

Summary:
Metastatic melanoma is an aggressive disease, despite recent improvements in therapy. Eradicating all melanoma cells even in drug-sensitive tumors is unsuccessful in patients because a subset of cells can transition to a slow-cycling state, rendering them resistant to most targeted therapy. It is still unclear what pathways define these subpopulations and promote this resistant phenotype. In the current study, we show that Wnt5A, a non-canonical Wnt ligand that drives a metastatic, therapy-resistant phenotype, stabilizes the half-life of p53 and uses p53 to initiate a slow-cycling state following stress (DNA damage, targeted therapy, and aging). Inhibiting p53 blocks the slow-cycling phenotype and sensitizes melanoma cells to BRAF/MEK inhibition. In vivo, this can be accomplished with a single dose of p53 inhibitor at the commencement of BRAF/MEK inhibitor therapy. These data suggest that taking the paradoxical approach of inhibiting rather than activating WTp53 may sensitize previously resistant metastatic melanoma cells to therapy.
Keywords: slow-cycling phenotype, tumor microenvironment, therapy resistance
Graphical Abstract
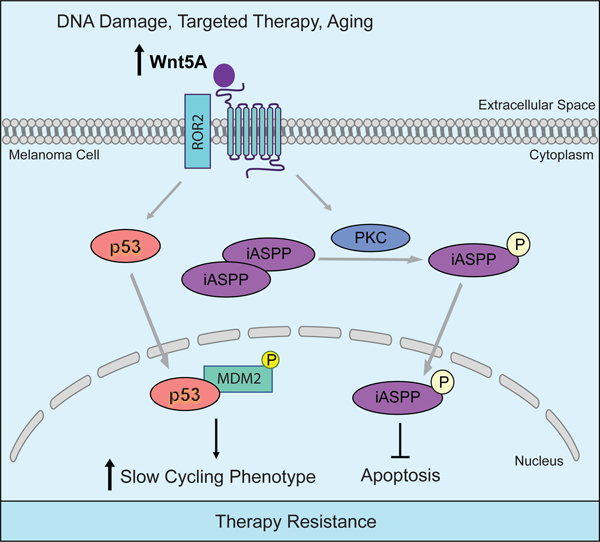
eToc Blurb:
Cancer cells adopt multiple ways to evade therapy, including adopting a slow-cycling phenotype to resist drugs that target rapidly proliferating cells. Webster et al. show by circumventing this slow-cycling state through inhibition of p53, cells can be sensitized to targeted therapies that affect proliferation pathways such as the MAPK pathway.
Introduction
Melanoma is a highly malignant disease that is subject to multiple forms of stress, including UV, and aging. As melanoma progresses, it undergoes a phenomenon known as “phenotype switching” where proliferation and invasion tend to be antagonistic so that cells that are more proliferative are less invasive, and cells that are more invasive are less proliferative. This is partially effected by changes in Wnt signaling (Arozarena and Wellbrock, 2017). Canonical Wnt signaling via β-catenin promotes a more proliferative phenotype where as non-canonical Wnt signaling drives a more invasive phenotype (Dissanayake et al., 2008; Webster et al., 2015a; Weeraratna et al., 2002). The non-canonical Wnt protein, Wnt5A drives invasion through degradation of β-catenin in an SIAH2-dependent mechanism (O’Connell et al., 2013; Webster and Weeraratna, 2013). This switch from canonical to non-canonical Wnt signaling, which promotes invasion, has also been linked with resistance to targeted therapy (Asiedu et al., 2014; O’Connell et al., 2013). Attempts to target metastatic melanoma cells have been relatively unsuccessful due to their highly plastic state (Ahmed and Haass, 2018; Roesch et al., 2013).
Recent studies have shown that slow-cycling populations drive a highly invasive cell state, that leads to increased tumor cell dissemination and metastasis (Ahmed and Haass, 2018). Perego et al. have shown that slow cycling cells are two to eight times more invasive compared to cycling cells (Perego et al., 2018). One of the three cell lines used to examine invasion of slow cycling cells in these studies was wild type for p53 (WM88). This cell line had the greatest difference in invasive capacity of the slow cycling cells, 8-fold more invasive. However, authors revealed no conclusion regarding mutational status and invasive ability of slow cycling cells in their study. Previous data from our lab suggest that Wnt5A drives a senescent-like phenotype in response to multiple types of stress. This phenotype is characterized by multiple hallmarks of senescence, yet the cells remain highly invasive and retain the ability to grow out as distant metastases in vivo while continuing to harbor senescent traits such as chromatin marks, senescence-associated heterochromatic foci, and PML bodies. (Webster et al., 2015b) This senescent-like phenotype is not p16 driven, as it occurs in cells that have high Wnt5A, and low p16, but which retain p21 and WTp53 expression.
New data from the cancer genome atlas suggest that p53 mutations in melanoma are not as infrequent as previously thought, and that 19% of melanoma harbor p53 mutations (Hodis et al., 2012; Zhang et al., 2016). However, over 80% of melanoma tumors retain WTp53 expression. In our current study we find that wild type p53 is frequently associated with high Wnt5A expression and show that in fact Wnt5A utilizes wild type p53 expression to drive a slow-cycling phenotype. This is enriched following multiple types of stress including aging and targeted therapy. These data suggest an alternate role for p53, as a driver of therapy resistance, while still in keeping with its role as a cell cycle regulator.
Results
Wnt5A requires wild type p53 to promote a slow cycling phenotype.
We first analyzed a panel of melanoma cells, that have been characterized as Wnt5A high or Wnt5A low expressing cell lines via protein and qPCR analysis (Fig. S1A), for slow-cycling populations (Webster et al., 2015b). The number of slow-cycling cells was determined by flow cytometry, measuring the retention of the dye CellTrace Violet, which is retained in less proliferative cell populations (Perego et al., 2018). We observed that melanoma cells expressing higher levels of Wnt5A had an increased percentage of cells exhibiting a slow-cycling phenotype (Fig. 1A, Fig. S1B). When we isolated these slow-cycling populations, we found that slow-cycling cells expressed significantly higher Wnt5A (Fig. 1B, Fig. S1C). Since slow cycling cells are only about six percent of the cell population, obtaining enough cells to analyze by Western proved difficult, therefore changes in Wnt5A expression were normalized to HSP90. In support of Wnt5A promoting a slow cycling phenotype, when we knocked down Wnt5A, we observed a decrease in the slow cycling population (Fig. 1C, Fig. S1D–E) suggesting that Wnt5A is a key driver of this slow-cycling phenotype. Since slow-cycling cells have been implicated in tumor maintenance, residual disease, and therapy resistance, we analyzed Wnt5A high melanoma cells for markers of a slow-cycling phenotype, therapy resistance, and cell cycle regulation. We found that melanoma cells expressing high levels of Wnt5A also express p53, p21, and JARID1B, which has previously been identified as a marker of slow cycling cells in melanoma (Fig. 1D, Fig. S1F–S1H) (11). We sequenced these cells for p53 mutations, and found them to be wild type for p53 (Webster et al., 2015b). Since p53 regulates p21, and we have previously observed that Wnt5A increases p21 expression following stress to induce a senescent-like phenotype, we analyzed slow-cycling cells for p53 and p21. We found that slow-cycling cells are enriched for both p53 expression (Fig. 1E, Fig. S1I) and p21 (Fig. S1I–S1J) when compared to cycling cells. To compare our observed slow-cycling cells with previously reported JARID1B associated slow-cycling populations, we analyzed the cell cycle profile of cycling and slow-cycling populations. We found that cells that express a slow-cycling phenotype spend more time in G2/M (Fig. S1K), which is a reported characteristic of cells in a slow-cycling state (Perego et al., 2018).
Figure 1. Wnt5A promotes a slow cycling phenotype through wild type p53.
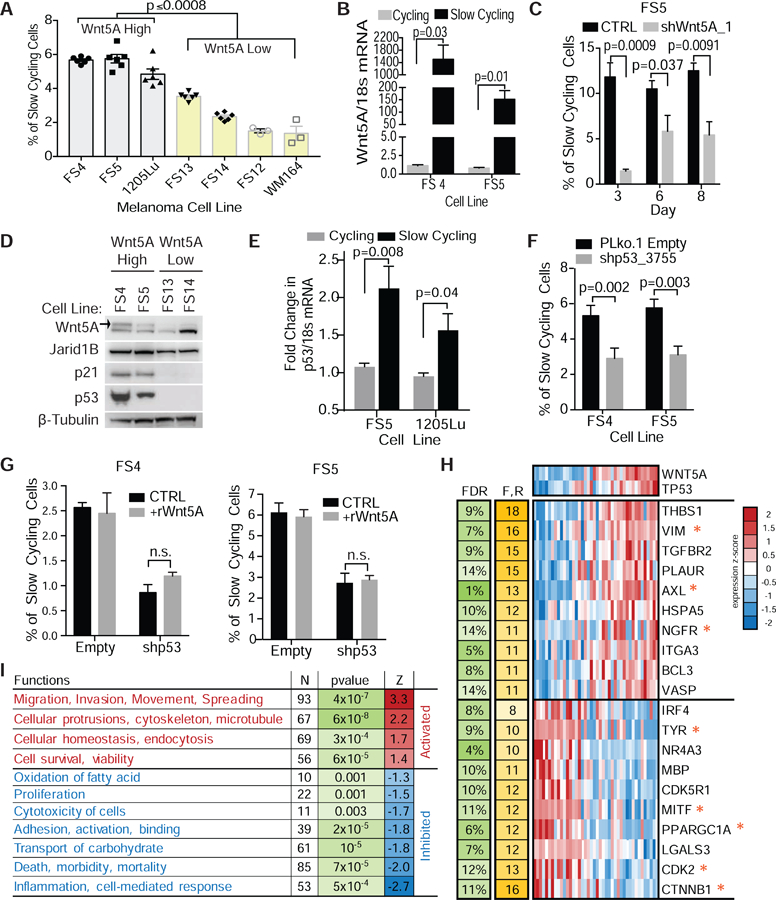
(A) FACS analysis of slow-cycling cell populations in invasive Wnt5A high (1205Lu, FS4, FS5) and proliferative Wnt5A low (yellow bars) (FS13, FS14, FS12, WM164) melanoma cells. (ANOVA, multiple comparisons) (B) qPCR analysis of slow-cycling and cycling populations for Wnt5A mRNA. (C) FACS analysis of changes in slow-cycling cell populations following knockdown of Wnt5A using shRNA at day 3, 6, and 8 in FS5 invasive melanoma cells. (D) Western blot analysis of markers of therapy resistance, slow-cycling phenotype, and cell cycle regulation in Wnt5A high and low human melanoma cells. Active glycosylated Wnt5A is marked with arrow. β-tubulin used as a loading control. (E) Slow-cycling and cycling populations were sorted by flow cytometry and analyzed by qPCR for p53 mRNA. (F) FACS analysis of slow-cycling cells following p53 KD. (G) FACS analysis of slow-cycling cells following addition of rWnt5A to p53 knock down melanoma cells. Data are represented as mean ± SEM. (H) Expression heatmap for top genes most involved in enriched function/regulators (F,R = number of functions/regulators in which the gene is involved). * genes associated with invasion, stem cells, therapy resistance and proliferation. (I) Enriched functions activated/inhibited in WNT5A/TP53 high vs low (N- number of genes in the function. Z = activation z-score predicted by IPA). Also see Figures S1–S2.
To determine if p53 was important for this slow-cycling phenotype, we knocked down p53 and analyzed changes in this population. We found that knocking down p53, in high Wnt5A expressing cell lines, decreased the number of cells exhibiting a slow-cycling phenotype (Fig. 1F). Addition of rWnt5A to these cells did not rescue the slow-cycling phenotype (Fig. 1G), suggesting that p53 is required for a Wnt5A induced slow-cycling phenotype. To evaluate the relationship between wild-type p53 and Wnt5A in melanoma, we analyzed human melanoma cell lines from the Cancer Cell Line Encyclopedia (Barretina et al., 2012) taking expression data from only wild type p53 samples. We found 137 such genes significantly positively correlated with p53 and Wnt5A at the same time, and 206 genes significantly negatively correlated (FDR<15%, Fig. S2A) (Storey and Tibshirani, 2003). Of the top 10 genes significantly positively correlated with p53 and Wnt5A several are associated with invasion, migration, and stem-like properties (AXL, VIM, NGFR) (Fig. 1H) (Müller et al., 2014; Radke et al., 2017; Rambow et al., 2018; Tirosh et al., 2016; Tsoi et al., 2018). Negatively correlated genes such as CTNB1, MITF, CDK2 and TYR, are known to be correlated with proliferation, supports the premise that Wnt5A and p53 decrease proliferation (Fig 1H) (Webster et al., 2015a). We then analyzed the gene list using Ingenuity Pathway Analysis in order to gain insight into possible regulatory mechanisms and cellular functions enriched among the significant genes. We found 11 functional categories (Fig. 1I) and 17 upstream regulators (TGFβ1, TWIST1, SNAI1) (Fig. S2B) significantly enriched in the gene set, suggesting that they are upregulated in Wnt5A high and p53 high expressing cells. Together, these data along with the pathways and regulators were identified as inhibited (MITF, proliferation) (Fig. 1H–I, Fig. S2B) in p53 and Wnt5A expressing cells, support a role for Wnt5A and p53 in driving a more invasive, less proliferative, therapy resistant phenotype.
Wnt5A increases the half-life of wild type p53.
The connection between Wnt5A, a marker of tumor progression and therapy resistance, and increased wild type p53 expression is surprising since p53 is generally thought of as a tumor suppressor due to its role in the induction of apoptosis. However, in melanoma, p53 is mutated in only about 20% of melanoma and recent data suggest that when p53 mutations do occur, they are not connected with other common mutations (BRAF, RAS, NF1) (Garman et al., 2017). To determine if there is a direct connection between Wnt5A and p53 expression in metastatic melanoma cells, we knocked down Wnt5A using shRNA (Fig. 2A). Our lab has previously shown that Wnt5A typically migrates as a doublet on a SDS Page gel and that the top band, highlighted using an arrow, is the glycosylated and active form of Wnt5A (Dissanayake and Weeraratna, 2008). We found that p53 expression was decreased in the Wnt5A knockdown cells. Knocking down Wnt5A also decreased the half-life of p53, suggesting that Wnt5A stabilizes the p53 protein (Fig. 2B, 2C). To determine the mechanism of increased p53 half-life, we analyzed expression of MDM2 and MDM4, which are ubiquitin ligases that regulate p53 via proteasomal degradation. There was no difference in basal expression of either MDM2 or MDM4 across multiple melanoma cell lines (Fig. 2D, Fig. S3A). Following DNA damage, induced by doxorubicin treatment, in Wnt5A high melanoma cells, we observed an increase in MDM2 and p53 expression (Fig. 2E), but not MDM4. Recently, it was discovered that phosphorylation of MDM2 at Ser-395 blocks the ability of MDM2 to export p53 from the nucleus (Hu et al., 2012; Maya et al., 2001). The ser395 phosphorylation site also promotes an interaction between p53 mRNA and the MDM2 protein, which increases the synthesis of the p53 protein after genotoxic stress (Gajjar et al., 2012). When we analyzed nuclear and cytoplasmic extracts, we found that p53 and p-MDM2ser395 are nuclear in melanoma cells with high Wnt5A expression (Fig. 2F). To determine if phosphorylation of MDM2 at serine395 increases the half-life of p53, we performed a proximity ligation assay using antibodies for p53 and p-MDM2ser395. We observed an increase in fluorescence upon treatment with rWnt5A suggesting that Wnt5A promotes the interaction of p53 and p-MDM2ser395 (Fig 2. G,H). We confirmed the interaction of p53 and pMDM2ser395 in the presence of Wnt5A by performing an immunoprecipitation using a p53 antibody where we pulled down pMDM2ser395 (Fig. S3B). These data suggest that Wnt5A increases p53 expression in part by negative regulation of MDM2.
Figure 2. Wnt5A regulates p53 expression via MDM2 phosphorylation.
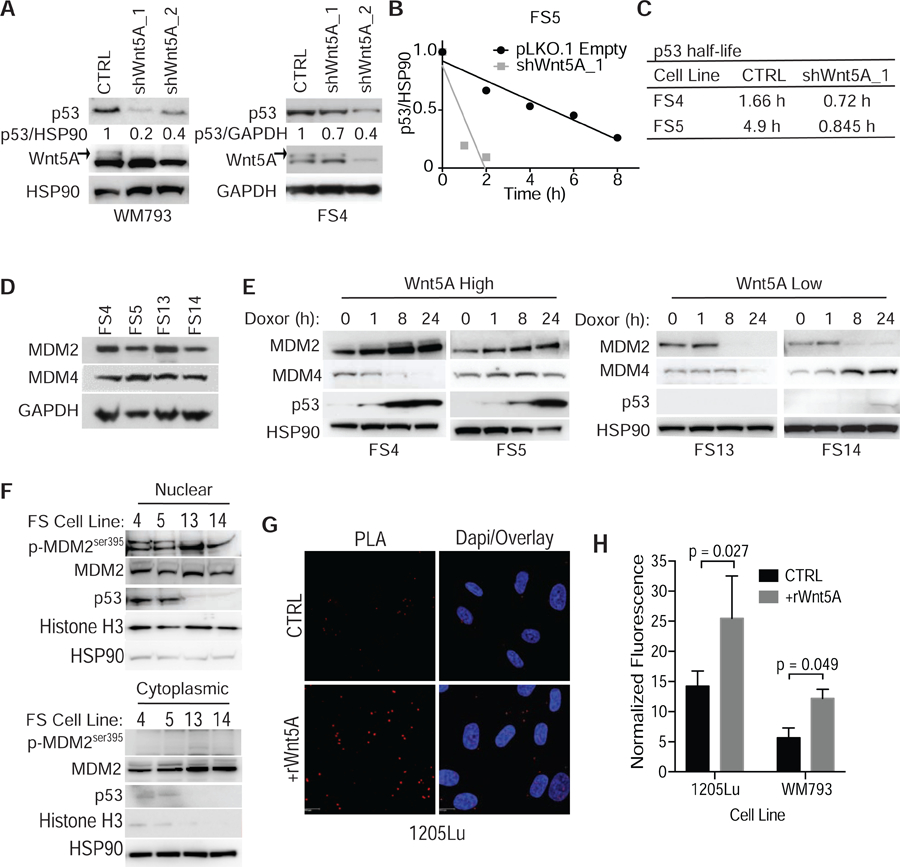
(A) Western blot analysis of p53 expression following knockdown of Wnt5A in invasive melanoma cells with shRNA. HSP90 was used as a loading control. (B) Quantification of change in p53 half-life following shRNA knockdown of Wnt5A assessed following treatment with cycloheximide. (C) Changes in half-life of p53 upon Wnt5A knockdown. (D) Western blot of proteins known to regulate p53 expression and function in melanoma. GAPDH used as a loading control. (E) Western blot analysis of MDM2, MDM4, and p53 expression in Wnt5A high (FS4 and FS5) and low (FS13 and FS14) melanoma cells following DNA damage induced by doxorubicin (1μg/mL). HSP90 used as a loading control. (F) Western blot analysis of nuclear and cytoplasmic fractions for p-MDM2ser395, total MDM2, and p53 expression. HSP90 and Histone H3 used as loading control for cytoplasmic and nuclear fractions, respectively. (G) Proximity ligation assay for the interaction of p53 and p-MDM2ser395 in 1205Lu melanoma cells treated with rWnt5A. (H) Quantification of increased p53-pMDM2ser395 interaction in proximity ligation assay in 1205Lu and WM793 cells +/− rWnt5A (200 ng/mL). Data are represented as mean ± SEM. Also see Figure S3.
To address why p53 high cells do not undergo apoptosis, but merely arrest (Webster et al., 2015b), we asked if p53-induced apoptosis is blocked in these cells via iASPP (inhibitor of apoptosis stimulating protein p53), as has been previously shown in melanoma (Lu et al., 2016). One possibility is that PKC, which is down stream of Wnt5A, may phosphorylate iASPP leading to nuclear localization and activation (Dissanayake et al., 2007; Lu et al., 2016). iASPP exists as a dimer in the cytoplasm, but when iASPP is phosphorylated, it is activated and moves into the nucleus as a monomer. When we analyzed basal expression of iASPP in Wnt5A high and low expressing cell lines we found no difference in expression (Fig. 3A). However, when we analyzed nuclear and cytoplasmic extracts in cells with high and low Wnt5A expression, we found that cells with higher expression of Wnt5A had increased nuclear iASPP (Fig. 3B), suggesting that iASPP is active in these cells. We also found iASPP expression increased in melanoma cells along with pPKC following DNA damage (Fig. 3C). The increase in expression and activation of iASPP correlates with decreased apoptosis (Fig. 3D), and decreased PUMA upregulation (Fig. S3C), following DNA damage in Wnt5A high cells. Upon iASPP knock down, there is an increase in apoptotic cells following doxorubicin induced DNA damage in Wnt5A high cells (Fig. S3D). We hypothesize that Wnt5A is promoting iASPP phosphorylation and activation via increased pPKC expression following DNA damage (Fig. 3E). To test this, we treated Wnt5A high cells with a PKC inhibitor and induced DNA damage. We found an increase in apoptotic cells compared to control in cells treated with PKC inhibitor prior to doxorubicin treatment (Fig. S3E), suggesting that PKC is playing a role in survival following DNA damage. Since we observed a greater increase in iASPP in Wnt5A high cells following DNA damage we hypothesized that there may be a link between iASPP and Wnt5A expression. When we knocked down Wnt5A (Fig. S3F), we observed a decrease in iASPP expression (Fig. 3F). These data suggest that Wnt5A is driving p53 expression to promote a slow cycling phenotype, while inhibiting p53-induced apoptosis via increased iASPP activation and expression.
Figure 3. Wild type p53 expression increases following DNA damage and targeted therapy.
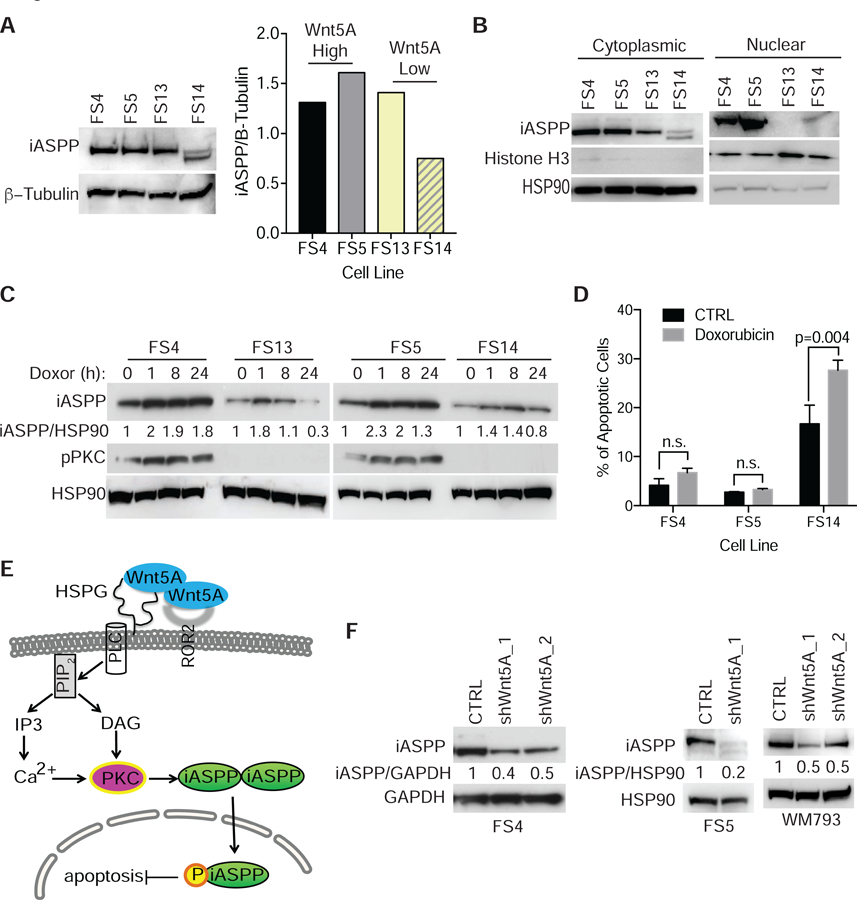
(A) Western blot analysis and quantification of basal iASPP expression in Wnt5A high (FS4 and FS5) and low (FS13 and FS14) expressing cells. (B) Western blot analysis of nuclear and cytoplasmic fractions from Wnt5A high (FS4, FS5) and Wnt5A low (FS13, FS14) melanoma cells for iASPP expression. (C) Western blot analysis of iASPP and pPKC in melanoma cells following DNA damage induced by doxorubicin (1μg/mL). (D) Quantification of apoptotic cells using AnnexinV and propidium iodide in Wnt5A high (FS4 and FS5) and Wnt5A low (FS14) cells following doxorubicin 1 ug/mL treatment. Data are represented as mean ± SEM. (E) Schematic of how Wnt5A may be activating iASPP in metastatic melanoma cells. (F) Western blot analysis of iASPP expression in melanoma cells following Wnt5A KD. Also see Figure S3.
Aged microenvironment increases Wnt5A, p53, and a slow cycling phenotype.
p53 is responsible for cellular response to multiple types of stress, including oxidative stress, which we have previously shown to be increased in an aging microenvironment. The aging microenvironment promotes an increase in reactive oxygen species (ROS). Aged dermal fibroblasts secrete Wnt5A and sFRP2, which act to suppress b-catenin and ultimately, APE1, a key redox effector in melanoma. Inhibiting either Wnt5A or sFRP2 or treating with antioxidants overcomes resistance to targeted therapy (Kaur et al., 2016). We hypothesized therefore that p53 might play a role in this observed age-induced resistance to therapy, downstream of Wnt5A activation. To test this hypothesis, we treated human melanoma cells with conditioned media from aged (³ 55 years) and young (£ 35 years) dermal fibroblasts. We found that conditioned media from aged fibroblasts decreased proliferation (Fig. S4A), increased the percentage of cells exhibiting a slow-cycling phenotype (Fig. 4A), and increased the expression of Wnt5A, p53 and p21 (Fig. S4B). We further analyzed the effect of conditioned media on melanoma cells and found that conditioned media from aged fibroblasts increased iASPP expression (Fig. S4C and S4D). To determine if an aged microenvironment promotes Wnt5A and p53 expression in vivo, we examined transplantable Yumm1.7 (BRAFV600E/CDKN2A−/−/PTEN−/−/p53WT) tumors from young (<9 wks) and aged (>52 wks) C57BL/6 mice. We found that tumors in aged mice had increased Wnt5A and p53 expression compared to tumors in young mice (Figure 4B and Fig. S4E). When we analyzed these tumors by IHC, we found that more cells in tumors from aged mice expressed p53 (Fig. 4C and 4D). To confirm that an aged microenvironment in vivo promotes a slow cycling phenotype, we stained Yumm1.7 cells tagged with GFP, with the membrane dye PKH26, which is a red membrane dye that is diluted as cells proliferate. Cells that retain the dye over time are characterized as slow-cycling (Perego et al., 2018). We injected these labeled cells subcutaneously into young and aged C57/BL6 mice (Fig. 4E). After five weeks, we analyzed the tumors for GFP positivity (GFP+), which identifies the Yumm1.7 melanoma cells, and for PKH26 (PKH26+), which identifies the slow-cycling cells. We found that tumors in the aged mice had a higher percent of GFP+ melanoma cells that were also PKH26+ (Fig. 4F), and that the intensity of PKH26 staining was higher in tumors from aged mice (Fig. 4G). These data suggest that an aged microenvironment promotes a slower-cycling phenotype. We next analyzed lung tissue from these mice for GFP+ cells that were also PKH26+. We found that the lungs from aged mice had more GFP+ cells that were PKH26+ (Fig. 4H), suggesting that the slower cycling cells may be more invasive in aged mice. These data suggest that the aged microenvironment, which promotes invasion and therapy resistance (Kaur et al., 2016), also promotes p53 expression and a slow-cycling phenotype.
Figure 4. Aged microenvironment promotes a slow cycling phenotype.
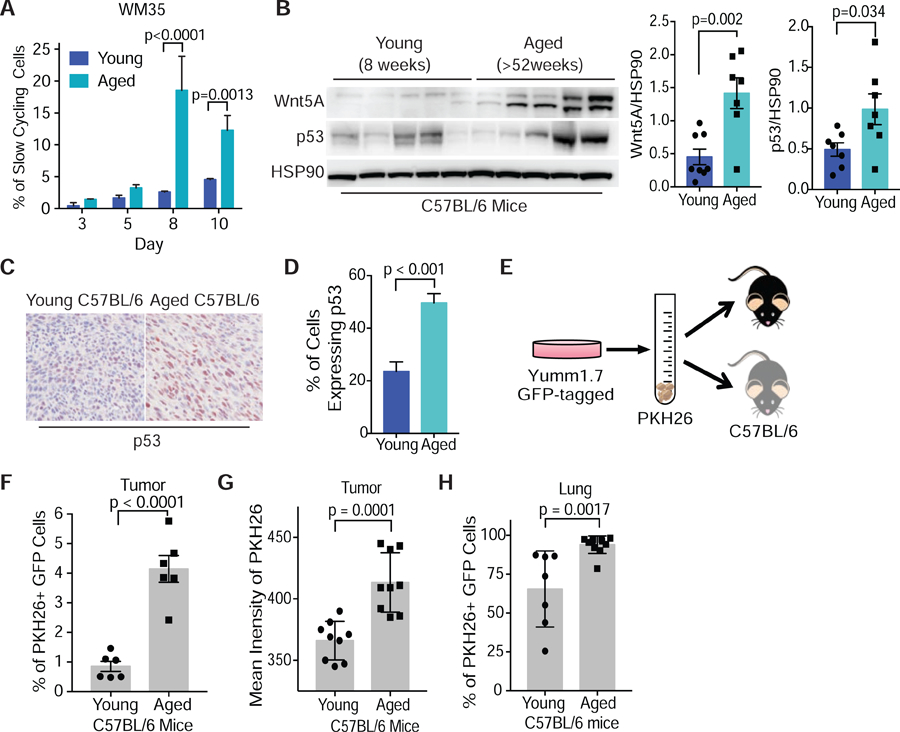
(A) Flow cytometry analysis of changes in slow-cycling populations following treatment of WM35 melanoma cells with conditioned media from young (25–35 yrs) and aged (55–65 yrs) fibroblasts (two-way ANOVA sidak’s multiple comparison test). (B) Western blot analysis of Wnt5A and p53 (also see Fig. S4E) expression in Yumm1.7 derived tumors grown in young (8 wks) and aged (>52 wks) C57BL/6 mice (unpaired two-tailed t-test). (C) IHC for p53 in mouse tumors from young and aged mice for p53. (D) Quantification of p53 positive cells using ImageJ in young and aged mouse tumors. (E) Yumm1.7 cells labelled with GFP were stained with a membrane dye, PKH26, and injected into young (8 wks) and aged (>52 wks) C57BL/6 mice (2.5x105cells/mouse). (F) At week 5 post injection the tumors were analyzed for GFP+ cells that were PHK26+ by flow cytometry. (G) Flow cytometry analysis of intensity of PKH26 in GFP+ cells in tumors from young and aged tumor bearing mice. (H) Analysis of lungs from young and aged tumor bearing mice for cells that are GFP+ and PKH26+. Data are represented as mean ± SEM. Also see Figure S4.
Inhibition of wild type p53 promotes sensitivity to targeted therapy.
By increasing p53 and iASPP concomitantly, Wnt5A is able to promote a slow-cycling phenotype, which has been shown previously to contribute to therapy resistance in melanoma cells (Ahn et al., 2017; O’Connell et al., 2013). Since we observed that the aged tumor microenvironment promotes p53 expression and we have previously shown that the aged microenvironment drives therapy resistance (Kaur et al., 2016), we asked if inhibiting p53 would promote sensitivity to therapies currently used in the clinic. We injected aged (>52 wks) C57BL/6 mice subcutaneously with Yumm1.7 mouse melanoma cells. Once the tumors were established (150 mm3), we separated the mice into treatment groups and started treatment with BRAF/MEKi chow (day 22). Twenty-four hours after mice were started on the combination therapy (day 23), we dosed a single intra-tumoral dose of 2 mg/kg of pifithrin-α, a p53 inhibitor that only affects cells that express p53 (Komarov et al., 1999). After 8 days of treatment (day 30 of the experiment), all the mice on the BRAF/MEKi treatment showed tumor regression (Fig. 5A). However, 13 days after starting BRAF/MEKi treatment (day 35), the tumors of aged mice that received only the BRAF/MEKi treatment began to relapse (Fig. 5A). Surprisingly, the aged mice that received a single dose of pifithrin-α remained sensitive to the BRAF/MEKi combination therapy (Fig. 5A). This suggests that a single-dose inhibition of p53 at the time of therapy is sufficient to sensitize human melanoma cells to BRAF/MEKi combination therapy presumably by driving them out of a slow-cycling phenotype such that they can be targeted by the BRAF/MEKi combination therapy. To confirm that pifithrin-α decreases p53 expression and inhibits the slow-cycling phenotype, we treated melanoma cells with pifithrin-α and analyzed p53 and p21 expression as well as changes in the number of slow-cycling cells. We found that pifithrin-α decreases both p53 and p21 expression (Fig 5B) as well as the number of slow-cycling cells (Fig 5C) in Wnt5A high melanoma cells. To further confirm that pifithrin-α blocks p53 expression following stress, melanoma cells were treated with doxorubicin in the presence and absence of pifithrin-α. Pifithrin-α inhibited the upregulation of p53 and p21 following DNA damage induced by doxorubicin (Fig. 5D). Melanoma cells were also treated with BRAF/MEKi therapy in combination with pifithrin-α. Pifithrin-α inhibits p53 expression following BRAF/MEKi treatment (Fig. S5A). Together, these data suggest that Wnt5A regulates p53 expression to promote a slow-cycling phenotype in order to survive multiple types of stress. These data suggest that inhibiting p53 at the early stages of BRAF/MEKi combination therapy may be an effective way of targeting slow-cycling populations, that are linked with therapy resistance.
Figure 5. p53 promotes therapy resistance.
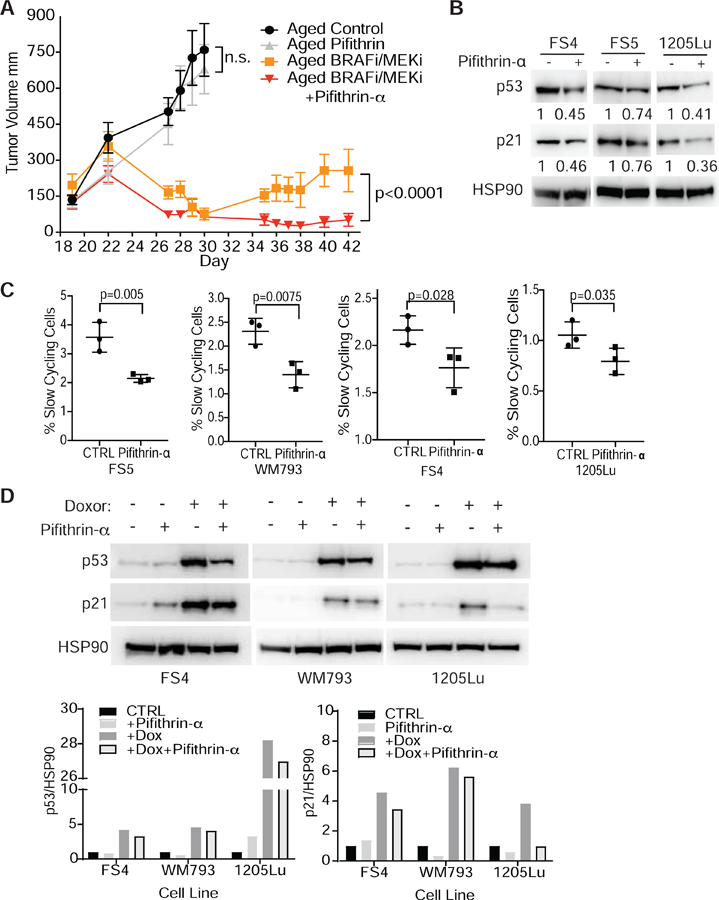
(A) Tumor volume of aged (>52 wks) C57BL/6 mice injected with Yumm1.7 cells labeled with mCherry. At day 22, mice were treated with +/− PLX4720 (200mg/kg)/PD0325901(7mg/kg) and on day 23 mice were treated +/− a single intra-tumoral dose of pifithrin-α (2mg/kg). (E) IHC of p53 expression in a patient tumor pre- and post-treatment with BRAF/MEKi (Trametanib/Debrafenib). (B) Western blot analysis of p53 and p21 expression in human melanoma cells treated with 5µM pifithrin-α. (C) FACS analysis of slow cycling populations in human melanoma cell populations day 5 following treatment with 5µM pifithrin-α (one-tailed unpaired t-test analysis) (also see Fig S5C). (D) Western of Wnt5A high melanoma cell lines treated with +/− 5 μM pifithrin-α +/− 1 μg/mL doxorubicin. Quantification of changes in p53 and p21 following treatment with doxorubicin and doxorubicin combined with pifithrin-α. Data are represented as mean ± SEM. Also see Figure S5.
Wild type p53 expression increases following therapy.
Inhibiting p53 and slow cycling cells early during treatment prolonged sensitivity to BRAF/MEKi therapy. To further evaluate the role of wild type p53 in therapy response we knocked down p53 in Yumm1.7 mouse cells using shRNA (Fig. 6A) (Dickins et al., 2005), and injected these cells intradermally into aged C57/BL6 mice. Once the tumors were established, the mice were given either control or BRAF/MEKi chow. Surprisingly around day 30 of treatment both the empty vector control and p53 KD tumors started to become resistant to therapy (Fig. 6B). To determine if p53 was still knocked down in the tumors, we analyzed the tumors by IHC for p53 expression. We found that the p53 KD tumors treated with BRAF/MEKi expressed high levels of p53 (Fig. 6C, Fig. S6A). This suggested that cells retaining WT p53 may have been selected for during therapy. To determine if p53 is increased following BRAF/MEKi therapy in other models, we analyzed PDX tumors with wild type p53 following BRAF/MEKi combination or BRAFi therapy (Krepler et al., 2017). We observed an increase in p53 and p21 positive cells following both BRAF/MEKi combination and BRAFi therapy (Fig. 6D–E, Fig S6B). When we analyzed these tumors for Wnt5A expression, we found that PDX tumors (supplemental table 1) treated with either BRAF/MEKi combination (Fig. S6C) or BRAFi (Fig. S6D) also had increased Wnt5A expression following treatment, further linking Wnt5A and p53 with resistance to BRAF/MEKi therapy. We then asked if p53 expressing cells are increased in patients following therapy. We were able to obtain and analyzed three sets of patient tumors pre- and post- BRAF/MEKi combination therapy. In this small sample set, we found that the patient that had p53 positive cells prior to treatment showed an increase in the number of p53 positive cells following therapy (Fig. 6F). These data suggest that melanoma cells expressing wild type p53 exhibit a survival advantage, and that inhibiting p53 may promote sensitivity to therapy.
Figure 6. Wild type p53 is increased following therapy.
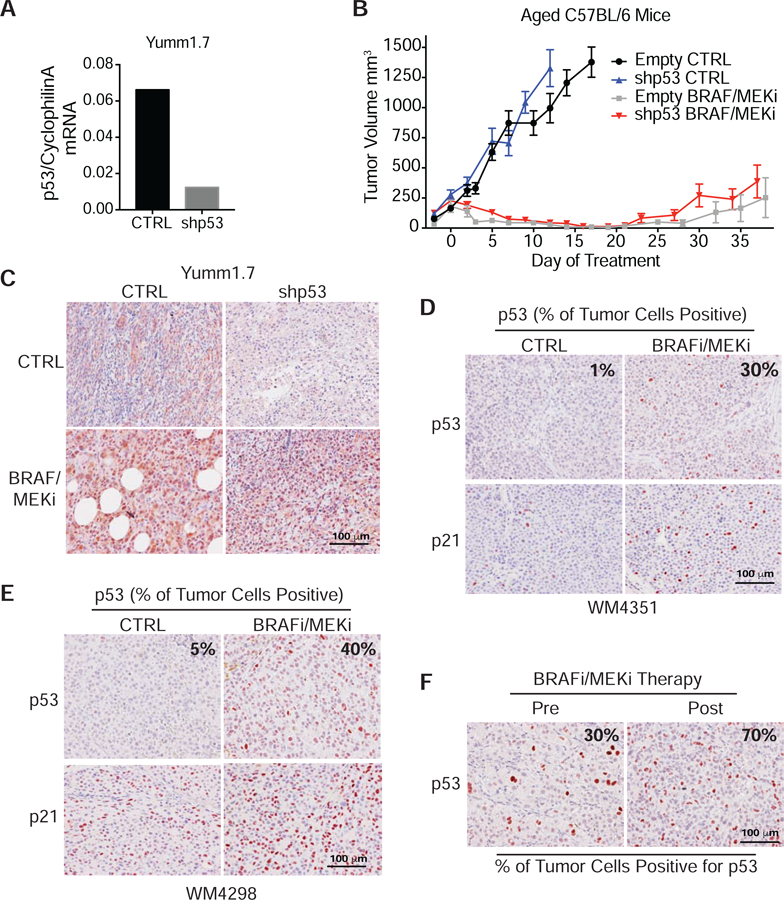
(A) p53 was knocked down in Yumm1.7 cells using shRNA. Cyclophilin A used as a loading control. (B) Tumor volume of aged (>52 wks) C57BL/6 mice injected with Yumm1.7 shp53 KD and control cells. Mice were treated with +/− PLX4720 (200mg/kg) /PD0325901(7mg/kg). Data are represented as mean ± SEM. (C) IHC for p53 expression in p53 KD and control tumors from mice treated with +/− BRAF/MEKi (PLX4720/PD0325901). IHC for p53 and p21 expression in WT p53 PDX tumors WM4351 (D) and WM4298 (E) from mice treated with BRAF/MEKi therapy compared to control. (F) IHC for p53 in patient tumor before and after BRAF/MEKi therapy. Also see Figure S6 and Table S1.
Discussion
In this study, we show for the first time that the non-canonical Wnt signaling molecule Wnt5A can stabilize the p53 protein, affecting its half-life in the cell. This results in the ability of Wnt5A to drive a slow-cycling phenotype by utilizing wild type p53 in melanoma. Induction of this slow-cycling phenotype may promote tumor cell heterogeneity, plasticity, and survival following multiple types of stress. Our results point to the potential for a novel Wnt-p53-Wnt feedback loop, as p53 has been shown to suppress canonical Wnt signaling in numerous studies, by promoting its degradation (Levina et al., 2004; Liu et al., 2001; Sadot et al., 2001). Canonical Wnt signaling, via β-catenin, has been shown in melanoma to drive a highly proliferative but less invasive phenotype, and has also been shown to sensitize melanoma cells to BRAF inhibitors (O’Connell et al., 2013). We and others have shown that Wnt5A signals to degrade β-catenin, via SIAH2 (O’Connell et al., 2013; Topol et al., 2003). SIAH2 is a target of p53 (Chan et al., 2011; Grishina et al., 2012) and our finding that Wnt5A stabilizes p53 points to one more way in which Wnt5A can signal to degrade β-catenin. This switch from a highly proliferative to a highly invasive state is known as a “phenotype switch” and Wnt signaling plays a critical role in driving the switch (Eichhoff et al., 2011; O’Connell et al., 2013).
Given the fact that p53 is mutated in a large percentage of many cancers, resulting in a loss of its activity, strategies to date have focused largely on the development of p53 activators. In melanoma, WTP53 is present in over 80% of melanomas, but it is largely inactivated. In NRAS mutant melanoma with wild type p53, upregulation of MITF leads to an increase in Bcl-2, which blocks p53 induced apoptosis (Najem et al., 2017). However, we have shown that Wnt5A decreases MITF expression (Dissanayake et al., 2008), and here we show Wnt5A promotes p53 and p21 expression. p53 appears to be functional in our Wnt5A high cells, as observed by an increase in p21 expression and cycle arrest following DNA damage (data not shown). Previously, resistance to radiotherapy and DNA damaging agents in melanoma cells with wild type p53 has been linked with inactivation of DNA damage signaling pathways; however, this correlated with an absence of cell cycle arrest (Satyamoorthy et al., 2000). Loss of cell cycle regulation can occur with mutations on both p53 alleles. Cells retaining one WT p53 allele have been shown to respond to drug by undergoing growth arrest (Livingstone et al., 1992). Perez et al. has shown using colon cancer cells that a WT p53 allele is often silenced via copy-neutral mechanisms in the presence of p53 mutation on the second allele. In our melanoma cell lines with high Wnt5A, p53 and p21 are functional, which may suggest that both alleles need to be wild type for our observed phenotype (Perez et al., 2016).
Survival of Wnt5A high melanoma cells appears to be due in part to the nuclear expression of iASPP, which inhibits the ability of WT p53 to perform its pro-apoptotic functions (Lu et al., 2016). We show however, that p53 is still able to regulate and slow down the cell cycle, which has the unfortunate effect of driving cells into a state where they can resist therapy. Resistance to therapy has also been linked with WT p53 in breast cancer. In breast cancer, WT p53 promotes cell cycle arrest and senescence in response to chemotherapy instead of the desired cell death. WT p53 also correlates with poor response and overall survival following chemotherapy in breast cancer patients (Jackson et al., 2012; Ungerleider et al., 2018)). Where wild type p53 results in arrest or senescence following chemotherapy instead of the desired cell death, inhibiting p53 in combination with chemotherapy may increase response and survival following therapy. Treating tumors with a p53 inhibitor during the early stage of BRAF/MEK inhibitor treatment, promoted sensitivity to targeted therapy.
It is interesting to note that resistance to BRAF/ MEK inhibitors is not always due to mutations in the signaling pathway. Instead, adaptive resistance, which may encompass a variety of changes from MITF alterations (Najem et al., 2017; Smith et al., 2016), to expression of NGFR (Fallahi-Sichani et al., 2017), and phenotype switching (Ramsdale et al., 2015; Su et al., 2017) appears predominant in the generation of therapy resistant populations of melanoma. It has been shown through single cell sequencing that melanoma cells have a profound transcriptional variability, that is semi-coordinated, and results in an epigenetic reprogramming that leads to therapy resistance (Rambow et al., 2018). In addition, we have shown that secreted microenvironmental factors can also promote resistance to targeted therapy. As described above, Wnt5A is an architect of the phenotype switch to an invasive, but slow-cycling state, and it is induced by stress. We have shown that serum starvation, aging, and treatment of melanoma cells with irradiation or targeted therapy can increase Wnt5A expression (Behera et al., 2017; Kaur et al., 2016; Webster et al., 2015b). Similar changes in the microenvironment also increase wild type p53 expression, which we show drives a slow-cycling phenotype. Slow-cycling populations are increased during continuous drug treatment resulting in a subpopulation of cells with stem-like behavior that maintain phenotypic plasticity. It was recently shown that BRAF/MEK combination therapy enriches multiple therapy resistant populations including a neural crest stem cell (NCSC) phenotype (NGFR+, MITFlow), characterized by NGFR expression and driven by RXRG signaling, which retains the ability to proliferate in the absence of therapeutic stress (Rambow et al., 2018). Here we show that NGFR, which is increased in the previously described NCSC phenotype (NGFR+, MITFlow) population, is also increased in Wnt5A and p53 expressing cells. In breast cancer, expression of wild type p53 induces senescence and drives resistance to chemotherapy (Jackson et al., 2012). In multiple types of cancer, pathways that drive metastasis have been linked with therapy resistance and a stem cell like phenotype. We propose that targeting these slow-cycling cells by inhibiting p53 may help to overcome therapy resistance in melanoma.
Our data suggest, that at least in melanoma, strategies designed to activate p53 may lead to therapy resistance. As a therapeutic strategy, inhibiting WT p53 has its obvious drawbacks, as normal cells could easily be affected. However, in the absence of stress most normal cells have lower p53 expression (Rogel et al., 1985), which is temporal and we show that improved response to therapy can be achieved by a single dose of p53 inhibitor, such that unwanted consequences of sustained p53 inhibition can be avoided.
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Marie Webster (websterm@mlhs.org). All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement. Human shRNA (TRCN0000062716, TRCN0000062717, TRCN0000063755) used in these studies were obtained under MTA from the Broad Institute. More information about these shRNA can be found at https://www.broadinstitute.org/rnai-consortium/rnai-consortium-shrna-library
EXPERIMENTAL MODELS AND SUBJECT DETAIL
In vivo animal studies.
All mouse studies were conducted using male C57BL6 mice. The young mice were 6–8 wks and the aged mice were 43–52 weeks (Charles River, Taconic). All animal experiments were approved by the Institutional Animal Care and Use Committee (protocols 112503 and 112510). The animals were housed in a facility accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care. Aged mice were singly housed and young mice were housed in groups of four or five.
Cell lines.
FS cell lines were maintained in RPMI 1640 (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum, 100 U/ml penicillin and streptomycin and 4 mM L-glutamine. WM793, 1205Lu, and WM164 were maintained in MCDB153 (Sigma, St Louis, MO)/ L-15 (Cellgro, Manassas, VA) (4:1 ratio) supplemented with 2% FBS and 1.6 mM CaCl2 (Tu2% media). Yumm1.7 cells were maintained in DMEM/F12 with 10% fetal bovine serum, and 100 U/ml penicillin and streptomycin (Meeth et al., 2016). All cells were cultured at 37°C in 5% CO2. Cell culture supernatants were mycoplasma tested using a Lonza MycoAlert assay at the University of Pennsylvania Cell Center Services.
Treatments.
Cells were treated with 200 ng/mL of recombinant Wnt5A (R&D Systems: cat. No. 645WN010CF) every 48 hours. Doxorubicin was used at a final concentration of 1 μg/mL and pifithrin-α (Selleckchem, cat. No S2929) was used at a final concentration of 5 μM for 72 hours unless otherwise stated. Cells were treated with 50 μg/mL of cycloheximide for 2, 4, 6, and 8 hrs. Conditioned media was prepared as previously described (ref) Treatment chow for the in vivo experiments was prepared by Research Diets (NJ). Control chow (AIN-76, research diets) and treatment chow (AIN-76, research diets) containing 200mg/kg PLX4720 (SelleckChem cat. no. S1152) and 7 mg/kg PD0325901(Selleckchem, cat no. S1036) was given to young (8 wks) or aged (> 43 wks) C57BL6 mice starting at day 22.
METHOD DETAILS
Analysis of slow cycling cells by flow cytometry.
For in vitro studies, cells were stained using the CellTrace Violet cell proliferation kit (cat C34557) following the manufacturer’s protocol. Briefly, 1x106 cells were stained using 5μM CellTrace Violet in 1% FCS in PBS for 40 min at 37°C. The cells were then washed two times in PBS, an aliquot was removed for day zero analysis, and the cells were plated into six well plates. On Day 3, 5, 6 or 8, media was removed, cells were washed with PBS, and 200 uL TrypLE was added to each well. The plate was incubated at 37°C 5% CO2 for 5 min. The TrypLE was quenched with equal volume of media and the cells were transferred to FACS tubes for analysis. For Western blot and qPCR analysis, slow-cycling and cycling cells were sorted using MoFlowAstriosEQ. For in vivo studies, Yumm1.7-GFP cells were stained using PKH26 (cat PKH26GL, Sigma) following the manufacturer’s protocol. Fluorescence dilution was measured as indicated by flow cytometry (LSR14, Becton Dickinson, Franklin Lakes, NJ, USA) and analyzed using FlowJo Software v10.2 (FlowJo, LLC, Ashland, OR, USA). Stained cells were normalized to unstained controls at each time point.
siRNA Transfection.
Cells were transfected with high performance (HP)-validated negative control (CTRL) (AllStars negative control, Qiagen cat. SI03650318), and Wnt5A siRNA (Qiagen, Venlo, the Netherlands) using Lipofectamine (Invitrogen) for 48–72h as previously described (9).
Cell Sorting.
Cells were prepared for slow cycling analysis as described above. Cells were resuspended at a concentration of ~ 1.0 x 106 cells/500 μL 1% FBS/RPMI. Slow-cycling and cycling cells were then sorted on a BD ARIAII.
Western Blot Analysis
Western blotting was performed as previously described (9). Briefly, total protein lysate of 50 μg was run on 4% to 12% NuPAGE Bis-Tris gels (Invitrogen, cat. No. NW041222BOX), transferred onto PVDF membrane using an iBlot 2 transfer system (Life Technologies). The PVDF membrane was then blocked in either 5% milk or BSA in TBST. All primary antibodies were diluted in 5% milk or BSA/TBST at 4°C overnight. The membranes were washed 4x 10 min with TBST and probed with the corresponding horseradish peroxidase (HRP)-conjugated secondary antibody (0.2 – 0.02 μg/mL anti-mouse, anti-mouse, or streptavidin). Proteins were visualized using Luminata Crescendo (Millipore, WBLUR0100) on an ImageQuant 4000 scanner and quantified using ImageJ (RRID: SCR_003070).
Nuclear and cytoplasmic extraction.
Cells were assayed according to the manufacturer’s protocol for nuclear and cytoplasmic extraction (NE-PER, Thermo Fisher Scientific, Product #78833). Nuclear and cytoplasmic lysates were stored at −80°C and analyzed by western blot as described above.
Immunohistochemistry.
Mouse tumors were paraffin embedded and sectioned. Paraffin embedded sections were rehydrated through a xylene and alcohol series, rinsed in H2O and washed in PBS. The slides were then fixed with 10% formalin for 10 min and washed in PBS. Antigen retrieval was performed using target retrieval buffer (Vector Labs, Burlingame, CA) and steamed for 20 min. Samples were then blocked in a peroxidase blocking buffer (Thermo Scientific) for 15 min, followed by Protein block (Thermo Scientific) for 5 min, and incubated in appropriate primary antibody diluted in antibody diluent (S0809, Dako) at 4°C overnight in a humidified chamber. Following washing in PBS, samples were incubated in biotinylated anti-rabbit or polyvalent secondary antibody (Thermo Scientific) followed by streptavidin-HRP solution at room temperature for 20 min. Samples were then washed in PBS and incubated in 3-Amino-9-Ethyl-l-Carboazole (AEC) chromogen for up to 10 min. Finally, samples were washed in H2O, incubated in Mayers hematoxylin for 1 min, rinsed in cold H2O, and mounted in Aquamount.
Lentiviral infection.
All shRNA was obtained from the TRC shRNA library available through the Molecular Screening Facility at The Wistar Institute. Lentiviral production was carried out as described in the protocol developed by the TRC library (Broad Institute). Briefly, 293T cells were co-transfected with Wnt5A (TRCN0000062716, TRCN0000062717) or p53 (TRCN0000063755) shRNA vector and lentiviral packaging plasmids (pCMV-dR8.74psPAX2, pMD2.G). The supernatant containing virus was harvested at 36 and 60 hours, combined and filtered through a 0.45µm filter. For transduction, the cells were layered overnight with lentivirus containing 8 µg/ml polybrene. The cells were allowed to recover for 48 hours and then selected using 1µg/ml puromycin.
In vivo assays.
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) and conducted at the Wistar Institute, an Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC) accredited facility. Yumm1.7 mCherry or GFP cells were injected subcutaneously into 8 weeks old or >43 weeks old C57/Bl6 mice (Charles River). Tumors were measured using digital calipers and tumor volumes were calculated using the formula 0.5 x (length x width)2. For slow-cycling experiments, mice were euthanized at week 5 and tumors and lungs were harvested for further analysis. For therapy resistant experiments, once tumors reached ~250 mm3 mice were randomized into treatment groups. BRAF/MEKi chow or Control chow was given to the mice on day 22. Treatment chow for the in vivo experiments was prepared by Research Diets (NJ). A single dose of 2 mg/kg pifithrin-α (2.5% DMSO in PBS) or PBS control was injected intratumorally on day 23. Initial toxicity was observed in two out of ten aged mice, within 24 hours of injection of pifithrin-α. Over the course of the experiment no other toxicity was observed in the group treated with BRAF/MEKi combination and pifithrin-α and across all treatment groups (Fig. S5B), however more studies need to be performed in order to determine potential toxicity of pifithrin-α. When the young control tumors reached 1250 mm3 all the mice from the control groups were euthanized, the mice in the treatment groups were euthanized at day 44, and the tumors and lungs were harvested for further analysis.
Quantitative real-time PCR.
RNA was extracted using Trizol (Invitrogen, cat 15596018), chloroform and RNeasy Mini kit (Qiagen, cat 74106) as previously (9). cDNA was prepared using cDNA synthesis kit (Bio-Rad, cat. No. 1708891). Gene expression was quantified using SYBR green method of qPCR on an ABI StepOnePlus sequence detection system using fast conditions. Samples were normalized against the 18s gene using Universal 18s primers (ThermoFisher, AM1718). Expression was calculated using the standard curve method according to the manufacturer’s protocol (Perkin Elmer, Waltham, MA, USA). Primers used were: WNT5A forward, 5′ ATT CTT GGT GGT CGC TAG GTA 3′; WNT5A reverse, 5′ CGC CTT CTC CGA TGT ACT GC 3′; TP53 forward, 5’ GTG GAA GGA AAT TTG CGT GT3’; TP53 reverse, 5’ CCA GTG TGA TGA TGG TGA GG3’; KDM5B forward1, 5’ CCATAGCCGAGCAGACTGG3’; KDM5B reverse1, 5’ GGA TAC GTG GCG TAA AAT GAA GT3’; KDM5B forward2, 5’ CAA TGC TGT GGA CCT GTA TGT; KDM5B reverse2, 5’ TAC GGA GGG TAT AGT CCC TGG3’; PUMA forward, 5’ GTC CCC TGC CAG ATT TGT G3’; PUMA reverse, 5’AGA GGC CGC AGG ACA CTG3’; TP53 F mouse AAG ATC CGC GGG CGT AA3’; TP53 R mouse 5’ CAT CCT TTA ACT CTA AGG CCT CAT TC3’; Cyclophilin A F 3’ GGG TTC CTC CTT TCA CAG AA5’; Cyclophilin A R 3’ GAT GCC AGG ACC TGT ATG CT5’.
Cell cycle analysis.
Cell distribution was measuring by flow cytometry as previously described (9). Briefly, slow-cycling and cycling populations were sorted as described above. Once the slow-cycling and cycling populations were isolated, the cells were centrifuged at 1,000 rpm for 3 min. The cells were then fixed using a permeabilization kit (Biolegend, cat No. 424401), stained with propidium iodide as previously described and analyzed by flow cytometry (BD LSRII 14 color).
Protein half-life determination.
Cells were plated in a 6 well plate and 16 hours later cells were treated with cycloheximide. The protein was collected as stated above for Western blot analysis. The density of the bands for p53 and HSP90 at each time point were quantified using ImageJ. The band density for p53 was normalized to HSP90 at each time point and time zero was normalized to one. These values were then graphed versus time and analyzed for linear regression. Using the linear equation, the time (x) was calculated when y = 0.5.
Proximity ligation assay.
To detect interaction or association of proteins by PLA, Duolink in situ starter kit (Sigma, DUO92101) was used, and the manufacturer’s protocol was followed exactly as described to perform these studies. PLA signals were quantified using ImageJ software.
Immunoprecipitation.
Cells were grown on 100 mm dishes. Cells were removed using a cell scraper with media still on the cells. The cells/media was transferred to a 50 mL conical and store on ice. The cells were centrifuged at 2000 rpm for 5 min, supernatant was removed, and washed with PBS. Cells were resuspended in 200 μL of NP-40 lysis buffer, sonicated for 10 sec and incubated on ice for 20 min. The resulting lysate was centrifuged at 13,000 rpm for 10 min. Supernatant was transferred to a new tube and protein was quantified using Qubit protein assay kit (ThermoFisher, cat Q33211). A total of 1000 μg of whole-cell lysate was precleared by Protein G Agarose (16–266, 50% slurry Millipore) and incubated with 2 μg of antibody (OP43A Anti-p53 (Ab-6) (Pantropic) Mouse mAb (DO-1) Agarose Conjugate) or IgG control for 30 min at 4°C. Proteins were eluted using 2X Laemmli sample buffer (62.5 mmol/L Tris-HCL at pH6.8, 25% glycerol, 2% SDS, 0.01% bromophenol blue) and subjected to Western blot analysis as described earlier.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data from the Cancer Cell Encyclopedia was downloaded from cbioportal. Expression for all genes were tested for significant association with p53 and Wnt5A using linear multivariate linear regression with p53 and Wnt5A as regression factors. Nominal pvalues were corrected for multiple testing using Storey et al. (11) method and results that had both positive or both negative regression coefficients that passed FDR<15% threshold were considered significant. Significant genes were analyzed for enrichments with Ingenuity Pathway Analysis using “Diseases & Functions” and “Upstream Analysis” options. Only Results with p<0.005 (p<0.0005 for regulators) with at least 10 genes and predicted activation z-score of at least 1.2 were considered. Additional filtering for functions to remove cell line and tissue specific results was applied and functions were grouped by categories and best p-value and Z-score was reported along with individual function results. For tumor microarray samples, Fisher’s exact test was used to determine the association between age group and the level of p53. For in vitro studies where two groups were compared, a Student t-test was used for two-group comparison. ANOVA or Holm-Sidak’s adjusted P values was used for multiple comparisons. For in vivo studies of tumor volume, ANOVA was used to determine significance. Data was represented as ± SEM.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Wnt5A | R&D systems | Cat # BAF645 |
| Jarid1B | Novus Biologicals | Cat # NB100-97821 |
| p21 (human) | Cell Signaling | Cat # 2947S |
| p53 Ab-6 (human) | Millipore | Cat # OP43 |
| β-tubulin | Cell Signaling | Cat # 2146S |
| HSP90 | Cell Signaling | Cat # 4877S |
| MDM2 | Calbiochem | Cat # OP115-100UG |
| MDM4(X) | Abcam | ab49993 |
| iASPP | Abcam | ab34898 |
| GAPDH (14C10) | Cell Signaling | Cat # 2118S |
| p-MDM2ser395 | Abgent | AP3579a |
| Histone H3 | Cell Signaling | 9715 |
| p53 (mouse) | Abcam | Ab31333 |
| Phospho-PKC α/β II (Thr638/641) | Cell Signaling | 9375 |
| p21 (mouse) [EPR18021] | Abcam | Ab188224 |
| Bacterial and Virus Strains | ||
| JM109 High Efficiency Competent Cells | Promega Corp. | Cat# L2001 |
| Biological Samples | ||
| Patient-derived xenografts (PDX) PFFE blocks | Herlyn Lab at Wistar | WM4351, WM4298, WM4070 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| rWnt5A | R&D | 645-WN-010 |
| PLX4720 | SelleckChem | S1152 |
| PD0325901 | SelleckChem | S1036 |
| G06983 | Sigma | G1918-500UG |
| AIN-76A Rodent Diet | Research Diets | D10001i |
| Critical Commercial Assays | ||
| Qubit protein assay kit | ThermoFisher | cat Q33211 |
| Duolink in situ starter kit | Sigma | DUO92101 |
| Transcription Factor Fixation/Permeabilization Buffer Set | Biolegend | 424401 |
| cDNA synthesis kit | Bio-Rad | Cat. 1708891 |
| nuclear and cytoplasmic extraction NE-PER | Fisher | Product # 78833 |
| Experimental Models: Cell Lines | ||
| FS4 | Webster et al. 2015 | N/A |
| FS5 | Webster et al. 2015 | N/A |
| FS13 | Webster et al. 2015 | N/A |
| FS14 | Webster et al. 2015 | N/A |
| Yumm1.7 | Meeth et al., 2016 | N/A |
| Yumm1.7 mCherry | Kaur et al., 2016 | N/A |
| Yumm1.7 EmGFP | This paper | N/A |
| Yumm.17 shp53_E | This paper | N/A |
| 1205Lu, WM164 | Rockland Immunochemicals | |
| WM793 | Herlyn Lab, Wistar Institute | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL6 young male mice (6–8 weeks) | Charles River | 556 |
| C57BL6 young male mice (6–8 weeks) | Taconic | B6-M |
| C57BL6 aged mice (43–52 weeks) retired male breeders | Charles River | 24107819 |
| C57BL6 aged mice (43–52 weeks) retired male breeders | Taconic | B6-RB-M |
| Oligonucleotides | ||
| Primers for Wnt5A, TP53, KDM5B, PUMA, Cyclophilin A, see Table S2 | This paper | NA |
| Quantum RNA Universal 18S Internal Standard | ThermoFisher | Cat# AM1718 |
| Recombinant DNA | ||
| pLU-EF1-MCS-mCherry | This paper | |
| pLV[Exp]-Puro-EF1A>EmGFP | Vector Labs | Cat# VB171103-1159dvu |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Graphpad Prism 7 | https://www.graphpad.com/data-analysis-resource-center/#guides | https://www.graphpad.com/scientific-software/prism/ |
Highlights.
Wnt5A increases the half-life of wild type p53 to promote a slow cycling phenotype.
Multiple types of stress increase Wnt5A and p53 expression in metastatic melanoma.
Inhibiting p53 sensitizes melanoma cells to BRAF/MEK inhibitor therapy.
Acknowledgments:
We thank the outstanding Core Facilities of the Wistar Institute, supported by P30CA010815. We thank F. Chen, F. Keeney, and J. Faust. Funding: A.T.W, A.K., F.A., and G.A., are supported by R01CA174746, and A.T.W., M.F. and S.M.D are supported by R01CA207935. A.N., Q.L., W.X., C.Z., L.S., X.X., G.K., R.K.A., T.M. and A.T.W. are supported by P01 CA114046. A.T.W., R.K.A., L.M.S., and M.H. are also supported by P50 CA174523. M.R.W. is supported by K99 CA208012-01. M.H., L.M.S., G.C.K., T.C.M., X.X. are supported by the Tara Miller Foundation. M.E.M is supported by R01 CA102184. M.E.M. and A.T.W are supported by P01 CA114046. A.T.W. is also supported by a Melanoma Research Alliance/ L’Oréal Paris-USA Women in Science Team Science Award, an Established Investigator Award from the Melanoma Research Foundation, P30 CA00697356, and R01CA207935. Core facilities used in this grant are supported by P30CA010815.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests: The authors declare no competing interests.
DATA AND CODE AVAILABILITY
All data is available in the main text or the supplementary materials.
References and Notes:
- Ahmed F, and Haass NK (2018). Microenvironment-Driven Dynamic Heterogeneity and Phenotypic Plasticity as a Mechanism of Melanoma Therapy Resistance. Front Oncol 8, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn A, Chatterjee A, and Eccles MR (2017). The Slow Cycling Phenotype: A Growing Problem for Treatment Resistance in Melanoma. Mol Cancer Ther 16, 1002–1009. [DOI] [PubMed] [Google Scholar]
- Arozarena I, and Wellbrock C (2017). Targeting invasive properties of melanoma cells. FEBS J 284, 2148–2162. [DOI] [PubMed] [Google Scholar]
- Asiedu MK, Beauchamp-Perez FD, Ingle JN, Behrens MD, Radisky DC, and Knutson KL (2014). AXL induces epithelial-to-mesenchymal transition and regulates the function of breast cancer stem cells. Oncogene 33, 1316–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D,. et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behera R, Kaur A, Webster MR, Kim S, Ndoye A, Kugel CH, Alicea GM, Wang J, Ghosh K, Cheng P, et al. (2017). Inhibition of Age-Related Therapy Resistance in Melanoma by Rosiglitazone-Mediated Induction of Klotho. Clinical Cancer Research 23, 3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan P, Möller A, Liu MC, Sceneay JE, Wong CS, Waddell N, Huang KT, Dobrovic A, Millar EK, O’Toole SA, et al. (2011). The expression of the ubiquitin ligase SIAH2 (seven in absentia homolog 2) is mediated through gene copy number in breast cancer and is associated with a basal-like phenotype and p53 expression. Breast Cancer Res 13, R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickins RA, Hemann MT, Zilfou JT, Simpson DR, Ibarra I, Hannon GJ, and Lowe SW (2005). Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat Genet 37, 1289–1295. [DOI] [PubMed] [Google Scholar]
- Dissanayake SK, Olkhanud PB, O’Connell MP, Carter A, French AD, Camilli TC, Emeche CD, Hewitt KJ, Rosenthal DT, Leotlela PD, et al. (2008). Wnt5A regulates expression of tumor-associated antigens in melanoma via changes in signal transducers and activators of transcription 3 phosphorylation. Cancer Res 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dissanayake SK, Wade M, Johnson CE, O’Connell MP, Leotlela PD, French AD, Shah KV, Hewitt KJ, Rosenthal DT, Indig FE, et al. (2007). The Wnt5A/protein kinase C pathway mediates motility in melanoma cells via the inhibition of metastasis suppressors and initiation of an epithelial to mesenchymal transition. J Biol Chem 282, 17259–17271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dissanayake SK, and Weeraratna AT (2008). Detecting PKC phosphorylation as part of the Wnt/calcium pathway in cutaneous melanoma. Methods Mol Biol 468, 157–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichhoff OM, Weeraratna A, Zipser MC, Denat L, Widmer DS, Xu M, Kriegl L, Kirchner T, Larue L, Dummer R, et al. (2011). Differential LEF1 and TCF4 expression is involved in melanoma cell phenotype switching. Pigment Cell Melanoma Res 24, 631–642. [DOI] [PubMed] [Google Scholar]
- Fallahi-Sichani M, Becker V, Izar B, Baker GJ, Lin JR, Boswell SA, Shah P, Rotem A, Garraway LA, and Sorger PK (2017). Adaptive resistance of melanoma cells to RAF inhibition via reversible induction of a slowly dividing de-differentiated state. Mol Syst Biol 13, 905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajjar M, Candeias MM, Malbert-Colas L, Mazars A, Fujita J, Olivares-Illana V, and Fåhraeus R (2012). The p53 mRNA-Mdm2 interaction controls Mdm2 nuclear trafficking and is required for p53 activation following DNA damage. Cancer Cell 21, 25–35. [DOI] [PubMed] [Google Scholar]
- Garman B, Anastopoulos IN, Krepler C, Brafford P, Sproesser K, Jiang Y, Wubbenhorst B, Amaravadi R, Bennett J, Beqiri M, et al. (2017). Genetic and Genomic Characterization of 462 Melanoma Patient-Derived Xenografts, Tumor Biopsies, and Cell Lines. Cell Rep 21, 1936–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishina I, Debus K, García-Limones C, Schneider C, Shresta A, García C, Calzado MA, and Schmitz ML (2012). SIAH-mediated ubiquitination and degradation of acetyl-transferases regulate the p53 response and protein acetylation. Biochim Biophys Acta 1823, 2287–2296. [DOI] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, et al. (2012). A landscape of driver mutations in melanoma. Cell 150, 251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Feng Z, and Levine AJ (2012). The Regulation of Multiple p53 Stress Responses is Mediated through MDM2. Genes Cancer 3, 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson JG, Pant V, Li Q, Chang LL, Quintas-Cardama A, Garza D, Tavana O, Yang P, Manshouri T, Li Y, et al. (2012). p53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell 21, 793–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur A, Webster MR, Marchbank K, Behera R, Ndoye A, Kugel CH, Dang VM, Appleton J, O’Connell MP, Cheng P, et al. (2016). sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature 532, 250–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, and Gudkov AV (1999). A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 285, 1733–1737. [DOI] [PubMed] [Google Scholar]
- Krepler C, Sproesser K, Brafford P, Beqiri M, Garman B, Xiao M, Shannan B, Watters A, Perego M, Zhang G, et al. (2017). A Comprehensive Patient-Derived Xenograft Collection Representing the Heterogeneity of Melanoma. Cell Rep 21, 1953–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levina E, Oren M, and Ben-Ze’ev A (2004). Downregulation of beta-catenin by p53 involves changes in the rate of beta-catenin phosphorylation and Axin dynamics. Oncogene 23, 4444–4453. [DOI] [PubMed] [Google Scholar]
- Liu J, Stevens J, Rote CA, Yost HJ, Hu Y, Neufeld KL, White RL, and Matsunami N (2001). Siah-1 mediates a novel beta-catenin degradation pathway linking p53 to the adenomatous polyposis coli protein. Mol Cell 7, 927–936. [DOI] [PubMed] [Google Scholar]
- Livingstone LR, White A, Sprouse J, Livanos E, Jacks T, and Tlsty TD (1992). Altered cell cycle arrest and gene amplification potential accompany loss of wild-type p53. Cell 70, 923–935. [DOI] [PubMed] [Google Scholar]
- Lu M, Breyssens H, Salter V, Zhong S, Hu Y, Baer C, Ratnayaka I, Sullivan A, Brown NR, Endicott J, et al. (2016). Restoring p53 Function in Human Melanoma Cells by Inhibiting MDM2 and Cyclin B1/CDK1-Phosphorylated Nuclear iASPP. Cancer Cell 30, 822–823. [DOI] [PubMed] [Google Scholar]
- Maya R, Balass M, Kim ST, Shkedy D, Leal JF, Shifman O, Moas M, Buschmann T, Ronai Z, Shiloh Y, et al. (2001). ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev 15, 1067–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeth K, Wang JX, Micevic G, Damsky W, and Bosenberg MW (2016). The YUMM lines: a series of congenic mouse melanoma cell lines with defined genetic alterations. Pigment Cell Melanoma Res 29, 590–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller J, Krijgsman O, Tsoi J, Robert L, Hugo W, Song C, Kong X, Possik PA, Cornelissen-Steijger PD, Geukes Foppen MH, et al. (2014). Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat Commun 5, 5712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najem A, Krayem M, Salès F, Hussein N, Badran B, Robert C, Awada A, Journe F, and Ghanem GE (2017). P53 and MITF/Bcl-2 identified as key pathways in the acquired resistance of NRAS-mutant melanoma to MEK inhibition. Eur J Cancer 83, 154–165. [DOI] [PubMed] [Google Scholar]
- O’Connell MP, Marchbank K, Webster MR, Valiga AA, Kaur A, Vultur A, Li L, Herlyn M, Villanueva J, Liu Q, et al. (2013). Hypoxia induces phenotypic plasticity and therapy resistance in melanoma via the tyrosine kinase receptors ROR1 and ROR2. Cancer Discov 3, 1378–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perego M, Maurer M, Wang JX, Shaffer S, Müller AC, Parapatics K, Li L, Hristova D, Shin S, Keeney F, et al. (2018). A slow-cycling subpopulation of melanoma cells with highly invasive properties. Oncogene 37, 302–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez RE, Shen H, Duan L, Kim RH, Kim T, Park NH, and Maki CG (2016). Modeling the Etiology of p53-mutated Cancer Cells. J Biol Chem 291, 10131–10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radke J, Roßner F, and Redmer T (2017). CD271 determines migratory properties of melanoma cells. Sci Rep 7, 9834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambow F, Rogiers A, Marin-Bejar O, Aibar S, Femel J, Dewaele M, Karras P, Brown D, Chang YH, Debiec-Rychter M, et al. (2018). Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell. [DOI] [PubMed]
- Ramsdale R, Jorissen RN, Li FZ, Al-Obaidi S, Ward T, Sheppard KE, Bukczynska PE, Young RJ, Boyle SE, Shackleton M, et al. (2015). The transcription cofactor c-JUN mediates phenotype switching and BRAF inhibitor resistance in melanoma. Sci Signal 8, ra82. [DOI] [PubMed] [Google Scholar]
- Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, Körbel C, Laschke MW, Gimotty PA, Philipp SE, et al. (2013). Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 23, 811–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogel A, Popliker M, Webb CG, and Oren M (1985). p53 cellular tumor antigen: analysis of mRNA levels in normal adult tissues, embryos, and tumors. Mol Cell Biol 5, 2851–2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadot E, Geiger B, Oren M, and Ben-Ze’ev A (2001). Down-regulation of beta-catenin by activated p53. Mol Cell Biol 21, 6768–6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satyamoorthy K, Chehab NH, Waterman MJ, Lien MC, El-Deiry WS, Herlyn M, and Halazonetis TD (2000). Aberrant regulation and function of wild-type p53 in radioresistant melanoma cells. Cell Growth Differ 11, 467–474. [PubMed] [Google Scholar]
- Smith MP, Brunton H, Rowling EJ, Ferguson J, Arozarena I, Miskolczi Z, Lee JL, Girotti MR, Marais R, Levesque MP, et al. (2016). Inhibiting Drivers of Non-mutational Drug Tolerance Is a Salvage Strategy for Targeted Melanoma Therapy. Cancer Cell 29, 270–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, and Tibshirani R (2003). Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 100, 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Wei W, Robert L, Xue M, Tsoi J, Garcia-Diaz A, Homet Moreno B, Kim J, Ng RH, Lee JW, et al. (2017). Single-cell analysis resolves the cell state transition and signaling dynamics associated with melanoma drug-induced resistance. Proc Natl Acad Sci U S A 114, 13679–13684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topol L, Jiang X, Choi H, Garrett-Beal L, Carolan PJ, and Yang Y (2003). Wnt-5a inhibits the canonical Wnt pathway by promoting GSK-3-independent beta-catenin degradation. J Cell Biol 162, 899–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, Wong DJL, Atefi M, Shirazi R, Wang X, et al. (2018). Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 33, 890–904.e895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungerleider NA, Rao SG, Shahbandi A, Yee D, Niu T, Frey WD, and Jackson JG (2018). Breast cancer survival predicted by TP53 mutation status differs markedly depending on treatment. Breast Cancer Res 20, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster MR, Kugel CH, and Weeraratna AT (2015a). The Wnts of change: How Wnts regulate phenotype switching in melanoma. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 1856, 244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster MR, and Weeraratna AT (2013). A Wnt-er migration: the confusing role of β-catenin in melanoma metastasis. Sci Signal 6, pe11. [DOI] [PubMed] [Google Scholar]
- Webster MR, Xu M, Kinzler KA, Kaur A, Appleton J, O’Connell MP, Marchbank K, Valiga A, Dang VM, Perego M, et al. (2015b). Wnt5A promotes an adaptive, senescent-like stress response, while continuing to drive invasion in melanoma cells. Pigment Cell & Melanoma Research 28, 184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeraratna AT, Jiang Y, Hostetter G, Rosenblatt K, Duray P, Bittner M, and Trent JM (2002). Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell 1. [DOI] [PubMed] [Google Scholar]
- Zhang T, Dutton-Regester K, Brown KM, and Hayward NK (2016). The genomic landscape of cutaneous melanoma. Pigment Cell Melanoma Res 29, 266–283. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.