

Abstract
Drug regulators around the world make decisions about drug approvability based on qualitative benefit-risk analysis. In this work, a quantitative benefit-risk analysis approach captures regulatory decision-making about new drugs to treat multiple myeloma (MM). MM assessments have been based on endpoints such as time to progression (TTP), progression-free survival (PFS), and objective response rate (ORR) which are different than benefit-risk analysis based on overall survival (OS). Twenty-three FDA decisions on MM drugs submitted to FDA between 2003 and 2016 were identified and analyzed. The benefits and risks were quantified relative to comparators (typically the control arm of the clinical trial) to estimate whether the median benefit-risk was positive or negative. A sensitivity analysis was demonstrated using ixazomib to explore the magnitude of uncertainty. FDA approval decision outcomes were consistent and logical using this benefit-risk framework.
Regulatory decisions about drug approvability are benefit-risk assessments (as documented by representative quotations in Supplementary Table 1). Benefit-risk approaches1,2 have the potential to facilitate stakeholder decision-making throughout the drug development lifecycle, and thereby support the accessibility of safe and effective medicines to patients.3–5 As part of the Prescription Drug User Fee Act (PDUFA V) and US Food and Drug Administration Safety and Innovation Act (FDASIA), the FDA committed to implement a structured benefit-risk approach in assessments of new drug applications (NDAs) and biological license applications (BLAs). In prior work, a quantitative benefit-risk analysis approach was proposed6 and used for analysis of regulatory decision-making in non-small cell lung cancer (NSCLC).7
In this work, that approach is applied to multiple myeloma (MM). Analyzing benefit-risk for MM requires extension beyond the analysis applied to NSCLC. In particular, the NSCLC analysis used overall survival (OS) as a common metric of benefit. In MM, FDA decisions pertaining to drug approval8 are mostly based on non-OS metrics. Instead, they are based on metrics of time to progression (TTP), progression-free survival (PFS), and/or overall response rate (ORR). Although these metrics are not the only possible measures of benefit, they are commonly used by regulatory reviewers.
MM represents about 13% of hematological cancers and is the second most common hematologic malignancy.9,10 Estimated median OS for newly diagnosed patients was less than 3 years in the year 2000 and exceeds 6 years in 2016.11
APPROACH
Candidate treatments and regulatory decisions
Regulators often communicate their decisions to approve or not approve a drug in terms of benefits and risks (see Supplementary Table 1 for representative quotations from regulatory review documents). A major focus of this work was to develop understanding of the extent to which benefit and/or risk analysis align with recent decisions made by the FDA. The candidate treatments considered in this work are indicated for MM. This analysis was based on publicly available details of clinical trials submitted to the FDA between 2003 and 2016 in support of applications for New Drug Applications (NDAs) or Biologics License Applications (BLAs). Details of these cases are tabulated in Supplementary Table 2 and are depicted in Supplementary Figure 1.
Twenty-one pivotal trials (23 decisions because of two trials with multiple drug arms for comparison) were identified spanning both first-line and nonfirst-line MM treatments as shown in Table 1. The clinical trial information was available either within the Medical Reviews11–22 or in publications.23–40 Although FDA decisions are intended to be based on the totality of evidence, the focus of this work is on the pivotal trials, given their relative importance in decision-making.
Table 1.
Estimated Benefits and Risks of MM for First-line Drugs and Non-First-line Drugs
| Estimated benefit (delay in progression) [months] | Estimated risk [months] | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Drug product | Control | Drug product | Control | ||||||
| Median (LCL, UCL) | Median (LCL, UCL) | Median (LCL, UCL) | Median (LCL, UCL) | ||||||
| First Line | |||||||||
| Thai idomide + Dexamethasone* | 1L | 2006 | Tha+Dex-lL-06* | ORR | Accelerated Approval | 12.82 (9.56, 16.07) | 7.45 (4.35, 10.56) | 1.17 (0.83, 1.51) | 1.36(0.91, 1.8) |
| Bortezomib+MP* | 1L | 2008 | Bor+MP-lL-08* | TTP | Approved | 20.9 (17.6, 24.7) | 15 (14.1, 17.9) | 4.09 (3.01, 5.16) | 5.19 (3.82, 6.55) |
| Thalidomide + Dexamethasone* | 1L | 2008 | Tha+Dex-lL-08* | TTP | Accelerated Approval | 24.43 (15.47, 26.51) | 7.08(6.93, 9.11) | 3.04(2.16, 3.92) | 2.3 (1.5, 3.11) |
| Lenalidomide+Dexamethasone (RD)* | 1L | 2014 | Len+Dex-1L-14* | PFS | Approved | 25.5 (20.7, 29.4) | 21.2 (19.3, 23.2) | 5.49 (4.45, 6.52) | 4.72 (3.65, 5.8) |
| Non-first Line | |||||||||
| Bortezomib* | 3L | 2003 | Bor-3 L-03* | ORR | Accelerated Approval | 6.6 (4.38, 8.82) | 1.9 (1.9, 2.2) | 0.18 (0.13,0.25) | 0.56(0.26, 0.85) |
| Bortezomib* | 2L | 2005 | Bor-2 L-05* | TTP | Approved | 6.2 (4.9, 6.9) | 3.5 (2.8, 4.2) | 1.26(1.06, 1.45) | 0.95 (0.75, 1.15) |
| Liposomal Doxorubicin+ Bortezomib* | 2L | 2007 | Dox+Bor-2L-07* | TTP | Approved | 9.3 (8.33, 11.27) | 6.5(5.67, 7.23) | 0.55 (0.24, 0.86) | 0.51(0.21, 0.82) |
| Lenalidomide+Dexamethasone* | 2L | 2007 | Len+Dex-2L-MM09–07* | TTP | Approved | 11.1(8.68, 13.52) | 4.7(2.71, 6.69) | 0.81(0.65,0.99) | 0.6 (0.47, 0.83) |
| Lenalidomide+Dexamethasone* | 2L | 2007 | Len+Dex-2L-MM10–07* | TTP | Approved | 13.1(10.23, 21.4) | 5.03 (4.15, 5.18) | 0.77 (0.58, 0.96) | 1.08 (0.56, 1.6) |
| Carfilzomib* | 3L | 2012 | Car-3 L-12* | ORR | Accelerated Approval | 3.7 (2.8, 4.6) | 1.9 (1.9, 2.2) | 0.25 (0.18, 0.32) | 0.56(0.26, 0.85) |
| Carfilzomib+Lenolidomide+ Low Dose Dexamethasone* | 2L | 2014 | Car+Len+Dex-2L-14* | PFS | Approved | 26.3 (23.3, 30.5) | 17.6(15,20.6) | 1.49 (1.12, 1.86) | 1.83 (1.19, 2.47) |
| Panobinostat+Bortezomi b+ Dexamethasone* | 2L | 2014 | Pan + Bor+Dex-2L-14* | PFS | Indication Refined | 9.9 (8.3, 11.3) | 7.7 (6.9, 8.5) | 3.21(2.25,4.16) | 1.48 (0.91, 2.05) |
| Panobinostat+Bortezomib+ Dexamethasone* | 3L | 2015 | Pan + Bor+Dex-3L-15* | PFS | Accelerated Approval | 10.6 (7.6, 13.8) | 5.8 (4.4, 7.1) | 2.31(1.05,3.56) | 1.25 (0.41, 2.08) |
| Carfilzomib+Dexamethosone* | 2L | 2016 | Car+Dex-2L-16* | PFS | Approved | 18.7 (15.6, 19.96) | 9.4 (8.4, 10.4) | 1.24 (0.90, 1.58) | 1.39 (0.90, 1.87) |
| Pomalidomide* | 3L | 2012 | Pom-3 L-12* | ORR | Accelerated Approval | 2.5 (1.9, 3.7) | 1.9 (1.9, 2.2) | 0.62 (0.36, 0.88) | 0.56(0.26, 0.85) |
| Pomalidomide + dexamethasone* | 3L | 2012 | Pom+ Dex-3 L-12* | ORR | Not Used | 3.8 (3.2, 4.9) | 1.9 (1.9, 2.2) | 0.62 (0.36, 0.89) | 0.56(0.26, 0.85) |
| Pomalidomide + Dexamethasone (Low Dose)* | 3L | 2014 | Pom + Dex-3L-14* | PFS | Approved | 4(3.6, 4.7) | 1.9 (1.9, 2.2) | 0.38 (0.26, 0.5) | 0.56(0.26, 0.85) |
| Daratumab 16 mg* | 4L | 2015 | Dar-4L-MMY-15* | ORR | Accelerated Approval | 5.29 (2.37, 8.21) | 1.9 (1.9, 2.2) | 0.23 (0.12, 0.34) | 0.56(0.26, 0.85) |
| Daratumumab 16 mg* | 4L | 2015 | Dar-4L-GEN-15* | ORR | Accelerated Approval | 7.49 (2.6, 12.37) | 1.9 (1.9, 2.2) | 0.23 (0.12, 0.34) | 0.56 (0.26, 0.85) |
| Daratumumab+Bortezomib+Dexamethasone* | 2L | 2016 | Dar+Bor+Dex-2L-16* | PFS | Approved | 23.41(21.84, 24.98) | 7.2 (6.2, 7.9) | 2.21 (1.26, 3.16) | 2.41 (1.24, 3.58) |
| Daratumumab+Lenalidomide+Dexamethasone* | 2L | 2016 | Dar+Len + Dex-2L-16* | PFS | Approved | 26.78(25.78, 27.79) | 18.4(13.9, 20.07) | 3.39 (2.11, 4.68) | 3.23 (1.9, 4.56) |
| Elotuzumab+LD* | 2L | 2015 | Elo+Len + Dex-2L-15* | PFS, ORR | Approved | 19.4(16.6, 22.2) | 14.9 (12.1, 17.2) | 2.6(1.95,3.25) | 3.31 (2.31, 4.32) |
| Ixazomib + LD* | 2L | 2015 | lxa + Len + Dex-2L-15* | PFS | Approved | 20.6 (17, 22.04) | 14.7 (12.9, 17.6) | 1.53 (0.94, 2.13) | 2.2 (1.4, 2.99) |
indicates a pivotal trial.
Confidence intervals are 95% confidence intervals. Date field shows year of publication of the reference. In the “Design” field in the table above “H-H” means Head-to-Head trial; “A-O” means Add-on trial; “SA” means Single Arm trial. Panobinostat plus Bortezomib plus Dexamethasone analyses of 2014 and 2015 are both based on the same trial 2308 (as mentioned in the Study column in the table above). However, the 2015 analysis is for a subpopulation.
Benefit, risk, and uncertainty
Benefits.
A benefit is a desirable (or positive) effect of a therapy (e.g., increase in duration of life and/or in quality of life6,7). A benefit typically corresponds to an increase in the area-under-the-curve (AUC) associated with quality of life profile over time for a patient.
Unlike most solid and some hematologic malignancies, where progression/response of disease is usually determined by observed changes in tumor size, MM response is usually determined by changes in a blood biomarker that myeloma cells secrete (monoclonal protein) and percentage of monoclonal plasma cells observed in the bone marrow.41
The 21 pivotal trials are described below and are depicted in Supplementary Figure 1:
- ORR as the primary endpoint: Of the six pivotal trials where ORR was the primary endpoint, two were randomized trials, out of which one did not isolate the treatment effect of the experimental agent. The remaining four were single-arm trials. The MM-002 trial had pomalidomide in both of its arms, so each arm was analyzed as if it were a single arm trial.
- Hypothetical control arms for single-arm trials: The controls used for each trial are listed in Table 1. The dexamethasone control for pomalidomide plus dexamethasone in trial MM-00316 was used as the hypothetical control for all single-arm trials, because MM-003 was the only randomized trial in the third or higher line of therapy (consistent with the experimental drugs) and MM-003 used a common therapy as its control arm (dexamethasone).
- Experimental drug arms:
- Three of the six trials reported TTP or PFS as a secondary endpoint. For these, median TTP or PFS was used directly from the trial.
- Three out of the six trials did not report PFS or TTP as a secondary endpoint. For these, PFS benefit was estimated using an ORR to PFS correlation that is described later in this section.
- PFS or TTP as the primary endpoint: There were control arms in all 15 pivotal trials where PFS (nine trials) or TTP (six trials) was the primary endpoint.
- Control arms: The primary benefit considered is delay in progression (PFS or TTP), which is generally not significantly impacted by crossover.42 Therefore, PFS or TTP of control arms are used as reported from the trials.
- Experimental drug arms: The above 15 pivotal trials together contained 17 experimental drug arms. Fifteen of these trial arms reported PFS or TTP as the primary endpoint, and median PFS or TTP was used as reported from the trial. In two of these arms, median PFS was not reached at the time of reporting of results.
The linear correlation between ORR and PFS (number of points = 12; R2 = 0.85) was determined using data from all experimental drug arms (weighted based on number of patients) that had ORR or PFS as the primary endpoint and reported both ORR and PFS. This correlation was used to estimate PFS in trials where PFS was not reported and ORR was reported. The full list of these trials is shown in Supplementary Table 2 (with pivotal trials designated by an asterisk). The resulting data used to generate the correlation are shown in the scatterplot of Supplementary Figure 3a(i). Similar correlation approaches were considered without weighted averages (Supplementary Figure 3a(ii) with 12 points, R2 = 0.88) and with control arms included (Supplementary Figure 3a(iii) with 22 points, R2 = 0.75). Similarly, correlations between ORR and OS, and between PFS and OS, were determined using each of these approaches (see Supplementary Figure 3b,c) with the data from weighted average experimental drug arms used to estimate OS in trials where OS was not reported.
Risks.
Harm connotes an undesirable (or negative) effect (e.g., headache, dizziness). For a population, the relative importance of harm is dependent on the severity and frequency of that harm. As in prior work6,7 and similar to the CTCAE severity grades, harms are categorized into three types: death; nonfatal serious adverse events; and common adverse events. Each of these categories can be associated with a percent reduction in area under the quality of life vs. time curve, where the percent reductions used in this work are the same as in prior work6,7 along with previously highlighted limitations43–45 such that impact on the quality of life of an untreated patient ranges from 100% reduction for death to 10% reduction for nonfatal serious adverse events, and 0.5% for common adverse events.
Specific to the use of TTP and PFS, the time horizon used to quantify any deaths that occur in the clinical trial is the remaining life expectancy of the patient in the control arm of that clinical trial (which is either reported in the clinical trial results or is estimated OS from a relevant correlation described in the Benefits section). The time horizon used to quantify the seriousness of nonfatal events is the duration of treatment in the corresponding arm of that clinical trial. As with the prior work, reported adverse events (or harms) for all patients in each trial were aggregated into an overall measure of median risk.
An example of risk assessment for ixazomib is shown in Supplementary Table 3. The assessment of the ixazomib arm compared to the control arm uses delay in disease progression as benefit. For risk, the seriousness of death due to toxicity is taken as the estimated OS in the control arm, which is 39.81 months. The number of deaths in the ixazomib arm due to toxicity has been estimated to be 9 out of the 360 patients in that arm (2.5% of patients in that arm). Applying the normal approximation of the binomial distribution (as described in the Uncertainty section, below), the upper and lower confidence limits of the proportion of death due to toxicity in the ixazomib arm are estimated to be 4.1% and 0.9%, respectively. The risk of death is the product of the seriousness (39.81 months) and occurrence of death (2.5%) and is thus estimated to be 1.0 months in the ixazomib arm. Similarly, the upper and lower confidence limits of the risk of death in the ixazomib arm are estimated as 1.6 and 0.35 months, respectively.
The seriousness of nonfatal serious adverse events is estimated as 10% of the duration of treatment (13 months). The seriousness of common adverse events is estimated as 0.5% of the duration of treatment multiplied by the estimated number of common adverse events per patient. Since it is assumed that a patient who experiences a nonfatal serious adverse event can also undergo common adverse events, the seriousness associated with adverse events, both nonfatal serious and common, is estimated as the sum of the seriousness of nonfatal serious and common adverse events. Thus, the seriousness of the adverse events experienced by a patient who undergoes nonfatal serious adverse events in the ixazomib arm is estimated as (0.10+2.3*0.005)*13 months = 1.5 months. The proportion of patients undergoing nonfatal serious adverse events in the ixazomib arm is estimated as 40%. Thus, the risk of adverse events in a patient undergoing nonfatal serious adverse events in the ixazomib arm is estimated as 1.45*0.4 = 0.6 months.
The seriousness of a common adverse event is estimated, as described above, in the ixazomib arm as (2.3*0.005)*13 = 0.15 months. The proportion of patients who experience only common adverse events is the fraction that did not die due to toxicity and who did not undergo serious adverse events. In this case that is 100% − 2.5% − 40% = 57.5% in the ixazomib arm. Thus, the risk of adverse events in a patient undergoing only common adverse events = 0.15 months*0.575 = 0.1 months in the ixazomib arm.
Thus, the total risk for a patient in the ixazomib arm = 1 + 0.6 + 0.1 = 1.7 months. The total risk in the ixazomib arm adjusted for difference in the duration of treatment with respect to the control = total risk * (duration of treatment in control arm)/(duration of treatment in the ixazomib arm) = 1.7*12/13 = 1.6 months. Since the above adjustment is a normalization by the treatment duration in the control arm, such an adjustment is required only in the ixazomib arm and not the control arm. Similarly, the total risk for a patient in the control (lenalidomide in combination with dexamethasone) arm is estimated as 2.2 months.
For analyses of decisions based on OS estimates taken as the benefit, the following changes are made in the risk assessment described above (for both the experimental drug and control arms): the estimated OS in the control arm is used as the potential duration of treatment in the nonfatal adverse events; and the total risk adjusted for potential duration of treatment is obtained by multiplying the total risk by the ratio of estimated OS of the control arm to the actual duration of treatment in the clinical trial. Thus, for the analysis of decisions based on estimated OS taken as the benefit, the seriousness of adverse events in patients who undergo nonfatal serious adverse events in the ixazomib arm is estimated as (0.10+2.3*0.005)*39.81 = 4.4 months, and in the control arm as (0.10+1.9*0.005)*39.81 = 4.4 months; the seriousness of adverse events in patients who undergo only common adverse events is estimated as (2.3*0.005)*39.81 = 0.5 months in the ixazomib arm and (1.9*0.005)*39.81 = 0.4 months in the control arm; and the risk adjusted for duration of treatment in the ixazomib arm is estimated as (39.81/13)*2.03 = 6.2 months in the ixazomib arm and (39.81/12)*2.15 = 7.1 months in the control arm.
Note that the intrinsic risk of birth defects associated with certain MM drugs are not reflected in the risk assessments, because Risk Evaluation and Mitigation Strategy (REMS) have already been applied to certain classes of drugs.
Uncertainty.
Uncertainty can be an important factor in review of clinical trial data, as described in prior work.6,7 The uncertainty associated with the estimation of OS from ORR or PFS, and the estimation of PFS from ORR, is particularly relevant here. In cases where confidence intervals were not reported in the clinical trial reports for ORR, the normal approximation method for 95% confidence interval was used, as shown below:
where n is the sample size and p is the proportion of interest.
For example, the ORR of the Thalidomide+Dexamethasone arm of study E1A0014 is 51.5%. Since the upper and lower confidence limits of the ORR are not available in the corresponding reference,14 these have been estimated using the above equations as follows:
The corresponding confidence limits in the estimated OS in the Thalidomide+Dexamethasone arm can then be estimated by multiplying those confidence limits by the equation relating ORR to estimate OS, resulting in confidence intervals of 67.6 months around the estimated OS of 18.9 months.
SUMMARY AND DISCUSSION
The estimated benefit, as measured by median TTP or PFS as described in the Approach section and the corresponding hazard ratio, associated with each of the MM drugs along with their corresponding confidence intervals, are shown in Table 1 for the pivotal trials.
The calculated overall risk, as measured by a weighted summation of individual harms reported as described in prior work,6,7 associated with each of the MM drugs and corresponding confidence intervals, are shown in Table 1. The risk associated with each of the severity groups (fatalities, nonfatal serious adverse events, and common adverse events) is listed in Supplementary Table 3.
In general, the experimental set-ups and the control arms are not described in detail in the tables and are potentially significantly different between the different trials.
Benefits, risks, and decisions: By line of treatment
A graphical representation of the benefit, risk, and decisions associated with the MM drugs are shown in Figure 1 for (a) first-line treatments and (b) subsequent lines of treatment. Additional graphs categorized based on primary endpoint are shown in Supplementary Figure 2a (trials with PFS as primary endpoint), Supplementary Figure 2b (trials with TTP as primary endpoint), Supplementary Figure 2c (trials with ORR as primary endpoint). Supplementary Figure 5a,b shows benefit as measured by median PFS and TTP compared to benefit as measured by hazard ratio. A similar association is observed between the two measures of benefit (median PFS/TTP and hazard ratio) in the separation of approvals and nonapprovals.
Figure 1.
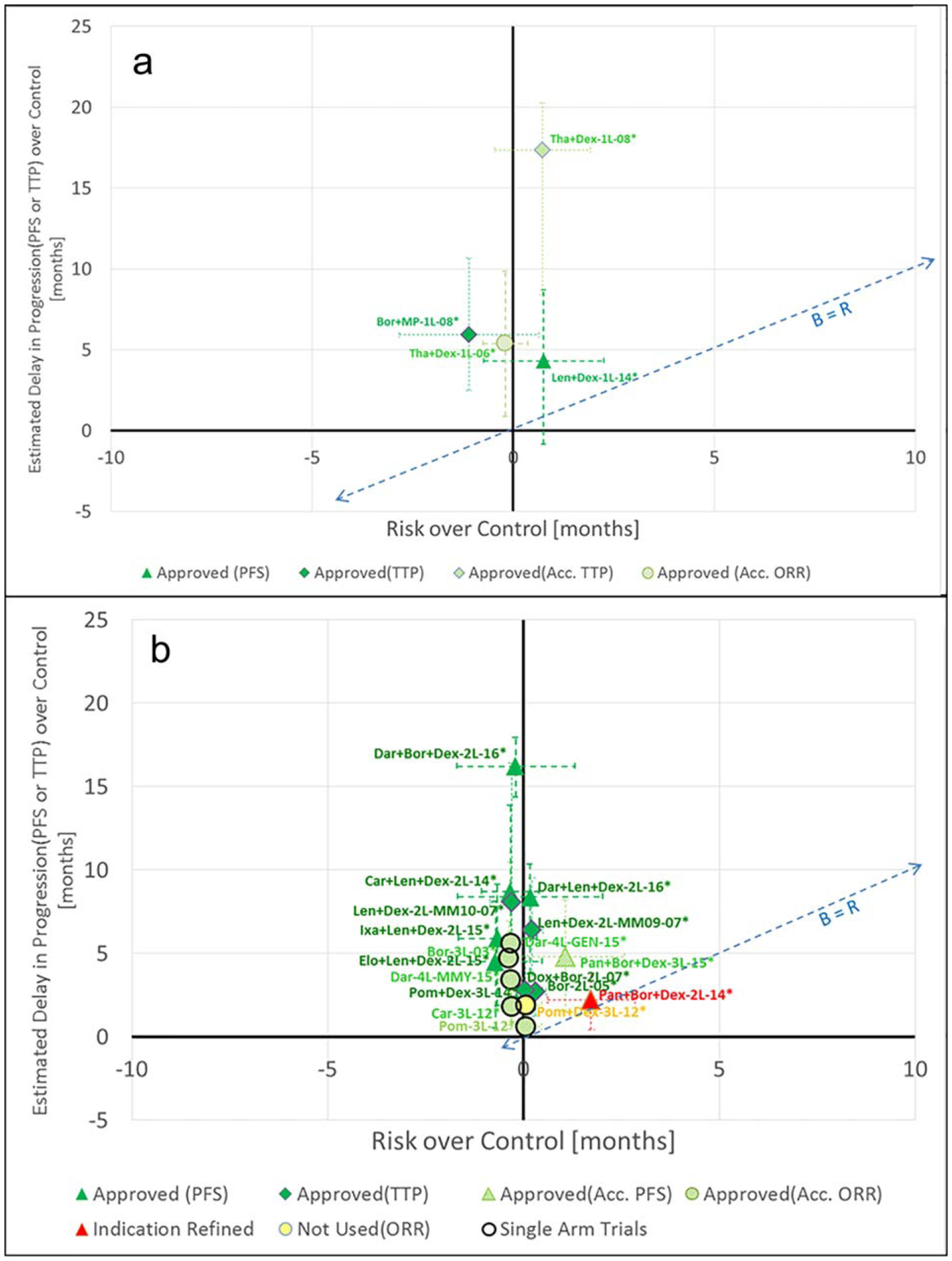
Analysis of benefits and risks (a). First-line (b). Nonfirst-line. The naming convention for the points in the chart above is as follows: NNN-LL QQQQ-YY; where: NNN represents the first three letters in the name of the drug (e.g., “Len” for lenalidomide; LL represents the line of treatment (e.g., 1L indicating lenalidomide used as the first-line of treatment); QQQQ (if needed) is a description of a relevant subpopulation and/or specific pivotal trial; YY represents the year associated with the study/decision pertaining to the data point. The PFS improvement shown for the point Pan+Bor+Dex-2L-14* in Figure 1b is based on an assessment by an Independent Review Committee (IRC).
The angled dotted lines in Figure 1a and in Figure 1b are lines drawn at the benefit equal to risk values. The approved drugs were found to be distinguishable in median benefit minus risk compared to their control arms. Within their confidence intervals, the approved drugs exceed the benefit equal to risk line, meaning that they demonstrated positive benefit-risk. The confidence intervals of the drug that was not approved fall below this line, meaning that it did not demonstrate positive benefit-risk with a high level of confidence based on the analysis in this work. This is consistent with the use of benefit-risk as a criterion for regulatory decision-making and is consistent with the benefit-risk results in the NSCLC analysis.7
Among the nonfirst-line treatments (Figure 1b), panobinostat (2L, 2014) is the drug that was not approved for its original indication of relapsed refractory MM. It was subsequently approved for use in a targeted population of patients who have received at least two prior therapies, including bortezomib and an immuno-modulatory agent. Panobinostat plus bortezomib and dexamethasone for second-line treatment of MM is a case where a 4-month improvement in median PFS over the control was not sufficient to overcome the potential risk of toxicity.18 This could be explained by a combination of multiple factors, including the following18: benefit not outweighing risk, or uncertainty in benefit due to missing response data. See Supplementary Table 1 for the relevant quotations from the FDA medical review.
The primary endpoint of the pivotal trial for lenalidomide plus dexamethasone was PFS. In that trial, lenalidomide in combination with dexamethasone demonstrated a 4-month PFS advantage over the control, but the lower confidence interval of median PFS over control fell below the zero net benefit line. Despite this characteristic of median PFS, the hazard ratio for PFS was statistically significant. The improvement in median PFS over control may have been affected by nonproportionate hazards in the Kaplan-Meier curves for PFS. The experimental drug arm demonstrated a 10-month OS advantage over the control.31 This advantage in OS may have been taken into account in the regulatory decision to approve lenalidomide in combination with dexamethasone as a first-line treatment for MM.
Nonfirst-line drugs are shown in Figure 1b. Similar to the first-line drugs, the drugs that were approved by the FDA were found to have confidence intervals of their median PFS benefit-risk above the parity line. This is indicative of the approved drugs offering statistically significant incremental benefit over incremental risk.
Benefit-risk and decision analysis example: Ixazomib in combination with lenalidomide and dexamethasone (first-line)
Use of the benefit-risk analysis described above to understand decision-making for a drug is illustrated for ixazomib plus lenalidomide plus dexamethasone. Figure 2 shows the details of the benefit-risk balance of ixazomib plus lenalidomide plus dexamethasone based on clinical trial C16010.46 Detailed computations for this clinical trial are shown in Supplementary Table 5. Details of how the existing FDA Benefit-Risk Summary Assessment1 is updated with the benefit-risk approach used in this work (updates from this work are noted with italicized text) are shown in Supplementary Table 4.
Figure 2.
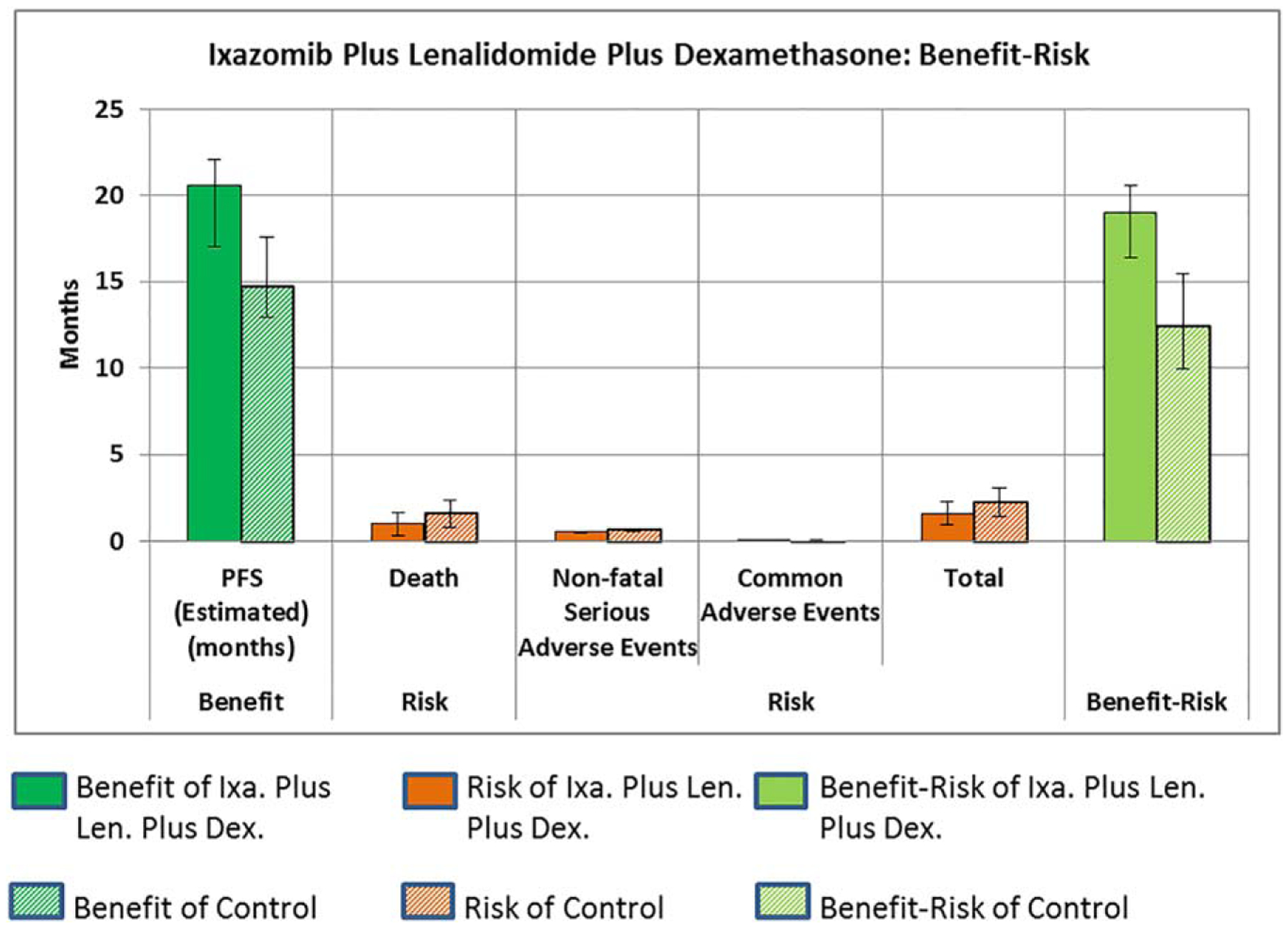
Benefit-risk of ixazomib plus lenalidomide plus dexamethasone.
Sensitivity analysis of benefit-risk over control for ixazomib in combination with lenalidomide and dexamethasone.
The approach to sensitivity of the benefit-risk of a drug is as discussed in prior work.6,7
The factors considered in the sensitivity analysis, their range of variation, and associated rationale based on information available46 is described in Table 2. The ranges of values of parameters are intended to reflect the potential effects of variation in many parameters specific to patient and trial variation. The resulting sensitivity analysis assessing the impact on overall benefit-risk of ixazomib plus lenalidomide plus dexamethasone over control is depicted in Figure 3. The sensitivity analysis of ixazomib plus lenalidomide plus dexamethasone, within the ranges of variation specified for the parameters of the analysis, results in a positive net benefit minus risk (over control).
Table 2.
Sensitivity analysis of benefit-risk over control: ixazomib plus lenalidomide plus dexamethasone
| Range of variation | Basis of selection of range | |||||
|---|---|---|---|---|---|---|
| Low | Expected | High | Factor | Detail | ||
| Benefit | Estimated PFS improvement [months] | 4.42 | 5.90 | 7.38 | Population heterogeneity | Variation in estimated PFS of Ixazomib and Control corresponding to ± 50% variation in proportion of population with no prior stem cell transplant (± 0.45 months for Ixazomib and ± 1.41 months for Control) |
| Risk Death | Severity [months] | 36.86 | 39.81 | 42.76 | Natural history | Variation in life expectancy for Control corresponding to ± 50% variation in proportion of population with stem cell transplant (± 2.95 months) |
| Occurrence relative to control | −0.037 | −0.014 | 0.009 | Exposure | Attributability to treatment (± 50% of occurrence adjusted for treatment duration) | |
| −0.037 | −0.014 | 0.009 | Attributability | Attributability to treatment (± 50% of occurrence adjusted for treatment duration) | ||
| Nonfatal serious AEs | Severity [months] | 0.88 | 1.32 | 2.62 | Estimation of seriousness | Variation of ± 1/2 grade of severity of AE |
| Occurrence relative to control | −0.347 | −0.047 | 0.253 | Exposure | Attributability to treatment (± 50% of occurrence adjusted for treatment duration) | |
| −0.347 | −0.047 | 0.253 | Attributability | Attributability to treatment (± 50% of occurrence adjusted for treatment duration) | ||
| Common AEs | Severity [months] | 0.1045 | 0.1479 | 0.2779 | Estimation of seriousness | Variation of ± 1/2 grade of severity of AE |
| Occurrence relative to control | −1.000 | −1.680 | −1.227 | Exposure | Attributability to treatment (± 50% of occurrence adjusted for treatment duration) | |
| −1.000 | −1.680 | −1.295 | Attributability | Attributability to treatment (± 50% of occurrence adjusted for treatment duration) | ||
Figure 3.
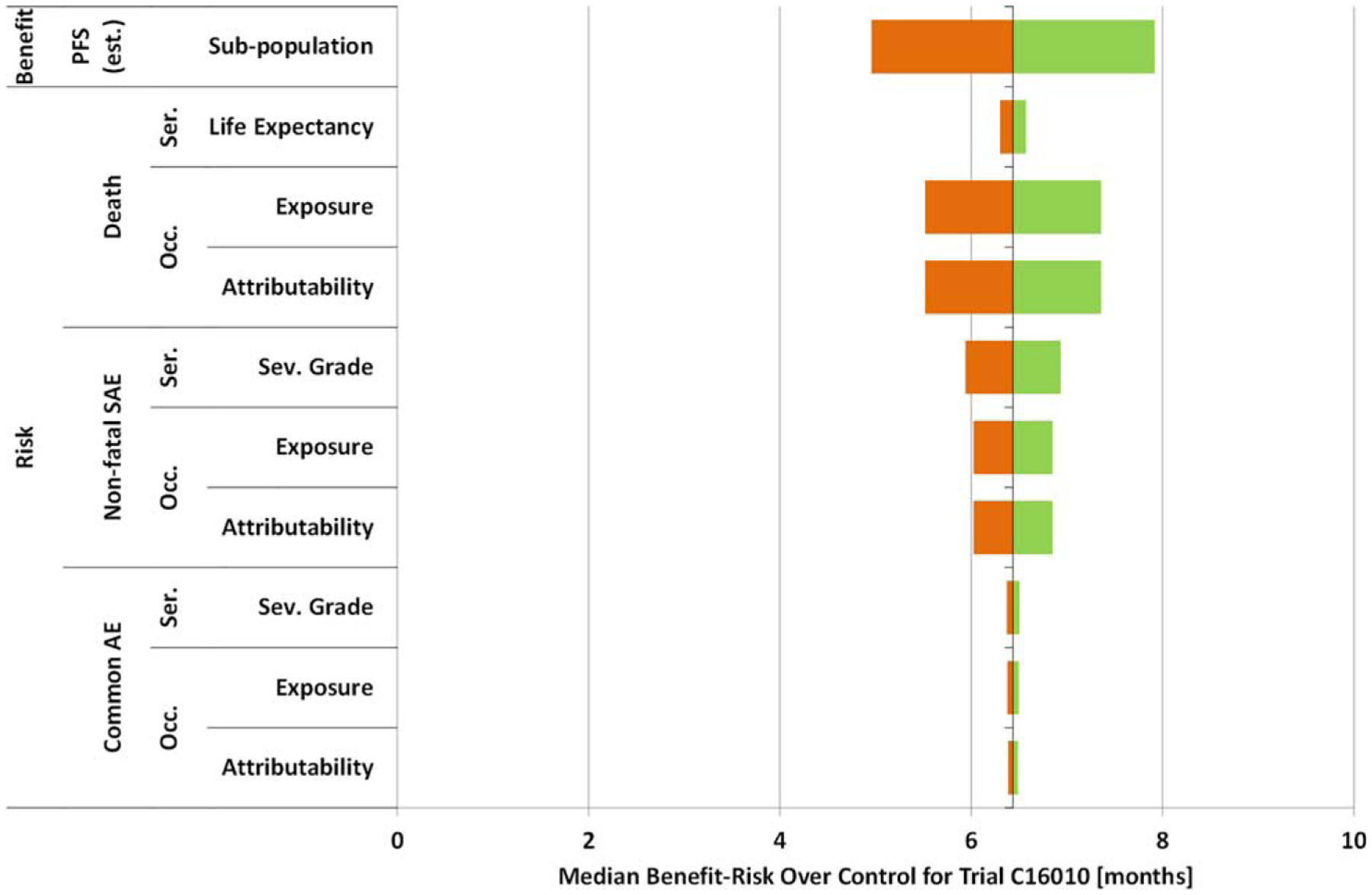
Sensitivity analysis of benefit-risk over control: ixazomib plus lenalidomide plus dexamethasone.
Evolution of benefit minus risk from MM treatments
In an attempt to connect directly to final patient outcomes, an estimated OS-based benefit minus risk assessment of MM therapies was also conducted. This used correlations between the endpoints mentioned previously (ORR, PFS, TTP) and OS. Each value of ORR, PFS, or TTP was converted to an equivalent estimated OS. Supplementary Tables 6, 7 show where the benefit and risk considered are based on estimated OS. Supplementary Table 8 shows an associated risk analysis with the risks also scaled to the estimated OS for each trial. When the benefit considered is estimated OS, the risk assessment is calculated as in prior work,6,7 wherein: the risk is based on nonfatal serious adverse events and common adverse events (timing of death is not counted again in the risk) and the time horizon for quantifying risk is the estimated OS of the control arm of the trial. Supplementary Figure 4a,b shows resulting estimated OS and risk for first-line and nonfirst-line drugs, respectively. The results shown in Supplementary Figure 4a,b are in alignment with the PFS- or TTP-based analysis as shown in Figure 1a,b.
Figure 4 shows the evolution of the estimated median OS minus risk for treatment outcome of MM drugs over time. The blue line is the maximum estimated median OS minus risk for first-line MM patients for a given year based on survival data available at the time the clinical trials were analyzed (not based on long-term follow-up) and therefore may be different from other estimations of OS. Step increases in the blue line correspond to improvements within a given line of treatment. For example, the Bor-3L-03 drug arm has 14.6 months of estimated OS minus risk, which is an increase of 8.7 months over the estimated OS minus risk of the third-line treatment (5.9 months) prior to 2003 (assumed to be the same as that of dexamethasone control of the experimental drug arm of pomalidomide plus dexamethasone in the MM003 trial). So the gain of 8.7 months is added to the blue line. As another example, Tha+Dex-1L-08 has 52.7 months of estimated OS minus risk, which is not surpassed by that of any other trial in 2008 considered in this analysis nor by the estimated OS minus risk shown by the blue line prior to 2008 (46 months). Therefore, the estimated OS minus risk of Tha+Dex-1L-08 is taken as the new level of the blue line at 2008. Overall, there is a significant increase in estimated OS benefit minus risk (from about 2.5 years to 7.5 years) from 2001 to 2016. A similar chart for estimated median OS (without subtracting risk) is shown in Supplementary Figure 6.
Figure 4.
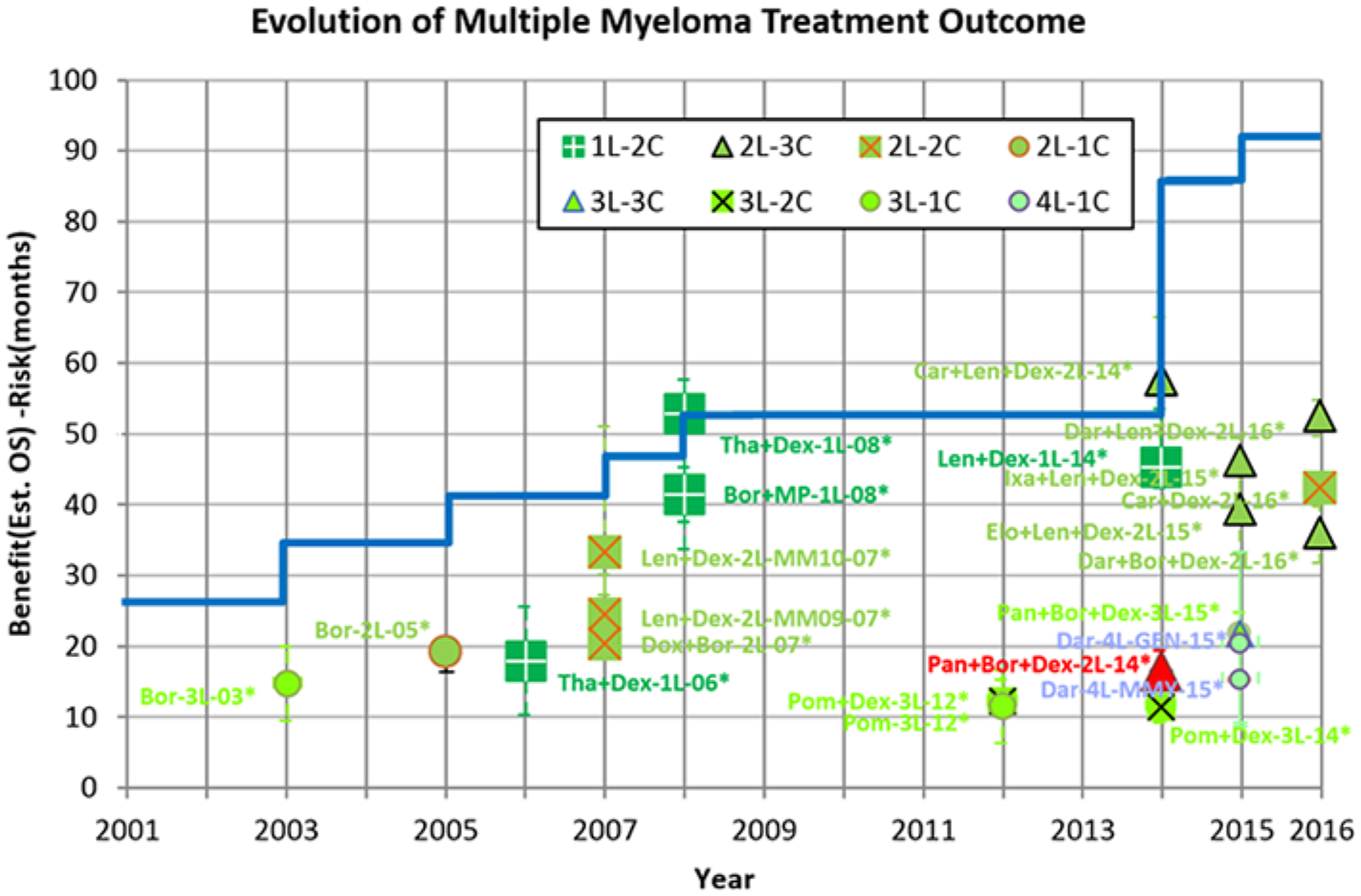
Evolution of estimated clinical outcome of OS benefit-risk over time for MM patients. The starting point for the blue line at year 2001 is taken as the estimated median OS of newly diagnosed MM patients prior to 199647 with the ratio of risk to benefit the same as that of thalidomide plus dexamethasone.30
The benefit-risk approach applied in prior work6,7 was extended to capture regulatory decision-making for MM drugs, with a focus on use of biomarkers as the metric of benefit. When trial data were transformed into common metrics for benefit and risk, FDA approval decisions in this disease were found to have consistent benefit-risk logic.
Limitations of this approach are discussed elsewhere.6,7,48 Limitations especially applicable to this work include those pertaining to: estimations of PFS and OS based on correlations; effect of crossover on OS; and use of median time-to-event metrics (e.g., median PFS). An additional limitation is the use of the two similar but not identical metrics of PFS and TTP. However, these metrics reflect those used in the evaluation of clinical trials.
The limitation of using median PFS as benefit is exemplified by the assessment of lenalidomide plus dexamethasone (first-line),31 wherein nonproportionate hazards in the Kaplan-Meier curves of PFS are likely to have affected the median PFS over the control. The panobinostat 2L case illustrates the impact of a combination of potentially high-risk and systematic uncertainty about benefit (apparently some missing response data) on the regulatory decision.
Modern combination therapies seem to have enabled a significant increase in median overall survival since the start of the 21st century.48,49 This could be further characterized through the use of natural history studies. If regulatory decisions had to wait for OS results prior to approval of MM drugs, the estimated improvement in median OS minus risk shown in Figure 4 is unlikely. Greater use of biomarkers such as minimal residual disease (MRD)48,50,51 that can be used in place of currently established clinical trial endpoints for drug approval may be beneficial to further speed drug development and availability of new treatments of MM.
FDA approval decision outcomes considered were found to embody a consistent benefit-risk logic. The availability of new treatments has enabled a significant increase in median estimated overall survival minus risk since the start of the 21st century.
Supplementary Material
ACKNOWLEDGMENTS
The opinions expressed in the article are those of the individual contributors and not necessarily those of the U.S. Food and Drug Administration. This work is partially funded by the U.S. Food and Drug Administration. Dr. Landgren thanks Memorial Sloan Kettering Core Grant, Core Grant (P30 CA008748) for grant support.
Footnotes
Additional Supporting Information may be found in the online version of this article.
CONFLICT OF INTEREST
The authors declared no conflicts of interest.
References
- 1.Frey P Benefit-Risk Framework (U.S. Food and Drug Administration, CDER, Silver Spring, MD, 2012). [Google Scholar]
- 2.U.S. Food and Drug Administration. Structured Approach to Benefit-Risk Assessment in Drug Regulatory Decision-Making. Washington, DC; 2013. [Google Scholar]
- 3.U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Health Statistics Health, United States, 2010: With Special Feature on Death and Dying. Hyattsville, MD; 2011. [Google Scholar]
- 4.Morris S, Rosenblatt M, Orloff J, Lewis-Hall F & Waldstreicher J The PCAST report: impact and implications for the pharmaceutical industry. Clin. Pharmacol. Ther 94, 300–302 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Woodcock J The PCAST Report on Pharmaceutical Innovation: implications for the FDA. Clin. Pharmacol. Ther 94, 297–300 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Raju GK, Gurumurthi K & Domike R Benefit-risk analysis for decision-making: an approach. Clin. Pharmacol. Ther 100, 654–671 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Raju GK et al. A Benefit-risk analysis approach to capture regulatory decision-making: non-small cell cancer. Clin. Pharmacol. Ther 100, 672–684 (2016). [DOI] [PubMed] [Google Scholar]
- 8.U.S. Department of Health and Human Services, U.S. Food and Drug Administration Expedited Programs for Serious Conditions — Drugs and Biologics. Washington, DC; 2013. [Google Scholar]
- 9.Moreau P, Attal M & Facon T Frontline therapy of multiple myeloma. Blood 125, 3076–3084 (2015). [DOI] [PubMed] [Google Scholar]
- 10.American Cancer Society. Survival rates by stage for multiple myeloma. [Online]. 2016 [cited 2016 Feb 16]. Available from: <http://www.cancer.org/cancer/multiplemyeloma/detailedguide/mulitiple-myeloma-survival-rates>
- 11.U.S. Food and Drug Administration. Drug Approval Package; Empliciti (Elotuzumab): Company: Bristol-Myers Squibb; Application No.: 761035Orig1s000; Approval Date; 11/30/2015. [Online]. 2016. [cited 2016 Feb 16]. Available from: <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/761035Orig1s000TOC.cfm>
- 12.U.S. Food and Drug Administration. Drug Approval Package; Velcade (Bortezomib) Injection; Company: Millenium Pharmaceuticals; Application No.: 021602; Approval Date: 5/13/2003 [Online]. 2003. [cited 2015 June 9]. Available from: <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2003/21602_Velcade.cfm>
- 13.U.S. Food and Drug Administration. Drug Approval Package; Revlimid (Lenalidomide) Capsules; Company: Celgene; Application No.: 021880/S-001; Approval Date: 06/29/2006. [Online]. 2006. [cited 2015 June 25]. Available from: <http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Label_ApprovalHistory#apphist>
- 14.U.S. Food and Drug Administration. Drug Approval Package; Thalomid (Thalidomide) Capsules; Company: Celgene; Application No.: 021430; Approval Date: 05/25/2006. [Online]. 2008. [cited 2015 June 23]. Available from: <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2006/021430s000TOC.cfm> [Google Scholar]
- 15.U.S. Food and Drug Administration. Drug Approval Package; Pomalyst (Pomalidomide) Capsules; Company: Celgene Corporation; Application No.: 0204026; Approval Date: 02/08/2013. [Online]. 2013. [cited 2015 June 23]. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/204026Orig1s000TOC.cfm [Google Scholar]
- 16.Kane RC, Farrell AT, Sridhara R & Pazdur R United States Food and Drug Administration approval summary: bortezomib for the treatment of progressive multiple myeloma after one prior therapy. Clin. Cancer Res 12, 10 (2006). [DOI] [PubMed] [Google Scholar]
- 17.U.S. Food and Drug Administration. Drug Approval Package; Kyprolis (Carfilzomib) for Injection; Company: Onyx Pharmaceuticals; Application No.: 0202714; Approval Date: 07/20/2012. [Online]. 2012. [cited 2015 June 23]. Available from: <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/202714Orig1s000TOC.cfm>
- 18.U.S. Food and Drug Administration. Summary Minutes of the Oncologic Drug Advistory Comittee Meeting. [Online]. 2014. [cited 2016 10 13]. Available from: <http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM430826.pdf>
- 19.U.S. Food and Drug Administration. Drug Approval Package; Darzalex Daratumumab injection; Company: Janssen Biotech; Application No.: 761036; Approval Date: 11/16/2015. [Online]. [cited 2016 October 22]. Available from: <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/761036Orig1s000TOC.cfm>
- 20.U.S. Food and Drug Administration. Drug Approval Package; Farydak (Panobinostat) Capsules; Company: Novartis Pharmaceuticals; Application No.: 0205353; Approval Date: 02/13/2015. [Online]. 2015. [cited 2015 June 25]. Available from: <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/205353Orig1s000TOC.cfm>
- 21.European Medicines Agency. Pomalidomide Celgene: International non-proprietary name: pomalidomide. [Online]. 2013. [cited 2016 Dec 20]. Available from: <www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public-assessment_report/human/002682/WC500147721.pdf>
- 22.European Medicines Agency. Assessment Report. [Online]. 2015. [cited 2016 Dec 20]. Available from: <www.ema.europa/edu/docs/en_GB/document_library/EPAR_-_Assessment_Report_-_Variation/human/000717/WC500184943.pdf>
- 23.NIH US National Libary of Science. DailyMed. [Online]. 2016. [cited 2016 Dec 20]. Available from: <https://dailymed.nlm.nih.gov/dailymed/druginfo.cfm?setid=2b25ef01-5c9e-11e1-b86c-0800200c9a66>
- 24.NIH US National Library of Medicine. Label: Revlimid-lenalidomide capsule. [Online]. January [cited Dec 20 2016]. Available from: <https://dailymed.nlm.nih.gov/dailymed/druginfo.cfm?setid=5fa97bf5-28a2-48f1-8955-f56012d296be>
- 25.NIH US National Library of Medicine. DailyMed. [Online]. March [cited Dec 20 2016]. Available from: <https://dailymed.nlm.nih.gov/dailymed/druginfo.cfm?setid=21d9c619-7e94-49e2-ac41-31e9ea96554a>
- 26.NIH US National Library of Science. DailyMed. [Online]. 2015. [cited 2016 Dec 20]. Available from: <https://dailymed.nlm.nih.gov/dailymed/druginfo.cfm?setid=2eda833b-1357-4ed4-a093-194524fcb061>
- 27.US National Institutes of Health. ClinicalTrials.gov. [Online]. 2015. [cited 2016 Dec 20]. Available from: <https://clinicaltrials.gov/ct2/show/results/NCT00424047?term=NCT00424047&rank=1>
- 28.US National Institutes of Health. ClinicalTrials.gov. [Online]. 2009. [cited 2016 Dec 20]. Available from: <https://clinicaltrials.gov/ct2/show/results/NCT00056160?term=NCT00056160&RANK=1>
- 29.Dimopoulos M et al. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N. Engl. J. Med 357, 2023–2032 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Rajkumar SV et al. Multicenter, randomized, double-blind, placebo-controlled study of thalidomide plus dexamethasone compared with dexamethasone as initial therapy for newly diagnosed multiple myeloma. J. Clin. Oncol 26, 13 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benboubker L et al. Lenalidomide and dexamethasone in transplant-ineligible patients with myeloma. N. Engl. J. Med 371, 906–917 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Richardson PG et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N. Engl. J. Med 348, 2609–2617 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Richardson PG et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N. Engl. J. Med 352, 2487–2495 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Miguel JS et al. Pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM-003): a randomized, open-label, phase 3 trial. Lancet Oncol. 14, 1055–1066 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Stewart AK et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N. Engl. J. Med 372, 142–152 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Weber DM et al. Lenolidomide plus dexamethasone for relapsed multiple myeloma in North America. N. Engl. J. Med 357, 2133–2142 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Orlowski RZ et al. Randomized phase III study of pegylated liposomal doxorubicin plus bortezomib compared with bortezomib alone in relapsed or refractory multiple myeloma: combination therapy improves time to progression. J. Clin. Oncol 25, 25 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Palumbo A et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N. Engl. J. Med 375, 754–766 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Dimopoulos M et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N. Engl. J. Med 375, 1319–1331 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Dimopoulos MA et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study Lancet Oncol. 17, 27–38 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Kumar S et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 17, e328–346 (2016). [DOI] [PubMed] [Google Scholar]
- 42.U.S. Food and Drug Administration. Guidance for industry: Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics. [Online]. 2007. [cited 2016 October 24]. Available from: <http://www.fda.gov/downloads/Drugs/./Guidances/ucm071590.pdf> [Google Scholar]
- 43.Basch E The missing voice of patients in drug-safety reporting. N. Engl. J. Med 362, 865–869 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thanarajasingam G, Hubbard JM, Sloan JA & Grothey A The imperative for a new approach to toxicity analysis in oncology clinical trials. J. Natl. Cancer Inst 107, 1–4 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Sivendran S et al. Adverse event reporting in cancer clinical trial publications. J. Clin. Oncol 32, 83 (2014). [DOI] [PubMed] [Google Scholar]
- 46.U.S. Food and Drug Administration. Drug Approval Package; Ninlaro (ixazomib) capsules; Company; Millennium Pharmaceuticals; Application No.; 208462; Approval Date: 11/20/2015. [Online]. 2015 [cited 2016 Feb 16]. Available from: <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/208462Orig1s000TOC.cfm> [Google Scholar]
- 47.Kumar S et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 111, 2516–2520 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Landgren O, Devlin S, Boulad M & Mailankody S Role of MRD status in relation to clinical outcomes in newly diagnosed multiple myeloma patients: a meta-analysis. Bone Marrow Transpl. 1–5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rajkumar S & Kyle R Progress in myeloma — a monoclonal breakthrough. N. Engl. J. Med 375, 14 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Gormley NJ et al. Regulatory perspective on minimal residual disease flow cytometry testing in multiple myeloma. Cytom. Part B Clin. Cytom 90B, 73–80 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Munshi N et al. Association of minimal residue disease with superior survival outcomes in patients with multiple myeloma: a meta-analysis. JAMA Oncol. 2016. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.