

Abstract
Cancer cachexia, characterized by continuous muscle wasting, is a key determinant of cancer‐related death; however, there are few medical treatments to combat it. Myostatin (MSTN)/growth differentiation factor 8 (GDF‐8), which is a member of the transforming growth factor‐β family, is secreted in an inactivated form noncovalently bound to the prodomain, negatively regulating the skeletal muscle mass. Therefore, inhibition of MSTN signaling is expected to serve as a therapeutic target for intractable muscle wasting diseases. Here, we evaluated the inhibitory effect of peptide‐2, an inhibitory core of mouse MSTN prodomain, on MSTN signaling. Peptide‐2 selectively suppressed the MSTN signal, although it had no effect on the activin signal. In contrast, peptide‐2 slightly inhibited the GDF‐11 signaling pathway, which is strongly related to the MSTN signaling pathway. Furthermore, we found that the i.m. injection of peptide‐2 to tumor‐implanted C57BL/6 mice alleviated muscle wasting in cancer cachexia. Although peptide‐2 was unable to improve the loss of heart weight and fat mass when cancer cachexia model mice were injected with it, peptide‐2 increased the gastrocnemius muscle weight and muscle cross‐sectional area resulted in the enhanced grip strength in cancer cachexia mice. Consequently, the model mice treated with peptide‐2 could survive longer than those that did not undergo this treatment. Our results suggest that peptide‐2 might be a novel therapeutic candidate to suppress muscle wasting in cancer cachexia.
Keywords: cancer cachexia, Lewis lung carcinoma, mice model, muscle wasting, myostatin
Peptide‐2 selectively suppressed the myostatin signal. The i.m. injection of peptide‐2 into tumor‐implanted C57BL/6 mice attenuated muscle wasting in cachexia model mice. The model mice treated with peptide‐2 could survive longer than those that did not undergo this treatment.

1. INTRODUCTION
Cancer cachexia is a multifactorial, severely debilitating syndrome that causes weight loss due to loss of skeletal muscle mass. 1 , 2 , 3 In cancer patients, the main symptoms associated with cachexia are asthenia, physical illness, and fatigue. Muscle wasting and weight loss are suitable indicators of the patient’s quality of life as they correlate with the patient’s prognosis. 2 In advanced stages of cancer, more than 50% of the patients exhibit cachexia. Furthermore, nearly 20% of the cases of cancer‐related mortality exhibit cachexia rather than tumor burden. 4 , 5 However, there are few medical treatments available to treat cancer cachexia.
The members of the transforming growth factor‐β (TGF‐β) family, myostatin (MSTN, also known as growth differentiation factor 8 [GDF‐8]), activin A, and GDF‐11 are known to negatively regulate skeletal muscle mass in mammals. 6 , 7 The loss of MSTN function in mice, 8 dogs, 9 cattle, 10 and humans is known to induce an increase in the skeletal muscle mass. The inhibition of MSTN with neutralizing Abs increases the muscle mass and the MSTN null mutations lead to skeletal muscle hypertrophy in mammals. 9 , 11 , 12 Thus, the nature of MSTN is conserved beyond mammalian species. Similar to other TGF‐β members, MSTN is initially synthesized as a precursor, which then undergoes proteolytic cleavage to become a mature ligand. The mature ligand initially binds to the activin type II receptor‐B (ActRIIB) Ser/Thr kinase receptor, and then to one of the type I receptors, ie activin receptor‐like kinase 4 (ALK4 or ActRIB) or ALK5 (TβRI). 13 Subsequently, the type I receptor phosphorylates Smad2 and Smad3 to regulate transcriptional activities. The MSTN precursor is known to bind to a mature MSTN and inhibits its function. 14 , 15 In our previous study, we identified an inhibitory core peptide composed of 24 amino acids in the mouse MSTN prodomain, termed peptide‐2. Interestingly, the muscle mass in mice increased when peptide‐2 was given i.m., suggesting that peptide‐2 could hold promise for treating muscle wasting diseases. 14
Recently, it has been reported that inhibition of MSTN and activin signals alleviates cachexia in mice with tumors. The ActRIIB is known to bind to MSTN GDF‐11 with a high affinity, in addition to activin. The expression of a dominant‐negative ActRIIB in mice results in skeletal muscle hypertrophy, suggesting that ActRIIB plays an important role in muscle growth. 16 Furthermore, the treatment of mice with the ActRIIB dominant‐negative receptor (ActRIIB‐Fc) as a ligand trapper resulted in prolonged survival associated with alleviation of muscle wasting. 17 In this study, we examined whether peptide‐2 alleviates cachexia symptoms in mice implanted with Lewis lung carcinoma (LLC).
2. MATERIALS AND METHODS
2.1. Cell culture
C2C12, HepG2, 293T, COS7, and LLC cells were cultured in DMEM (Nacalai Tesque) containing 15% (for C2C12 cells) or 10% FCS (Invitrogen), 1× MEM non‐essential amino acids (Nacalai Tesque), and 100 U/mL penicillin/streptomycin (PS; Wako). To induce myoblast differentiation, C2C12 cells were cultured in DMEM containing 2% horse serum (Cosmo Bio), 1× MEM non‐essential amino acids, and PS.
2.2. Myostatin inhibitors
Synthesis of peptide‐2 was described previously. 15 The peptide‐2 was reconstituted in saline. SB431542 (Sigma‐Aldrich) was reconstituted in DMSO.
2.3. Dominant‐negative ActRIIB
One day before transfection, COS7 cells were seeded at 1.0 × 106 cells/well in a 10‐cm dish. COS7 cells were transfected with CSII‐EF‐ctrl‐Fc or CSII‐EF‐ActRIIB‐Fc expressing plasmid with 2.5 mg/mL polyethyleneimine (PEI; Polyscience). After 36 hours of transfection, cell lysates were immunoprecipitated with Protein G sepharose 4B (Invitrogen) overnight and used for the experiment.
2.4. Transcriptional reporter assay
HepG2 cells were seeded at 7.5 × 104 cells/well in 24‐well plates 1 day before transfection. The cells were transfected with the indicated reporter construct and pCH110 (GE Healthcare Bioscience) using 2.5 mg/mL PEI. Twenty‐four hours later, media were changed to 0.3% FCS containing DMEM. Where indicated, 10 ng/mL MSTN, 10 ng/mL GDF‐11, 5 ng/mL activin A, or 5 ng/mL TGF‐β was added to the wells after cells were pretreated with peptide‐2 or SB431542 for 1 hour. 18 , 19 All the ligands were purchased from Wako. Subsequently, cells were cultured for 8 hours with or without ligands. The cell lysates were prepared to measure the luciferase and β‐galactosidase activities. In all experiments, β‐galactosidase activity was measured to normalize for transfection efficiency. Each transfection was carried out in triplicate and repeated at least twice. Values are presented as the means ± SD (n = 3).
2.5. Animal studies
All animal experimental procedures were approved by the President of the Tokyo University of Pharmacy and Life Sciences after review by the Institutional Animal Care and Use Committee (permission numbers L18‐003 and L19‐017) and carried out according to the Tokyo University of Pharmacy and Life Sciences Animal Experimentation Regulations. Male C57BL/6J mice (8‐12 weeks old; 20‐24 g) were purchased from Oriental Bio, and the mice were fed a standard laboratory diet and housed in a temperature‐controlled room under specific pathogen‐free conditions. A combination anesthetic was prepared with 0.3 mg/kg medetomidine, 4.0 mg/kg midazolam, and 5.0 mg/kg butorphanol. For the cancer cachexia model, LLC cells (5 × 105 cells in 100 µL saline) were s.c. injected around the caudal side of the thigh of C57BL/6J mice 20 after they were anesthetized by i.p. injection. After injection of the cells, the weight of each mouse was measured every week. For ethical reasons, the end‐point of the survival curve was set 40 days after transplantation. Many of the mice reached natural death, but the remaining mice were killed when they showed signs of morbidity defined by the experimental animal guideline (eg emaciation, lethargy or failure to respond to gentle stimuli, hypothermia).
To measure total 4‐limb grip strength in mice, we used a digital force gauge. 21 Mice were acclimatized on wire mesh for 5 minutes, then the mouse tail was gently pulled backward in a horizontal fashion for as long as the mouse could grasp the mesh. The force at the time when the mouse released the mesh was recorded as the peak tension. Each mouse was tested 3 times with a 1‐2 minute break between tests.
2.6. Western blot analysis
Western blot analysis was carried out as described previously. 22 Briefly, samples from gastrocnemius muscle (approximately 50 mg) were homogenized in TNE buffer (10 mmol/L Tris [pH 7.4], 150 mmol/L NaCl, 1 mmol/L EDTA, 1% NP‐40, 1 mmol/L PMSF, 5 μg/mL leupeptin, 100 U/mL aprotinin, 2 mmol/L sodium vanadate, 40 mmol/L NaF, and 20 mmol/L β‐glycerophosphate), and centrifuged at 10 000 g for 5 minutes at 4°C. Protein concentration was assayed using the DC Protein Assay Kit (Bio‐Rad Laboratories). The samples were boiled for 10 minutes in sample buffer, separated by SDS‐PAGE, and transferred to Ultra Cruz Nitrocellulose Pure Transfer Membrane (Santa Cruz Biotechnology). The membranes were probed with the indicated Abs. Primary Abs were detected with HRP‐conjugated goat anti‐rabbit or anti‐mouse IgG Ab (GE Healthcare) with chemiluminescent substrate (Thermo Fisher Scientific).
2.7. Histology and immunofluorescence
The muscle tissues were surgically removed and embedded into frozen section compound (Leica Camera). Fresh‐frozen sections (5 μm) were cut with a CM1850 cryostat (Leica), mounted on Cryofilm (Leica), and fixed in 100% ethanol then in 4% paraformaldehyde. The films were washed 3 times with PBS, permeabilized with 0.1% polyoxyethylene (10) octylphenyl ether (Wako) for 10 minutes, and blocked with blocking reagent (PerkinElmer) for 1 hour at 37°C. Rabbit anti‐myogenin (1:200) Abs in blocking reagent were added and incubated overnight at 4°C. The films were washed 3 times with PBS and then incubated with Alexa 488‐conjugated goat anti‐mouse IgG (Molecular Probes) Ab at 1:200 for 1 hour at room temperature. After the nuclei were stained with 2 μg/mL DAPI for 10 minutes, the films were mounted with mounting medium (Dako). A BZ‐9000 fluorescence microscope (Keyence) was used to visualize the fluorescence. To determine the diameter of C2C12 myotubes, phalloidin‐positive myotubes were measured by ImageJ.
2.8. RNA isolation and RT‐PCR
Total RNA was isolated using the ReliaPrep RNA Cell Miniprep System (Promega). Reverse transcription was carried out with the PrimeScript II 1st strand cDNA synthesis kit (Takara Bio). Reverse transcription‐PCR was carried out with BlendTaq (Toyobo). The following primer sets were used to amplify myogenin, MylpF, and β‐actin cDNAs: 5′‐TGAATGCAACTCCCACAGC‐3′ and 5′‐CAGACATATCCTCCACCGTG‐3′ for myogenin, 23 5′‐AGGATGTGATCACTGGAGC‐3′ and 5′‐TGAGAGATGGAGCGGCTAGAAGC‐3′ for MylpF, and 5′‐TGAACCCTAAGGCCAACCGTG‐3′ and 5′‐GCTCATAGCTCTTCTCCAGGG‐3′ for β‐actin.
2.9. Statistical analysis
Data are expressed as mean ± SD unless otherwise mentioned. Significance was assessed using Student’s t test. Probability values below .05, .01, and .001 were considered significant.
3. RESULTS
3.1. Peptide‐2 is an MSTN‐specific inhibitor
In our previous study, we identified an MSTN‐inhibiting peptide, termed peptide‐2, composed of 24 amino acids from the mouse MSTN prodomain, and showed that peptide‐2 has a high affinity for MSTN, through the surface plasmon resonance assay. 14 We undertook a luciferase assay using the (SBE)4‐luc reporter 24 to characterize the inhibitory effect of peptide‐2 on TGF‐β family signaling in hepatocellular carcinoma HepG2 cells. The cells were stimulated with each ligand that was preincubated with peptide‐2 or SB‐431542, which is a kinase inhibitor for ALK4, ALK5, and ALK7 18 (Figure 1A). Eight hours later, the luciferase activity was measured. SB‐431542 effectively blocked MSTN, GDF‐11, activin, and TGF‐β‐induced reporter activities, whereas peptide‐2 significantly inhibited MSTN‐induced reporter activity and suppressed GDF‐11‐induced reporter activity by up to approximately 67%. When we examined the inhibitory effects of peptide‐2 on Smad2 and Smad3 nuclear translocation following MSTN stimulation, it was observed that peptide‐2 completely interfered with MSTN‐induced Smad2 nuclear accumulation, similar to SB‐431542 (Figure 1B).
FIGURE 1.
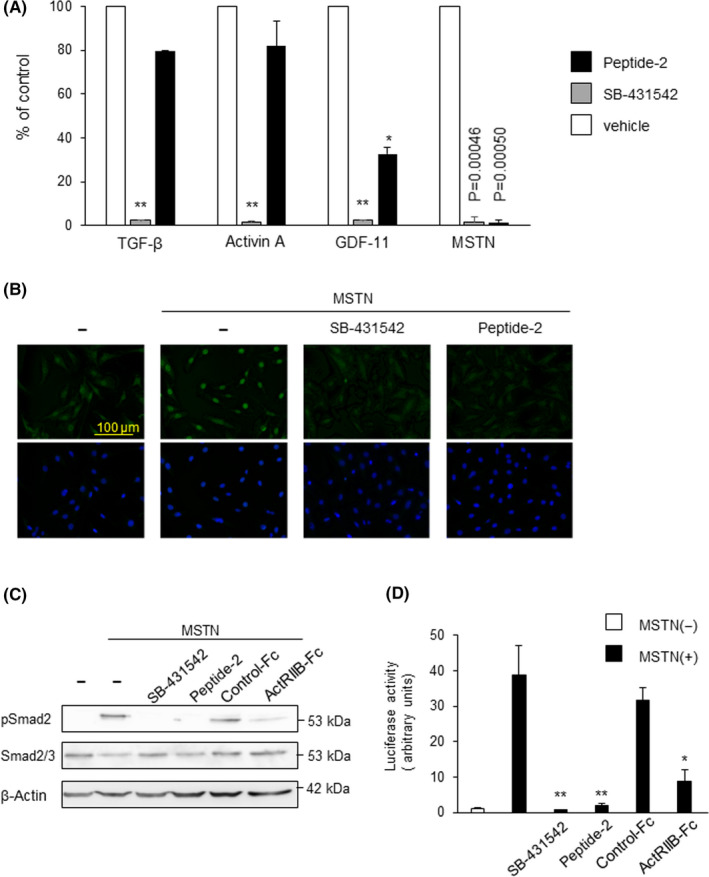
Peptide‐2 is a selective inhibitor of myostatin (MSTN). A, Peptide‐2 significantly inhibited the activity of (SBE)4‐luc following MSTN stimulation in HepG2 cells. HepG2 cells were cotransfected with a (SBE)4‐luc reporter construct and pCH110 as an internal marker and stimulated with transforming growth factor‐β (TGF‐β; 5 ng/mL), activin A (10 ng/mL), growth differentiation factor‐11 (GDF‐11; 10 ng/mL), or MSTN (10 ng/mL) for 8 h. Luciferase values were normalized for transfection efficiency. These ligands were preincubated with peptide‐2 (30 nmol/L) or SB‐431542 (10 µmol/L) and were added to the cell. Inhibition efficiency was shown as % of control. All values represent mean ± SD (n = 3). B, Smad2 and Smad3 nuclear localization induced by MSTN were inhibited by peptide‐2. Immunofluorescence staining for Smad2 and Smad3 (Green) in C2C12 cells. Peptide‐2 (30 nmol/L) or SB‐431542 (10 µmol/L) was preincubated with MSTN for 1 h, and added to C2C12 cells for 1 h before staining. Nuclei were counterstained with DAPI (blue). A BZ‐9000 fluorescence microscope was used to visualize the fluorescence. C, Peptide‐2 and the activin type II receptor‐B dominant‐negative receptor (ActRIIB‐Fc) inhibit MSTN‐induced Smad2 phosphorylation. SB‐431542, peptide‐2 (30 nmol/L), ActRIIB‐Fc (0.146 nmol/L), or control‐Fc were preincubated with MSTN for 1 h and added to HepG2 cells for 1 h. These cells were lysed and subjected to western blot analysis using anti‐phospho‐Smad2 (upper panel), anti‐Smad2 Ab (middle panel), and β‐actin (lower panel). Each expression was normalized using the intensity of the band corresponding to β‐actin. Inducibility was calculated relative to the value for cells in the absence of TGF‐β. D, Peptide‐2 significantly inhibited the activity of (CAGA)12‐luc upon MSTN stimulation in HepG2 cells. SB‐431542 (10 µmol/L), peptide‐2 (30 nmol/L), ActRIIB‐FC (0.146 nmol/L), or control‐Fc (0.146 nmol/L) were preincubated with MSTN (10 ng/mL) for 1 h, and added to HepG2 cells for 1 h. HepG2 cells were cotransfected with (SBE)4‐luc reporter construct and pCH110 as an internal marker and stimulated with MSTN for 8 h. All values represent mean ± SD (n = 3). **P < .01; *P < .05
Activin type II receptor‐B lacking an intracellular domain is known to act as a dominant‐negative receptor. 17 When we compared the inhibitory ability of MSTN signaling between peptide‐2 (dissociation constant= 35.9 nmol/L, IC50 = 4.1 µmol/L) 14 and ActRIIB‐Fc (IC50 = 1.1 nmol/L), 25 the phosphorylation of Smad2 was almost equally inhibited by both peptide‐2 and ActRIIB‐Fc (Figure 1C). We then carried out a luciferase assay using the (CAGA)12‐luc reporter. 26 HepG2 cells transfected with the (CAGA)12‐luc reporter plasmid were stimulated with MSTN that was preincubated with either peptide‐2 or ActRIIB‐Fc for 1 hour. As seen in Figure 1D, peptide‐2 was able to inhibit MSTN‐induced transcriptional activity similar to ActRIIB‐Fc (Figure 1D).
3.2. Peptide‐2 inhibits the effect of MSTN and promotes C2C12 myoblast differentiation
We next examined whether peptide‐2 potentiated the differentiation of C2C12 myoblasts through its inhibitory activity for MSTN signaling. C2C12 myoblast cells differentiate into syncytial myotubes in 2% horse serum. 27 When cell fusions and formation of small myofibers were detected 2 days after the substitution of 15% FCS to 2% horse serum (data not shown), we investigated the transcripts of basic helix‐loop‐helix transcriptional factors, myogenin and MylpF, which play an important role in myotube differentiation. 27 , 28 Both the mRNA expressions increased with the differentiation of C2C12 myoblasts, whereas MSTN decreased the mRNA expressions. However, preincubation of MSTN with peptide‐2 was able to restore these mRNA expressions similar to SB‐431542 (Figure 2A). Furthermore, muscle syncytia and stress fiber formation induced by horse serum were inhibited by MSTN, whereas pretreatment of MSTN with either peptide‐2 or SB431542 improved muscle syncytia and stress fiber formation (Figure 2B). These data were confirmed by measurement of the diameter of C2C12 myotubes (Figure 2C).
FIGURE 2.
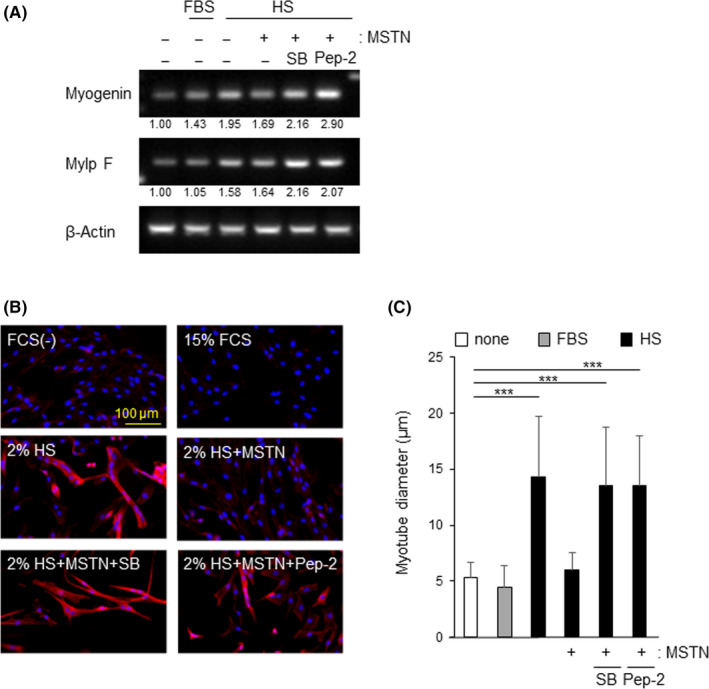
Peptide‐2 inhibited the effect of myostatin (MSTN) and promoted C2C12 myoblast differentiation. A, Expression of myotube differentiation genes. C2C12 cells were cultured with 15% FCS or 2% horse serum (HS) containing medium for 2 d. Cells were stimulated with 10 ng/mL MSTN with or without SB‐431541 (SB) (10 µmol/L) or peptide‐2 (30 nmol/L). Total RNA from these cells was subjected to RT‐PCR for myogenin (upper panel), MylpF (middle panel), and β‐actin mRNAs (bottom panel). B, C2C12 cells cultured in growth medium (15% FCS) reached 90% confluence, and the culture media were switched to differentiation medium (2% HS) for 2 d. These cells were stained with TRITC‐phalloidin (red) and DAPI (blue) to counterstain cell nuclei. The BZ‐9000 fluorescence microscope was used visualize the fluorescence. C, Diameters of C2C12 myotubes was analyzed. Thirty myotubes per experiment were counted over 2 independent experiments, and a representative plot is displayed. Significance was compared to serum‐free control. ***P < .001. Data presented are the mean ± SD
3.3. Peptide‐2 prolongs survival in cancer cachexia model mice
To analyze the therapeutic effect of peptide‐2 on cancer cachexia, LLC cells were inoculated s.c. into the back of C57BL/6 mice. Subsequently, peptide‐2 was injected i.m. into mouse gastrocnemius muscles a total of 3‐4 times (ie 4, 11, 18, and 26 days after cell injection) (Figure 3A). Peptide‐2 was also i.m. injected into cachexia model mice and changes in survival rate were analyzed. Compared to the saline treatment, the i.m. injection of peptide‐2 significantly prolonged the survival time over the period studied (Figure 3B). For ethical reasons, the analysis was carried out 3 weeks after the transplantation and changes in body weight were assessed. During the experimental period, the body weight of the tumor‐bearing mice was increased due to the progression of tumor growth (Figure 3C‐E, Table S1). The body weight after subtracting the tumor weight at the end‐point significantly decreased by approximately 10% in the LLC‐inoculated groups compared to the mice without LLC implantation (Figure 3F, Table S1). There was no significant difference in body or tumor weight (Figure 3E) between the mice treated with peptide‐2 and those not treated with peptide‐2. These results suggest that peptide‐2 might prolong survival in tumor‐bearing mice.
FIGURE 3.
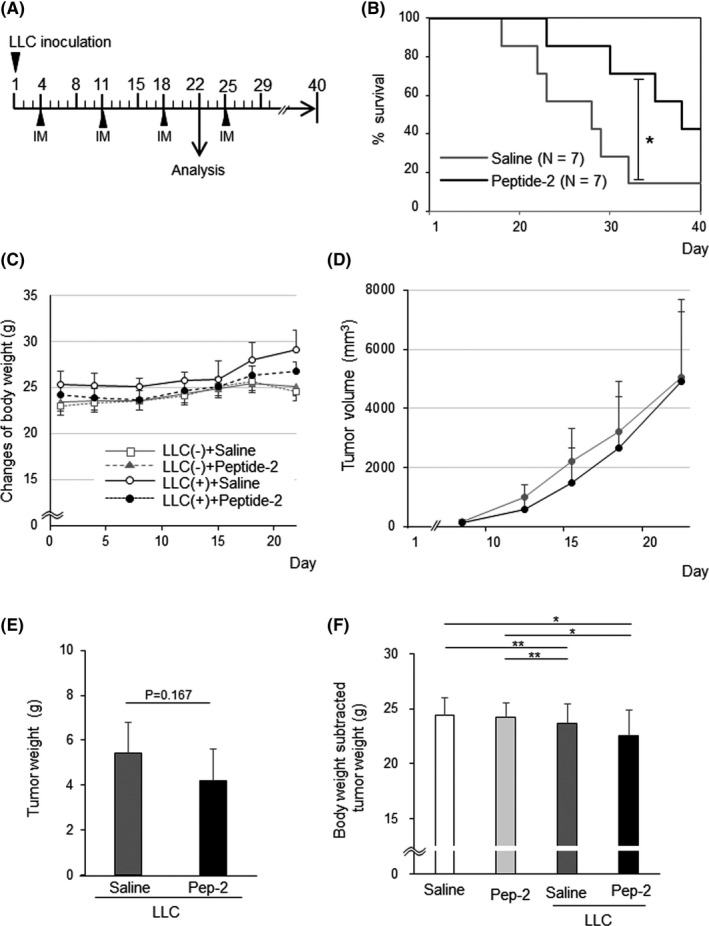
Peptide‐2 prolongs survival in cancer cachexia model mice. A, Schematic diagram of the experimental schedule. Peptide‐2 or saline was injected i.m. to the mouse gastrocnemius muscles of both legs a total of 3 times, at 4, 11, 18, and 25 d after cell injection. B, Survival rate of Lewis lung carcinoma (LLC)‐bearing mice with peptide‐2 or saline treatment was analyzed (χ2 test, P = .03, power = 0.991). C, Changes in body weights in control and LLC tumor‐bearing mice. Both groups of mice received peptide‐2 or saline at indicated days. Body mass following LLC inoculation was monitored every week at 1, 8, 15, and 22 d after inoculation (n = 6). Representative data are shown. Raw data are shown in Table S1A. D, Tumor volume estimated from width of tumors in LLC‐bearing mice with peptide‐2 or saline treatment. Tumor size was serially measured from above the skin. Tumor volumes were calculated using the formula: length × width ×width × 0.5. Data are expressed as mean ± SD (n = 6). E, Peptide‐2 (Pep‐2) treatment did not alter LLC tumor growth (P = .20) (n = 6). Representative data are shown. F, Body weight of mice excluding tumor weight at day 22. LLC tumor‐bearing mice had significant weight loss compared to controls. **P < .01; *P < .05. Raw data of statistical results are shown in Table S1B
3.4. Peptide‐2 prevent cancer‐induced muscle wasting
When we compared the gross muscles of the hind limbs, muscle wasting was evident in the LLC‐bearing mice at 3 weeks after the transplantation (Figure 4A). However, muscle mass was restored by peptide‐2 treatment in cachexia model mice, which had increased gastrocnemius muscle mass compared to the vehicle‐treated mice (Figure 4B, Table S2). The effect of peptide‐2 was localized at the site of injection, and there was no effect on gastrocnemius muscle when peptide‐2 was injected into the thigh muscle (Figure S1). As skeletal muscle wasting is a characteristic of cancer cachexia, 29 the intracellular protein degradation in gastrocnemius was analyzed. As shown in Figure 4C, LLC‐transplanted mice showed a markedly decreased protein concentration of the gastrocnemius, which was not ameliorated by peptide‐2 treatment. Other cancer cachexia parameters such as decreased heart weight (Figure 4C,D, Tables S3 and S4) and abdominal s.c. fat (Figure 4E, Table S5) did not improve with peptide‐2 treatment. These results indicate that peptide‐2 treatment partially alleviated LLC tumor‐induced cachexia symptoms.
FIGURE 4.
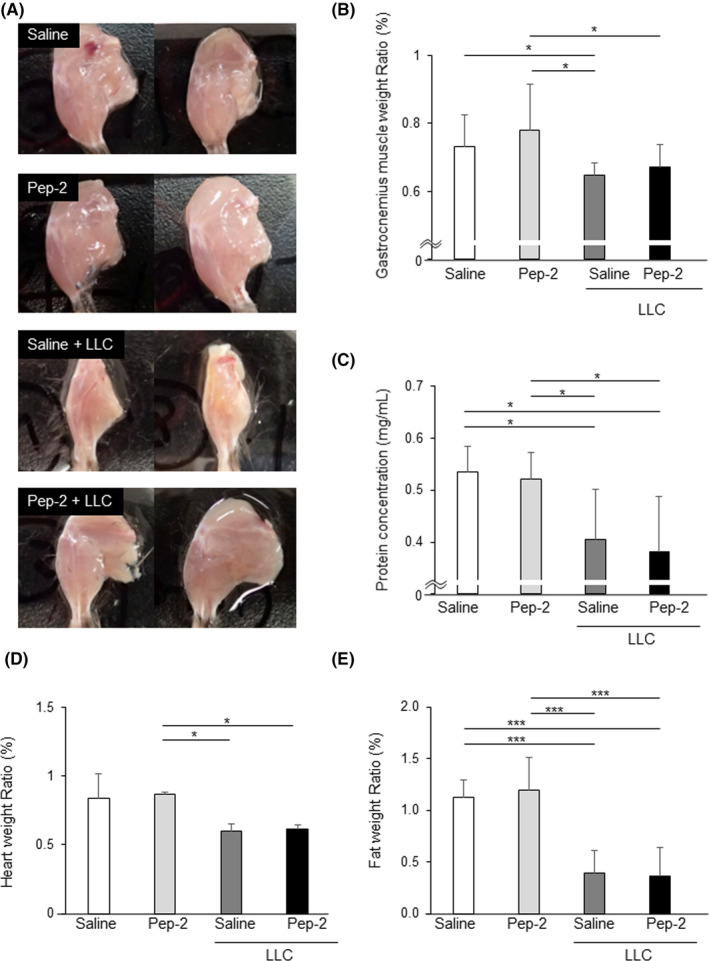
Peptide‐2 attenuates muscle wasting induced by cancer cachexia. Peptide‐2 (Pep‐2) or saline was injected i.m. in both legs of control or Lewis lung carcinoma (LLC)‐transplanted mice. A, Photographs of legs of control and cancer cachexia mice. B, Gastrocnemius muscle weight/body weight (n = 6). C, Protein concentration of gastrocnemius muscle (n = 6). D, E, Heart weight/body weight (n = 3‐4) (D) and fat weight/body weight (n = 6) (E) at 22 d after LLC implantation. The weight of each tissue was normalized to the body weight at day 1. ***P < .001; *P < .05. All measurements and statistical analyses are shown in Table [Link], [Link], [Link], [Link]
Muscle atrophy is characterized by a decrease in the muscle fiber area. Accordingly, a cross‐section of the gastrocnemius muscles of these mice was prepared and stained with the dystrophin Ab and compared to the fiber areas (Figure 5A). In the LLC‐transplanted mice, the average muscle fiber area decreased by approximately 25% and fiber atrophy was predominant. In the whole muscle fiber comparison, there were no significant differences between saline‐ or peptide‐2‐treated cachexia mice (Figure 5B). However, when the fiber size was expressed in terms of frequency distribution, peptide‐2‐treated cachexia mice showed enlarged fibers compared to the saline‐treated mice (Figure 5C). In addition, the grip strength of mice treated with peptide‐2 was increased compared to the controls. The LLC‐bearing mice showed decreased grip strength, but peptide‐2 treatment resulted in functional muscle recovery in tumor‐bearing mice with levels more than the control.
FIGURE 5.
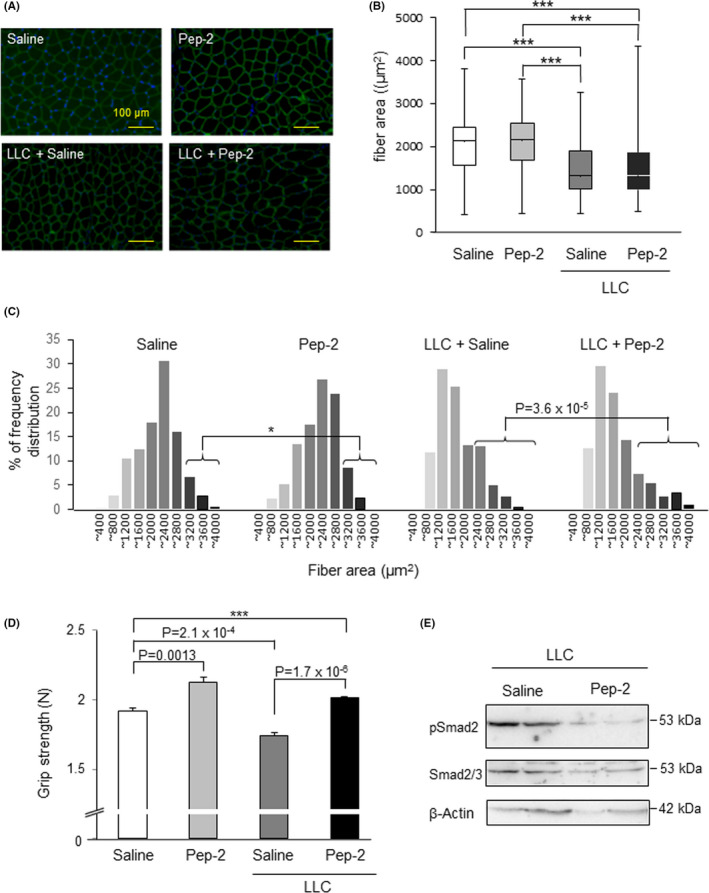
Peptide‐2 (Pep‐2) improves muscle fiber area and strength in cancer cachexia model mice. A, Representative immunofluorescence analysis of dystrophin‐stained gastrocnemius sections from indicated groups. Cryosections of gastrocnemius muscles from healthy control and cancer cachexia model mice treated with saline or Pep‐2 were stained with antidystrophin Ab (green) and DAPI (blue) to counterstain cell nuclei. Sections were imaged using a fluorescence microscope. B, Images were analyzed using a BZ‐H3A software, which automatically identifies individual areas surrounded by the muscle fiber membrane. Box and whisker plots showed the distribution of muscle fiber cross‐sectional area from 3 identical images for each group. The horizontal line in the box indicates the median values, the bottom and top edges indicate the 25th and 75th percentile, respectively. The whiskers extend to the most extreme bottom and top, indicating the minimum and maximum, respectively. In cancer cachexia model mice, the mean area of muscle fibers decreased significantly (***P < .001) indicating muscle atrophy, but treatment with Pep‐2 did not improve the mean value. C, Frequency distribution plots of the area of gastrocnemius muscle fiber. Plots are expressed as a percentage of the total number of fibers. Peptide‐2 increased the area of large muscle fibers in healthy mice and increased the proportion of thick muscle fibers in cachectic mice. D, Grip strength analysis of control and cachexic mice treated with Pep‐2 or saline. Each mouse was tested 5 times and the maximum force demonstrated by each animal was recorded (n = 4). Specific P values are shown in the figure. *P < .05. E, Intramuscular injection of Pep‐2 inhibited Smad2 phosphorylation in gastrocnemius muscle. The gastrocnemius muscle of cancer cachexia model mice treated with saline or Pep‐2 were analyzed. Smad2 phosphorylation was analyzed at 22 d after tumor cell inoculation. These muscles were lysed and subjected to western blot analysis using anti‐phospho‐Smad2 (upper panel), anti‐Smad2 Ab (middle panel), and β‐actin (lower panel) (n = 2)
Next, to investigate whether the dramatic improvement detected in cancer cachexia‐affected mice was mediated by the MSTN/Smad2 signaling pathway, western blot analysis of the gastrocnemius muscle was carried out. As shown in Figure 5E, saline‐treated cachexia mice showed Smad2 phosphorylation, which was suppressed by treatment with peptide‐2. These results suggested that peptide‐2 inhibited MSTN signaling and that it was partially resistant to cancer cachexia in vivo.
4. DISCUSSION
Cancer cachexia seen in patients with advanced cancer is a multifactorial metabolic disorder characterized by severe skeletal muscle atrophy rather than fat loss. 2 , 30 Therefore, counteracting muscle atrophy seems to be one of the key therapies for cancer cachexia as it is not possible for patients with cancer cachexia to regain muscle mass through nutrient intake. 31 Although MSTN and activin A generally inhibit muscle growth, both these cytokines induce catabolism by promoting loss of muscle proteins in case of skeletal muscle disorders such as sarcopenia and cachexia. 17 , 32 , 33 Therefore, the inhibition of MSTN and activin A signaling is considered as a therapeutic target for these skeletal muscle disorders. To date, ActRIIB‐Fc, a soluble form of MSTN/activin A receptor type II receptor, a decoy receptor, and neutralizing Abs for ActRIIB 34 or MSTN have been reported to show therapeutic effects on muscle wasting associated with cachexia in several tumor‐bearing mice. However, clinical trials of the MSTN mAb LY2495655 did not improve overall survival of pancreatic cancer with cachexia. 35 Golan et al reported that the reactivity was stronger in precachectic patients than cachectic patients. Therefore, MSTN inhibitors could be beneficial for preventing muscle loss and maintaining quality of life, not for reversing muscle wasting. 36
In our previous study, we identified the minimum inhibitory core region composed of only 24 amino acid‐long peptides in the MSTN precursor, termed peptide‐2, and reported that i.m. injection in the hind limbs of mice increased muscle mass. 14 To verify the clinical application of peptide‐2 in muscle wasting disease, we confirmed the therapeutic effects of cancer cachexia when peptide‐2 was given to mice implanted with LLCs. Myostatin inhibitors such as Abs, 37 , 38 , 39 decoy receptors 17 and siRNAs, 40 have been effective on muscles throughout the body. However, we were able to show the therapeutic effect on the restoration of local gastrocnemius muscle mass to extend duration of their life. It is reported that maintenance of leg muscle mass can prevent patients with sarcopenia from becoming bedridden. 39 Thus, prevention of muscle wasting might be essential for improving the quality of life.
It is important to develop highly selective drugs for clinical applications. The ActRIIB‐Fc binds to MSTN, GDF‐11, and activin A. Peptide‐2 is a specific inhibitor of MSTN as it does not block activin A signaling in addition to its marginal inhibition of GDF‐11 signaling (Figure 1A), due to the high homology between GDF‐11 and MSTN. As GDF‐11 can suppress the growth and differentiation of skeletal muscles such as MSTN, the inhibitory effect of peptide‐2 on GDF‐11 is preferable to improve muscle wasting in cancer cachexia.
Intramuscular injection of peptide‐2 slightly reduced gastrocnemius muscle weight in cancer cachexia mice (Figure 3B). In addition, peptide‐2 improved the survival of LLC‐implanted mice during the period examined (Figure 3B). Lewis lung carcinoma cells are known to produce lung metastasis, but none of the mice showed dyspnea during the experimental period. Furthermore, when comparing the lung metastasis 22 days after transplantation, there was no significant difference in size or number of metastases between the 2 groups (data not shown); therefore, it is unlikely that death occurred due to lung metastasis. Similar to anti‐MSTN neutralizing Abs, peptide‐2 shows a therapeutic effect on muscle atrophy. 40 , 41 , 42 In the current study, peptide‐2 increased the area of thick muscle fibers and gastrocnemius muscle weight in healthy mice, in addition to counteracting muscle atrophy in LLC‐implanted mice. Skeletal muscle fiber hypertrophy and recovery of muscle mass led to increase in grip strength when peptide‐2 was given to LLC‐implanted mice (Figure 5D). It is generally known that maximum force and muscle cross‐sectional area (CSA) are strongly related. The grip strength of mice with muscle atrophy would be considered to be restored by peptide‐2 because their muscle CSA increased (Figure 5C). Alleviation of muscle wasting could help mice with muscle atrophy to ingest food and water, and improved their quality of life.
Peptide‐2 did not affect heart mass in the cancer cachexia model mice, although the inhibition of the ActRIIB pathway by ActRIIB‐Fc restored the heart mass in mice implanted with C26 cells. 17 As peptide‐2 was locally injected into mice, peptide‐2 might not reach the myocardium. It is reported that MSTN expression in adult mice is dominant in the skeletal muscle and less in the myocardium. 43 Thus, another possibility is that activin A and GDF‐11 might influence the myocardium as ActRIIB can be bound by activin A and GDF‐11, apart from MSTN. This will need to be elucidated in further experiments. The reduced weight of the primary tumor pointed towards an impact of peptide‐2 on tumor growth/progression. The effect of peptide‐2 on muscle weight is much less than ActRIIB decoy receptor 17 and anti‐MSTN mAb. 42 In general, classic peptide drugs have less significance in specificity and efficacy than Ab drugs; however, structural modifications such as cyclization can increase the activity and stability of the classical peptides. The overwhelming advantage over Ab drugs is the low manufacturing cost.
Collectively, the peptide‐2 that perturbs MSTN signaling might serve a therapeutic drug for patients with muscle wasting diseases, such as cancer cachexia and sarcopenia, because of its ability to restore their muscle mass. One of the greatest advantages is that peptide‐2 has 15 times fewer amino acids than ActRIIB‐Fc, thereby reducing the cost of synthesizing the protein. However, it will need to be improved further as a highly effective therapeutic agent in terms of various other factors such as prolongation of its half‐life and reduction of its dosage.
DISCLOSURE
The authors have no conflicts of interest.
Supporting information
Figure S1
Table S1
Table S2
Table S3
Table S4
Table S5
ACKNOWLEDGMENTS
This research was supported by the Japanese Ministry of Education, Culture, Sports, Science, and Technology (17K08794 to FI), the Core‐to‐Core program “Cooperative International Framework in TGF‐β Family Signaling” of the Japan Society for the Promotion of Science (FI and TW), and MEXT‐Supported Program for the Strategic Research Foundation at Private Universities (2015‐2019 to FI and YH). We thank Dr S. Itoh for his marvelous advice and thank Mr H. Hinata and Dr T. Watanabe for their support. We would like to thank Editage for English language editing.
Ojima C, Noguchi Y, Miyamoto T, et al. Peptide‐2 from mouse myostatin precursor protein alleviates muscle wasting in cancer‐associated cachexia. Cancer Sci. 2020;111:2954–2964. 10.1111/cas.14520
REFERENCES
- 1. Evans WJ, Morley JE, Argiles J, et al. Cachexia: a new definition. Clin Nutr. 2008;27:793‐799. [DOI] [PubMed] [Google Scholar]
- 2. Fearon K, Arends J, Baracos V. Understanding the mechanisms and treatment options in cancer cachexia. Nat Rev Clin Oncol. 2013;10:90‐99. [DOI] [PubMed] [Google Scholar]
- 3. Cole CL, Kleckner IR, Jatoi A, Schwarz EM, Dunne RF. The role of systemic inflammation in cancer‐associated muscle wasting and rationale for exercise as a therapeutic intervention. JCSM Clin Rep. 2018;3:e00065. [PMC free article] [PubMed] [Google Scholar]
- 4. Tan BH, Fearon KC. Cachexia: prevalence and impact in medicine. Curr Opin Clin Nutr Metab Care. 2008;11:400‐407. [DOI] [PubMed] [Google Scholar]
- 5. Muscaritoli M, Bossola M, Aversa Z, Bellantone R, Rossi Fanelli F. Prevention and treatment of cancer cachexia: new insights into an old problem. Eur J Cancer. 2006;42:31‐41. [DOI] [PubMed] [Google Scholar]
- 6. Latres E, Mastaitis J. Activin A more prominently regulates muscle mass in primates than does GDF8. Nat Commun. 2017;8:15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Walker RG, Poggioli T, Katsimpardi L, et al. Biochemistry and biology of GDF11 and myostatin: similarities, differences, and questions for future investigation. Circ Res. 2016;118:1125‐1142; discussion 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Iskenderian A, Liu N, Deng Q, et al. Myostatin and activin blockade by engineered follistatin results in hypertrophy and improves dystrophic pathology in mdx mouse more than myostatin blockade alone. Skelet Muscle. 2018;8:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mosher DS, Quignon P, Bustamante CD, et al. A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet. 2007;3:e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grobet L, Martin LJ, Poncelet D, et al. A deletion in the bovine myostatin gene causes the double‐muscled phenotype in cattle. Nat Genet. 1997;17:71‐74. [DOI] [PubMed] [Google Scholar]
- 11. McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF‐beta superfamily member. Nature. 1997;387:83‐90. [DOI] [PubMed] [Google Scholar]
- 12. Schuelke M, Wagner KR, Stolz LE, et al. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004;350:2682‐2688. [DOI] [PubMed] [Google Scholar]
- 13. Morikawa M, Derynck R, Miyazono K. TGF‐beta and the TGF‐beta family: context‐dependent roles in cell and tissue physiology. Cold Spring Harb Perspect Biol. 2016;8:a021873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takayama K, Noguchi Y, Aoki S, et al. Identification of the minimum peptide from mouse myostatin prodomain for human myostatin inhibition. J Med Chem. 2015;58:1544‐1549. [DOI] [PubMed] [Google Scholar]
- 15. Ohsawa Y, Takayama K, Nishimatsu S, et al. The inhibitory core of the myostatin prodomain: its interaction with both type I and II membrane receptors, and potential to treat muscle atrophy. PLoS One. 2015;10:e0133713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fakhfakh R, Michaud A, Tremblay JP. Blocking the myostatin signal with a dominant negative receptor improves the success of human myoblast transplantation in dystrophic mice. Mol Ther. 2011;19:204‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou X, Wang JL, Lu J, et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell. 2010;142:531‐543. [DOI] [PubMed] [Google Scholar]
- 18. Inman GJ, Nicolas FJ, Callahan JF, et al. SB‐431542 is a potent and specific inhibitor of transforming growth factor‐beta superfamily type I activin receptor‐like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002;62:65‐74. [DOI] [PubMed] [Google Scholar]
- 19. Laping NJ, Grygielko E, Mathur A, et al. Inhibition of transforming growth factor (TGF)‐beta1‐induced extracellular matrix with a novel inhibitor of the TGF‐beta type I receptor kinase activity: SB‐431542. Mol Pharmacol. 2002;62:58‐64. [DOI] [PubMed] [Google Scholar]
- 20. Deboer MD. Animal models of anorexia and cachexia. Expert Opin Drug Discov. 2009;4:1145‐1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Castro B, Kuang S. Evaluation of muscle performance in mice by treadmill exhaustion test and whole‐limb grip strength assay. Bio Protoc. 2017;7:e2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Itoh F, Itoh S, Adachi T, et al. Smad2/Smad3 in endothelium is indispensable for vascular stability via S1PR1 and N‐cadherin expressions. Blood. 2012;119:5320‐5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tanaka S, Kawahara E, Nakagawa T. Myogenic cell response to muscle contraction with short electrical stimulation. J Phys Ther Sci. 2015;27:2349‐2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jonk LJ, Itoh S, Heldin CH, ten Dijke P, Kruijer W. Identification and functional characterization of a Smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor‐beta, activin, and bone morphogenetic protein‐inducible enhancer. J Biol Chem. 1998;273:21145‐21152. [DOI] [PubMed] [Google Scholar]
- 25. Lee SJ, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci USA. 2001;98:9306‐9311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF beta‐inducible elements in the promoter of human plasminogen activator inhibitor‐type 1 gene. EMBO J. 1998;17:3091‐3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sabourin LA, Rudnicki MA. The molecular regulation of myogenesis. Clin Genet. 2000;57:16‐25. [DOI] [PubMed] [Google Scholar]
- 28. Jin W, Peng J, Jiang S. The epigenetic regulation of embryonic myogenesis and adult muscle regeneration by histone methylation modification. Biochem Biophys Rep. 2016;6:209‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Penna F, Ballaro R, Beltra M, De Lucia S, Garcia Castillo L, Costelli P. The skeletal muscle as an active player against cancer cachexia. Front Physiol. 2019;10:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miller BS, Ignatoski KM, Daignault S, et al. Worsening central sarcopenia and increasing intra‐abdominal fat correlate with decreased survival in patients with adrenocortical carcinoma. World J Surg. 2012;36:1509‐1516. [DOI] [PubMed] [Google Scholar]
- 31. Fearon K, Strasser F, Anker SD, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 2011;12:489‐495. [DOI] [PubMed] [Google Scholar]
- 32. Zimmers TA, Davies MV, Koniaris LG, et al. Induction of cachexia in mice by systemically administered myostatin. Science. 2002;296:1486‐1488. [DOI] [PubMed] [Google Scholar]
- 33. Han HQ, Zhou X, Mitch WE, Goldberg AL. Myostatin/activin pathway antagonism: molecular basis and therapeutic potential. Int J Biochem Cell Biol. 2013;45:2333‐2347. [DOI] [PubMed] [Google Scholar]
- 34. Hatakeyama S, Summermatter S, Jourdain M, Melly S, Minetti GC, Lach‐Trifilieff E. ActRII blockade protects mice from cancer cachexia and prolongs survival in the presence of anti‐cancer treatments. Skelet Muscle. 2016;6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jameson GS, Von Hoff DD, Weiss GJ, et al. Safety of the antimyostatin monoclonal antibody LY2495655 in healthy subjects and patients with advanced cancer. J Clin Oncol. 2012;30:2516.22614993 [Google Scholar]
- 36. Golan T, Geva R, Richards D, et al. LY2495655, an antimyostatin antibody, in pancreatic cancer: a randomized, phase 2 trial. J Cachexia Sarcopenia Muscle. 2018;9:871‐879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Murphy KT, Chee A, Gleeson BG, et al. Antibody‐directed myostatin inhibition enhances muscle mass and function in tumor‐bearing mice. Am J Physiol Regul Integr Comp Physiol. 2011;301:R716‐R726. [DOI] [PubMed] [Google Scholar]
- 38. Mosler S, Relizani K, Mouisel E, Amthor H, Diel P. Combinatory effects of siRNA‐induced myostatin inhibition and exercise on skeletal muscle homeostasis and body composition. Physiol Rep. 2014;2:e00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Iolascon G, Di Pietro G, Gimigliano F, et al. Physical exercise and sarcopenia in older people: position paper of the Italian Society of Orthopaedics and Medicine (OrtoMed). Clin Cases Miner Bone Metab. 2014;11:215‐221. [PMC free article] [PubMed] [Google Scholar]
- 40. Whittemore LA, Song K, Li X, et al. Inhibition of myostatin in adult mice increases skeletal muscle mass and strength. Biochem Biophys Res Comm. 2003;300:965‐971. [DOI] [PubMed] [Google Scholar]
- 41. Cohn RD, Liang HY, Shetty R, Abraham T, Wagner KR. Myostatin does not regulate cardiac hypertrophy or fibrosis. Neuromuscul Disord. 2007;17:290‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Smith RC, Cramer MS, Mitchell PJ, et al. Myostatin neutralization results in preservation of muscle mass and strength in preclinical models of tumor‐induced muscle wasting. Mol Cancer Ther. 2015;14:1661‐1670. [DOI] [PubMed] [Google Scholar]
- 43. Takehara‐Kasamatsu Y, Tsuchida K, Nakatani M, et al. Characterization of follistatin‐related gene as a negative regulatory factor for activin family members during mouse heart development. J Med Invest. 2007;54:276‐288. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Table S1
Table S2
Table S3
Table S4
Table S5