

Abstract
Chemoresistance is a hallmark of cancer stem cells (CSCs). To develop novel therapeutic strategies that target CSCs, we established osteosarcoma‐initiating (OSi) cells by introducing the c‐Myc gene into bone marrow stromal cells derived from Ink4a/Arf KO mice. These OSi cells include bipotent committed cells (similar to osteochondral progenitor cells) with a high tumorigenic activity as well as tripotent cells (similar to mesenchymal stem cells) of low tumorigenicity. We recently showed that the tripotent OSi cells are highly resistant to chemotherapeutic agents, and that depolymerization of the actin cytoskeleton in these cells induces their terminal adipocyte differentiation and suppresses their tumorigenicity. We here provide an overview of modulation of actin cytoskeleton dynamics associated with terminal adipocyte differentiation in osteosarcoma as well as discuss the prospects for new therapeutic strategies that target chemoresistant CSCs by inducing their differentiation.
Keywords: actin, adipocyte, cancer stem cell, differentiation, osteosarcoma
This review article describes an overview of modulation of actin cytoskeleton dynamics associated with terminal adipocyte differentiation in osteosarcoma. We also discuss the prospects for new therapeutic strategies that target chemoresistant cancer stem cells by inducing their differentiation.
1. INTRODUCTION
Tumors are heterogeneous tissues comprised of various tumor cell types, including cancer stem cells (CSCs), which manifest self‐renewal capacity and pluripotency, as well as progenitor cells and more differentiated cells, with a hierarchy similar to that of normal tissues. 1 , 2 Given that tumors are maintained by CSCs and that these cells are able to survive conventional anticancer treatments and to evade immune responses, thereby giving rise to therapeutic resistance, disease recurrence, and metastasis, CSCs are thought to be an attractive target for novel therapies aimed at eliminating cancer cells and eradicating cancer. 3 In addition to agents that directly target CSCs for elimination, drugs that induce senescence or terminal differentiation in these cells or that trigger their conversion to other cell types that are sensitive to conventional anticancer treatments have the potential to achieve cancer eradication. 2 Recent evidence has indeed suggested that the plasticity of CSCs can be exploited by differentiation therapy to achieve depletion of the CSC pool. Differentiation therapy has long been considered a potential approach to suppression of tumorigenesis through conversion of undifferentiated cancer cells of high malignancy into differentiated cells of low tumorigenicity. In this review, we address the concept of induction of differentiation in CSCs, including its mechanisms and relevance to potential novel treatment strategies.
2. CANCER STEM CELLS AND TUMOR HETEROGENEITY
Intratumoral heterogeneity among tumor cells within a given tumor is apparent at both phenotypic and functional levels as a result of genetic alterations, environmental influences, and reversible changes in cellular properties. 4 Heterogeneity of tumor tissue has been accounted for in recent years by a hierarchy‐based model in which CSCs have the ability both to self‐renew and to give rise to differentiated tumor cells and are responsible for the overall organization of a tumor. 2 , 5 , 6
Cancer stem cells are a subpopulation of tumor cells that possess high tumorigenic activity and represent the top of a hierarchical organization similar to that of normal tissues. Cancer stem cells are thought to arise either from normal tissue stem cells through the acquisition of malignant properties due to mutations or changes in gene expression or from the spontaneous dedifferentiation of tumor cells. 7 , 8 The tumor microenvironment has also been implicated in both the generation and maintenance of CSCs. 6 , 9 Ever since the first experimental identification of CSCs, the CSC‐based hierarchical model has been a major topic of debate in the field of cancer biology as a result of uncertainties concerning the properties of these cells, such as their defining cell surface markers as well as their frequency and plasticity. 10 , 11 , 12 , 13 , 14 We believe that the introduction of the concept of CSCs has led to significant advances in cancer research, having provided a better understanding of tumor heterogeneity, as well as served as the basis for the development of novel therapeutic strategies that target subpopulations of tumor cells with a high tumorigenic potential.
Chemoresistance is a key characteristic of CSCs. Much evidence thus now supports the existence of CSCs that are highly resistant to classical anticancer drugs in comparison with other cancer cells. Cancer stem cells are thought to play a key role in disease relapse after termination of anticancer treatment, 11 , 15 and targeting of CSCs at the top of the hierarchical organization of tumors is a key potential strategy for eradication of cancer tissue.
3. TRANSDIFFERENTIATION THERAPY FOR NONCANCER DISEASES
Transdifferentiation is defined as the conversion of one mature somatic cell type to another, and many cell types have been found to be capable of this process. 16 , 17 Forced expression of lineage‐specific transcription factors has been shown to induce the transdifferentiation of fibroblasts into neurons, 18 cardiomyocytes, 19 or endothelial cells. 20 , 21 In addition to transcription factors, microRNAs have been found to trigger transdifferentiation through posttranscriptional regulation. 22
Disorders of differentiation underlie some pathological conditions and noncancer diseases, and transdifferentiation‐based therapies that correct differentiation abnormalities are thus under development. Strategies for the induction of therapeutic transdifferentiation that do not rely on viral vectors for overexpression of transcription factors are under investigation for ischemic heart disease. 23 An approach based on the activation of innate immune signaling by stimulation of Toll‐like receptors (TLRs) has thus been found to be effective for the conversion of fibroblasts into cardiomyocytes. Activation of such signaling through TLRs and the transcription factor nuclear factor‐κB triggers epigenetic changes such as DNA demethylation and chromatin modification that affect cellular plasticity. For orthopedic disorders such as osteoporosis and bone dysplasia, transdifferentiation of myoblasts or adipocytes into osteoblasts also has the potential to provide a novel therapy. 17 , 24 Many studies have now provided a proof‐of‐concept for therapeutic transdifferentiation.
The production of differentiated cells from embryonic stem cells, induced pluripotent stem cells, or dedifferentiated cells has also been thought to be a potential effective strategy for differentiation therapy. Although transdifferentiation was originally defined as a process by which differentiated cells undergo a change in lineage without going through an intermediate cell stage, it remains to be determined whether transdifferentiation does actually occur without passage through progenitor cell or undifferentiated cell stages.
4. CANCER STEM CELL DIFFERENTIATION OR CONVERSION
Similar to normal tissue stem cells, some CSCs might also have the potential to differentiate into cell lineages other than the original lineage from which the tumor arose. 2 Such plasticity of CSCs is considered to be a promising therapeutic target. 7 The concept of CSC differentiation therapy encompasses induction of terminal differentiation in CSCs or of their conversion into nonstem cells that are sensitive to conventional anticancer treatments. Differentiation therapy has thus been under consideration as a means to suppress tumorigenesis through conversion of undifferentiated cancer cells of high malignancy into differentiated cells with low tumorigenicity. Cancer stem cell differentiation therapy could attenuate the malignant potential of a tumor or suppress its aggressive behavior. It also offers a therapeutic strategy to deplete the CSC pool and eradicate cancer.
All‐trans retinoic acid has been established as an effective treatment for patients with acute promyelocytic leukemia. It elicits terminal granulocytic differentiation in the leukemia cells by impairing transcriptional repression of genes necessary for differentiation. 25 Although much research has been done, limited evidence is available at present regarding the applicability of such a strategy to solid tumors. 26 , 27 , 28 A better understanding of the mechanisms underlying CSC differentiation or conversion as well as improvements in cellular reprogramming technology that increase the efficiency and quality and reduce the risk of this promising therapeutic approach will benefit many patients in the future.
5. ESTABLISHMENT OF INDUCED CSCS
To develop novel therapeutic strategies that target chemoresistant CSCs, we set out to isolate subpopulations of tumor cells that acquire chemoresistance for study as CSCs. We established induced CSCs (iCSCs) from mouse somatic stem or progenitor cells of various tissues by forced expression of a set of defined factors. 2 Retroviral transduction with oncogenic driver genes such as Myc or Ras has thus given rise to several types of iCSCs capable of forming tumors after transplantation in recipient mice. We have established iCSC mouse models of osteosarcoma, 29 choriocarcinoma, 30 glioblastoma, 31 ovarian cancer, 32 and leukemia‐lymphoma. 33 These iCSCs possess the abilities to undergo both self‐renewal and differentiation, and the iCSC‐derived tumors in mice show phenotypes similar to those of the corresponding human cancers, including excessive growth, heterogeneity, and invasive or metastatic abilities.
6. OSTEOSARCOMA CSC MODEL
Osteosarcoma is the most common primary malignant bone cancer in childhood and adolescence 34 and comprises heterogeneous cell types including CSC‐like cells, progenitor cells, and differentiated cells. 35 Although chemotherapy regimens based on doxorubicin or cisplatin have improved the survival rate of individuals with osteosarcoma, 20% to 30% of patients are refractory to these conventional treatments. 36 , 37 , 38 The mechanisms underlying the survival of chemoresistant cells and which types of tumor cells are responsible for such resistance are unclear.
We have established osteosarcoma‐initiating (OSi) cells by introducing the gene for c‐Myc into bone marrow stromal cells of Ink4a/Arf KO mice. Mice injected with these OSi cells develop lethal osteosarcoma with undifferentiated and osteo‐ or chondro‐like differentiated areas. 29 These OSi cells include AX cells, which have a bilineage (osteogenic and chondrogenic) differentiation potential similar to that of osteochondro progenitor cells, as well as AO cells, which have a trilineage (adipogenic, osteogenic, and chondrogenic) differentiation potential similar to that of mesenchymal stem cells (MSCs).
Mesenchymal stem cells are pluripotent cells that have the ability to give rise to osteoblasts, chondrocytes, skeletal muscle cells, and adipocytes. 39 The differentiation fate of a particular lineage is determined by growth factors, ECM proteins, cytokines, and cell‐to‐cell contact, 40 , 41 with differentiation being executed through the action of various transcription factors (Figure 1). The differentiation of MSCs into preadipocytes during adipogenesis is induced by the action of insulin, dexamethasone, cyclic AMP, and bone morphogenetic protein 4. 24 Transcription factors that contribute to adipocyte differentiation include CCAAT/enhancer binding protein‐α (C/EBPα), C/EBPβ, and peroxisome proliferator‐activated receptor‐γ (PPARγ).
FIGURE 1.
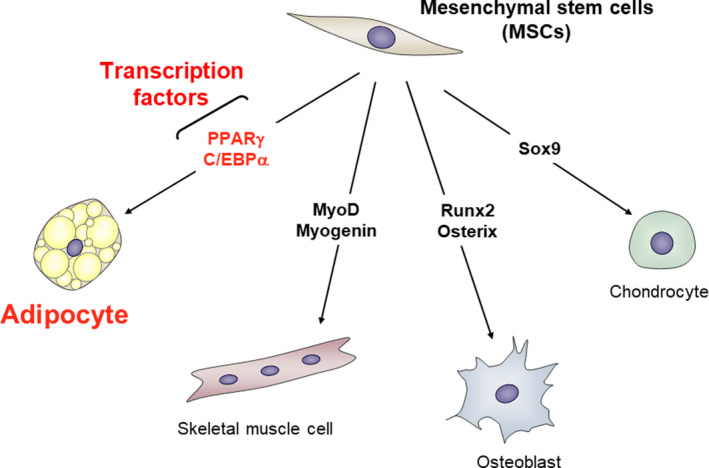
Defined transcription factors regulate cellular differentiation. Cellular differentiation is strictly regulated by defined transcription factors. Mesenchymal stem cells (MSCs) differentiate into adipocytes, skeletal muscle cells, osteoblasts, and chondrocytes. Adipocyte differentiation is directly regulated by the transcription factors CCAAT/enhancer binding protein‐α (C/EBPα) and peroxisome proliferator‐activated receptor‐γ (PPARγ), the latter of which controls the expression of various adipocyte‐specific genes that leads to terminal adipocyte differentiation. Skeletal muscle cell differentiation is regulated by MyoD and myogenin, osteoblast differentiation by Runx2 and Osterix, and chondrocyte differentiation by Sox9
We found that AX cells in our OSi model of osteosarcoma possess a higher tumorigenic activity than do AO cells, whereas AO cells have a higher efflux capacity compared with AX cells. AO cells, unlike AX cells, are also highly resistant to conventional anticancer drugs (doxorubicin and cisplatin). 42 In fact, when AO cells were transplanted into mice, they were observed to lose their ability to differentiate into adipocytes as tumors grow and convert into AX‐like cells. 29 Considering these properties—that proliferation is slower than AX, resistant to treatment, and tripotent differentiation ability like MSC—it can be proposed that AO cells are bona fide CSCs. In addition, AO cells express PPARγ, a master regulator of adipogenesis, at a high level. These observations suggest that the high intrinsic efflux capacity and the adipogenic differentiation potential of AO cells are related to their chemoresistance and CSC‐like features. Consistent with this notion, we found that the proportion of AO‐like cancer cells expressing PPARγ is increased in human recurrent osteosarcoma tissue compared with primary tumor tissue. The induction of terminal adipocyte differentiation might thus provide a novel approach to suppression of chemoresistant AO‐like CSCs in osteosarcoma.
7. ACTIN CYTOSKELETON DYNAMICS AND ADIPOCYTE DIFFERENTIATION
Mechanical or physical stimuli, such as matrix stiffness and cytoskeletal tension, regulate cell fate determination. 43 Mesenchymal stem cells thus differentiate into osteoblasts when seeded on rigid matrices, into myoblasts when grown on matrices with intermediate stiffness, and into adipocytes when cultured on soft matrices. 44 Mesenchymal stem cells also differentiate into osteoblasts when allowed to flatten and spread by plating on large islands, whereas they differentiate into adipocytes when they are confined to a round shape by plating on small islands. 45 Moreover, when MSCs were patterned into 3 different geometric shapes and cultured with a mixture of adipogenic and osteogenic media, cells with a flower shape characterized by large convex curves along each edge preferentially adopted an adipogenic lineage, those with a pentagonal shape characterized by straight lines for each edge differentiated into both adipocytes and osteoblasts, and those with a star shape characterized by concave edges and sharp points at the vertices favored an osteogenic fate. 46 Such mechanical cues have been found to influence the activity of RhoA–ROCK (Rho‐kinase) intracellular signaling, which in turn regulates dynamics of the actin cytoskeleton, 47 , 48 and ROCK‐induced cytoskeletal tension and downstream effects have been found to regulate the lineage commitment of MSCs. 44 , 45 , 46 , 49
Signaling by the small GTPase RhoA and the kinase ROCK negatively regulates adipocyte differentiation of MSCs or preadipocytes. 50 , 51 However, some studies have suggested that this regulation of adipogenesis is attributable to a cytoskeleton‐independent action of RhoA‐ROCK signaling. 52 , 53 We attempted to clarify the role of RhoA‐ROCK signaling in adipocyte differentiation. 50 We found that, before induction of adipogenesis, dedifferentiated fat (DFAT) cells, a preadipocyte cell line derived from isolated mature adipocytes, 54 manifest a fibroblastic morphology with well‐developed actin stress fibers associated with a high level of RhoA activity. Exposure of the cells to an adipogenic cocktail resulted in the rapid depolymerization of actin stress fibers as a result of downregulation of RhoA activity, with this effect then allowing expression of PPARγ and consequent adipogenic differentiation. We also recently found that Rac1, which is a downstream effector of the insulin‐PI3K signaling pathway, promotes the formation of adipocyte‐associated cortical actin structures, which is essential for completion of adipocyte differentiation, 55 suggesting that actin cytoskeleton dynamics play a key role in regulation of the overall adipocyte differentiation program.
8. REGULATION OF ADIPOCYTE DIFFERENTIATION BY ACTIN CYTOSKELETON DYNAMICS
Molecular mechanisms by which dynamics of the actin cytoskeleton directly affect cell differentiation through the control of gene expression have been identified. 56 , 57 The transcriptional coregulators Yes‐associated protein (YAP) and transcriptional coactivator with PDZ binding motif (TAZ) have been identified as mechanotransducers that mediate the effects of actin cytoskeleton dynamics or ECM stiffness, with RhoA‐ROCK signaling being essential for the nuclear translocation and function of these factors. 57 , 58 Yes‐associated protein and TAZ were found to be localized to the nucleus and active in MSCs subjected to strong mechanical forces by culture on a large adhesive area or on a rigid matrix, resulting in the induction of osteogenic differentiation. 59 In contrast, YAP and TAZ are inactive and localized to the cytoplasm in MSCs subjected to weak mechanical forces by culture on a small adhesive area or a soft matrix, resulting in the induction of adipogenic differentiation. 59
Megakaryoblastic leukemia 1 (MKL1, also known as MAL or MRTF‐A) is a transcriptional coregulator of serum response factor, and the binding of MKL1 to monomeric (globular, or G) actin produced as a result of the depolymerization of filamentous (F) actin prevents its translocation to the nucleus and thereby inhibits its transcriptional function. 60 , 61 We examined whether the control of MKL1 translocation by actin cytoskeleton dynamics contributes to the regulation of adipocyte differentiation. The induction of adipocyte differentiation in multipotent DFAT cells was associated with the rapid depolymerization of F‐actin, resulting in an increase in the G‐actin concentration. Although MKL1 was exclusively localized to the nucleus before adipogenic induction, it was predominantly cytoplasmic after such induction. We also examined the effects of the actin‐depolymerizing agents latrunculin A (LatA), which increases the concentration of G‐actin, 60 , 62 and swinholide A (SwinA), which increases the concentration of a dimeric form of actin that does not interact with MKL1. 60 , 62 , 63 Both LatA and SwinA induced depolymerization of F‐actin stress fibers and increased the cellular abundance of G‐actin monomers or actin dimers, respectively. Whereas LatA induced the cytoplasmic sequestration of MKL1 and the expression of PPARγ, SwinA did not alter the nuclear localization of MKL1 and did not induce PPARγ expression, suggesting that MKL1 contributes to suppression of the expression of PPARγ in the nucleus and that the increase in the cytoplasmic abundance of G‐actin triggers adipocyte differentiation by preventing the nuclear translocation of MKL1 (Figure 2). Megakaryoblastic leukemia 1 has also been shown to inhibit brown adipogenesis through a direct inhibitory interaction with PPARγ, 64 suggesting that disruption of such interaction between MKL1 and PPARγ might also be important for initiation of adipocyte differentiation. Given that both LatA and SwinA reduced mechanical force by inducing the depolymerization of F‐actin stress fibers, but that the expression of PPARγ was induced only by LatA, we propose that the negative regulation of MKL1 by monomeric G‐actin rather than inactivation of YAP and TAZ is key to the initiation of adipocyte differentiation. However, given that YAP‐TAZ and MKL1 physically interact with each other and function in a mutually dependent manner, 65 the inhibition of YAP‐TAZ signaling might also play a role in the induction of adipogenesis by depolymerization of F‐actin stress fibers. Our findings indicate that MKL1 functions as a gatekeeper that controls adipocyte differentiation.
FIGURE 2.
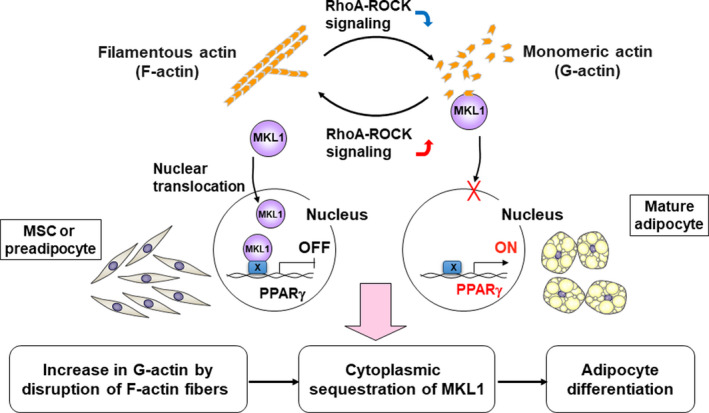
Regulation of megakaryoblastic leukemia 1 (MKL1) through actin cytoskeleton dynamics drives adipocyte differentiation. During adipogenesis, cell morphology changes markedly from fibroblastic (mesenchymal stem cells [MSCs] or preadipocytes) to spherical (adipocytes) in association with a dynamic change in the actin cytoskeleton characterized by the depolymerization of F‐actin stress fibers and the formation of cortical F‐actin structures. This remodeling of the actin cytoskeleton drives peroxisome proliferator‐activated receptor‐γ (PPARγ)‐mediated adipocyte differentiation in MSCs or preadipocytes through inhibition of the transcriptional regulator MKL1. In undifferentiated cells, MKL1 is localized predominantly to the nucleus, where it suppresses PPARγ gene expression. The inactivation of RhoA‐Rho‐kinase (ROCK) signaling induces the depolymerization of F‐actin stress fibers and the consequent cytoplasmic sequestration of MKL1 by monomeric G‐actin, resulting in initiation of PPARγ gene expression and terminal adipocyte differentiation
Mechanical forces have also been shown to regulate gene expression through global chromatin remodeling. 66 , 67 Growth of cells on rigid substrates promotes actin polymerization, which in turn induces nuclear localization of MKL1 as well as cytosolic localization of histone deacetylase 3 (HDAC3). In contrast, growth of cells on soft substrates promotes actin depolymerization, which induces cytosolic localization of MKL1 as well as nuclear localization of HDAC3. The mechanism by which dynamics of the actin cytoskeleton drive adipocyte differentiation might thus involve not only MKL1‐mediated transcriptional regulation but also epigenetic modification. In addition, MICAL‐2, an atypical actin‐regulatory protein, induces depolymerization of nuclear actin through redox modification of methionine, and the resulting oxidized G‐actin is unable to bind to MKL1, resulting in nuclear retention of and consequent transcriptional regulation by MKL1. 61 , 68 , 69 , 70 Redox‐mediated actin remodeling is thus another potential means for control of adipocyte differentiation through regulation of MKL1 in a RhoA‐ and ROCK‐independent manner.
9. PROPOSED DIFFERENTIATION THERAPY TARGETED TO OSTEOSARCOMA STEM CELLS
On the basis of our observations, we believe that differentiation therapy has the potential to be effective against chemoresistant cancer, including solid tumors. In our osteosarcoma model, AO cells show a high level of resistance to conventional chemotherapeutic agents such as doxorubicin, whereas AX cells are eliminated by doxorubicin treatment. 42 We also found that the clinically administered ROCK inhibitor fasudil elicited terminal adipocyte differentiation in AO cells through negative regulation of MKL1 associated with actin cytoskeleton dynamics. 42 Fasudil treatment attenuated tumorigenesis by AO cells, 42 indicating that the conversion of these bona fide CSCs into terminally differentiated adipocytes results in tumor suppression. Given that human osteosarcoma tumors are composed of heterogeneous cell types, including chemoresistant stemlike tumor cells, we propose a novel therapeutic option for such tumors (Figure 3): treatment with doxorubicin to eliminate AX‐type chemosensitive tumor cells, followed by treatment with fasudil to convert the remaining AO‐type chemoresistant cells into terminally differentiated adipocytes. The combination of conventional chemotherapy and differentiation therapy might also provide a potential treatment approach for other types of heterogeneous solid tumors that include chemoresistant CSCs.
FIGURE 3.
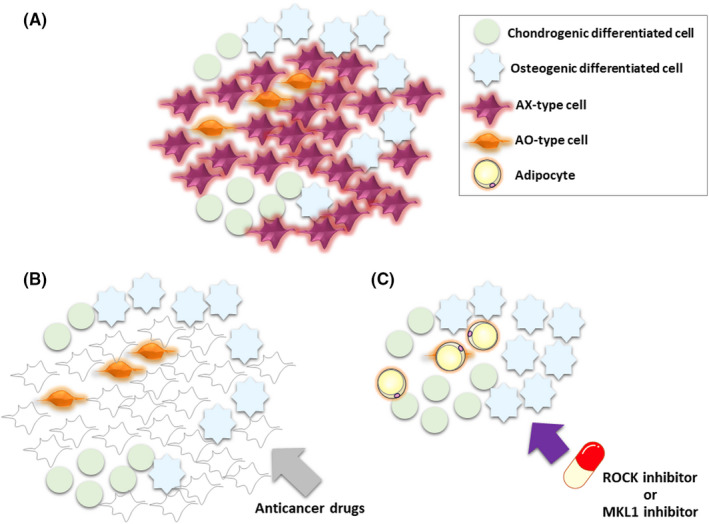
Proposed differentiation therapy for osteosarcoma cancer stem cells. Osteosarcoma is a heterogeneous tissue that includes AX‐type cells, AO‐type cells, chondrogenic differentiated cells, and osteogenic differentiated cells (A). We propose a differentiation‐based therapeutic strategy for osteosarcoma in which treatment with conventional anticancer drugs eliminates AX‐type tumor cells (B) and then treatment with a Rho‐kinase (ROCK) or megakaryoblastic leukemia 1 (MKL1) inhibitor induces terminal adipocyte differentiation of AO‐type chemoresistant stemlike cells through modulation of the actin cytoskeleton (C)
10. CONCLUSION
Our findings suggest a new therapeutic strategy for heterogeneous osteosarcoma based on the combination of conventional chemotherapy and the induction of cell differentiation‐conversion through modulation of actin cytoskeleton dynamics in chemoresistant osteosarcoma stem cells. The clinical application of such a therapeutic strategy will be facilitated by the development of quantitative biomarkers for assessment of the effects of differentiation inducers or the establishment of imaging systems to monitor the differentiation state of tumor cells in the human body.
CONFLICT OF INTEREST
Hideyuki Saya serves as a consultant to RBI and received research grants from Daiichi‐Saynkyo, Esai, Nihon Noyaku, and JSR.
Arima Y, Nobusue H, Saya H. Targeting of cancer stem cells by differentiation therapy. Cancer Sci. 2020;111:2689–2695. 10.1111/cas.14504
REFERENCES
- 1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 2. Sugihara E, Saya H. Complexity of cancer stem cells. Int J Cancer. 2013;132:1249‐1259. [DOI] [PubMed] [Google Scholar]
- 3. Sato R, Semba T, Saya H, Arima Y. Stem cells and epithelial‐mesenchymal transition in cancer: biological implications and therapeutic targets. Stem Cells. 2016;34:1997‐2007. [DOI] [PubMed] [Google Scholar]
- 4. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10:717‐728. [DOI] [PubMed] [Google Scholar]
- 6. Lawson DA, Kessenbrock K, Davis RT, Pervolarakis N, Werb Z. Tumor heterogeneity and metastasis at single‐cell resolution. Nat Cell Biol. 2018;20:1349‐1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shekhani MT, Jayanthy AS, Maddodi N, Setaluri V. Cancer stem cells and tumor transdifferentiation: implications for novel therapeutic strategies. Am J Stem Cells. 2013;2:52‐61. [PMC free article] [PubMed] [Google Scholar]
- 8. Huang Z, Wu T, Liu AY, Ouyang G. Differentiation and transdifferentiation potentials of cancer stem cells. Oncotarget. 2015;6:39550‐39563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fessler E, Dijkgraaf FE, De Sousa E, Melo F, Medema JP. Cancer stem cell dynamics in tumor progression and metastasis: is the microenvironment to blame? Cancer Lett. 2013;341:97‐104. [DOI] [PubMed] [Google Scholar]
- 10. Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012;21:283‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23:1124‐1134. [DOI] [PubMed] [Google Scholar]
- 12. da Silva‐Diz V, Lorenzo‐Sanz L, Bernat‐Peguera A, Lopez‐Cerda M, Muñoz P. Cancer cell plasticity: impact on tumor progression and therapy response. Semin Cancer Biol. 2018;53:48‐58. [DOI] [PubMed] [Google Scholar]
- 13. Gupta PB, Pastushenko I, Skibinski A, Blanpain C, Kuperwasser C. Phenotypic plasticity: driver of cancer initiation, progression, and therapy resistance. Cell Stem Cell. 2019;24:65‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fumagalli A, Oost KC, Kester L, et al. Plasticity of Lgr5‐negative cancer cells drives metastasis in colorectal cancer. Cell Stem Cell. 2020;26:569‐578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kleppe M, Levine RL. Tumor heterogeneity confounds and illuminates: assessing the implications. Nat Med. 2014;20:342‐344. [DOI] [PubMed] [Google Scholar]
- 16. Merrell AJ, Stanger BZ. Adult cell plasticity in vivo: de‐differentiation and transdifferentiation are back in style. Nat Rev Mol Cell Biol. 2016;17:413‐425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lin DPL, Carnagarin R, Dharmarajan A, Dass CR. Transdifferentiation of myoblasts into osteoblasts—possible use for bone therapy. J Pharm Pharmacol. 2017;69:1661‐1671. [DOI] [PubMed] [Google Scholar]
- 18. Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Südhof TC, Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035‐1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ieda M, Fu JD, Delgado‐Olguin P, et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 2010;142:375‐386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ginsberg M, James D, Ding BS, et al. Efficient direct reprogramming of mature amniotic cells into endothelial cells by ETS factors and TGFβ suppression. Cell. 2012;151:559‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Margariti A, Winkler B, Karamariti E, et al. Direct reprogramming of fibroblasts into endothelial cells capable of angiogenesis and reendothelialization in tissue‐engineered vessels. Proc Natl Acad Sci USA. 2012;109:13793‐13798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang H, Li X, Gao S, Sun X, Fang H. Transdifferentiation via transcription factors or microRNAs: current status and perspective. Differentiation. 2015;90:69‐76. [DOI] [PubMed] [Google Scholar]
- 23. Lee J, Sayed N, Hunter A, et al. Activation of innate immunity is required for efficient nuclear reprogramming. Cell. 2012;151:547‐558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lin DPL, Dass CR. Transdifferentiation of adipocytes to osteoblasts: potential for orthopaedic treatment. J Pharm Pharmacol. 2018;70:307‐319. [DOI] [PubMed] [Google Scholar]
- 25. Huang ME, Ye YC, Chen SR, et al. Use of all‐trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood. 1988;72:567‐572. [DOI] [PubMed] [Google Scholar]
- 26. de Thé H. Differentiation therapy revisited. Nat Rev Cancer. 2018;18:117‐127. [DOI] [PubMed] [Google Scholar]
- 27. Basu‐Roy U, Han E, Rattanakorn K, et al. PPARγ agonists promote differentiation of cancer stem cells by restraining YAP transcriptional activity. Oncotarget. 2016;7:60954‐60970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ishay‐Ronen D, Christofori G. Targeting cancer cell metastasis by converting cancer cells into fat. Cancer Res. 2019;79:5471‐5475. [DOI] [PubMed] [Google Scholar]
- 29. Shimizu T, Ishikawa T, Sugihara E, et al. c‐MYC overexpression with loss of Ink4a/Arf transforms bone marrow stromal cells into osteosarcoma accompanied by loss of adipogenesis. Oncogene. 2010;29:5687‐5699. [DOI] [PubMed] [Google Scholar]
- 30. Kobayashi Y, Shimizu T, Naoe H, et al. Establishment of a choriocarcinoma model from immortalized normal extravillous trophoblast cells transduced with HRASV12. Am J Pathol. 2011;179:1471‐1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sampetrean O, Saga I, Nakanishi M, et al. Invasion precedes tumor mass formation in a malignant brain tumor model of genetically modified neural stem cells. Neoplasia. 2011;13:784‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Motohara T, Masuko S, Ishimoto T, et al. Transient depletion of p53 followed by transduction of c‐Myc and K‐Ras converts ovarian stem‐like cells into tumor‐initiating cells. Carcinogenesis. 2011;32:1597‐1606. [DOI] [PubMed] [Google Scholar]
- 33. Sugihara E, Shimizu T, Kojima K, et al. Ink4a and Arf are crucial factors in the determination of the cell of origin and the therapeutic sensitivity of Myc‐induced mouse lymphoid tumor. Oncogene. 2012;31:2849‐2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kansara M, Teng MW, Smyth MJ, Thomas DM. Translational biology of osteosarcoma. Nat Rev Cancer. 2014;14:722‐735. [DOI] [PubMed] [Google Scholar]
- 35. Brown HK, Tellez‐Gabriel M, Heymann D. Cancer stem cells in osteosarcoma. Cancer Lett. 2017;386:189‐195. [DOI] [PubMed] [Google Scholar]
- 36. Chou AJ, Geller DS, Gorlick R. Therapy for osteosarcoma: where do we go from here? Paediatr Drugs. 2008;10:315‐327. [DOI] [PubMed] [Google Scholar]
- 37. Gorlick R. Current concepts on the molecular biology of osteosarcoma. Cancer Treat Res. 2009;152:467‐478. [DOI] [PubMed] [Google Scholar]
- 38. Isakoff MS, Bielack SS, Meltzer P, Gorlick R. Osteosarcoma: current treatment and a collaborative pathway to success. J Clin Oncol. 2015;33:3029‐3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Berendsen AD, Olsen BR. Osteoblast‐adipocyte lineage plasticity in tissue development, maintenance and pathology. Cell Mol Life Sci. 2014;71:493‐497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Scadden DT. The stem‐cell niche as an entity of action. Nature. 2006;441:1075‐1079. [DOI] [PubMed] [Google Scholar]
- 41. Gattazzo F, Urciuolo A, Bonaldo P. Extracellular matrix: a dynamic microenvironment for stem cell niche. Biochim Biophys Acta. 2014;1840:2506‐2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Takahashi N, Nobusue H, Shimizu T, et al. ROCK inhibition induces terminal adipocyte differentiation and suppresses tumorigenesis in chemoresistant osteosarcoma cells. Cancer Res. 2019;79:3088‐3099. [DOI] [PubMed] [Google Scholar]
- 43. Ivanovska IL, Shin JW, Swift J, Discher DE. Stem cell mechanobiology: diverse lessons from bone marrow. Trends Cell Biol. 2015;25:523‐532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677‐689. [DOI] [PubMed] [Google Scholar]
- 45. McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell. 2004;6:483‐495. [DOI] [PubMed] [Google Scholar]
- 46. Kilian KA, Bugarija B, Lahn BT, et al. Geometric cues for directing the differentiation of mesenchymal stem cells. Proc Natl Acad Sci USA. 2010;107:4872‐4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ridley AJ, Hall A. The small GTP‐binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389‐399. [DOI] [PubMed] [Google Scholar]
- 48. Riento K, Ridley AJ. ROCKs: multifunctional kinases in cell behavior. Nat Rev Mol Cell Biol. 2003;4:446‐456. [DOI] [PubMed] [Google Scholar]
- 49. Settleman J. Tension precedes commitment—even for a stem cell. Mol Cell. 2004;14:148‐150. [DOI] [PubMed] [Google Scholar]
- 50. Nobusue H, Onishi N, Shimizu T, et al. Regulation of MKL1 via actin cytoskeleton dynamics drives adipocyte differentiation. Nat Commun. 2014;5:3368. [DOI] [PubMed] [Google Scholar]
- 51. Chen Q, Shou P, Zheng C, et al. Fate decision of mesenchymal stem cells: adipocytes or osteoblasts ? Cell Death Differ. 2016;23:1128‐1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sordella R, Jiang W, Chen GC, et al. Modulation of Rho GTPase signaling regulates a switch between adipogenesis and myogenesis. Cell. 2003;113:147‐158. [DOI] [PubMed] [Google Scholar]
- 53. Noguchi M, Hosoda K, Fujikura J, et al. Genetic and pharmacological inhibition of Rho‐associated kinase II enhances adipogenesis. J Biol Chem. 2007;282:29574‐29583. [DOI] [PubMed] [Google Scholar]
- 54. Nobusue H, Endo T, Kano K. Establishment of a preadipocyte cell line derived from mature adipocytes of GFP transgenic mice and formation of adipose tissue. Cell Tissue Res. 2008;332:435‐446. [DOI] [PubMed] [Google Scholar]
- 55. Kunitomi H, Oki Y, Onishi N, et al. The insulin‐PI3K‐Rac1 axis contributes to terminal adipocyte differentiation through regulation of actin cytoskeleton dynamics. Genes Cells. 2020;25:165‐174. [DOI] [PubMed] [Google Scholar]
- 56. Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol. 2010;11:353‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Totaro A, Panciera T, Piccolo S. YAP/TAZ upstream signals and downstream responses. Nat Cell Biol. 2018;20:888‐899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Panciera T, Azzolin L, Cordenonsi M, Piccolo S. Mechanobiology of YAP and TAZ in physiology and disease. Nat Rev Mol Cell Biol. 2017;18:758‐770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dupont S, Morsut L, Aragona M, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179‐183. [DOI] [PubMed] [Google Scholar]
- 60. Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell. 2003;113:329‐342. [DOI] [PubMed] [Google Scholar]
- 61. Hyrskyluoto A, Vartiainen MK. Regulation of nuclear actin dynamics in development and disease. Curr Opin Cell Biol. 2020;64:18‐24. [DOI] [PubMed] [Google Scholar]
- 62. Lyubimova A, Bershadsky AD, Ben‐Ze'ev A. Autoregulation of actin synthesis responds to monomeric actin levels. J Cell Biochem. 1997;65:469‐478. [PubMed] [Google Scholar]
- 63. Bubb MR, Spector I, Bershadsky AD, Korn ED. Swinholide A is a microfilament disrupting marine toxin that stabilizes actin dimers and severs actin filaments. J Biol Chem. 1995;270:3463‐3466. [DOI] [PubMed] [Google Scholar]
- 64. Rosenwald M, Efthymiou V, Opitz L, Wolfrum C. SRF and MKL1 independently inhibit brown adipogenesis. PLoS One. 2017;12:e0170643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Foster CT, Gualdrini F, Treisman R. Mutual dependence of the MRTF‐SRF and YAP‐TEAD pathways in cancer‐associated fibroblasts is indirect and mediated by cytoskeletal dynamics. Genes Dev. 2017;31:2361‐2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jain N, Iyer KV, Kumar A, Shivashankar GV. Cell geometric constraints induce modular gene‐expression patterns via redistribution of HDAC3 regulated by actomyosin contractility. Proc Natl Acad Sci USA. 2013;110:11349‐11354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Uhler C, Shivashankar GV. Regulation of genome organization and gene expression by nuclear mechanotransduction. Nat Rev Mol Cell Biol. 2017;18:717‐727. [DOI] [PubMed] [Google Scholar]
- 68. Hung RJ, Yazdani U, Yoon J, et al. Mical links semaphorins to F‐actin disassembly. Nature. 2010;463:823‐827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hung RJ, Pak CW, Terman JR. Direct redox regulation of F‐actin assembly and disassembly by Mical. Science. 2011;334:1710‐1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lundquist MR, Storaska AJ, Liu TC, et al. Redox modification of nuclear actin by MICAL‐2 regulates SRF signaling. Cell. 2014;156:563‐576. [DOI] [PMC free article] [PubMed] [Google Scholar]