

Abstract
Antibody-dependent enhancement (ADE) is a mechanism by which the pathogenesis of certain viral infections is enhanced in the presence of sub-neutralizing or cross-reactive non-neutralizing antiviral antibodies. In vitro modelling of ADE has attributed enhanced pathogenesis to Fcγ receptor (FcγR)-mediated viral entry, rather than canonical viral receptor-mediated entry. However, the putative FcγR-dependent mechanisms of ADE overlap with the role of these receptors in mediating antiviral protection in various viral infections, necessitating a detailed understanding of how this diverse family of receptors functions in protection and pathogenesis. Here, we discuss the diversity of immune responses mediated upon FcγR engagement and review the available experimental evidence supporting the role of FcγRs in antiviral protection and pathogenesis through ADE. We explore FcγR engagement in the context of a range of different viral infections, including dengue virus and SARS-CoV, and consider ADE in the context of the ongoing SARS-CoV-2 pandemic.
Subject terms: Viral infection, Translational immunology
Antibody-dependent enhancement (ADE) has been described as a mechanism that contributes to the pathogenesis of dengue virus infection. Limited evidence also suggests that it can also occur in other viral infections. Here, the authors explore the history of the ADE phenomenon, discuss the diversity of Fc effector functions and consider its potential relevance in the context of SARS-CoV-2 infection.
Introduction
Amid the ongoing COVID-19 pandemic, efforts to actively vaccinate the general population against the SARS-CoV-2 virus in the context of poorly neutralizing and waning immunity have renewed interest in the phenomenon of antibody-dependent enhancement (ADE). This property of antibodies attributes enhanced disease pathogenesis in specific instances of viral infection to the presence of sub-neutralizing titres of antiviral host antibodies. In cases of ADE, rather than contributing to antiviral immunity, pre-existing antibodies facilitate viral entry and subsequent infection of host cells, leading to both increased infectivity and virulence.
ADE was first clearly described in dengue virus (DENV) infection by Halstead et al. in 1973 (refs1,2), although earlier epidemiological evidence had identified that two specific Thai patient populations, specifically first-time infected infants born to immune mothers and children suffering from secondary infection, were associated with an increased incidence of dengue haemorrhagic fever and dengue shock syndrome — two patient groups that ostensibly had pre-existing antibodies to DENV3. It was not until years later that studies proposed models of ADE, identified optimal conditions for in vitro ADE and quantified antibody titres permissive for ADE4–9. Numerous studies have since identified Fcγ receptors (FcγRs), surface receptors on immune cells that recognize the Fc portion of IgG and trigger a wide array of downstream effector functions, as the key mediators of ADE in dengue pathogenesis, as they allow for the internalization of multimeric virus-bound IgG and subsequent productive infection. Furthermore, FcγR-bearing immune cells susceptible to DENV ADE may lack canonical viral entry receptors for DENV, thereby constituting a unique mode of viral pathogenesis.
ADE is particularly relevant in the context of pre-existing immunity — gained either through previous infection or vaccination that results in circulating antibodies to viral antigens — and is carefully considered when designing both active and passive immunization strategies in an effort to prevent exacerbation of disease. However, little is known about the detailed cellular mechanisms of ADE, their interplay and potential redundancy with protective antibody mechanisms and the extent to which the principles of anti-DENV ADE may apply to the pathogenesis of various other viral infections. Due to the surge in interest and concern regarding ADE and the chief role for FcγRs in both antiviral and ADE mechanisms, this Review examines FcγR structure, function and signalling in both protection and pathogenesis, particularly in the context of the COVID-19 global pandemic.
Fcγ receptor structure and function
Whereas the Fab domain of an IgG molecule binds to viral epitopes and can neutralize the virus by blocking entry, fusion or maturation, the engagement of the IgG Fc domain with members of the FcγR family is responsible for triggering the effector cell responses critical for host protection against infection. The affinity and binding specificity of the Fc domain for different FcγRs are determined by differences in the primary amino acid sequence of the IgG subclasses (IgG1–IgG4 in humans) as well as by the structure and composition of the Fc-associated glycan structure10–15. These two determinants drive Fc domain diversification, resulting in IgG Fc domains with different capacities for engaging and activating the various members of the FcγR family expressed by effector leukocytes16.
Canonical, type I FcγRs are broadly classified as activating or inhibitory, depending on the signalling properties of their intracellular domains. In humans, activating FcγRs include FcγRI, FcγRIIa, FcγRIIc and FcγRIIIa, which contain immunoreceptor tyrosine activating motifs (ITAMs) either in the ligand-binding receptor α-chain in the case of FcγRIIa and FcγRIIc or in the associated FcR γ-chain for FcγRI and FcγRIIIa. ITAMs are necessary for receptor expression, surface assembly and signalling (Fig. 1). By contrast, FcγRIIb represents the sole inhibitory FcγR, mediating signalling activity through an immunoreceptor tyrosine inhibitory motif (ITIM) present in its cytoplasmic region. In contrast to activating or inhibitory FcγRs, FcγRIIIb is expressed as a GPI-anchored protein and is therefore incapable of signal transduction; however, FcγRIIIb still has the capacity to transduce activation signals following receptor crosslinking, mainly by associating and acting synergistically with activating receptors such as FcγRIIa17–20.
Fig. 1. Overview of the FcγR family and expression patterns among human effector leukocytes.

Canonical, type I Fcγ receptors (FcγRs) are broadly categorized into activating or inhibitory based on the presence of immunoreceptor tyrosine activating motif (ITAM) or immunoreceptor tyrosine inhibitory motif (ITIM) signalling in their intracellular domains. The majority of effector leukocytes co-express combinations of activating and inhibitory FcγRs and their opposing signalling activities determine the outcome of IgG-mediated inflammation. Although FcγRs are expressed abundantly among the various leukocyte subsets, inflammatory cues and differentiation status modulate the expression of the various FcγRs, thereby altering the responsiveness of effector leukocytes to IgG-mediated signalling. +, constitutive expression; –, no expression; #, inducible expression; *, expression depends on FCGR2C allelic status; NK, natural killer.
FcγRs are broadly expressed on the surface of both lymphoid and myeloid cells, although the distribution of different FcγRs is unique to each cell type; for example, B cells express FcγRIIb as their sole FcγR, whereas natural killer cells exclusively express the activating receptor FcγRIIIa. Most other immune cells express a combination of different FcγRs, pairing activating and inhibitory receptors to achieve balanced cellular responses (Fig. 1). FcγR surface expression is modulated by cytokines in a manner through which pro-inflammatory cytokines generally increase expression of activating FcγRs over their inhibitory counterparts, whereas anti-inflammatory signals downregulate activating FcγRs and enhance FcγRIIb expression16. Promoter polymorphisms and copy number variation in FcγR genes can also influence the expression levels of FcγRs on the surface of effector leukocytes, acting as an additional determinant for IgG-mediated signalling21.
Fcγ receptor effector activities
Fcγ receptor signalling
Despite the structural differences between FcγR family members, all activating FcγRs are characterized by the same sequence of signal transduction events. With the exception of FcγRI, which can engage monomeric IgG with high affinity, FcγRs exhibit low affinity for IgGs and can only interact with multimeric IgG immune complexes or opsonized cells, generated during an infectious challenge. Despite the high concentration of circulating IgG in serum, FcγRs on immune cells are incapable of crosslinking in the absence of a pathogenic trigger, thereby preventing inappropriate effector cell activation. Such interactions cause receptor clustering and aggregation, which in turn leads to the phosphorylation of ITAM domains22–25 — tandem YxxI/L motifs — by SRC family kinases, such as LYN, LCK, HCK and FGR, and the recruitment and activation of SYK family kinases23,24,26–30. A crucial step in this phosphorylation cascade is the activation of PI3K by SYK, which in turn recruits pleckstrin homology domain-expressing proteins such as BTK, GAB2 and phosphoinositide-specific phospholipase Cγ (PLCγ). These proteins help to generate inositol triphosphate (IP3) for the mobilization of intracellular Ca2+ from the endoplasmic reticulum and diacylglycerol (DAG) for the activation of protein kinase C (PKC)31. Taken together, these intracellular biochemical changes — including the subsequent activation of the Rho GTPases CDC2, RAC1 and RAC2, and actin polymerization mediated by ARP2/3 and WASP proteins — leads to phagocytosis of IgG complexes and receptor internalization32. In addition to these early events, several signalling pathways — including the MEK and MAP family kinases and the RAS pathway — also become activated, leading to the expression of pro-inflammatory cytokines and chemokines with direct and indirect effects on cellular survival and differentiation33–35 (Fig. 2). All of these signalling events are counterbalanced by the regulatory activity of FcγRIIb, which is mediated by the recruitment of phosphatases to its ITIM domain following receptor crosslinking and phosphorylation by SRC family kinases36–38. ITIM-recruited phosphatases, such as SHIP1 and SHP2, promote the hydrolysis of phosphatidylinositol 3,4,5-triphosphate (PIP3) on the inner leaflet of the plasma membrane to phosphatidylinositol 4,5-biphosphate (PIP2), which in turn inhibits the recruitment and activation of PLCγ and the tyrosine kinase BTK36,39,40. Because the majority of effector leukocytes co-express activating FcγRs and FcγRIIb, the outcome of FcγR-mediated signalling represents a fine balance between the opposing functions of these receptors.
Fig. 2. Downstream signalling events induced upon crosslinking of activating FcγRs by IgG immune complexes.

Multimeric IgG immune complexes engage multiple Fcγ receptors (FcγRs) through low-affinity, high-avidity interactions (step 1). Receptor crosslinking upon IgG immune complex binding triggers phosphorylation (P) of their immunoreceptor tyrosine activating motifs (ITAMs), which in turn leads to the activation of kinases of the SYK and SRC family (step 2), as well as activation of the protein kinase C (PKC) pathway, resulting in a rapid increase in intracellular Ca2+ levels following activation of Ca2+ channels (step 3). Kinase activation also leads to actin remodelling (step 4), which is critical for receptor internalization and phagocytosis of the IgG immune complex. At later stages, cellular activation is associated with activation of specific transcription factors such as p38 and Jun amino-terminal kinases (JNK) (step 5) that drive the expression and release of pro-inflammatory cytokines and chemokines (for example, tumour necrosis factor (TNF), IL-1β and IL-8) (step 6) that shape immune responses and alter the effector function, migration and survival of leukocytes.
Respiratory burst and degranulation
Intracellular signals transduced upon activating FcγR crosslinking ultimately lead to cellular activation; however, the precise biological consequences are diverse and differ substantially among the various effector leukocytes, contributing differentially to protection against viral infections. In granulocytes such as neutrophils, basophils and eosinophils, activation of SYK and SRC kinases following FcγR crosslinking leads to the assembly of the NADPH-dependent oxidase complex in the plasma membrane and the membranes of phagosomes, promoting the generation of reactive oxygen species and reactive nitrogen species with potent antimicrobial and cytotoxic activity41–46. FcγR-mediated PKC activation and the elevation in intracellular Ca2+ levels trigger the rapid mobilization and release of granule contents, including serine proteases, leukotrienes, proteins with antimicrobial activity, such as lysozyme and lactoferrin, and antimicrobial peptides such as α-defensins47–53. For example, HNP1, an α-defensin found in neutrophils and other immune cell populations, exhibits antiviral activity by interfering with the gp120–CD4 interaction essential for HIV viral fusion54,55. Therefore, in addition to FcγR-mediated phagocytosis of opsonized virions, signalling through activating FcγRs has a significant impact on granulocyte function, eliciting effector responses that represent a major immune mechanism for efficient and rapid protection against viral infection. Similarly, crosslinking of FcγRIIIa on natural killer cells triggers cellular activation and degranulation. The release of natural killer granule contents, including perforin and granzymes, in close proximity to IgG-coated cells induces the formation of pores on the target cell membrane and stimulates pro-apoptotic pathways that ultimately lead to cell death29,56. This lytic process is referred to as antibody-dependent cellular cytotoxicity and helps to eliminate infected cells, thereby limiting viral load and subsequent virus propagation.
Phagocytosis and regulation of immune cell differentiation
Crosslinking of FcγRs on phagocytes such as neutrophils, dendritic cells, monocytes and macrophages induces phagocytosis of IgG-opsonized virions and infected cells that harbour actively replicating virus in a process referred to as antibody-dependent cellular phagocytosis. In this process, engulfed virions or cells are degraded by acidification of the phagosome and digestion by lysosomal enzymes. In addition to phagocytosis, FcγR crosslinking has pleiotropic effects on leukocyte function; for example, on antigen-presenting cells such as dendritic cells, phagocytosis of IgG immune complexes by activating FcγRs is associated with enhanced endosomal maturation and lysosomal fusion, facilitating antigen processing and presentation on MHC class II molecules57–60. Additionally, dendritic cell maturation is tightly regulated by the opposing signalling activity of FcγRIIa and FcγRIIb — the two FcγRs expressed by these cells. Skewing the balance of dendritic cell FcγRIIa and FcγRIIb has a marked impact on cell maturation and the development of T cell immunity; for example, conditional genetic deletion or antibody-mediated blockade of FcγRIIb ligand-binding activity on dendritic cells results in augmented IgG immune complex-mediated cell maturation, characterized by upregulated expression of MHC class II and co-stimulatory molecules, as well as enhanced antigen presentation and T cell activation61–65.
FcγR-mediated signalling has a profound impact on monocyte and macrophage function, representing a key determinant of macrophage polarization. In the absence of inflammatory stimuli, engagement of activating FcγRs on these cells is associated with the production of pro-inflammatory cytokines and chemokines including IL-8, tumour necrosis factor (TNF) and IL-1β (refs66,67). By contrast, activating FcγR signalling in tandem with stimulation of Toll-like receptors (TLRs) such as TLR4 induces a specific polarization phenotype in non-polarized macrophages known as the M2b or ‘regulatory’ phenotype, which is characterized by a unique cytokine expression profile and increased migratory and phagocytic activity68–70. The opposing effects of activating FcγR signalling on monocyte and macrophage function in the presence or absence of additional microbial stimuli have been well characterized in vivo in mouse strains deficient in FcγRIIb. In models of mAb-mediated protection against influenza infection and pneumococcal peritonitis, Fcgr2b–/– mice exhibit significantly less severe disease, improved microbial clearance and faster infection resolution when compared with wild-type mice, whereas overexpression of FcγRIIb is associated with increased mortality upon challenge with Streptococcus pneumoniae71–73. By contrast, in autoimmune models of IgG immune complex-induced shock, arthritis and alveolitis, FcγRIIb deficiency is generally associated with a more severe disease phenotype, as FcγRIIb-deficient macrophages have a lower threshold for cellular activation and pro-inflammatory cytokine expression69,74–76. The studies described above highlight how the balance of activating and inhibitory FcγRs regulates macrophage polarization and dendritic cell maturation and function, and has important biological effects on innate effector responses and adaptive immunity.
Antibody-dependent enhancement
Antiviral Fc effector functions — protective or pathogenic
Engagement of activating FcγRs by antiviral antibodies and downstream signalling is associated with diverse protective activities that result in the rapid elimination of opsonized virions and infected cells through phagocytic and cytotoxic mechanisms, as well as the induction of adaptive immune responses through the modulation of dendritic cell function67 (Fig. 3). A large body of evidence from in vivo experimental systems and genetic association studies shows that a major component of the antiviral activity of IgG antibodies is attributed to their capacity to engage and activate specific FcγR pathways on specific immune cell populations that are critical for mediating Fc effector functions67,73,77–84. Indeed, there are numerous instances where even the most potent neutralizing mAbs are significantly compromised in their ability to confer antiviral protection in vivo when Fc–FcγR interactions are abrogated; conversely, mAbs with poor neutralizing activity in in vitro assays can provide robust antiviral protection in vivo, suggesting that antiviral protection of these mAbs is dependent on activating FcγR engagement67,73,77–82,84.
Fig. 3. Diversity of FcγR-mediated antiviral effector functions.
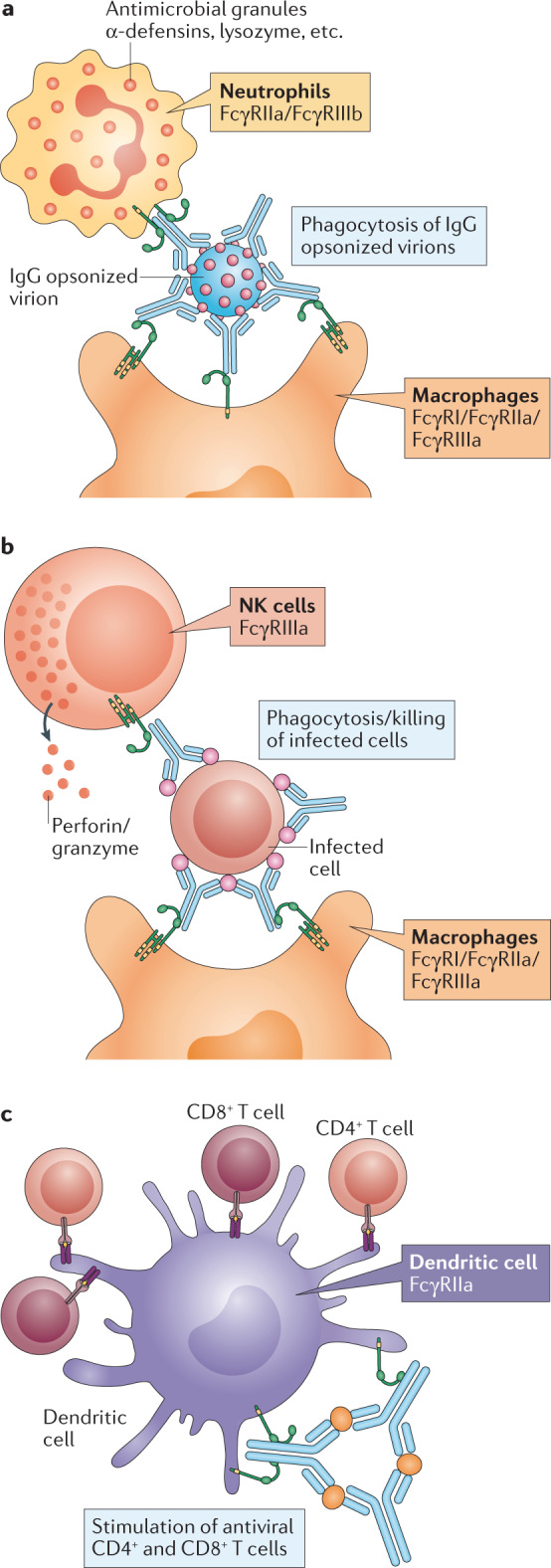
Fc–Fcγ receptor (FcγR) interactions drive pleiotropic effector functions that limit viral replication and provide potent antiviral protection. a | clearance of IgG opsonized virions. b | Cytotoxic elimination and phagocytic clearance of IgG-coated infected cells by natural killer (NK) cells and macrophages. c | Crosslinking of FcγRs on dendritic cells accelerates phagolysosome fusion and maturation, leading to more efficient antigen processing and presentation to CD8+ and CD4+ T cells, which in turn confer antiviral activities. All of these functions are key components of the host antiviral response and are mediated by the specific engagement and signalling of specific activating FcγRs (indicated in the figure) on specific leukocyte subsets.
The above findings highlight the discrepancy between in vitro neutralizing potency and in vivo protective activity, suggesting that in vitro assays rarely predict the full in vivo function of antiviral mAbs. This is exemplified in studies assessing the capacity of antiviral antibodies to mediate ADE. ADE refers to a phenomenon by which antiviral antibodies promote viral infection of host cells by exploiting the phagocytic FcγR pathway5,85. This phenomenon has been studied in the context of flaviviruses — particularly in the context of DENV infection5,86.
Dengue virus
A pathogenic role for antibodies was suggested when epidemiological studies reported that patients with particularly severe DENV disease were associated with prior infection with a different DENV serotype and pre-existing, non-neutralizing anti-DENV IgG antibodies9,87. These findings and subsequent studies led to a model by which IgG antibodies mediate infection of FcγR-expressing cells through increased uptake of virus–IgG complexes in an FcγR-dependent manner2,5,7. At sub-neutralizing titres, anti-DENV antibodies complex with the DENV virion and attach to the surface of FcγR-expressing leukocytes, utilizing the phagocytic FcγR pathway for entry5,88. Mechanistic studies have determined that activating FcγRs — specifically, FcγRIIa and FcγRIIIa — promote ADE during DENV infection, whereas FcγRIIb acts as a negative regulator for this process89,90. Upon internalization into the cell, antibody–virus complexes are sequestered to phagolysosomes, where low pH conditions trigger conformational changes to the structure of the DENV envelope protein (E), promoting viral fusion and infection. As DENV has the ability to productively replicate in FcγR-expressing myeloid cells, this process of viral entry leads to enhanced viral replication and leukocyte dysfunction, thereby contributing to disease pathogenesis. Given the lack of high-affinity, specialized entry receptors for DENV and its capacity to infect a diverse array of cell types in vitro — including endothelial, myeloid and epithelial cells — from a range of species, including humans, non-human primates and mosquitoes91,92, FcγR-mediated entry and infection of leukocytes likely represents a key immune evasion mechanism for DENV, accounting for increased susceptibility to severe symptomatic disease in individuals with prior immune history. In agreement with these in vitro observations, a pathogenic role for antibodies in DENV has been demonstrated in vivo in mouse and non-human primate disease models using polyclonal IgG isolated from symptomatic patients with DENV disease or using monoclonal anti-DENV IgG7,93–97. Engineering of the Fc domain of these antibodies to abrogate Fc–FcγR binding diminished their pathogenic activity, confirming the requirements for FcγR engagement in conferring DENV disease pathogenesis81. Further, studies in clinical cohorts determined that patients with severe symptomatic DENV disease have elevated serum levels of Fc glycoforms with enhanced affinity for FcγRIIIa88 and increased allelic frequency of the high-affinity single-nucleotide polymorphism of FcγRIIa98, highlighting the role of activating FcγRs in modulating DENV disease susceptibility.
ADE-associated viruses
ADE has been demonstrated in vitro for IgG antibodies to several diverse viruses, including influenza99–103, HIV104,105 and Ebola virus106–108, and under specific conditions (narrow range of IgG concentrations, specific cell lines, presence or not of complement), IgG antibodies promote in vitro infection of FcγR-expressing leukocytes to an extent comparable with that observed for DENV. However, studies in animal models and clinical studies have failed to support a pathogenic role for antibodies in these infections. Indeed, extensive experimental evidence from diverse viral infection models strongly supports that Fc–FcγR interactions are often critical for the antiviral function of IgG antibodies67. Numerous studies in mouse and non-human primate models of HIV infection have shown that both neutralizing and non-neutralizing anti-HIV mAbs depend on FcγR engagement for their antiviral activity78,80,81,84,109. Indeed, one of the few correlates of protection seen in the RV144 HIV vaccine trial was the level of non-neutralizing IgG antibodies capable of engaging FcγRs and inducing Fc effector functions110,111. Similarly, high-affinity allelic variants of FcγRIIa are associated with protection against progression to AIDS in HIV-infected individuals112. Similar results showing the importance of Fc–FcγR interactions and the relative absence of ADE effects were observed for several anti-Ebola virus and anti-influenza mAbs in animal disease models. Administration of neutralizing antibodies at sub-neutralizing doses113 or non-neutralizing mAbs capable of engaging and activating FcγR pathways was not associated with enhanced disease pathogenesis or IgG-mediated tissue inflammation73,77,79,82,112,114–117. By contrast, for many of these mAbs, FcγR engagement was required for their antiviral potency, as loss of their FcγR binding capacity was associated with significantly reduced protective activity73,77,79. In addition, the clinical evaluation and therapeutic use of anti-Ebola virus, anti-HIV or anti-influenza mAbs has not been associated with adverse events or increased susceptibility to disease118–124. This is despite the fact that these mAbs, expressed as the human IgG1 isotype, are capable of interacting with activating FcγRs, and subject to glycoengineering (afucosylation) have increased affinity for the activating FcγRIIIa, as in the case of the anti-Ebola virus mAb cocktail ZMApp. These findings highlight the discrepancy between in vitro ADE assays and in vivo experimental systems; although under specific non-physiological in vitro conditions IgG antibodies can allow the infection of FcγR-expressing cells that are normally non-permissive for infection, in vivo antibody administration has never been shown to be associated with enhanced viral replication, accelerated disease pathogenesis or uncontrolled IgG-mediated inflammation for these viral infections.
Vaccine-associated enhanced respiratory disease
In addition to promoting infection of non-permissive cell types in vitro, Fc–FcγR interactions have been proposed to have a pathogenic role in the context of vaccination against respiratory pathogens such as respiratory syncytial virus (RSV) and influenza. This phenomenon, termed vaccine-associated enhanced respiratory disease (VAERD), was first described in paediatric populations and associated with vaccination with inactivated measles virus or RSV125,126. Mechanistic studies in the context of RSV vaccination have established that formalin-inactivated RSV immunogens largely elicit IgG responses characterized by IgG molecules with poor neutralizing activity, due to the aberrant conformation of specific RSV antigens on the formalin-inactivated virion. Studies in mice determined that, following RSV challenge, vaccine-elicited, non-neutralizing IgG antibodies form immune complexes that induce lung tissue damage through activation of the complement pathway following deposition in the lung127. In addition, vaccination with formalin-inactivated RSV followed by RSV infection elicits inappropriate airway inflammation characterized by aberrant CD4+ T cell responses and expression of type 2 T helper (TH2) cytokines, which contribute to lung injury128–130.
VAERD has been demonstrated for several influenza vaccine candidates in ferrets and pigs and was characterized predominantly by non-neutralizing IgG antibody responses, excessive complement activation and deposition of immune complexes to lung tissue131–134. Additionally, clinical evidence from the 2009 influenza pandemic suggested that deceased patients were commonly characterized by increased complement fixation and deposition of immune complexes in the alveolar space, consistent with a VAERD-like mechanism potentiating lethal acute lung injury135. Although complement-mediated pathways have been implicated as a key component of VAERD pathogenesis, there is insufficient evidence to suggest a pathogenic role for Fc–FcγR interactions in driving VAERD and, consequently, acute lung injury. Indeed, several in vivo studies have failed to demonstrate any pathogenic activity of passively administered mAbs against RSV G proteins or F proteins136–139. In contrast to vaccine-elicited, non-neutralizing polyclonal anti-RSV IgG antibodies, anti-RSV mAbs exhibit minimal pathogenic activity, induce anti-inflammatory responses and protect mice from lethal RSV challenge136. Their protective activity was shown to rely on Fc–FcγR interactions, as subclass switching from IgG to IgA was associated with a significant reduction in their in vivo potency137 whereas Fc glycoengineering to enhance FcγRIIIa binding resulted in improved antiviral activity140. In addition to data from animal disease models, the capacity of passively administered IgG antibodies to protect against RSV infection without pathological consequences is demonstrated by the extensive clinical use of polyclonal RSV immune globulin (RespiGam; MedImmune) or anti-RSV mAbs such as palivizumab (Synagis; MedImmune) as a means of prophylaxis against RSV disease in children.
Despite the previously reported association between non-neutralizing antibodies and exacerbated influenza131–135, no studies have definitively supported a pathogenic role for FcγRs in driving acute lung injury and increasing susceptibility to severe disease. Instead, engagement of activating FcγRs by mAbs that target distinct epitopes on influenza virus haemagglutinin (HA) and neuraminidase (NA) drives potent antiviral protection in both prophylactic and therapeutic settings73,79,82. Even non-neutralizing mAbs — which are thought to be a major driver for VAERD — exhibit minimal pathogenic activity and confer FcγR-dependent protection against a lethal influenza challenge without eliciting uncontrolled or excessive lung inflammation79,82. Consistent with the lack of a pathogenic role for activating FcγRs, genetic association studies have not demonstrated a correlation between the high-affinity FcγRIIa allele (H131) and susceptibility to severe pneumonia or mortality in influenza-infected patients141,142. Interestingly, single-nucleotide polymorphisms in CD55 and C1QBP — key genes of the complement pathway — were associated with increased risk of mortality in hospitalized influenza patients, suggesting a potential pathogenic role for complement142. Additionally, studies on the mechanisms of VAERD disease pathogenesis revealed that VAERD is characterized by inappropriate airway inflammation due to a strong vaccine-elicited, TH2 cell-biased immune response and excessive production of TH2 cytokines, which exacerbates tissue damage and delays the clearance of infected cells129,131–134. These data suggest that VAERD represents a clinical syndrome characterized by a generalized dysregulation of lung immunity rather than an IgG-mediated pathology due to excessive production of non-neutralizing IgG responses. Although the precise mechanisms that drive VAERD pathogenesis have not been fully elucidated, such mechanisms are fundamentally different to those that drive mAb-mediated protection, which reflect the synergistic activity of Fab-mediated antigen recognition, as well as Fc-mediated engagement and tightly regulated activation of specific FcγR pathways.
ADE in coronavirus infection
Prior reports have suggested the potential for IgG antibodies to coronaviruses, such as SARS-CoV and MERS-CoV, to confer pathogenic activities through ADE and VAERD-like mechanisms. IgG antibodies to the spike (S) protein of SARS-CoV and MERS-CoV have the capacity to mediate ADE, facilitating the infection of cell types that are commonly non-permissive for infection143–147. However, the mechanism of ADE mediated by mAbs in vitro against SARS-CoV differs significantly from the well-established mechanisms that govern ADE in DENV infection. For example, DENV ADE relies on activating FcγRs such as FcγRIIa and FcγRIIIa89,90, whereas ADE mediated by SARS-CoV mAbs is dependent primarily on the inhibitory FcγRIIb and has been shown to cause preferential infection of B cell lines in vitro146,147. DENV exploits the FcγR pathway because of the lack of a specialized high-affinity entry receptor for DENV; however, as SARS-CoV can bind with high affinity to its entry receptor ACE2, it is questionable whether the virus utilizes low-affinity FcγRs such as FcγRIIb for infection within the lung microenvironment. In contrast to DENV infections, FcγR-expressing cells such as macrophages cannot sustain productive SARS-CoV infection, as these cell types are not permissive for viral replication145; therefore, if SARS-CoV does infect leukocytes through FcγRs in the lung microenvironment, the impact on viral dynamics during the course of infection is likely to be inconsequential. Although some discussions of ADE in coronavirus infection have noted a correlation between high anti-SARS-CoV IgG titres with disease severity as potential evidence for ADE, these studies are missing a causal link between IgG and enhanced disease148. Further, although inactivated SARS-CoV and MERS-CoV vaccine candidates have been proposed to induce VAERD in non-human primates and mice, respectively, previous studies on this topic reported contrasting findings149–151. Passive transfer of IgG antibodies from deceased SARS patients has been shown to induce acute lung injury in SARS-CoV infected non-human primates152, an effect attributed to skewed macrophage activation caused by pro-inflammatory cytokine production upon FcγR crosslinking. However, this assumption was based on in vitro experimental systems using isolated monocytes, which are clearly not predictive of in vivo conditions — especially as acute viral infections are characterized by strong interferon, TNF and IL-6 responses. On the other hand, some studies in mice have shown that vaccination with individual structural proteins of SARS-CoV and subsequent infection can induce transcriptional upregulation of pro-inflammatory TH1 and TH2 cytokines and CCL2 and CCL3 chemokines in lung tissue, while downregulating anti-inflammatory cytokines such as IL-10 and transforming growth factor-β (TGFβ)153. Whether this effect is specifically due to FcγR-mediated enhancement of viral entry into specific cell types permissive for productive infection remains unknown. Consistent with the lack of a pathogenic role for anti-SARS-CoV IgG antibodies, genetic association studies in SARS-CoV patient cohorts with variable disease severity demonstrated increased frequency of the low-affinity allele of FcγRIIa (R131) in deceased and hospitalized patients, whereas the high-affinity allele (H131) was associated with reduced disease and mortality risk, indicating that the activating FcγRIIa has minimal pathogenic potential and may actively contribute to protection against SARS-CoV infection and disease through interactions with IgG antibodies154.
SARS-CoV-2
For the reasons discussed above, the biological relevance of reports of in vitro SARS-CoV ADE remains unknown, and in vivo studies show enhancement of disease without clearly implicating FcγR-mediated ADE in pathogenesis. The picture of coronavirus ADE is further complicated in light of recent findings from SARS-CoV-2 studies that show no evidence of ADE. Recent preclinical evaluation studies of inactivated vaccine candidates on SARS-CoV-2 in mice, rats and non-human primates demonstrated the induction of protective IgG responses, without evidence for IgG-mediated pathology or increased susceptibility to VAERD155. Although small (mouse, hamster, ferret) and large (non-human primate) animal models of SARS-CoV-2 infection have been described156–158, sequence variability in FcγR-coding genes — as well as substantial interspecies differences in FcγR structure and function — limits our ability to interpret data from diverse animal models on the mechanisms of protection by IgG antibodies159,160. Such differences represent a major translational barrier for the evaluation of human IgG antibody activities in vivo, thereby limiting our understanding of the FcγR mechanisms that contribute to antiviral immunity. Recently developed transgenic mouse strains humanized for all classes of FcγRs represent a unique platform for the preclinical evaluation of human mAb-based therapeutics and vaccine-elicited IgGs161,162. FcγR humanized mice should address limitations associated with interspecies differences in FcγR biology between humans and other mammalian species and could be used to dissect precisely the FcγR mechanisms by which anti-SARS-CoV-2 antibodies confer protection and further address whether anti-SARS-CoV-2 antibodies mediate ADE.
In preclinical studies, passive transfer of convalescent plasma to critically ill patients with COVID-19 had an acceptable safety profile and was not associated with accelerated disease, indicating that IgG antibodies — even given under conditions that favour VAERD, such as a high dose and low neutralizing:non-neutralizing Ab ratio — do not have pathogenic consequences following administration and instead offer meaningful clinical benefits163. Finally, how pre-existing immunity to SARS-CoV may influence the response to SARS-CoV-2 infection has been explored recently164. Whereas anti-SARS-CoV antibodies were cross-reactive with SARS-CoV-2 S protein, they were unable to neutralize the heterologous virus, therefore approximating the conditions that favour ADE as described for DENV. Whether this has pathological consequences in vivo through a mechanism of ADE remains unknown and should be explored in further studies.
Future directions for studying Fc receptors in the context of SARS-CoV-2
As the study of anti-SARS-CoV-2 antibody responses progresses, careful characterization of the Fc domain structure and allelic distribution of FcγR genetic variants in patients with symptomatic and asymptomatic SARS-CoV-2 infection is expected to provide novel insights into the contribution of Fc–FcγR interactions to protection from, or susceptibility to, symptomatic disease. Likewise, rigorous assessment of the role of FcγR pathways in antibody-mediated protection from infection is critically needed for the development of vaccine and mAb-based therapeutic strategies for the effective control of COVID-19. Although high-throughput in vitro assays have been developed and are systematically used to interrogate Fc effector function of antiviral antibodies, any findings should be interpreted with caution, as such artificial in vitro assays and experimental systems fail to recapitulate the unique complexity and diversity of the FcγR-expressing cells that infiltrate the lung parenchyma during SARS-CoV-2 infection. For example, cell lines that are commonly used to assess antibody-dependent cellular cytotoxicity, phagocytosis or ADE express a limited set of human FcγRs and their FcγR expression pattern and levels differ substantially from those of FcγR-bearing leukocytes present at infectious sites16. Additionally, pseudovirus-based or bead-based assays for evaluating phagocytosis do not accurately replicate the structural and functional properties of SARS-CoV-2 antigens and fail to take into account unique attributes of SARS-CoV-2 viral entry and replication. Therefore, evaluation of the Fc effector function of anti-SARS-CoV-2 mAbs and vaccine-elicited IgG antibodies necessitates the use of well-defined, biologically relevant in vivo models of infection. Selective engagement of specific activating FcγRs on distinct leukocyte types with reduced inhibitory receptor engagement and complement activation is expected to mediate rapid clearance of opsonized virions and cytotoxic elimination of SARS-CoV-2-infected cells, leading to efficient control of viral replication and limiting tissue damage and inappropriate inflammatory responses. In addition to these innate immune effects, selective FcγR engagement on dendritic cells could help stimulate cytotoxic antiviral CD8+ T cell responses, which are commonly suppressed during severe SARS-CoV-2 infection as a result of excessive viral replication and uncontrolled recruitment of monocytes leading to TH2 cell-biased airway inflammation and acute lung damage165. Such selective FcγR engagement could be accomplished through the use of glycoengineered or protein-engineered Fc domain variants that exhibit unique FcγR binding properties. This approach has previously seen success in generating mAbs with preferable binding properties for treating other viral diseases (Table 1).
Table 1.
FcγR, FcRn and C1q binding profile of Fc-engineered variants of mAbs in clinical use or testing
| Fc variant | FcγRIIa | FcγRIIIa | FcγRIIb | C1q | FcRn | Example mAbs (antigen) | ||
|---|---|---|---|---|---|---|---|---|
| H131 | R131 | V158 | F158 | |||||
| N297A | – | – | – | – | – | – | ▪ | Atezolizumaba (PDL1), clazakizumab (IL-6), TRX518 (GITR) |
| L234A/L235A | – | – | – | – | – | – | ▪ | Spesolimab (IL-36R), teplizumab (CD37) |
| L234F/L325E/P331S | – | – | – | – | – | – | ▪ | Durvalumaba (PDL1), anifrolumab (IFNα/βR1) |
| Afucosylated | ▪ | ▪ | ↑↑ | ↑↑↑ | ▪ | ▪ | ▪ | Mogamulizumaba (CCR4), obinutuzumaba (CD20), benralizumaba (IL-5Rα), ublituximab (CD20), palivizumab-N (RSV), bemarituzumab (FGFR2b), cusatuzumab (CD70), gatipotuzumab (MUC1), ifabotuzumab (EPHA3) |
| M428L/N434S | ▪ | ▪ | ▪ | ▪ | ▪ | ▪ | ↑↑ | VRC01LS (HIV), 10-1074-LS (HIV), 3BNC117-LS (HIV), PGT121.414.LS (HIV), VIR-2482 (influenza) |
| M252Y/S254T/T256E | ↓↓ | ↓↓ | ↓↓ | ↓↓ | ↓↓ | ▪ | ↑↑ | MEDI8897 (RSV), BOS161721 (IL-21), MEDI4893 (Staphylococcus aureus) |
| S239D/K274Q/Y296F/Y300F/L309V/I332E/A339T/V397M | ↑↑ | ↑↑ | ↑↑ | ↑↑↑ | ↑↑ | ? | ▪ | Tafasitamab (CD19), talacotuzumab (CD123) |
| P247I/A339Q | ? | ? | ↑↑ | ↑↑ | ? | ? | ? | Ocaratuzumab (CD20) |
| L235V/F243L/R292P/Y300L/P392L | ▪ | ↓ | ↑↑ | ↑↑ | ↓ | ↑↑ | ▪ | Margetuximab (HER2) |
| S267E | ▪ | ↑↑ | ↓ | ↓ | ↑↑ | ↑ | ▪ | APX005M (CD40) |
| S267E/L328F | ↓↓ | ↑↑↑ | – | – | ↑↑↑ | ↑ | ▪ | Obexelimab (CD19), XmAb7195 (IgE) |
| G237D/P238D/H268D/P271G/A330R | – | ↓ | – | – | ↑↑↑↑ | – | ▪ | 2141-V11 (CD40) |
| G236A/S239D/A330L/I332E/M428L/N434S | ↑↑↑ | ↑↑↑ | ↑↑ | ↑↑↑ | ↑ | – | ↑↑ | Elipovimab (HIV) |
| G236A/A330L/I332E/M428L/N434S | ↑↑ | ↑↑ | ↑↑ | ↑↑ | ↓ | – | ↑↑ | VIR-3434 (HBV) |
Note that the data for afucosylated Fc variants include data from mAbs enriched for afucosylated glycoforms and the binding affinities shown are dependent on the abundance of afucosylated glycoforms. –, no detectable binding; ▪, no change; ↓, reduced affinity compared with wild-type human IgG1; ↑, increased affinity compared with wild-type human IgG1; ?, no data available; CCR4, C-C chemokine receptor type 4; EPHA3, EPH receptor A3; FcγR, Fcγ receptor; FcRn, neonatal Fc receptor; FGFR2b, fibroblast growth factor receptor 2b; GITR, glucocorticoid-induced tumour necrosis factor; HBV, hepatitis B virus; HER2, human epidermal growth factor receptor (also known as ERBB2); IFNα/βR1, interferon-α/β receptor 1; MUC1, mucin 1; RSV, respiratory syncytial virus. aAntibodies in clinical use.
Concluding remarks
FcγR-mediated effector functions are diverse and complex. However, advances in Fc domain engineering, the availability of animal strains that recapitulate the unique features of human FcγR physiology138 and our extensive knowledge of the specific FcγR pathways that drive protective innate and adaptive antiviral immunity will help further the development of novel antibody-based therapeutics that can confer potent and durable protection against infection without inducing ADE or VAERD. Over the past decade, numerous mAbs against neoplastic and infectious diseases that are currently in the clinic or in clinical testing have been engineered to exhibit altered FcγR and neonatal Fc receptor (FcRn) binding profiles in an attempt to optimize efficacy, increase therapeutic potency, extend the half-life or minimize inappropriate leukocyte activation (Table 1). Past experience in the development and use of Fc-engineered mAbs could guide the development of anti-SARS-CoV-2 mAbs to result in superior therapeutic efficacy through selective activation of specific FcγR pathways on distinct leukocyte types.
Acknowledgements
The authors thank all members of the Laboratory of Molecular Genetics and Immunology for discussions. They acknowledge support from the Rockefeller University and the National Institute of Allergy and Infectious Diseases (R01AI129795, R01AI145870, R01AI137276 and U19AI111825). The content of this Review is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health (NIH).
Author contributions
S.B. and A.G. researched data for the article. J.V.R. and S.B. made a substantial contribution to the discussion of content. All authors equally contributed to the writing and the review/editing of the manuscript before submission.
Competing interests
J.V.R is a consultant and member of the Scientific Advisory Board of Vir Biotechnology, Inc. S.B. and A.G. declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Halstead SB, Chow J, Marchette NJ. Immunologic enhancement of dengue virus replication. Nat. New Biol. 1973;243:24–25. [PubMed] [Google Scholar]
- 2.Halstead SB, Shotwell H, Casals J. Studies on the pathogenesis of dengue infection in monkeys. II. Clinical laboratory responses to heterologous infection. J. Infect. Dis. 1973;128:15–22. doi: 10.1093/infdis/128.1.15. [DOI] [PubMed] [Google Scholar]
- 3.Halstead SB, Nimmannitya S, Cohen SN. Observations related to pathogenesis of dengue hemorrhagic fever. IV. Relation of disease severity to antibody response and virus recovered. Yale J. Biol. Med. 1970;42:311–328. [PMC free article] [PubMed] [Google Scholar]
- 4.Halstead SB, O’Rourke EJ. Antibody enhanced dengue virus infection in primate leukocytes. Nature. 1977;265:739–741. doi: 10.1038/265739a0. [DOI] [PubMed] [Google Scholar]
- 5.Halstead SB, O’Rourke EJ. Dengue viruses and mononuclear phagocytes. I. Infection enhancement by non-neutralizing antibody. J. Exp. Med. 1977;146:201–217. doi: 10.1084/jem.146.1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Halstead SB, O’Rourke EJ, Allison AC. Dengue viruses and mononuclear phagocytes. II. Identity of blood and tissue leukocytes supporting in vitro infection. J. Exp. Med. 1977;146:218–229. doi: 10.1084/jem.146.1.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Halstead SB. In vivo enhancement of dengue infection with passively transferred antibody. J. Infect. Dis. 1979;140:527–533. doi: 10.1093/infdis/140.4.527. [DOI] [PubMed] [Google Scholar]
- 8.Porterfield JS. Antibody-dependent enhancement of viral infectivity. Adv. Virus Res. 1986;31:335–355. doi: 10.1016/s0065-3527(08)60268-7. [DOI] [PubMed] [Google Scholar]
- 9.Katzelnick LC, et al. Antibody-dependent enhancement of severe dengue disease in humans. Science. 2017;358:929–932. doi: 10.1126/science.aan6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sondermann P, Pincetic A, Maamary J, Lammens K, Ravetch JV. General mechanism for modulating immunoglobulin effector function. Proc. Natl Acad. Sci. USA. 2013;110:9868–9872. doi: 10.1073/pnas.1307864110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferrara C, et al. Unique carbohydrate–carbohydrate interactions are required for high affinity binding between FcγRIII and antibodies lacking core fucose. Proc. Natl Acad. Sci. USA. 2011;108:12669–12674. doi: 10.1073/pnas.1108455108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krapp S, Mimura Y, Jefferis R, Huber R, Sondermann P. Structural analysis of human IgG–Fc glycoforms reveals a correlation between glycosylation and structural integrity. J. Mol. Biol. 2003;325:979–989. doi: 10.1016/s0022-2836(02)01250-0. [DOI] [PubMed] [Google Scholar]
- 13.Shields RL, et al. High resolution mapping of the binding site on human IgG1 for FcγRI, FcγRII, FcγRIII, and FcRn and design of IgG1 variants with improved binding to the FcγR. J. Biol. Chem. 2001;276:6591–6604. doi: 10.1074/jbc.M009483200. [DOI] [PubMed] [Google Scholar]
- 14.Sondermann P, Kaiser J, Jacob U. Molecular basis for immune complex recognition: a comparison of Fc-receptor structures. J. Mol. Biol. 2001;309:737–749. doi: 10.1006/jmbi.2001.4670. [DOI] [PubMed] [Google Scholar]
- 15.Sondermann P, Huber R, Oosthuizen V, Jacob U. The 3.2-A crystal structure of the human IgG1 Fc fragment–FcγRIII complex. Nature. 2000;406:267–273. doi: 10.1038/35018508. [DOI] [PubMed] [Google Scholar]
- 16.Bournazos S, Wang TT, Dahan R, Maamary J, Ravetch JV. Signaling by antibodies: recent progress. Annu. Rev. Immunol. 2017;35:285–311. doi: 10.1146/annurev-immunol-051116-052433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou MJ, Brown EJ. CR3 (Mac-1, αMβ2, CD11b/CD18) and FcγRIII cooperate in generation of a neutrophil respiratory burst: requirement for FcγRIII and tyrosine phosphorylation. J. Cell. Biol. 1994;125:1407–1416. doi: 10.1083/jcb.125.6.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marois L, Paré G, Vaillancourt M, Rollet-Labelle E, Naccache PH. FcγRIIIb triggers raft-dependent calcium influx in IgG-mediated responses in human neutrophils. J. Biol. Chem. 2011;286:3509–3519. doi: 10.1074/jbc.M110.169516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Green JM, Schreiber AD, Brown EJ. Role for a glycan phosphoinositol anchor in Fcγ receptor synergy. J. Cell. Biol. 1997;139:1209–1217. doi: 10.1083/jcb.139.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coxon A, et al. FcγRIII mediates neutrophil recruitment to immune complexes. a mechanism for neutrophil accumulation in immune-mediated inflammation. Immunity. 2001;14:693–704. doi: 10.1016/s1074-7613(01)00150-9. [DOI] [PubMed] [Google Scholar]
- 21.Bournazos S, Woof JM, Hart SP, Dransfield I. Functional and clinical consequences of Fc receptor polymorphic and copy number variants. Clin. Exp. Immunol. 2009;157:244–254. doi: 10.1111/j.1365-2249.2009.03980.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duchemin AM, Ernst LK, Anderson CL. Clustering of the high affinity Fc receptor for immunoglobulin G (FcγRI) results in phosphorylation of its associated γ-chain. J. Biol. Chem. 1994;269:12111–12117. [PubMed] [Google Scholar]
- 23.Jouvin MH, et al. Differential control of the tyrosine kinases Lyn and Syk by the two signaling chains of the high affinity immunoglobulin E receptor. J. Biol. Chem. 1994;269:5918–5925. [PubMed] [Google Scholar]
- 24.Swanson JA, Hoppe AD. The coordination of signaling during Fc receptor-mediated phagocytosis. J. Leukoc. Biol. 2004;76:1093–1103. doi: 10.1189/jlb.0804439. [DOI] [PubMed] [Google Scholar]
- 25.Unkeless JC, Shen Z, Lin CW, DeBeus E. Function of human FcγRIIA and FcγRIIIB. Semin. Immunol. 1995;7:37–44. doi: 10.1016/1044-5323(95)90006-3. [DOI] [PubMed] [Google Scholar]
- 26.Durden DL, Liu YB. Protein–tyrosine kinase p72syk in FcγRI receptor signaling. Blood. 1994;84:2102–2108. [PubMed] [Google Scholar]
- 27.Durden DL, Kim HM, Calore B, Liu Y. The FcγRI receptor signals through the activation of hck and MAP kinase. J. Immunol. 1995;154:4039–4047. [PubMed] [Google Scholar]
- 28.Eiseman E, Bolen JB. Engagement of the high-affinity IgE receptor activates src protein-related tyrosine kinases. Nature. 1992;355:78–80. doi: 10.1038/355078a0. [DOI] [PubMed] [Google Scholar]
- 29.Selvaraj P, Carpén O, Hibbs ML, Springer TA. Natural killer cell and granulocyte Fcγ receptor III (CD16) differ in membrane anchor and signal transduction. J. Immunol. 1989;143:3283–3288. [PubMed] [Google Scholar]
- 30.Pignata C, et al. FcγRIIIA-mediated signaling involves src-family lck in human natural killer cells. J. Immunol. 1993;151:6794–6800. [PubMed] [Google Scholar]
- 31.Botelho RJ, et al. Localized biphasic changes in phosphatidylinositol-4,5-bisphosphate at sites of phagocytosis. J. Cell Biol. 2000;151:1353–1368. doi: 10.1083/jcb.151.7.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoppe AD, Swanson JA. Cdc42, Rac1, and Rac2 display distinct patterns of activation during phagocytosis. Mol. Biol. Cell. 2004;15:3509–3519. doi: 10.1091/mbc.E03-11-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sánchez-Mejorada G, Rosales C. Fcγ receptor-mediated mitogen-activated protein kinase activation in monocytes is independent of Ras. J. Biol. Chem. 1998;273:27610–27619. doi: 10.1074/jbc.273.42.27610. [DOI] [PubMed] [Google Scholar]
- 34.Bracke M, Coffer PJ, Lammers JW, Koenderman L. Analysis of signal transduction pathways regulating cytokine-mediated Fc receptor activation on human eosinophils. J. Immunol. 1998;161:6768–6774. [PubMed] [Google Scholar]
- 35.Aramburu J, Azzoni L, Rao A, Perussia B. Activation and expression of the nuclear factors of activated T cells, NFATp and NFATc, in human natural killer cells: regulation upon CD16 ligand binding. J. Exp. Med. 1995;182:801–810. doi: 10.1084/jem.182.3.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pearse RN, et al. SHIP recruitment attenuates FcγRIIB-induced B cell apoptosis. Immunity. 1999;10:753–760. doi: 10.1016/s1074-7613(00)80074-6. [DOI] [PubMed] [Google Scholar]
- 37.Amigorena S, et al. Cytoplasmic domain heterogeneity and functions of IgG Fc receptors in B lymphocytes. Science. 1992;256:1808–1812. doi: 10.1126/science.1535455. [DOI] [PubMed] [Google Scholar]
- 38.Muta T, et al. A 13-amino-acid motif in the cytoplasmic domain of FcγRIIB modulates B-cell receptor signalling. Nature. 1994;368:70–73. doi: 10.1038/368070a0. [DOI] [PubMed] [Google Scholar]
- 39.Ono M, Bolland S, Tempst P, Ravetch JV. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor FcγRIIB. Nature. 1996;383:263–266. doi: 10.1038/383263a0. [DOI] [PubMed] [Google Scholar]
- 40.Ono M, et al. Deletion of SHIP or SHP-1 reveals two distinct pathways for inhibitory signaling. Cell. 1997;90:293–301. doi: 10.1016/s0092-8674(00)80337-2. [DOI] [PubMed] [Google Scholar]
- 41.Hampton MB, Kettle AJ, Winterbourn CC. Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood. 1998;92:3007–3017. [PubMed] [Google Scholar]
- 42.Martyn KD, Kim MJ, Quinn MT, Dinauer MC, Knaus UG. p21-activated kinase (Pak) regulates NADPH oxidase activation in human neutrophils. Blood. 2005;106:3962–3969. doi: 10.1182/blood-2005-03-0859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suh CI, et al. The phosphoinositide-binding protein p40phox activates the NADPH oxidase during FcγIIA receptor-induced phagocytosis. J. Exp. Med. 2006;203:1915–1925. doi: 10.1084/jem.20052085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamauchi A, et al. Rac2-deficient murine macrophages have selective defects in superoxide production and phagocytosis of opsonized particles. J. Immunol. 2004;173:5971–5979. doi: 10.4049/jimmunol.173.10.5971. [DOI] [PubMed] [Google Scholar]
- 45.Nathan CF, Brukner LH, Silverstein SC, Cohn ZA. Extracellular cytolysis by activated macrophages and granulocytes. I. Pharmacologic triggering of effector cells and the release of hydrogen peroxide. J. Exp. Med. 1979;149:84–99. doi: 10.1084/jem.149.1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nathan CF, Silverstein SC, Brukner LH, Cohn ZA. Extracellular cytolysis by activated macrophages and granulocytes. II. Hydrogen peroxide as a mediator of cytotoxicity. J. Exp. Med. 1979;149:100–113. doi: 10.1084/jem.149.1.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sørensen O, Arnljots K, Cowland JB, Bainton DF, Borregaard N. The human antibacterial cathelicidin, hCAP-18, is synthesized in myelocytes and metamyelocytes and localized to specific granules in neutrophils. Blood. 1997;90:2796–2803. [PubMed] [Google Scholar]
- 48.Cowland JB, Johnsen AH, Borregaard N. hCAP-18, a cathelin/pro-bactenecin-like protein of human neutrophil specific granules. FEBS Lett. 1995;368:173–176. doi: 10.1016/0014-5793(95)00634-l. [DOI] [PubMed] [Google Scholar]
- 49.Egesten A, Breton-Gorius J, Guichard J, Gullberg U, Olsson I. The heterogeneity of azurophil granules in neutrophil promyelocytes: immunogold localization of myeloperoxidase, cathepsin G, elastase, proteinase 3, and bactericidal/permeability increasing protein. Blood. 1994;83:2985–2994. [PubMed] [Google Scholar]
- 50.Fouret P, et al. Expression of the neutrophil elastase gene during human bone marrow cell differentiation. J. Exp. Med. 1989;169:833–845. doi: 10.1084/jem.169.3.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Owen CA, Campbell MA, Boukedes SS, Campbell EJ. Inducible binding of bioactive cathepsin G to the cell surface of neutrophils. A novel mechanism for mediating extracellular catalytic activity of cathepsin G. J. Immunol. 1995;155:5803–5810. [PubMed] [Google Scholar]
- 52.Panyutich AV, Hiemstra PS, van Wetering S, Ganz T. Human neutrophil defensin and serpins form complexes and inactivate each other. Am. J. Respir. Cell Mol. Biol. 1995;12:351–357. doi: 10.1165/ajrcmb.12.3.7873202. [DOI] [PubMed] [Google Scholar]
- 53.Gabay JE, Almeida RP. Antibiotic peptides and serine protease homologs in human polymorphonuclear leukocytes: defensins and azurocidin. Curr. Opin. Immunol. 1993;5:97–102. doi: 10.1016/0952-7915(93)90087-9. [DOI] [PubMed] [Google Scholar]
- 54.Furci L, Sironi F, Tolazzi M, Vassena L, Lusso P. α-Defensins block the early steps of HIV-1 infection: interference with the binding of gp120 to CD4. Blood. 2007;109:2928–2935. doi: 10.1182/blood-2006-05-024489. [DOI] [PubMed] [Google Scholar]
- 55.Daher KA, Selsted ME, Lehrer RI. Direct inactivation of viruses by human granulocyte defensins. J. Virol. 1986;60:1068–1074. doi: 10.1128/jvi.60.3.1068-1074.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vivier E, et al. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331:44–49. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Amigorena S, Salamero J, Davoust J, Fridman WH, Bonnerot C. Tyrosine-containing motif that transduces cell activation signals also determines internalization and antigen presentation via type III receptors for IgG. Nature. 1992;358:337–341. doi: 10.1038/358337a0. [DOI] [PubMed] [Google Scholar]
- 58.Bergtold A, Desai DD, Gavhane A, Clynes R. Cell surface recycling of internalized antigen permits dendritic cell priming of B cells. Immunity. 2005;23:503–514. doi: 10.1016/j.immuni.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 59.Hoffmann E, et al. Autonomous phagosomal degradation and antigen presentation in dendritic cells. Proc. Natl Acad. Sci. USA. 2012;109:14556–14561. doi: 10.1073/pnas.1203912109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bonnerot C, et al. syk protein tyrosine kinase regulates Fc receptor gamma-chain-mediated transport to lysosomes. EMBO J. 1998;17:4606–4616. doi: 10.1093/emboj/17.16.4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boruchov AM, et al. Activating and inhibitory IgG Fc receptors on human DCs mediate opposing functions. J. Clin. Invest. 2005;115:2914–2923. doi: 10.1172/JCI24772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dhodapkar KM, et al. Selective blockade of inhibitory Fcγ receptor enables human dendritic cell maturation with IL-12p70 production and immunity to antibody-coated tumor cells. Proc. Natl Acad. Sci. USA. 2005;102:2910–2915. doi: 10.1073/pnas.0500014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Diaz de Ståhl T, Heyman B. IgG2a-mediated enhancement of antibody responses is dependent on FcRγ+ bone marrow-derived cells. Scand. J. Immunol. 2001;54:495–500. doi: 10.1046/j.1365-3083.2001.01000.x. [DOI] [PubMed] [Google Scholar]
- 64.Kalergis AM, Ravetch JV. Inducing tumor immunity through the selective engagement of activating Fcγ receptors on dendritic cells. J. Exp. Med. 2002;195:1653–1659. doi: 10.1084/jem.20020338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Regnault A, et al. Fcγ receptor-mediated induction of dendritic cell maturation and major histocompatibility complex class I-restricted antigen presentation after immune complex internalization. J. Exp. Med. 1999;189:371–380. doi: 10.1084/jem.189.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bournazos S, Ravetch JV. Fcγ receptor pathways during active and passive immunization. Immunol. Rev. 2015;268:88–103. doi: 10.1111/imr.12343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bournazos S, DiLillo DJ, Ravetch JV. The role of Fc–FcγR interactions in IgG-mediated microbial neutralization. J. Exp. Med. 2015;212:1361–1369. doi: 10.1084/jem.20151267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dhodapkar KM, et al. Selective blockade of the inhibitory Fcγ receptor (FcγRIIB) in human dendritic cells and monocytes induces a type I interferon response program. J. Exp. Med. 2007;204:1359–1369. doi: 10.1084/jem.20062545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Clynes R, et al. Modulation of immune complex-induced inflammation in vivo by the coordinate expression of activation and inhibitory Fc receptors. J. Exp. Med. 1999;189:179–185. doi: 10.1084/jem.189.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sutterwala FS, Noel GJ, Clynes R, Mosser DM. Selective suppression of interleukin-12 induction after macrophage receptor ligation. J. Exp. Med. 1997;185:1977–1985. doi: 10.1084/jem.185.11.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brownlie RJ, et al. Distinct cell-specific control of autoimmunity and infection by FcγRIIb. J. Exp. Med. 2008;205:883–895. doi: 10.1084/jem.20072565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Clatworthy MR, Smith KG. FcγRIIb balances efficient pathogen clearance and the cytokine-mediated consequences of sepsis. J. Exp. Med. 2004;199:717–723. doi: 10.1084/jem.20032197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.DiLillo DJ, Tan GS, Palese P, Ravetch JV. Broadly neutralizing hemagglutinin stalk-specific antibodies require FcγR interactions for protection against influenza virus in vivo. Nat. Med. 2014;20:143–151. doi: 10.1038/nm.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Clynes R, Dumitru C, Ravetch JV. Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritis. Science. 1998;279:1052–1054. doi: 10.1126/science.279.5353.1052. [DOI] [PubMed] [Google Scholar]
- 75.Yuasa T, et al. Deletion of Fcγ receptor IIB renders H-2b mice susceptible to collagen-induced arthritis. J. Exp. Med. 1999;189:187–194. doi: 10.1084/jem.189.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. FcR γ chain deletion results in pleiotrophic effector cell defects. Cell. 1994;76:519–529. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 77.Bournazos S, DiLillo DJ, Goff AJ, Glass PJ, Ravetch JV. Differential requirements for FcγR engagement by protective antibodies against Ebola virus. Proc. Natl Acad. Sci. USA. 2019;116:20054–20062. doi: 10.1073/pnas.1911842116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lu CL, et al. Enhanced clearance of HIV-1-infected cells by broadly neutralizing antibodies against HIV-1 in vivo. Science. 2016;352:1001–1004. doi: 10.1126/science.aaf1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.DiLillo DJ, Palese P, Wilson PC, Ravetch JV. Broadly neutralizing anti-influenza antibodies require Fc receptor engagement for in vivo protection. J. Clin. Invest. 2016;126:605–610. doi: 10.1172/JCI84428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bournazos S, et al. Broadly neutralizing anti-HIV-1 antibodies require Fc effector functions for in vivo activity. Cell. 2014;158:1243–1253. doi: 10.1016/j.cell.2014.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Halper-Stromberg A, et al. Broadly neutralizing antibodies and viral inducers decrease rebound from HIV-1 latent reservoirs in humanized mice. Cell. 2014;158:989–999. doi: 10.1016/j.cell.2014.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.He W, et al. Alveolar macrophages are critical for broadly-reactive antibody-mediated protection against influenza A virus in mice. Nat. Commun. 2017;8:846. doi: 10.1038/s41467-017-00928-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Leon PE, et al. Optimal activation of Fc-mediated effector functions by influenza virus hemagglutinin antibodies requires two points of contact. Proc. Natl Acad. Sci. USA. 2016;113:E5944–E5951. doi: 10.1073/pnas.1613225113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Horwitz JA, et al. Non-neutralizing antibodies alter the course of HIV-1 Infection in vivo. Cell. 2017;170:637–648. doi: 10.1016/j.cell.2017.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Halstead SB. Immune enhancement of viral infection. Prog. Allergy. 1982;31:301–364. [PubMed] [Google Scholar]
- 86.Halstead SB. Neutralization and antibody-dependent enhancement of dengue viruses. Adv. Virus Res. 2003;60:421–467. doi: 10.1016/s0065-3527(03)60011-4. [DOI] [PubMed] [Google Scholar]
- 87.Salje H, et al. Reconstruction of antibody dynamics and infection histories to evaluate dengue risk. Nature. 2018;557:719–723. doi: 10.1038/s41586-018-0157-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang TT, et al. IgG antibodies to dengue enhanced for FcγRIIIA binding determine disease severity. Science. 2017;355:395–398. doi: 10.1126/science.aai8128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chan KR, et al. Ligation of Fcγ receptor IIB inhibits antibody-dependent enhancement of dengue virus infection. Proc. Natl Acad. Sci. USA. 2011;108:12479–12484. doi: 10.1073/pnas.1106568108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thulin NK, et al. Maternal anti-dengue IgG fucosylation predicts susceptibility to dengue disease in infants. Cell Rep. 2020;31:107642. doi: 10.1016/j.celrep.2020.107642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Diamond MS, Edgil D, Roberts TG, Lu B, Harris E. Infection of human cells by dengue virus is modulated by different cell types and viral strains. J. Virol. 2000;74:7814–7823. doi: 10.1128/jvi.74.17.7814-7823.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Diamond MS, Pierson TC. Molecular insight into dengue virus pathogenesis and its implications for disease control. Cell. 2015;162:488–492. doi: 10.1016/j.cell.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pinto AK, et al. Defining new therapeutics using a more immunocompetent mouse model of antibody-enhanced dengue virus infection. mBio. 2015;6:e01316–01415. doi: 10.1128/mBio.01316-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Orozco S, et al. Characterization of a model of lethal dengue virus 2 infection in C57BL/6 mice deficient in the α/β interferon receptor. J. Gen. Virol. 2012;93:2152–2157. doi: 10.1099/vir.0.045088-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zellweger RM, Prestwood TR, Shresta S. Enhanced infection of liver sinusoidal endothelial cells in a mouse model of antibody-induced severe dengue disease. Cell Host Microbe. 2010;7:128–139. doi: 10.1016/j.chom.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Balsitis SJ, et al. Lethal antibody enhancement of dengue disease in mice is prevented by Fc modification. PLoS Pathog. 2010;6:e1000790. doi: 10.1371/journal.ppat.1000790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Goncalvez AP, Engle RE, St Claire M, Purcell RH, Lai CJ. Monoclonal antibody-mediated enhancement of dengue virus infection in vitro and in vivo and strategies for prevention. Proc. Natl Acad. Sci. USA. 2007;104:9422–9427. doi: 10.1073/pnas.0703498104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mohsin SN, et al. Association of FcγRIIa polymorphism with clinical outcome of dengue infection: first insight from Pakistan. Am. J. Trop. Med. Hyg. 2015;93:691–696. doi: 10.4269/ajtmh.15-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ochiai H, et al. Infection enhancement of influenza A NWS virus in primary murine macrophages by anti-hemagglutinin monoclonal antibody. J. Med. Virol. 1992;36:217–221. doi: 10.1002/jmv.1890360312. [DOI] [PubMed] [Google Scholar]
- 100.Tamura M, Webster RG, Ennis FA. Antibodies to HA and NA augment uptake of influenza A viruses into cells via Fc receptor entry. Virology. 1991;182:211–219. doi: 10.1016/0042-6822(91)90664-w. [DOI] [PubMed] [Google Scholar]
- 101.Ochiai H, Kurokawa M, Kuroki Y, Niwayama S. Infection enhancement of influenza A H1 subtype viruses in macrophage-like P388D1 cells by cross-reactive antibodies. J. Med. Virol. 1990;30:258–265. doi: 10.1002/jmv.1890300406. [DOI] [PubMed] [Google Scholar]
- 102.Ochiai H, Kurokawa M, Hayashi K, Niwayama S. Antibody-mediated growth of influenza A NWS virus in macrophage-like cell line P388D1. J. Virol. 1988;62:20–26. doi: 10.1128/jvi.62.1.20-26.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gotoff R, et al. Primary influenza A virus infection induces cross-reactive antibodies that enhance uptake of virus into Fc receptor-bearing cells. J. Infect. Dis. 1994;169:200–203. doi: 10.1093/infdis/169.1.200. [DOI] [PubMed] [Google Scholar]
- 104.Trischmann H, Davis D, Lachmann PJ. Lymphocytotropic strains of HIV type 1 when complexed with enhancing antibodies can infect macrophages via FcγRIII, independently of CD4. AIDS Res. Hum. Retroviruses. 1995;11:343–352. doi: 10.1089/aid.1995.11.343. [DOI] [PubMed] [Google Scholar]
- 105.Laurence J, Saunders A, Early E, Salmon JE. Human immunodeficiency virus infection of monocytes: relationship to Fc-γ receptors and antibody-dependent viral enhancement. Immunology. 1990;70:338–343. [PMC free article] [PubMed] [Google Scholar]
- 106.Takada A, Feldmann H, Ksiazek TG, Kawaoka Y. Antibody-dependent enhancement of Ebola virus infection. J. Virol. 2003;77:7539–7544. doi: 10.1128/JVI.77.13.7539-7544.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Takada A, Watanabe S, Okazaki K, Kida H, Kawaoka Y. Infectivity-enhancing antibodies to Ebola virus glycoprotein. J. Virol. 2001;75:2324–2330. doi: 10.1128/JVI.75.5.2324-2330.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kuzmina NA, et al. Antibody-dependent enhancement of Ebola virus infection by human antibodies isolated from survivors. Cell Rep. 2018;24:1802–1815. doi: 10.1016/j.celrep.2018.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hessell AJ, et al. Fc receptor but not complement binding is important in antibody protection against HIV. Nature. 2007;449:101–104. doi: 10.1038/nature06106. [DOI] [PubMed] [Google Scholar]
- 110.Yates NL, et al. Vaccine-induced Env V1–V2 IgG3 correlates with lower HIV-1 infection risk and declines soon after vaccination. Sci. Transl Med. 2014;6:228ra239. doi: 10.1126/scitranslmed.3007730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Haynes BF, et al. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N. Engl. J. Med. 2012;366:1275–1286. doi: 10.1056/NEJMoa1113425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Forthal DN, et al. FcγRIIa genotype predicts progression of HIV infection. J. Immunol. 2007;179:7916–7923. doi: 10.4049/jimmunol.179.11.7916. [DOI] [PubMed] [Google Scholar]
- 113.Rao GK, et al. In vivo assessment of antibody-dependent enhancement of influenza B infection. Toxicol. Sci. 2019;169:409–421. doi: 10.1093/toxsci/kfz053. [DOI] [PubMed] [Google Scholar]
- 114.Gunn BM, et al. A role for Fc function in therapeutic monoclonal antibody-mediated protection against Ebola virus. Cell Host Microbe. 2018;24:221–233. doi: 10.1016/j.chom.2018.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.He W, et al. Epitope specificity plays a critical role in regulating antibody-dependent cell-mediated cytotoxicity against influenza A virus. Proc. Natl Acad. Sci. USA. 2016;113:11931–11936. doi: 10.1073/pnas.1609316113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Corti D, et al. Protective monotherapy against lethal Ebola virus infection by a potently neutralizing antibody. Science. 2016;351:1339–1342. doi: 10.1126/science.aad5224. [DOI] [PubMed] [Google Scholar]
- 117.Corti D, et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science. 2011;333:850–856. doi: 10.1126/science.1205669. [DOI] [PubMed] [Google Scholar]
- 118.Caskey M, et al. Antibody 10-1074 suppresses viremia in HIV-1-infected individuals. Nat. Med. 2017;23:185–191. doi: 10.1038/nm.4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Scheid JF, et al. HIV-1 antibody 3BNC117 suppresses viral rebound in humans during treatment interruption. Nature. 2016;535:556–560. doi: 10.1038/nature18929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bar KJ, et al. Effect of HIV antibody VRC01 on viral rebound after treatment interruption. N. Engl. J. Med. 2016;375:2037–2050. doi: 10.1056/NEJMoa1608243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lynch RM, et al. Virologic effects of broadly neutralizing antibody VRC01 administration during chronic HIV-1 infection. Sci. Transl Med. 2015;7:319ra206. doi: 10.1126/scitranslmed.aad5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gaudinski MR, et al. Safety, tolerability, pharmacokinetics, and immunogenicity of the therapeutic monoclonal antibody mAb114 targeting Ebola virus glycoprotein (VRC 608): an open-label phase 1 study. Lancet. 2019;393:889–898. doi: 10.1016/S0140-6736(19)30036-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hershberger E, et al. Safety and efficacy of monoclonal antibody VIS410 in adults with uncomplicated influenza A infection: results from a randomized, double-blind, phase-2, placebo-controlled study. EBioMedicine. 2019;40:574–582. doi: 10.1016/j.ebiom.2018.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ali SO, et al. Evaluation of MEDI8852, an anti-Influenza A monoclonal antibody, in treating acute uncomplicated Influenza. Antimicrob. Agents Chemother. 2018;62:e00694–e00718. doi: 10.1128/AAC.00694-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kim HW, et al. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am. J. Epidemiol. 1969;89:422–434. doi: 10.1093/oxfordjournals.aje.a120955. [DOI] [PubMed] [Google Scholar]
- 126.Fulginiti VA, Eller JJ, Downie AW, Kempe CH. Altered reactivity to measles virus. Atypical measles in children previously immunized with inactivated measles virus vaccines. JAMA. 1967;202:1075–1080. doi: 10.1001/jama.202.12.1075. [DOI] [PubMed] [Google Scholar]
- 127.Polack FP, et al. A role for immune complexes in enhanced respiratory syncytial virus disease. J. Exp. Med. 2002;196:859–865. doi: 10.1084/jem.20020781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ruckwardt TJ, Morabito KM, Graham BS. Immunological lessons from respiratory syncytial virus vaccine development. Immunity. 2019;51:429–442. doi: 10.1016/j.immuni.2019.08.007. [DOI] [PubMed] [Google Scholar]
- 129.Graham BS, et al. Priming immunization determines T helper cytokine mRNA expression patterns in lungs of mice challenged with respiratory syncytial virus. J. Immunol. 1993;151:2032–2040. [PubMed] [Google Scholar]
- 130.Openshaw PJM, Chiu C, Culley FJ, Johansson C. Protective and harmful immunity to RSV infection. Annu. Rev. Immunol. 2017;35:501–532. doi: 10.1146/annurev-immunol-051116-052206. [DOI] [PubMed] [Google Scholar]
- 131.Rajao DS, et al. Heterologous challenge in the presence of maternally-derived antibodies results in vaccine-associated enhanced respiratory disease in weaned piglets. Virology. 2016;491:79–88. doi: 10.1016/j.virol.2016.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rajão DS, Loving CL, Gauger PC, Kitikoon P, Vincent AL. Influenza A virus hemagglutinin protein subunit vaccine elicits vaccine-associated enhanced respiratory disease in pigs. Vaccine. 2014;32:5170–5176. doi: 10.1016/j.vaccine.2014.07.059. [DOI] [PubMed] [Google Scholar]
- 133.Skowronski DM, et al. Randomized controlled ferret study to assess the direct impact of 2008-09 trivalent inactivated influenza vaccine on A(H1N1)pdm09 disease risk. PLoS ONE. 2014;9:e86555. doi: 10.1371/journal.pone.0086555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kobinger GP, et al. Assessment of the efficacy of commercially available and candidate vaccines against a pandemic H1N1 2009 virus. J. Infect. Dis. 2010;201:1000–1006. doi: 10.1086/651171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Monsalvo AC, et al. Severe pandemic 2009 H1N1 influenza disease due to pathogenic immune complexes. Nat. Med. 2011;17:195–199. doi: 10.1038/nm.2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Caidi H, et al. Anti-respiratory syncytial virus (RSV) G monoclonal antibodies reduce lung inflammation and viral lung titers when delivered therapeutically in a BALB/c mouse model. Antivir. Res. 2018;154:149–157. doi: 10.1016/j.antiviral.2018.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Jacobino SR, et al. Reformatting palivizumab and motavizumab from IgG to human IgA impairs their efficacy against RSV infection in vitro and in vivo. MAbs. 2018;10:453–462. doi: 10.1080/19420862.2018.1433974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cortjens B, et al. Broadly reactive anti-respiratory syncytial virus G antibodies from exposed individuals effectively inhibit infection of primary airway epithelial cells. J. Virol. 2017;91:e02357–e02416. doi: 10.1128/JVI.02357-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Collarini EJ, et al. Potent high-affinity antibodies for treatment and prophylaxis of respiratory syncytial virus derived from B cells of infected patients. J. Immunol. 2009;183:6338–6345. doi: 10.4049/jimmunol.0901373. [DOI] [PubMed] [Google Scholar]
- 140.Hiatt A, et al. Glycan variants of a respiratory syncytial virus antibody with enhanced effector function and in vivo efficacy. Proc. Natl Acad. Sci. USA. 2014;111:5992–5997. doi: 10.1073/pnas.1402458111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Maestri A, et al. The His131Arg substitution in the FCGR2A gene (rs1801274) is not associated with the severity of influenza A(H1N1)pdm09 infection. BMC Res. Notes. 2016;9:296. doi: 10.1186/s13104-016-2096-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chatzopoulou F, Gioula G, Kioumis I, Chatzidimitriou D, Exindari M. Identification of complement-related host genetic risk factors associated with influenza A(H1N1)pdm09 outcome: challenges ahead. Med. Microbiol. Immunol. 2019;208:631–640. doi: 10.1007/s00430-018-0567-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wan Y, et al. Molecular mechanism for antibody-dependent enhancement of coronavirus entry. J. Virol. 2020;94:e02015–e02019. doi: 10.1128/JVI.02015-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wang SF, et al. Antibody-dependent SARS coronavirus infection is mediated by antibodies against spike proteins. Biochem. Biophys. Res. Commun. 2014;451:208–214. doi: 10.1016/j.bbrc.2014.07.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yip MS, et al. Antibody-dependent infection of human macrophages by severe acute respiratory syndrome coronavirus. Virol. J. 2014;11:82. doi: 10.1186/1743-422X-11-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jaume M, et al. Anti-severe acute respiratory syndrome coronavirus spike antibodies trigger infection of human immune cells via a pH- and cysteine protease-independent FcγR pathway. J. Virol. 2011;85:10582–10597. doi: 10.1128/JVI.00671-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kam YW, et al. Antibodies against trimeric S glycoprotein protect hamsters against SARS-CoV challenge despite their capacity to mediate FcγRII-dependent entry into B cells in vitro. Vaccine. 2007;25:729–740. doi: 10.1016/j.vaccine.2006.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lee N, et al. Anti-SARS-CoV IgG response in relation to disease severity of severe acute respiratory syndrome. J. Clin. Virol. 2006;35:179–184. doi: 10.1016/j.jcv.2005.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Luo F, et al. Evaluation of antibody-dependent enhancement of SARS-CoV infection in Rhesus macaques immunized with an inactivated SARS-CoV vaccine. Virol. Sin. 2018;33:201–204. doi: 10.1007/s12250-018-0009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wang Q, et al. Immunodominant SARS coronavirus epitopes in humans elicited both enhancing and neutralizing effects on infection in non-human primates. ACS Infect. Dis. 2016;2:361–376. doi: 10.1021/acsinfecdis.6b00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Agrawal AS, et al. Immunization with inactivated Middle East respiratory syndrome coronavirus vaccine leads to lung immunopathology on challenge with live virus. Hum. Vaccin. Immunother. 2016;12:2351–2356. doi: 10.1080/21645515.2016.1177688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Liu L, et al. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight. 2019;4:e123158. doi: 10.1172/jci.insight.123158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Yasui F, et al. Prior immunization with severe acute respiratory syndrome (SARS)-associated coronavirus (SARS-CoV) nucleocapsid protein causes severe pneumonia in mice infected with SARS-CoV. J. Immunol. 2008;181:6337–6348. doi: 10.4049/jimmunol.181.9.6337. [DOI] [PubMed] [Google Scholar]
- 154.Yuan FF, et al. Influence of FcγRIIA and MBL polymorphisms on severe acute respiratory syndrome. Tissue Antigens. 2005;66:291–296. doi: 10.1111/j.1399-0039.2005.00476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gao Q, et al. Rapid development of an inactivated vaccine candidate for SARS-CoV-2. Science. 2020;369:77–81. doi: 10.1126/science.abc1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sia SF, et al. Pathogenesis and transmission of SARS-CoV-2 in golden hamsters. Nature. 2020;583:834–838. doi: 10.1038/s41586-020-2342-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Rogers TF, et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science. 2020 doi: 10.1126/science.abc7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Cleary SJ, et al. Animal models of mechanisms of SARS-CoV-2 infection and COVID-19 pathology. Br. J. Pharmacol. 2020 doi: 10.1111/bph.15143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bournazos S. IgG Fc receptors: evolutionary considerations. Curr. Top. Microbiol. Immunol. 2019;423:1–11. doi: 10.1007/82_2019_149. [DOI] [PubMed] [Google Scholar]
- 160.Bournazos S, DiLillo DJ, Ravetch JV. Humanized mice to study FcγR function. Curr. Top. Microbiol. Immunol. 2014;382:237–248. doi: 10.1007/978-3-319-07911-0_11. [DOI] [PubMed] [Google Scholar]
- 161.Smith P, DiLillo DJ, Bournazos S, Li F, Ravetch JV. Mouse model recapitulating human Fcγ receptor structural and functional diversity. Proc. Natl Acad. Sci. USA. 2012;109:6181–6186. doi: 10.1073/pnas.1203954109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lee CH, et al. An engineered human Fc domain that behaves like a pH-toggle switch for ultra-long circulation persistence. Nat. Commun. 2019;10:5031. doi: 10.1038/s41467-019-13108-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Joyner M, et al. Early safety indicators of COVID-19 convalescent plasma in 5,000 patients. J. Clin. Invest. 2020 doi: 10.1101/2020.05.12.20099879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lv H, et al. Cross-reactive antibody response between SARS-CoV-2 and SARS-CoV infections. Cell Rep. 2020;31:107725. doi: 10.1016/j.celrep.2020.107725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Channappanavar R, et al. Dysregulated type I interferon and inflammatory monocyte–macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe. 2016;19:181–193. doi: 10.1016/j.chom.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]