

Abstract
Death‐associated protein kinase 1 (DAPK) is a calcium/calmodulin kinase that plays a vital role as a suppressor gene in various cancers. Yet its role and target gene independent of p53 is still unknown in hepatocellular carcinoma (HCC). In this study, we discovered that DAPK suppressed HCC cell migration and invasion instead of proliferation or colony formation. Using a proteomics approach, we identified DEAD‐box helicase 20 (DDX20) as an important downstream target of DAPK in HCC cells and critical for DAPK‐mediated inhibition of HCC cell migration and invasion. Using integrin inhibitor RGD and GTPase activity assays, we discovered that DDX20 suppressed HCC cell migration and invasion through the CDC42‐integrin pathway, which was previously reported as an important downstream pathway of DAPK in cancer. Further research using cycloheximide found that DAPK attenuates the proteasomal degradation of DDX20 protein, which is dependent on the kinase activity of DAPK. Our results shed light on new functions and regulation for both DAPK and DDX20 in carcinogenesis and identifies new potential therapeutic targets for HCC.
Keywords: DEAD‐box helicase 20, death‐associated protein kinase, hepatocellular carcinoma, invasion, migration, proteasome degradation
This work identifies DEAD‐box helicase 20 (DDX20) as a new downstream target of death‐associated protein kinase 1 (DAPK). DAPK suppresses hepatocellular carcinoma cell migration and invasion, but not proliferation or colony formation, through DDX20.
Abbreviations
- Cdc42
Cell division control protein 42
- CHX
cycloheximide
- DAPK
death‐associated protein kinase 1
- DDX20
DEAD‐box helicase 20
- DOX
doxycycline
- FDR
false discovery rate
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- IHC
immunohistochemical
- MEF
mouse embryonic fibroblast
- MG132
carbobenzoxy‐Leu‐Leu‐leucinal
- NF‐κB
nuclear factor‐κB
- SMA
spinal muscular atrophy
- SRB
sulforhodamine B
- TSC2
tuberous sclerosis 2
1. INTRODUCTION
Death‐associated protein kinase 1 was initially identified as a mediator of γ‐interferon‐induced apoptosis in 1995. 1 , 2 Later, DAPK was found to suppress cancer transformation and metastasis by activating the p53 pathway. 3 , 4 Moreover, DAPK acts as a powerful inhibitor of cell polarization in the process of directional migration through suppression of integrin β1. 5 In our previous study, we discovered that DAPK activates mTORC1 by phosphorylating TSC2, 6 , 7 suggesting DAPK might also participate in protein translation and cell survival pathways. In 2015, Jing et al reported that DAPK promotes p53‐negative breast cancer cell proliferation through phosphorylation of TSC2 at Ser939, 8 which was consistent with our previous discoveries and indicated that the function of DAPK in cancer might vary depending on the cancer type and genetic background.
Hepatocellular carcinoma is one of the leading cancers with unfavorable prognosis. Surgical resection is the main method for the treatment of HCC, but recurrence and metastasis often occur after resection of the tumor. The 5‐year survival rate of HCC patients is very low, ranging from 24% to 50%. 9 According to the World Cancer Report 2014 released by the WHO, the number of new HCC cases and deaths in China in 2012 accounted for more than half of the world’s new cases and deaths. There is an urgent need for identifying new diagnosis and therapeutic targets for HCC. In clinical samples, both Matsumoto et al and our team reported that higher DAPK levels in cancer tissues correlated with better patient survival, 10 , 11 suggesting DAPK might act as a tumor suppressor in HCC and potentially be a therapeutic target.
So far there have no studies on the cellular function of DAPK in HCC. Recently it was reported that WT p53 is required for the maintenance of glycosylation and growth of HCC cells, 12 suggesting p53 acts as an oncogene and DAPK might exert its tumor suppressive role independent of p53 in HCC. Further research is required to elucidate the molecular function of DAPK in HCC.
2. MATERIALS AND METHODS
2.1. Cell lines
Hep3B and PLC/PRF/5 cells were cultured in Eagle’s minimum essential medium (Biological Industries [BI]), and HEK‐293T cells were cultured in DMEM (BI) supplemented with 10% FBS (BI), 100 U/mL penicillin, and 100 μg/mL streptomycin (1% penicillin/streptomycin) (BBI Life Sciences) at 37°C in a humidified atmosphere containing 5% CO2. Transient transfection was carried out using VitalGENE‐II In Vitro DNA Transfection Kit (Biocanaan) according to the manufacturer’s instructions. Cells were treated with CHX at 10 μg/mL (Sigma‐Aldrich) for 4 hours, chloroquine at 1 μg/mL (Sigma‐Aldrich) for 24 hours, and MG132 at 1 μg/mL (Sigma‐Aldrich) for 6 hours. The DAPK kinase activity inhibitor TC‐DAPK6 (MedChem Express) was used at 100 nmol/L for 24 hours. RGD (MedChem Express) was used at 100 μmol/L for 24 hours.
2.2. Other assays
Vector construction, lentivirus infection, MEF generation, confocal microscopy, Cdc42 activity detection, western blot analysis, Co‐Immunoprecipitation, protein preparation, iTRAQ labeling, PCR, and real‐time PCR are described in detail in Document S1. The relative reagents and resources are listed in Table S1, and sequences of primers or shRNAs or siRNAs for the target genes are sorted individually in Tables S2‐S4 in Document S1.
2.3. Proliferation assay
The cells were diluted in the culture media with or without DOX (Sigma) at a concentration of 2 × 104 cells/mL. Then 200 μL diluted cells were seeded into each well of a 96‐well plate. Cell proliferation was then determined by sulforhodamine B assay 13 (Sigma‐Aldrich) or with the CCK‐8 (TransGen Biotech) assay as instructed by the manufacturer. The absorbance was determined at 510 or 450 nm with the multifunctional enzyme marking instrument (Bio‐Tek) after another 0, 24, 48, or 72 hours. The “0 hour” was the time when all seeded cells adhered to the plate.
2.4. Colony formation
For colony formation assay, cells were plated at a density of 500 cells/well into 6‐well plates. After cells were cultured for 24 hours, media were changed with fresh media containing 800 ng/mL DOX in inducible groups and without DOX in control groups. Cells were left to grow for 10 days, and the media were changed every 5 days. After fixation with 70% methanol for 30 minutes, cells were washed with 70% PBS and left to dry naturally before being dyed with gentian violet (Beyotime) for 40 minutes.
2.5. Wound healing assay
The cells (1.5 × 105) with or without DOX were seeded onto 6‐well plates and cultured until being confluent. Wounds were generated using a sterile 10‐μL pipette tip (Biosharp). The culture media were then replaced with serum‐free media with or without DOX. The cells were cultured for an additional 48 hours. Wound closure was photographed with an inversion phase contrast microscope (Carl Zeiss). Multiple views of each well were documented, and 3 independent experiments were carried out.
2.6. Invasion assays
For the Transwell invasion assay, the cells (1 × 104) were resuspended in serum‐free DMEM with or without DOX and seeded in the interior of the insert chamber (Corning) with the Matrigel‐coated membrane. Then 500 μL complete medium was added to the bottom of each well. The plate was incubated for 24 hours; the media and the cells remaining in the top chambers were then removed. After fixation and staining with 0.1% crystal violet (Beyotime), the cells that had invaded to the lower membrane of the inserts were captured under an inverted phase contrast microscope (Carl Zeiss).
2.7. Immunohistochemical staining and analysis
Immunohistochemical assay was carried out as described previously. 14 The intensity and percentage of positive cells from 5 fields in each sample were determined independently by 2 experienced pathologists blinded to the clinical and pathologic data. Staining intensity was assessed using a 4‐point scale (0, undetectable; 1, weak; 2, moderate; and 3, strong). Percentage of positively stained cells was examined as 1 of 4 categories: 1, 0%–25% cells stained; 2, 26%–50% stained; 3, 51%–75% stained; and 4, 76%–100% stained. Protein expression was calculated by adding the values of the intensity and percentage scores together. The tissue microarray LivH180Su07 containing 90 pairs of tissue samples and HLivH180Su14 containing 90 pairs of tissue samples were commercially bought from Shanghai Outdo Biotech Company (http://www.superchip.com.cn). The DAPK1 expression was considered high for IHC scores of 3‐7 and low for scores of 0‐2. The DDX20 expression was considered high for scores of 4‐7 and low for scores of 0‐3. The relationship between the protein expression (low or high) and patient overall survival was analyzed using Kaplan‐Meier analysis and assessed for significance using the log‐rank test.
2.8. Statistical analysis
Data were analyzed using Prism 6.0 software (GraphPad Software). Results are presented as the mean ± SD of 3 independent experiments. The difference between the means were tested by one‐way ANOVA testing or Student’s t test and P < .05 was considered to indicate a statistically significant difference. For proteomic analysis, proteins identification from the iTRAQ experiments was carried out using Proteome Discoverer (version 1.4; Thermo Fisher Scientific) against a human database provided by The Universal Protein Resource (http://www.uniprot.org/uniprot; released 10 April 2014 with 20 264 entries). A decoy database search strategy was adopted to estimate the FDR for peptide identification. The proteins were assembled using the parsimony method and accepted if the peptide FDR was less than 1% and protein probability higher than 99.0%. To identify proteins whose expression was significantly altered in Hep3B‐sDK cells following DOX induction (compare to Hep3B‐Scr cells), we set a threshold of iTRAQ ratios to define differentially expressed proteins. The proteins were considered to be differentially expressed if the iTRAQ ratio was greater than 1.5 or less than 0.67 (P < .05, paired t test) in cells with DOX induction compared to cells without. The proteins that were differentially expressed in Hep3B‐Scr cells were ruled out from the differentially expressed protein list of the Hep3B‐sDK cells.
3. RESULTS
3.1. Death‐associated protein kinase 1 suppresses HCC cell migration and invasion, but not proliferation or colony formation, through DDX20
In order to investigate the cellular function of DAPK in HCC, we established stable‐inducible HCC cell lines that can downregulate or upregulate DAPK levels following DOX induction using Tet‐On inducible expression system 15 (Figure S1A,B). After confirming their successful establishment (Figure 1A,B), the stable‐inducible Hep3B cells were subjected to a series of functional experiments. Knockdown of endogenous DAPK did not significantly affect cell proliferation of either viable cells (Figure 1C,D) or total cells (Figure 1E,F), nor colony formation (Figure 1G). However, Hep3B cell migration (Figure 1H) and invasion (Figure 1I) were significantly upregulated when DAPK levels dropped. Consistent with this observation, overexpression of DAPK suppressed Hep3B cell migration (Figure 1J) and invasion (Figure 1K). In order to rule out the possibility that the suppression of Hep3B cell migration and invasion comes from an off‐target effect from the DAPK shRNA we used in these experiments, another DAPK shRNA was employed to create stable‐inducible Hep3B cell lines for functional assays and further confirmed our conclusion (Figure S1C‐I). Moreover, similar results were observed in another HCC cell line PLC/PRF/5 (Figure S2). These results suggested the physiological function of DAPK is mainly related to suppression of HCC cell migration and invasion.
FIGURE 1.
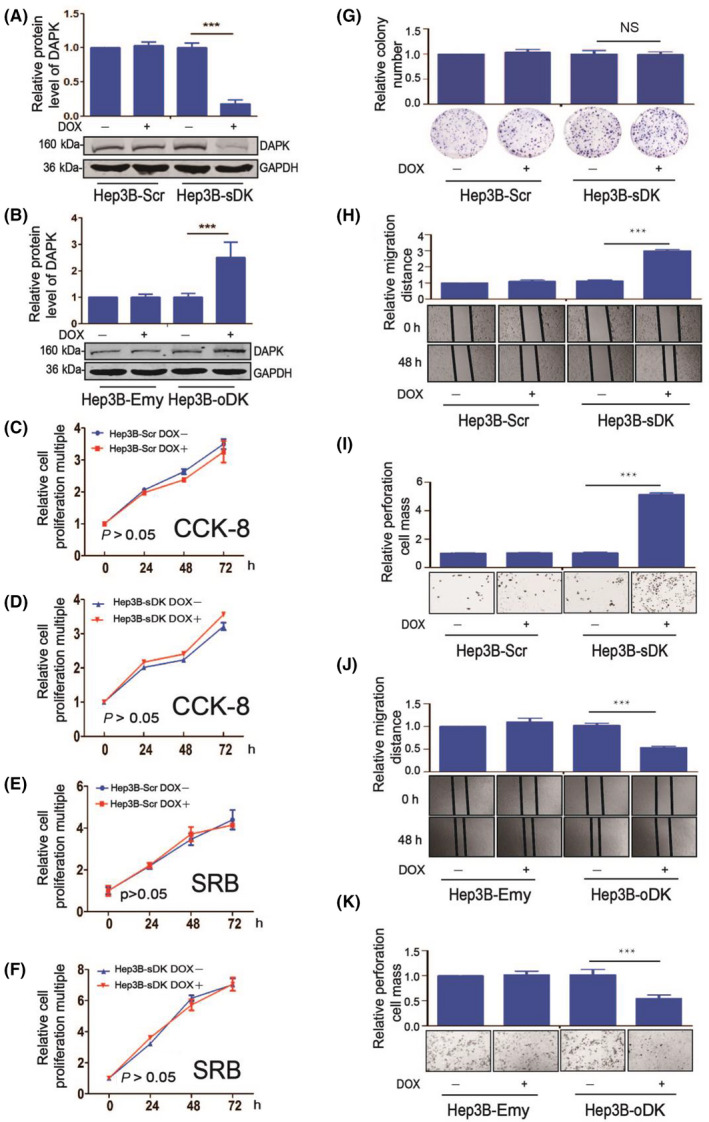
Death‐associated protein kinase 1 (DAPK) suppresses Hep3B cell migration and invasion, but not proliferation or colony formation. A, DAPK protein levels in the Tet‐inducible DAPK knockdown Hep3B cell lines (Hep3B‐Scr and Hep3B‐sDK) with or without doxycycline (DOX) induction. B, DAPK protein levels in the Tet‐inducible DAPK overexpression Hep3B cell lines (Hep3B‐Emy and Hep3B‐oDK) with or without DOX induction. C‐F, Proliferation of the Hep3B‐Scr and Hep3B‐sDK cell lines with or without DOX induction using both CCK‐8 (C,D) and SRB (E,F) detection assays. G‐I, Colony formation (G), migration (H), and invasion (I) of Hep3B‐Scr and Hep3B‐sDK cell lines with or without DOX induction. J, K, Migration (J) and invasion (K) of Hep3B‐Emy and Hep3B‐oDK cell lines with or without DOX induction. For statistical analysis of colony formation, migration, and invasion, the values (colony numbers for colony formation, migration distance for migration, and colony numbers through the Transwell for invasion) of cells with DOX induction were divided by that of cells without DOX induction, and the values of Hep3B‐Scr or Hep3B‐Emy cells without DOX induction were normalized as 1. A t test was used to evaluate the statistical significance of these experiments. ***P < .001. NS, not significant (P > .05). [Correction added on 02 August 2020, after first online publication: The missing two statistical charts were corrected in parts A and G of figure 1]. [Correction added on 05 August 2020, after first online publication: The missing two statistical charts were corrected in parts A and G of figure 1]
Canonically DAPK is considered to exert its tumor suppressor function through the p53 signaling pathway. However, Hep3B and PLC/PRF/5 cells were p53‐null or mutant, 16 indicating there exists an alternative signaling pathway to mediate the function of DAPK in HCC. Therefore, we carried out a proteomics experiment to search potential downstream pathways of DAPK using our stable‐inducible Hep3B cells (Figure S3A,B). When the DAPK protein level dropped, on total the levels of 15 proteins increased and 21 proteins decreased (Figure S3C). DEAD‐box helicase 20, also known as DP103/Gemin3, which decreased by approximately 40% when DAPK was knocked down in Hep3B cells and was reported to suppress hepatocarcinogenesis, 17 , 18 attracted our attention. First, we verified the proteomics results in our stable‐inducible Hep3B and PLC/PRF/5 cell lines using western blot and confirmed that downregulation of DAPK led to a decrease in DDX20 protein levels (Figure 2A,B), whereas upregulation led to an increase (Figure 2C,D). Then we examined the protein levels of DAPK and DDX20 in MEFs from a DAPK KO‐first transgenic mouse model (Figures 2E and S4). The DDX20 protein level in the WT MEFs was significantly higher than that in the DAPK KO MEFs, further confirming the positive correlation between DAPK and DDX20 protein levels (Figure 2F). In HCC patient samples, the DAPK protein levels were lower in tumor tissues in comparison to the adjacent normal tissues (Figure 2G), whereas the DDX20 protein levels were higher (Figure 2H). There was a correlation between the protein levels of DAPK and DDX20 in the tumor tissues (Figure 2I), supporting our discoveries in HCC cell lines. Considering the strong relationship between virus infection and HCC in China, we also detected the correlation of the expression of these 2 genes with HBV and HCV infection in this patient cohort. The HBV infection had no statistically significant effect on the protein level of either DAPK or DDX20 (Figure S5). The number of patients infected with HCV was only 2, which is too small, so we did not analyze the correlation between these 2 genes and HCV infection.
FIGURE 2.
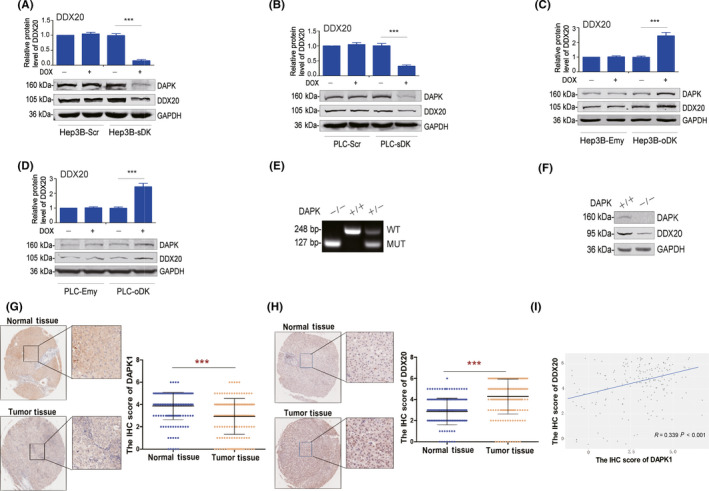
Death‐associated protein kinase 1 (DAPK) positively regulates the protein levels of DEAD‐box helicase 20 (DDX20). A‐D, Protein levels of DAPK and DDX20 in Hep3B‐Scr and Hep3B‐sDK (A), PLC‐Scr and PLC‐sDK (B), Hep3B‐Emy and Hep3B‐oDK (C), and PLC‐Emy and PLC‐oDK (D) cells with or without doxycycline (DOX) induction. E, Genotype identification of DAPK WT (+/+), heterozygote (+/−), and KO (−/−) mice using PCR. F, Protein levels of DAPK and DDX20 in mouse embryonic fibroblasts from DAPK WT (+/+) and KO (−/−) mice. G, H, Protein levels of DAPK (G) and DDX20 (H) in 180 pairs of liver cancer tissues and adjacent normal tissues were determined using an immunohistochemical (IHC)‐based tissue microarray. Representative images of DAPK and DDX20 staining were taken at a magnification of ×40 or ×200. I, The analysis of the possible correlation between DAPK and DDX20 protein levels in liver cancer tissues based on the IHC‐based tissue microarray was carried out using ggplot2 package in R. The scatter plot was set as “disturbance” where the data overlaps. ***P < .001. MUT, mutant
Next, we set out to investigate whether DDX20 was involved in the tumor suppressor function of DAPK. When DDX20 was overexpressed, knockdown of DAPK could no longer stimulate the invasion (Figure 3A,B) or migration (Figure 3C,D) of Hep3B cells. Consistently, overexpression of DAPK could not suppress Hep3B cell invasion (Figure 3E,F) or migration (Figure 3G,H) when DDX20 was knocked down using siRNA. Similar results were observed in PLC/PRF/5 cells (Figure 3I‐P), suggesting DAPK regulates HCC cell migration and invasion through DDX20.
FIGURE 3.
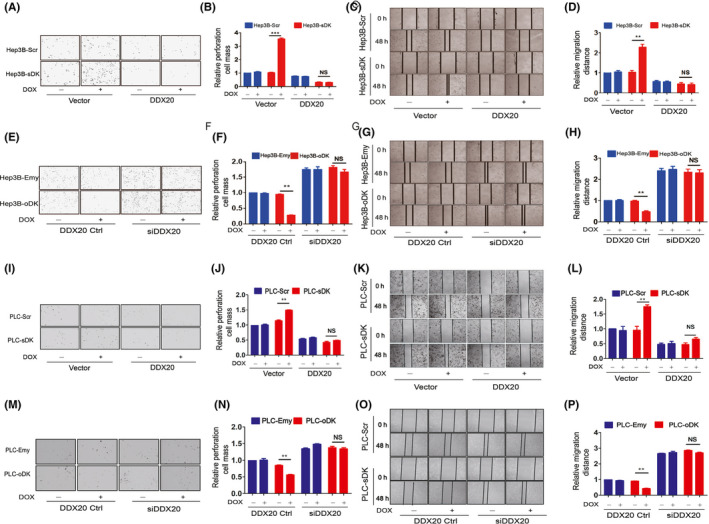
Death‐associated protein kinase 1 regulates migration and invasion of Hep3B and PLC/PRF5 cells through DEAD‐box helicase 20 (DDX20). A‐D, Invasion (A, B) and migration (C, D) of Hep3B‐Scr and Hep3B‐sDK cells when DDX20 was overexpressed. Hep3B‐Scr and Hep3B‐sDK cells were transiently transfected with DDX20 plasmids or empty plasmids, and cultured in media with or without doxycycline (DOX). The values of the Hep3B‐Scr cells transfected with empty plasmids and without DOX induction were normalized as 1 in the statistical analyses of invasion (B) and migration (D). E‐H, Invasion (E, F) and migration (G, H) of Hep3B‐Emy and Hep3B‐oDK cells when DDX20 was knocked down. Hep3B‐Emy and Hep3B‐oDK cells were transiently transfected with DDX20 siRNA or control siRNA (Ctrl), and cultured in media with or without DOX. The values of the Hep3B‐Emy cells transfected with control siRNA and without DOX induction were normalized as 1 in the statistical analyses of invasion (F) and migration (H). I‐L, Invasion (I, J) and migration (K, L) of PLC‐Scr and PLC‐sDK cells when DDX20 was overexpressed. PLC‐Scr and PLC‐sDK cells were transiently transfected with DDX20 plasmids or empty plasmids, and cultured in media with or without DOX. The values of the PLC‐Scr cells transfected with empty plasmids and without DOX induction were normalized as 1 in the statistical analyses of invasion (J) and migration (L). M‐P, Invasion (M, N) and migration (O, P) of PLC‐Emy and PLC‐oDK cells when DDX20 was knocked down. PLC‐Emy and PLC‐oDK cells were transiently transfected with DDX20 siRNA or control siRNA and cultured in media with or without DOX. The values of the Hep3B‐Emy or PLC‐Emy cells transfected with control siRNA and without DOX induction were normalized as 1 in the statistical analyses of invasion (N) and migration (P). **P < .01. ***P < .001. NS, not significant (P > .05).
Multiple signaling pathways were reported to mediate cell migration and invasion. In order to investigate the downstream signaling pathway of the DAPK‐DDX20 axis, we first examined the protein levels of several classical metastasis‐related proteins, such as E‐cadherin, MMP9, MMP20, and Cdc42, and did not observe any significant changes (Figure 4A,B). It has been published before that DAPK regulates breast cancer cell migration through the Cdc42‐integrin pathway. 5 Therefore, we used an integrin inhibitor, RGD, to investigate its impact on DAPK function in HCC cells and discovered that RGD completely blocked the effect of DAPK knockdown on Hep3B cell invasion (Figure 4C,D). In addition, although it did not change the Cdc42 protein level, knockdown of DAPK significantly upregulated the GTPase activity of Cdc42 (Figure 4E,F), which was blocked by overexpression of DDX20 (Figure 4G,H), suggesting that DAPK regulates Cdc42 activity through DDX20.
FIGURE 4.
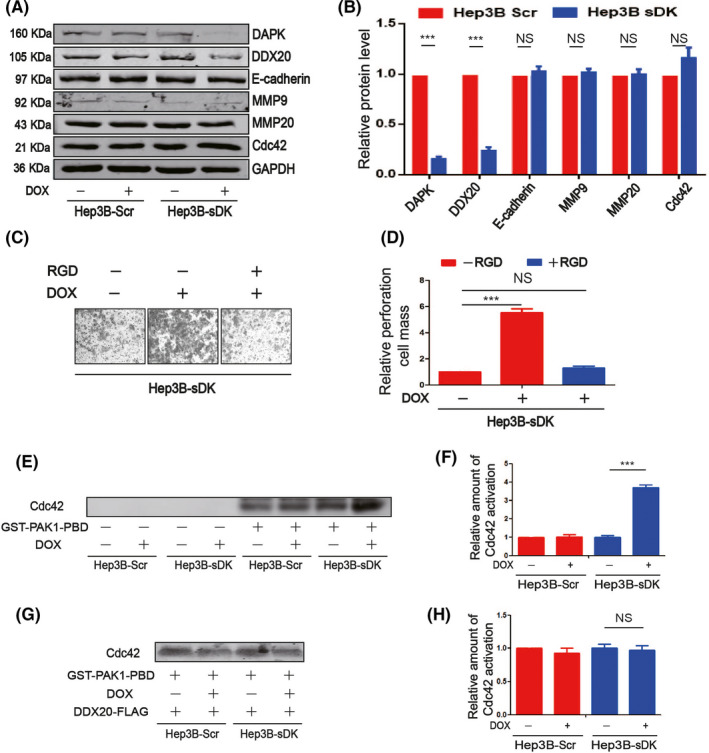
Death‐associated protein kinase 1 (DAPK) regulates hepatocellular carcinoma cell invasion through the integrin pathway. A, B, Protein levels of DAPK, DEAD‐box helicase 20 (DDX20), E‐cadherin, MMP9, MMP20, and Cdc42 in Hep3B‐Scr and Hep3B‐sDK cell lines with or without doxycycline (DOX) induction. C, D, Detection of the cell invasion ability of Hep3B‐sDK cells exposed to DOX and RGD. The values of the Hep3B‐sDK cells without DOX and RGD induction were normalized as 1 in the statistical analyses of invasion. E, F, Analysis of Cdc42 activity in Hep3B‐Scr and Hep3B‐sDK cells with or without DOX induction. G, H, Analysis of Cdc42 activity in Hep3B‐Scr and Hep3B‐sDK cells with or without DOX induction following DDX20 plasmid transfection. **P < .01, ***P < .001. NS, not significant (P > .05).
3.2. Death‐associated protein kinase 1 attenuates proteasomal degradation of DDX20 protein
In order to investigate the molecular mechanisms underlying the correlation between DAPK and DDX20, we first examined the mRNA levels of DDX20 when DAPK levels were altered in HCC cells. No significant DDX20 mRNA changes were observed when DAPK was downregulated or upregulated in either Hep3B (Figure 5A,B) or PLC/PRF/5 cells (Figure 5C,D), suggesting DAPK might regulate DDX20 levels at protein level. Using proteasome inhibitor MG132 and lysosome inhibitor chloroquine, we found that both exogenous (Figure 5E,F) and endogenous (Figure 5G,H) DDX20 proteins were mainly degraded through proteasomes. In addition, HA‐DAPK significantly increased the stability of DDX20‐FLAG when cotransfected (Figure 6A‐C), indicating DAPK could positively regulate DDX20 protein levels by attenuating the proteasomal degradation of DDX20 protein. This was further supported by the observation that when endogenous DAPK was knocked down or overexpressed, the stability of endogenous DDX20 protein was downregulated (Figure 6D‐F) or upregulated (Figure 6G‐I).
FIGURE 5.
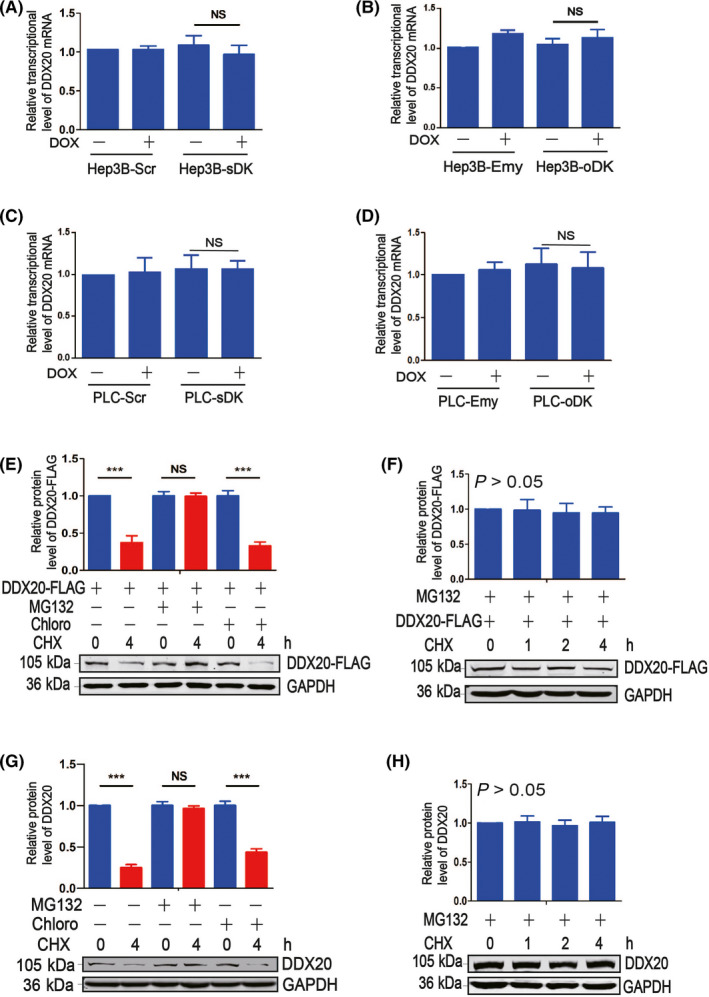
Death‐associated protein kinase 1 regulates DEAD‐box helicase 20 (DDX20) expression at protein level. A, B, mRNA levels of DDX20 in Hep3B‐Scr and Hep3B‐ sDK (A) and Hep3B‐Emy and Hep3B‐oDK (B) cells with or without doxycycline (DOX) induction. C, D, mRNA levels of DDX20 PLC‐Scr and PLC‐ sDK (C) and PLC‐Emy and PLC‐oDK (D) cells with or without DOX induction. E‐H, Protein levels of DDX20‐FLAG or DDX20 in 293T cells (E, F) and Hep3B cells (G, H) exposed to MG132, chloroquine (Chloro), and/or cycloheximide (CHX). **P < .01, ***P < .001. NS, not significant (P > .05).
FIGURE 6.
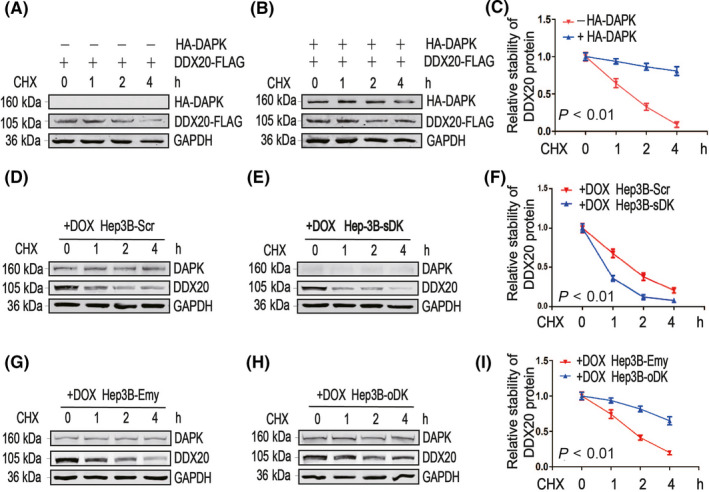
Death‐associated protein kinase 1 (DAPK) stabilizes DEAD‐box helicase 20 (DDX20) protein. A‐C, DDX20‐FLAG plasmids were cotransfected with empty plasmids (A) or HA‐DAPK plasmids (B) into 293T cells exposed to cycloheximide (CHX) for the indicated time (0, 1, 2 and 4 h). Protein levels of DDX20‐FLAG and HA‐DAPK were detected by western blot (A, B). Relative Quantitative analysis of the protein stability of DDX20‐FLAG is shown in (C). D‐I, Hep3B‐Scr (D), Hep3B‐sDK (E), Hep3B‐Emy (G), and Hep3B‐oDK (H) cells were exposed to doxycycline (DOX) and CHX for the indicated time (0, 1, 2 and 4 h), and protein levels of DAPK and DDX20 were detected by western blot. Quantitative analysis of the protein stability of DDX20 is shown in (F) and (I).
3.3. Death‐associated protein kinase 1 regulates DDX20 through kinase activity
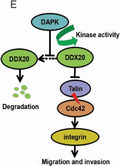
As DAPK can improve DDX20 protein stability, it is important to investigate whether the kinase activity of DAPK is required for its regulation towards DDX20. Compared with WT HA‐DAPK (Figure 7A), the kinase dead HA‐DAPK (K42A) mutant could no longer upregulate the DDX20‐FLAG level (Figure 7B). Consistently, the endogenous DDX20 protein level dropped when Hep3B cells were exposed to DAPK kinase inhibitor TC‐DAPK6 (Figure 7C), suggesting DAPK might regulate DDX20 through phosphorylation. Further experiments indicated that HA‐DAPK (K42A) could not alter the level of DDX20‐FLAG (1‐244) (Figure 7D), which indicated that the potential phosphorylation sites of DDX20 regulated by DAPK reside in this domain.
FIGURE 7.
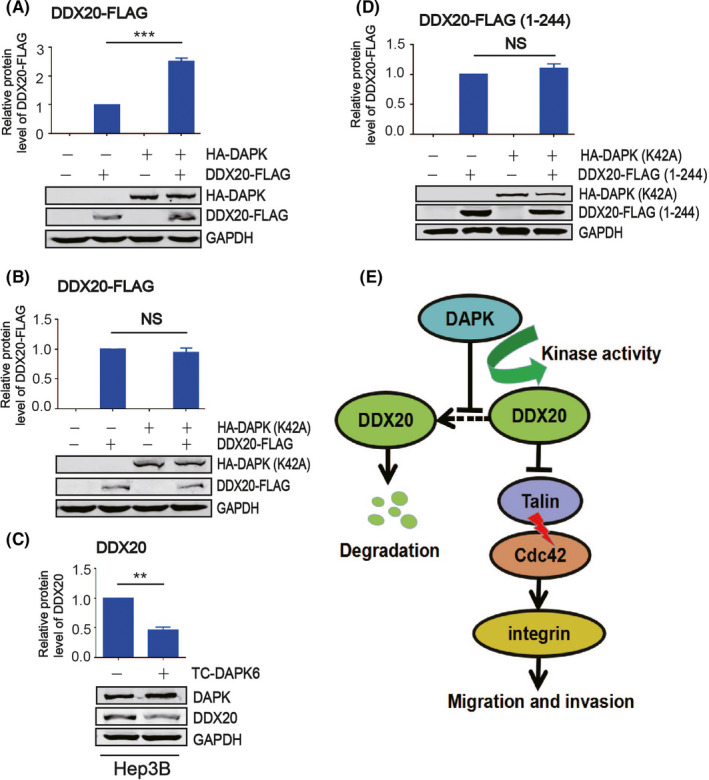
Death‐associated protein kinase 1 (DAPK) regulates DEAD‐box helicase 20 (DDX20) through kinase activity. A, B, Protein levels of DDX20‐FLAG when cotransfected with HA‐DAPK (A) or HA‐DAPK (K42A) (B) plasmids in 293T cells. C, Protein levels of DAPK and DDX20 in Hep3B cells with or without TC‐DAPK6 (a DAPK inhibitor). D, HA‐DAPK (K42A) and DDX20‐FLAG (1‐244) plasmids were separately transfected or cotransfected into 293T cells. Protein levels of HA‐DAPK (K42A) and DDX20‐FLAG (1‐244) were examined by western blot. E, Pattern diagram of signal path. **P < .01, ***P < .001. NS, not significant (P > .05).
4. DISCUSSION
In this work, we used 2 systems to establish the DAPK‐inducible knockdown and overexpression cell lines. One is the lentivirus Tet‐pLKO‐puro (Addgene) system, and the other is the pTRE3G‐puro (Clontech) double plasmid system. Interestingly, the basic migration and invasion of the control cell lines infected with scramble shRNA or transfected with empty vector were different (Figures 1H‐K and S2F‐H). This could be due to the differential impact of the 2 systems on cells. However, the effect of altering DAPK levels was consistent in both knockdown and overexpression cell lines, suggesting DAPK does suppress cell migration and invasion in HCC cells.
DDX20 is a multifunctional DEAD‐box RNA helicase and a core member of the survival motor neuron complex, the key function of which is the assembly of small nuclear ribonucleic acid proteins in spliceosomes. 19 It is also a component of the RNA‐induced silencing complex and regulates the process of miRNA maturation. 20 , 21 In addition, DDX20 is found to modulate some transcription factors such as steroidogenic factor 1 and early growth response protein 2 through its nonconserved C‐terminal domain. 22 , 23 , 24 Our study builds up a link between DAPK and DDX20 pathways, which will bring many potential research directions for both proteins. For example, DDX20 and its downstream miRNAs were found to be critical in SMA pathogenesis. 21 , 25 Extracellular signal‐regulated kinase, which is a known interacting partner of DAPK, 26 was also found to be involved in the pathophysiology of SMA. 27 It is possible that DAPK also participates in the development of SMA, and is worthy of further investigation.
In cancer, DDX20 showed contradictory roles. In breast cancer, DDX20 was identified as an oncogene that promotes metastasis by elevation of MMP9 levels and activation of the NF‐κB pathway. 28 A similar oncogenic function of DDX20 was also reported in prostate cancer. 29 In HCC, DDX20 was identified as a tumor suppressor by suppressing NF‐κB pathway through microRNA‐140. 18 Interestingly, DAPK showed similar contradictory roles in breast cancer 8 and HCC, suggesting DDX20 could be a key determinate of the physiological function of DAPK in cancer. Of note, in both breast cancer and HCC, DDX20 was reported to exert its function through the NF‐κB pathway. Several studies have reported the inhibitory function of DAPK towards the NF‐κB pathway in T cells, 30 U251 cells, 31 and OVCAR‐3 cells, 32 the mechanisms of which are still unclear. Our results indicate that DAPK could regulate the NF‐κB pathway through DDX20 and the discrepancy of the DDX20 and DAPK functions in cancer might be due to the differential effect of the NF‐κB pathway in different types of cancer.
In the HCC patient tumor samples, the correlation value between DAPK and DDX20 proteins was only 0.339 (Figure 2I), which was not as strong as the observations from the inducible cell lines. Considering the aberrant regulation of DAPK and DDX20 transcriptionally in cancer, 19 , 33 it is likely that the proteasomal degradation of DDX20 protein only contributed partially to its levels in tumor samples. Moreover, knockdown of DDX20 by siRNA was able to stimulate HCC cell migration and invasion (Figures 3 and S6) without simultaneous alteration of the DAPK level, suggesting DDX20 is likely to regulate HCC cell migration and invasion physiologically in these cells. Whether these physiological functions of DDX20 rely on DAPK is still unclear.
In conclusion, this study identifies DDX20 as a new downstream target of DAPK that mediates the tumor suppressor function of DAPK in HCC. Death‐associated protein kinase 1 inhibits the proteasomal degradation of DDX20, which is dependent on the kinase activity of DAPK (Figure 7E). Our results shed light on new functions and regulation for both DAPK and DDX20 in carcinogenesis and provides new therapeutic targets for HCC cell metastasis.
5. ACKNOWLEDGMENTS
This work was supported by the International S&T Cooperation Program of China (2016YFE0121900), the Scientific Research Innovation Team Construction Program of Fujian Normal University (IRTL1702), and the United Fujian Provincial Health and Education Project Tackling the Key Research (WKJ2016‐2‐27).
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest with the contents of this article. The authors have declared that no competing interest exists.
Supporting information
Document S1
Document S2
Huang Y, Wang C, Li K, et al. Death‐associated protein kinase 1 suppresses hepatocellular carcinoma cell migration and invasion by upregulation of DEAD‐box helicase 20. Cancer Sci. 2020;111:2803–2813. 10.1111/cas.14499
Huang, Wang, Li, Ye, and Shen contributed equally to this work.
Contributor Information
Xiaolong Liu, Email: xiaoloong.liu@gmail.com.
Jun Peng, Email: pjunlab@hotmail.com.
Yao Lin, Email: yaolin@fjnu.edu.cn.
REFERENCES
- 1. Shiloh R, Bialik S, Kimchi A. The DAPK family: a structure‐function analysis. Apoptosis. 2014;19(2):286‐297. [DOI] [PubMed] [Google Scholar]
- 2. Deiss LP, Feinstein E, Berissi H, Cohen O, Kimchi A. Identification of a novel serine/threonine kinase and a novel 15‐kD protein as potential mediators of the gamma interferon‐induced cell death. Genes Dev. 1995;9(1):15‐30. [DOI] [PubMed] [Google Scholar]
- 3. Raveh T, Droguett G, Horwitz MS, DePinho RA, Kimchi A. DAP kinase activates a p19ARF/p53‐mediated apoptotic checkpoint to suppress oncogenic transformation. Nat Cell Biol. 2001;3(1):1‐7. [DOI] [PubMed] [Google Scholar]
- 4. Inbal B, Cohen O, Polak‐Charcon S, et al. DAP kinase links the control of apoptosis to metastasis. Nature. 1997;390(6656):180‐184. [DOI] [PubMed] [Google Scholar]
- 5. Kuo JC, Wang WJ, Yao CC, Wu PR, Chen RH. The tumor suppressor DAPK inhibits cell motility by blocking the integrin‐mediated polarity pathway. J Cell Biol. 2006;172(4):619‐631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lin Y, Henderson P, Pettersson S, Satsangi J, Hupp T, Stevens C. Tuberous sclerosis‐2 (TSC2) regulates the stability of death‐associated protein kinase‐1 (DAPK) through a lysosome‐dependent degradation pathway. FEBS J. 2011;278(2):354‐370. [DOI] [PubMed] [Google Scholar]
- 7. Stevens C, Lin Y, Harrison B, et al. Peptide combinatorial libraries identify TSC2 as a death‐associated protein kinase (DAPK) death domain‐binding protein and reveal a stimulatory role for DAPK in mTORC1 signaling. J Biol Chem. 2009;284(1):334‐344. [DOI] [PubMed] [Google Scholar]
- 8. Zhao J, Zhao D, Poage GM, et al. Death‐associated protein kinase 1 promotes growth of p53‐mutant cancers. J Clin Invest. 2015;125(7):2707‐2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394‐424. [DOI] [PubMed] [Google Scholar]
- 10. Matsumoto H, Nagao M, Ogawa S, et al. Prognostic significance of death‐associated protein‐kinase expression in hepatocellular carcinomas. Anticancer Res. 2003;23(2B):1333‐1341. [PubMed] [Google Scholar]
- 11. Li L, Guo L, Wang Q, et al. DAPK1 as an independent prognostic marker in liver cancer. PeerJ. 2017;5:e3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim J, Yu L, Chen W, et al. Wild‐Type p53 Promotes Cancer Metabolic Switch by Inducing PUMA‐Dependent Suppression of Oxidative Phosphorylation. Cancer Cell. 2019;35(2): 191‐203.e8. [DOI] [PubMed] [Google Scholar]
- 13. Lin Y, Richards FM, Krippendorff BF, et al. Paclitaxel and CYC3, an aurora kinase A inhibitor, synergise in pancreatic cancer cells but not bone marrow precursor cells. Br J Cancer. 2012;107(10):1692‐1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shen A, Chen Y, Liu L, et al. EBF1‐Mediated Upregulation of Ribosome Assembly Factor PNO1 Contributes to Cancer Progression by Negatively Regulating the p53 Signaling Pathway. Cancer Res. 2019;79(9):2257‐2270. [DOI] [PubMed] [Google Scholar]
- 15. Tremblay P, Meiner Z, Galou M, et al. Doxycycline control of prion protein transgene expression modulates prion disease in mice. Proc Natl Acad Sci U S A. 1998;95(21):12580‐12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mitry RR, Sarraf CE, Havlik R, Habib NA. Detection of adenovirus and initiation of apoptosis in hepatocellular carcinoma cells after Ad‐p53 treatment. Hepatology. 2000;31(4):885‐889. [DOI] [PubMed] [Google Scholar]
- 17. Takata A, Otsuka M, Yoshikawa T, et al. A miRNA machinery component DDX20 controls NF‐kappaB via microRNA‐140 function. Biochem Biophys Res Commun. 2012;420(3):564‐569. [DOI] [PubMed] [Google Scholar]
- 18. Takata A, Otsuka M, Yoshikawa T, et al. MicroRNA‐140 acts as a liver tumor suppressor by controlling NF‐kappaB activity by directly targeting DNA methyltransferase 1 (Dnmt1) expression. Hepatology. 2013;57(1):162‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Curmi F, Cauchi RJ. The multiple lives of DEAD‐box RNA helicase DP103/DDX20/Gemin3. Biochem Soc Trans. 2018;46(2):329‐341. [DOI] [PubMed] [Google Scholar]
- 20. Murashov AK, Chintalgattu V, Islamov RR, et al. RNAi pathway is functional in peripheral nerve axons. FASEB Journal. 2007;21(3):656‐670. [DOI] [PubMed] [Google Scholar]
- 21. Borg R, Cauchi RJ. GEMINs: potential therapeutic targets for spinal muscular atrophy? Front Neurosci. 2014;8:325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yan X, Mouillet JF, Ou Q, Sadovsky Y. A novel domain within the DEAD‐box protein DP103 is essential for transcriptional repression and helicase activity. Mol Cell Biol. 2003;23(1):414‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ou Q, Mouillet JF, Yan X, Dorn C, Crawford PA, Sadovsky Y. The DEAD box protein DP103 is a regulator of steroidogenic factor‐1. Mol Endocrinol. 2001;15(1):69‐79. [DOI] [PubMed] [Google Scholar]
- 24. Gillian AL, Svaren J. The Ddx20/DP103 dead box protein represses transcriptional activation by Egr2/Krox‐20. J Biol Chem. 2004;279(10):9056‐9063. [DOI] [PubMed] [Google Scholar]
- 25. O'Hern PJ, do Carmo G, Brecht J, et al. Decreased microRNA levels lead to deleterious increases in neuronal M2 muscarinic receptors in Spinal Muscular Atrophy models. eLife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stevens C, Lin Y, Sanchez M, et al. A germ line mutation in the death domain of DAPK‐1 inactivates ERK‐induced apoptosis. J Biol Chem. 2007;282(18):13791‐13803. [DOI] [PubMed] [Google Scholar]
- 27. Hensel N, Baskal S, Walter LM, Brinkmann H, Gernert M, Claus P. ERK and ROCK functionally interact in a signaling network that is compensationally upregulated in Spinal Muscular Atrophy. Neurobiol Dis. 2017;108:352‐361. [DOI] [PubMed] [Google Scholar]
- 28. Shin EM, Hay HS, Lee MH, et al. DEAD‐box helicase DP103 defines metastatic potential of human breast cancers. J Clin Invest. 2014;124(9):3807‐3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen W, Zhou P, Li X. High expression of DDX20 enhances the proliferation and metastatic potential of prostate cancer cells through the NF‐kappaB pathway. Int J Mol Med. 2016;37(6):1551‐1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chuang YT, Fang LW, Lin‐Feng MH, Chen RH, Lai MZ. The tumor suppressor death‐associated protein kinase targets to TCR‐stimulated NF‐kappa B activation. J Immunol. 2008;180(5):3238‐3249. [DOI] [PubMed] [Google Scholar]
- 31. Wu B, Yao H, Wang S, Xu R. DAPK1 modulates a curcumin‐induced G2/M arrest and apoptosis by regulating STAT3, NF‐kappaB, and caspase‐3 activation. Biochem Biophys Res Commun. 2013;434(1):75‐80. [DOI] [PubMed] [Google Scholar]
- 32. Yoo HJ, Byun HJ, Kim BR, Lee KH, Park SY, Rho SB. DAPk1 inhibits NF‐kappaB activation through TNF‐alpha and INF‐gamma‐induced apoptosis. Cell Signal. 2012;24(7):1471‐1477. [DOI] [PubMed] [Google Scholar]
- 33. Huang Y, Chen L, Guo L, Hupp TR, Lin Y. Evaluating DAPK as a therapeutic target. Apoptosis. 2014;19(2):371‐386. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1
Document S2