

Abstract
Characterized by the expansion of somatic mutations in the hematopoietic lineages of aging individuals, clonal hematopoiesis of indeterminate potential (CHIP) is a common condition that increases the risk of developing hematological malignancies and cardiovascular disease (CVD). The presence of CHIP-associated mutations in hematopoietic stem and progenitor cells (HSPCs) suggests that these mutations may alter the functions of the diverse hematopoietic lineages, many of which influence the pathogenesis of CVD. Inflammation may be a potential pathogenic mechanism, linking both CVD and hematological malignancy. However, it remains unknown whether CHIP-associated CVD and hematological malignancy are features of a common disease spectrum. The contributions of CHIP-associated mutations to both CVD and hematological malignancy underscore the importance of stem cell biology in pathogenesis and treatment. This review discusses possible mechanisms underlying the contributions of multiple hematopoietic lineages to CHIP-associated CVD and the putative pathogenic links between CHIP-associated CVD and hematological malignancy.
Keywords: hematopoietic stem and progenitor cells, cardiovascular disease, clonal hematopoiesis of indeterminate potential, hematological malignancy, inflammation
Graphical Abstract
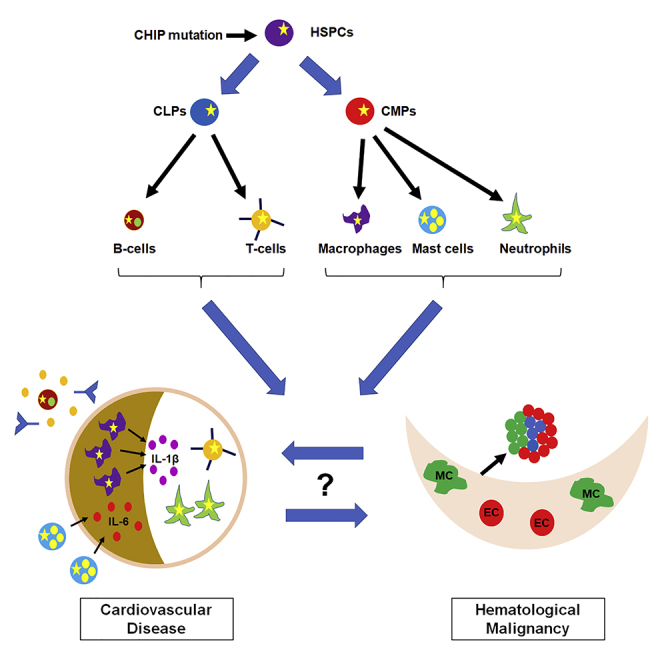
In this review article, Kapur and colleague discuss mechanisms by which clonal hematopoiesis of indeterminate potential (CHIP) contributes to cardiovascular disease. Specifically, the authors address the possibilities that CHIP-associated mutations in hematopoietic stem and progenitor cells affect multiple hematopoietic lineages and that CHIP-associated cardiovascular disease and hematological malignancy may represent features of a common disease process.
Main Text
Introduction
With age, individuals acquire somatic mutations in their hematopoietic stem and progenitor cells (HSPCs), which produce the various blood cell lineages, including monocytes, mast cells, neutrophils, megakaryocytes, B cells, and T cells (Jaiswal and Ebert, 2019). Some of these mutations can promote the competitive advantage and expansion of specific leukocyte clones in a process called clonal hematopoiesis (CH) (Jaiswal and Ebert, 2019). The presence of CH in the context of aging is called age-related clonal hematopoiesis (ARCH), which describes the existence of any stem cell subpopulation or clone (Busque et al., 2018). When these clones harbor mutations in preleukemic genes at a variant allele frequency (VAF) of 2% or greater, they define a novel condition called clonal hematopoiesis of indeterminate potential (CHIP), in which seemingly healthy individuals are at an increased risk of developing hematological disease (Jaiswal et al., 2014). Depending on the depth of DNA sequencing, CHIP can be detected in as high as 95% of adults, making it a very common but poorly understood phenomenon (Young et al., 2016). Epigenetic regulators, including the Tet methylcytosine dioxygenase 2 (TET2), DNA methyltransferase 3A (DNMT3A), and associated sex combs-like 1 (ASXL1) genes, are commonly mutated in CHIP (Jaiswal and Ebert, 2019). In leukemia, mutations in these genes are often present in HSPCs early in the development of hematological malignancy (Jan et al., 2012). However, the scope of the pathogenesis and the clinical implications of CHIP have not yet been fully characterized.
Remarkably, recent studies have also linked CHIP to an increased risk of cardiovascular and cerebrovascular disease (CVD), including atherosclerosis, myocardial infarction, and ischemic stroke (Jaiswal et al., 2014, 2017). This correlation suggests that preleukemic mutations in hematopoietic lineages can predispose individuals to CVD and that CVD may be an important clinical manifestation of CHIP. Recently, CHIP has been linked to inferior outcomes in heart failure patients (Dorsheimer et al., 2019). The magnitude of the VAF correlated with the severity of the clinical outcome, suggesting an important role for the expansion of clones containing CHIP-associated mutations in CVD pathogenesis (Dorsheimer et al., 2019). The effects of CHIP-associated mutations on atherosclerosis and heart failure, but not cerebrovascular disease, have been confirmed in mouse models. Transplantation of Tet2-deficient bone marrow (BM) into the Low-density lipoprotein receptor (Ldlr)-deficient mouse model of atherosclerosis susceptibility accelerated atherosclerosis via pro-inflammatory macrophages (Fuster et al., 2017; Jaiswal et al., 2017). Similarly, mice transplanted with Tet2-deficient BM exhibited worse cardiac remodeling in heart failure models (Sano et al., 2018a, 2018b). Expression of mutant Janus kinase 2 (JAK2), which is also mutated in CHIP, in myeloid cells exacerbated heart failure by promoting inflammation and cardiac remodeling (Sano et al., 2019). Notably, transplant of Tet2-deficient BM into non-irradiated wild-type mice increased fibrosis and hypertrophy, indicating that loss of Tet2 in BM may not only exacerbate existing CVD but may also directly cause CVD (Wang et al., 2020).
CHIP also affects the outcomes of cardiac procedures, implicating it in multiple manifestations of CVD. The presence of CHIP-associated mutations increased all-cause mortality after transcatheter aortic valve implantation (Mas-Peiro et al., 2020). Remarkably, CVD is also observed in patients undergoing hematopoietic stem cell transplantation (HSCT). Similar to CHIP patients, HSCT patients exhibit coronary heart disease and heart failure (Armenian and Chow, 2014; Armenian et al., 2012). Notably, HSCT patients developing CVD tended to be older, and conditioning regimens included total body irradiation, chest irradiation, and anthracycline treatment (Armenian et al., 2018). It is not yet known if CVD in HSCT patients can be attributed to CHIP-associated mutations. However, if so, it would suggest that HSPCs carrying CHIP-associated mutations have intrinsic pathogenic features that enable them to promote CVD in a new environment. HSPCs bearing CHIP-associated mutations can preserve cell-autonomous features under stressful conditions, such as transplantation, but additional studies are needed to investigate this possibility in the context of CVD (Yu et al., 2016).
Collectively, these studies support a broad role for CHIP-associated mutations in CVD, affecting multiple stages in its pathogenesis. They indicate that CHIP is an important novel risk factor for CVD in seemingly healthy individuals, but much remains unknown regarding the mechanisms underlying its contributions to CVD. The breadth of CHIP's effects is consistent with the diverse functions of the hematopoietic cell types, many of which play significant roles in CVD (Swirski and Nahrendorf, 2018). The presence of CHIP-associated mutations in HSPCs suggests that these mutations have the potential to influence multiple cell types involved in CVD as well as cause hematological malignancy, providing a possible link between these pathologies (Figure 1). However, these processes are only beginning to be explored. It is not yet known if mutant HSPCs or their differentiated progeny contribute to or cause CHIP-associated CVD. Two important unanswered questions regarding CHIP are (1) whether CHIP-associated mutations alter the functions of multiple hematopoietic lineages and (2) whether CHIP-associated CVD and hematological malignancies represent a common disease process. The objective of this review is to discuss support for these notions and to provide a rationale for their further investigation.
Figure 1.

Putative Contributions of Hematopoietic Lineages to CVD and Potential Links between CHIP-Associated CVD and Hematological Diseases
HSPCs give rise to the many hematopoietic cell lineages. CHIP mutations (yellow star) in HSPCs are transmitted to mature hematopoietic lineages, many of which play roles in the different stages of CVD by modulating the inflammatory response. The roles of CHIP-associated mutations in these different cell types have not yet been explored. CHIP-associated mutations have been linked to both CVD and hematological malignancy, but it is not yet known if these two pathologies represent a common disease process or distinct events. LT-HSPCs, long-term HSPCs; ST-HSPCs, short-term HSPCs; CLPs, common lymphoid progenitors; CMPs, common myeloid progenitors; MC, mesenchymal cell; EC, endothelial cell.
CVD Is a Complex Process Involving Multiple Cell Types and Stages
CVD is a dynamic process involving multiple stages (Libby et al., 2019a; Libby and Kobold, 2019). Atherosclerosis is characterized by the accumulation of lipid plaques in vessels, and its progression increases the risk for myocardial infarction, which can cause heart failure (Swirski and Nahrendorf, 2013). Importantly, many types of leukocytes play key and distinct roles throughout CVD (Swirski and Nahrendorf 2013, 2018). During the stages of CVD, these cell types interact to promote either inflammation or repair (Tabas and Lichtman, 2017; Tourki and Halade, 2017). The most abundant leukocytes in atherosclerotic lesions, macrophages are involved in both the promotion and regression of inflammation (Swirski and Nahrendorf, 2013). Neutrophils modulate monocyte recruitment and oxidative stress, while platelets facilitate monocyte entry into plaques and promote thrombus formation (Swirski and Nahrendorf, 2013). B cells and T cells can either promote or inhibit atherosclerosis (Swirski and Nahrendorf, 2013). In heart failure, B cells influence the myocardial leukocyte pool and affect myocardial mass and left ventricular contractility (Adamo et al., 2020). CD8+ T cells attenuate inflammation and promote scar formation, and their loss increases the numbers of neutrophils and macrophages, indicating cooperation among hematopoietic lineages in CVD (Ilatovskaya et al., 2019). Mast cells destabilize plaques, attract monocytes and neutrophils from the BM to the injured heart, and affect fibrosis (Lagraauw et al., 2019; Legere et al., 2019; Swirski and Nahrendorf, 2018). Neutrophils also aid in macrophage polarization into distinct subtypes, highlighting the complex contributions of different hematopoietic cell subtypes to CVD (Swirski and Nahrendorf, 2018). The involvement of multiple hematopoietic lineages throughout the various stages of CVD provides many opportunities for CHIP to influence CVD; however, the roles of these cell types in the context of CHIP are only beginning to be investigated.
Inflammation May Be a Key Mechanism for CHIP-Associated CVD
Many of the hematopoietic lineages contribute to the inflammatory response, and recent studies suggest that inflammation may play a critical role in the pathogenesis of CHIP-associated CVD (Fuster et al., 2017; Sano et al., 2018a, 2018b; Wang et al., 2020). The accelerated atherosclerosis observed in Ldlr-deficient mice transplanted with Tet2-deficient BM is attributed to pro-inflammatory macrophages secreting increased interleukin-1β (IL-1β) via NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3), a key inflammatory mediator that cleaves IL-1β into its active form (Fuster et al., 2017). TET2 regulates the expression of NLRP3 and IL-1β as well as NLPR3-mediated cleavage of IL-1β; consequently, Tet2 inactivation increases IL-1β levels (Fuster et al., 2017). Elevated expression of CXC motif chemokine ligand (Cxcl) 1, Cxcl2, Cxcl3, platelet factor 4 (Pf4), and IL-6 was also observed, indicating that the inflammatory response in CHIP-associated CVD may be complex (Jaiswal et al., 2017). In heart failure, loss of Tet2 prevented recovery and promoted inflammation, indicating that inflammation may contribute to CHIP-associated CVD in different disease contexts and stages (Sano et al., 2018a, 2018b). Inhibition of the NLRP3 inflammasome reduced fibrosis and hypertrophy and normalized cardiac functions, suggesting that macrophages play a similar role in both atherosclerosis and heart failure in mice (Sano et al, 2018b). These results are consistent with previous studies showing that TET2 prevents the expression of inflammatory genes and that tumor necrosis factor alpha (TNF-α) promotes the expansion of Tet2-deficient HSPCs in vitro (Cull et al., 2017; Abegunde et al., 2018). Similarly, inactivation of Dnmt3a in mice promotes mast cell activation, supporting the notion that inflammation may be a key mechanism underlying CHIP-associated CVD (Leoni et al., 2017). In a model of CHIP-associated CVD in which mice transplanted with Tet2- or Dnmt3a-deficient BM cells were stimulated with angiotensin II, loss of either Tet2 or Dnmt3a caused cardiac and renal dysfunction, but differences in CVD kinetics and cytokine and chemokine expression were detected (Sano et al., 2018a). As the inflammatory response in CVD involves a balance of resolving and non-resolving factors, the increased inflammation due to CHIP may impede the ability to effectively resolve inflammation, leading to an exacerbation of CVD (Tourki and Halade, 2017). Notably, inflammation plays a differential role in subtypes of heart failure (Schiattarella et al., 2020). Consequently, CHIP may have distinct implications for different types of heart failure.
As Tet2 loss promotes a pro-inflammatory macrophage phenotype, it is possible that specific subpopulations of macrophages exacerbate CHIP-associated CVD. Macrophages consist of diverse subpopulations and exhibit distinct functions during the different stages of CVD pathogenesis and progression (Bajpai et al., 2018; Peled and Fisher, 2014). These changes in cell-type subpopulations and the stage of pathogenesis may have significant implications for signaling. For example, while inhibition of IL-1β reduces atherosclerotic plaque size, it is protective at later stages of atherosclerosis (Gomez et al., 2018). While IL-1 receptor signaling appears to be important for CHIP-associated atherosclerosis in mice, it remains unclear whether CHIP-associated CVD is exclusively IL-1-dependent or whether additional inflammatory pathways contribute. Elevated serum IL-6, IL-8, monocyte chemoattractant protein 1 (MCP1 or CCL2), and TNF-α have been reported in CHIP patients, supporting a complex or heterogeneous inflammatory response (Cook et al., 2019). Notably, IL-6 deficiency reduces the risk of CHIP-associated CVD, indicating a key role for IL-6 (Bick et al., 2020). The cell types responsible for producing these other cytokines are not yet known. Collectively, these studies suggest that inflammation and macrophages play critical roles in CHIP-associated CVD; however, multiple inflammatory pathways and hematopoietic cell types may promote pathogenesis (Table 1).
Table 1.
Significant Studies Investigating CHIP-Associated CVD
| Study | Hematopoietic Cell Types | Key Findings |
|---|---|---|
| Jaiswal et al. (2014) | N/A | Increased risks of hematologic cancer, all-cause mortality, coronary heart disease, and ischemic stroke were observed in individuals with CHIP. Mutations in DNMT3A, TET2, and ASXL1 constituted most of the CHIP variants in this study. |
| Montagner et al. (2016) | Mast cells | TET2 regulated mast cell differentiation. Upon IgE and antigen stimulation, Tet2-deficient mast cells produced reduced levels of the mast cell cytokines IL-6, TNF-α, and IL-13. |
| Cull et al. (2017) | Macrophages | TET2 prevented inflammation and was highly expressed during macrophage differentiation. Tet2 loss led to increased Il-1β, Il-6, and Arginase 1 during later stages of LPS stimulation of BM-derived macrophages. |
| Fuster et al. (2017) | Macrophages | In Ldlr-deficient mice transplanted with Tet2-deficient BM cells, pro-inflammatory Tet2-deficient macrophages increased the secretion of IL-1β via the NLRP3 inflammasome. TET2 regulated expression of the NLRP3. inflammasome. Inhibition of the NLRP3 inflammasome impaired the growth of atherosclerotic plaques. |
| Jaiswal et al. (2017) | N/A | Increased risks of coronary heart disease, coronary-artery calcification, and early-onset myocardial infarction were observed in individuals with CHIP. Ldlr-deficient mice transplanted with BM from Tet2-deficient mice exhibited accelerated atherosclerosis. Increased RNA and protein expression of IL-1β, IL-6, CXCL1, CXCL2, CXCL3, and PF-4 was measured in these transplanted mice. Increased expression of IL-8 was detected in the plasma of individuals with CHIP. |
| Leoni et al. (2017) | Mast cells | DNMT3A attenuated mast cell responses to acute and chronic stimuli. Loss of Dnmt3a was associated with increased sensitivity to stimuli, increased cytokine release, and enhanced degranulation capacity. |
| Abegunde et al. (2018) | HSPCs | Chronic TNF-α exposure promoted a clonogenic advantage for murine Tet2-deficient and human TET2-mutant HSPCs in vitro. The expansion of these HSPCs coincided with resistance to apoptosis and myeloid skewing. This study suggests an important role for HSPCs carrying CHIP-associated mutations, in addition to mature hematopoietic lineages. |
| Buscarlet et al. (2018) | N/A | Patients with CHIP-associated mutations in TET2 or DNMT3A exhibited different hematopoietic lineages. TET2 mutations correlated with greater myeloid populations and no multipotent lineages, whereas DNMT3A mutations coincided with multipotent lineages. The kinetics of clonal expansion differed with distinct CHIP-associated mutations. |
| Cai et al. (2018) | HSPCs and myeloid cells | Increased Tet2-deficient mature myeloid cells and HSPCs were detected in response to inflammatory stress, leading to enhanced production of inflammatory cytokines, such as IL-6. The long non-coding RNA Morrbid was increased in response to IL-6. Inhibition of SHP2 or STAT3 or loss of Morrbid inhibited CH of Tet2-deficient HSPCs. |
| Sano et al. (2018a) | Macrophages | In response to angiotensin II, mice lacking either Tet2 or Dnmt3a developed cardiac hypertrophy, cardiac and renal fibrosis, and reduced cardiac function. The kinetics of clonal expansion differed in Tet2- and Dnmt3a-deficient mice. Increased expression of Il-1β, Il-6, and Ccl5 was detected in an LPS-stimulated Tet2-deficient macrophage cell line, whereas elevation of Cxcl1, Cxcl2, Il-6, and Ccl5 was observed in an LPS-stimulated Dnmt3a-deficient macrophage cell line. |
| Sano et al. (2018b) | Macrophages | Tet2-deficient hematopoietic or myeloid cells exacerbated cardiac remodeling and function and correlated with increased IL-1β expression in transaortic constriction and chronic ischemia models of heart failure. Inhibition of the NLRP3 inflammasome improved cardiac measurements and alleviated heart failure development. |
| Wolach et al. (2018) | Neutrophils | CH for JAK2V617F, the most common driver of myeloproliferative neoplasm, correlated with an increased occurrence of thrombosis. Mice expressing JAK2V617F exhibited increased NET formation and thrombosis, which were alleviated by the JAK2 inhibitor ruxolitinib. |
| Cook et al. (2019) | N/A | Elevated expression of IL-6, MCP1/CCL2, and TNF-α was detected in individuals with CHIP. |
| Dorsheimer et al. (2019) | N/A | Patients with CHIP often had a history of hypertension. Mutations in TET2 or DNMT3A were associated with worse heart failure outcomes. |
| Mudersbach et al. (2019) | N/A | In response to TNF-α stimulation of endothelial cells, increased methylation was detected at sites in the ACE promoter in a DNMT3A- and DNMT3B-dependent manner. |
| Sano et al. (2019) | Myeloid cells | Mice with myeloid expression of the JAK2V617F mutation demonstrated increased cardiac inflammation and dysfunction after permanent left anterior descending artery ligation and transverse aortic constriction. |
| Bick et al. (2020) | N/A | Individuals with large CHIP clones (VAF > 10%) that also carry an IL-6 receptor p.Asp358Ala mutation exhibited fewer CVD events. |
| Mas-Peiro et al. (2020) | T cells and myeloid cells | Patients with aortic valve stenosis undergoing transcatheter aortic valve implantation and carrying TET2 or DNMT3A mutations experienced worse outcomes. Patients with TET2 mutations showed increased pro-inflammatory non-classical monocytes, whereas patients with DNMT3A mutations exhibited pro-inflammatory T cell polarization. |
| Veninga et al. (2020) | Platelets | Mutations in different CHIP-associated genes were linked to differences in platelet number and risk of thrombosis and bleeding. |
| Wang et al. (2020) | Myeloid cells | Transplantation of Tet2-deficient BM cells into non-irradiated recipient mice resulted in cardiac dysfunction, hypertrophy, and fibrosis. Macrophages isolated from the heart exhibited upregulation of cytokines, cytokine receptors, and interferon-related genes and downregulation of genes involved in regulation of cell differentiation and neurogenesis. |
ACE, angiotensin-converting enzyme; ASXL1, Associated sex combs-like 1; BM, bone marrow; CHIP, clonal hematopoiesis of indeterminate potential; CCL2, C-C motif chemokine ligand 2; Ccl5, C-C motif chemokine ligand 5; CXCL1, CXC motif chemokine ligand 1; CXCL2, CXC motif chemokine ligand 2; CXCL3, CXC motif chemokine ligand 3; DNMT3A, DNA methyltransferase 3a; DNMT3B, DNA methyltransferase 3b; HSPCs, hematopoietic stem and progenitor cells; IgE, immunoglobulin E; IL-1β, interleukin-1β; IL-6, interleukin-6; IL-8, interleukin-8; IL-13, interleukin-13; JAK2, Janus-associated kinase 2; Ldlr, low-density lipoprotein receptor; LPS, lipopolysaccharide; MCP1, monocyte chemoattractant protein 1; NET, neutrophil extracellular trap; NLRP3, NOD-, LRR-, and pyrin domain-containing protein 3; PF-4, platelet factor 4; SHP2, Src homology phosphatase 2; STAT3, signal transducer and activator of transcription 3; TET2, Tet methylcytosine dioxygenase 2; TNF-α, tumor necrosis factor-α; VAF, variant allele frequency; N/A, not applicable.
Additional Hematopoietic Lineages May Contribute to CHIP-Associated CVD
In addition to macrophages, many other hematopoietic lineages participate in CVD. Consequently, they may be altered by CHIP and may modify the pathogenesis and outcomes of CHIP-associated CVD. For example, perivascular mast cells release pro-inflammatory cytokines, such as IL-6 and interferon-γ, and facilitate atherosclerotic plaque destabilization (Bot et al., 2007; Lagraauw et al., 2019; Sun et al., 2007). Loss of Dnmt3a in mast cells increases cytokine release, enhances degranulation capacity, and increases sensitivity to stimuli (Leoni et al., 2017). Levels of mast cells correlate with an increased risk of adverse cardiovascular (CV) events, supporting an important clinical role for mast cells in CVD (Willems et al., 2013). Consequently, CHIP-associated genes may also promote the inflammatory response in CVD via mast cells. CHIP-associated mutations were recently associated with differential effects on platelet number and risk for thrombosis and bleeding, and pro-inflammatory lymphoid lineages have been detected in CHIP patients with CVD (Mas-Peiro et al., 2020, Veninga et al., 2020). Furthermore, individuals exhibiting CH for the JAK2V617F mutation have an increased occurrence of thrombosis associated with neutrophil extracellular traps (NETs), indicating another hematopoietic lineage that can contribute to CHIP-associated CVD (Wolach et al., 2018). In this context, this increased risk of thrombosis does not appear to be associated with CV risk factors, inferring that CHIP alone may increase the risk of CVD. Additional studies are needed to elucidate the functions of these different hematopoietic cell types in CHIP-associated CVD.
Differences in the distribution and functions of hematopoietic lineages may influence the manifestation of inflammation at different stages of pathogenesis and may have important implications for disease heterogeneity and the treatment of CHIP-associated CVD. For example, mutations in TET2 may lead to an accumulation of pro-inflammatory macrophages, whereas mutations in JAK2 and DNMT3A may be characterized by prominent responses by neutrophils, mast cells, and T cells (Figure 2). These changes may influence the ability to resolve inflammation. Single-cell RNA sequencing has identified functional subtypes of these cell types, further complicating the potential implications of CHIP-associated mutations (Winkels et al., 2018). In addition, cell-type-specific functions of the NLRP3 inflammasome have been reported in myocardial infarction, providing an opportunity for multiple cell types to modulate this pathway (Takahashi, 2019).
Figure 2.

Different CHIP-Associated Mutations May Have Differential Effects on the Distinct Hematopoietic Lineages That Contribute to CVD
(A) Many different hematopoietic lineages contribute to CVD. In atherosclerosis, pro-inflammatory macrophages (purple) accelerate plaque formation. Mast cells (blue) and neutrophils (green) promote thrombosis via IL-6 and neutrophil extracellular traps, respectively. B cells (brown) secrete antibodies and cytokines. Polarized T cells (orange) facilitate inflammation. All of these cell types have the potential to carry CHIP-associated mutations (yellow star) and to alter CHIP-associated CVD.
(B) Different CHIP-associated mutations may influence the cell types involved in and the pathogenesis of CHIP-associated CVD at different stages. Loss of TET2, JAK2, and DNMT3A has been associated with macrophages, neutrophils, and mast cells and T cells, respectively.
Different CHIP-associated genes differentially affect the various hematopoietic lineages, biasing the distribution of these lineages toward specific cell populations. For example, inactivation of the Tet2 gene promotes a myeloid shift and can alter mast cell differentiation and proliferation (Montagner et al., 2016, Buscarlet et al., 2018). In CHIP patients undergoing transcatheter aortic valve implantation for degenerative aortic valve stenosis, DNMT3A mutations were associated with pro-inflammatory T cells, whereas TET2 mutations correlated with pro-inflammatory non-classical monocytes, suggesting that different mutations may influence the hematopoietic lineages that can contribute to CHIP-associated CVD (Mas-Peiro et al., 2020). While both the T cells and monocytes were pro-inflammatory, it is not yet known if they promote inflammation via a common mechanism. CHIP-associated mutations have been detected in lymphoid lineages; however, the implications of lymphoid lineages for CHIP-associated CVD have not yet been explored (Boettcher et al., 2020; Buscarlet et al., 2018). Significantly, the penetrance of CHIP-associated mutations in different hematopoietic lineages depends on the specific gene carrying the mutation. In CHIP patients carrying single DNMT3A mutations, 41% of the peripheral mononuclear cells (PMNs) were multipotent, while 36% and 23% were myeloid and myelolympho-B cells, respectively. The VAFs in the myelolympho-B and multipotent lineages were 10% or greater, while the VAF in the myeloid lineage was less than 10% (Buscarlet et al., 2018). In contrast, CHIP patients bearing single TET2 mutations exhibited 54% of PMNs from the myeloid lineage and 46% from the myelolympho-B lineage. The VAFs were approximately 10% in both the myeloid and myelolympho-B lineages (Buscarlet et al., 2018). In HSCT donors with CHIP and their recipients, granulocytes, monocytes, B cells, and T cells demonstrated mean VAFs of 0.08, 0.083, 0.028, and 0.007, respectively (Boettcher et al., 2020). These studies support the notion that different CHIP-associated mutations may differentially affect the distribution of the affected hematopoietic cell types and their functions in CVD. These mutation-specific and cell-type differences may have important implications for the clinical presentations, prognoses, and latencies of and the therapeutic development for CHIP-associated CVD.
Epigenetic Regulators May Drive the Distributions of Hematopoietic Lineages and May Contribute to CHIP-Associated CVD
As epigenetic regulators are frequently mutated in CHIP, epigenetic modulation may contribute to the pathogenesis of CHIP-associated CVD (Jaiswal et al., 2017). Mutations in and loss of CHIP-associated epigenetic regulators in HSPCs differentially affect the resulting hematopoietic lineages, supporting an important role for stem cells in the pathogenesis of CHIP-associated diseases (Buscarlet et al., 2018). Inactivation of Tet2 or Asxl1 in HSPCs leads to more myeloid phenotypes, whereas loss of Dnmt3a in these cells causes both myeloid and lymphoid disease, indicating that these epigenetic regulators can affect the availability of hematopoietic lineages (Abdel-Wahab et al., 2013; Li et al., 2011; Mayle et al., 2015). However, the effects of these CHIP-associated genes on the distribution of hematopoietic lineages, disease severity, latency, and penetrance of CHIP-associated CVD are not yet known.
In addition to skewing the differentiation of HPSCs, epigenetic regulation may also influence the contributions of mature hematopoietic cell types to CHIP-associated CVD as epigenetic regulation contributes to atherosclerosis, heart failure, and inflammation (Rizzacasa et al., 2019). For example, differences in DNA methylation detected in atherosclerotic and normal aortas may correlate with disease progression, and loss of histone deacetylase 3 exacerbates atherosclerosis (Zampetaki et al., 2010; Valencia-Morales Mdel et al., 2015). Exercise can modulate epigenetic factors in a model of atherosclerosis, indicating that epigenetic differences may alter the ability to respond to environmental influences that affect CVD (Frodermann et al., 2019). Many of these studies report epigenetic changes in endothelial cells or vascular smooth muscle cells; however, less is known about the effects of epigenetic modification in hematopoietic lineages on CVD.
Many opportunities exist for epigenetic regulators to alter the functions of mature hematopoietic lineages. For example, TET2 regulates the expression of NLRP3 and IL-1β (Fuster et al., 2017). Previously, our group demonstrated that the long-noding RNA Morrbid is increased in Tet2-deficient cells challenged with lipopolysaccharide and that loss of Morrbid ameliorates the inflammatory phenotype of Tet2-deficient cells, broadening the impact of epigenetic regulation in CH and suggesting that multiple epigenetic regulators may be important in CH (Cai et al., 2018). Notably, Dnmt3a is upregulated in response to altered blood flow and can modulate the expression levels of angiotensin-converting enzyme (ACE), which regulates blood pressure, in endothelial cells in response to inflammation (Figure 4) (Davies et al., 2014; Mudersbach et al., 2019). Cardiac mast cells also secrete renin, an upstream regulator of angiotensin, and the release of renin can contribute to various manifestations of CVD (Mackins et al., 2006; Reid et al., 2007). These observations suggest a role for mast cells in blood pressure regulation and the ability of CHIP-associated genes to influence blood pressure. Notably, CHIP patients more frequently exhibit hypertension; however, the mechanisms are not understood (Dorsheimer et al., 2019). These epigenetic changes may modify the distributions and functions of the hematopoietic lineages that modulate CHIP-associated CVD. These studies suggest that there are multiple possible mechanisms by which epigenetic factors can affect CHIP-associated CVD.
Figure 4.

Model for Altered Hematopoiesis and the Development of Hematological Malignancy in Response to CHIP-Associated CVD
Loss of CHIP-associated genes is associated with an increased release of cytokines from HSPCs and mature hematopoietic lineages. These cytokines may promote expansion of the mutant clones and CVD. Increased inflammation and blood pressure are associated with CVD. Both of these changes can alter the BM microenvironment, which contributes to hematopoiesis. These stresses on the BM microenvironment may aberrantly alter hematopoiesis to promote hematological malignancy and may create genotoxic stress that facilitates the acquisition of additional mutations. MC, mesenchymal cell; EC, endothelial cell.
Inflammation and CVD Can Influence Hematopoiesis
While epigenetic regulation may impact the hematopoietic lineages contributing to CHIP-associated CVD, inflammation may also shape the distributions of these cell types as cytokines play a critical role in regulating hematopoiesis (Fuster et al., 2017). Associated with stresses, such as infection, inflammation mobilizes HSPCs and influences differentiation patterns to bolster the immune system. For example, the NLRP3 inflammasome, interferon-γ, and IL-1 promote the mobilization of HSPCs (de Bruin et al., 2014; Lenkiewicz et al., 2019; Mantovani et al., 2019). However, the timing of the inflammatory response may be critically important as chronic inflammation is associated with HSPC exhaustion and damage (Capitano, 2019; Pietras et al., 2016). In addition, different cytokines may exert differential effects depending on the context and the other inflammatory mediators present, both of which may be altered by CHIP (Mantovani et al., 2019). We and others have shown that Tet2-deficient HSPCs secrete cytokines and that inflammation promotes the expansion of these HSPCs, suggesting that, in addition to mature hematopoietic lineages, HSPCs carrying CHIP-associated mutations may actively contribute to the inflammation observed in CHIP (Abegunde et al., 2018; Cai et al., 2018). These findings are consistent with the observation that transplantation of Tet2-deficient BM is sufficient to cause CHIP-associated CVD (Wang et al., 2020).
In addition to promoting the expansion of clones carrying CHIP-associated mutations, inflammation can also alter the BM microenvironment (Leimkuhler and Schneider, 2019). Inflammation and the BM microenvironment change with age, influencing the context in which CHIP-associated inflammation occurs (Ho et al., 2019; Kovtonyuk et al., 2016). As loss of either Tet2 or Dnmt3a is implicated in inflammation, HSPCs carrying CHIP-associated mutations and/or the presence of CHIP-associated CVD may promote an inflammatory state that can alter the BM microenvironment, leading to more permanent changes in hematopoiesis and a propensity to promote HSPC damage. It has been proposed that, in CVD, mature hematopoietic cells and HSPCs create a reinforcing feedforward system in which the inflammation from one fuels the other (Chavakis et al., 2019). This notion may also apply in CHIP. Elevated levels of cytokines and chemokines have been detected in the serum of CHIP patients, indicating that these patients exhibit systemic inflammation (Cook et al., 2019). However, changes in the BM microenvironment of patients with CHIP or CHIP-associated CVD have not yet been investigated.
CVD can also increase hematopoietic activity, identifying a means of communication between the CV and hematopoietic systems (Al-Sharea et al., 2019a, 2019b). Ischemic stroke and atherosclerosis stimulate HSPCs (Courties et al., 2015; van der Valk et al., 2017). HSCTs have been evaluated to treat infarction and ischemic damage as HSPCs can home to injured heart tissues (Kavanagh and Kalia, 2011; Vagnozzi et al., 2020). In addition, exercise alleviates CV inflammation by modulating hematopoiesis (Frodermann et al., 2019). The existence of these effects implies the possibility of a system in which the hematopoietic and CV systems may reciprocally influence disease progression in CHIP. Hypertension leads to unstable atherosclerotic lesions due to stimulation of hematopoiesis (Al-Sharea et al., 2019b). Remarkably, stress-induced hypertension causes depletion of HSPCs and reduction of osteoblasts and sinusoidal endothelial cells in the BM microenvironment, suggesting that hypertension may also promote remodeling of the BM microenvironment (Al-Sharea et al, 2019b). As DNMT3A can regulate ACE expression, mutations in DNMT3A may lead to an elevation in blood pressure that modulates the BM microenvironment (Figure 4) (Mudersbach et al., 2019). It remains to be seen if loss of DNMT3A function in hematopoietic cells can dysregulate ACE expression. Importantly, as CHIP is predominant in older individuals, CHIP-associated diseases will also occur in an older CV system, which is characterized by increased cardiac leukocytes, including monocyte-derived cardiac macrophages, granulocytes, and CD8+ T cells (Swirski and Nahrendorf, 2018). These aging-associated changes in the CV system may also have important implications for CHIP-associated CVD. Collectively, these observations raise the possibility that CHIP-associated mutations may trigger a cascade of events that alters hematopoiesis and/or the BM microenvironment via inflammation and cardiac physiology.
CHIP-Associated CVD and Hematological Malignancy May Represent a Progression of the Same Disease Process
The communication between the CV and hematopoietic systems supports a potential interaction between these systems in CHIP-associated CVD. Increased reports of CVD in patients with hematological malignancies further suggest that these pathologies may coexist or represent a spectrum of clinical presentations. For example, patients with acute leukemia exhibit increased left ventricle masses and volumes and decreased global longitudinal strain before chemotherapy, demonstrating an inherent CV deficit in patients with hematological malignancies (Assuncao et al., 2017). In addition, patients with hematological malignancies can exhibit CVD and CV toxicities to chemotherapy, and these toxicities appear to increase with age, suggesting that these patients may also be more susceptible to CV injury (Dickerson et al., 2019; Malato et al., 2010). It is currently unclear if CHIP contributes to these pathologies; however, a more thorough understanding of the role of CHIP in CVD may help identify patients who will be more susceptible to the development of CVD and CV toxicities. An increased incidence of thrombosis associated with JAK2V617F mutations was observed in patients with myeloproliferative neoplasms, demonstrating the possibility that cancer and CVD can coexist (Wolach et al., 2018). Patients with myeloproliferative disease and increased thrombotic events also have an increased risk of secondary cancers (De Stefano et al., 2020). Together, these studies support a potential link between CVD and oncogenesis. However, while CHIP is associated with an increased risk of both hematological malignancies and CVD, it is not yet known if these pathologies occur in the same individuals, if they represent a spectrum of disease progression, or if they are distinct events. Consequently, there are several possible outcomes associated with CHIP (Figure 3). Individuals may carry CHIP-associated mutations, but the mutant clones maintain their size and fail to contribute to pathology. If the mutant clones expand, they may lead to CVD, hematological malignancy, or both. Longitudinal studies of CHIP patients are needed to fully understand the progression of CHIP-associated diseases and the contributions of different clinical presentations.
Figure 3.

Potential Outcomes for the Evolution of CHIP-Associated CVD and Hematological Malignancy from HSPCs Carrying CHIP-Associated Mutations
Mutant clones (red) can expand or be maintained. As CHIP has been linked to both CVD and hematological malignancy, expansion of mutant clones may lead to CVD alone, hematological malignancy alone, or a combination of CVD and hematological malignancy. The acquisition of additional mutations (green) is usually needed to promote the evolution of CH to hematological malignancy and often requires an inciting event (lightning bolt). It is not yet known if CVD and hematological malignancy represent progression of the same disease process or distinct events. However, the inflammation from CHIP-associated CVD may drive the acquisition of additional mutations and the progression to hematological malignancy.
Commonalities exist between the pathogenesis of CVD and cancer, including cell proliferation, apoptosis, inflammation, leukocyte infiltration, and remodeling of the extracellular matrix (Libby and Kobold, 2019; Tapia-Vieyra et al., 2017). These similarities and the ability of the CV and hematological systems to communicate may allow these conditions to coexist and evolve simultaneously. In particular, inflammation may serve as a unifying driver of pathogenesis for CVD and hematological malignancy as it is an important factor in both of these pathologies. TNF-α promotes survival of HSPCs and myeloid regeneration, and IL-6 facilitates the development of chronic myeloid leukemia, indicating that inflammation can promote leukemogenesis (Reynaud et al., 2011; Yamashita and Passegue, 2019). Additional mutations are often required for the development of leukemia and are obtained during clonal evolution of HSPCs (Sanden et al., 2020). In preleukemic scenarios, inflammation can promote genotoxic stress and disease progression, suggesting that inflammation may create an environment in which HSPCs carrying CHIP-associated mutations can acquire additional mutations and facilitate leukemogenesis (Zambetti et al., 2016).
A potential explanation for the coexistence of CHIP-associated CVD and hematological malignancy is that raised blood pressure from increased expression of ACE or renin or cytokines from HSPCs or mature hematopoietic lineages may act as selective pressures that facilitate the remodeling of the BM microenvironment (Figure 4). Interferon-γ from the BM can reduce the size of atherosclerotic plaques in Ldlr-deficient mice, indicating that signals from the BM can modulate CVD (Niwa et al., 2004). In addition, the vascular niche, a component of the BM microenvironment, plays an important role in hematopoiesis and leukemogenesis; however, it is not known if CHIP-associated CVD alters the vascular niche (Duarte et al., 2018; Sasine et al., 2017). If so, changes in the vascular niche due to CHIP-associated CVD may link CVD and hematological malignancy in CHIP. These changes in the BM microenvironment may create a biological context conducive to leukemogenesis and genotoxic stress (Figure 4). It is also compelling to consider the ways in which changes in clonal dynamics may modify the CVD presentation. Further studies are needed to investigate these interactions and the potential co-occurrence of CHIP-associated CVD and hematological malignancies. Collectively, these studies suggest the existence of a significant relationship between the CV and hematopoietic systems and underscore the importance of studying the consequences of this relationship for CHIP.
CHIP Has Implications for the Treatment of CVD
Current studies indicate anti-inflammatory agents, such as inhibitors of IL-1β and the NLRP3 inflammasome, as potential treatments for CHIP-associated CVD (Fuster et al., 2017). As the inflammatory response can manifest as a complex and dynamic cascade, the timing of treatment with anti-inflammatory therapeutics may be critical. Different cytokines, inflammatory pathways, and hematopoietic lineages may play important roles at different times during the inflammatory response and during CVD and may represent potential therapeutic targets. Presently, these anti-inflammatory agents appear to target mature hematopoietic lineages, but it is intriguing to consider whether HSPCs bearing CHIP-associated mutations could be directly targeted. Our group has shown that inhibition of inflammation can impede the expansion of HSPCs bearing CHIP-associated mutations, suggesting that inflammation also contributes to the pathogenic functions of mutant HSPCs in CHIP (Cai et al., 2018). It is not yet known if blocking the expansion of these mutant HSPCs can ameliorate CHIP-associated CVD. Inhibiting the differentiation and function of specific hematopoietic lineages and targeting aberrant proteins encoded by genes bearing CHIP-associated mutations may also be valuable approaches. For example, the JAK2 inhibitor ruxolitinib effectively inhibits NET formation and decreases thrombosis in mice (Wolach et al., 2018), indicating that directly targeting the mutant proteins associated with CHIP may abrogate the altered hematopoietic cell types contributing to CVD.
The clinical relevance of screening CVD patients for CHIP is currently unclear (Libby et al., 2019b). If anti-inflammatory agents can prevent CHIP-associated CVD, then screening for CHIP-associated mutations could identify patients who may benefit from prophylactic anti-inflammatory treatment. However, patients presenting with cardiac symptoms likely already have active disease, emphasizing the importance of identifying therapies that can treat existing disease. An important question in understanding the clinical relevance of CHIP-associated CVD is whether it differs from traditional CVD. Screening for CHIP-associated CVD would be most advantageous if it could be used to guide treatment decisions. While current studies indicate that inflammation may be a key mechanism by which CHIP contributes to CVD, inflammation also plays an important role in CVD in general. For example, canakinumab, an antibody against IL-1β, is also efficacious in traditional CVD (Ridker et al., 2017). The observation that transplantation of Tet2-deficient BM can induce CVD suggests that CHIP-associated mutations may be sufficient to cause CVD in the absence of additional cardiac risk factors (Wang et al., 2020). Larger epidemiological studies are needed to more fully understand the clinical consequences of CHIP-associated CVD, the ways in which it differs from other types of CVD, and the mechanisms by which it interacts with other CVD risk factors. This knowledge will promote the development of therapeutics that can specifically target this patient population.
The screening of specific CHIP-associated mutations may also have significant implications for the clinical manifestation, disease progression, and cell types involved. Understanding the effects of specific mutations may be able to promote a precision medicine approach to treating CHIP-associated CVD in the future. However, as the presence of different comorbidities can also influence CH, the manifestation of CHIP-associated CVD may be affected by these comorbidities (Dragoljevic et al., 2018). This possibility may complicate the utility of genetic screening for predicting prognosis and guiding treatment. In considering potential therapies for CHIP-associated CVD, it is important to also consider the ability of these therapies to modulate the leukemic potential of preleukemic HSPCs carrying CHIP-associated mutations. Treatment with anti-inflammatory agents may alter the distribution of cytokines influencing hematopoiesis, potentially modifying the resulting hematopoietic lineages and promoting leukomogenesis. Collectively, these studies indicate the complexities of treating CHIP-associated CVD and underscore the importance of future studies to better understand its pathogenesis and to facilitate the development of novel therapies.
Conclusions
CHIP is a common novel risk factor for both CVD and hematological malignancy. Many unanswered questions remain regarding its underlying mechanisms. Inflammation may play a critical role in the pathogenesis of CHIP-associated CVD; however, additional studies are needed to fully understand its complexity and impact. Furthermore, it is not yet known if inflammation is a common mechanism for CHIP-associated mutations. As many different hematopoietic lineages contribute to CVD, it is likely that CHIP-associated mutations in HSPCs are transmitted to multiple hematopoietic cell types and that these different cell types can influence clinical presentation and disease progression. These differences may be affected by specific CHIP-associated mutations and may have important implications for disease heterogeneity and treatment. Inflammation may also serve as a key link between CVD and hematological malignancy. The relationship between CVD and cancer development in CHIP patients remains unexplored but represents a fascinating new area in cardio-oncology, highlighting compelling links between the CV and hematological systems. Further elucidation of the disease characteristics unique to CHIP-associated CVD will facilitate the discovery of new therapies and improve the clinical care of CHIP patients.
Author Contributions
S.S.B. and R.K. designed the concept and revised the manuscript. S.S.B. wrote the manuscript. R.K. supervised the project. Both authors reviewed and approved the final version of the manuscript.
Acknowledgments
This work was supported in part by grants from the NIH (R01-CA134777, R01-HL140961, R01-HL146137, and R01-CA173852 to R.K.) and by funds to R.K. from the Riley Children’s Foundation and by an NIH T32 fellowship and a Cancer Biology Training Program Fellowship (DeVault Foundation) from the Melvin and Bren Simon Cancer Center at Indiana University School of Medicine to S.S.B.
References
- Abdel-Wahab O., Gao J., Adli M., Dey A., Trimarchi T., Chung Y.R., Kuscu C., Hricik T., Ndiaye-Lobry D., LaFave L.M. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J. Exp. Med. 2013;210:2641–2659. doi: 10.1084/jem.20131141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abegunde S.O., Buckstein R., Wells R.A., Rauh M.J. An inflammatory environment containing TNFalpha favors Tet2-mutant clonal hematopoiesis. Exp. Hematol. 2018;59:60–65. doi: 10.1016/j.exphem.2017.11.002. [DOI] [PubMed] [Google Scholar]
- Adamo L., Rocha-Resende C., Lin C.Y., Evans S., Williams J., Dun H., Li W., Mpoy C., Andhey P.S., Rogers B.E., Lavine K. Myocardial B cells are a subset of circulating lymphocytes with delayed transit through the heart. JCI Insight. 2020;5:e134700. doi: 10.1172/jci.insight.134700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Sharea A., Lee M.K.S., Purton L.E., Hawkins E.D., Murphy A.J. The haematopoietic stem cell niche: a new player in cardiovascular disease? Cardiovasc. Res. 2019;115:277–291. doi: 10.1093/cvr/cvy308. [DOI] [PubMed] [Google Scholar]
- Al-Sharea A., Lee M.K.S., Whillas A., Michell D.L., Shihata W.A., Nicholls A.J., Cooney O.D., Kraakman M.J., Veiga C.B., Jefferis A.M., Jackson K. Chronic sympathetic driven hypertension promotes atherosclerosis by enhancing hematopoiesis. Haematologica. 2019;104:456–467. doi: 10.3324/haematol.2018.192898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armenian S.H., Chow E.J. Cardiovascular disease in survivors of hematopoietic cell transplantation. Cancer. 2014;120:469–479. doi: 10.1002/cncr.28444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armenian S.H., Sun C.L., Vase T., Ness K.K., Blum E., Francisco L., Venkataraman K., Samoa R., Wong F.L., Forman S.J., Bhatia S. Cardiovascular risk factors in hematopoietic cell transplantation survivors: role in development of subsequent cardiovascular disease. Blood. 2012;120:4505–4512. doi: 10.1182/blood-2012-06-437178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armenian S.H., Yang D., Yang D., Teh J.B., Atencio L.C., Gonzales A., Wong F.L., Leisenring W.M., Forman S.J., Nakamura R., Chow E.J. Prediction of cardiovascular disease among hematopoietic cell transplantation survivors. Blood Adv. 2018;2:1756–1764. doi: 10.1182/bloodadvances.2018019117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assuncao B., Handschumacher M.D., Brunner A.M., Yucel E., Bartko P.E., Cheng K.H., Campos O., Fathi A.T., Tan T.C., Scherrer-Crosbie M. Acute leukemia is associated with cardiac alterations before chemotherapy. J. Am. Soc. Echocardiogr. 2017;30:1111–1118. doi: 10.1016/j.echo.2017.07.016. [DOI] [PubMed] [Google Scholar]
- Bajpai G., Schneider C., Wong N., Bredemeyer A., Hulsmans M., Nahrendorf M., Epelman S., Kreisel D., Liu Y. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 2018;24:1234–1245. doi: 10.1038/s41591-018-0059-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bick A.G., Pirruccello J.P., Griffin G.K., Gupta N., Gabriel S., Saleheen D., Libby P., Kathiresan S., Natarajan P. Genetic interleukin 6 signaling deficiency attenuates cardiovascular risk in clonal hematopoiesis. Circulation. 2020;141:124–131. doi: 10.1161/CIRCULATIONAHA.119.044362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettcher S., Wilk C.M., Singer J., Beier F., Burcklen E., Beisel C., Ventura Ferreira M.S., Gourri E., Gassner C., Frey B.M., Schanz U. Clonal hematopoiesis in donors and long-term survivors of related allogeneic hematopoietic stem cell transplantation. Blood. 2020;135:1548–1559. doi: 10.1182/blood.2019003079. [DOI] [PubMed] [Google Scholar]
- Bot I., de Jager S.C., Zernecke A., Lindstedt K.A., van Berkel T.J., Weber C., Biessen E.A. Perivascular mast cells promote atherogenesis and induce plaque destabilization in apolipoprotein E-deficient mice. Circulation. 2007;115:2516–2525. doi: 10.1161/CIRCULATIONAHA.106.660472. [DOI] [PubMed] [Google Scholar]
- Buscarlet M., Provost S., Zada Y.F., Bourgoin V., Mollica L., Dubé M.P., Busque L. Lineage restriction analyses in CHIP indicate myeloid bias for TET2 and multipotent stem cell origin for DNMT3A. Blood. 2018;132:277–280. doi: 10.1182/blood-2018-01-829937. [DOI] [PubMed] [Google Scholar]
- Busque L., Buscarlet M., Mollica L., Levine R.L. Concise review: age-related clonal hematopoiesis: stem cells tempting the devil. Stem Cells. 2018;36:1287–1294. doi: 10.1002/stem.2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z., Kotzin J.J., Ramdas B., Chen S., Nelanuthala S., Palam L.R., Pandey R., Mali R.S., Liu Y., Kelley M.R., Sandusky G. Inhibition of inflammatory signaling in Tet2 mutant preleukemic cells mitigates stress-induced abnormalities and clonal hematopoiesis. Cell Stem Cell. 2018;23:833–849.e5. doi: 10.1016/j.stem.2018.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capitano M.L. Toll-like receptor signaling in hematopoietic stem and progenitor cells. Curr. Opin. Hematol. 2019;26:207–213. doi: 10.1097/MOH.0000000000000511. [DOI] [PubMed] [Google Scholar]
- Chavakis T., Mitroulis I., Hajishengallis G. Hematopoietic progenitor cells as integrative hubs for adaptation to and fine-tuning of inflammation. Nat. Immunol. 2019;20:802–811. doi: 10.1038/s41590-019-0402-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook E.K., Izukawa T., Young S., Rosen G., Jamali M., Zhang L., Johnson D., Bain E., Hilland J., Ferrone C.K., Buckstein J. Comorbid and inflammatory characteristics of genetic subtypes of clonal hematopoiesis. Blood Adv. 2019;3:2482–2486. doi: 10.1182/bloodadvances.2018024729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courties G., Herisson F., Herisson F., Sager H.B., Heidt T., Ye Y., Wei Y., Sun Y., Severe N., Dutta P. Ischemic stroke activates hematopoietic bone marrow stem cells. Circ. Res. 2015;116:407–417. doi: 10.1161/CIRCRESAHA.116.305207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull A.H., Snetsinger B., Buckstein R., Wells R.A., Rauh M.J. Tet2 restrains inflammatory gene expression in macrophages. Exp. Hematol. 2017;55:56–70.e13. doi: 10.1016/j.exphem.2017.08.001. [DOI] [PubMed] [Google Scholar]
- Davies P.F., Manduchi E., Stoeckert C.J., Jimenez J.M., Jiang Y.-Z. Emerging topic: flow-related epigenetic regulation of endothelial phenotype through DNA methyation. Vascul Pharmacol. 2014;62:88–93. doi: 10.1016/j.vph.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin A.M., Voermans C., Nolte M.A. Impact of interferon-gamma on hematopoiesis. Blood. 2014;124:2479–2486. doi: 10.1182/blood-2014-04-568451. [DOI] [PubMed] [Google Scholar]
- De Stefano V., Ghirardi A., Masciulli A., Carobbio A., Palandri F., Vianelli N., Rossi E., Betti S., Di Veroli A., Iurlo A., Cattaneo D. Arterial thrombosis in Philadelphia-negative myeloproliferative neoplasms predicts second cancer: a case-control study. Blood. 2020;135:381–386. doi: 10.1182/blood.2019002614. [DOI] [PubMed] [Google Scholar]
- Dickerson T., Wiczer T., Waller A., Philippon J., Porter K., Haddad D., Guha A., Rogers K.A., Bhat S., Byrd J.C., Woyach J.A. Hypertension and incident cardiovascular events following ibrutinib initiation. Blood. 2019;134:1919–1928. doi: 10.1182/blood.2019000840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsheimer L., Assmus B., Rasper T., Ortmann C.A., Ecke A., Abou-El-Ardat K., Schmid T., Brüne B., Wagner S., Serve H., Hoffmann J. Association of mutations contributing to clonal hematopoiesis with prognosis in chronic ischemic heart failure. JAMA Cardiol. 2019;4:25–33. doi: 10.1001/jamacardio.2018.3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragoljevic D., Westerterp M., Veiga C.B., Nagareddy P., Murphy A.J. Disordered haematopoiesis and cardiovascular disease: a focus on myelopoiesis. Clin. Sci. (Lond.) 2018;132:1889–1899. doi: 10.1042/CS20180111. [DOI] [PubMed] [Google Scholar]
- Duarte D., Hawkins E.D., Akinduro O., Ang H., De Filippo K., Kong I.Y., Haltalli M., Ruivo N., Straszkowski L., Vervoort S.J., McLean C. Inhibition of endosteal vascular niche remodeling rescues hematopoietic stem cell loss in AML. Cell Stem Cell. 2018;22:64–77.e66. doi: 10.1016/j.stem.2017.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frodermann V., Rohde D., Courties G., Severe N., Schloss M.J., Amatullah H., McAlpine C.S., Cremer S., Hoyer F.F., Ji F., van Koeverden I.D. Exercise reduces inflammatory cell production and cardiovascular inflammation via instruction of hematopoietic progenitor cells. Nat. Med. 2019;25:1761–1771. doi: 10.1038/s41591-019-0633-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster J.J., MacLauchlan S., Zuriaga M.A., Polackal M.N., Ostriker A.C., Chakraborty R., Wu C.L., Sano S., Muralidharan S., Rius C., Vuong J. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355:842–847. doi: 10.1126/science.aag1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez D., Baylis R.A., Durgin B.G., Newman A.A., Alencar G.F., Mahan S., Hilaire C.S., Müller W., Waisman A., Francis S.E., Pinteaux E. Interleukin-1beta has atheroprotective effects in advanced atherosclerotic lesions of mice. Nat. Med. 2018;24:1418–1429. doi: 10.1038/s41591-018-0124-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho Y.H., Del Toro R., Rivera-Torres J., Rak J., Korn C., García-García A., Macías D., González-Gómez C., Del Monte A., Wittner M., Waller A.K. Remodeling of bone marrow hematopoietic stem cell niches promotes myeloid cell expansion during premature or physiological aging. Cell Stem Cell. 2019;25:407–418.e6. doi: 10.1016/j.stem.2019.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilatovskaya D.V., Pitts C., Clayton J., Domondon M., Troncoso M., Pippin S., DeLeon-Pennell K.Y. CD8(+) T-cells negatively regulate inflammation post-myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2019;317 doi: 10.1152/ajpheart.00112.2019. H581–h596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S., Ebert B.L. Clonal hematopoiesis in human aging and disease. Science. 2019;366:eaan4673. doi: 10.1126/science.aan4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S., Fontanillas P., Flannick J., Manning A., Grauman P.V., Mar B.G., Lindsley R.C., Mermel C.H., Burtt N., Chavez A., Higgins J.M. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014;371:2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S., Natarajan P., Silver A.J., Gibson C.J., Bick A.G., Shvartz E., McConkey M., Gupta N., Gabriel S., Ardissino D., Baber U. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 2017;377:111–121. doi: 10.1056/NEJMoa1701719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan M., Snyder T.M., Corces-Zimmerman M.R., Vyas P., Weissman I.L., Quake S.R., Majeti R. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci. Transl Med. 2012;4:149ra118. doi: 10.1126/scitranslmed.3004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanagh D.P., Kalia N. Hematopoietic stem cell homing to injured tissues. Stem Cell Rev. Rep. 2011;7:672–682. doi: 10.1007/s12015-011-9240-z. [DOI] [PubMed] [Google Scholar]
- Kovtonyuk L.V., Fritsch K., Fritsch K., Feng X., Manz M.G., Takizawa H. Inflamm-aging of hematopoiesis, hematopoietic stem cells, and the bone marrow microenvironment. Front. Immunol. 2016;7:502. doi: 10.3389/fimmu.2016.00502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagraauw H.M., Wezel A., van der Velden D., Kuiper J., Bot I. Stress-induced mast cell activation contributes to atherosclerotic plaque destabilization. Sci. Rep. 2019;9:2134. doi: 10.1038/s41598-019-38679-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legere S.A., Haidl I.D., Legare J.F., Marshall J.S. Mast cells in cardiac fibrosis: new insights suggest opportunities for intervention. Front. Immunol. 2019;10:580. doi: 10.3389/fimmu.2019.00580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leimkuhler N.B., Schneider R.K. Inflammatory bone marrow microenvironment. Hematol. Am Soc Hematol Educ Program. 2019;2019:294–302. doi: 10.1182/hematology.2019000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenkiewicz A.M., Adamiak M., Thapa A., Bujko K., Pedziwiatr D., Abdel-Latif A.K., Kucia M., Ratajczak J., Ratajczak M.Z. The Nlrp3 inflammasome orchestrates mobilization of bone marrow-residing stem cells into peripheral blood. Stem Cell Rev. Rep. 2019;15:391–403. doi: 10.1007/s12015-019-09890-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoni C., Montagner S., Rinaldi A., Bertoni F., Polletti S., Balestrieri C., Monticelli S. Dnmt3a restrains mast cell inflammatory responses. Proc. Natl. Acad. Sci. U S A. 2017;114 doi: 10.1073/pnas.1616420114. E1490–e1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Cai X., Cai C.L., Wang J., Zhang W., Petersen B.E., Yang F.C., Xu M. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. 2011;118:4509–4518. doi: 10.1182/blood-2010-12-325241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P., Buring J.E., Badimon L., Hansson G.K., Dean eld J., Bittencourt M.S., Tokgözoğlu, Lewis E.F. Atherosclerosis. Nat. Rev. Dis. Primers. 2019;5:56. doi: 10.1038/s41572-019-0106-z. [DOI] [PubMed] [Google Scholar]
- Libby P., Sidlow R., Lin A.E., Gupta D., Jones L.W., Moslehi J., Zeiher A., Jaiswal S., Schulz C., Blankstein R., Bolton K.L. Clonal hematopoiesis: crossroads of aging, cardiovascular disease, and cancer: JACC review topic of the week. J. Am. Coll. Cardiol. 2019;74:567–577. doi: 10.1016/j.jacc.2019.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P., Kobold S. Inflammation: a common contributor to cancer, aging, and cardiovascular diseases-expanding the concept of cardio-oncology. Cardiovasc. Res. 2019;115:824–829. doi: 10.1093/cvr/cvz058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackins C.J., Kano S., Seyedi N., Schäfer U., Reid A.C., Machida T., Silver R.B., Levi R. Cardiac mast cell-derived renin promotes local angiotensin formation, norepinephrine release, and arrhythmias in ischemia/reperfusion. J. Clin. Invest. 2006;116:1063–1070. doi: 10.1172/JCI25713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malato A., Saccullo G., Fazio G., Vergara B., Raso S., Paolo Guarneri G., Russo A., Abbadessa V., Siragusa S. Drug-related cardiotoxicity for the treatment of haematological malignancies in elderly. Curr. Pharm. Des. 2010;16:2872–2879. doi: 10.2174/138161210793176446. [DOI] [PubMed] [Google Scholar]
- Mantovani A., Dinarello C.A., Molgora M., Garlanda C. Interleukin-1 and related cytokines in the regulation of inflammation and immunity. Immunity. 2019;50:778–795. doi: 10.1016/j.immuni.2019.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mas-Peiro S., Hoffmann J., Fichtlscherer S., Dorsheimer L., Rieger M.A., Dimmeler S., Vasa-Nicotera M., Zeiher A.M. Clonal haematopoiesis in patients with degenerative aortic valve stenosis undergoing transcatheter aortic valve implantation. Eur. Heart J. 2020;41:933–939. doi: 10.1093/eurheartj/ehz591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayle A., Yang L., Rodriguez B., Zhou T., Chang E., Curry C.V., Challen G.A., Li W., Wheeler D., Rebel V.I., Goodell M.A. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood. 2015;125:629–638. doi: 10.1182/blood-2014-08-594648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagner S., Leoni C., Emming S., Della Chiara G., Balestrieri C., Barozzi I., Piccolo V., Togher S., Ko M., Rao A., Natoli G. TET2 regulates mast cell differentiation and proliferation through catalytic and non-catalytic activities. Cell Rep. 2016;15:1566–1579. doi: 10.1016/j.celrep.2016.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudersbach T., Siuda D., Kohlstedt K., Fleming I. Epigenetic control of the angiotensin-converting enzyme in endothelial cells during inflammation. PLoS One. 2019;14:e0216218. doi: 10.1371/journal.pone.0216218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa T., Wada H., Ohashi H., Iwamoto N., Ohta H., Kirii H., Fujii H., Saito K., Seishima M. Interferon-gamma produced by bone marrow-derived cells attenuates atherosclerotic lesion formation in LDLR-deficient mice. J. Atheroscler. Thromb. 2004;11:79–87. doi: 10.5551/jat.11.79. [DOI] [PubMed] [Google Scholar]
- Peled M., Fisher E.A. Dynamic aspects of macrophage polarization during atherosclerosis progression and regression. Front. Immunol. 2014;5:579. doi: 10.3389/fimmu.2014.00579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras E.M., Mirantes-Barbeito C., Fong S., Loeffler D., Kovtonyuk L.V., Zhang S., Lakshminarasimhan R., Chin C.P., Techner J.M., Will B., Nerlov C. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol. 2016;18:607–618. doi: 10.1038/ncb3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid A.C., Silver R.B., Levi R. Renin: at the heart of the mast cell. Immunol. Rev. 2007;217:123–140. doi: 10.1111/j.1600-065X.2007.00514.x. [DOI] [PubMed] [Google Scholar]
- Reynaud D., Pietras E., Barry-Holson K., Mir A., Binnewies M., Jeanne M., Sala-Torra O., Radich J.P., Passegué E. IL-6 controls leukemic multipotent progenitor cell fate and contributes to chronic myelogenous leukemia development. Cancer Cell. 2011;20:661–673. doi: 10.1016/j.ccr.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker P.M., Everett B.M., Thuren T., MacFadyen J.G., Chang W.H., Ballantyne C., Fonseca F., Nicolau J., Koenig W., Anker S.D. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017;377:1119–1131. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- Rizzacasa B., Amati F., Romeo F., Novelli G., Mehta J.L. Epigenetic modification in coronary atherosclerosis: JACC review topic of the week. J. Am. Coll. Cardiol. 2019;74:1352–1365. doi: 10.1016/j.jacc.2019.07.043. [DOI] [PubMed] [Google Scholar]
- Sanden C., Lilljebjorn H., Pietras C.O., Henningsson R., Saba K.H., Landberg N., Thorsson H., von Palffy S., Peña-Martinez P., Högberg C., Rissler M. Clonal competition within complex evolutionary hierarchies shapes AML over time. Nat. Commun. 2020;11:579. doi: 10.1038/s41467-019-14106-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano S., Oshima K., Wang Y., Katanasaka Y., Sano M., Walsh K. CRISPR-mediated gene editing to assess the roles of Tet2 and Dnmt3a in clonal hematopoiesis and cardiovascular disease. Circ. Res. 2018;123:335–341. doi: 10.1161/CIRCRESAHA.118.313225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano S., Oshima K., Wang Y., MacLauchlan S., Katanasaka Y., Sano M., Zuriaga M.A., Yoshiyama M., Goukassian D., Cooper M.A., Fuster J.J. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1beta/NLRP3 inflammasome. J. Am. Coll. Cardiol. 2018;71:875–886. doi: 10.1016/j.jacc.2017.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano S., Wang Y., Yura Y., Sano M., Oshima K., Yang Y., Katanasaka Y., Min K.D., Matsuura S., Ravid K. JAK2 (V617F)-mediated clonal hematopoiesis accelerates pathological remodeling in murine heart failure. JACC Basic Transl. Sci. 2019;4:684–697. doi: 10.1016/j.jacbts.2019.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasine J.P., Yeo K.T., Chute J.P. Concise review: paracrine functions of vascular niche cells in regulating hematopoietic stem cell fate. Stem Cells Transl. Med. 2017;6:482–489. doi: 10.5966/sctm.2016-0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiattarella G.G., Sequeira V., Ameri P. Distinctive patterns of inflammation across the heart failure syndrome. Heart Fail Rev. 2020 doi: 10.1007/s10741-020-09949-5. [DOI] [PubMed] [Google Scholar]
- Sun J., Sukhova G.K., Wolters P.J., Yang M., Kitamoto S., Libby P., MacFarlane L.A., Mallen-St Clair J., Shi G.P. Mast cells promote atherosclerosis by releasing proinflammatory cytokines. Nat. Med. 2007;13:719–724. doi: 10.1038/nm1601. [DOI] [PubMed] [Google Scholar]
- Swirski F.K., Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339:161–166. doi: 10.1126/science.1230719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swirski F.K., Nahrendorf M. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat. Rev. Immunol. 2018;18:733–744. doi: 10.1038/s41577-018-0065-8. [DOI] [PubMed] [Google Scholar]
- Tabas I., Lichtman A.H. Monocyte-macrophages and T cells in atherosclerosis. Immunity. 2017;47:621–634. doi: 10.1016/j.immuni.2017.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M. Cell-specific roles of NLRP3 inflammasome in myocardial infarction. J. Cardiovasc. Pharmacol. 2019;74:188–193. doi: 10.1097/FJC.0000000000000709. [DOI] [PubMed] [Google Scholar]
- Tapia-Vieyra J.V., Delgado-Coello B., Mas-Oliva J. Atherosclerosis and cancer; a resemblance with far-reaching implications. Arch. Med. Res. 2017;48:12–26. doi: 10.1016/j.arcmed.2017.03.005. [DOI] [PubMed] [Google Scholar]
- Tourki B., Halade G. Leukocyte diversity in resolving and nonresolving mechanisms of cardiac remodeling. FASEB J. 2017;31:4226–4239. doi: 10.1096/fj.201700109R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagnozzi R.J., Maillet M., Sargent M.A., Khalil H., Johansen A.K., Schwanekamp J.A., York A.J., Huang V., Nahrendorf M., Sadayappan S., Molkentin J.D. An acute immune response underlies the benefit of cardiac stem cell therapy. Nature. 2020;577:405–409. doi: 10.1038/s41586-019-1802-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia-Morales Mdel P., Zaina S., Heyn H., Carmona F.J., Varol N., Sayols S., Condom E., Ramírez-Ruz J., Gomez A., Moran S., Lund G. The DNA methylation drift of the atherosclerotic aorta increases with lesion progression. BMC Med. Genomics. 2015;8:7. doi: 10.1186/s12920-015-0085-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Valk F.M., Kuijk C., Verweij S.L., Stiekema L.C., Kaiser Y., Zeerleder S., Nahrendorf M., Voermans C., Stroes E.S. Increased haematopoietic activity in patients with atherosclerosis. Eur. Heart J. 2017;38:425–432. doi: 10.1093/eurheartj/ehw246. [DOI] [PubMed] [Google Scholar]
- Veninga A., De Simone I., Heemskerk J.W., ten Cate H., van der Meijden P.E. Clonal haematopoietic mutations linked to platelet traits and the risk of thrombosis or bleeding. Haematologica. 2020 doi: 10.3324/haematol.2019.235994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Yura Y., Ke Z., Sano M., Oshima K., Ogawa H., Horitani K., Min K.D., Miura-Yura E., Kour A. Tet2-mediated clonal hematopoiesis in nonconditioned mice accelerates age-associated cardiac dysfunction. JCI Insight. 2020;5:e135204. doi: 10.1172/jci.insight.135204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems S., Vink A., Bot I., Quax P.H., de Borst G.J., de Vries J.P., van de Weg S.M., Moll F.L., Kuiper J., Kovanen P.T., de Kleijn D.P. Mast cells in human carotid atherosclerotic plaques are associated with intraplaque microvessel density and the occurrence of future cardiovascular events. Eur. Heart J. 2013;34:3699–3706. doi: 10.1093/eurheartj/eht186. [DOI] [PubMed] [Google Scholar]
- Winkels H., Ehinger E., Ghosheh Y., Wolf D., Ley K. Atherosclerosis in the single-cell era. Curr. Opin. Lipidol. 2018;29:389–396. doi: 10.1097/MOL.0000000000000537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolach O., Sellar R.S., Martinod K., Cherpokova D., McConkey M., Chappell R.J., Silver A.J., Adams D., Castellano C.A., Schneider R.K., Padera R.F. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl Med. 2018;10:eaan8292. doi: 10.1126/scitranslmed.aan8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M., Passegue E. TNF-alpha coordinates hematopoietic stem cell survival and myeloid regeneration. Cell Stem Cell. 2019;25:357–372.e7. doi: 10.1016/j.stem.2019.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young A.L., Challen G.A., Challen G.A., Birmann B.M., Druley T.E. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun. 2016;7:12484. doi: 10.1038/ncomms12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu V.W.C., Yusuf R.Z., Oki T., Wu J., Saez B., Wang X., Cook C., Baryawno N., Ziller M.J., Lee E., Gu H. Epigenetic memory underlies cell-autonomous heterogeneous behavior of hematopoietic stem cells. Cell. 2016;167:1310–1322.e7. doi: 10.1016/j.cell.2016.10.045. [DOI] [PubMed] [Google Scholar]
- Zambetti N.A., Ping Z., Ping Z., Chen S., Kenswil K.J., Mylona M.A., Sanders M.A., Hoogenboezem R.M., Bindels E.M., Adisty M.N. Mesenchymal inflammation drives genotoxic stress in hematopoietic stem cells and predicts disease evolution in human pre-leukemia. Cell Stem Cell. 2016;19:613–627. doi: 10.1016/j.stem.2016.08.021. [DOI] [PubMed] [Google Scholar]
- Zampetaki A., Zeng L., Margariti A., Xiao Q., Li H., Zhang Z., Pepe A.E., Wang G., Habi O., deFalco E. Histone deacetylase 3 is critical in endothelial survival and atherosclerosis development in response to disturbed flow. Circulation. 2010;121:132–142. doi: 10.1161/CIRCULATIONAHA.109.890491. [DOI] [PubMed] [Google Scholar]