

Genomes are complexly organized in three-dimensional space. The intricate spatial architecture of the genome is reflected in prominent topological features such as blocks of heterochromatin and euchromatin, chromatin domains and chromatin loops. It seems intuitive that these major structural elements would play critical roles in the control of genome function and gene activity, and observations on individual genes have provided evidence for this hypothesis. However, in the current issue, Ghavi-Helm et al.1 have probed this structure–function relationship more globally and have found a disconnect between architectural features of genomes and gene activity, suggesting that topology may be less of a driver of gene activity than is commonly assumed.
The architectural features that have most consistently been linked to genome function are chromatin loops and domains2,3. Loops typically range in size from local interactions of thousands of base pairs in length to rarer long-range interactions over millions of base pairs (ref. 2) and are thought to affect gene regulation primarily by bringing enhancers into the proximity of their target promoters. Loop formation often occurs in the context of chromatin domains a few hundred kilobases in size, which are commonly referred to as topologically associating domains (TADs) and are demarcated by boundary elements defined by binding sites for architectural chromatin proteins3. A proposed function for TADs is that they facilitate promoter–enhancer contacts within a domain by insulating promoters from off-target enhancers in adjacent TADs. In support of this model, several examples have been described in which disruption of TAD boundaries leads to the formation of ectopic loops and consequent aberrant gene expression4,5.
A balanced approach
Rather than relying on individual model genes to probe the structure–function relationship, Ghavi-Helm et al.1 set out to investigate the role of chromatin topology by using a more systematic genome-wide approach. They used an elegant and powerful strategy in Drosophila in which heterozygous animals were generated carrying a wild-type set of chromosomes and a set of balancer chromosomes that contain extensive rearrangements over large portions of the genome. The beauty of this system is that it allows the behavior of the two alleles to be experimentally distinguished. These strains were used to systematically map allele-specific chromatin topology and relate it to the expression of the corresponding alleles (Fig. 1). These experiments yielded a number of informative and important observations.
Fig. 1 |. Using balancer chromosomes to probe the relationship between genome architecture and function.
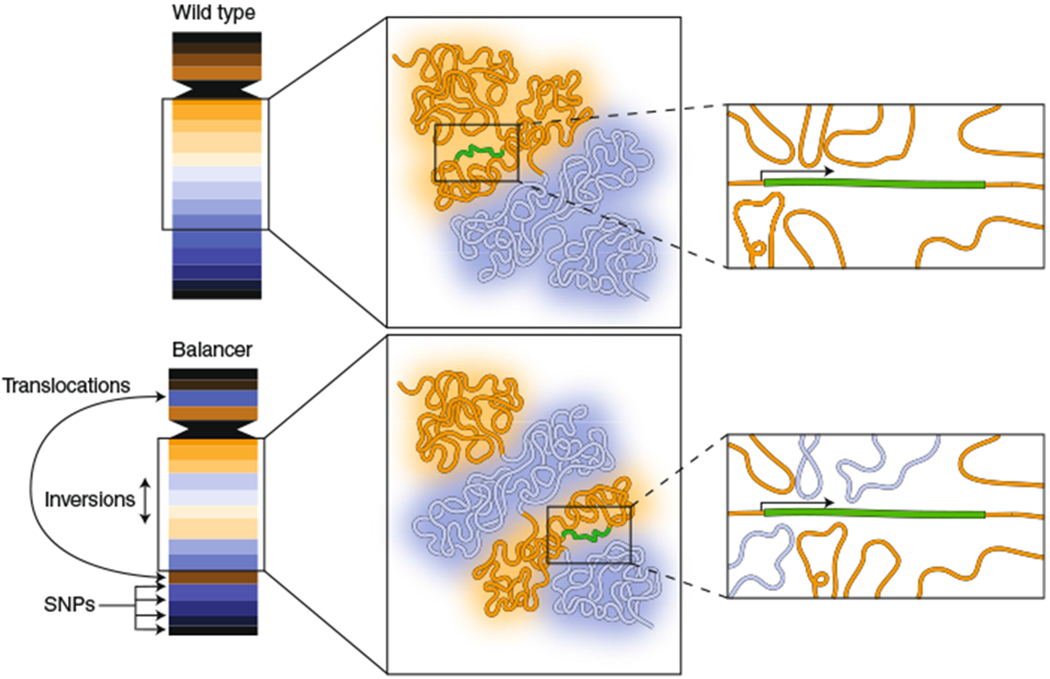
Balancer chromosomes contain inversions, deletions and translocations, thus making them topologically distinct from wild-type chromosomes. They also contain single-nucleotide variants, enabling capture of allele-specific expression data from RNA-seq. Ghavi-Helm et al.1 took advantage of the presence of wild-type and balancer chromosomes in the same nucleus to compare the effects of altered genome topology on gene expression. The activity of many genes was found to be insensitive to the chromatin topology in their environment. As an example, an internal inversion of two chromatin domains (blue, orange) may expose a gene (green) to distinct chromatin interactions on the balancer chromosome, yet its activity is unaffected. The results may suggest that multiple enhancers, often located in a single chromatin domain, act coordinately on a single promoter and that the effect of a given enhancer can be compensated for by other enhancers, even though it may originate from a distinct chromatin domain.
A key finding was that, despite widespread structural variation, less than 10% of tested genes showed altered expression levels. Even disruption of TADs or changes in their size affected only 12% of nearby genes. These results suggest that many genes are generally impervious to the large-scale topological features of their surroundings.
The instances in which genes were differentially regulated between wild-type and balancer chromosomes were also informative. In support of a role for genome structure in determining function, these genes did have more altered topological interactions near their promoters than genes whose expression was not affected. Strikingly, however, changes in local chromatin topology were not predictive of changes in gene expression, thus suggesting that structure alone does not determine activity.
One caveat to this approach is that balancer chromosome re-arrangements may have been selected for events that do not alter the expression of critical genes, or for mutations that compensate for the disruption of endogenous enhancers. However, the authors show that balancer chromosomes in general are not subject to strong selective pressures, thus arguing against this potentially confounding factor in the analysis.
Function drives structure
The general tenor of these observations is that genome topology is not a good predictor, or major driver, of gene activity. One possibility is that topological features may arise as a consequence of sequence features and gene activity, which then may stabilize or modulate the activity of a gene. Alternatively, and not exclusively, topological features may regulate genes in a collaborative fashion, with several enhancers acting on a single promoter by forming multiple interactions (Fig. 1). Each interaction may be unlikely in a given cell at a given time and may influence expression in a largely stochastic fashion. These models are consistent with recent observations on the heterogeneity of genome organization, suggesting that most chromatin-chromatin interactions occur with only low frequency in a cell population and that TADs reflect the average of many different chromatin configurations present in individual cells rather than a pervasive structure found in every cell6–8. These interpretations align well with the realization that the output of gene expression itself entails stochastic underlying processes such as cycles of transcriptional bursts and transcriptional silence9.
The findings by Ghavi-Helm et al.1 challenge the view that TADs provide a strongly insulated environment that limits the ability of enhancers to interact only with promoters within the same TAD. In line with a less restrictive model of TAD function, single-cell observations suggest that inter-TAD interactions are relatively frequent, as compared with intra-TAD interactions, and TAD boundaries themselves are fluid7,8, thus explaining the ability of enhancers to interact with their promoters even after rearrangement of the TAD structure, as observed by Ghavi-Helm et al.1 (Fig. 1).
The results reported here also highlight our surprisingly incomplete understanding of enhancer–promoter interactions10. Although the traditional view has been that a single enhancer physically interacts with its cognate promoter in a directed fashion, these new results suggest that other models that incorporate redundancy and cooperativity between multiple enhancers to cumulatively drive gene expression should be considered (Fig. 1).
A key step in clarifying the structure–function relationship in the genome will be to directly relate topological features of chromatin to gene activity with single-cell resolution and in real time. Recent visualization of promoter–enhancer interactions in live Drosophila cells indeed suggests not only that sustained proximity of an enhancer to its cognate promoter is required for activation but also that transcription shapes and stabilizes promoter–enhancer contacts11,12. These results are in line with the view that function drives structure in the genome at least as much as structure drives function.
The results presented by Ghavi-Helm et al.1 are important and provocative. Do they undermine the suggested role of genome organization on function? Not at all. They are simply a reminder that we should be careful not to jump to overly generalized conclusions on the basis of individual case studies. As these findings illustrate, it is not just the topological organization of genomes but also its effect on gene expression that is complex.
References
- 1.Ghavi-Helm Y et al. Nat. Genet 10.1038/s41588-019-0462-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dekker J & Misteli T Cold Spring Harb. Perspect. Biol 7, 019356 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dixon JR, Gorkin DU & Ren B Mol. Cell 62, 668–680 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lupiáñez DG et al. Cell 161, 1012–1025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nora EP et al. Nature 485, 381–385 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cattoni DI et al. Nat. Commun 8, 1753 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bintu B et al. Science 362, eaau1783 (2018).30361340 [Google Scholar]
- 8.Finn EH et al. Cell 176, 1502–1515.e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Symmons O & Raj A Mol. Cell 62, 788–802 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halfton MS Trends Genet. 35, 93–103 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen H et al. Nat. Genet 50, 1296–1303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heinz S et al. Cell 174, 1522–1536.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]