

Abstract
One pivotal factor that leads to multidrug resistance (MDR) is the overexpression of ABCG2. Therefore, tremendous effort has been devoted to the search of effective reversal agents to overcome ABCG2‐mediated MDR. CC‐671 is a potent and selective inhibitor of both TTK (human protein kinase monopolar spindle 1 [hMps1]) and CDC like kinase 2 (CLK2). It represents a new class of cancer therapeutic drugs. In this study, we show that CC‐671 is an effective ABCG2 reversal agent that enhances the efficacy of chemotherapeutic drugs in ABCG2‐overexpressing lung cancer cells. Mechanistic studies show that the reversal effect of CC‐671 is primarily attributed to the inhibition of the drug efflux activity of ABCG2, which leads to an increased intracellular level of chemotherapeutic drugs. In addition, CC‐671 does not alter the protein expression or subcellular localization of ABCG2. The computational molecule docking analysis suggests CC‐671 has high binding affinity to the drug‐binding site of ABCG2. In conclusion, we reveal the interaction between CC‐671 and ABCG2, providing a rationale for the potential combined use of CC‐671 with ABCG2 substrate to overcome MDR.
Keywords: ABCG2, CC‐671, dual TTK/CLK2 inhibitor, lung cancer, multidrug resistance
CC‐671 is an effective ABCG2 reversal agent that enhances the efficacy of chemotherapeutic drugs in ABCG2‐overexpressing lung cancer cells.

1. INTRODUCTION
Chemotherapy and targeted therapy are the main strategies for cancer treatment. One major obstacle that limits the efficacy of cancer therapy is the development of drug resistance. 1 , 2 In some cases, failure of traditional and targeted cancer therapy can be attributed to increased drug efflux, which leads to reduced intracellular drug concentration, resulting in drug resistance. 3 Multidrug resistance (MDR) describes the acquired resistance of cancer cells to a range of anticancer drugs that have distinct mechanism of actions. One pivotal factor that can lead to MDR is the overexpression of ATP‐binding cassette (ABC) transporters. The most common ABC transporters include ABCB1 (P‐glycoprotein/P‐gp, multidrug resistance 1/MDR1), 4 ABCG2 (breast cancer resistance protein/BCRP, mitoxantrone resistance/MXR), 5 , 6 and ABCC1 (MDR‐associated protein 1/MRP1). 7 By utilizing the energy derived from ATP hydrolysis, ABC transporters can extrude substrates from the cells against the direction of their concentration gradients. 8 , 9 Therefore, they have critical functions in maintaining different physiological processes but also efflux anticancer drugs, making them well‐known mediators of MDR. 10 , 11 In the clinical setting, ABCB1 and ABCG2 have been shown to interfere with the response of various conventional and targeted therapeutic drugs, such as paclitaxel, 12 doxorubicin, 13 topotecan, 14 gefitinib, 15 and imatinib. 16
ABCG2 is expressed in a wide range of tissues and localized on the membrane of several major physiological barriers, including the blood‐brain barrier 17 and placenta. 18 It has been suggested that ABCG2 exerts the protective role by reducing the cellular accumulation of toxins. 19 , 20 However, ABCG2 expression can affect the pharmacokinetic parameters of anticancer drugs. In lung cancer cells, ABCG2 expression is correlated with cancer cell side population formation. 21 ABCG2 is suggested to be a cancer stem cell marker that predicts the survival of patients with non‐small cell lung cancer (NSCLC). 22 , 23 Clinical study revealed that high exposure to erlotinib and sunitinib is associated with ABCG2 overexpression in lung cancer patients. 24 Therefore, both preclinical and clinical evidence have shown that ABCG2 expression might correlate with the outcome of cancer treatment.
Given the important role ABC transporter plays in mediating MDR, great effort has been devoted to the search for effective reversal agents. Many drugs in clinical use were identified as reversal agents, such as tepotinib, 25 ulixertinib, 26 and midostaurin 27 , 28 for reversing ABCB1‐mediated MDR, and olmutinib, 29 dacomitinib, 30 and quizartinib 31 for reversing ABCG2‐mediated MDR.
CC‐671, a potent and selective inhibitor of both monopolar spindle 1 kinase, also known as TTK, and CDC like kinase 2 (CLK2), showed significant anticancer effect in a large panel of cancer cell lines. 32 The anticancer activity of CC‐671 is mainly attributed to TTK inhibition. In addition, CLK2 was shown to act as an oncogene in breast cancer and inhibition of CLK2 can lead to apoptosis induction. The dual inhibitory mechanism represents a new class of cancer therapy, particularly in triple negative breast cancer patients. 33
In the course of searching MDR‐reversing agents, we found that CC‐671 can effectively antagonize ABCG2‐mediated MDR. Our study indicates the significant reversal effect of CC‐671 and therefore provides a potential combined treatment strategy to overcome MDR.
2. MATERIALS AND METHODS
2.1. Chemicals
CC‐671 was kindly provided by Chemietek. Ko143 was obtained from Enzo Life Sciences. All other materials were bought from Sigma Chemical Company unless stated otherwise.
2.2. Cell lines
The NSCLC cell line NCI‐H460 and its mitoxantrone‐resistant subline NCI‐H460/MX20, A549 and its mitoxantrone‐resistant subline A549/MX10, as well as the human epidermoid carcinoma KB‐3‐1 and its colchicine‐resistant subline KB‐C2 were maintained in DMEM containing 10% FBS. NCI‐H460/MX20 cells were cultured in the presence of 20 nmol/L mitoxantrone, 34 A549/MX10 cells were cultured in the presence of 10 nmol/L mitoxantrone, 34 and KB‐C2 cells were cultured in the presence of 2 µg/mL colchicine. 35 HEK293 cells stably transfected with either an empty vector pcDNA3.1 or a pcDNA3.1 vector containing a full length ABCG2 were maintained in DMEM containing 10% FBS. Transfected cell sublines were selected with medium containing 2 mg/mL G418. 36 The drug‐selected resistant cell lines were allowed to grow in drug‐free complete medium at least 14 days before experiment.
2.3. Cellular proliferation assay
The cytotoxicity of CC‐671 and other chemotherapeutic agents were determined by MTT colorimetric assay as previously described. 37 Briefly, cells were diluted to 5000‐6000 cells per well, seeded evenly into 96‐well plates, and incubated overnight to allow cell attachment. Subsequently, cells were incubated with drugs for 3 days. At the end of the assay period, cell viability was assessed by adding MTT solution. After 4 hours of incubation with MTT, DMSO was added to dissolve the produced formazan. The light absorbance at 570 nm was measured using the Fisherbrand accuSkan GO UV/Vis Microplate Spectrophotometer (Thermo Fisher Scientific).
2.4. Immunoblot analysis
Total proteins were extracted from NCI‐H460 and NCI‐H460/MX20 cells after incubating with CC‐671 for different time periods. The protein concentrations of the lysates were determined by Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). The samples were evaluated by SDS‐PAGE and immunoblotting using the following primary Abs: anti‐ABCG2 (BCRP) Ab, clone BXP‐21, and GAPDH loading control mAb (Thermo Fisher Scientific), followed by immunoblotting with HRP‐conjugated rabbit anti‐mouse IgG secondary Ab (Cell Signaling Technology). Pierce ECL Western blotting substrate (Thermo Fisher Scientific) was then used to detect the protein bands. The results were quantified and analyzed using ImageJ software.
2.5. Immunofluorescence assay
Cells were trypsinized and seeded into 24‐well plates at the density of 1 × 105 cells per well and incubated overnight. The cells were incubated with 1 μmol/L CC‐671 for up to 3 days. After the treatment period, the cells were fixed with 4% formaldehyde, permeabilized by 0.25% Triton X‐100, and blocked with 6% BSA (VWR Chemicals). Subsequently, the cells were incubated with anti‐ABCG2 (BCRP) Ab (1:1000) at 4°C overnight. The following day, cells were incubated with Alexa Fluor 488 conjugated goat anti‐mouse IgG cross‐adsorbed secondary Ab (Thermo Fisher Scientific) at room temperature for 2 hours. The nuclei were stained by incubating with DAPI solution. Images were taken with a Nikon TE‐2000S fluorescence microscope (Nikon Instruments).
2.6. Mitoxantrone accumulation assay
A FACSort flow cytometer was used to undertake the accumulation assay and FlowJo software V10 was used to analyze the data. To prepare the samples, NCI‐H460, NCI‐H460/MX20, A549, and A549/MX10 cells were separated into different treatment groups with 2 × 105 cells per group. Cells were incubated with Ko143 or CC‐671 for 2 hours. Subsequently, the fluorescent substrate mitoxantrone (5 μmol/L) was added into each treatment group with or without Ko143 or CC‐671. At the end, cells were further incubated for 2 hours then centrifuged at 500 g, followed by resuspension in 300 μL PBS with 0.5% BSA prior to analysis.
2.7. Evaluation of ABCG2 ATPase activity
The PREDEASY ATPase Kits (Tebu‐Bio) was selected to carry out the ATPase assay according to the vendor’s protocol with modifications as previously described. 25 In short, a series concentration of CC‐671 with or without Na3VO4 − was mixed with the ABCG2 membrane suspension. The mixtures were incubated at 37°C for 5 minutes, followed by the addition of 5 mmol/L Mg2+ATP to initiate the reaction. The inorganic phosphate released during the reaction period was colorimetrically determined by spectrophotometry. The changes of relative light units were determined by comparing the Na3VO4 − treated group with the corresponding CC‐671 treated groups.
2.8. In silico molecular docking analysis
Maestro version 11.1 software (Schrödinger) was used to undertake the computational molecular docking analysis. The protein model used in this study is the recently published human ABCG2 cryogenic Electron Microscopy (cryo‐EM) structure model (PDB: 6ETI) from the RCSB Protein Data Bank. 38 The detailed protocol was as previously described. 39 The result of the docking scores was calculated and demonstrated as kcal/mol.
2.9. Statistical analysis
All experiments were carried out in triplicate and repeated at least 3 times. Statistical analysis was undertaken using GraphPad Prism 7.0 software. Data are presented as a mean ± SD and one‐way ANOVA were applied. A difference was considered significant at P less than .05.
3. RESULTS
3.1. Antiproliferative effect of CC‐671 on parental and drug‐resistant cells
The antiproliferative effect of CC‐671 was determined in various cell lines. In this study, we used KB‐3‐1 and its ABCB1‐overexpressing subline KB‐C2, NCI‐H460 and its ABCG2‐overexpressing subline NCI‐H460/MX20, and A549 and its ABCG2‐overexpressing subline A549/MX10 to investigate the reversal effect of CC‐671 in cancer cells. HEK293 gene‐transfected cells were selected to further validate the results. The cell viability curves are presented in Figure 1. In order to circumvent the additive toxicity of CC‐671 and chemotherapeutic drugs, nontoxic concentrations (0.1‐1 μmol/L) were selected based on the viability curves to undertake the following reversal studies.
FIGURE 1.
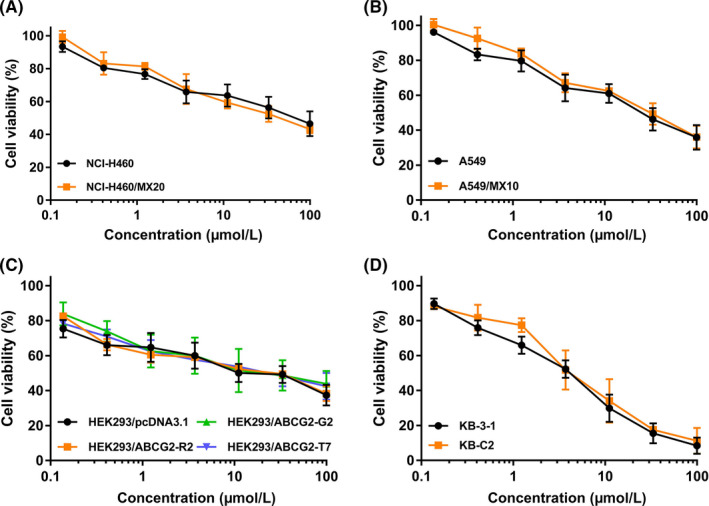
Cytotoxicity of CC‐671 in drug‐sensitive and ABCG2‐overexpressing cell lines. Cell viability curves for (A) NCI‐H460 and NCI‐H460/MX20 cells, (B) A549 and A549/MX10 cells, (C) HEK293/pcDNA3.1, HEK293/ABCG2‐WT, HEK293/ABCG2‐R482G, and HEK293/ABCG2‐R482T cells, and (D) KB‐3‐1 and KB‐C2 cells. Data are expressed as mean ± SD from a representative of 3 independent experiments. *P < .05 vs. control group
3.2. CC‐671 resensitized ABCG2‐overexpressing cells to ABCG2 substrate drugs
Reversal studies were carried out by combining CC‐671 with ABCG2 substrates mitoxantrone or topotecan. The data, as presented in Table 1, showed that CC‐671 can sensitize drug‐resistant NCI‐H460/MX20 cells in a concentration‐dependent manner without affecting the drug‐sensitive NCI‐H460 cells. When using the highest reversal concentration at 1 μmol/L, the resistance‐fold of mitoxantrone and topotecan in NCI‐H460/MX20 was decreased from 125.13‐fold to 3.34‐fold and from 69.12‐fold to 5.58‐fold, respectively. CC‐671 also sensitized A549/MX10 cells to mitoxantrone and topotecan, with the resistance‐fold decreased from 76.78‐fold to 4.61‐fold and from 40.03‐fold to 4.74‐fold, respectively. By undertaking reversal studies in 2 pairs of NSCLC cell lines, we confirmed that CC‐671 can reverse ABCG2‐mediated MDR in cancer cells. Of note, the reversal effect of CC‐671 at 1 μmol/L was comparable to the well‐established ABCG2 inhibitor Ko143. Furthermore, the reversal effect was observed when CC‐671 was used in combination with ABCG2 substrates mitoxantrone or topotecan but not with the nonsubstrate drug cisplatin, suggesting that CC‐671 might interact with the ABCG2 transporter instead of with specific drugs. To further validate CC‐671 is an ABCG2 reversal agent, MTT assay was carried out in ABCG2 gene‐transfected HEK293 cells. Cells transfected with empty vector pcDNA3.1 does not express any ABC transporters; cells transfected with WT or mutant (R482G/R482T) ABCG2 gene overexpress WT or mutant ABCG2 protein. Consistently, the cytotoxicity of CC‐671 was not affected by overexpression of WT or mutant ABCG2, as shown in Figure 1D. Due to the relatively high toxicity in HEK293 cells, we undertook combined studies using 0.1 and 0.3 μmol/L CC‐671. A similar reversal effect was observed in HEK293 cell lines (Table 2). In both WT and mutant ABCG2‐overexpressing HEK293 cells, CC‐671 was able to significantly reverse the mitoxantrone or topotecan resistance. In contrast, CC‐671 failed to antagonize ABCB1‐mediated MDR, as shown in Table 3.
TABLE 1.
Reversal effect of CC‐671 in non‐small cell lung cancer cells overexpressing ABCG2 transporter
| Treatment | IC50 value ± SD a (μmol/L, resistance‐fold b ) | |||
|---|---|---|---|---|
| NCI‐H460 | NCI‐H460/MX20 | A549 | A549/MX10 | |
| Mitoxantrone | 0.011 ± 0.003 (1.00) | 1.437 ± 0.326 (125.13) | 0.026 ± 0.009 (1.00) | 2.401 ± 0.486 (76.78) |
| + CC‐671 0.1 μmol/L | 0.014 ± 0.006 (1.20) | 0.081 ± 0.006 (7.09)* | 0.027 ± 0.004 (1.07) | 0.633 ± 0.255 (20.24)* |
| + CC‐671 0.3 μmol/L | 0.013 ± 0.004 (1.16) | 0.053 ± 0.004 (4.63)* | 0.021 ± 0.005 (0.83) | 0.252 ± 0.064 (8.06)* |
| + CC‐671 1 μmol/L | 0.012 ± 0.002 (1.07) | 0.038 ± 0.002 (3.34)* | 0.025 ± 0.008 (0.96) | 0.144 ± 0.032 (4.61) * |
| + Ko143 1 μmol/L | 0.014 ± 0.007 (1.19) | 0.044 ± 0.003 (3.87)* | 0.028 ± 0.007 (1.10) | 0.181 ± 0.040 (5.78) * |
| Topotecan | 0.043 ± 0.025 (1.00) | 3.003 ± 0.641 (69.12) | 0.035 ± 0.003 (1.00) | 1.401 ± 0.486 (40.03) |
| + CC‐671 0.1 μmol/L | 0.053 ± 0.008 (1.23) | 0.504 ± 0.115 (11.61)* | 0.033 ± 0.004 (0.94) | 0.531 ± 0.147 (15.17) * |
| + CC‐671 0.3 μmol/L | 0.068 ± 0.024 (1.56) | 0.411 ± 0.095 (9.46)* | 0.032 ± 0.007 (0.92) | 0.326 ± 0.034 (9.31) * |
| + CC‐671 1 μmol/L | 0.064 ± 0.018 (1.46) | 0.242 ± 0.040 (5.58)* | 0.041 ± 0.011 (1.17) | 0.166 ± 0.029 (4.74) * |
| + Ko143 1 μmol/L | 0.059 ± 0.028 (1.37) | 0.218 ± 0.088 (5.01)* | 0.028 ± 0.004 (0.82) | 0.210 ± 0.021 (6.06) * |
| Cisplatin | 3.730 ± 0.834 (1.00) | 5.198 ± 2.412 (1.39) | 4.730 ± 0.534 (1.00) | 3.991 ± 0.187 (0.84) |
| + CC‐671 1 μmol/L | 3.581 ± 0.388 (0.96) | 5.106 ± 1.334 (1.37) | 4.987 ± 0.561 (0.95) | 4.847 ± 0.406 (1.02) |
| + Ko143 1 μmol/L | 3.067 ± 0.947 (0.82) | 4.028 ± 0.581 (1.08) | 4.991 ± 0.287 (1.06) | 4.067 ± 0.347 (0.86) |
IC50 values are represented as mean ± SD of at least 3 independent experiments carried out in triplicate.
Calculated by dividing the IC50 values of substrates in the presence or absence of inhibitor by the IC50 of parental cells without inhibitor.
P < .05 vs. control group without reversal agent.
TABLE 2.
Reversal effect of CC‐671 in HEK293 transfected cells overexpressing ABCG2 transporter
| Treatment | IC50 value ± SD a (μmol/L, resistance‐fold b ) | |||
|---|---|---|---|---|
| pcDNA3.1 | ABCG2‐WT | ABCG2‐R482G | ABCG2‐R482T | |
| Mitoxantrone | 0.018 ± 0.002 (1.00) | 0.590 ± 0.102 (32.42) | 0.417 ± 0.147 (22.95) | 1.342 ± 0.263 (73.81) |
| + CC‐671 0.1 μmol/L | 0.022 ± 0.006 (1.05) | 0.179 ± 0.026 (9.85)* | 0.111 ± 0.042 (6.11)* | 0.545 ± 0.321 (29.98)* |
| + CC‐671 0.3 μmol/L | 0.024 ± 0.004 (1.13) | 0.035 ± 0.014 (1.92)* | 0.035 ± 0.018 (1.92)* | 0.124 ± 0.036 (6.85) * |
| + Ko143 0.3 μmol/L | 0.020 ± 0.009 (0.79) | 0.022 ± 0.012 (1.22)* | 0.032 ± 0.005 (1.75)* | 0.021 ± 0.008 (1.15) * |
| Topotecan | 0.104 ± 0.030 (1.00) | 2.628 ± 1.319 (25.30) | 4.856 ± 1.482 (46.73) | 6.107 ± 0.656 (58.77) |
| + CC‐671 0.1 μmol/L | 0.121 ± 0.036 (1.16) | 0.706 ± 0.228 (6.79)* | 0.977 ± 0.348 (9.40)* | 2.971 ± 0.131 (28.59)* |
| + CC‐671 0.3 μmol/L | 0.155 ± 0.064 (1.49) | 0.466 ± 0.067 (4.48)* | 0.343 ± 0.170 (3.30)* | 0.277 ± 0.090 (2.67) * |
| + Ko143 0.3 μmol/L | 0.124 ± 0.040 (1.19) | 0.275 ± 0.140 (2.64)* | 0.130 ± 0.019 (1.26)* | 0.332 ± 0.125 (3.20) * |
| Cisplatin | 8.247 ± 1.125 (1.00) | 12.853 ± 0.083 (1.56) | 7.605 ± 1.288 (0.92) | 7.895 ± 0.795 (0.95) |
| + CC‐671 0.3 μmol/L | 8.759 ± 3.243 (1.06) | 11.564 ± 2.415 (1.40) | 11.752 ± 2.171 (1.43) | 9.351 ± 1.636 (1.13) |
| + Ko143 0.3 μmol/L | 6.234 ± 0.137 (0.76) | 11.758 ± 2.923 (1.43) | 10.050 ± 2.756 (1.22) | 7.325 ± 1.124 (0.89) |
IC50 values are represented as mean ± SD of at least 3 independent experiments carried out in triplicate.
Calculated by dividing the IC50 values of substrates in the presence or absence of inhibitor by the IC50 of parental cells without inhibitor.
P < .05 vs. control group without reversal agent.
TABLE 3.
Reversal effect of CC‐671 in cancer cells overexpressing ABCB1 transporter
| Treatment | IC50 value ± SD a (μmol/L, resistance‐fold b ) | |
|---|---|---|
| KB‐3‐1 | KB‐C2 | |
| Vincristine | 0.035 ± 0.014 (1.00) | 3.814 ± 1.595 (107.46) |
| + CC‐671 0.3 μmol/L | 0.035 ± 0.015 (0.99) | 1.826 ± 0.261 (51.46) |
| + Verapamil 0.3 μmol/L | 0.034 ± 0.019 (0.95) | 0.571 ± 0.072 (16.09)* |
| Cisplatin | 2.041 ± 0.165 (1.00) | 2.845 ± 0.877 (1.39) |
| + CC‐671 0.3 μmol/L | 1.826 ± 0.115 (0.89) | 2.651 ± 0.555 (1.30) |
| + Verapamil 0.3 μmol/L | 1.664 ± 0.258 (0.82) | 3.043 ± 0.728 (1.49) |
IC50 values are represented as mean ± SD of at least 3 independent experiments carried out in triplicate.
Calculated by dividing the IC50 values of substrates in the presence or absence of inhibitor by the IC50 of parental cells without inhibitor.
P < .05 vs. control group without reversal agent.
3.3. CC‐671 increased intracellular accumulation of mitoxantrone in ABCG2‐overexpressing cells
To further investigate the reversal mechanism, mitoxantrone accumulation assay was carried out to evaluate whether CC‐671 can hinder the substrate efflux function of ABCG2. By detecting the fluorescence intensity of mitoxantrone using flow cytometry, it allows direct quantification of the intracellular amount of mitoxantrone. As CC‐671 can enhance the cytotoxicity of ABCG2 substrate drugs, we postulated that CC‐671 might hinder the drug efflux process and increase drug accumulation. As shown in Figure 2, CC‐671 was able to elevate the intracellular level of mitoxantrone, in a concentration‐dependent manner, in ABCG2‐overexpressing NCI‐H460/MX20 and A549/MX10 cells but not in the parental cells. These results suggested the reversal effect, at least in part, can be attribute to ABCG2 substrate efflux inhibition.
FIGURE 2.
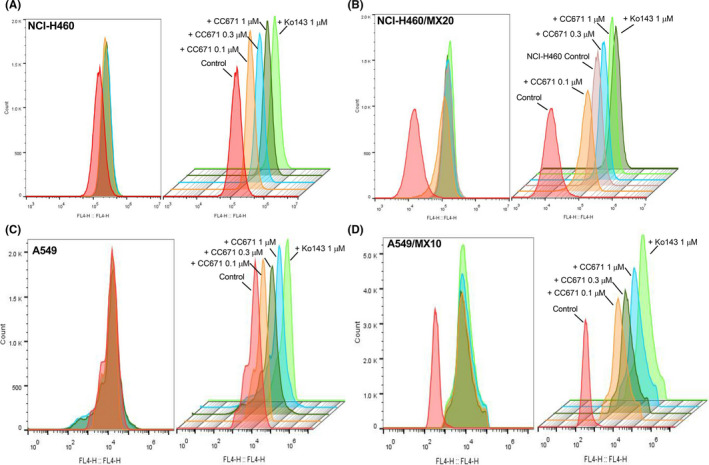
Effect of CC‐671 on intracellular accumulation of mitoxantrone in drug‐sensitive and drug‐resistant cells. A, NCI‐H460 cells. B, NCI‐H460/MX20 cells. C, A549 cells. D, A549/MX10 cells
3.4. CC‐671 had no significant effect on ABCG2 protein expression or its cell surface localization
We explored 2 possible mechanisms by which CC‐671 might exert the reversal effect: alteration of ABCG2 protein expression and/or cell membrane localization. Using western blot analysis, we confirmed ABCG2 expression levels remain unchanged after cells were incubated with CC‐671 for up to 72 hours (Figure 3A,B). However, the immunofluorescent assay suggested that ABCG2 transporter of H460/MX20 was localized on the cell membrane and showed no trend of internalization after incubation with CC‐671 (Figure 3C). In addition, the localization of ABCG2 has no detectable change in A549/MX10 cells with or without CC‐671 treatment (data not shown). Whether CC‐671 can affect ABCG2 expression levels or localization after long‐term treatment warrants further investigation.
FIGURE 3.
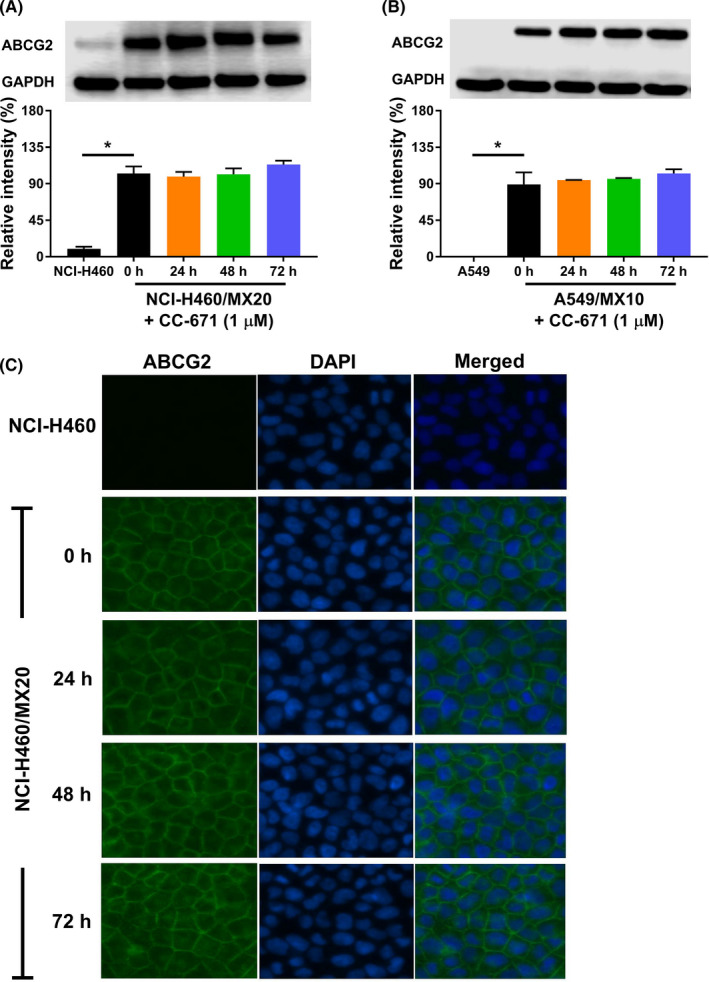
Effect of CC‐671 on ABCG2 protein expression and localization. A, B, Effect of CC‐671 on the expression level of ABCG2 in (A) NCI‐H460 and NCI‐H460/MX20 cells and (B) A549 and A549/MX10 cells. C, Cell surface localization of ABCG2 in NCI‐H460/MX20 cells incubated with 1 μmol/L CC‐671 for up to 72 h. Data are expressed as mean ± SD from a representative of three independent experiments. *P < .05 versus the control group
3.5. CC‐671 stimulated ABCG2 ATPase activity
Learning that CC‐671 can inhibit the efflux function of ABCG2, the ABCG2 ATPase assay was carried out to investigate the mechanism of efflux inhibition. The ABCG2‐mediated ATP hydrolysis in ABCG2 overexpressing membranes was measured after incubation with serial concentrations of CC‐671 (0‐10 μmol/L). According to the result (Figure 4), CC‐671 can stimulate the activity of ABCG2 ATPase in a concentration‐dependent manner. The ABCG2 ATPase activity reached a maximum of 341.3% of the basal activity and the stimulatory effect reached half maximal effective concentration at 0.49 μmol/L, indicating CC‐671 might be a substrate of ABCG2.
FIGURE 4.
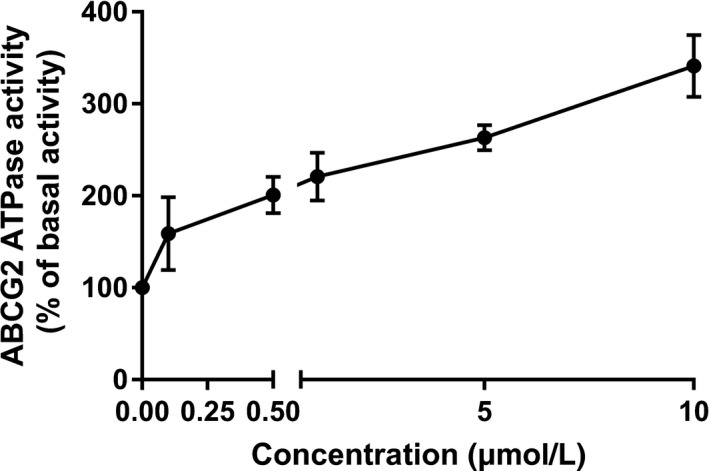
Effect of CC‐671 on ABCG2 ATPase activity. CC‐671 (0‐10 μmol/L) stimulated the ATPase activity of ABCG2 transporter. Data are expressed as mean ± SD from a representative of 3 independent experiments
3.6. CC‐671 showed high binding affinity with ABCG2 in molecular docking analysis
The molecular docking analysis was undertaken to simulate the interactions between CC‐671 and human ABCG2 protein model. The highest Glide score of CC‐671 was −12.318 kcal/mol, which indicates the binding free energy of the ligand. As shown in Figure 5A, the optimal scoring pose of CC‐671 was mainly stabilized in the ligand‐binding cavity of ABCG2 transmembrane domain through hydrophobic interactions with nearby residues. Two hydrogen bonds were formed between the nitrogen of the methylbenzamide group of CC‐671 and Asn436 of ABCG2 chain A, and the nitrogen in the oxazole ring of CC‐671 and Asn436 of ABCG2 chain B. One π‐π stacking interaction was observed between the benzene ring of benzoxazole of CC‐671 and the side chain of PHE432 of ABCG2 chain B. Figure 5B shows that CC‐671 was stabled in ABCG2 transmembrane domain with surrounding residues including Val401, Leu405, Phe432, Thr435, Asn436, Phe439, Ser440, Thr542, Val546, Met549 of ABCG2 chain A, and Leu405, Phe431, Phe432, Thr435, Asn436, Phe439, Ser440, Thr542, Val546, and Met549 of ABCG2 chain B.
FIGURE 5.
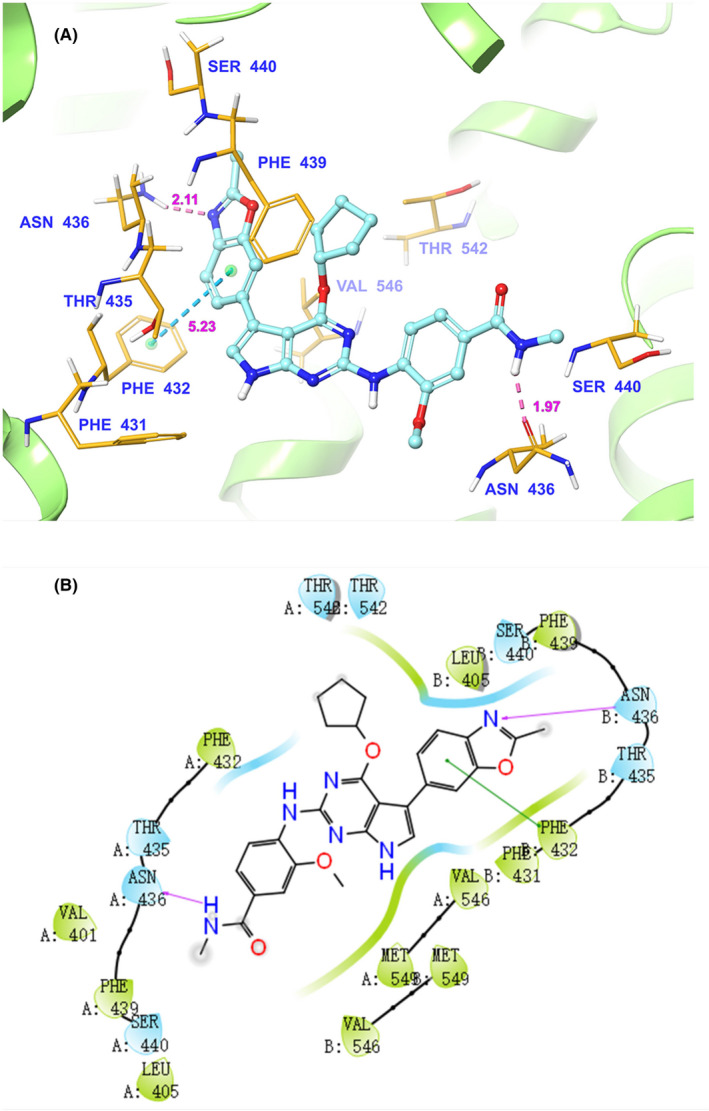
Computational molecular docking binding of CC‐671 to the human ABCG2 model. The binding mode of CC‐671 to the human ABCG2 model predicted by induce‐fit docking. A, Docked conformation of CC‐671 (ball and rod model) is shown within the ABCG2 drug‐binding cavity, with the atoms colored as follows: carbon, cyan; hydrogen, white; oxygen, red; nitrogen, blue. The ABCG2 structure is in the ribbon diagram in light green. Important amino acid residues are described (rods model) with the same color scheme as above for all atoms but carbon atoms in orange. Dotted blue lines represent hydrogen‐bonding interactions, whereas dotted azure lines represent π‐π stacking interactions. The values of the correlation distances are indicated in Å. B, The 2‐D schematic diagram of ligand‐receptor interaction between CC‐671 and the human ABCG2 model. Amino acids within 4 Å are indicated as colored bubbles, polar residues are depicted as light blue, and hydrophobic residues are depicted as green. Pink arrows denote H‐bonds and dark green lines denote π‐π stacking aromatic interactions
4. DISCUSSION
One major obstacle that hinders the efficacy of cancer treatment is the development of MDR. ABCG2 has long been characterized as a major MDR contributor that can pump out a wide range of anticancer drugs including mitoxantrone, doxorubicin, topotecan, and some tyrosine kinase inhibitors. 40 , 41 Therefore, a large number of investigations have been undertaken in an effort to search for effective reversal agents targeting ABCG2 to overcome MDR. 42 , 43 , 44 , 45 In the present study, we investigated the possibility of using CC‐671, a dual inhibitor of TTK/CLK2, to reverse ABCG2‐mediated MDR.
To explore the interaction between CC‐671 and ABCG2, an MTT assay was carried out to determine the cytotoxicity of CC‐671 in both parental and drug‐resistant cells. It was observed that the antiproliferative effect of CC‐671 was not affected by overexpression of ABCG2 and nontoxic concentration can be equally achieved in both parental and drug‐resistant cells. Subsequently, reversal studies were undertaken by combining CC‐671, as a potential reversal agent, with an ABCG2 substrate, mitoxantrone or topotecan. We found that CC‐671, at nanomolar concentrations, was able to sensitize ABCG2‐overexpressing NSCLC cells to mitoxantrone or topotecan, suggesting CC‐671 could be an effective reversal agent of ABCG2‐mediated MDR.
It has been suggested that ABCG2 polymorphism can alter the drug efflux profile and the most well‐established residue to be involved in the transportation of ABCG2 substrates is arginine 482 (R482) in the transmembrane domain. 36 , 46 , 47 R482 was confirmed as the WT sequence, and R482G and R482T were identified as mutant variants of ABCG2. Previous work has shown that the R482 mutation can affect to the substrate specificity of ABCG2. 48 Most of the ABCG2 substrates can be effectively transported by both WT and mutant ABCG2, such as mitoxantrone, doxorubicin, or Hoechst 33342. However, rhodamine 123 and daunorubicin can be transported only by mutant ABCG2, whereas methotrexate can be transported only by WT ABCG2. 36 , 49 The mutants R482G and R482T were shown to render cancer cells with higher resistance to several substrates than the WT ABCG2, suggesting a more active efflux activity in the mutants. 50 Recently, it was reported that venetoclax can inhibit the efflux function of WT ABCG2 but not mutant ABCG2, suggesting that the R482 mutants might also determine the effect of reversal agents. 51 Therefore, it is important to investigate whether CC‐671 can effectively inhibit substrate efflux in both WT and mutant ABCG2. By undertaking MTT assays in ABCG2‐transfected cell lines, we confirmed that CC‐671 can effectively reverse both WT and mutant ABCG2‐mediated MDR. Of note, the reversal effect was not observed when CC‐671 was combined with cisplatin, which is not a substrate drug of ABCG2. As the reversal effect was reproducible in both drug‐selected and gene‐transfected ABCG2‐overexpressing cells, it provides evidence to show the reversal mechanism of CC‐671 is indeed targeting the ABCG2 transporter. In addition, we also evaluated the reversal effect of CC‐671 on ABCB1‐mediated MDR. CC‐671 maintained the same cytotoxicity in ABCB1‐overexpressing cells as in parental cells but showed no significant reversal effect at the highest nontoxic concentration. Therefore, these results suggest that CC‐671 can be a potential reversal agent that selectively antagonizes ABCG2‐mediated MDR. Future studies could focus on other MDR‐associated ABC transporters, such as ABCC1 7 and ABCC10, 52 to further evaluate the specificity of CC‐671 as a reversal agent.
Subsequently, we investigated several mechanisms by which CC‐671 might act to reverse MDR: alteration of ABCG2 protein expression level, cell surface localization of ABCG2, and/or drug efflux function of ABCG2. ABCG2 downregulation has been reported as a reversal mechanism for several agents. 44 , 53 , 54 Using western blot analysis, we found no significant change of ABCG2 protein expression level after cells were incubated with CC‐671 in 2 drug‐resistant lung cancer cell lines for up to 72 hours. In contrast, ABCG2 is a membrane‐bound efflux pump that localized on the cell surface to pump out its substrates. A possible reversal mechanism is to translocate ABCG2 into the cytoplasm, which might inactivate the efflux function of ABCG2. To investigate this potential mechanism, immunofluorescence assay was carried out to visualize the localization of ABCG2 after CC‐671 treatment. Our result showed that the expression of ABCG2 remained on the cell surface of H460/MX20 cells after 72 hours of CC‐671 treatment. These results suggest that neither alteration of ABCG2 expression level nor ABCG2 cell surface localization may be responsible for the reversal effect. It should be noted that the question whether CC‐671 can affect ABCG2 expression or cell surface localization after long‐term treatment remained inconclusive and deserves further investigation.
To evaluate whether CC‐671 can interfere with the efflux function of ABCG2, mitoxantrone accumulation assay was carried out. Because mitoxantrone is a substrate of ABCG2, the intracellular accumulation of mitoxantrone was significantly lower in 2 mitoxantrone‐resistant lung cancer cell lines than in their parental cell lines, as a large proportion of mitoxantrone was pumped out from the resistant cells. CC‐671, when combined with mitoxantrone, was able to significantly increase the intracellular accumulation of mitoxantrone only in drug‐resistant cells in a concentration‐dependent manner. Therefore, the substrate efflux process was attenuated in resistant cells after treatment with CC‐671. As ABCG2 overexpression is the major factor that leads to decreased mitoxantrone accumulation in resistant cells, this result provides strong evidence to confirm the reversal effect of CC‐671 is due to inhibition of ABCG2 efflux function. Previous studies have shown that reversal agents that inhibit substrate efflux can be either ATPase inhibitors that hinder ATP hydrolysis and energy production, or ATPase stimulators that act as transported substrates and stimulate the ATPase activity of ABCG2. Therefore, we measured the ABCG2‐mediated ATPase hydrolysis in the presence of CC‐671. The data showed that CC‐671 can stimulate the activity of ABCG2 ATPase activity in a concentration‐dependent manner, indicating CC‐671 can interact with ABCG2 transporter. Combining the results from our mechanistic studies, we postulated CC‐671 can reverse drug resistance be interacting with ABCG2 at the drug‐binding site, thereby inhibiting the substrate efflux process.
Computational molecular docking analysis has been extensively applied in the field of structural molecular biology, which provides a useful tool to predict the interaction between the small molecules and protein binding sites. 55 As we hypothesized CC‐671 can bind to the drug‐binding cavity of ABCG2, we undertook molecular docking to further validate our hypothesis. The cryo‐EM structure of ABCG2 bound with a well‐known ABCG2 inhibitor, Ko143, was utilized to investigate the interaction between reversal agents and ABCG2 transporter. 38 The docking scores are predicted values of the free energy of protein‐ligand binding, and a higher absolute value represents a higher affinity. CC‐671 obtained a docking score of −12.318 kcal/mol. Furthermore, the docking score of CC‐671 is comparable to other reported ABCG2 reversal agents such as selonsertib (−11.094 kcal/mol), 56 voruciclib (−10.304 kcal/mol), 57 and ZM323881 (−11.509 kcal/mol). 58 Of note, the molecular docking is not meant to be an accurate affinity predictor, thereby the bound conformation might not represent the actual binding situation 59 and is therefore used as a reference in this study.
Overall, our study reveals a new reversal agent of ABCG2 and provides the rational basis of using CC‐671 to overcome ABCG2‐mediated MDR in cancer patients.
CONFLICT OF INTEREST
The authors have no conflict of interest.
ACKNOWLEDGMENTS
The authors would like to thank Chemietek for kindly provided CC‐671 compound. We would like to thank Drs. Robert W. Robey and Susan E. Bates (NCI, NIH, Bethesda, MD, USA) for providing NCI‐H460, NCI‐H460/MX20, A549, and A549/MX10 cell lines. We thank Dr Shin‐Ichi Akiyama (Kagoshima University, Kagoshima, Japan) for the gift of KB‐3‐1 and KB‐C2 cell lines. We would like to thank Dr Tanaji T. Talele (St. John’s University, Queens, NY, USA) for providing the computational resource for the molecular docking analysis. This work was supported by The Science and Technology Foundation of Shenzhen (JCYJ20170412155231633 and JCYJ20180305164128430), the Shenzhen Economic and Information Committee “Innovation Chain and Industry Chain” integration special support plan project (20180225112449943), the Shenzhen Public Service Platform on Tumor Precision Medicine and Molecular Diagnosis, the Shenzhen Cell Therapy Public Service Platform.
Wu Z‐X, Yang Y, Wang G, et al. Dual TTK/CLK2 inhibitor, CC‐671, selectively antagonizes ABCG2‐mediated multidrug resistance in lung cancer cells. Cancer Sci. 2020;111:2872–2882. 10.1111/cas.14505
Contributor Information
Lizhu Lin, Email: lizhulin26@yahoo.com.
Zhe‐Sheng Chen, Email: chenz@stjohns.edu.
Chang Zou, Email: zou.chang@szhospital.com.
REFERENCES
- 1. Kartal‐Yandim M, Adan‐Gokbulut A, Baran Y. Molecular mechanisms of drug resistance and its reversal in cancer. Crit Rev Biotechnol. 2016;36(4):716‐726. [DOI] [PubMed] [Google Scholar]
- 2. Ni R, Zhu J, Xu Z, Chen Y. A self‐assembled pH/enzyme dual‐responsive prodrug with PEG deshielding for multidrug‐resistant tumor therapy. J Mater Chem B. 2020;8(6):1290‐1301. [DOI] [PubMed] [Google Scholar]
- 3. Begicevic RR, Falasca M. ABC transporters in cancer stem cells: beyond chemoresistance. Int J Mol Sci. 2017;18(11):2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Endicott JA, Ling V. The biochemistry of P‐glycoprotein‐mediated multidrug resistance. Annu Rev Biochem. 1989;58:137‐171. [DOI] [PubMed] [Google Scholar]
- 5. Robey RW, Medina‐Pérez WY, Nishiyama K, et al. Overexpression of the ATP‐binding cassette half‐transporter, ABCG2 (Mxr/BCrp/ABCP1), in flavopiridol‐resistant human breast cancer cells. Clin Cancer Res. 2001;7(1):145‐152. [PubMed] [Google Scholar]
- 6. Doyle LA, Yang W, Abruzzo LV, et al. A multidrug resistance transporter from human MCF‐7 breast cancer cells. Proc Natl Acad Sci USA. 1998;95(26):15665‐15670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cole SP, Bhardwaj G, Gerlach JH, et al. Overexpression of a transporter gene in a multidrug‐resistant human lung cancer cell line. Science. 1992;258(5088):1650‐1654. [DOI] [PubMed] [Google Scholar]
- 8. Vasiliou V, Vasiliou K, Nebert DW. Human ATP‐binding cassette (ABC) transporter family. Hum Genomics. 2009;3(3):281‐290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP‐dependent transporters. Nat Rev Cancer. 2002;2(1):48‐58. [DOI] [PubMed] [Google Scholar]
- 10. Han SH, Kim JW, Kim M, et al. Prognostic implication of ABC transporters and cancer stem cell markers in patients with stage III colon cancer receiving adjuvant FOLFOX‐4 chemotherapy. Oncol Lett. 2019;17(6):5572‐5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Toyoda Y, Takada T, Suzuki H. Inhibitors of Human ABCG2: from technical background to recent updates With clinical implications. Front Pharmacol. 2019;10:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Björn N, Jakobsen Falk I, Vergote I, Gréen H. ABCB1 variation affects myelosuppression, progression‐free survival and overall survival in Paclitaxel/Carboplatin‐treated ovarian cancer patients. Basic Clin Pharmacol Toxicol. 2018;123(3):277‐287. [DOI] [PubMed] [Google Scholar]
- 13. Tecza K, Pamula‐Pilat J, Lanuszewska J, Butkiewicz D, Grzybowska E. Pharmacogenetics of toxicity of 5‐fluorouracil, doxorubicin and cyclophosphamide chemotherapy in breast cancer patients. Oncotarget. 2018;9(10):9114‐9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Maliepaard M, van Gastelen MA, de Jong LA, et al. Overexpression of the BCRP/MXR/ABCP gene in a topotecan‐selected ovarian tumor cell line. Cancer Res. 1999;59(18):4559‐4563. [PubMed] [Google Scholar]
- 15. Elkind NB, Szentpétery Z, Apáti Á, et al. Multidrug transporter ABCG2 prevents tumor cell death induced by the epidermal growth factor Receptor inhibitor Iressa (ZD1839, Gefitinib). Cancer Res. 2005;65(5):1770‐1777. [DOI] [PubMed] [Google Scholar]
- 16. Burger H, van Tol H, Boersma AWM, et al. Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood. 2004;104(9):2940‐2942. [DOI] [PubMed] [Google Scholar]
- 17. Nagy I, Tóth B, Gáborik Z, Erdo F, Krajcsi P. Membrane transporters in physiological barriers of pharmacological importance. Curr Pharm Des. 2016;22(35):5347‐5372. [DOI] [PubMed] [Google Scholar]
- 18. Szilagyi JT, Vetrano AM, Laskin JD, Aleksunes LM. Localization of the placental BCRP/ABCG2 transporter to lipid rafts: role for cholesterol in mediating efflux activity. Placenta. 2017;55:29‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cleophas MC, Joosten LA, Stamp LK, Dalbeth N, Woodward OM, Merriman TR. ABCG2 polymorphisms in gout: insights into disease susceptibility and treatment approaches. Pharmgenomics Pers Med. 2017;10:129‐142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Krishnamurthy P, Ross DD, Nakanishi T, et al. The stem cell marker Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. J Biol Chem. 2004;279(23):24218‐24225. [DOI] [PubMed] [Google Scholar]
- 21. Ho MM, Ng AV, Lam S, Hung JY. Side population in human lung cancer cell lines and tumors is enriched with stem‐like cancer cells. Cancer Res. 2007;67(10):4827‐4833. [DOI] [PubMed] [Google Scholar]
- 22. Yoh K, Ishii G, Yokose T, et al. Breast cancer resistance protein impacts clinical outcome in platinum‐based chemotherapy for advanced non‐small cell lung cancer. Clin Cancer Res. 2004;10(5):1691‐1697. [DOI] [PubMed] [Google Scholar]
- 23. Ota S, Ishii G, Goto K, et al. Immunohistochemical expression of BCRP and ERCC1 in biopsy specimen predicts survival in advanced non‐small‐cell lung cancer treated with cisplatin‐based chemotherapy. Lung Cancer. 2009;64(1):98‐104. [DOI] [PubMed] [Google Scholar]
- 24. Fujita KI, Hirose T, Kusumoto S, et al. High exposure to erlotinib and severe drug‐induced interstitial lung disease in patients with non‐small‐cell lung cancer. Lung Cancer. 2014;86(1):113‐114. [DOI] [PubMed] [Google Scholar]
- 25. Wu ZX, Teng QX, Cai CY, et al. Tepotinib reverses ABCB1‐mediated multidrug resistance in cancer cells. Biochem Pharmacol. 2019;166:120‐127. [DOI] [PubMed] [Google Scholar]
- 26. Ji N, Yang Y, Lei ZN, et al. Ulixertinib (BVD‐523) antagonizes ABCB1‐ and ABCG2‐mediated chemotherapeutic drug resistance. Biochem Pharmacol. 2018;158:274‐285. [DOI] [PubMed] [Google Scholar]
- 27. Ji N, Yang Y, Cai C‐Y, et al. Midostaurin reverses ABCB1‐mediated multidrug resistance, an in vitro study. Front Oncol. 2019;9:514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hsiao SH, Lusvarghi S, Huang YH, Ambudkar SV, Hsu SC, Wu CP. The FLT3 inhibitor midostaurin selectively resensitizes ABCB1‐overexpressing multidrug‐resistant cancer cells to conventional chemotherapeutic agents. Cancer Lett. 2019;445:34‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang W, Fan YF, Cai CY, et al. Olmutinib (BI1482694/HM61713), a novel epidermal growth factor receptor tyrosine kinase inhibitor, reverses ABCG2‐mediated multidrug resistance in cancer cells. Front Pharmacol. 2018;9:1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guo X, To KKW, Chen Z, et al. Dacomitinib potentiates the efficacy of conventional chemotherapeutic agents via inhibiting the drug efflux function of ABCG2 in vitro and in vivo. J Exp Clin Cancer Res. 2018;37(1):31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li J, Kumar P, Anreddy N, et al. Quizartinib (AC220) reverses ABCG2‐mediated multidrug resistance: In vitro and in vivo studies. Oncotarget. 2017;8(55):93785‐93799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Riggs JR, Nagy M, Elsner J, et al. The discovery of a dual TTK protein kinase/CDC2‐like kinase (CLK2) inhibitor for the treatment of triple negative breast cancer initiated from a phenotypic screen. J Med Chem. 2017;60(21):8989‐9002. [DOI] [PubMed] [Google Scholar]
- 33. Zhu D, Xu S, Deyanat‐Yazdi G, et al. Synthetic lethal strategy identifies a potent and selective TTK and CLK1/2 inhibitor for treatment of triple‐negative breast cancer with a compromised G(1)‐S checkpoint. Mol Cancer Ther. 2018;17(8):1727‐1738. [DOI] [PubMed] [Google Scholar]
- 34. Robey RW, Honjo Y, van de Laar A, et al. A functional assay for detection of the mitoxantrone resistance protein, MXR (ABCG2). Biochim Biophys Acta. 2001;1512(2):171‐182. [DOI] [PubMed] [Google Scholar]
- 35. Akiyama S, Fojo A, Hanover JA, Pastan I, Gottesman MM. Isolation and genetic characterization of human KB cell lines resistant to multiple drugs. Somat Cell Mol Genet. 1985;11(2):117‐126. [DOI] [PubMed] [Google Scholar]
- 36. Robey RW, Honjo Y, Morisaki K, et al. Mutations at amino‐acid 482 in the ABCG2 gene affect substrate and antagonist specificity. Br J Cancer. 2003;89(10):1971‐1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen ZS, Mutoh M, Sumizawa T, et al. Reversal of heavy metal resistance in multidrug‐resistant human KB carcinoma cells. Biochem Biophys Res Commun. 1997;236(3):586‐590. [DOI] [PubMed] [Google Scholar]
- 38. Jackson SM, Manolaridis I, Kowal J, et al. Structural basis of small‐molecule inhibition of human multidrug transporter ABCG2. Nat Struct Mol Biol. 2018;25(4):333‐340. [DOI] [PubMed] [Google Scholar]
- 39. Wu ZX, Yang Y, Teng QX, et al. Tivantinib, A c‐Met inhibitor in clinical trials, is susceptible to ABCG2‐mediated drug resistance. Cancers (Basel). 2020;12(1):E186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stacy AE, Jansson PJ, Richardson DR. Molecular pharmacology of ABCG2 and its role in Chemoresistance. Mol Pharmacol. 2013;84(5):655‐669. [DOI] [PubMed] [Google Scholar]
- 41. Khot MI, Downey CL, Armstrong G, et al. The role of ABCG2 in modulating responses to anti‐cancer photodynamic therapy. Photodiagn Photodyn Ther. 2020;29:101579. [DOI] [PubMed] [Google Scholar]
- 42. Toyoda Y, Takada T, Suzuki H. Inhibitors of human ABCG2: from technical background to recent updates with clinical implications. Front Pharmacol. 2019;10:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cai CY, Zhai H, Lei ZN, et al. Benzoyl indoles with metabolic stability as reversal agents for ABCG2‐mediated multidrug resistance. Eur J Med Chem. 2019;179:849‐862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang GN, Zhang YK, Wang YJ, et al. Epidermal growth factor receptor (EGFR) inhibitor PD153035 reverses ABCG2‐mediated multidrug resistance in non‐small cell lung cancer: in vitro and in vivo. Cancer Lett. 2018;424:19‐29. [DOI] [PubMed] [Google Scholar]
- 45. Xu L, Huang J, Liu J, et al. CM082 enhances the efficacy of chemotherapeutic drugs by inhibiting the drug efflux function of ABCG2. Mol Ther Oncolytics. 2020;16:100‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Alqawi O, Bates S, Georges E. Arginine482 to threonine mutation in the breast cancer resistance protein ABCG2 inhibits rhodamine 123 transport while increasing binding. Biochem J. 2004;382(Pt 2):711‐716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ejendal KFK, Diop NK, Schweiger LC, Hrycyna CA. The nature of amino acid 482 of human ABCG2 affects substrate transport and ATP hydrolysis but not substrate binding. Protein Sci. 2006;15(7):1597‐1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ozvegy‐Laczka C, Koblos G, Sarkadi B, Varadi A. Single amino acid (482) variants of the ABCG2 multidrug transporter: major differences in transport capacity and substrate recognition. Biochim Biophys Acta. 2005;1668(1):53‐63. [DOI] [PubMed] [Google Scholar]
- 49. Chen Z‐S, Robey RW, Belinsky MG, et al. Transport of Methotrexate, Methotrexate Polyglutamates, and 17β‐Estradiol 17‐(β‐<span class="sc">d</span>‐glucuronide) by ABCG2: effects of acquired mutations at R482 on methotrexate transport. Cancer Res. 2003;63(14):4048‐4054. [PubMed] [Google Scholar]
- 50. Honjo Y, Hrycyna CA, Yan Q‐W, et al. Acquired mutations in the %3cstrong%3e%3cem%3eMXR/BCRP/ABCP%3c/em%3e%3c/strong%3e gene alter substrate specificity in MXR/BCRP/ABCP‐overexpressing cells. Cancer Res. 2001;61(18):6635. [PubMed] [Google Scholar]
- 51. Wang JQ, Li JY, Teng QX, et al. Venetoclax, a BCL‐2 inhibitor, enhances the efficacy of chemotherapeutic agents in wild‐type ABCG2‐overexpression‐mediated MDR cancer cells. Cancers (Basel). 2020;12(2):466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Oguri T, Ozasa H, Uemura T, et al. MRP7/ABCC10 expression is a predictive biomarker for the resistance to paclitaxel in non‐small cell lung cancer. Mol Cancer Ther. 2008;7(5):1150‐1155. [DOI] [PubMed] [Google Scholar]
- 53. Nakanishi T, Shiozawa K, Hassel BA, Ross DD. Complex interaction of BCRP/ABCG2 and imatinib in BCR‐ABL‐expressing cells: BCRP‐mediated resistance to imatinib is attenuated by imatinib‐induced reduction of BCRP expression. Blood. 2006;108(2):678‐684. [DOI] [PubMed] [Google Scholar]
- 54. Zhang L, Li Y, Wang Q, et al. The PI3K subunits, P110α and P110β are potential targets for overcoming P‐gp and BCRP‐mediated MDR in cancer. Mol Cancer. 2020;19(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Morris GM, Lim‐Wilby M. Molecular docking. Methods Mol Biol. 2008;443:365‐382. [DOI] [PubMed] [Google Scholar]
- 56. Ji N, Yang Y, Cai C‐Y, et al. Selonsertib (GS‐4997), an ASK1 inhibitor, antagonizes multidrug resistance in ABCB1‐ and ABCG2‐overexpressing cancer cells. Cancer Lett. 2019;440‐441:82‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gupta P, Zhang YK, Zhang XY, et al. Voruciclib, a potent CDK4/6 inhibitor, antagonizes ABCB1 and ABCG2‐mediated multi‐drug resistance in cancer cells. Cell Physiol Biochem. 2018;45(4):1515‐1528. [DOI] [PubMed] [Google Scholar]
- 58. Zhang YK, Zhang XY, Zhang GN, et al. Selective reversal of BCRP‐mediated MDR by VEGFR‐2 inhibitor ZM323881. Biochem Pharmacol. 2017;132:29‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ramírez D, Caballero J. Is it reliable to take the molecular docking top scoring position as the best solution without considering available structural data? Molecules. 2018;23(5):1038. [DOI] [PMC free article] [PubMed] [Google Scholar]