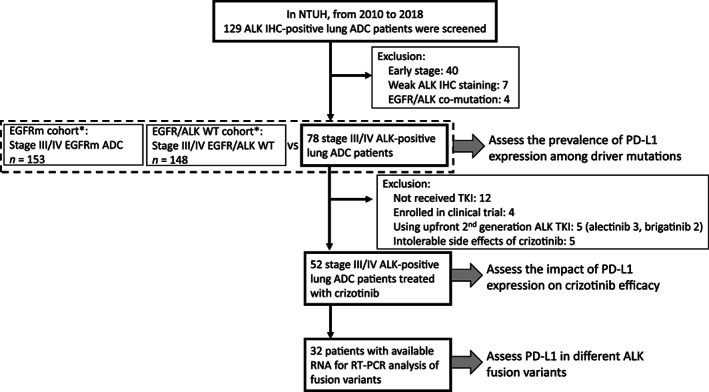
CONSORT diagram of the study. *: The EGFRm and EGFR/ALK WT cohort have been reported in our previous work [17].
Abbreviations: ADC, adenocarcinoma; ALK, anaplastic lymphoma kinase; EGFR, epidermal growth factor receptor; IHC; immunohistochemistry; NTUH, National Taiwan University Hospital; PD‐L1, programmed death‐ligand 1; RT‐PCR, reverse transcriptase‐polymerase chain reaction; TKI, tyrosine kinase inhibitor; WT, wild type.
The demographic data and clinical outcomes were collected by chart review or phone contact. During the treatment course of crizotinib, patients received chest computed tomography (CT) at least every 3 months. The treatment response was defined according to RECIST version 1.1. PFS started on the first day of crizotinib and ended on the day of death or disease progression documented by CT imaging studies. OS was defined as the period from the start of crizotinib to patient death. The patient survival was censored if there was no disease progression (PFS) or death (OS) at last follow‐up. The data cutoff date was September 30, 2019.
IHC Staining of ALK and PD‐L1
Tumor ALK expression was detected by IHC staining using the ALK (D5F3) assay with a Ventana BenchMark XT automated staining instrument (Ventana, AZ) according to the manufacturer's instructions. Tumor PD‐L1 expression was detected by IHC staining using the PD‐L1 Clone 22C3 pharmDx Kit and the Automated Link 48 platform (Dako, Carpinteria, CA). The degree of PD‐L1 expression was described by counting the cells with positive membrane staining within 100 viable cancer cells, which is known as the tumor proportion score (TPS) [28]. The TPS was then divided into three categories (TPS 0%, 1%–49%, and 50%–100%) based on previous clinical trials that adopted 22C3 antibodies as a companion diagnostic test [7, 29]. PD‐L1 was tested in the pretreatment tumors of all patients receiving crizotinib, and all these analyses were provided by Dr. Yih‐Leong Chang, Chen‐Tu Wu, and other pathologists at NTUH.
Detection of ALK Fusion Variants
We analyzed cancer specimens for EML4‐ALK variants with RNA reverse transcriptase‐polymerase chain reaction (RT‐PCR) [30], as previously described. In brief, we extracted RNA from patients’ tissue specimens for RT‐PCR amplification by using a OneStep RT‐PCR Kit (Qiagen, Hilden, Germany) with the following primers: 5′‐TGGCTGATGTTTTGAGGCGT‐3′ (forward, on exon 2 of EML4), 5′‐AGAGCCCACACCTGGGAAAG‐3′ (forward, on exon 13 of EML4), 5′‐CCACACAGACGGGAATGAAC‐3′ (forward, on exon 18 of EML4), and 5′‐AGCAAAGCAGTAGTTGGGGT‐3′ (reverse, on exon 20 of ALK). The PCR conditions were as follows: 50°C for 30 minutes, 95°C for 15 minutes, followed by 40 cycles of 94°C for 50 seconds, 60°C for 50 seconds, and 72°C for 60 seconds, and finally 72°C for 10 minutes. RT‐PCR amplicons were then purified and sequenced using Sanger sequencing conducted in both sense and antisense directions.
Immunofluorescence‐Based Multiplex Staining
Four‐micrometer‐thick sections were prepared and stained using the PerkinElmer Opal 7 Solid Tumor Immunology Kit according to the manufacturer's directions. We scanned the slides by the PerkinElmer Polaris system and analyzed images by inForm software (PerkinElmer, Hopkinton, MA) [31]. Five antibodies recognizing CD20, CD4, CD8, CD68, and forkhead box P3 (foxP3) were used to identify five types of tumor‐infiltrating immune cells, including CD20+ B cells, CD4+ T cells, CD8+ T cells, CD68+ macrophages, and regulatory T cells (CD4+ and foxP3+). Anticytokeratin antibodies plus 4′,6‐diamidino‐2‐phenylindole were adopted for tumor cell identification. The extent of cell infiltration was recorded as cell density (cells per square millimeter).
Statistical Analysis
Differences in PD‐L1 expression among ALK‐positive patients, EGFRm cohort, and EGFR/ALK WT cohort were analyzed by the chi‐square test. The comparison of PD‐L1 expression in different ALK fusion variants were conducted by Mann‐Whitney U test. PFS was calculated using Kaplan‐Meier survival plots, whereas the log‐rank test was used for comparisons. The prognostic factors associated with PFS were analyzed by Cox regression methods, and variables with p < .05 in univariate analysis were added to the final multivariate Cox regression model. All tests were two‐sided, and statistical significance was set at p < .05. All statistical analyses were performed with PASW Statistics 25 (IBM Corporation, Armonk, NY).
Results
Patient Demographics, Clinical Characteristics, and Prevalence of PD‐L1 Expression
The demographic data of the ALK‐positive patients, EGFRm cohort, and EGFR/ALK WT cohort are shown in supplemental online Table 1, with the study CONSORT diagram in Figure 1. Compared with EGFR/ALK wild‐type patients, there were more young (median age: 58 years), never‐smoking (74.4%), and female patients (53.8%) in the ALK‐positive cases, approximating the pattern of previously described ALK‐positive populations. In patients treated with crizotinib (Table 1), most had stage IV disease (43/52, 82.7%) and a fair performance status (Eastern Cooperative Oncology Group 0–1: 45/52, 86.5%). Because crizotinib has been reimbursed by our National Health Insurance program as a first‐line therapy since November 2017, only approximately half the patients received crizotinib as a first‐line therapy (27/52, 51.9%).
Table 1.
Demographic data of patients with ALK‐positive adenocarcinoma receiving crizotinib
| Characteristic | ALK‐positive cases (n = 52) |
|---|---|
| Age, median (range), years | 55.1 (32–89) |
| Male gender, n (%) | 26 (50) |
| Smoking, n (%) | 14 (26.9) |
| Stage, IIIB/IV | 9/43 |
| ECOG, 0–1/2–4 | 45/7 |
| Metastasis, n (%) | |
| Brain | 13 (25) |
| Liver | 9 (17.3) |
| Bone | 20 (38.5) |
| Line of crizotinib, 1st line/≥2nd line | 27/25 |
Abbreviations: ALK, anaplastic lymphoma kinase; ECOG, Eastern Cooperative Oncology Group.
Compared with EGFR/ALK wild‐type tumors, ALK‐positive tumors had a lower extent of PD‐L1 expression (ALK vs. EGFR ALK WT in TPS 0%: 59% vs. 38.5%; TPS 1%–49%: 20.5% vs. 37.2%; TPS ≥50%: 20.5% vs. 24.3%, p = .009; Fig. 2A), which was similar to the PD‐L1 expression distribution in EGFR‐mutated patients. In 32 of 52 patients who had available RNA for fusion partner analysis, V3 (16 of 32) was the most common variant in our cohort, followed by V1 (10 of 32), V2 (3 of 32), V5 (2 of 32), and one case of V6. The distribution was similar to that described in previously reported literature [32]. We found that cases with short fusion variants (V3 and V5) had higher PD‐L1 expression than cases with other long variants (Fig. 2B). The detailed fusion partners identified in our study are listed in supplemental online Table 2, among whom part of the patients (18 of 32) have been reported in another of our previous works for ALK fusion variants [33]. Furthermore, we assessed the association between PD‐L1 expression and metastatic sites. ALK‐positive ADC with PD‐L1 ≥50% were more likely to have liver metastases (Fig. 2C), whereas there was no significant difference in brain and bone metastases regarding PD‐L1 expression.
Figure 2.
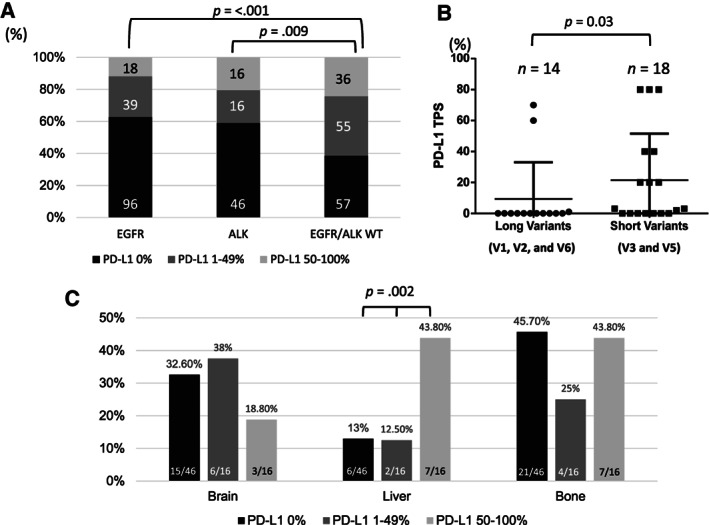
PD‐L1 expression in ALK‐positive adenocarcinoma (ADC). (A): Compared with EGFR/ALK WT tumors, PD‐L1 expression was lower in ALK‐positive lung ADC (p = .009), whereas EGFR‐mutant ADC exhibited significantly lower PD‐L1 expression (p < .001). (B): The difference in PD‐L1 expression between ALK tumors with short variants and long variants. Tumors with short variants (V3 and V5) tended to have higher PD‐L1 expression (p = .03). (C): The association of PD‐L1 and metastatic sites in patients with ALK‐positive ADC. Tumors with PD‐L1 ≥50% were more likely to have liver metastases.
Abbreviations: ALK, anaplastic lymphoma kinase; EGFR, epidermal growth factor receptor; PD‐L1, programmed death‐ligand 1; TPS, tumor proportion score; V, variant; WT, wild type.
Pretreatment PD‐L1 Is Associated with the ORR and PFS of Patients Treated with Crizotinib
The median follow‐up time for this cohort was 33.6 months (95% confidence interval: 17.7–49.5 months). The objective response rate (ORR) among patients with TPS 0%, 1%–49%, and ≥50% were 60.7%, 38.5%, and 36.4%, respectively (p = .007; Fig. 3A). Univariate analysis showed that PD‐L1 TPS 0% (vs. TPS ≥1%) was associated with prolonged PFS, whereas brain, liver, and bone metastases were poor prognostic factors. Using a multivariate Cox proportional hazards model, we found that PD‐L1 0% and bone metastases were independent prognostic factors (Table 2). Kaplan‐Meier analysis revealed that the median PFS of patients with PD‐L1 TPS 0%, 1%–49%, and ≥50% were 11.8, 6.5, and 4.0 months, respectively (Fig. 3B, 3C). The survival plots of crizotinib‐relevant PFS are illustrated in Fig. 3D. Overall, we found that pretreatment tumor PD‐L1 expression was correlated with the efficacy of crizotinib in ALK‐positive lung ADC, as well as the relevant clinical outcomes.
Figure 3.
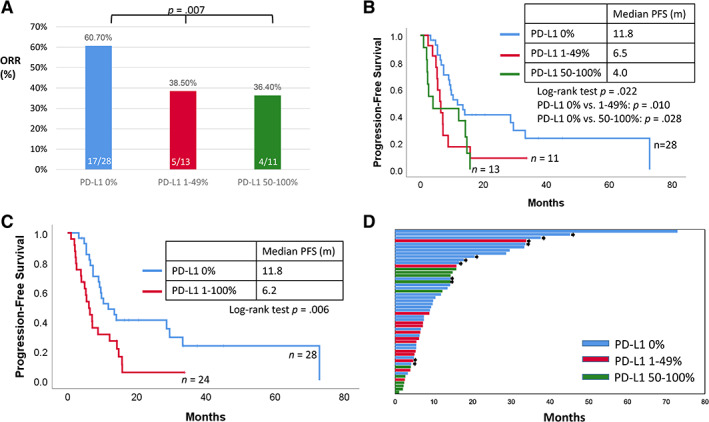
The impact of PD‐L1 on the outcomes of anaplastic lymphoma kinase‐positive patients treated with crizotinib. (A): The objective response rate in patients with PD‐L1 tumor proportion score (TPS) 0%, 1%–49%, and 50%–100% was 60.7%, 38.5%, and 36.4%, respectively. (B): The Kaplan‐Meier plot shows that the PFS of patients with PD‐L1 TPS 0%, 1%–49%, and 50%–100% was 11.8, 6.5, and 4.0 months (p = .022), respectively. (C): The Kaplan‐Meier plot shows that the PFS of patients with PD‐L1 TPS 0% and 1%–100% was 11.8 and 6.2 months (p = .006), respectively. (D): The survival plot illustrating the PFS of each patient. The bar colors of blue, red, and green refer to PD‐L1 TPS 0%, 1%–49%, and 50%–100%, respectively. The black arrowhead refers to the ongoing treatment of crizotinib at last follow‐up.
Abbreviations: ORR, objective response rate; PD‐L1, programmed death‐ligand 1; PFS, progression‐free survival.
Table 2.
Factors associated with the progression‐free survival of patients with anaplastic lymphoma kinase‐positive lung adenocarcinoma treated with crizotinib
| Characteristic | HR (95% CI) | p value | Adjusted HR (95% CI) | p value |
|---|---|---|---|---|
| Age ≥65 vs <65, years | 0.925 (0.407–2.104) | .852 | ||
| Male vs. female sex | 0.744 (0.397–1.392) | .355 | ||
| Smoker vs. nonsmoker | 1.039 (0.518–2.083) | .914 | ||
| Stage IIIB vs. IV | 2.448 (1.127–5.317) | .024 | 1.625 (0.717–3.682) | .244 |
| ECOG 2–4 vs. 0–1 | 0.729 (0.257–2.068) | .552 | ||
| Line of therapy ≥2nd line vs. 1st line | 0.531 (0.278–1.013) | .055 | ||
| PD‐L1 expression | ||||
| <50% vs. ≥50% | 0.522 (0.250–1.089) | .083 | ||
| 0% vs. ≥1% | 0.421 (0.224–0.793) | .007 | 0.322 (0.160–0.650) | .002 |
| Metastases | ||||
| Brain (positive vs. negative) | 2.404 (1.191–4.854) | .014 | 1.693 (0.725–3.957) | .224 |
| Liver (positive vs. negative) | 1.944 (0.892–4.236) | .095 | 2.197 (0.942–5.125) | |
| Bone (positive vs. negative) | 2.820 (1.479–5.376) | .002 | 2.813 (1.345–5.885) | .006 |
Abbreviations: CI, confidence interval; ECOG, Eastern Cooperative Oncology Group; HR, hazard ratio; PD‐L1, programmed death‐ligand 1; PFS, progression‐free survival.
The Association of PD‐L1 and OS in ALK‐Positive and EGFR/ALK Wild‐Type Patients
We further analyzed the association of PD‐L1 and OS in ALK‐positive patients treated with crizotinib and compared the results with those in the EGFR/ALK WT cohort, among whom 127 of 148 patients received follow‐ups at our hospital and were feasible for OS analysis (supplemental online Fig. 1A). In the crizotinib‐treated ALK‐positive cases, the median OS was 50.0 and 49.6 months in patients with PD‐L1 0% and 50%–100%, whereas the median OS was not reached in patients with PD‐L1 1%–49%. However, there was no significant difference in OS statistically (supplemental online Fig. 1B). In the EGFR/ALK WT cohort, the OS still did not differ in each group of patients with different PD‐L1 expression (supplemental online Fig. 1C). Thus, our results suggested that PD‐L1 might be a predictive biomarker of crizotinib PFS rather than a prognostic biomarker of OS.
Otherwise, we performed an exploratory analysis of the association of PD‐L1 and other anticancer therapies beyond crizotinib in ALK‐positive patients. Of the 52 patients, 33 received second‐generation ALK TKIs (19 were alectinib, 12 were ceritinib, and 2 were brigatinib) after failure of crizotinib, and 22 patients received pemetrexed‐containing regimens (19 were pemetrexed/platinum doublets, 3 were pemetrexed only). The results are illustrated in supplemental online Figure 2. In this exploratory analysis, PD‐L1 did not correlate with PFS of pemetrexed‐containing chemotherapy or second‐generation ALK TKIs. However, the case number was limited. Whether PD‐L1 is associated with efficacy of chemotherapy or ALK inhibitors other than crizotinib needs further large‐scale studies to clarify. Of note, no patients received immunotherapy during their treatment courses in our cohort.
Immune Microenvironments Explored by Multiplex Immunofluorescence Staining
Using multiplex staining with the Opal7 Immunology Kit, we analyzed the immune microenvironments of three cases with different PD‐L1 expression levels showing different responses to crizotinib (Fig. 4). Case A was a 43‐year‐old male patient who had ALK‐positive lung ADC with fusion partner V1 detected by RT‐PCR. The tumor had negative PD‐L1 expression (TPS 0%) and clinically responded well to crizotinib. Multiplex IHC staining of his tumor revealed abundant immune cell infiltration (Fig. 4A, 4D), including CD20+ B cells, CD8+ T cells, and CD4+ T cells. Case B was a 71‐year‐old female patient with ALK‐positive lung ADC. She received first‐line crizotinib treatment and experienced a partial response 3 months later. The tumor also showed negative PD‐L1 expression, and multiplex IHC revealed abundant immune cell infiltrations similar to case A (Fig. 4B, 4D). Case C was a 55‐year‐old male patient who had ALK‐positive lung ADC with fusion partner V3. The tumor showed high PD‐L1 expression (PD‐L1 TPS 80%) and primary resistance to crizotinib (disease progression at first image follow‐up). Multiplex IHC staining of the tumor revealed less infiltration of CD20+ B cells, CD8+ T cells, and CD4+ T cells (Fig. 4C, 4D). The baseline characteristics, efficacy of crizotinib, and treatment courses of the three cases are summarized in Figure 4D–4H.
Figure 4.
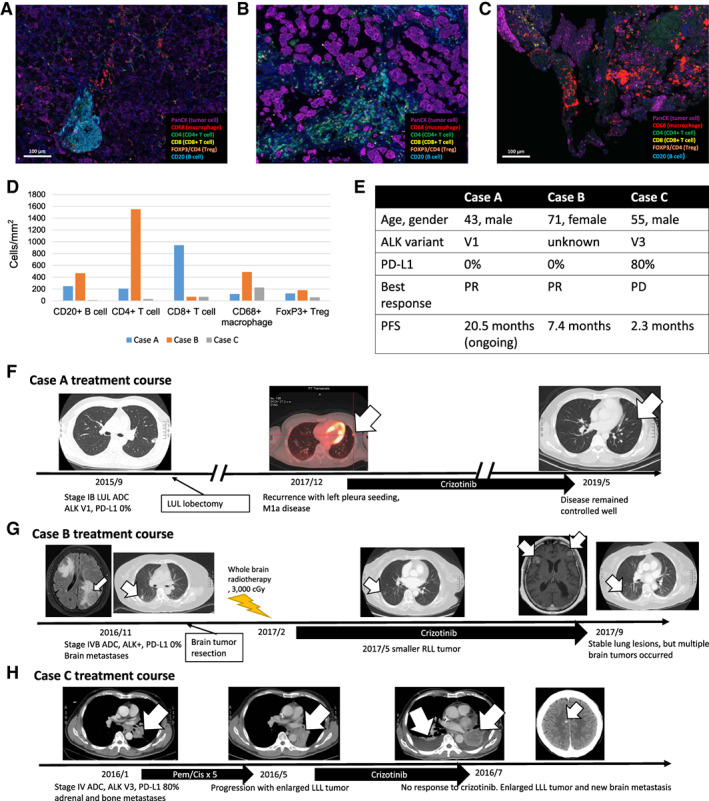
Fluorescence‐based multiplex immunohistochemical staining of three cases showing the spatial distributions and cell densities of different immune cells in the tumor microenvironments. The specific colors refer to individual markers (4′,6‐diamidino‐2‐phenylindole: blue; CD4: green; CD20: cyan; CD8: yellow; FoxP3: orange; CD68: red; and pancytokeratin for cancer cells: magenta). (A): The illustration of a case (case A) with a good response to crizotinib showing abundant immune cell infiltration, including CD20+ B cells and CD4+ and CD8+ T cells. (B): The illustration of case B with a partial response to crizotinib showing a high extent of immune cell infiltration. (C): The illustration of case C with a poor response to crizotinib showing a relatively lower extent of immune cell infiltration. (D): The cell densities (cells per square millimeter) of each immune cell in the three cases. (E): Brief summaries of the demographic characteristics and treatment outcomes of the three cases. (F): Illustration of the treatment courses of case A, who had sustained a response to crizotinib at the last data cutoff. (G): Illustration of the treatment courses of case B, who had partial response to crizotinib and a PFS of 7.4 months. (H): Illustration of the treatment courses of case C, who developed primary resistance to crizotinib. The arrow indicates the targeted lesions in the three cases.
Abbreviations: ADC, adenocarcinoma; ALK anaplastic lymphoma kinase; Cis, cisplatin; LLL, left lower lobe; LUL, left upper lobe; Pem, pemetrexed; PD, progressive disease; PD‐L1, programmed death‐ligand 1; PFS, progression‐free survival; PR, partial response; RLL, right lower lobe.
Discussion
In addition to PD‐L1 being a biomarker and druggable target of immunotherapy, researchers are currently also interested in the impacts of PD‐L1 on targeted therapies. Our study found that PD‐L1 expression was lower in ALK‐positive tumors than in wild‐type EGFR/ALK tumors. Among different fusion partners, tumors with short fusion variants tended to have higher PD‐L1 expression. Negative PD‐L1 expression in pretreatment tumors was associated with longer PFS in ALK‐positive patients receiving crizotinib. To our knowledge, this is the first study demonstrating the association between PD‐L1, ALK fusion variants, and clinical outcomes in patients with ALK‐positive ADC treated with targeted therapy.
The prevalence of PD‐L1 expression in each driver oncogene in NSCLC has drawn much attention in recent years. A number of studies using different IHC antibodies have shown that PD‐L1 is expressed at a lower level in EGFR mutant ADC than in wild‐type tumors [10, 11, 12, 13, 14, 15, 16], which was demonstrated in a recent meta‐analysis [34]. However, the extent of PD‐L1 expression in other driver mutations yielded varied results. BRAF [35] and MET exon 14 skipping mutations [36] were the two driver genes associated with higher PD‐L1 expression and better responses to PD‐1 blockade treatments [5], probably resulting from their association with cigarette smoking and higher tumor mutation burdens. Unlike EGFR mutations, the analyses of PD‐L1 expression in ALK‐positive tumors yielded inconclusive results in previous reports [37, 38, 39, 40] and meta‐analyses [34]. Several issues may account for the differences. First, the adopted IHC antibodies to detect PD‐L1 expression varied among each study. Recently, the harmonization Blueprint project demonstrated that Dako 28‐8, 22C3, and Ventana SP263 had similar staining intensities of PD‐L1 in tumor cells [41], which would compensate for the antibody‐related biases. We used the Dako 22C3 antibody to score PD‐L1 expression, which was the complementary diagnostic test for pembrolizumab in PD‐L1‐positive cases [7, 29]. Second, the heterogeneous patient populations spanning different ethnicities, cancer stages, and smoking statuses may confound the results of PD‐L1 analysis. Finally, most of the studies contained a limited number of ALK‐positive cases. Because the ALK fusion gene is found in only 5% of NSCLC [18, 19], its rarity is usually an inevitable obstacle hampering the validity of ALK‐related studies. In the recent large U.K. national study (10,005 cases) [39] and the global EXPRESS trial (2,617 cases) [40] examining PD‐L1 expression in NSCLC by the Dako 22C3 kit, there were only 56 and 74 ALK‐positive cases, respectively. In our study, we enrolled 78 purely Eastern Asian patients with advanced‐stage ALK‐positive ADC, which may better represent the appearance in this population and mitigate the disadvantages of patient heterogeneities.
Another important finding in our study was that lung ADC tumors with short fusion variants (V3 and V5) tended to exhibit higher PD‐L1 expression. The fusion variants of EML4‐ALK rearrangements depend on the EML4 exon fusing with ALK exon 20, and variants 1 and 3 were the most common fusion partners discovered in previous studies [42]. The major impact of fusion variants was considered to be their different sensitivities to ALK inhibitors [32]. Owing to the lack of tandem atypical beta‐propeller domain, ALK variants V3 and V5 were regarded as “short variants” and had a more stable structure with a longer half‐life, resulting in stronger oncogenic signaling in vitro [43]. Clinically, ALK V3 tumors also showed a more aggressive behavior and an unfavorable response to ALK inhibitors compared with other variants [44]. The mechanisms that regulate PD‐L1 expression may be a clue to interpret the link between ALK variants and PD‐L1. In addition to being elicited by adaptive resistance, PD‐L1 can also be upregulated by oncogene activation in in vitro cell line experiments, including EGFR [45], ALK [46], and other oncogenes. Thus, the stronger oncogenic signaling of ALK short variants may be capable of conferring a higher downstream PD‐L1 protein translation than that of other variants. However, not all the tumors with short variants exhibited high PD‐L1 expression in our study, in which 7 of 16 V3 tumors still showed negative of PD‐L1 IHC staining, suggesting that the in vivo expression of PD‐L1 was a more complex issue. In the tumor microenvironments, whether PD‐L1 expression results from immune pressure or oncogene activation may be determined by the existence of surrounding immune cells. Using multiplex IHC, we found that PD‐L1 was highly expressed in a case with an ALK V3 tumor, which was associated with a smaller extent of immune cell infiltration than the ALK V1 tumor with negative PD‐L1 expression (Fig. 4). Because there are only three index cases, large‐scale prospective studies of multiplex IHC may reveal a more distinct picture of immune microenvironments in ALK‐positive lung cancer.
We demonstrated a potential predictive value of PD‐L1 in patients with ALK‐positive lung ADC treated with an ALK TKI. Previously, many researchers have investigated the impact of PD‐L1 on EGFR‐mutant ADC [10, 11, 12, 13, 14, 15, 16], which yielded inconsistent results. Bai et al. [47] conducted a meta‐analysis and found that PD‐L1 was neither a predictive nor a prognostic biomarker for TKI efficacies in EGFR‐mutant ADC; however, some of these studies [12, 14, 16] used laboratory‐developed IHC antibodies not harmonized with the Dako or Ventana systems. Recently, five larger studies showed that high PD‐L1 expression was correlated with poor outcomes in EGFR‐mutant patients treated with EGFR TKIs using Dako 22C3, SP142, and SP263. In both biological and clinical aspects, EGFR and ALK share many similar characteristics, including an enrichment in female and nonsmoking patients, remarkable responses to small‐molecule targeted therapies, and most importantly, the capability to activate downstream PD‐L1 [45, 46]. Our ALK study was concordant with recent findings in EGFR‐mutant ADC that PD‐L1 might be a poor factor of TKI‐related outcomes. Thus, the association of PD‐L1 and TKI efficacy in EGFR‐ and ALK‐driven lung ADC implicated that an underlying molecular or immune mechanism could impact the TKI efficacy in oncogene‐driven tumors. Besides EGFR and ALK, several oncogenes have been reported to upregulate PD‐L1 expression, including Myc, hypoxia inducible factor‐1 alpha [48], phosphatase and tensin homolog loss [49], mitogen‐activated protein kinase (MAPK) [50], and Kirsten rat sarcoma (KRAS) [48]. Thus, high PD‐L1 expression in ALK‐positive tumors may reflect either a stronger ALK signaling or activation of other oncogenes beyond ALK, both of which could result in a worse response of crizotinib. Su et al. found that EGFR mutant ADC with PD‐L1 ≥ 50% carried a higher rate of de novo resistance to EGFR TKI. In their study, 1 and 4 of 15 high‐PD‐L1 patients had KRAS mutation and MET amplification, respectively, in addition to EGFR mutation [10], supporting the hypothesis that PD‐L1 was probably resulted from a bypass oncogene activation. On the other hand, immune mechanisms might also predispose tumors to develop TKI resistance, which has been demonstrated in a melanoma model. For example, Smith et al. [51] showed that the immune microenvironment could play an important role in the resistance to MAPK inhibitors in BRAF‐mutant melanoma. Tumor‐associated macrophages were upregulated and led to drug resistance after MAPK inhibition, which could be reversed by relevant IκB kinase inhibitors [51]. In terms of lung cancer, Yoshiya et al. had divided tumor microenvironments of EGFR‐mutant ADC to four categories according to the extent of PD‐L1+ tumor cell/CD8+ T cell, and they displayed that PD‐L1+/CD8+ patients carried the shortest PFS of EGFR TKI [52]. However, the underlying immune mechanisms mediating TKI resistance in oncogene‐driven lung cancer remained unclear. Recently, Feng et al. demonstrated a negative impact of myeloid‐derived suppressor cell on EGFR TKI response, probably via its derived tumor‐associated macrophages through activating noncanonical NF‐κB pathway [53]. To further explore underlying molecular and immune mechanisms accounting for high PD‐L1‐associated poor TKI efficacy, researchers may need to survey both genetic events using NGS, and immune microenvironments using multiplex IHC, in paired tissue samples before and after ALK TKI treatments. Collectively, our results suggested that high PD‐L1 expression in ALK‐positive ADC might represent a high‐risk group with poor response to first‐generation crizotinib, resulted from either a genetic or an immune mechanism. The next issue to address will be whether a more powerful ALK inhibitor [21, 22] or a combination therapy is more suitable in patients with ALK‐positive ADC with high PD‐L1 expression, necessitating a well‐conducted clinical trial to verify.
There were some limitations in our study. First, we did not address the impact of PD‐L1 on frontline second‐generation ALK inhibitors, which has become a standard of care in treatment‐naïve ALK‐positive patients. Because alectinib and ceritinib had not been reimbursed in our country until 2019, to enroll a sufficient number of cases treated with second‐generation ALK inhibitors was difficult at this time. In our exploratory analysis, we found that PD‐L1 had a nonsignificant association with PFS of second‐generation ALK inhibitors after failure of crizotinib. However, the results could not achieve a meaningful conclusion because of limited cases and more tumor heterogeneities emerging at crizotinib resistance. Thus, whether PD‐L1 is associated with PFS of second‐generation TKIs needs to be confirmed by further studies. Otherwise, we did not evaluate the impact of PD‐L1 on acquired resistance to crizotinib, which could be resulted from bypass mechanisms or kinase domain mutations. This issue may be resolved in the future when NGS is more available and less time‐consuming.
Conclusion
We found that PD‐L1 expression tended to be higher in ALK tumors with short fusion variants and might be associated with a greater risk of crizotinib failure. Thus, testing PD‐L1 expression in ALK‐positive lung cancer may provide additional information to predict clinical outcomes. Future studies to investigate the association of PD‐L1 with newer‐generation ALK inhibitors and to unravel the underlying immune mechanisms mediating TKI resistance will further improve the precision medicine in ALK‐positive lung cancer.
Author Contributions
Conception/design: Ching‐Yao Yang, Yih‐Leong Chang, Chen‐Tu Wu, Jin‐Yuan Shih
Provision of study material or patients: Ching‐Yao Yang, Wei‐Yu Liao, Chao‐Chi Ho, Kuan‐Yu Chen, Tzu‐Hsiu Tsai, Chia‐Lin Hsu, Yi‐Nan Liu, Kang‐Yi Su, Yih‐Leong Chang, Chen‐Tu Wu, Bin‐Chi Liao, Chia‐Chi Hsu, Wei‐Hsun Hsu, Jih‐Hsiang Lee, Chia‐Chi Lin, Jin‐Yuan Shih, James Chih‐Hsin Yang, Chong‐Jen Yu
Collection and/or assembly of data: Ching‐Yao Yang, Yih‐Leong Chang, Chen‐Tu Wu, Jin‐Yuan Shih
Data analysis and interpretation: Ching‐Yao Yang, Yi‐Nan Liu, Yih‐Leong Chang, Chen‐Tu Wu, Chia‐Chi Hsu, Jin‐Yuan Shih
Manuscript writing: Ching‐Yao Yang, Yih‐Leong Chang, Chen‐Tu Wu, Jin‐Yuan Shih
Final approval of manuscript: Ching‐Yao Yang, Wei‐Yu Liao, Chao‐Chi Ho, Kuan‐Yu Chen, Tzu‐Hsiu Tsai, Chia‐Lin Hsu, Yi‐Nan Liu, Kang‐Yi Su, Yih‐Leong Chang, Chen‐Tu Wu, Bin‐Chi Liao, Chia‐Chi Hsu, Wei‐Hsun Hsu, Jih‐Hsiang Lee, Chia‐Chi Lin, Jin‐Yuan Shih, James Chih‐Hsin Yang, Chong‐Jen Yu
Disclosures
Chao‐Chi Ho: AstraZeneca (RF), Boehringer Ingelheim, Eli Lilly, Roche/Genentech/Chugai, Merck Sharp & Dohme, Pfizer, Novartis, Bristol‐Myers Squibb, Ono Pharmaceutical (ET); Kuan‐Yu Chen: AstraZeneca, Roche, Novartis, Pfizer, Merck Sharp & Dohme, Bristol‐Myers Squibb, Boehringer Ingelheim (H), Merck Sharp & Dohme, Chugai Pharmaceutical, Pfizer (travel expenses); Chia‐Chi Lin: BeiGene, Eli Lilly, Pfizer (C/A), Novartis, Roche (H), Novartis, Takeda (SAB); Jin‐Yuan Shih: AstraZeneca, Roche, Boehringer Ingelheim, Pfizer, Novartis, Bristol‐Myers Squibb, Merck Sharp & Dohme, Eli Lilly (H), AstraZeneca, Roche, Boehringer Ingelheim, Novartis, Merck Sharp & Dohme, AbbVie, Chugai Pharmaceutical (C/A); James Chih‐Hsin Yang: Boehringer Ingelheim, Novartis, AstraZeneca, Roche/Genentech, Eli Lilly, Merck Sharp & Dohme Oncology, Merck Serono, Celgene, Bayer, Pfizer, Ono Pharmaceutical, Bristol‐Myers Squibb, Yuhan, Hansoh, Blueprint Medicines, Daiichi Sankyo, Amgen, Takeda Oncology, Incyte (C/A), Boehringer Ingelheim, Roche, Merck Sharp & Dohme, AstraZeneca, Novartis, Bristol‐Myers Squibb, Ono Pharmaceutical, Takeda Oncology, Eli Lilly, Pfizer (H). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Supplemental Figures
Supplemental Tables
Acknowledgments
We thank all the patients and their families and caregivers for participating in this study. We also thank Hsin‐Hui Tsai and Pei‐Shu Ho for their masterful skills in performing multiplex IHC testing. This study was funded by the National Taiwan University Hospital (NTUH 106‐N3715 and NTUH 108‐S4190) and in part supported by National Taiwan University (NTU107L9014 and NTU‐108L901403) and the Ministry of Science and Technology [107‐2314‐B‐002‐275‐MY3 and 108‐3017‐F‐002‐004].
Disclosures of potential conflicts of interest may be found at the end of this article.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact Commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
Contributor Information
Yih‐Leong Chang, Email: ntuhylc@gmail.com.
Chen‐Tu Wu, Email: ipohdamu@gmail.com.
Jin‐Yuan Shih, Email: jyshih@ntu.edu.tw.
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30. [DOI] [PubMed] [Google Scholar]
- 2. Lynch TJ, Bell DW, Sordella R et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2004;350:2129–2139. [DOI] [PubMed] [Google Scholar]
- 3. Soda M, Choi YL, Enomoto M et al. Identification of the transforming EML4‐ALK fusion gene in non‐small‐cell lung cancer. Nature 2007;448:561–566. [DOI] [PubMed] [Google Scholar]
- 4. Bergethon K, Shaw AT, Ou SH et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol 2012;30:863–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mazieres J, Drilon A, Lusque A et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann Oncol 2019;30:1321–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee CK, Man J, Lord S et al. Checkpoint inhibitors in metastatic EGFR‐mutated non‐small cell lung cancer‐A meta‐analysis. J Thorac Oncol 2017;12:403–407. [DOI] [PubMed] [Google Scholar]
- 7. Herbst RS, Baas P, Kim DW et al. Pembrolizumab versus docetaxel for previously treated, PD‐L1‐positive, advanced non‐small‐cell lung cancer (KEYNOTE‐010): A randomised controlled trial. Lancet 2016;387:1540–1550. [DOI] [PubMed] [Google Scholar]
- 8. Mok TSK, Wu YL, Kudaba I et al. Pembrolizumab versus chemotherapy for previously untreated, PD‐L1‐expressing, locally advanced or metastatic non‐small‐cell lung cancer (KEYNOTE‐042): A randomised, open‐label, controlled, phase 3 trial. Lancet 2019;393:1819–1830. [DOI] [PubMed] [Google Scholar]
- 9. Wang Z, Duan J, Cai S et al. Assessment of blood tumor mutational burden as a potential biomarker for immunotherapy in patients with non‐small cell lung cancer with use of a next‐generation sequencing cancer gene panel. JAMA Oncol 2019;5:696–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Su S, Dong ZY, Xie Z et al. Strong programmed death ligand 1 expression predicts poor response and de novo resistance to EGFR tyrosine kinase inhibitors among NSCLC patients with EGFR mutation. J Thorac Oncol 2018;13:1668–1675. [DOI] [PubMed] [Google Scholar]
- 11. Soo RA, Kim HR, Asuncion BR et al. Significance of immune checkpoint proteins in EGFR‐mutant non‐small cell lung cancer. Lung Cancer 2017;105:17–22. [DOI] [PubMed] [Google Scholar]
- 12. Lin C, Chen X, Li M et al. Programmed death‐ligand 1 expression predicts tyrosine kinase inhibitor response and better prognosis in a cohort of patients with epidermal growth factor receptor mutation‐positive lung adenocarcinoma. Clin Lung Cancer 2015;16:e25–e35. [DOI] [PubMed] [Google Scholar]
- 13. Yoneshima Y, Ijichi K, Anai S et al. PD‐L1 expression in lung adenocarcinoma harboring EGFR mutations or ALK rearrangements. Lung Cancer 2018;118:36–40. [DOI] [PubMed] [Google Scholar]
- 14. D'Incecco A, Andreozzi M, Ludovini V et al. PD‐1 and PD‐L1 expression in molecularly selected non‐small‐cell lung cancer patients. Br J Cancer 2015;112:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hsu KH, Huang YH, Tseng JS et al. High PD‐L1 expression correlates with primary resistance to EGFR‐TKIs in treatment naive advanced EGFR‐mutant lung adenocarcinoma patients. Lung Cancer 2019;127:37–43. [DOI] [PubMed] [Google Scholar]
- 16. Tang Y, Fang W, Zhang Y et al. The association between PD‐L1 and EGFR status and the prognostic value of PD‐L1 in advanced non‐small cell lung cancer patients treated with EGFR‐TKIs. Oncotarget 2015;6:14209–14219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou C, Wu YL, Chen G et al. Final overall survival results from a randomised, phase III study of erlotinib versus chemotherapy as first‐line treatment of EGFR mutation‐positive advanced non‐small‐cell lung cancer (OPTIMAL, CTONG‐0802). Ann Oncol 2015;26:1877–1883. [DOI] [PubMed] [Google Scholar]
- 18. Jordan EJ, Kim HR, Arcila ME et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov 2017;7:596–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu L, Liu J, Shao D et al. Comprehensive genomic profiling of lung cancer using a validated panel to explore therapeutic targets in East Asian patients. Cancer Sci 2017;108:2487–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Solomon BJ, Mok T, Kim DW et al. First‐line crizotinib versus chemotherapy in ALK‐positive lung cancer. N Engl J Med 2014;371:2167–2177. [DOI] [PubMed] [Google Scholar]
- 21. Gadgeel S, Peters S, Mok T et al. Alectinib versus crizotinib in treatment‐naive anaplastic lymphoma kinase‐positive (ALK+) non‐small‐cell lung cancer: CNS efficacy results from the ALEX study. Ann Oncol 2018;29:2214–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Soria JC, Tan DSW, Chiari R et al. First‐line ceritinib versus platinum‐based chemotherapy in advanced ALK‐rearranged non‐small‐cell lung cancer (ASCEND‐4): A randomised, open‐label, phase 3 study. Lancet 2017;389:917–929. [DOI] [PubMed] [Google Scholar]
- 23. Xing P, Wang S, Hao X et al. Clinical data from the real world: Efficacy of crizotinib in Chinese patients with advanced ALK‐rearranged non‐small cell lung cancer and brain metastases. Oncotarget 2016;7:84666–84674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yoshida T, Oya Y, Tanaka K et al. Clinical impact of crizotinib on central nervous system progression in ALK‐positive non‐small lung cancer. Lung Cancer 2016;97:43–47. [DOI] [PubMed] [Google Scholar]
- 25. Heuckmann JM, Balke‐Want H, Malchers F et al. Differential protein stability and ALK inhibitor sensitivity of EML4‐ALK fusion variants. Clin Cancer Res 2012;18:4682–4690. [DOI] [PubMed] [Google Scholar]
- 26. Lin C, Shi X, Yang S et al. Comparison of ALK detection by FISH, IHC and NGS to predict benefit from crizotinib in advanced non‐small‐cell lung cancer. Lung Cancer 2019;131:62–68. [DOI] [PubMed] [Google Scholar]
- 27. Yang CY, Liao WY, Ho CC et al. Association between programmed death‐ligand 1 expression, immune microenvironments, and clinical outcomes in epidermal growth factor receptor mutant lung adenocarcinoma patients treated with tyrosine kinase inhibitors. Eur J Cancer 2020;124:110–122. [DOI] [PubMed] [Google Scholar]
- 28. Garon EB, Rizvi NA, Hui R et al. Pembrolizumab for the treatment of non‐small‐cell lung cancer. N Engl J Med 2015;372:2018–2028. [DOI] [PubMed] [Google Scholar]
- 29. Reck M, Rodriguez‐Abreu D, Robinson AG et al. Pembrolizumab versus chemotherapy for PD‐L1‐positive non‐small‐cell lung cancer. N Engl J Med 2016;375:1823–1833. [DOI] [PubMed] [Google Scholar]
- 30. Lin YT, Yu CJ, Yang JC et al. Anaplastic lymphoma kinase (ALK) kinase domain mutation following ALK inhibitor(s) failure in advanced ALK positive non‐small‐cell lung cancer: Analysis and literature review. Clin Lung Cancer 2016;17:e77–e94. [DOI] [PubMed] [Google Scholar]
- 31. Mezheyeuski A, Bergsland CH, Backman M et al. Multispectral imaging for quantitative and compartment‐specific immune infiltrates reveals distinct immune profiles that classify lung cancer patients. J Pathol 2018;244:421–431. [DOI] [PubMed] [Google Scholar]
- 32. Yoshida T, Oya Y, Tanaka K et al. Differential crizotinib response duration among ALK fusion variants in ALK‐positive non‐small‐cell lung cancer. J Clin Oncol 2016;34:3383–3389. [DOI] [PubMed] [Google Scholar]
- 33. Lin YT, Liu YN, Shih JY. The impact of clinical factors, alk fusion variants, and bim polymorphism on crizotinib‐treated advanced EML4–ALK rearranged non‐small cell lung cancer. Front Oncol 2019;9:880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lan B, Ma C, Zhang C et al. Association between PD‐L1 expression and driver gene status in non‐small‐cell lung cancer: A meta‐analysis. Oncotarget 2018;9:7684–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dudnik E, Peled N, Nechushtan H et al. BRAF mutant lung cancer: Programmed death ligand 1 expression, tumor mutational burden, microsatellite instability status, and response to immune check‐point inhibitors. J Thorac Oncol 2018;13:1128–1137. [DOI] [PubMed] [Google Scholar]
- 36. Sabari JK, Leonardi GC, Shu CA et al. PD‐L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann Oncol 2018;29:2085–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Koh J, Go H, Keam B et al. Clinicopathologic analysis of programmed cell death‐1 and programmed cell death‐ligand 1 and 2 expressions in pulmonary adenocarcinoma: Comparison with histology and driver oncogenic alteration status. Mod Pathol 2015;28:1154–1166. [DOI] [PubMed] [Google Scholar]
- 38. Zhang Y, Wang L, Li Y et al. Protein expression of programmed death 1 ligand 1 and ligand 2 independently predict poor prognosis in surgically resected lung adenocarcinoma. Onco Targets Ther 2014;7:567–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Evans M, O'Sullivan B, Hughes F et al. The clinicopathological and molecular associations of PD‐L1 expression in non‐small cell lung cancer: Analysis of a series of 10,005 cases tested with the 22C3 assay. Pathol Oncol Res 2020;26:79–89. [DOI] [PubMed] [Google Scholar]
- 40. Dietel M, Savelov N, Salanova R et al. Real‐world prevalence of programmed death ligand 1 expression in locally advanced or metastatic non‐small‐cell lung cancer: The global, multicenter EXPRESS study. Lung Cancer 2019;134:174–179. [DOI] [PubMed] [Google Scholar]
- 41. Shaw AT, Kim DW, Nakagawa K et al. Crizotinib versus chemotherapy in advanced ALK‐positive lung cancer. N Engl J Med 2015;373:1582. [DOI] [PubMed] [Google Scholar]
- 42. Sasaki T, Rodig SJ, Chirieac LR et al. The biology and treatment of EML4‐ALK non‐small cell lung cancer. Eur J Cancer 2010;46:1773–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Woo CG, Seo S, Kim SW et al. Differential protein stability and clinical responses of EML4‐ALK fusion variants to various ALK inhibitors in advanced ALK‐rearranged non‐small cell lung cancer. Ann Oncol 2017;28:791–797. [DOI] [PubMed] [Google Scholar]
- 44. Christopoulos P, Endris V, Bozorgmehr F et al. EML4‐ALK fusion variant V3 is a high‐risk feature conferring accelerated metastatic spread, early treatment failure and worse overall survival in ALK(+) non‐small cell lung cancer. Int J Cancer 2018;142:2589–2598. [DOI] [PubMed] [Google Scholar]
- 45. Chen N, Fang W, Zhan J et al. Upregulation of PD‐L1 by EGFR activation mediates the immune escape in EGFR‐driven NSCLC: Implication for optional immune targeted therapy for NSCLC patients with EGFR mutation. J Thorac Oncol 2015;10:910–923. [DOI] [PubMed] [Google Scholar]
- 46. Ota K, Azuma K, Kawahara A et al. Induction of PD‐L1 expression by the EML4‐ALK oncoprotein and downstream signaling pathways in non‐small cell lung cancer. Clin Cancer Res 2015;21:4014–4021. [DOI] [PubMed] [Google Scholar]
- 47. Bai Y, Chen X, Hou L et al. PD‐L1 expression and its effect on clinical outcomes of EGFR‐mutant NSCLC patients treated with EGFR‐TKIs. Cancer Biol Med 2018;15:434–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Guo R, Li Y, Bai H et al. KRAS mutants to regulate PD‐L1 expression through NF‐B and HIF‐1α pathways in non‐small cell lung cancer cells. J Clin Oncol 2017;35(suppl 15):e20049. [Google Scholar]
- 49. Xu C, Fillmore CM, Koyama S et al. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD‐L1 expression. Cancer Cell 2014;25:590–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sumimoto H, Takano A, Teramoto K et al. RAS‐mitogen‐activated protein kinase signal is required for enhanced PD‐L1 expression in human lung cancers. PLoS One 2016;11:e0166626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Smith MP, Sanchez‐Laorden B, O'Brien K et al. The immune microenvironment confers resistance to MAPK pathway inhibitors through macrophage‐derived TNFalpha. Cancer Discov 2014;4:1214–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Matsumoto Y, Sawa K, Fukui M et al. Impact of tumor microenvironment on the efficacy of epidermal growth factor receptor‐tyrosine kinase inhibitors in patients with EGFR‐mutant non‐small cell lung cancer. Cancer Sci 2019;110:3244–3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Feng PH, Yu CT, Chen KY et al. S100a9(+) MDSC and TAM‐mediated EGFR‐TKI resistance in lung adenocarcinoma: The role of RELB. Oncotarget 2018;9:7631–7643. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Supplemental Figures
Supplemental Tables