

Abstract
The bone marrow is a complex tissue in which heterogeneous populations of stromal cells interact with hematopoietic cells to dynamically respond to organismal needs in defense, hemostasis, and oxygen delivery. Physiologic challenges modify stromal/hematopoietic cell interactions to generate changes in blood cell production. When either stroma or hematopoietic cells are impaired, the system distorts. The distortions associated with myeloid malignancy are reviewed here and may provide opportunities for therapeutic intervention.
Introduction
The bone marrow (BM) microenvironment plays a crucial role in supporting normal hematopoiesis lifelong and is composed of multiple different cell types including endothelial, perivascular, mesenchymal stromal, osteolineage, neuronal, and hematopoietic cells.1-4 Primary alterations in the BM microenvironment would be predicted to alter homeostatic balance and foster hematologic disease. Less intuitive, but now experimentally supported, is the evidence that malignant cells can alter their local microenvironment and create an “unhealthy” niche advantaging abnormal cells at the expense of normal hematopoiesis. The underlying mechanisms, by which these pathologic exchanges occur are being defined and emerging from them are therapeutic strategies for diseases of hematopoiesis.
Niche changes can initiate myeloid dysplasia and neoplasia
Clinical observations such as donor-derived leukemia in transplant recipients, altered marrow stromal morphology in myelodysplasia, some myeloproliferative disorders, and HIV disease have raised the issue that the BM microenvironment can contribute to hematologic disease. The first experimental evidence came with retinoic acid receptor γ-deficient (RARγ−/−) mice inducing a myeloproliferative state when transplanted with wild-type (WT) hematopoietic cells.5 Elevated levels of tumor necrosis factor-α in RARγ−/− mice partially contributed to the observed phenotype. Similarly, conditional deletion of mind bomb 1, an E3 ubiquitin ligase regulating endocytosis of Notch ligands, resulted in lethal myeloproliferative neoplasm (MPN)-like disease in mice receiving WT BM.6 These studies indicate the potential for dysregulated stroma to drive myeloid cell production to pathologic levels. In some cases, concurrent genetic abnormalities were required in both the stroma and hematopoietic cells, such as for the retinoblastoma gene.7 Despite the evident abnormal expansion of myeloid cells, it is unclear whether this is due to malignant transformation or simply overexpansion of normal blood cells in response to excessive levels of proliferative cytokines. Furthermore, the cellular identity of the environmental source(s) responsible for the observed phenotypes was unknown.
Subsequent studies revealed that specific BM populations can initiate hematopoietic disease, including malignancies. Conditional deletion of the RNA-processing endonuclease enzyme Dicer 1 in primitive osterix (Osx)-expressing osteolineage cells resulted in an myelodysplastic syndrome (MDS)–like disease, which in some cases developed into acute myeloid leukemia (AML) (Figure 1).8 MDS/AML was induced by specific cells in the BM microenvironment, since deletion of Dicer1 in mature osteocalcin-expressing osteolineage cells did not result in a hematopoietic phenotype. The abnormal niche cells were both necessary and sufficient to induce the MDS-like state because transplantation of hematopoietic cells from Dicer1-deficient mice in WT environment completely reverted observed cytopenia and cellular dysplasia. Conversely, Dicer1-deficient mice transplanted with WT BM cells developed leukopenia, anemia, and thrombocytopenia. Interestingly, the AML that developed in some animals was transplantable, but only if the host environment was Dicer1 deficient. The AML cells had acquired multiple genetic mutations that did not include Dicer1, so these cells did not just represent cells with ectopic deletion of the targeted gene. Rather, the Dicer1 depleted niche fostered outgrowth of mutated hematopoietic cells and some degree of cooperativity between the abnormal microenvironment and the abnormal hematopoietic cells persisted to maintain the leukemia. These data support the interesting possibility that the multihit hypothesis of cancer first proposed by Knudson9 does not depend on the mutations all occurring in the same cell. The initiating mutational event may be within the niche, leading to niche driven oncogenesis.
Figure 1.
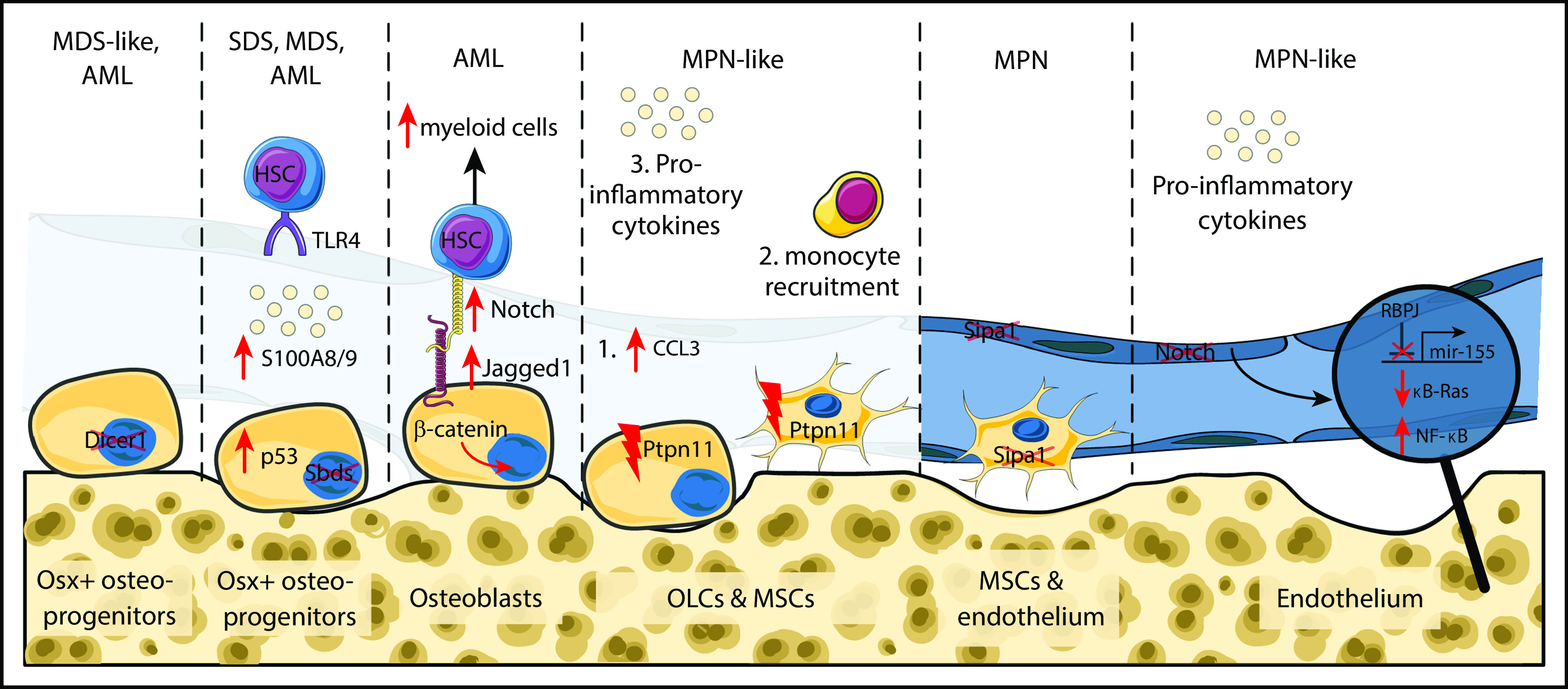
Schematic overview of cellular and molecular alterations in the bone marrow microenvironment leading to hematopoietic malignancies. Mice with conditional deletion of the RNA-processing endonuclease Dicer1 in osterix but not osteocalcin-expressing osteolineage cells developed MDS-like disease and AML. Similarly, deletion of the Sbds gene from osterix-expressing (Osx+) cells augmented p53 levels followed by elevated secretion of S100A8 and A9 proinflammatory cytokines. S100A8/9 bind to toll-like receptor 4 and altered physiological properties of HSCs. Mice with constitutively active β-catenin protein in osteoblasts manifested expansion of myeloid cells and development of AML. Activated osteoblasts upregulated Jagged1 expression on their cell surface which augments Notch signaling and shifts differentiation potential of HSCs. Osteolineage and mesenchymal stroma cells with activating mutations of tyrosine phosphatase non-receptor type 11 (Ptpn11) resulted in elevated levels of the chemokine CCL3, subsequent monocyte recruitment and secretion of proinflammatory cytokines that activated HSCs and caused MPN-like disease. MPN was also developed upon deletion of the signal-induced proliferation-associated gene 1 (Sipa1) from mesenchymal stroma and endothelial cells. Endothelial cells with abrogated canonical Notch signaling resulted in development of MPN-like disease. Activation of canonical Notch signaling results in proteolytic cleavage of the Notch intracellular domain and its translocation to the nucleus to activate transcription of Notch target genes via binding to the transcription factor recombination signal binding protein for immunoglobulin κ J region (RBPJ). In the absence of Notch signals, RBPJ acts as transcriptional repressor. Deletion of RBPJ upregulated mir-155, which by targeting an inhibitor of NF-κb signaling (κB-Ras1) resulted in NF-κB activation followed by elevated levels of inflammatory cytokines, including granulocyte colony-stimulating factor and tumor necrosis factor-α. This again elevated numbers of immature myeloid cells. MSCs, mesenchymal stroma cells; OLCs, osteolineage cells; SDS, Shwachman-Diamond syndrome. This figure was created using SMART Servier Medical Art Web site.
Other studies have supported this concept in different models. Osteoblasts expressing a constitutively active form of β-catenin shift the differentiation program of hematopoietic stem and progenitor cells (HSPCs) toward the myeloid lineage leading to AML.10 Activated osteoblasts upregulated Notch ligand jagged 1 triggering upregulation of Notch pathway in HSPCs, whereas Notch inhibition prevented leukemogenesis.10 It is important to note that elevated β-catenin levels have been observed in one-third of human AML patients. Interestingly, the effects of a specific alteration in the BM microenvironment can significantly vary depending on the biology of the underlying disease. For example, constitutive activation of parathyroid hormone receptor in osteoblasts accelerates MLL-AF9-driven AML, but delays progression of BCR-ABL1 chronic myeloid leukemia (CML)-like MPN.11
Osteolineage cells are not the only BM cell type implicated in leukemogenesis. Loss of canonical Notch signaling in endothelial cells leads to myeloproliferative-like disease, through constitutive activation of mir-155 and NF-κB signaling (Figure 1).12 Increased NF-κB signaling in BM endothelial and mesenchymal cells result in increased levels of proinflammatory cytokines (mainly granulocyte colony-stimulating factor and tumor necrosis factor-α), leading to expansion of immature myeloid cells and lethal myeloproliferative-like disease.12 In another study, activating mutations of the protein tyrosine phosphatase nonreceptor type 11 (Ptpn11) in mesenchymal stromal cells and osteoprogenitors, but not mature osteoblasts or endothelial cells, has also been shown to induce an MPN-like state (Figure 1). Mechanistically, elevated levels of the CCL3 chemokine triggered monocyte production of proinflammatory cytokines that activated hematopoietic stem cells (HSCs), with the hematopoietic phenotype being reversible with CCL3 antagonists.13
Deletion of signal-induced proliferation-associated gene 1 (Sipa1) expressed in mouse and human BM mesenchymal and endothelial cells also results in MPN.14 Similar effects have been observed when healthy donor cells are transplanted in Sipa1−/− recipients, suggesting again that alterations in the BM microenvironment are sufficient to drive disordered growth. Studying preleukemic malignancies, such as the Shwachman-Diamond syndrome, provided additional support for the role of BM in inducing hematological disorders by generating a genotoxic environment. Shwachman-Diamond syndrome is caused by mutations in the Sbds gene and is characterized by high probability to develop into MDS and AML. Sbds deletion from Osx+ BM mesenchymal cells led to increased p53 levels and secretion of the proinflammatory factors S100A8 and S100A9. Subsequently, S100A8/9 caused DNA damage and mitochondrial dysfunction in mouse and human HSPCs through binding to their canonical receptor (toll-like receptor 4).15 Notably, Sbds deletion from hematopoietic cells did not cause genotoxic/oxidative damage in HSPCs nor development of MDS/AML. In humans, S100A8/9+ patients exhibit a significantly shorter progression-free survival. This suggests that activation of the p53-S100A8/9 axis in BM mesenchymal cells has a predictive value in MDS progression. Recent studies provided evidence that non-genetic changes can also foster myeloid cell expansion. For example, premature or physiological aging remodels the BM microenvironment,16-19 resulting in lymphoid deficiency and myeloid skewing of HSC lineage output.20 Collectively, these data illustrate that genetic and nongenetic alterations in BM components may be involved in the initiation and progression of myeloid malignancies.3,21
Although the models used to demonstrate niche contributions to myeloid malignancies are mostly artificial, the principle that they illustrate is compelling: the niche–hematopoietic interface dictates the health of the hematopoietic system. Changes in the niche driving malignancy are likely to be extremely rare. Acquired niche alterations facilitating mutant myeloid cell outgrowth are the more likely scenario in human disease.
Malignant myeloid cell-mediated remodeling of the BM niche
The homeostatic regulation of blood production is mediated through reciprocal interactions between hematopoietic cells and the BM microenvironment. Healthy blood cells have been reported to feed back to niche cells.22 Malignant cells appear to have similar potential, with several studies illustrating their ability to remodel different BM components to favor disease progression at the expense of normal hematopoiesis.
Alterations in osteolineage cells or osteo-primed MSCs have been reported in several hematologic conditions. Using an inducible mouse to model chronic phase of human CML, Schepers and colleagues reported significant accumulation of endosteal osteoblasts and myelofibrotic tissue, without affecting MSC numbers.23 Through CCL3/THPO secretion and cell–cell mediated interactions, BCR-ABL+ CML cells modulated MSC differentiation toward functionally modified OLCs. Profiling of abnormal OLCs revealed elevated levels of transforming growth factor-β and interleukin-1 (IL-1)/tumor necrosis factor-α inflammatory signaling, and downregulation of key hematopoietic factors (Scf, Cxcl12, Angpt1, Tfgβ1). Those events seem to be CML-phase dependent because blast-crisis CML driven by simultaneous expression of BCR-ABL and Nup98/HoxA9 fusion proteins was associated with reduced bone-lining osteoblasts and bone mass.24 Interestingly, osteoclasts were not the main drivers of this phenotype because inhibition of osteoclast-mediated bone resorption only partially rescued bone loss. Remodeling of the BM microenvironment was also evident in MDS, with human MDS cells switching the molecular profile of healthy MSCs toward signatures associated with MDS patient-derived MSCs.25 This included induction of cytokines such as leukemia inhibitory factor25 previously reported to be significantly elevated in MDS patient samples.26
In the context of AML, infiltration of MLL-AF9 AML cells led to significant decrease of endosteal mature osteoblasts and mineralized trabecular bone.27 Through β2-adrenergic receptor signaling, AML damaged tyrosine hydroxylase sympathetic nerve fibers leading to marked increase of osteo-primed Nestin-GFP+ MSCs. Conversely, the number of NG2+ periarteriolar cells decreased leading to impaired HSC function.27,28 Recent advances in single-cell RNA sequencing shed light on the cellular and molecular alterations following AML infiltration at tissue-wide scale. Baryawno and colleagues identified 17 distinct BM populations at steady state, including 3 endothelial (sinusoids, arteries, and arterioles), 2 osteolineage (OLC1 and OLC2), 5 fibroblastic, 1 mesenchymal (LepR-MSC), and 5 chondrocytic clusters.29 AML caused significant alterations in the cellular composition of the BM microenvironment. A marked decrease of osteolineage committed LepR-MSCs showed that AML cells compromised osteogenic differentiation of MSCs. This is consistent with the reported decrease of bone formation and osteoblast numbers in AML patients.30
The adipogenic differentiation of LepR-MSCs may also be impaired associated with downregulation of several adipocytic-related genes (including Adipoq) and PPARγ signaling components.29 This is in line with previous studies linking AML with defective adipogenesis and impaired myeloid/erythroid cell production. Using xenograft models, 2 independent studies revealed that human AML samples disrupt de novo adipogenesis and shift MSC differentiation toward OLCs.31,32 BM adipocytes may serve as an important energy and lipid source for AML cells by inducing triglyceride lipolysis. Blocking fatty-acid transfer between adipocytes and AML cells was shown to reduce AML cell survival and delayed disease lethality.33 Notably, adipocytes have been implicated in chemoresistance by metabolizing and sequestering chemotherapeutic agents, thus protecting malignant cells.34,35
In addition to the impaired osteogenic and adipocytic differentiation of MSCs and OLCs, AML reduces expression of key hematopoietic cytokines (CXCL12, SCF) from endothelial cells.29 BM endothelial cells are critical components of the HSC niche and have been shown to undergo morphological and structural alterations during hematologic disease, including AML27 and CML.36 BM biopsies from AML patients revealed increased angiogenesis and microvessel formation.37,38 Interestingly, AML cells seem to actively induce angiogenesis through secretion of vascular endothelial growth factor.39 The ability of AML cells to alter BM vasculature is further supported by extensive vascular remodeling in patient-derived xenografts40 and upregulation of nitric oxide synthase 3 (Nos3) and nitric oxide (NO). Elevated NO levels lead to increased vascular leakiness associated with therapy resistance.35,41 Live imaging of animals with AML has also revealed striking vascular effects.42 AML cells clustered near the endosteum and were associated with gradual loss of endosteal endothelial and osteoblastic cells, whereas central vessels were unaffected. The disordered endosteal regions had compromised ability to support normal hematopoiesis. The small molecule, deferoxamine, was capable of rescuing the loss of normal HSC and improved animal survival. These findings strongly argue for the endosteal vasculature as a key participant in the pathology of myeloid leukemia (the changes were not seen with acute lymphoid leukemia): so much so that it may be a therapeutic target.
The presence of malignant myeloid cells also affects the immune microenvironment of the bone marrow. In particular, myeloid-derived suppressor cells (MDSCs) appear to be induced.43 These are a population of CD33+CD11b+HLA DRlow/neg cells with the potential to induce T-cell tolerance,44 recruit T regulatory cells (Tregs) and impair natural killer (NK) function through metabolic and cytokine perturbations of the microenvironment.45,46 MDSCs expand when exposed to AML cells in vitro through extracellular vesicle transfer of MUC1.47 They thereby foster an environment in which immune clearance can be impaired and, indeed, are found in greater abundance in AML patients with high levels of residual disease after chemotherapy.48 They thus represent a potential microenvironment target.
The malignant BM microenvironment as a therapeutic target
The reciprocal interaction between malignant cells and their local BM microenvironment may contribute to myeloid disease initiation and progression. Current treatments mostly focus on selective elimination of malignant cells via cytotoxic chemotherapy. However, targeting the complex interactions with the marrow microenvironment represents a complementary therapeutic opportunity.
Targeting key BM populations and signals
Leukemia cell infiltration affects a number of BM niche populations and in so doing is thought to advantage abnormal over normal hematopoietic cells. It follows that targeting the niche should alter the leukemia and that has been tested in several studies. Krevvata and colleagues used a compound that boosts osteoblast numbers, by inhibiting tryptophan hydroxylase 1 required for synthesis of the osteoblast-suppressing hormone, duodenal serotonin (LP533401).30 Osteoblast preservation delayed engraftment of AML and lymphoblastic leukemia and prolonged overall survival.30 In vivo administration of the PPARγ agonist GW1929 was also tested, with the hypothesis that it might preserve adipocytes. GW1929 increased both the number and size of BM adipocytes, whereas leukemic cells were decreased and disease progression was delayed.31
The previously mentioned studies of Duarte and colleagues from the Lo Celso laboratory documented alterations in both endosteal endothelial and osteoblastic cells with AML progression.42 They elegantly demonstrated the gradual degradation of stromal elements as the disease progressed and that such changes were accompanied by reduction of normal HSPC in the BM. Remarkably, these abnormalities were rescued simply by using the iron chelator, deferoxamine. This agent inhibits ferroptosis, but can also protect endothelial cells by enhancing HIF-1a stability through inhibiting prolyl-hydroxylase critical for its degradation. In vivo deferoxamine treatment preserved HSC numbers in the BM of AML-infiltrated mice and improved the ability of the niche to support HSC.42 This was evident even in the presence of AML, suggesting that it can alter the competitive balance in favor of normal cells. This niche targeted therapy may be particularly interesting in settings of minimal residual disease, where competitive relationships may impact the kinetics of relapse.
The same study used a genetic model to enhance endothelial function and test its effect on chemotherapy for AML. In the setting of inducible, endothelial-specific enhanced Notch signaling, AML was more effectively killed and animal survival was improved. Interestingly, that was achieved with a chemotherapy drug commonly used in patients.42 Others have shown that mice lacking expression of the NO synthase (Nos3) had delayed AML engraftment and slower relapse kinetics following AraC chemotherapy.40 Further, co-administration of NOS inhibitors and cytarabine was superior to chemotherapy alone in delaying leukemia progression and preserving numbers and function of HSCs, via reduced vascular permeability.40 Similarly, Cxcl12 deletion from mesenchymal cells targeted by Prx1-Cre decreased quiescence of CML cells and increased their sensitivity to tyrosine kinase inhibitor (TKI) treatments (nilotinib).36 In fact, TKI treatment alone has been reported to enhance quiescence and therapy resistance of CML cells, by upregulating mir-126 levels supplied by neighboring endothelial cells.49 Supplementing TKI treatment with mir-126 oligonucleotide inhibitor significantly improved mouse survival and reduced CML leukemia-initiating cells (LICs).49
The function and lineage output of normal HSCs can also be controlled by niche-derived noradrenergic signaling, with β2-adrenergic receptor (AR) signals promoting and β3-AR inhibiting myeloid cell production, respectively.20 Chronic administration of β3-AR agonists restored lineage-output balance in physiologically or prematurely aged mice.18,20 In addition, β3-AR agonist treatment has been shown to protect components of the BM niche and control mutant HSC expansion in murine MPN models.50 The opposing effects of β2/β3-AR niche signaling can be further exploited in the context of different hematologic malignancies; however, further studies will be required. These results suggest potential strategies for translation to patients.
Targeting adherence interactions between malignant cells and the BM microenvironment
Selective targeting of cross-talk factors involved in the interaction between malignant cells and their surrounding microenvironment offers another alternative. Infiltrating and chemoresistant AML and CML cells show high expression of several cell-surface receptors that facilitate physical interactions with heterologous niche cells.35,51,52 These include CXCR4, CD44, and very late antigen 4 (VLA4), known mediators of lodgment and engraftment in the BM niche.53,54 The CXCL12/CXCR4 axis is disrupted by Plerixafor (AMD3100) and BL-8040, and can lead to dislodgement of leukemic cells and render them more vulnerable to cytotoxic or pro-apoptotic chemotherapy in mice.55-57 A phase 1/2 clinical trial testing plerixafor and combination chemotherapy (mitoxantrone, etoposide, and cytarabine) reported encouraging remission rates.58
Adhesion of AML cells to BM vasculature has been associated with quiescence59 and chemotherapy evasion, thus a number of antagonists targeting this interaction have been developed.60 Early clinical assessment of a phase 1/2 clinical study combining E-selectin inhibitor (GMI-1271) with induction chemotherapy also reported encouraging response rates in relapsed or refractory AML patients.61 AML adhesion to endothelial and Cxcl12-expresssing cells is also mediated through the interaction of the integrin very late antigen-4 (VLA4) with vascular cell adhesion molecule 1 (VCAM1). VLA4 activation has been associated with patient survival in AML60,62 and VLA4/VCAM1 interaction activates phosphatidylinositol 3-kinase/Akt and NF-κB signaling affecting chemoresistance.63,64 VLA4 also mediates AML cell adhesion to fibronectin (FN)-expressing cells and extracellular matrix fibers. Given that the VLA4/FN axis promotes survival of AML cells following chemotherapy via phosphatidylinositol 3-kinase/Akt signaling, developing efficient methods to block this interaction might have beneficial effects.
Targeting immune responses
The potential of immune cells to treat hematologic malignancies was experimentally demonstrated in 1956. In a seminal study, Barnes and Loutit reported the elimination of leukemia cells in murine recipients receiving allogeneic BM transplantation after irradiation.65 That provided the basis for the concept of a graft-versus-leukemia effect, which has been exploited in clinical practice since the mid-1970s.66 It was validated when T-cell-depleted BM grafts resulted in inefficient leukemia clearance and higher relapse rates67,68 and made myeloid leukemias among the first cancers treated by immunotherapy. In homeostasis, immune cells are important components of the HSC niche. Tregs provide an immune privileged environment that may protect HSCs from autoimmunity and excessive inflammation.69,70 They may also contribute to the limited effect of immune modulators in treating myeloid malignancies. Despite the known immune responsiveness of AML, it has been difficult to gain traction against the disease with immune therapies. There are a number of factors contributing to that,71 including the suppressive Tregs and MDSCs, a population that impairs T-effector cells through metabolic and cytokine modifications.45,46 Leukemia cell heterogeneity is 1 of those factors. Lack of NKG2DL expression in LICs enabled them to evade NK-mediated immune clearance, whereas NKG2DL+ bulk AML cells were targeted.72 Interestingly, selective pharmacological NKG2DL induction in LICs could be achieved and resulted in suppressed leukemogenesis.72
Immunologic response is dampened through checkpoint proteins like programmed cell death protein-1 (PD-1) and cytotoxic T-lymphocyte antigen-4. Malignant myeloid cells express and upmodulate them in the context of leukemia progression and response to treatment.35,73,74 This enables myeloid leukemic cells to elude cytotoxic T cell–mediated cell death.75-78 Administration of checkpoint inhibitors have reduced AML burden and delayed disease progression in murine models.79 Unfortunately, a number of clinical trials using single76,77 or multiple immune checkpoint inhibitors alone78,80 or in combination with chemotherapy and hypomethylating agents74,81 have had disappointing results to date. Checkpoint inhibitors in patients who had relapsed after hematopoietic transplantation were tested, with evidence of some long-term remissions, but also increased graft-versus-host disease immune-related adverse events.82
As an alternative to checkpoint inhibitors, vaccines have also been explored in AML. These include patient-derived AML cells fused with autologous dendritic cells (DCs). Among patients achieving relapse after chemotherapy, serial vaccination with the fused DCs resulted in a sustained increase in numbers and reactivity of AML-specific cytotoxic CD8+ T cells.83 Twelve of the 17 patients remain in remission at a median of 57 months, though a comparative trial will be needed to define the true benefit of the approach. Other vaccine strategies are early in development, including a biomaterial-based approach with a hydrogel containing factors that recruit and induce DC proliferation.84 This subcutaneously injected material could be loaded with an antigen or simply been present during chemotherapy-induced AML cell death (and presumed antigen loading of DCs) to achieve a robust cytotoxic T-cell response, which was AML sterilizing. The acquired immunity could be transferred to recipient mice by BM transplantation.84
Bispecific antibodies that bind targets on AML cells and T cells are another encouraging approach. Bispecific T-cell engagers have fragments that recognize the CD3 receptor expressed in most T cells and an epitope expressed by malignant cells. NK cells can also be recruited through fragments against CD16 (bispecific killer cell engagers). This method works well in B-acute lymphoblastic leukemia (blinatumomab, a CD3/CD19 bispecific T-cell engager) and is US Food and Drug Administration approved in that setting.85 Targeting AML cells with bispecific antibodies against CD123 (IL-3r), CD33, and WT1 are ongoing.86 These antigens are not limited to malignant cells and some substantial toxicities have been seen. However, the use of targeting antibodies with either bispecificity or as antibody–drug conjugates is still early in development in AML and are ultimately anticipated to yield useful new drugs. A related approach is the use of antibodies blocking the “don’t eat me” CD47 and signal regulatory protein α, either alone or fused to a targeting antibody such as anti-CD123.87-89 These have been shown to enhance antibody-dependent cell-mediated cytotoxicity and, for the anti-CD47 antibody, achieved impressive clinical remission rates. Some bispecific antibodies, such as those targeting CD33 and CD3 have the potential to also reduce MDSC and may enhance their effect by virtue of it.90 Multiple other efforts to impair MDSCs are underway in solid tumors, but have not been sufficiently encouraging to test in myeloid malignancies.91
An alternative method involves engineered immune cells with improved tumor recognition and cytotoxic activity. Induced pluripotent stem-cell derived NKs cells (iNKs) have been genetically modified to express a high-affinity, noncleavable CD16 receptor for enhanced cytotoxic activity against tumor cells. Malignant cell recognition is achieved either through monoclonal antibody tumor labeling recognized by hnCD16 (FT516)92 or expression of chimeric antigen receptor by iNKs (FT596).93 To circumvent limitations associated with NKs’ short lifespan and inability to expand post infusion in vivo, iNKs also expressed a fusion of IL-15 and IL-15 receptor α (IL-15R) obviating the need for continuous IL15 treatment.93 After promising results in lymphoblastic leukemia and lymphoma, this approach is currently in phase 1 clinical trial.
The field of modulating the immune milieu of the BM microenvironment is clearly burgeoning with multiple distinctive approaches rapidly moving through clinical testing. If the results in other immune responsive cancers is any guide, the future for this approach positively affecting the lives of patients with myeloid malignancies is high.
Future directions
Despite extensive efforts over past decades, treatment of (refractory) myeloid malignancies remains a massive challenge. Most of those applied approaches remain focused on targeting the malignant cell itself and combinatorial chemotherapy has been the mainstay of treatment. However, it is likely that we will soon see the added dimension of microenvironment-directed agents in the combined agent approach. The immune responsiveness of myeloid malignancies has long been known and guided allotransplantation. Newer, more precise ways of enhancing immune targeting are rapidly moving to the clinic. These will almost certainly give us new biologic or cell therapies that take advantage of the immune microenvironment, in which the malignant cells reside. Whether therapies leveraging what we are learning about stromal-malignant cell interactions in the bone marrow will follow is at this point still unknown. However, advances in single-cell transcriptomic,29,94,95 proteomic profiling,96,97 BM imaging,42,98-100 and spatial transcriptomics101 are providing an unprecedented understanding of tissue biology, and the BM is a leading focus of that effort. If the systems biology of tissues does lead us to new therapeutics, it is likely to be pioneered through myeloid malignancies.
Acknowledgments
The authors are grateful to David Sykes and Nick van Gastel for their thoughtful comments on the manuscript. We apologize to all authors whose work could not be cited due to space limitations.
This work was supported by the Human Frontier Science Program long-term fellowship (K.D.K.), Swiss National Science Foundation postdoctoral mobility fellowship (K.D.K.), the Gerald and Darlene Jordan Professor of Medicine Chair (D.T.S.), the Harvard Stem Cell Institute (D.T.S.), and the National Institutes of Health, National Cancer Institute (CA194596 and CA193461) (D.T.S.).
Footnotes
Requests for data sharing should be e-mailed to the corresponding author, david_scadden@harvard.edu.
Authorship
Contribution: K.D.K. and D.T.S. prepared the manuscript.
Conflict-of-interest disclosure: D.T.S. is a director and equity holder of Agios Pharmaceuticals, Magenta Therapeutics, Editas Medicines, ClearCreekBio, and Life-VaultBio; he is a founder of Fate Therapeutics and Magenta Therapeutics and a consultant to FOG Pharma and VCanBio. K.D.K. declares no competing financial interests.
Correspondence: David T. Scadden, 185 Cambridge St, Massachusetts General Hospital, Harvard Medical School, Boston, MA 02114; e-mail: david_scadden@harvard.edu.
References
- 1.Kfoury Y, Scadden DT. Mesenchymal cell contributions to the stem cell niche. Cell Stem Cell. 2015;16(3):239-253. [DOI] [PubMed] [Google Scholar]
- 2.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505(7483):327-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pinho S, Frenette PS. Haematopoietic stem cell activity and interactions with the niche. Nat Rev Mol Cell Biol. 2019;20(5):303-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kokkaliaris KD. Dissecting the spatial bone marrow microenvironment of hematopoietic stem cells. Curr Opin Oncol. 2020;32(2):154-161. [DOI] [PubMed] [Google Scholar]
- 5.Walkley CR, Olsen GH, Dworkin S, et al. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell. 2007;129(6):1097-1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim YW, Koo BK, Jeong HW, et al. Defective Notch activation in microenvironment leads to myeloproliferative disease. Blood. 2008;112(12):4628-4638. [DOI] [PubMed] [Google Scholar]
- 7.Walkley CR, Shea JM, Sims NA, Purton LE, Orkin SH. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell. 2007;129(6):1081-1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raaijmakers MHGP, Mukherjee S, Guo S, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464(7290):852-857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knudson AG., Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68(4):820-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kode A, Manavalan JS, Mosialou I, et al. Leukaemogenesis induced by an activating β-catenin mutation in osteoblasts. Nature. 2014;506(7487):240-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krause DS, Fulzele K, Catic A, et al. Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nat Med. 2013;19(11):1513-1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L, Zhang H, Rodriguez S, et al. Notch-dependent repression of miR-155 in the bone marrow niche regulates hematopoiesis in an NF-κB-dependent manner. Cell Stem Cell. 2014;15(1):51-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dong L, Yu WM, Zheng H, et al. Leukaemogenic effects of Ptpn11 activating mutations in the stem cell microenvironment. Nature. 2016;539(7628):304-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao P, Dolinska M, Sandhow L, et al. Sipa1 deficiency-induced bone marrow niche alterations lead to the initiation of myeloproliferative neoplasm. Blood Adv. 2018;2(5):534-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zambetti NA, Ping Z, Chen S, et al. Mesenchymal inflammation drives genotoxic stress in hematopoietic stem cells and predicts disease evolution in human pre-leukemia. Cell Stem Cell. 2016;19(5):613-627. [DOI] [PubMed] [Google Scholar]
- 16.Kusumbe AP, Ramasamy SK, Itkin T, et al. Age-dependent modulation of vascular niches for haematopoietic stem cells [published correction appears in Nature. 2016;539:314]. Nature. 2016;532(7599):380-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guidi N, Sacma M, Ständker L, et al. Osteopontin attenuates aging-associated phenotypes of hematopoietic stem cells. EMBO J. 2017;36(7):840-853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maryanovich M, Zahalka AH, Pierce H, et al. Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche [published correction appears in Nat Med. 2019;25(4):701]. Nat Med. 2018;24(6):782-791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frisch BJ, Hoffman CM, Latchney SE, et al. Aged marrow macrophages expand platelet-biased hematopoietic stem cells via interleukin1B. JCI Insight. 2019;5(10):124213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ho Y-H, Del Toro R, Rivera-Torres J, et al. Remodeling of bone marrow hematopoietic stem cell niches promotes myeloid cell expansion during premature or physiological aging. Cell Stem Cell. 2019;25(3):407-418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Witkowski MT, Kousteni S, Aifantis I. Mapping and targeting of the leukemic microenvironment. J Exp Med. 2020;217(2):e20190589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou BO, Ding L, Morrison SJ. Hematopoietic stem and progenitor cells regulate the regeneration of their niche by secreting Angiopoietin-1. eLife. 2015;4:e05521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schepers K, Pietras EM, Reynaud D, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell. 2013;13(3):285-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frisch BJ, Ashton JM, Xing L, Becker MW, Jordan CT, Calvi LM. Functional inhibition of osteoblastic cells in an in vivo mouse model of myeloid leukemia. Blood. 2012;119(2):540-550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Medyouf H, Mossner M, Jann JC, et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell. 2014;14(6):824-837. [DOI] [PubMed] [Google Scholar]
- 26.Wetzler M, Estrov Z, Talpaz M, et al. Leukemia inhibitory factor in long-term adherent layer cultures: increased levels of bioactive protein in leukemia and modulation by IL-4, IL-1□, and TNF-□. Cancer Res. 1994;54(7):1837-1842. [PubMed] [Google Scholar]
- 27.Hanoun M, Zhang D, Mizoguchi T, et al. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell. 2014;15(3):365-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kunisaki Y, Bruns I, Scheiermann C, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. 2013;502(7473):637-643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baryawno N, Przybylski D, Kowalczyk MS, et al. A cellular taxonomy of the bone marrow stroma in homeostasis and leukemia. Cell. 2019;177(7):1915-1932.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krevvata M, Silva BC, Manavalan JS, et al. Inhibition of leukemia cell engraftment and disease progression in mice by osteoblasts. Blood. 2014;124(18):2834-2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boyd AL, Reid JC, Salci KR, et al. Acute myeloid leukaemia disrupts endogenous myelo-erythropoiesis by compromising the adipocyte bone marrow niche. Nat Cell Biol. 2017;19(11):1336-1347. [DOI] [PubMed] [Google Scholar]
- 32.Battula VL, Le PM, Sun JC, et al. AML-induced osteogenic differentiation in mesenchymal stromal cells supports leukemia growth. JCI Insight. 2017;2(13):e90036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shafat MS, Oellerich T, Mohr S, et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood. 2017;129(10):1320-1332. [DOI] [PubMed] [Google Scholar]
- 34.Sheng X, Parmentier J-H, Tucci J, et al. Adipocytes sequester and metabolize the chemotherapeutic daunorubicin. Mol Cancer Res. 2017;15(12):1704-1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Behrmann L, Wellbrock J, Fiedler W. Acute myeloid leukemia and the bone marrow niche - take a closer look. Front Oncol. 2018;8:444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agarwal P, Isringhausen S, Li H, et al. Mesenchymal niche-specific expression of Cxcl12 controls quiescence of treatment-resistant leukemia stem cells [published correction appears in Cell Stem Cell. 2020;26(10:123]. Cell Stem Cell. 2019;24(5):769-784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Padró T, Ruiz S, Bieker R, et al. Increased angiogenesis in the bone marrow of patients with acute myeloid leukemia. Blood. 2000;95(8):2637-2644. [PubMed] [Google Scholar]
- 38.Hussong JW, Rodgers GM, Shami PJ. Evidence of increased angiogenesis in patients with acute myeloid leukemia. Blood. 2000;95(1):309-313. [PubMed] [Google Scholar]
- 39.Fiedler W, Graeven U, Ergün S, et al. Vascular endothelial growth factor, a possible paracrine growth factor in human acute myeloid leukemia. Blood. 1997;89(6):1870-1875. [PubMed] [Google Scholar]
- 40.Passaro D, Di Tullio A, Abarrategi A, et al. Increased vascular permeability in the bone marrow microenvironment contributes to disease progression and drug response in acute myeloid leukemia. Cancer Cell. 2017;32(3):324-341.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Azzi S, Hebda JK, Gavard J. Vascular permeability and drug delivery in cancers. Front Oncol. 2013;3:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duarte D, Hawkins ED, Akinduro O, et al. Inhibition of endosteal vascular niche remodeling rescues hematopoietic stem cell loss in AML. Cell Stem Cell. 2018;22(1):64-77.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ostrand-Rosenberg S, Fenselau C. Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol. 2018;200(2):422-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagaraj S, Schrum AG, Cho H-I, Celis E, Gabrilovich DI. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J Immunol. 2010;184(6):3106-3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Movahedi K, Guilliams M, Van den Bossche J, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111(8):4233-4244. [DOI] [PubMed] [Google Scholar]
- 46.Schouppe E, Mommer C, Movahedi K, et al. Tumor-induced myeloid-derived suppressor cell subsets exert either inhibitory or stimulatory effects on distinct CD8+ T-cell activation events. Eur J Immunol. 2013;43(11):2930-2942. [DOI] [PubMed] [Google Scholar]
- 47.Pyzer AR, Stroopinsky D, Rajabi H, et al. MUC1-mediated induction of myeloid-derived suppressor cells in patients with acute myeloid leukemia. Blood. 2017;129(13):1791-1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun H, Li Y, Zhang ZF, et al. Increase in myeloid-derived suppressor cells (MDSCs) associated with minimal residual disease (MRD) detection in adult acute myeloid leukemia. Int J Hematol. 2015;102(5):579-586. [DOI] [PubMed] [Google Scholar]
- 49.Zhang B, Nguyen LXT, Li L, et al. Bone marrow niche trafficking of miR-126 controls the self-renewal of leukemia stem cells in chronic myelogenous leukemia. Nat Med. 2018;24(4):450-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arranz L, Sánchez-Aguilera A, Martín-Pérez D, et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature. 2014;512(7512):78-81. [DOI] [PubMed] [Google Scholar]
- 51.Krause DS, Lazarides K, von Andrian UH, Van Etten RA. Requirement for CD44 in homing and engraftment of BCR-ABL-expressing leukemic stem cells. Nat Med. 2006;12(10):1175-1180. [DOI] [PubMed] [Google Scholar]
- 52.Winkler IG, Barbier V, Pattabiraman DR, et al. Vascular Niche E-selectin protects acute myeloid leukaemia stem cells from chemotherapy [abstract]. Blood. 2014;124(21):620. Abstract 616. [Google Scholar]
- 53.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12(10):1167-1174. [DOI] [PubMed] [Google Scholar]
- 54.Korn C, Méndez-Ferrer S. Myeloid malignancies and the microenvironment. Blood. 2017;129(7):811-822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nervi B, Ramirez P, Rettig MP, et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood. 2009;113(24):6206-6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abraham M, Klein S, Bulvik B, et al. The CXCR4 inhibitor BL-8040 induces the apoptosis of AML blasts by downregulating ERK, BCL-2, MCL-1 and cyclin-D1 via altered miR-15a/16-1 expression. Leukemia. 2017;31(11):2336-2346. [DOI] [PubMed] [Google Scholar]
- 57.Forte D, Krause DS, Andreeff M, Bonnet D, Méndez-Ferrer S. Updates on the hematologic tumor microenvironment and its therapeutic targeting. Haematologica. 2019;104(10):1928-1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Uy GL, Rettig MP, Motabi IH, et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood. 2012;119(17):3917-3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cogle CR, Goldman DC, Madlambayan GJ, et al. Functional integration of acute myeloid leukemia into the vascular niche. Leukemia. 2014;28(10):1978-1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang A, Zhong H. Roles of the bone marrow niche in hematopoiesis, leukemogenesis, and chemotherapy resistance in acute myeloid leukemia. Hematology. 2018;23(10):729-739. [DOI] [PubMed] [Google Scholar]
- 61.DeAngelo DJ, Liesveld JL, Jonas BA, et al. A phase I/II study of GMI-1271, a novel E-selectin antagonist, in combination with induction chemotherapy in relapsed/refractory and elderly previously untreated acute myeloid leukemia; results to date [abstract]. Blood. 2016;128(22):4049. Abstract 616. [Google Scholar]
- 62.Becker PS, Kopecky KJ, Wilks AN, et al. Very late antigen-4 function of myeloblasts correlates with improved overall survival for patients with acute myeloid leukemia. Blood. 2009;113(4):866-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Matsunaga T, Fukai F, Miura S, et al. Combination therapy of an anticancer drug with the FNIII14 peptide of fibronectin effectively overcomes cell adhesion-mediated drug resistance of acute myelogenous leukemia. Leukemia. 2008;22(2):353-360. [DOI] [PubMed] [Google Scholar]
- 64.Jacamo R, Chen Y, Wang Z, et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-κB mediates chemoresistance. Blood. 2014;123(17):2691-2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barnes DWH, Loutit JF. Treatment of murine leukaemia with x-rays and homologous bone marrow. II. Br J Haematol. 1957;3(3):241-252. [DOI] [PubMed] [Google Scholar]
- 66.Mathe G, Amiel JL, Schwarzenberg L, et al. Successful allogenic bone marrow transplantation in man: chimerism, induced specific tolerance and possible anti-leukemic effects. Blood. 1965;25(2):179-196. [PubMed] [Google Scholar]
- 67.Champlin RE. T-cell depletion for bone marrow transplantation: effects on graft rejection, graft-versus-host disease, graft-versus-leukemia, and survival. Cancer Treat Res. 1990;50:99-111. [DOI] [PubMed] [Google Scholar]
- 68.Horowitz MM, Gale RP, Sondel PM, et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood. 1990;75(3):555-562. [PubMed] [Google Scholar]
- 69.Fujisaki J, Wu J, Carlson AL, et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature. 2011;474(7350):216-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hirata Y, Furuhashi K, Ishii H, et al. CD150high bone marrow Tregs maintain hematopoietic stem cell quiescence and immune privilege via adenosine. Cell Stem Cell. 2018;22(3):445-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Epperly R, Gottschalk S, Velasquez MP. A bump in the road: how the hostile AML microenvironment affects CAR T cell therapy. Front Oncol. 2020;10:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Paczulla AM, Rothfelder K, Raffel S, et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion [published correction appears in Nature. 2019;572(7770):E19]. Nature. 2019;572(7768):254-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Giannopoulos K. Targeting immune signaling checkpoints in acute myeloid leukemia. J Clin Med. 2019;8(2):236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stahl M, Goldberg AD. Immune checkpoint inhibitors in acute myeloid leukemia: novel combinations and therapeutic targets. Curr Oncol Rep. 2019;21(4):37. [DOI] [PubMed] [Google Scholar]
- 75.Krönig H, Kremmler L, Haller B, et al. Interferon-induced programmed death-ligand 1 (PD-L1/B7-H1) expression increases on human acute myeloid leukemia blast cells during treatment. Eur J Haematol. 2014;92(3):195-203. [DOI] [PubMed] [Google Scholar]
- 76.Berger R, Rotem-Yehudar R, Slama G, et al. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res. 2008;14(10):3044-3051. [DOI] [PubMed] [Google Scholar]
- 77.Davids MS, Kim HT, Bachireddy P, et al. ; Leukemia and Lymphoma Society Blood Cancer Research Partnership . Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med. 2016;375(2):143-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kikushige Y, Shima T, Takayanagi S, et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell. 2010;7(6):708-717. [DOI] [PubMed] [Google Scholar]
- 79.Zhou Q, Munger ME, Highfill SL, et al. Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood. 2010;116(14):2484-2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Andrews LP, Marciscano AE, Drake CG, Vignali DAA. LAG3 (CD223) as a cancer immunotherapy target. Immunol Rev. 2017;276(1):80-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Daver N, Garcia-Manero G, Basu S, et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: a nonrandomized, open-label, phase II study. Cancer Discov. 2019;9(3):370-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Davids MS, Kim HT, Costello CL, et al. A phase I/Ib study of nivolumab for relapsed hematologic malignancies after allogeneic hematopoietic cell transplantation (alloHCT). Blood. 2018;132(suppl 1). Abstract 705. [Google Scholar]
- 83.Rosenblatt J, Stone RM, Uhl L, et al. Individualized vaccination of AML patients in remission is associated with induction of antileukemia immunity and prolonged remissions. Sci Transl Med. 2016;8(368):368ra171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shah NJ, Najibi AJ, Shih T-Y, et al. A biomaterial-based vaccine eliciting durable tumour-specific responses against acute myeloid leukaemia. Nat Biomed Eng. 2020;4(1):40-51. [DOI] [PubMed] [Google Scholar]
- 85.Kantarjian H, Stein A, Gökbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836-847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Velasquez MP, Bonifant CL, Gottschalk S. Redirecting T cells to hematological malignancies with bispecific antibodies. Blood. 2018;131(1):30-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Majeti R, Chao MP, Alizadeh AA, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138(2):286-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Veillette A, Chen J. SIRPα-CD47 immune checkpoint blockade in anticancer therapy. Trends Immunol. 2018;39(3):173-184. [DOI] [PubMed] [Google Scholar]
- 89.Tahk S, Schmitt S, Augsberger CP, et al. Evaluation of a bifunctional Sirpα-CD123 fusion antibody for the elimination of acute myeloid leukemia stem cells [abstract]. Blood. 2019;134(suppl 1):2544. Abstract 604. [Google Scholar]
- 90.Jitschin R, Saul D, Braun M, et al. CD33/CD3-bispecific T-cell engaging (BiTE®) antibody construct targets monocytic AML myeloid-derived suppressor cells. J Immunother Cancer. 2018;6(1):116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fleming V, Hu X, Weber R, et al. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front Immunol. 2018;9:398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bjordahl R, Zhu H, Rogers P, et al. FT516, an off-the-shelf engineered NK cell therapeutic product for universal anti-tumor targeting strategy in combination with monoclonal antibodies [abstract] Cancer Res. 2019;79 Abstract 3191.31641034 [Google Scholar]
- 93.Goodridge JP, Mahmood S, Zhu H, et al. FT596: translation of first-of-kind multi-antigen targeted off-the-shelf CAR-NK cell with engineered persistence for the treatment of B cell malignancies. Blood. 2019;134(suppl 1). Abstract 301. [Google Scholar]
- 94.Tikhonova AN, Dolgalev I, Hu H, et al. The bone marrow microenvironment at single-cell resolution [published correction appears in Nature. 2019;572(7767):E6]. Nature. 2019;569(7755):222-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.van Galen P, Hovestadt V, Wadsworth Ii MH, et al. Single-cell RNA-seq reveals AML hierarchies relevant to disease progression and immunity. Cell. 2019;176(6):1265-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Severe N, Karabacak NM, Gustafsson K, et al. Stress-induced changes in bone marrow stromal cell populations revealed through single-cell protein expression mapping. Cell Stem Cell. 2019;25(4):570-583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zeng Z, Konopleva M, Andreeff M. Single-cell mass cytometry of acute myeloid leukemia and leukemia stem/progenitor cells. Methods Mol Biol. 2017;1633:75-86. [DOI] [PubMed] [Google Scholar]
- 98.Coutu DL, Kokkaliaris KD, Kunz L, Schroeder T. Three-dimensional map of nonhematopoietic bone and bone-marrow cells and molecules. Nat Biotechnol. 2017;35(12):1202-1210. [DOI] [PubMed] [Google Scholar]
- 99.Acar M, Kocherlakota KS, Murphy MM, et al. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature. 2015;526(7571):126-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gomariz A, Helbling PM, Isringhausen S, et al. Quantitative spatial analysis of haematopoiesis-regulating stromal cells in the bone marrow microenvironment by 3D microscopy. Nat Commun. 2018;9(1):2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Baccin C, Al-Sabah J, Velten L, et al. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat Cell Biol. 2020;22(1):38-48. [DOI] [PMC free article] [PubMed] [Google Scholar]