

Abstract
Ubiquitin‐dependent protein degradation has been implicated in the control of various cellular processes such as cell cycle control, transcriptional regulation, DNA damage repair, and apoptosis, many of which are involved in the initiation, progression, metastasis, and drug resistance of cancers. E3 ubiquitin ligases are known to be the second most prevalent cancer‐related functional gene family next to protein kinases. Of these, FBXO22, an F‐box receptor subunit of SCF E3 ligase, has recently been proposed to play a critical role in multiple aspects related to cancer development and therapy response. Firstly, FBXO22 is a key regulator of senescence induction through ubiquitylation of p53 for degradation. FBXO22 also acts as a molecular switch for the antagonistic and agonistic actions of selective estrogen receptor modulators (SERM) and determines the sensitivity of breast cancer to SERM by ubiquitylating KDM4B complexed with unliganded or SERMs‐bound estrogen receptor (ER). Furthermore, FBXO22 binds to Bach1, a pro‐metastatic transcription factor, suppressing Bach1‐driven metastasis of lung adenocarcinoma, and loss of FBXO22 facilitates metastasis. These findings, as well as other reports, unveiled strikingly important roles of FBXO22 in cancer development and therapeutic strategy. In this review, we summarize recent findings of how FBXO22 regulates major cancer suppression pathways.
Keywords: Bach1, F‐box protein, KDM4, p53, ubiquitin
Fbxo22 plays strikingly important roles in cancer development and therapeutic strategy through the regulation of p53, KDM4B, and Bach1.
Abbreviations
- ChIP
chromatin immunoprecipitation
- CRL3
Cullin3‐RING ubiquitin ligase
- Cullin
F‐box containing complex
- EBAG9
Estrogen Receptor Binding Site Associated Antigen 9
- Fbxl
F‐box, and leucine‐rich repeat proteins
- Fbxo22
F‐box only protein 22
- Fbxw
F‐box, and WD repeat domain containing
- GREB1
Growth Regulating Estrogen Receptor Binding 1
- KDM
histone lysine demethylase
- Keap1
Kelch‐like ECH‐associated protein 1
- Maf
musculoaponeurotic fibrosarcoma
- Nrf2
NF‐E2‐related factor 2
- pRB
Retinoblastoma protein
- Skp
SCF complex
1. INTRODUCTION
F‐box proteins contain at least one F‐box domain, a motif consisting of approximately 50 amino acids, through which F‐box proteins bind to SCF ubiquitin ligase complexes. 1 , 2 The SCF complex is composed of S‐phase kinase‐associated protein 1 (SKP1), RING of cullin 1 (ROC1; also called RBX1), Cullin 1 (CUL1), and variable F‐box proteins that determine substrate specificity. 3 F‐box proteins are divided into 3 subclasses depending on the presence of specific substrate recognition domains, such as Fbxw which contains WD40 repeat domains, 4 Fbxl which contains leucine‐rich repeat, 5 and Fbxo which contains various domains that are not fully characterized. Thirty‐seven F‐box proteins were designated as F‐box only (Fbxo) proteins within putative F‐box proteins in the human genome. 1 , 2 In most cases, Fbxo proteins contain the F‐box domain in their N‐terminus and various types of protein interaction domains in their C‐terminus, the latter of which mediate substrate bindings. Recently, several lines of studies have begun to uncover some interesting physiological functions of Fbxo proteins, which are attributed to uncharacterized protein interaction domains. For example, FBXO22 has been reported to be involved in tumorigenesis, metastasis, and senescence. 6 , 7 , 8 , 9 , 10 , 11 , 12 In this review, we focus on our discussion on the recent biochemical and biomedical evidence showing the tumor suppressor or oncogenic roles for FBXO22.
2. FBXO22 IS CRITICAL FOR INDUCTION OF SENESCENCE‐ASSOCIATED PHENOTYPES
Cellular senescence was first discovered as the inability of cultured human cells to proliferate indefinitely. 13 Later on, several lines of evidence revealed that senescence was also triggered by diverse genotoxic stimuli including telomere dysfunction, activated oncogenes, reactive oxygen species, and DNA damage. 14 , 15 Senescence is now believed to play a critical role in the suppression of tumorigenesis as well as geriatric changes in various organs due to its inherent nature to permanently cease cell proliferation. 16 , 17 Thus, one important hallmark of senescence is the inability of cells to proliferate in response to any physiological mitotic stimuli. Induction of cellular senescence requires functional p53 and pRB family proteins, which frequently undergo oncogenic mutations in a majority of human cancers. 18 , 19 These notions are strongly supported by the fact that cellular senescence is bypassed by viral oncoproteins inhibiting either p53 or pRB family proteins. 20 However, the precise roles of these tumor suppressors in the process of senescence were incompletely understood. We have uncovered the molecular mechanisms of permanent cell cycle arrest in which p53 activation at G2 phase plays an essential role in the senescence process. 21 , 22 Thus, these observations strongly suggest that pathways or proteins regulating the amount and the timing of p53 activation upon senescent inducing stimuli are the key factors for senescence regulation.
Another hallmark of senescence is senescence‐associated secretory phenotypes (SASP), a robust secretion of numerous growth factors, cytokines, proteases, and other proteins. 23 Such effects support various pathophysiological phenotypes in age‐related diseases, such as chronic inflammation, disruption of tissue architecture, and growth stimulation. The permanent cessation of cell proliferation and SASP are considered 2 hallmarks of senescence and are often coordinately induced. However, their mechanisms do not always overlap. For example, p38MAPK is critically required for SASP, but activated p53 restrains function of p38MAPK. 24 Thus, there appear to be some missing links that more clearly explain the antagonistic effects of p53 on the induction of the 2 representative hallmarks of senescence.
To address the above issues, we first tried to identify genes that are predominantly expressed in senescent cells, and whose product could regulate the activity of p53. 11 Global gene expression analysis revealed that FBXO22 was markedly induced in senescent cells. Interestingly, FBXO22 is upregulated at the late phase of the senescent process in a p53‐dependent manner. ChIP‐seq analysis confirmed that p53 was recruited to the transcription start site of the FBXO22 gene upon genotoxic stress. Although activation of p53 is critically required for the induction of senescence, it should be downregulated at the late phase of the senescent process for SASP induction. We found that FBXO22 is essential for this downregulation of p53. SCFFBXO22 specifically ubiquitylates methylated p53 complexed with KDM4A for degradation at the late phase of senescence. FBXO22 binds to p53 and KDM4A through its FIST‐N and FIST‐C domains, respectively. Formation of a ternary complex between FBXO22, methylated p53, and KDM4A facilitates ubiquitylation of p53 by SCF complexes. In this case, KDM4A is likely to act as a scaffold independent of its demethylase activity. Importantly, downregulation of methylated p53 by SCFFBXO22 is required for the induction of p16 and SASP, the former of which is critical for permanent cessation of cell proliferation upon any mitogen stimuli (Figure 1). SCFFBXO22 as a ubiquitin ligase for p53 is confirmed by in vivo experiments showing marked accumulation of p53 in all organs tested from FBXO22 knockout mice. Taken together, the results suggest that FBXO22 serves as a key regulator of senescence induction through the creation of a negative feedback loop with p53. Formation of this negative feedback loop between p53 and its ubiquitin ligases such as mdm2 25 and FBXO22 might be a common mechanism by which transcriptional activity of p53 could be finely tuned.
FIGURE 1.
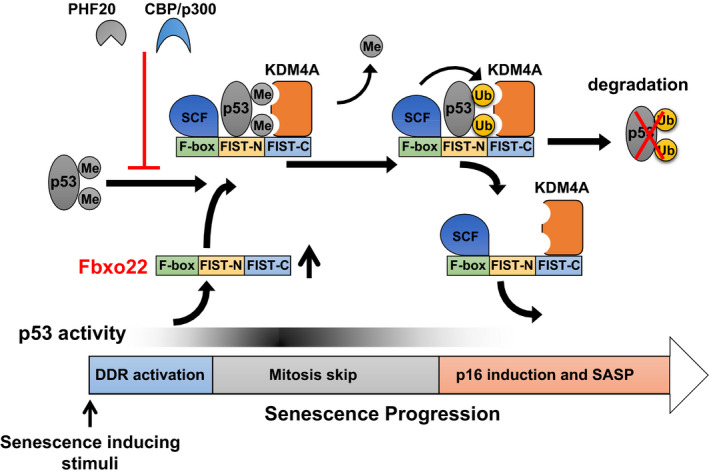
Regulation of p53 activity by FBXO22 during senescence. All known senescence‐inducing stimuli activate p53 through DNA damage responses (DDR). Activated p53 then transcriptionally induces FBXO22. An increase in FBXO22 complexed with SCF and KDM4A targets methylated p53 for ubiquitylation. Acetylation of p53 by CBP/p300 and PHF20 binding to methylated p53 suppresses SCF‐FBXO22‐KDM4A mediated ubiquitylation of p53. Ubiquitylated p53 is degraded through a proteasome pathway. Downregulation of p53 activity at the late phase of senescence triggers the induction of p16 and SASP, the former being essential for durable cessation of cell proliferation and the latter causing acceleration in aging phenotypes through induction of tissue microinflammation
3. FBXO22 DETERMINES SENSITIVITY OF BREAST CANCERS TO ENDOCRINE TREATMENT
Approximately 90 000 women are annually affected with breast cancer in Japan. Up to 70% of the cases are estrogen receptor‐α (ER)‐positive, Human Epidermal Growth Factor Receptor Type 2 (HER2)‐negative luminal type breast cancer, that are considered to be relatively non‐aggressive and sensitive to hormone therapy. Patients with the luminal breast cancer, especially for pre‐menopausal women, are treated with selective estrogen receptor modulators (SERMs) such as tamoxifen (TAM) as standard adjuvant therapy after surgery. 26 , 27 However, substantial patients treated with TAM undergo a relapse within 15 y 28 , 29 due to de novo resistance or acquired resistance. There exist various mechanisms for the hormone therapy resistance that have been extensively investigated for decades. However, these mechanisms are still insufficient to explain the entire picture of the endocrine resistance.
Estrogen‐bound ER regulates transcription of various downstream target genes through the recruitment of coactivators or corepressors. 29 , 30 , 31 This action is synergized by interplay between transactivation function 1 (AF1) and transactivation function 2 (AF2). 32 , 33 SERMs block estrogen action (antagonistic action) in many tissues, but in some tissues, they act like estrogen (agonistic action). 34 , 35 This selective modulation has been reported to be regulated by a complete blockade of AF2 function in a context specific manner. 36 , 37 Previously, it has been reported that steroid receptor coactivator 3 (SRC‐3) and HER2 in breast cancer cells convert SERMs actions from an antagonistic to agonistic. 38 However, given that overexpression of coactivators does not suppress the antagonistic action of SERMs 39 and that some ER‐positive breast cancers show a resistance to SERMs independent of HER2, there should be unidentified mechanisms underlying cofactor interactions with ER, modulating SERMs actions.
To identify the factor(s) that determines the antagonistic activity of SERMs, we first tried to uncover the mechanisms underlying regulation of ER signaling and to clarify the complexity of such signaling pathways that are interconnected or that converge into each other. 9 KDM4B is essential for ER‐mediated transcription. 40 , 41 , 42 , 43 Therefore, we speculated that the amount of KDM4B complexed with ER might play a key role in cofactor dynamics on ER. ER forms a complex with KDM4B and SRC‐3 after estrogen treatment. This complex dissociated after TAM treatment and ER then often formed a complex with N‐CoR‐HDAC3 repressors. Suppression of proteasome‐dependent protein degradation almost completely abrogated these cofactor dynamics on ER. Given that KDM4B depletion dissociated coactivators from estrogen‐bound ER, selective degradation of KDM4B complexed with ER might trigger cofactor dynamics on ER.
We then found that FBXO22 regulated the level of KDM4B protein. FBXO22 formed a ternary complex with ER and KDM4B through its FIST‐N and FIST‐C domains, respectively. Importantly, ER binding to FBXO22 was dependent on the types of ligands. ER predominantly formed a complex with unliganded or SERMs‐bound ER, but not E2‐bound ER. As the level of KDM4B was drastically decreased by co‐expression of FBXO22 and ER, but not by single expression of either one, formation of the ternary complex might facilitate KDM4B ubiquitylation by SCFFBXO22. Indeed, in vivo ubiquitylation assays under denaturing conditions revealed that SCFFBXO22 induced ubiquitylation of KDM4B and that this ubiquitylation was enhanced by co‐expression of ER in a dose‐dependent manner.
Given that proteasomal degradation was a prerequisite for cofactor switching from coactivators to corepressors on ER in response to SERMs treatment, SCFFBXO22‐dependent KDM4B ubiquitylation for degradation might play a critical role in the determination of the antagonistic activity of SERMs. In line with the role of SCFFBXO22 in the regulation of KDM4B complexed with unliganded or TAM‐bound ER, we found that FBXO22 is essential for antagonistic activity of SERMs in ER‐positive breast cancer cells. The FBXO22‐dependent antagonistic activity of SERMs appeared to be elicited by the AF1 domain of ER. It should be noted that requirement of FBXO22 in antagonistic activity against ER signaling was specific to SERMs, but not to selective ER down‐regulators (SERDs), such as fulvestrant.
In agreement with the essential role of FBXO22 in the antagonistic activity of SERMs, mapping of genome‐wide ER‐ and SRC‐3‐binding events by ChIP‐Seq analysis revealed that FBXO22 is prerequisite for TAM‐mediated SRC‐3 release from almost all ER‐SRC3‐bound genomic regions. In an in vivo xenograft model using NOD‐SCID mice, even in the presence of TAM, mice inoculated with T47D cells lacking FBXO22 showed progressive tumor growth, whereas mice inoculated with control T47D cells did not. These results suggest that FBXO22 is essential for the antagonistic activity of TAM both in vitro and in vivo through selective degradation of KDM4B complexed with unliganded or TAM‐bound ER (Figure 2).
FIGURE 2.
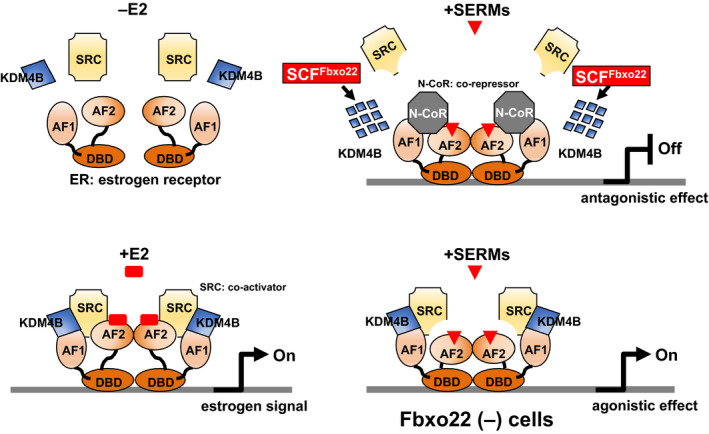
FBXO22 acts as a molecular switch for the antagonistic and agonistic actions of SERM. The antagonistic action of SERM requires dissociation of SRC from the ER that triggers substitution of SRC by N‐CoR on the ER. Ubiquitination and degradation of KDM4B by FBXO22 is essential for this substitution. The ER is comprised of AF1 and AF2 domains, which bind to SRC and N‐CoR, and also a DNA binding domain. AF2 binds to SRC in a ligand (estrogen)‐dependent manner, whereas the binding of AF1 to SRC is ligand‐independent. SERM trigger dissociation of SRC from AF2 regardless of the state of KDM4B. However, SERM‐induced dissociation of SRC from AF1 and subsequent substitution with N‐CoR required KDM4B degradation by FBXO22. Thus, FBXO22‐deficient cells retained estrogen signaling in the presence of SERM. ChIP‐sequence analyses using next generation sequencing revealed that SRC release by tamoxifen required FBXO22 on almost all ER/SRC‐bound enhancers and promoters
In addition to the effect on the sensitivity of breast cancer cells to SERM, FBXO22 deficiency may also contribute to a resistance to aromatase inhibitors, because dissociation of SRC from ER under E2‐depleted conditions also requires FBXO22 and degradation of KDM4B. 9 In response to E2‐deprivation, SRC was released from ER accompanied by KDM4B degradation in the similar fashion to the response to SERM treatment, whereas N‐CoR recruitment was not observed in this case. However, SRC and KDM4B remained attached with ER in cells depleted of FBXO22, and consequently EBAG9 and GREB1, downstream transcriptional target of ER, remained expressed at high level without E2. This supports that FBXO22 physiologically controls shutdown of ER‐mediated estrogen signals.
Based on such a critical role of FBXO22 on estrogen signaling, we hypothesized that FBXO22 deficiency may lead to a poor outcome of breast cancer due to a resistance to endocrine therapies. To test this, we performed immunohistochemical analysis of a set of 163 primary ER‐positive/HER2‐negative T2 breast cancer specimens to determine FBXO22 levels and analyzed its impact on prognosis. Outstandingly, tumors negative for FBXO22 expression showed significantly reduced relapse‐free survival (RFS) compared with tumors positive for FBXO22 (Figure 3). This significant difference was not affected by other clinicopathological variants, and preserved in separate cohorts of luminal A–like (low Ki‐67), node‐negative, grade‐1, and tamoxifen‐treated tumor cases. Multivariate survival analyses showed that the lack of FBXO22 was independently predictive of a poorer RFS, whereas Ki‐67 was not. The association of FBXO22 expression with RFS in ER‐positive/HER2‐negative breast cancer was further validated in another patient cohort at a different institution.
FIGURE 3.
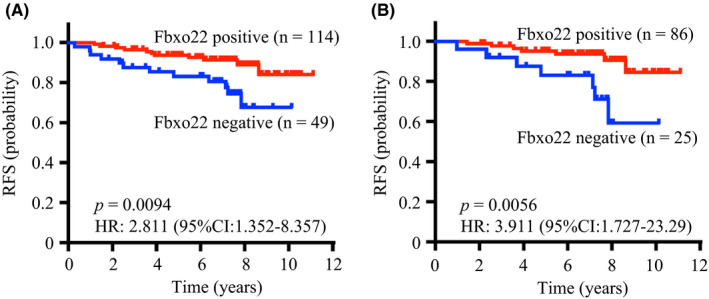
FBXO22 predicts an outcome in patients with ER‐positive/HER2‐negative breast cancer. A, Relapse‐free survival of FBXO22‐positive and FBXO22‐negative cases in all ER‐positive/HER2‐negative breast cancers. B, Relapse‐free survival of FBXO22‐positive and FBXO22‐negative cases in luminal A–like (low Ki‐67) breast cancers. P‐values and hazard ratio (HRs) were calculated using a log‐rank test
A biomarker to predict high‐risk groups of ER‐positive/HER2‐negative breast cancer has long been much needed, not only to treat such patients with additional therapy, but also to avoid unnecessary chemotherapy to low‐risk patients. Ki‐67 may currently represent such a marker. ER‐positive/HER2‐negative breast cancer has been subclassified as luminal A based on the Ki‐67 status to optimize adjuvant therapy. However, a considerable number of patients with low Ki‐67 luminal A–like breast cancer experience relapse after adjuvant hormone therapy, thus promoting efforts to seek better predictive markers. To overcome the problem, recent studies focused on multigene panels such as Oncotype DX, MammaPrint, and PAM50. However, these tests require a substantial length of time to come to diagnosis, and high costs. Therefore, FBXO22 immunohistochemical analysis may possibly be a faster and cheaper alternative to the multigene panel tests if its significance on hormone therapy sensitivity is further verified. In that regard, recent analyses of breast cancer prognosis with FBXO22 immunohistochemical analysis further validated its value. 8 , 10
4. FBXO22 REGULATES CANCER METASTASES
Metastases are one of the major causes of cancer‐related deaths. 44 Although intense efforts have been made to uncover their underlying mechanisms and identify effective therapeutic targets, progress in the treatment of metastatic cancer has been only minimal. Very recently, FBXO22 has been reported to play a key role in the metastatic process of various tumors in vivo. 6 , 8 Non‐small‐cell lung cancers (NSCLC) produce highly metastatic tumors. 45 , 46 , 47 However, the molecular basis underlying the metastasis of NSCLC is largely unclear. Recent genome‐wide analysis showed that a significant portion of metastatic NSCLC associates with mutations in either Keap1 or Nfe2/2, both of which result in the stabilization of Nrf2. 48 Keap1 is a substrate recognition subunit of a Cul3‐RING ubiquitin ligase CRL3 complex. 49 , 50 Under unperturbed conditions, Keap1‐CRL3 constitutively ubiquitylates Nrf2 for proteasomal degradation. Once cells sense oxidative stress, Nrf2 dissociates from Keap1‐CRL3 and is consequently stabilized. Transcriptional activity of Nrf2 is then executed by the formation of a heteromeric complex with a Maf on Maf recognition element (MARE). 51 Maf forms a complex with Bach1 to repress MAREs under unperturbed conditions. Upon oxidative stress, increased free heme binds to Bach1 through its heme regulatory motif (HRM), which triggers its proteasomal degradation, promoting the transcriptionally active Nrf2‐Maf complex. 52
In line with the frequent mutations in Keap1 in metastatic NSCLC, loss of Keap1 in mice caused a predisposition to lung adenocarcinoma (KrasLSL‐G12D/+; p53flox/flox), resulting in an increase in the incidence of metastasis. 6 Lung adenocarcinoma cells lacking Keap1 showed high cell migration in vitro and a metastatic ability in vivo. Gene Set Enrichment Analysis of RNA sequencing using these lung adenocarcinoma cells revealed that loss of Keap1 was associated with the Bach1 signature with the highest enrichment score. Indeed, the level of Bach1 was very high in Keap1‐mutated lung adenocarcinoma cells. Most intriguingly, loss of Keap1 led to an accumulation of Bach1 in a Nrf2‐induced heme oxiganase‐1 (Ho1)‐dependent manner. Increased Ho1 promoted degradation of free heme.
Hemin binding of Bach1 is reported to facilitate its ubiquitylation‐dependent degradation. 52 Although HOIL‐1 is involved in the ubiquitylation of Bach1 in a manner depending on its hemin binding, mass spectrometry analysis has identified FBXO22 as a heme‐dependent binding protein to Bach1. Indeed, FBXO22 depletion and overexpression results in the promotion of cell migration in vitro and the blockage of Bach1‐driven metastasis of lung adenocarcinoma, respectively. Thus, the results suggest that loss of FBXO22 in lung adenocarcinoma facilitates metastasis (Figure 4).
FIGURE 4.
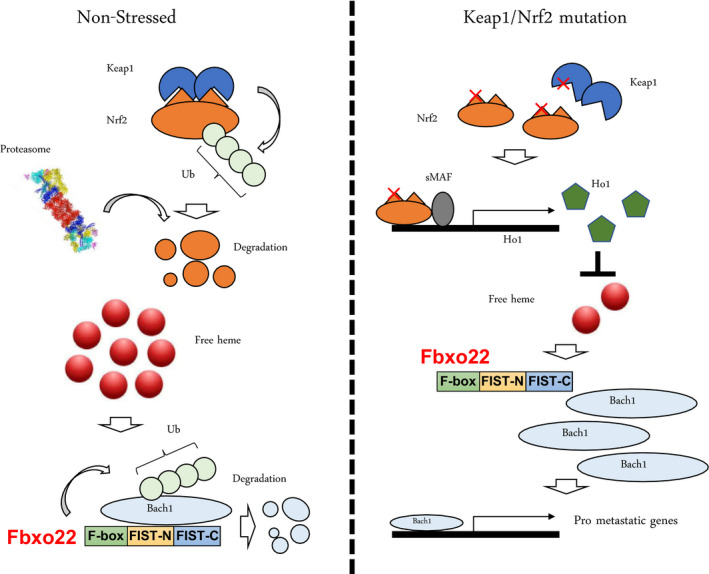
Loss of Keap1 and activation of Nrf2 enhance metastasis in NSCLC through acceleration of Bach1 transcriptional activity. Under non‐stressed and unperturbed conditions (left panel), Keap1 continuously ubiquitylates Nrf2 for proteasome‐dependent degradation. Increased levels of free heme enhance FBXO22‐mediated Bach1 ubiquitylation for degradation. Low levels of Bach1 suppress transcription of various pro‐metastatic genes. In the presence of a loss of mutation in Keap1 or a gain of mutation of Nrf2 (right panel), Nrf2 is stabilized and an increase in Nrf2/sMAF heterodimers transcriptionally induce Ho1. An increase in Ho1 reduces the levels of free heme, which in turn suppresses FBXO22‐mediated Bach1 ubiquitylation. An increase in Bach1 then transcriptionally induces various pro‐metastatic genes
In addition to lung adenocarcinomas, FBXO22 was also reported to be involved in the metastasis of breast cancers. 10 Oncogenic epithelial‐mesenchymal transition (EMT) is thought to be one of the main mechanisms underlying invasion and metastasis of breast cancers. EMT is regulated by a transcriptional network consisting of SNAIL, SLUG, ZEB1, ZEB2, and TWIST transcription factors. 53 , 54 Increased expression of SNAIL is correlated with a high incidence of metastasis and recurrence, and poor prognosis of breast cancers. 55 , 56 Activation of SNAIL sufficiently triggers EMT in breast cancers through downregulation of E‐cadherin expression, which is a known marker of EMT. 57 , 58 Intriguingly, FBXO22 was reported to ubiquitylate SNAIL for degradation. This degradation was dependent on the phosphorylation of SNAIL by GSK3β. 8 , 59 In agreement with GSK3β as a negative regulator of EMT, activation of Wnt signaling by suppression of GSK3β, is known to induce EMT. 60
FBXO22 was also reported to be involved in the metastasis of breast cancers through regulating the level of HDM2. 8 HDM2 is a RING‐type E3 ubiquitin ligase that target various oncogenic as well as anti‐oncogenic factors, such as p53. 61 Besides regulation of p53, HDM2 also functions in tumorigenesis by targeting various proteins. 62 For example, HDM2 was reported to target E‐cadherin for ubiquitylation, 63 regulating tumor cell invasiveness. Although the precise mechanism remains unknown, loss of FBXO22 promotes cell migration and invasion of human breast cancer cells in vitro. In line with this notion, using the mouse 4T1 breast tumor model, FBXO22 knock‐down facilitates metastasis of breast cancers in vivo. 8 Taken together with the results from multiple metastasis models, it is tempting to suggest that FBXO22 is an essential factor for regulating metastasis and invasion of cancers. Therefore, its activators represent an effective therapeutic strategy for prevention of cancer metastasis.
5. PERSPECTIVE
Several lines of evidence have suggested that mutation or aberrant expression of E3 ubiquitin ligases drives the initiation and progression of various types of cancers. Recent advances in FBXO22 research have clearly demonstrated that FBXO22 regulates tumorigenesis at multiple levels including initiation, hormone sensitivity, invasion, and metastasis of cancers. In normal cells, FBXO22 is likely to function as an anti‐tumor factor because senescence induction, suppression of hormone‐independent cell proliferation, and inhibition of cell migration are known to be involved in tumor suppressive mechanisms. Therefore, Fbxo22 might also regulate other anti‐tumor pathways, such as inhibition of anchorage‐independent growth, angiogenesis, and escape from immune systems. Although mutations or truncations in the FBXO22 gene are rare events in cancers, levels of FBXO22 expression vary even among the same types of cancers. The level of FBXO22 in cancer cells can be utilized as a predictive value for breast cancers, showing that a low level of FBXO22 in tumor tissues predicts a poorer outcome in ER‐positive/HER‐2‐negative breast cancers with high hazard ratios independently of other markers such as Ki‐67 and node status. Therefore, it would be of great interest to comprehensively analyze FBXO22 expression in various types of cancers and to clarify the relationship between the level of FBXO22 and patient outcome. Elucidation of mechanisms underlying the regulation of FBXO22 expression and identification of pathways that modulate the function of FBXO22 would also be important.
CONFLICT OF INTEREST
The authors declare that no conflict of interest exists.
ACKNOWLEDGMENTS
This work was supported by MEXT/JSPS KAKENHI under Grant Numbers JP19H05740, JP26250027, JP22118003, and JP16K15239, and by AMED under Grant Numbers JP17cm0106122, JP17fk0310111, and JP17gm5010001, as well as by Ono Medical Research Foundation, Princess Takamatsu Cancer Research Fund, and RELAY FOR LIFE JAPAN CANCER SOCIETY (M.N), by MEXT/JSPS KAKENHI under Grant Number 16H06148 (Y.J), and by MEXT/JSPS KAKENHI under Grant Numbers JP26290042, JP 24112005, and 17H03585, and by AMED under Grant Numbers JP16ck0106085h0003 (T.O).
Johmura Y, Harris AS, Ohta T, Nakanishi M. FBXO22, an epigenetic multiplayer coordinating senescence, hormone signaling, and metastasis. Cancer Sci. 2020;111:2718–2725. 10.1111/cas.14534
REFERENCES
- 1. Wang Z, Liu P, Inuzuka H, Wei W. Roles of F‐box proteins in cancer. Nat Rev Cancer. 2014;14(4):233‐247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nguyen KM, Busino L. The biology of F‐box proteins: the SCF family of E3 ubiquitin ligases. Adv Exp Med Biol. 2020;1217:111‐122. [DOI] [PubMed] [Google Scholar]
- 3. Zheng N, Zhou Q, Wang Z, Wei W. Recent advances in SCF ubiquitin ligase complex: clinical implications. Biochim Biophys Acta ‐ Rev Cancer. 2016;1866(1):12‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yeh CH, Bellon M, Nicot C. FBXW7: a critical tumor suppressor of human cancers. Mol Cancer. 2018;17(1):115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Matsushima N, Takatsuka S, Miyashita H, Kretsinger RH. Leucine rich repeat proteins: sequences, mutations, structures and diseases. Protein Pept Lett. 2018;26(2):108‐131. [DOI] [PubMed] [Google Scholar]
- 6. Lignitto L, LeBoeuf SE, Homer H, et al. Nrf2 activation promotes lung cancer metastasis by inhibiting the degradation of Bach1. Cell. 2019;178(2):316‐329.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhu XN, He P, Zhang L, et al. FBXO22 mediates polyubiquitination and inactivation of LKB1 to promote lung cancer cell growth. Cell Death Dis. 2019;10(7):486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bai J, Wu K, Cao MH, et al. SCFFBXO22 targets HDM2 for degradation and modulates breast cancer cell invasion and metastasis. Proc Natl Acad Sci U S A. 2019;116(24):11754‐11763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Johmura Y, Maeda I, Suzuki N, et al. Fbxo22‐mediated KDM4B degradation determines selective estrogen receptor modulator activity in breast cancer. J Clin Invest. 2018;128(12):5603‐5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sun R, Xie HY, Qian JX, et al. FBXO22 possesses both protumorigenic and antimetastatic roles in breast cancer progression. Cancer Res. 2018;78(18):5274‐5286. [DOI] [PubMed] [Google Scholar]
- 11. Johmura Y, Sun J, Kitagawa K, et al. Fbxo22‐KDM4A targets methylated p53 for degradation and regulates senescence. Nat Commun. 2016;7:10574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tian X, Dai S, Sun J, et al. F‐box protein FBXO22 mediates polyubiquitination and degradation of KLF4 to promote hepatocellular carcinoma progression. Oncotarget. 2015;6(26):22767‐22775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25(3):585‐621. [DOI] [PubMed] [Google Scholar]
- 14. Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24(22):2463‐2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8(9):729‐740. [DOI] [PubMed] [Google Scholar]
- 16. Campisi J, Robert L. Cell senescence: role in aging and age‐related diseases In: Aging: Facts and Theories, Interdiscip Top Gerontol 2014; 39:45‐61. 10.1159/00035889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene‐induced DNA damage model for cancer development. Science (80‐). 2008;319(5868):1352‐1355. [DOI] [PubMed] [Google Scholar]
- 18. Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9(10):749‐758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8(9):671‐682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shay JW, Pereira‐Smith OM, Wright WE. A role for both RB and p53 in the regulation of human cellular senescence. Exp Cell Res. 1991;196(1):33‐39. [DOI] [PubMed] [Google Scholar]
- 21. Johmura Y, Shimada M, Misaki T, et al. Necessary and sufficient role for a mitosis skip in senescence induction. Mol Cell. 2014;55(1):73‐84. [DOI] [PubMed] [Google Scholar]
- 22. Krenning L, Feringa FM, Shaltiel IA, van den Berg J, Medema RH. Transient activation of p53 in G2 phase is sufficient to induce senescence. Mol Cell. 2014;55(1):59‐72. [DOI] [PubMed] [Google Scholar]
- 23. Tchkonia T, Zhu Y, Van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123(3):966‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response‐independent regulator of the senescence‐associated secretory phenotype. EMBO J. 2011;30(8):1536‐1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hu W, Feng Z, Levine AJ. The regulation of multiple p53 stress responses is mediated through MDM2. Genes Cancer. 2012;3(3‐4):199‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Coates AS, Winer EP, Goldhirsch A, et al. Tailoring therapies–improving the management of early breast cancer: St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2015. Ann Oncol Off J Eur Soc Med Oncol. 2015;26(8):1533‐1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Osborne CK. Tamoxifen in the treatment of breast cancer. N Engl J Med. 1998;339(22):1609‐1618. [DOI] [PubMed] [Google Scholar]
- 28. Davies C, Pan H, Godwin J, et al. Long‐term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor‐positive breast cancer: ATLAS, a randomised trial. Lancet. 2013;381(9869):805‐816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Belandia B, Orford RL, Hurst HC, Parker MG. Targeting of SWI/SNF chromatin remodelling complexes to estrogen‐responsive genes. EMBO J. 2002;21(15):4094‐4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor‐regulated transcription. Cell. 2000;103(6):843‐852. [DOI] [PubMed] [Google Scholar]
- 31. McKenna NJ, Xu J, Nawaz Z, Tsai SY, Tsai MJ, O'Malley BW. Nuclear receptor coactivators: multiple enzymes, multiple complexes, multiple functions. J Steroid Biochem Mol Biol. 1999;69:3‐12. [DOI] [PubMed] [Google Scholar]
- 32. Kumar R, Thompson EB. The structure of the nuclear hormone receptors. Steroids. 1999;64(5):310‐319. [DOI] [PubMed] [Google Scholar]
- 33. Tora L, White J, Brou C, et al. The human estrogen receptor has two independent nonacidic transcriptional activation functions. Cell. 1989;59(3):477‐487. [DOI] [PubMed] [Google Scholar]
- 34. Smith CL, O'Malley BW. Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr Rev. 2004;25(1):45‐71. [DOI] [PubMed] [Google Scholar]
- 35. Nettles KW, Greene GL. Ligand control of coregulator recruitment to nuclear receptors. Annu Rev Physiol. 2005;67(1):309‐333. [DOI] [PubMed] [Google Scholar]
- 36. Tzukerman MT, Esty A, Santiso‐Mere D, et al. Human estrogen receptor transactivational capacity is determined by both cellular and promoter context and mediated by two functionally distinct intramolecular regions. Mol Endocrinol. 1994;8(1):21‐30. [DOI] [PubMed] [Google Scholar]
- 37. McInerney EM, Katzenellenbogen BS. Different regions in activation function‐1 of the human estrogen receptor required for antiestrogen‐ and estradiol‐dependent transcription activation. J Biol Chem. 1996;271(39):24172‐24178. [DOI] [PubMed] [Google Scholar]
- 38. Shou J, Massarweh S, Osborne CK, et al. Mechanisms of tamoxifen resistance: Increased estrogen receptor‐HER2/neu cross‐talk in ER/HER2‐positive breast cancer. J Natl Cancer Inst. 2004;96(12):926‐935. [DOI] [PubMed] [Google Scholar]
- 39. Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science (80‐). 2002;295(5564):2465‐2468. [DOI] [PubMed] [Google Scholar]
- 40. Shi L, Sun L, Li Q, et al. Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. Proc Natl Acad Sci U S A. 2011;108(18):7541‐7546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang J, Jubb AM, Pike L, et al. The histone demethylase JMJD2B is regulated by estrogen receptor α and hypoxia, and is a key mediator of estrogen induced growth. Cancer Res. 2010;70(16):6456‐6466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gaughan L, Stockley J, Coffey K, et al. KDM4B is a master regulator of the estrogen receptor signalling cascade. Nucleic Acids Res. 2013;41(14):6892‐6904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kawazu M, Saso K, Tong KI, et al. Histone demethylase JMJD2B functions as a co‐factor of estrogen receptor in breast cancer proliferation and mammary gland development. PLoS One. 2011;6(3):e17830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dillekås H, Rogers MS, Straume O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019;8(12):5574‐5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chuang CH, Greenside PG, Rogers ZN, et al. Molecular definition of a metastatic lung cancer state reveals a targetable CD109‐Janus kinase‐Stat axis. Nat Med. 2017;23(3):291‐300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Caswell DR, Chuang CH, Yang D, et al. Obligate progression precedes lung adenocarcinoma dissemination. Cancer Discov. 2014;4(7):781‐789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brady JJ, Chuang CH, Greenside PG, et al. An Arntl2‐driven secretome enables lung adenocarcinoma metastatic self‐sufficiency. Cancer Cell. 2016;29(5):697‐710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hammerman PS, Voet D, Lawrence MS, et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519‐525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jaramillo MC, Zhang DD. The emerging role of the Nrf2‐Keap1 signaling pathway in cancer. Genes Dev. 2013;27(20):2179‐2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Taguchi K, Yamamoto M. The KEAP1NRF2 system in cancer. Front Oncol. 2017;7(MAY):85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kimura M, Yamamoto T, Zhang J, et al. Molecular basis distinguishing the DNA binding profile of Nrf2‐Maf heterodimer from that of Maf homodimer. J Biol Chem. 2007;282(46):33681‐33690. [DOI] [PubMed] [Google Scholar]
- 52. Zenke‐Kawasaki Y, Dohi Y, Katoh Y, et al. Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol Cell Biol. 2007;27(19):6962‐6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Smith B, Bhowmick N. Role of EMT in metastasis and therapy resistance. J Clin Med. 2016;5(2):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Goossens S, Vandamme N, Van Vlierberghe P, Berx G. EMT transcription factors in cancer development re‐evaluated: Beyond EMT and MET. Biochim Biophys Acta ‐ Rev Cancer. 2017;1868(2):584‐591. [DOI] [PubMed] [Google Scholar]
- 55. Tran HD, Luitel K, Kim M, Zhang K, Longmore GD, Tran DD. Transient SNAIL1 expression is necessary for metastatic competence in breast cancer. Cancer Res. 2014;74(21):6330‐6340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Moody SE, Perez D, Pan TC, et al. The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell. 2005;8(3):197‐209. [DOI] [PubMed] [Google Scholar]
- 57. Batlle E, Sancho E, Francí C, et al. The transcription factor Snail is a repressor of E‐cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2(2):84‐89. [DOI] [PubMed] [Google Scholar]
- 58. Cano A, Pérez‐Moreno MA, Rodrigo I, et al. The transcription factor Snail controls epithelial‐mesenchymal transitions by repressing E‐cadherin expression. Nat Cell Biol. 2000;2(2):76‐83. [DOI] [PubMed] [Google Scholar]
- 59. Zhou BP, Deng J, Xia W, et al. Dual regulation of Snail by GSK‐3β‐mediated phosphorylation in control of epithelial‐mesenchymal transition. Nat Cell Biol. 2004;6(10):931‐940. [DOI] [PubMed] [Google Scholar]
- 60. Basu S, Cheriyamundath S, Ben‐Ze'ev A. Cell–cell adhesion: linking Wnt/β‐catenin signaling with partial EMT and stemness traits in tumorigenesis. F1000Research. 2018;7:1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Manfredi JJ. The Mdm2‐p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev. 2010;24(15):1580‐1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bohlman S, Manfredi JJ. p53‐independent effects of Mdm2. Subcell Biochem. 2014;85:235‐246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yang J‐Y, Zong CS, Xia W, et al. MDM2 promotes cell motility and invasiveness by regulating E‐cadherin degradation. Mol Cell Biol. 2006;26(19):7269‐7282. [DOI] [PMC free article] [PubMed] [Google Scholar]