

Abstract
Introduction
Amyloid beta (Aβ) deposition was identified to precede tau pathology and neurodegeneration in familial Alzheimer's disease (AD). But the divergence between sporadic and familial AD limits the extension of these findings to sporadic AD.
Methods
Longitudinal changes of biomarkers among different stages were assessed using linear mixed‐effects models. The slopes of the models were used to estimate rates of change to calculate the biomarker trajectories in sporadic AD.
Results
Cerebrospinal fluid (CSF) Aβ was estimated to decline 45.2 years (abnormal: 27.8 years) before dementia, and Aβ deposition seemed to increase 31.7 years (abnormal: 26.7 years) before dementia. It was estimated to take 29.0 years (CSF t‐tau), 12.2 years (memory), 11.6 years (hippocampus), 9.3 years (hypometabolism), and 6.1 years (cognition) to move from normal to dementia.
Discussion
The trajectory in sporadic AD is led by Aβ accumulation, followed by CSF t‐tau increase, memory deficits, brain atrophy, hypometabolism, and cognitive decline.
Keywords: amyloid beta, Alzheimer's disease, biomarkers, clinical, tau
1. BACKGROUND
Alzheimer's disease (AD) is a neurodegenerative disorder clinically characterized by early memory loss and progressing into dementia. 1 It has been documented that amyloid beta (Aβ) deposition, tau pathology, and neuronal degeneration precede clinical symptoms. 2 , 3 , 4 , 5 , 6 The long preclinical phase of AD is an opportunity to identify the changes in pathophysiological biomarkers that might improve diagnosis and prognosis, and offer the opportunity for prevention trials. 7 , 8 , 9 , 10 Establishment of a dynamic model of biomarkers of AD is important for improving the design of clinical treatment trials and developing new effective interventions.
To date, cross‐sectional and longitudinal data in autosomal dominant AD identified that Aβ deposition emerged as an upstream event in the pathogenesis of AD and was associated with downstream pathophysiologic changes (ie, tau pathology and neurodegeneration). 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 Aβ has been observed to aggregate and accumulate in the brain in the preclinical phase of AD until a critical threshold is reached, with a sigmoidal trajectory characterized by rapid progression and finally a plateau. 19 A significant portion of the published data on AD progression is based on autosomal dominant AD, and the extension to sporadic AD remains to be verified, a task complicated by mixed pathology due to non‐AD pathologic change and aging in elderly individuals. 1 , 20
The Alzheimer's Disease Neuroimaging Initiative (ADNI) observational study enrolled a large cohort of sporadic AD (preclinical AD, prodromal AD, and dementia due to AD) collecting longitudinal cerebrospinal fluid (CSF) proteins (Aβ, t‐tau, and p‐tau), neuroimaging measurements, and cognitive functions at assessments separated by approximately a year. 21 Here, we compared the levels and the change rates of in vivo biomarkers among the three stages (preclinical AD, prodromal AD, and dementia due to AD), and attempted to describe in this longitudinal study the clinical and biomarker trajectories in sporadic AD.
2. METHODS
2.1. ADNI dataset
ADNI is a large, multicenter, longitudinal neuroimaging study, launched in 2003 by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, the Food and Drug Administration, pharmaceutical companies, and nonprofit organizations. 21 The study was approved by the institutional review boards of all participating centers and written informed consent was obtained from all participants or authorized representatives after extensive description of the ADNI based on the 1975 Declaration of Helsinki. The participants in this study were from the ADNI database (http://adni.loni.usc.edu). Inclusion criteria for AD subjects included National Institute of Neurological and Communication Disorders/Alzheimer's Disease and Related Disorders Association criteria for probable AD (NINCDS–ADRDA) 22 with a Mini‐Mental State Examination (MMSE) score between 20 and 26, a global Clinical Dementia Rating (CDR) of 0.5 or 1, and a sum‐of‐boxes Clinical Dementia Rating (CDR‐SB) of 1.0 to 9.0. All amnestic mild cognitive impairment (aMCI) subjects fulfilled a MMSE score of 24 to 30 and a Memory Box score of at least 0.5. Participants who had any serious neurological disease other than possible AD, any history of brain lesions or head trauma, or psychoactive medication use were excluded from this study. Details of the ADNI cohort can be found online (http://adni.loni.usc.edu/wp-content/uploads/2010/09/ADNI_GeneralProceduresManual.pdf).
2.2. CSF proteins examination
The data regarding CSF proteins were obtained from “UPENN CSF Biomarker Master [ADNI1, GO, 2] Version 2016‐07‐05.csv” online (http://loni.usc.edu/). The detailed methods of measurement of CSF proteins test are described elsewhere. 23 Based on the National Institute on Aging‐Alzheimer's Association (NIA‐AA) criteria, a cutoff value was necessary to categorize the Aβ1‐42 concentrations into normal and abnormal levels. Based on Shaw et al., 24 , 25 a cutoff value for CSF Aβ1‐42 ≤192 pg/mL was used to identify an abnormal level.
2.3. Neuroimaging methods
The positron emission tomography (PET) imaging data with amyloid tracer, florbetapir (AV‐45), were from the UC Berkeley–AV45 analysis dataset (“UC Berkeley ‐ AV45 Analysis [ADNIGO, 2] Version 2020‐05‐12.csv”) available online (http://adni.loni.usc.edu/). The native‐space magnetic resonance imaging (MRI) scan for each participant was segmented with Freesurfer (version 4.5.0) to define cortical gray matter regions of interest (ROI; frontal, anterior/posterior cingulate, lateral parietal, lateral temporal) that make up a summary cortical ROI. A composite reference region was defined as reference region. Each florbetapir scan was applied to the corresponding MRI and the mean florbetapir uptake calculated within the cortical and reference region. Finally, florbetapir standard uptake value ratios (SUVRs) were created by averaging across the four cortical regions and dividing this cortical summary ROI by the reference region. A cutoff value of 0.79 defined positive or negative amyloid.
The MRI data were extracted from the dataset (“UCSF – cross‐Sectional FreeSurfer (5.1) [ADNI1, GO, 2] Version 2019‐11‐08.csv”), and measurements of cerebral metabolic fluorodeoxyglucose PET (FDG‐PET) were downloaded from the dataset (“UC Berkeley – FDG Analysis [ADNI1, GO, 2] Version 2020‐05‐28.csv”) in the ADNI dataset. A detailed description of the acquisition and processing of the imaging data from ADNI has been previously described. 26 In this study, we used hippocampal atrophy as a MRI‐related marker and cerebral metabolism rate for glucose (CMRgl) of the bilateral posterior cingulate as FDG‐PET markers for our analysis.
2.4. Cognitive assessment
To assess memory function, we used a composite memory score from the database “UW – Neuropsych Summary Scores [ADNI1, GO, 2] Version 2020‐03‐26.csv” available online (http://adni.loni.usc.edu/). This was a weighted score based on memory items in the Rey Auditory Verbal Learning Test (RAVLT), the Alzheimer's Disease Assessment Scale‐Cognitive (ADAS‐cog), the MMSE, and Logical Memory. In addition, the CDR‐SB score was used to evaluate general cognition and the CDR‐SB score was extracted from the merged ADNI database (“ADNIMERGE.csv”).
2.5. Statistical analysis
Based on the NIA‐AA criteria, the ADNI participants were divided into four groups. Because the Aβ markers have a higher specificity than did other markers in the diagnosis of AD, only Aβ markers (CSF Aβ or amyloid imaging) were used for categorization. 1 Dementia due to AD required meeting criteria for dementia and positive Aβ markers (either decreased Aβ1‐42 in CSF or elevated SUVR on amyloid PET). MCI due to AD required mild cognitive impairment and abnormal Aβ markers. Both preclinical AD and healthy controls required normal cognitive function, whereas preclinical AD also required positive Aβ markers.
Baseline differences between the four groups were analyzed using one‐way analysis of variance (ANOVA) for continuous variables and Pearson chi‐square analysis for categorical variables. The Tukey honestly significant difference (HSD) method was used to evaluate differences between two groups in post hoc analyses. A within‐subjects linear mixed‐effects model with data from at least two visits was used to assess how clinical and biomarkers changed over time in the four groups, with fixed effects of age, sex, time, years of education, and apolipoprotein E (APOE ε4) status, and random effects of random intercepts and slopes (see supporting information). The time intervals to progress to the next stage were estimated based on the differences between the mean values of different stages and the rates of change of variables in the longitudinal study. All models were fitted with the lmer function in the lme4 package in R, version 3.1.3. Estimates and upper and lower quartiles were based on parametric bootstrapping of the fitted model by use of the sim function in the arm package, with 10,000 replicates.
Research in context
Systematic review: We reviewed available English language literature in PubMed for the clinical and biomarker changes in Alzheimer's disease (AD). It has been well documented that amyloid beta (Aβ) deposition, tau pathology, and neuronal degeneration precede clinical symptoms in autosomal dominant AD. Because the divergence on pathogenesis between sporadic and familial AD significantly limits the extension of these findings to sporadic AD, the dynamic model of biomarkers in sporadic AD remains to be further characterized and is of great importance to clinical understanding and management.
Interpretation: Our findings identified that the trajectory of biomarkers in sporadic AD is led by Aβ accumulation; followed by CSF t‐tau increase, memory deficits, brain atrophy, hypometabolism; and last, cognition decline.
Future directions: Learning the trajectories of biomarkers in sporadic AD will be imperative to evaluating the development and progression of AD, and beneficial for screening participants in AD clinical trials.
3. RESULTS
3.1. Study participants
From the 1735 subjects in the ADNI database, 1215 subjects underwent CSF Aβ examination or amyloid imaging test (Figure 1). Based on the 2011 NIA‐AA criteria, 167 subjects who presented normal cognitive function and negative Aβ markers in CSF or amyloid imaging test were classified as healthy controls, and 133 patients with preclinical AD had normal cognitive function and abnormal Aβ burden. The MCI due to AD group (n = 451) showed mild cognitive impairment on cognition assessment and abnormal Aβ burden, and the dementia due to AD patients (n = 231) met dementia criteria on the cognitive assessment, and also had evidence of Aβ pathology in CSF or amyloid imaging.
FIGURE 1.
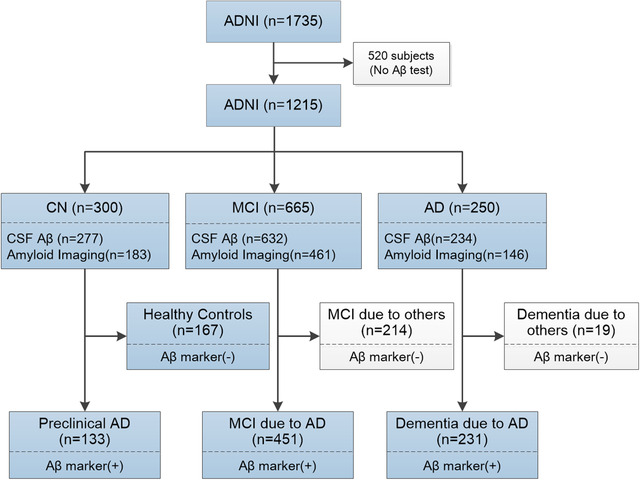
Classification of Alzheimer's Disease Neuroimaging Initiative subjects. Aβ, amyloid beta; AD, Alzheimer's disease; MCI, mild cognitive impairment; CSF, cerebrospinal fluid; Aβ marker including CSF Aβ and amyloid imaging
The demographic characteristics of the included subjects are listed in Table S1A in supporting information. The healthy controls, preclinical AD, MCI due to AD, and dementia due to AD patients significantly differed on age, sex, and education years. The frequency of APOE ε4 carriers was higher in dementia due to AD, MCI due to AD, and preclinical AD patients than in healthy controls. The four groups differed significantly on CSF proteins (Aβ1‐42, t‐tau, and p‐tau181) levels, Aβ deposition on amyloid imaging, hippocampal atrophy on MRI, cerebral hypometabolism on FDG‐PET, and cognitive assessment at baseline (P < .01; Table S1A).
3.2. CSF proteins
Our study included 521 subjects who underwent at least two CSF tests. The levels of CSF proteins and their annual rate of change among the groups are listed in Table S1A and Table S2B in supporting information. The difference in CSF Aβ1‐42 between preclinical AD patients and healthy controls was significantly larger than that between MCI due to AD and preclinical AD, and that between dementia due to AD and MCI due to AD (Figure 2A, Table S1B, and Table S3A–C in supporting information). It indicated that the decrease of Aβ1‐42 in CSF primarily occurred during the progression from normal cognition to preclinical AD (Figure 3A), and it was estimated to take 32.6 (25th–75th: 25.7–44.4) years, 5.2 (25th–75th: 3.8–8.5) years, and 7.4 (25th–75th: 6.4–8.9) years to change from healthy controls to preclinical AD, from preclinical AD to MCI due to AD, and from MCI due to AD to dementia due to AD, respectively (Figure 3A). Anchoring of the curve at the 192 pg/mL (cutoff value) showed that it may take 17.6 (25th–75th: 13.9–24.0) years to transition from 241.87 pg/mL (healthy controls) to below the cutoff of 192 pg/mL (Figure 2A). Both of these sets of evidence indicated the whole span of the CSF Aβ1‐42 to transition from normal level to dementia due to AD was 45.2 years, and CSF Aβ1‐42 was initially abnormal 27.8 years before the onset of dementia.
FIGURE 2.
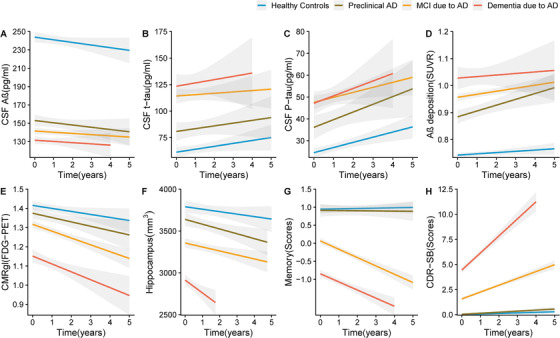
Biomarkers at baseline and in the longitudinal study. A, CSF Aβ. Lope (Dementia due to AD: –0.045, MCI due to AD: –1.26, Preclinical AD: –0.126, Healthy controls: –2.00, P = .617). B, CSF t‐tau. Lope (Dementia due to AD: 1.45, MCI due to AD: 2.93, Preclinical AD: 3.63, Healthy controls: 1.07, P =.558). C, CSF p‐tau. Lope (Dementia due to AD: 3.03, MCI due to AD: 3.94, Preclinical AD: 3.36, Healthy controls: 1.40, P =.458). D, Amyloid imaging (SUVR). Lope (Dementia due to AD: 0.009, MCI due to AD: 0.008, Preclinical AD: 0.010, Healthy controls: 0.003, P < .01). E, CMRgl (FDG‐PET). Lope (Dementia due to AD: –0.06, MCI due to AD: –0.03, Preclinical AD: –0.02, Healthy controls: ‐0.01, P < .01. F, Hippocampus. Lope (Dementia due to AD: –139.88, MCI due to AD: –98.12, Preclinical AD: –67.23, Healthy controls: –47.25, P =.02. G, Composite Memory. Lope (Dementia due to AD: –0.23, MCI due to AD: –0.16, Preclinical AD: –0.04, Healthy controls: –0.01, P < .01. H, CDR‐SB. Lope (Dementia due to AD: 1.8, MCI due to AD: 0.70, Preclinical AD: 0.15, Healthy controls: 0.06, P < .01). Aβ, amyloid beta; AD, Alzheimer's disease; CDR‐SB, Clinical Dementia Rating scale Sum of Boxes; CMRgl, cerebral metabolism rate for glucose on FDG‐PET (fluorodeoxyglucose‐positron‐emission tomography); CSF, cerebrospinal fluid; MCI, mild cognitive impairment; SUVR, standard uptake value ratios on amyloid imaging;.
FIGURE 3.
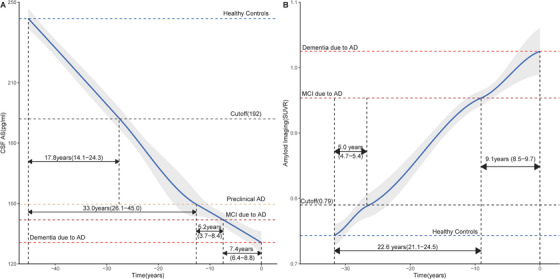
The dynamic change of Aβ markers in sporadic Alzheimer's disease. A, The change of Aβ in CSF. The time interval for CSF Aβ to transition from healthy controls to preclinical AD, from preclinical AD to MCI due to AD, and from MCI due to AD to dementia due to AD, was 32.6 (25th–75th: 25.7–44.4) years, 5.2 (25th–75th: 3.8–8.5) years, and 7.4 (25th‐=75th: 6.4–8.9) years, respectively. The time interval for CSF Aβ to transition from the level of healthy controls to cutoff value was 17.8 (25th–75th: 14.1–24.3) years. B, The change of Aβ deposition on amyloid imaging. The time interval to transition from healthy controls to MCI due to AD, and from MCI due to AD to dementia due to AD, was 22.6 (25th–75th: 21.1–24.5) years, and 9.1 (25th–75th: 8.5–9.7) years, respectively. The time interval for CSF Aβ to transition from the level of healthy controls to cutoff value was 5.0 (25th–75th: 4.7–5.4) years. Aβ, amyloid beta; AD, Alzheimer's disease; CSF, cerebrospinal fluid; MCI, mild cognitive impairment; SUVR, standard uptake value ratios on amyloid imaging.
We noted a significantly higher level of CSF tau (t‐tau and p‐tau181) in preclinical AD than in healthy controls, and a higher tau level in MCI due to AD than in the preclinical AD group (P < .05) (Table S4A‐C and Table S5A‐C in supporting information), but CSF tau in patients with dementia due to AD did not differ from that in patients with MCI due to AD (Figure 2B‐C, Table S1B). It suggested that the increased rate of CSF tau slowed as AD progressed. The time interval was about 23.4 (25th–75th: 18.8–31.1) years for CSF t‐tau to increase from normal level to the level of MCI due to AD, and 5.6 (25th–75th: 4.7–7.0) years from MCI due to AD to dementia due to AD (Figure S1 in supporting information). Thus, the whole span of CSF t‐tau to increase from normal level to dementia due to AD was about 29.0 years.
3.3. Amyloid imaging
There were 461 subjects with amyloid imaging who met our inclusion criteria. The SUVRs on amyloid imaging and their annual rates of change are listed in Tables S1A and S2A. We noted that the SUVR on amyloid imaging increased with disease progression (Figure 2D). Patients with preclinical AD had a faster rate of Aβ deposition than did healthy controls (P < .05; Table S2B, and Table S6A–C in supporting information). Anchoring of the curve at the 0.79 SUVR (cutoff value), it was estimated to take 5.0 (25th–75th: 4.7–5.4) years to progress from 0.74 SUVR (healthy controls) to 0.79 SUVR (Figure 3B). Furthermore, it was estimated to take 22.6 (25th–75th: 21.0–24.5) years to increase from 0.74 SUVR (healthy controls) to 0.95 SUVR (MCI due to AD), and 9.1 (25th–75th: 8.5–9.7.5) years to increase from 0.95 SUVR (MCI due to AD) to 1.02 SUVR (dementia due to AD). Thus, the entire time span of Aβ deposition to progress from healthy controls to dementia due to AD was 31.7 years, and SUVR on imaging became abnormal 26.7 years before the onset of symptoms.
3.4. Glucose metabolism
In the current study, 519 subjects underwent at least two FDG‐PET tests. The CMRgl on FDG‐PET and their annual rates of change are shown in Tables S1A and S2A. The CMRgl on FDG‐PET decreased with disease progression (Table S1B), and dementia due to AD patients showed the fastest rate of CMRgl decline among the four groups during follow‐up (P < .05; Figure 2E, Table S2B and Table S7A–C in supporting information). This suggested the decrease of cerebral metabolism accelerated as disease progressed. Furthermore, it was estimated to take 3.8 (25th–75th: 3.6–4.2) years to decline from 1.41 CMRgl (heathy controls) to 1.32 CMRgl (MCI due to AD), and 5.5 (25th–75th: 5.2–5.8) years to decline from 1.32 CMRgl to 1.16 CMRgl (dementia due to AD), which indicated it was estimated to take 9.3 years to decrease from normal levels to the level of dementia due to AD (Figure 2).
3.5. Hippocampal atrophy
In this study, 626 subjects were assessed at least twice using MRI. The size of the hippocampus and the annual rate of change are shown in Tables S1A and S2A. The volume of hippocampus decreased with disease progression (Table S1B), and patients with dementia due to AD had a higher hippocampal atrophy rate than did the other three groups in the longitudinal study (Figure 2F, Table S2B and Table S8A–C in supporting information). These results indicated the atrophy of the hippocampus accelerated as disease progressed. The time interval was 6.2 (25th–75th: 5.7–6.7) years for hippocampus decrease from 3836 mm3 (healthy controls) to 3431 mm3 (MCI due to AD), and 5.4 (25th–75th: 5.2–5.6) years to decrease from 3431 to 2947 mm3 (dementia due to AD), suggesting the whole span of the hippocampus to decrease from normal level to the level of dementia was 11.6 years (Figure 3).
3.6. Cognitive assessments
Here, 946 and 949 subjects received CDR‐SB and memory assessment at least twice, respectively. Patients with preclinical AD did not differ from healthy controls on CDR‐SB or memory assessment at baseline or on their change rates in follow‐up. We noted a worse performance on memory and cognition assessment in patients with dementia due to AD than those with MCI due to AD and preclinical AD (Table S1A–B). Similarly, patients with dementia due to AD had a faster rate of cognition decline than did patients with MCI due to AD and preclinical AD (Figure 2G–H, Table S2A‐B, Table S9A–C, and Table S10A–C in supporting information). Both of these results indicated that memory and cognitive function mostly declined during the MCI due to AD stage before the onset of dementia, and the decrease in memory and cognition accelerated as disease progressed. It was estimated to take 12.2 years for memory, and 6.1 years for cognition to change from the average level of healthy controls to that of dementia due to AD.
3.7. Combined model
Our study showed that Aβ deposition occurred first during the whole course of AD. It was estimated to take 45.2 years for CSF Aβ, and 31.7 years for amyloid imaging to change from the level of healthy controls to the level of dementia due to AD. Moreover, the Aβ biomarker began to be abnormal 27.8 years (for CSF) and 26.7 years (for amyloid imaging) before the onset of dementia. Then, CSF t‐tau started to increase, followed by memory impairment, hippocampal atrophy, and cerebral metabolism decline, and it was estimated to take 29.0 years for CSF t‐tau, 12.2 years for memory, 11.6 years for hippocampus volume, and 9.3 years for cerebral metabolism to change from normal levels to the levels of dementia due to AD. The general cognition declined with a time interval of 6.1 years for CDR‐SB to arrive at the level of dementia due to AD from the normal level (Figure 4).
FIGURE 4.
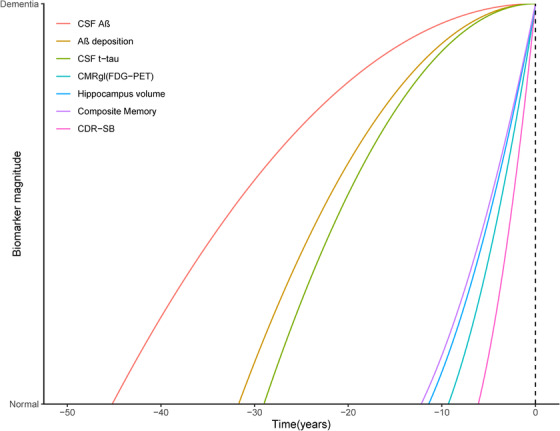
Sequence of biomarkers changes in Alzheimer's Disease Neuroimaging Initiative subjects. AD, Alzheimer's disease; CDR‐SB, Clinical Dementia Rating scale Sum of Boxes; CMRgl, cerebral metabolism rate for glucose on FDG‐PET (fluorodeoxyglucose‐positron‐emission tomography); CSF, cerebrospinal fluid; SUVR, standard uptake value ratios on amyloid imaging. Composite memory is a weighted score based on memory items in Rey Auditory Verbal Learning Test, the Alzheimer's Disease Assessment Scale‐Cognitive, the Mini‐Mental State Examination, and Logical Memory.
4. DISCUSSION
The 2011 NIA‐AA criteria for AD have incorporated biomarkers in the diagnosis of AD and expanded coverage of disease stages, from preclinical AD and MCI due to AD to dementia due to AD. 1 , 27 According to the new 2018 NIA‐AA framework for AD, 28 Aβ was the essential biomarker for the AD continuum. In the current study, we reclassified the included subjects with the Aβ assessment into four groups: healthy controls, preclinical AD, MCI due to AD, and dementia due to AD groups. The four groups differed significantly on the levels of clinical markers and biomarkers. In the dynamic model, CSF Aβ decreased first, followed by amyloid imaging, and the change rates of Aβ markers slowed down as AD progressed. When the levels of Aβ approached the threshold, CSF t‐tau was observed to increase; sequentially, memory function, hippocampal size, and cerebral metabolism began to decrease, and their change rates accelerated as the disease progressed. Finally, general cognition started to decline as dementia onset approached (Figure 5).
FIGURE 5.
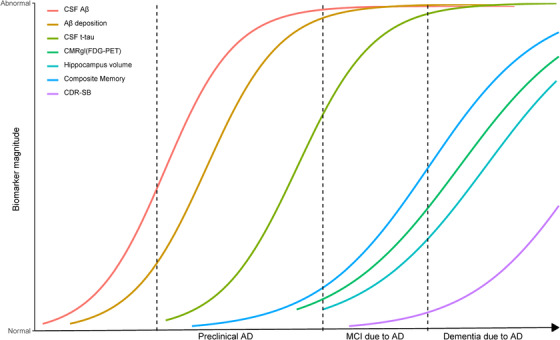
The trajectories of biomarkers in sporadic Alzheimer's disease. CDR‐SB, Clinical Dementia Rating scale Sum of Boxes; CMRgl, cerebral metabolism rate for glucose on FDG‐PET (fluorodeoxyglucose‐positron‐emission tomography); CSF, cerebrospinal fluid; SUVR, standard uptake value ratios on amyloid imaging. Composite memory is a weighted score based on memory items in Rey Auditory Verbal Learning Test, the Alzheimer's Disease Assessment Scale‐Cognitive, the MMSE and Logical Memory.
These findings identified the temporal occurrence of Aβ accumulation, CSF t‐tau increase, memory deficits, brain atrophy, hypometabolism, and cognitive decline in sporadic AD. Our study provided novel evidence for the hypothetical model of biomarkers based on sporadic AD pathological cascade established by Jack et al. 2 and Sperling et al. 7 Furthermore, we estimated the time interval for clinical and biomarkers to transition from normal level to the level of dementia due to AD based on the ADNI data. It may be useful to estimate the time interval for a patient with high risk for AD to archive the level of dementia, which was beneficial to assess disease severity, and screen the appropriate participants in clinical trials for AD.
Our current study supported Aβ deposition as the first phase of the natural history of sporadic AD. The time interval was 45.2 years for CSF Aβ and 31.7 years for amyloid imaging to reach the level of dementia from normal, respectively. This identified that CSF Aβ1‐42 decrease emerges prior to amyloid imaging, 29 which confirmed the findings that CSF Aβ1‐42 becomes abnormal before amyloid PET in sporadic AD, 30 and was consistent with the results from the longitudinal study in the Dominantly Inherited Alzheimer Network (DIAN) cohort. 13 Moreover, our study observed that the annual rate of change of CSF Aβ reduced as AD progressed, which supports Aβ accumulation progressing with a sigmoid‐shaped trajectory as a plausible course of sporadic AD. 19 , 31
Comparing our results to the findings from DIAN, 13 the dynamic changes of Aβ accumulation and tau pathology in sporadic AD were notably similar to those in autosomal dominant AD, suggesting that sporadic AD and familial AD may share similar basic mechanisms irrespective of the different pathogenic pathways. 13 Moreover, the trajectories of glucose metabolism and hippocampal atrophy in ADNI were comparable to those obtained from the data in DIAN, and earlier than that in the Australian Imaging, Biomarkers and Lifestyle (AIBL) study. 32 Inconsistency in data acquisition or analysis methods may account for some of the discrepancy. However, our analysis demonstrated that memory deficits occur before global cognitive impairment, 33 , 34 and even prior to glucose hypometabolism or brain atrophy, which was partially in agreement with the hypothetical model. 2 , 3 Aβ accumulation early in the disease affects hypoconnectivity in brain, but not brain atrophy or glucose hypometabolism. 35
The dynamic model of biomarkers must also be interpreted with caution. Unlike autosomal dominant AD, not all sporadic AD in the preclinical stage will progress into dementia. Thus, our findings represent population rather than individual trajectories, and it was essential to validate this model in an independent cohort. Moreover, the included participants only had a limited follow‐up and the time period may be too short to capture the whole course of AD. Follow‐up throughout the entire disease course would, of course, be ideal, but extremely challenging to achieve practically. Other vulnerabilities, such as age, education, genetic susceptibility, and lifestyle, may also contribute to the trajectories of biomarkers in sporadic AD development. 36 , 37 , 38 Furthermore, a positive amyloid test (CSF or PET) was needed to define AD based on the current evidence, but it limited us to make any inferences on where amyloid comes in the dynamic model of AD by making AD contingent on a positive amyloid test, and limits our claims about temporal ordering to other biomarkers and clinical tests.
5. CONCLUSIONS
In summary, the data from the ADNI cohort revealed the trajectory of biomarkers in sporadic AD with the sequence of Aβ accumulation, followed by CSF t‐tau, then memory deficits preceding hypometabolism and brain atrophy, and finally cognitive decline. This dynamic model provides insights into the progression of AD and facilitates the selection of participants and endpoints in clinical trials.
FUNDING INFORMATION
This study was supported by grants from the National Natural Science Foundation of China (91849126), the National Key R&D Program of China (2018YFC1314700), Shanghai Municipal Science and Technology Major Project (No.2018SHZDZX01) and ZHANGJIANG LAB, Tianqiao and Chrissy Chen Institute, and the State Key Laboratory of Neurobiology and Frontiers Center for Brain Science of Ministry of Education, Fudan University. Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. This study was also supported by grants from the National Natural Science Foundation of China (81471309) and the National Key R&D Program of China (2018YFC1314700, 2016YFC1305803).
CONFLICTS OF INTEREST
Jin‐Tai Yu serves as an associate editor‐in‐chief for Annals of Translational Medicine, and is senior editor for Journal of Alzheimer's Disease. Serge Gauthier has received clinical trial support from Lilly and Roche in DIAN‐TU, TauRx Therapeutics (TauRx), and Lundbeck; has been a data safety monitoring board (DSMB) member of ADCS, ATRI, API, and Eisai; and has been a scientific adviser to Affiris, Boehringer Ingelheim, Lilly, Roche, Servier, Sanofi, Schwabe, Takeda, and TauRx. All other authors declare no competing interests.
Supporting information
Supplementary Information
ACKNOWLEDGMENTS
The authors thank Prof. Clifford R. Jack Jr. and Prof. Randall J. Bateman for critical review.
Wang H‐F, Shen X‐N, Li J‐Q, et al. Clinical and biomarker trajectories in sporadic Alzheimer's disease: A longitudinal study. Alzheimer's Dement. 2020;12:e12095 10.1002/dad2.12095
Hui‐Fu Wang, Xue‐Ning Shen, and Jie‐Qiong Li contributed equally to this work.
Data used in preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at:
REFERENCES
- 1. Jack CR, Jr. , Bennett DA, Blennow K, et al. NIA‐AA Research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14(4):535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jack CR, Jr. , Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9(1):119‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jack CR, Jr. , Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12(2):207‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang HF, Tan L, Cao L, et al. Application of the IWG‐2 diagnostic criteria for Alzheimer's disease to the ADNI. J Alzheimers Dis. 2016;51(1):227‐236. [DOI] [PubMed] [Google Scholar]
- 5. Oxtoby NP, Young AL, Cash DM, et al. Data‐driven models of dominantly‐inherited Alzheimer's disease progression. Brain. 2018;141(5):1529‐1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xiong C, Jasielec MS, Weng H, et al. Longitudinal relationships among biomarkers for Alzheimer disease in the Adult Children Study. Neurology. 2016;86(16):1499‐1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):280‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sperling RA, Karlawish J, Johnson KA. Preclinical Alzheimer disease‐the challenges ahead. Nat Rev Neurol. 2013;9(1):54‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dubois B, Epelbaum S, Nyasse F, et al. Cognitive and neuroimaging features and brain beta‐amyloidosis in individuals at risk of Alzheimer's disease (INSIGHT‐preAD): a longitudinal observational study. Lancet Neurol. 2018;17(4):335‐346. [DOI] [PubMed] [Google Scholar]
- 10. Soldan A, Pettigrew C, Cai Q, et al. Hypothetical preclinical Alzheimer disease groups and longitudinal cognitive change. JAMA Neurol. 2016;73(6):698‐705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE. Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol. 2004;61(3):378‐384. [DOI] [PubMed] [Google Scholar]
- 12. Fagan AM, Xiong C, Jasielec MS, et al. Longitudinal change in CSF biomarkers in autosomal‐dominant Alzheimer's disease. Sci Transl Med. 2014;6(226):ra30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012;367(9):795‐804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Benzinger TL, Blazey T, Jack CR, Jr. , et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer's disease. Proc Natl Acad Sci U S A. 2013;110(47):E4502‐E4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fleisher AS, Chen K, Quiroz YT, et al. Associations between biomarkers and age in the presenilin 1 E280A autosomal dominant Alzheimer disease kindred: a cross‐sectional study. JAMA Neurol. 2015;72(3):316‐324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gordon BA, Blazey TM, Su Y, et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer's disease: a longitudinal study. Lancet Neurol. 2018;17(3):241‐250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jack CR, Jr. , Vemuri P, Wiste HJ, et al. Evidence for ordering of Alzheimer disease biomarkers. Arch Neurol. 2011;68(2):1526‐1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schindler SE, Li Y, Todd KW, et al. Emerging cerebrospinal fluid biomarkers in autosomal dominant Alzheimer's disease. Alzheimers Dement. 2019;15(5):655‐665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jack CR, Jr. , Wiste HJ, Lesnick TG, et al. Brain beta‐amyloid load approaches a plateau. Neurology. 2013;80(10):890‐896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Caroli A, Frisoni GB. The dynamics of Alzheimer's disease biomarkers in the Alzheimer's Disease Neuroimaging Initiative cohort. Neurobiol Aging. 2010;31(8):1263‐1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weiner MW, Veitch DP, Aisen PS, et al. The Alzheimer's Disease Neuroimaging Initiative: a review of papers published since its inception. Alzheimers Dement. 2013;9(5):e111‐e194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34(7):939‐944. [DOI] [PubMed] [Google Scholar]
- 23. Fortea J, Vilaplana E, Alcolea D, et al. Cerebrospinal fluid beta‐amyloid and phospho‐tau biomarker interactions affecting brain structure in preclinical Alzheimer disease. Ann Neurol. 2014;76(2):223‐230. [DOI] [PubMed] [Google Scholar]
- 24. Trojanowski JQ, Vandeerstichele H, Korecka M, et al. Update on the biomarker core of the Alzheimer's Disease Neuroimaging Initiative subjects. Alzheimers Dement. 2010;6(3):230‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shaw LM, Vanderstichele H, Knapik‐Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol. 2009;65(4):403‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang D, Wang Y, Zhou L, Yuan H, Shen D, Alzheimer's Disease Neuroimaging I . Multimodal classification of Alzheimer's disease and mild cognitive impairment. Neuroimage. 2011;55(3):856‐867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jack CR, Jr. , Albert MS, Knopman DS, et al. Introduction to the recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):257‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jack CR, Jr. , Bennett DA, Blennow K, et al. NIA‐AA Research Framework: Toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14(4):535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Palmqvist S, Mattsson N, Hansson O, Alzheimer's Disease Neuroimaging I. Cerebrospinal fluid analysis detects cerebral amyloid‐beta accumulation earlier than positron emission tomography. Brain. 2016;139(Pt 4):1226‐1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Palmqvist S, Mattsson N, Hansson O. Cerebrospinal fluid analysis detects cerebral amyloid‐beta accumulation earlier than positron emission tomography. Brain. 2016;139(Pt 4):1226‐1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yau WY, Tudorascu DL, McDade EM, et al. Longitudinal assessment of neuroimaging and clinical markers in autosomal dominant Alzheimer's disease: a prospective cohort study. Lancet Neurol. 2015;14(8):804‐813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Villemagne VL, Burnham S, Bourgeat P, et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol. 2013;12(4):357‐367. [DOI] [PubMed] [Google Scholar]
- 33. Sutphen CL, McCue L, Herries EM, et al. Longitudinal decreases in multiple cerebrospinal fluid biomarkers of neuronal injury in symptomatic late onset Alzheimer's disease. Alzheimers Dement. 2018;14(7):869‐879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Toledo JB, Xie SX, Trojanowski JQ, Shaw LM. Longitudinal change in CSF Tau and Abeta biomarkers for up to 48 months in ADNI. Acta Neuropathol. 2013;126(5):659‐670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McDade E, Wang G, Gordon BA, et al. Longitudinal cognitive and biomarker changes in dominantly inherited Alzheimer disease. Neurology. 2018;91(14):e1295‐e1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tan L, Yu JT, Zhang W, et al. Association of GWAS‐linked loci with late‐onset Alzheimer's disease in a northern Han Chinese population. Alzheimers Dement. 2013;9(5):546‐553. [DOI] [PubMed] [Google Scholar]
- 37. Li JQ, Tan L, Wang HF, et al. Risk factors for predicting progression from mild cognitive impairment to Alzheimer's disease: a systematic review and meta‐analysis of cohort studies. J Neurol Neurosurg Psychiatry. 2016;87(5):476‐484. [DOI] [PubMed] [Google Scholar]
- 38. Xu W, Tan L, Wang HF, et al. Meta‐analysis of modifiable risk factors for Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2015;86(12):1299‐1306. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information