

Key Points
superFVa, an engineered FVa variant, reduces bleeding associated with both FXa and FIIa inhibitors.
superFVa reduces bleeding because of its APC resistance, bypassing of the FV activation threshold, and prothrombinase enhancement.
Abstract
Control of bleeding with direct-acting oral anticoagulants (DOACs) remains an unmet clinical need. Activated superFactor V (superFVa) is an engineered activated protein C (APC)–resistant FVa variant with enhanced procoagulant activity resulting from an A2/A3 domain disulfide bond and was studied here for control of DOAC-induced bleeding. SuperFVa reversed bleeding induced by FXa inhibitors (rivaroxaban, apixaban), and the FIIa inhibitor dabigatran in BalbC mice. The blocking anti-protein C and APC [(A)PC] antibody SPC-54 also reduced FXa inhibitor induced bleeding similar to superFVa, whereas dabigatran-induced bleeding was not affected. This indicated that sufficient APC was generated to contribute to bleeding in the presence of FXa inhibitors, but not in the presence of dabigatran, suggesting that mechanisms contributing to bleeding differed for FXa and FIIa inhibitors. Despite different mechanisms contributing to bleeding, superFVa effectively reduced bleeding for all DOACs, indicating the versatility of superFVa’s properties that contribute to its universal prohemostatic effects for DOAC associated bleeding. Supported by thrombin generation assays on endothelial cells in normal plasma spiked with DOACs and patient plasma anticoagulated with DOACs, 3 complementary mechanisms were identified by which superFVa achieved DOAC class-independent prohemostatic efficiency. These mechanisms are resistance to inactivation by APC, overcoming the FV activation threshold, and maximizing the efficiency of the prothrombinase complex when the available FXa is increased by FVIIa-based prohemostatics. In summary, it is this versatility of superFVa that delineates it from other prohemostatic agents as a promising class-independent rescue agent in bleeding situations associated with DOACs.
Visual Abstract

Introduction
Direct-acting oral anticoagulants (DOACs) increasingly replace warfarin for treatment and prevention of venous thromboembolism or prevention of ischemic stroke.1-3 Anticoagulant therapy increases bleeding risk, requiring prohemostatic agents in case severe bleeding occurs. Bleeding rates in patients on DOACs reported from large clinical trials are ∼5% per year.4-9 “Real-world experience” data from the Dresden DOAC Registry and the Fushimi AF Registry are similar and demonstrate major bleeding in approximately 3% to 6% per year.10,11
Specific DOAC-reversal agents, idarucizumab (Praxbind, Boehringer-Ingelheim), a specific humanized monoclonal antibody against the direct thrombin inhibitor dabigatran (Pradaxa, Boehringer-Ingelheim),12-16 and coagulation factor Xa (recombinant) inactivated-zhzo (Andexanet Alfa, Andexxa, Portola Pharmaceuticals Inc.), a decoy for direct oral factor (F)Xa inhibitors (rivaroxaban, Bayer; apixaban, Bristol Meyers-Squibb; edoxaban, Daiichi-Sankyo), were approved by the US Food and Drug Administration for patients experiencing life-threatening bleeding.17-19 Both agents have proven efficacious in clinical trials for reversing the anticoagulant effects of DOACs, but their effect on clinical outcomes is less clear. Andexanet-alfa is a catalytically inactive FXa decoy20 shown to reverse the anticoagulant effects of FXa inhibitors by reduction of anti-FXa activity in healthy volunteers and patients,17-19,21 with hemostatic efficacy in the majority of patients.21 However, andexanet alfa has a boxed warning for thromboembolic risks, ischemic risks, cardiac arrest, and sudden death, with these adverse events occurring in up to 18% of patients in clinical trials.22 No conclusive data are published to gauge the contributions of idarucizumab to clinical hemostasis, but rapid reversal of anticoagulant effects has been shown.15,16,23
Additional agents are being developed for reversal of FXa inhibitors including ciraparantag (PER977; Amag Pharmaceuticals),24 which is a synthetic molecule that binds all DOACs.24,25 In healthy volunteers, ciraparantag demonstrated sustained reversal of anticoagulation after edoxaban administration based on visual inspection of whole blood clot formation.25 In this context, it is important to recognize that idarucizumab,12 andexanet-alfa,20 and ciraparantag24 are large molecules designed to absorb small molecular weight inhibitors to correct abnormal clotting parameters,26 and that these agents do not have intrinsic procoagulant properties. Hence, their efficacy and clinical utility to rescue severe bleeding situations without adding other procoagulants remains somewhat uncertain.
Here we propose activated superFactor V (superFVa), an engineered FVa-variant with improved stability, as a prohemostatic augmentation strategy rather than drug-absorbing strategy for reversal of DOAC-associated bleeding.27 SuperFVa normalizes hemostasis in other murine experimental bleeding models such as hemophilia or traumatic injury.27-29 Normal FVa enhances the rate of thrombin generation in the prothrombinase complex by approximately 10 000-fold,30 but is rapidly inactivated by activated protein C (APC). SuperFVa is resistant to APC inactivation because of mutations of 3 APC cleavage sites (Arg506/306/679Gln), and has enhanced specific activity because of an engineered disulfide bond (Cys609-Cys1691) between the A2 and A3 domains.27 SuperFVa’s ability to both enhance the DOAC-compromised prothrombinase complex and convey APC-resistance may portend a double advantage for inhibition of DOAC-associated bleeding. This may be important because it is increasingly recognized that APC contributes to bleeding in acute traumatic injury and in hemophilia.31-33
Materials and methods
Materials
Normal pooled human plasma (NHP) was purchased from George King Bio-Medical. The following reagents were used: rivaroxaban, apixaban, and dabigatran (all from Selleckchem), 4F-PCC (Kcentra; CSL-Behring), recombinant human (rh) FVIIa (NovoSeven, NovoNordisk), tissue factor (Dade Innovin, Dade Behring), corn trypsin inhibitor and thrombin (both from Enzyme Research Laboratories), thrombin calibrator (Diagnostica Stago), and Z-Gly-Gly-Arg-AMC (Bachem). Phosphatidylcholine (PC), phosphatidylserine (PS), and phosphatidylethanolamine (PE) were purchased from Avanti Lipids and phospholipid vesicles containing 40% PC, 20% PS, and 40% PE or 80% PC and 20% PS were prepared as described.34 Recombinant superFV was made and activated as described.27,35
Plasma assays
Thrombin generation on cells were performed on confluent EA.hy926 endothelial cells (ATCC) in black-clear bottom 96-well plates. Plasma was supplemented with corn trypsin inhibitor (1.45 μM) and mixed 1:1 with CaCl2 (final concentration [f.c.], 10 mM), anti-thrombomodulin (RTM96, Hycult), anti-endothelial protein C receptor (EPCR) (RCR-25236), anti-human protein C (C137), anti-mouse protein C (SPC-5438), or nonimmune rat control antibodies (all f.c., 50 μg/mL), z-Gly-Gly-Arg-AMC (f.c., 0.4 mM) and/or DOACs in Hepes buffered saline (20 mM Hepes, 147 mM NaCl, 3 mM KCl, pH 7.4) with 0.1% bovine serum albumin and 50 μL was transferred to each well. For DOAC patient plasma in the absence of cells, thrombin generation was determined under identical conditions but in the presence of 0.4 pM tissue factor (Innovin) and 10 μM phospholipid vesicles (PC/PS/PE 40/20/40).
Plasma thrombin generation assays were performed as described.39,40 The final reaction mixture contained 50% plasma, 0.725 µM corn trypsin inhibitor, 0.2 pM tissue factor, 4 µM PC/PS 80/20 vesicles, 0.5 mM Z-GGR-AMC, 7.6 mM CaCl2 in a total volume of 100 μL. Determination of thrombin peak height, lag time, and the endogenous thrombin potential (ETP), defined as the area under the curve, were performed as described.39,40
Animals
Animal research protocols were approved by the Institutional Animal Care and Use Committee of The Scripps Research Institute. BALB/c mice, aged ≥8 weeks, were used for experimentation. Dabigatran (BIBR953) was dissolved in acidified water (1 mM HCl) according to the manufacturer’s instructions and diluted to 0.1 mg/mL in sterile saline (Hospira Inc.) before use. Apixaban and rivaroxaban DOAC suspensions41 were made in 10% (v/v) glycerol, 10% (v/v) ethanol, 10% (v/v) PEG-400, and 70% (v/v) of a 5% dextrose solution. DOACs or vehicle (100 μ) were injected into the tail vein 30 minutes before tail transection. In some experiments, the anti-protein C antibody SPC-5438 was injected retro-orbitally (5 mg/kg in sterile saline; 100 µL) 2 hours before DOACs. SPC-54 blocks APC’s anticoagulant and cell signaling activities.38 Tail bleed assays were performed by cutting the distal portion of the tail at 2-mm diameter, after which the tail was immersed in a predefined volume of 0.9% NaCl at 37°C for 20 minutes as described.27 Blood loss was determined by hemoglobin content after red cell lysis with 2% acetic acid and was expressed in μL/g body weight assuming a hematocrit of 46%.27 BalbC mice were injected retro-orbitally within 5 minutes before the tail was cut with equal volumes (100 μL) of superFVa, rhFVIIa, or a combination of superFVa and rhFVIIa in sterile saline.
DOAC patient plasma
The patients taking DOACs for clinical indications were identified through the Scripps Anticoagulation Service. Blood was drawn during routine visits and citrated plasma was stored at −80°C. The protocol was approved by the Scripps institutional review board and subjects provided written informed consent. DOAC levels were determined using the DiXal and Hemoclot Thrombin-inhibitors kits with the appropriate reference plasmas (all from Hyphen BioMed).
Statistical analysis
Student t test, 1-way ANOVA with Holm-Sidak multiple comparisons test, or for bleeding, Kruskal-Wallis followed by a 2-tailed Mann-Whitney U test, were used to assess statistical significance (P ≤ .05) where appropriate.
Results
Bleed reduction by superFVa in normal wild-type mice treated with FXa and FIIa inhibitors
The ability of superFVa to reduce bleeding was studied for apixaban-, rivaroxaban- and dabigatran-induced bleeding. The optimal dose of apixaban42,43 (IV injection) to induce bleeding after tail transection was determined to be 8 mg/kg apixaban (mean blood loss apixaban 18.5 µL/g vs vehicle 3.3 µL/g; P ≤ .001) (supplemental Figure 1A). SuperFVa decreased apixaban-induced bleeding in a dose-dependent manner. Partial reduction and full reduction of mean blood loss from 18.5 µL/g to 8.1 µL/g and 4.3 µL/g was achieved at a dose of 0.16 mg/kg and 1.6 mg/kg superFVa, respectively (Figure 1A). Dosing for rivaroxaban was based on previously published evidence that rivaroxaban has a relatively weak affinity for rodent FXa and, therefore, high doses (40 mg/kg) were necessary to induce bleeding in rodent models.20,41,42,44 At 40 mg/kg, rivaroxaban induced meaningful bleeding and mean blood loss was significantly higher compared with vehicle-treated mice (13.8 vs 2.8 µL/g; P = .0001). SuperFVa decreased rivaroxaban-induced bleeding in a dose-dependent manner (Figure 1B). Partial bleed reduction was achieved at a dose of 0.08 mg/kg superFVa (mean blood loss 8.4 µL/g vs 13.8 µL/g in saline-injected mice; P ≤ .03). Doubling the dose of superFVa to 0.16 mg/kg further reduced bleeding to a level seen in untreated wild-type mice (6.1 µL/g vs 3.3 µL/g; P = .77) (Figure 1B). These findings suggested that doses of superFVa needed to achieve reasonable bleed reduction were ∼10-fold lower in rivaroxaban-treated mice compared with apixaban-treated mice. The optimal dose of dabigatran-induced bleeding (IV injection) after tail transection was determined to be 0.4 mg/kg dabigatran (supplemental Figure 1B). Treatment with superFVa resulted in effective bleed reduction of dabigatran-induced bleeding at 1.6 mg/kg superFVa (Figure 1C).
Figure 1.
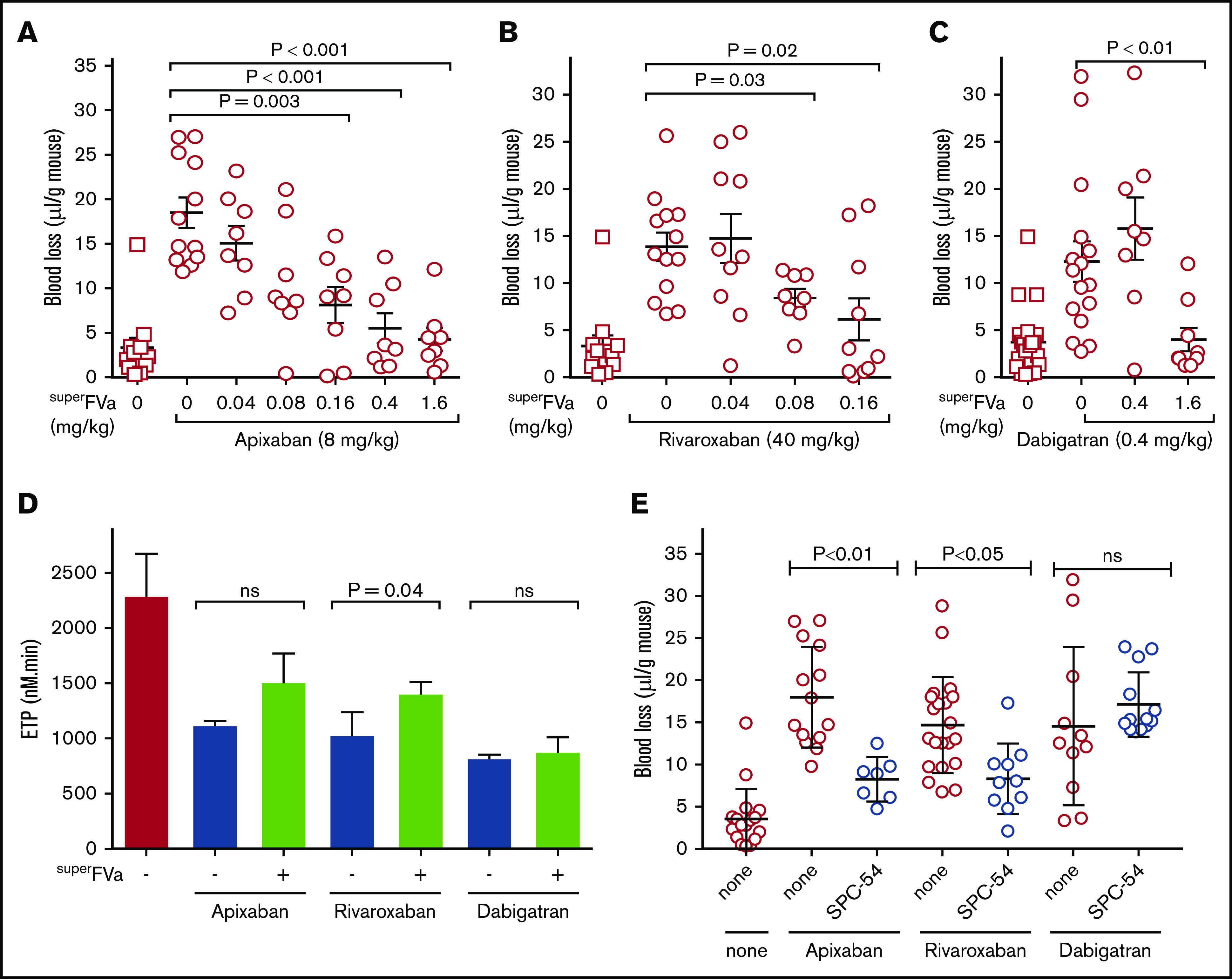
Inhibition of DOAC-induced bleeding by superFVa and an anti-protein C antibody. Mice were treated with apixaban (8 mg/kg) (A), rivaroxaban (40 mg/kg) (B), or dabigatran (0.4 mg/kg) (C) by IV tail vein injection and bleeding was measured for 20 minutes after tail clip. Blood loss was expressed in microliters of blood per gram (g) mouse. (D) Human plasma with and without apixaban, rivaroxaban (both 200 nM) or dabigatran (1 μM) were treated with superFVa (100 nM) and endogenous thrombin potential was assessed. (E) Mice were injected retro-orbitally with the blocking anti-protein C and APC [(A)PC] antibody SPC-54 (5 mg/kg) 2 hours before IV treatment with apixaban (8 mg/kg), rivaroxaban (40 mg/kg), or dabigatran (0.4 mg/kg). Bleeding was measured for 20 minutes after tail clip. Blood loss was expressed in microliters of blood per gram (g) mouse. Error bars represent SEM (n = 7-14 per group). P values were determined by Kruskal-Wallis followed by a 2-tailed Mann-Whitney U test and values ≤.05 were considered statistically significant.
The resistance of superFVa to inactivation by APC contributes to bleed reduction in vivo
Following a dose titration of rivaroxaban, apixaban, or dabigatran into NHP, a concentration of 200 nM for the FXa inhibitors and 1 μM for dabigatran resulted in ∼60% to 80% suppression of the ETP (supplemental Figure 2). These concentrations approximate peak plasma concentrations achieved in humans after oral administration,45-47 and were therefore used for in vitro experimentation in human plasma. Addition of superFVa to the DOAC-treated plasma resulted in a very modest increase of ETP for apixaban and rivaroxaban, whereas superFVa had no effect on the ETP in dabigatran-treated normal plasma (Figure 1D). The discrepancy between efficient bleed reduction by superFVa in the presence of DOACs in vivo and rather inefficient improvement of the ETP of thrombin generation in vitro may be explained by resistance of superFVa to APC. To test if APC generation contributes to DOAC-induced bleeding in vivo, despite potentially suboptimal activation of PC from inhibition of thrombin generation by FXa and FIIa inhibitors, a monoclonal antibody that inhibits all APC activities, SPC-54,38 was infused before tail clip. Inhibition of APC significantly reduced apixaban- and rivaroxaban-induced blood loss but had no effect on dabigatran-induced blood loss (Figure 1E). These observations suggest that APC contributed meaningfully to increase bleeding induced by FXa inhibitors, and that FXa inhibitors do not completely suppress thrombin generation, which is necessary to activate PC. These observations also suggest that inhibition of thrombin generation by dabigatran did not result in sufficient generation of APC, such that inhibition of APC generation could diminish bleeding.
Improvement of endothelial cell thrombin generation by superFVa
Thrombin generation assays were performed on adherent endothelial cells to better understand the contributions of APC to inhibition of thrombin generation in DOAC-treated normal plasma and the ability of superFVa to blunt APC’s anticoagulant effects. The endothelial cells in this assay provide both tissue factor and the phospholipid surface required for coagulation, in addition to thrombomodulin and EPCR required for the generation of APC. To validate the assay, thrombin generation on endothelial cells was first performed in the absence of DOACs. Inhibition of protein C, thrombomodulin, or EPCR by blocking antibodies (50 μg/mL) increased ETP values by ∼30%, whereas a control antibody had no effect (Figure 2A), indicating APC’s anticoagulant effect. Similarly, superFVa (100 nM) increased the ETP by ∼30% (Figure 2A). When DOACs were added to the endothelial thrombin generation assay, a consistent left-shift in the DOAC IC50 of seven- to eightfold was observed compared with thrombin generation in the absence of cells (Figure 2B). The addition of blocking antibodies to protein C, thrombomodulin or EPCR markedly improved the ETP in apixaban (Figure 2C), rivaroxaban (Figure 2D), but not in dabigatran (Figure 2E)-treated plasma, whereas a noninhibitory control antibody had no effect. Altogether, these observations revealed a major contribution of APC to the inhibition of thrombin generation in the presence of FXa inhibitors and uncovered a major difference between DOACs targeting FXa or FIIa with regard to the contributions of APC. SuperFVa (200 nM) improved thrombin generation on cells in DOAC-treated normal plasma for both the FXa and FIIa inhibitors (Figure 2C-E). Analysis of the thrombin generation curves revealed that superFVa not only increased the total thrombin generation (ETP value) but also shortened the lag time of the thrombin generation curve for apixaban-, rivaroxaban-, and especially dabigatran-treated plasma (Figure 2F). The shortening of the lag time appeared less with the blocking protein C antibody compared with superFVa. This observation may suggest that the activation of FV is a limiting factor, which may be overcome by the presence of a stabilized activated FVa.
Figure 2.
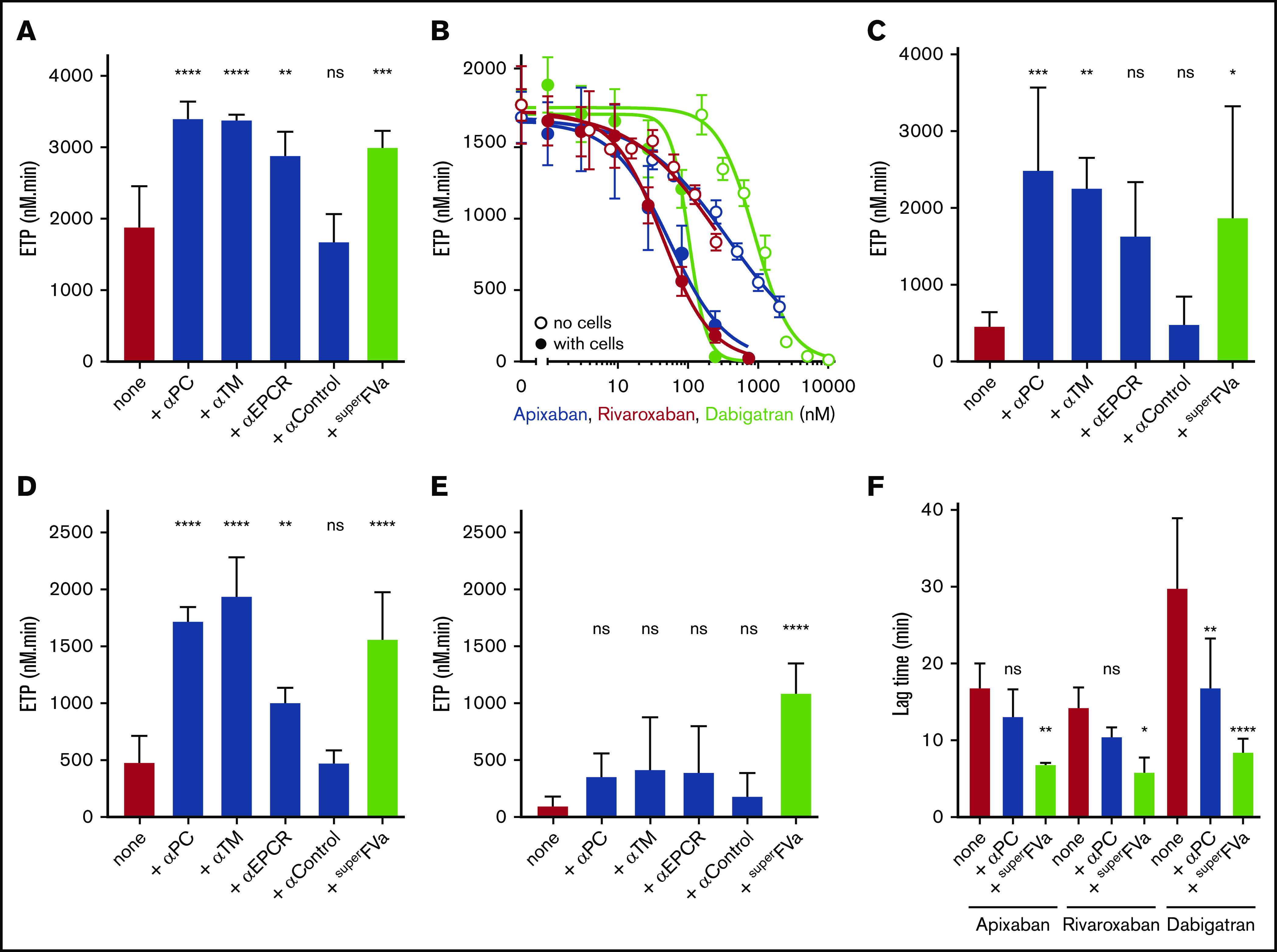
The role of superFVa and APC’s anticoagulant activity on the DOAC mediated inhibition of thrombin generation on endothelial cells. Thrombin generation was performed on EA.hy926 endothelial cells using human plasma in the absence of exogenously added tissue factor or phospholipids. (A) Thrombin generation on EA.hy926 endothelial cells in the presence of antibodies (α) against (A)PC (C1), TM, or EPCR (all 50 μg/mL) or superFVa (200 nM). (B) Thrombin generation in the presence and absence of EA.hy926 endothelial cells with different concentrations apixaban, rivaroxaban, or dabigatran. Thrombin generation in the absence of cells was initiated by 0.2 pM tissue factor and 4 μM phospholipid vesicles (PC/PS 80/20). (C) Panel A in the presence of apixaban (200 nM). (D) Panel A in the presence of rivaroxaban (200 nM). (E) Panel A in the presence of dabigatran (200 nM). (F) Lag time of thrombin generation on EA.hy926 endothelial cells was determined in the presence of all 3 direct oral anticoagulants (200 nM) with and without antibodies against (A)PC (50 μg/mL) or superFVa (200 nM). ns, not significant; TM, thrombomodulin.
SuperFVa improves thrombin generation on cells in patient plasma with DOACs
To further support the role of APC’s anticoagulant activity in DOAC-mediated inhibition of thrombin generation and to identify the mechanism(s) by which superFVa restores hemostasis, plasmas from patients taking DOACs (apixaban, rivaroxaban, or dabigatran) were tested side by side in the endothelial cell thrombin generation assay and in a regular thrombin generation assay without cells in the presence of tissue factor and phospholipid vesicles. Patient and sample characteristics are shown in supplemental Table 1 and supplemental Figure 3.
In plasma from patients on apixaban, ETP values for thrombin generation on cells improved significantly when APC’s anticoagulant effects were inhibited by a blocking protein C antibody or by superFVa (Figure 3A). In contrast, superFVa had no effect on ETP when thrombin generation was initiated by tissue factor in the absence of cells. SuperFVa also significantly reduced the lag time of the thrombin generation curve in plasma from patients on apixaban, regardless of whether endothelial cells were present (Figure 3B). The blocking antibody to protein C did not shorten lag times of thrombin generation, which suggests that the effect of superFVa on lag time is inherent to overcoming the activation of FV as the rate-limiting step, independent of its resistance to APC. Similar results were obtained in plasma samples from patients on rivaroxaban (Figure 3C-D). Notably, inhibition of APC’s anticoagulant activity or treatment with superFVa had little effect on the ETP with or without cells in plasma samples from patients on dabigatran (Figure 3E), but superFVa did reduce the lag time of initiation of thrombin generation in these plasmas (Figure 3E-F). ETP values and lag times were generally higher and more extended (Figure 3E-F) compared with samples from patients on apixaban or rivaroxaban (Figure 3A-D). Thus, resistance to inactivation by APC seems to be the main mechanisms by which superFVa improves thrombin generation in plasma samples from patients on apixaban or rivaroxaban, while overcoming the FV activation threshold seems to be the dominant mechanisms by which superFVa accelerates thrombin generation in plasma samples from patients on dabigatran.
Figure 3.

Improvement of endothelial thrombin generation in DOAC patient plasma by superFVa. Thrombin generation was performed in plasma from patients on chronic anticoagulation with DOACs in the presence of EA.hy926 endothelial cells (○) and in the absence of cells (□). Thrombin generation in the absence of cells was initiated by 0.4 pM tissue factor and 10 μM phospholipid vesicles (PC/PS/PE 40/20/40). Shown are the endogenous thrombin potential (ETP) (A,C,E) and the lag time (B,D,F) of thrombin generation in the presence or absence of antibodies (50 μg/mL) against (A)PC (αPC; C1) or superFVa (20 nM) of patients on chronic anticoagulation with apixaban (A-B), rivaroxaban (C-D) , and dabigatran (E-F).
Effects of superFVa and rhFVIIa on thrombin generation in NHP in the presence of FXa inhibitors
To determine the extent to which superFVa can enhance the efficiency of the prothrombinase complex, tissue factor-induced thrombin generation assays were performed in the absence of cells but in the presence of other prohemostatic molecules that are commonly used to arrest bleeding, such as rhFVIIa and KCENTRA (prothrombin complex concentrate [4F-PCC]). The rhFVIIa at 40 nM (2 µg/mL), which is the expected peak plasma concentration following IV administration in humans at the maximum approved dose for hemophilia patients with inhibitors (90 µg/kg), or superFVa at high concentration (400 nM) increased the ETP of NHP by 8% to 37% in the presence of either apixaban or rivaroxaban (200 nM) (Figure 4A-D). In addition, increasing concentrations of superFVa (6.25 to 400 nM) in combination with rhFVIIa (40 nM) enhanced ETP even further in the presence of apixaban or rivaroxaban, and restored ETP to 84% to 87% of normal. A plateau was reached at a concentration of 25 to 50 nM superFVa and 40 nM rhFVIIa, with no further increases in ETP with higher concentrations of superFVa (Figure 4B,D). Similarly, a dose-dependent increase in the peak height and reduction of lag time of the apixaban or rivaroxaban treated NHP was observed when superFVa was combined with rhFVIIa (40 nM) (supplemental Figure 4). Although effects on peak height appeared less pronounced compared with effects on the ETP, an approximate twofold increase over baseline was observed when the concentration of superFVa was increased to 100 to 400 nM in combination with rhFVIIa.
Figure 4.
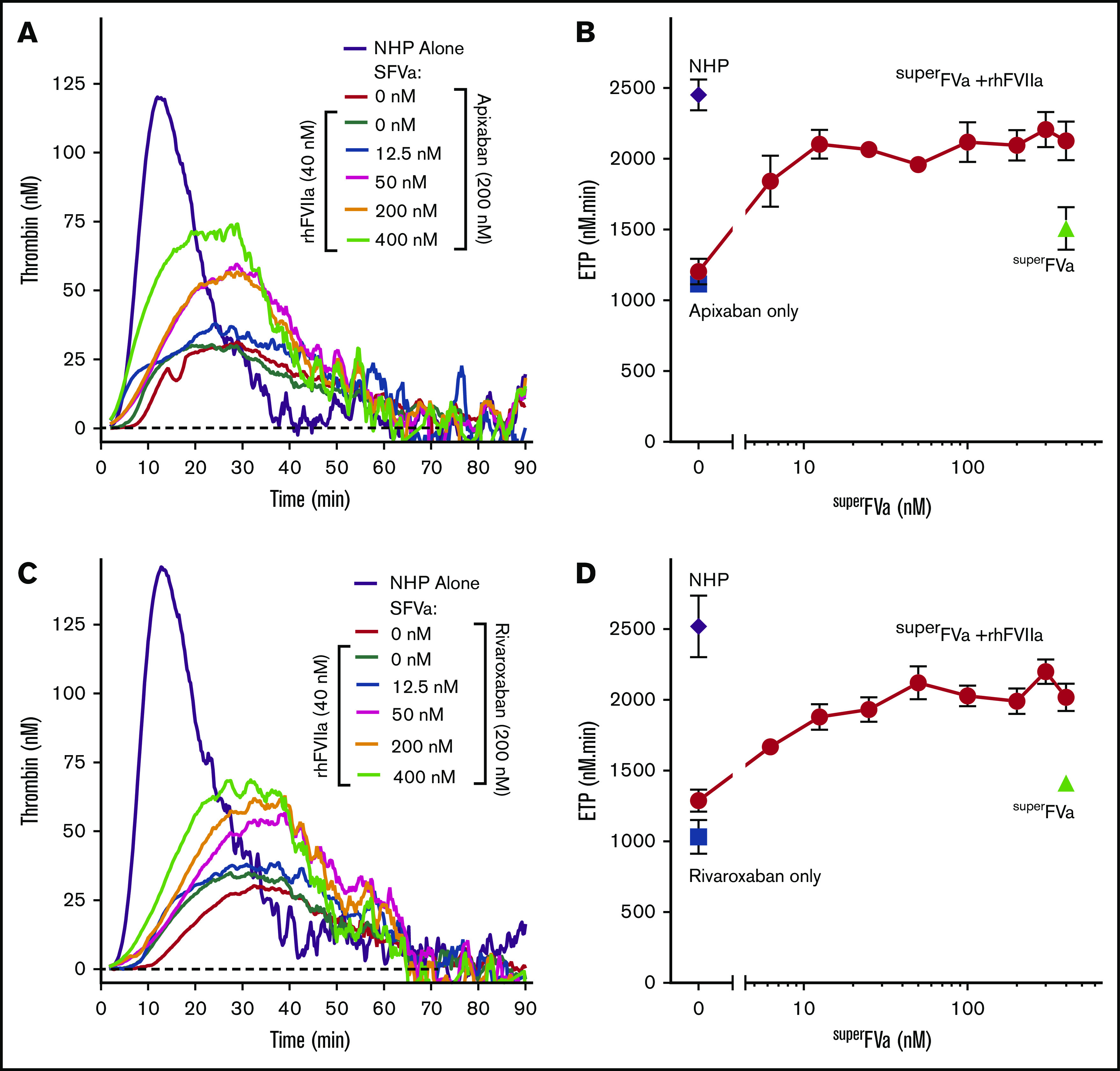
Effects of superFVa and rhFVIIa on thrombin generation in NHP spiked with FXa inhibitors. Thrombin generation was measured in NHP in the presence of the FXa inhibitors, apixaban or rivaroxaban. (A,C) Representative graphs showing the change in thrombin generation curves as the result of increasing concentrations of superFVa (0-400 nM) in the presence or absence of rhFVIIa (40 nM) in NHP spiked with apixaban (200 nM) (A) or rivaroxaban (200 nM) (C). (B,D) Enhancement of ETP by increasing concentrations of superFVa (0-400 nM) in the absence (Δ) or presence (○) of rhFVIIa (40 nM) in NHP spiked with apixaban (200 nM) (B) or rivaroxaban (200 nM) (D). Additional controls shown are NHP with (□) and without (◇) apixaban or rivaroxaban. (B,D) Peak height and lag time of are shown in supplemental Figure 4. Error bars represent standard error of the mean (n ≥ 3).
Effects of superFVa and 4F-PCC on thrombin generation in NHP in the presence of FXa inhibitors
4F-PCC is approved and effective for reversal of warfarin-induced bleeding48 and recommended off-label for DOAC-induced bleeding by some authorities.49 We compared the effect of 4F-PCC alone and in combination with superFVa using thrombin generation assays in NHP in the presence of either apixaban or rivaroxaban. When 4F-PCC was added to NHP at a concentration that approximates the expected plasma concentration (1.35 U/mL) after IV infusion of the highest recommended dose (50 U/kg), 4F-PCC did not increase thrombin generation suppressed by either apixaban or rivaroxaban at their therapeutic concentrations of ∼200 nM (Figure 5). However, in the presence of superFVa (50 nM), increasing concentrations of 4F-PCC (0.08 to 1.35 U/mL) restored the ETP of NHP treated with apixaban or rivaroxaban (200 nM) up to 110% to 114% of normal (NHP with 4F-PCC but no FXa inhibitors) (Figure 5A-B,D-E). Similarly, increasing concentrations of superFVa (1.25 to 400 nM) in the presence of 4F-PCC (1.35 U/mL) into NHP treated with either apixaban or rivaroxaban (200 nM) enhanced ETP up to approximately fourfold over baseline (Figure 5C,F) and restored ETP up to 105% to 125% of normal (NHP with 4F-PCC but no FXa inhibitors). Similar effects were observed for peak height and the reduction of the lag time in the presence of either apixaban (supplemental Figure 5) or rivaroxaban (supplemental Figure 6).
Figure 5.
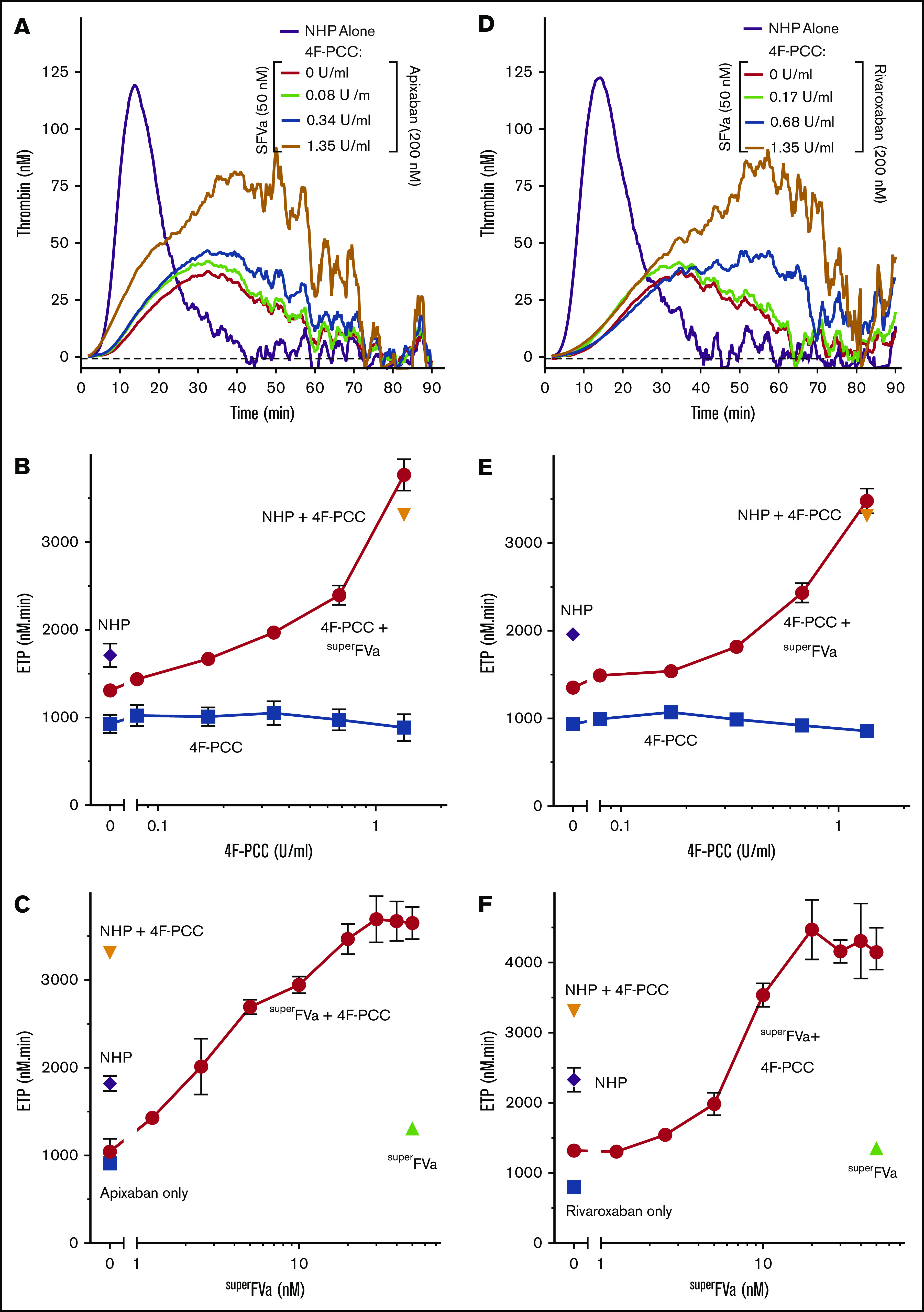
Effects of superFVa and 4F-PCC on thrombin generation in NHP spiked with FXa inhibitors. Thrombin generation was measured in NHP in the presence of the FXa inhibitors, apixaban or rivaroxaban. (A,D) Representative graphs showing the change in thrombin generation curves as the result of increasing concentrations of 4F-PCC (0-1.35 U/mL) in the presence or absence of superFVa (50 nM) in NHP spiked with apixaban (200 nM) (A) or rivaroxaban (200 nM) (D). (B,E) Enhancement of ETP by increasing concentrations of 4F-PCC (0-1.35 U/mL) in the absence (∎) or presence (○) of superFVa (50 nM) in NHP spiked with apixaban (200 nM) (B) or rivaroxaban (200 nM) (E). (C,F) Enhancement of ETP by increasing concentrations of superFVa (0-50 nM) in the absence (Δ) or presence (○) of 4F-PCC (1.35 U/mL) in NHP spiked with apixaban (200 nM) (C) or rivaroxaban (200 nM) (F). Peak height and lag time of (B-C) are shown in supplemental Figure 5 and of (E-F) in supplemental Figure 6. Additional controls shown are NHP with (□) and without (◇) apixaban or rivaroxaban or NHP with 1.35 U/mL 4F-PCC (∇). Error bars represent standard error of the mean (n ≥ 3).
Effects of superFVa and rhFVIIa on thrombin generation in NHP in the presence of dabigatran
A dose titration in NHP demonstrated that a concentration of 1 μM dabigatran suppressed the ETP by ∼80% (supplemental Figure 2E-F). This concentration was therefore used to investigate the potential to correct the ETP in NHP with addition of rhFVIIa (40 nM), superFVa at increasing concentrations (0.1 to 100 nM) alone, or in combination with rhFVIIa (40 nM) (supplemental Figure 7A). Neither superFVa nor rhFVIIa alone nor in combination corrected the ETP. However, a concentration-dependent reduction of time to peak thrombin generation was observed with superFVa, which was further enhanced to normal values in the presence of 40 nM rhFVIIa (supplemental Figure 7B).
Combinatorial effects of superFVa and rhFVIIa on DOAC-induced bleeding in vivo
Dose response titration of rhFVIIa showed that mean blood loss was significantly reduced at 6 mg/kg in apixaban-treated mice and at 0.1 mg/kg rhFVIIa in rivaroxaban-treated mice (supplemental Figure 8). Thus, similar to superFVa, much lower doses of rhFVIIa were needed to decrease bleeding induced by rivaroxaban compared with apixaban. To determine potential synergistic effects between superFVa and rhFVIIa to reduce apixaban- and rivaroxaban-induced bleeding, superFVa and rhFVIIa were combined at doses that individually only partially reduced bleeding. Indeed, the combination of superFVa and rhFVIIa provided an additional reduction of mean blood loss to ∼5 µL/g (all P < .01; n = 8-12 per group), which was similar to mean blood loss in untreated wild-type mice (3.3 µL/g; P = .85; n = 8-12 per group) (Figure 6A-B). Dabigatran-induced bleeding could not be reversed by high-dose rhFVIIa (4 mg/kg, ∼40-fold higher dose approved for human use) alone. However, the combination of rhFVIIa (4 mg/kg) with a dose of superFVa (0.4 mg/kg), previously shown to not affect dabigatran-induced bleeding (Figure 1C), resulted in partial bleed correction (Figure 6C).
Figure 6.

Reversal of DOAC-induced bleeding with superFVa and rhFVIIa in wild-type BalbC mice. Mice were treated with apixaban (8 mg/kg) (A), rivaroxaban (40 mg/kg) (B), and dabigatran (0.4 mg/kg) (C) by IV tail vein injection and bleeding was measured for 20 minutes after tail clip. Blood loss was expressed in microliters of blood per gram (g) mouse. Increasing doses of superFVa, rhFVIIa, or rhFVIIa in combination with superFVa were injected retro-orbitally 30 minutes after DOAC administration and 5 minutes before tail clip. Error bars represent SEM (n = 8-12 per group). P values were determined by Kruskal-Wallis followed by a 2-tailed Mann-Whitney U test and values ≤.05 were considered statistically significant.
In summary, these results indicated that superFVa was able to reverse bleeding associated with FXa and FIIa inhibitors in vivo and that effective bleed reduction could also be achieved by combination of low-dose superFVa and rhFVIIa that each individually provided only partially reduced bleeding.
Discussion
“FVa activity augmentation” by means of the engineered molecule superFVa is a novel way to improve coagulation and rescue bleeding. Previously, superFVa demonstrated efficient normalization of compromised hemostasis in hemophilic mice and hemophilic plasma with and without inhibitors27,29 and superFVa ablated traumatic bleeding in wild-type mice because of APC.27-29 Here, we report the efficacy of superFVa to reverse anticoagulation and bleeding associated with DOACs. At the optimized dose of each DOAC (rivaroxaban, apixaban, and dabigatran) for murine in vivo studies, blood loss following tail clip injury in wild-type mice was pronounced and comparable to blood loss previously observed in hemophilic mice or in wild-type mice treated with APC.27,28
SuperFVa efficiently reversed FXa inhibitor-induced bleeding in mice injected with the direct FXa inhibitors (rivaroxaban or apixaban) and the direct FIIa inhibitor dabigatran. The hemostatic efficacy of superFVa in vivo in the presence of FXa or FII inhibitors contrasted with only modest correction of thrombin generation in vitro by superFVa. This discrepancy arose as the anticoagulant contribution of APC is not routinely measured in coagulation assays because these assays typically do not include the cellular cofactors (eg, thrombomodulin, EPCR), required for protein C activation. This underlines the clinical paradigm that prediction of in vivo hemostasis based on in vitro routine coagulation assay results is imperfect. Indeed, the injection of an APC-inactivating antibody into mice in the presence of FXa inhibition, reduced bleeding to the same extent as superFVa, demonstrating that APC contributes substantially to bleeding with this class of DOACs. Thrombin generation studies performed on endothelial cells with human plasma spiked with DOACs, as well as plasma from patients chronically anticoagulated with DOACs and in the presence of antibodies against APC, thrombomodulin, or EPCR demonstrated that the prohemostatic effect of superFVa in the presence of FXa inhibition in vivo was also observed in vitro as long as the assay incorporated the anticoagulant contributions of APC. This implied that, despite suppression by FXa inhibitors, sufficient thrombin was generated to activate protein C to the extent that APC contributed to bleeding, and that the APC resistance of superFVa provides a major mechanism for the prohemostatic efficacy of superFVa in the presence of FXa-targeting DOACs.
However, superFVa also rescued bleeding in the presence of FIIa inhibition, whereas the APC-inactivating antibody did not reduce bleeding for this class of DOACs, indicating that additional mechanisms, independent of APC resistance, contributed to the hemostatic potential of superFVa. In thrombin generation assays, superFVa caused a universal shortening of the lag time, which was the most notable difference in the presence of FIIa inhibition. This shortening of the thrombin generation lag time was not observed in the presence of the APC-inactivating antibody, suggesting that this effect of superFVa was independent of its resistance to APC. Inherent to the lag time is overcoming the threshold for activation of FV as the rate-limiting step for FVa’s availability in the prothrombinase complex. Thus, overcoming the FV activation threshold provides a second major mechanism for the hemostatic effect of superFVa in the presence of DOACs; this likely is the predominant mechanism by which superFVa exerts its hemostatic effect in the face of FIIa-induced bleeding.
Notwithstanding, augmentation of prothrombinase is a third mechanism by which superFVa can improve hemostasis, as was demonstrated previously in hemophilia mice and plasma.27,29 However, effects of superFVa in standard thrombin generation assays in the presence of FXa-inhibition were modest, in line with the fact that superFVa is a nonenzymatic cofactor whose activity is regulated by the presence of functional FXa. In comparison, other prohemostatic products, such as rhFVIIa or 4F-PCC, induced similar modest effects on thrombin generation. In contrast, large improvement in all parameters of thrombin generation in the presence of FXa inhibitors were observed when superFVa and either rhFVIIa or 4F-PCC were combined at low concentrations. Presumably, the improved generation of FXa by the FVII-based prohemostatic permits more efficient enhancement of prothrombinase by superFVa resulting in a remarkable synergistic effect.
Recent studies demonstrated that superFVa did not show apparent signs of thrombogenicity in a model of pulmonary embolism in hemostasis competent wild-type mice.50 Although the human circulating half-life of superFVa is not yet known, superFVa has a short circulating half-life of 26 minutes in BalbC mice,50 and one may assume that it will be similarly short in humans. These properties appear advantageous for use of superFVa in bleeding situations in which patients suffer from underlying prothrombotic conditions, and where a rapid switch on and off effect is desired. Immunogenicity is always a potential concern of engineered recombinant proteins, but this risk seems relatively low in the current application where superFVa is administered as a single bolus followed by rapid clearance from the circulation (T1/2 = 26 minutes), and in silico modeling predicts that immunogenicity of superFVa is not noticeably increased compared with human FVa.50
In summary, the DOAC class-independent prohemostatic efficiency of superFVa can be explained by 3 different mechanisms. These are resisting inactivation by APC, overcoming the FV activation threshold, and maximizing the efficiency of the prothrombinase complex when the available FXa is increased by FVIIa-based prohemostatics.30,51 That the mechanisms of bleeding differ for FXa- and FIIa-targeting DOACs, and, that superFVa relies on different properties to improve hemostasis, is noteworthy. These observations are in agreement with a growing awareness that FXa- and FIIa-targeting DOACs affect downstream pathways differently, including the activation of protein C,52 regulation of fibrinolysis,53,54 and protease activated receptor signaling.55-57 Our results are also in line with the increasing recognition that disproportional APC generation may contribute to bleeding, as shown in acute traumatic coagulopathy and in hemophilia.27,32,58-60 Thus, the versatility of prohemostatic effectiveness delineates superFVa as a promising class-independent rescue agent in bleeding situations associated with DOACs.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors kindly thank Kenji Fukudome (Saga Medical School, Saga, Japan) for the anti-EPCR antibody RCR-252 and Jose Fernandez (The Scripps Research Institute) for the anti-(A)PC antibodies (C1 and SPC-54).
This work was funded by grant support from an Early Career Development Award from Bayer Hemophilia (A.v.D. and V.B.) and the National Institutes of Health, National Heart, Lung, and Blood Institute HL133728 (J.H.G.), HL142975 (J.H.G. and L.O.M.), and HL104165 and HL130678 (L.O.M.).
Footnotes
Requests for data sharing should be e-mailed to one of the corresponding authors, Annette von Drygalski at avondrygalski@ucsd.edu or Laurent O. Mosnier at lmosnier@scripps.edu.
Authorship
Contribution: A.v.D. and V.B. contributed equally to design of the research, experiments, and manuscript drafting; J.H.G. contributed to concept and supported the experimental work; T.J.C. and A.J.G. performed superFVa purification; A.J.G. also provided experimental guidance; P.M.A. and D.J.E. provided DOAC patient plasmas; L.O.M. designed the concept and research, performed experiments, provided project oversight, experimental guidance, and manuscript writing; and all authors critically reviewed the manuscript and approved it in its final version.
Conflict-of-interest disclosure: A.v.D. has received honoraria for participating in scientific advisory board panels, consulting, and speaking engagements for Takeda, Biomarin, Bioverativ/Sanofi, Novo Nordisk, and Uniqure, and reports research support from Pfizer and Sanofi. The University of California San Diego and The Scripps Research Institute hold intellectual property rights related to superFVa on which A.v.D., A.J.G., J.H.G., and L.O.M. are listed as inventors. A.v.D., A.J.G., and L.O.M. are founders of Hematherix LLC, a biotech company that is developing superFVa therapy for bleeding complications. A.v.D. and L.O.M. are members of the board of directors of Hematherix LLC. The remaining authors declare no competing financial interests.
Correspondence: Annette von Drygalski, Department of Molecular Medicine, IMM-315, The Scripps Research Institute, 10550 N Torrey Pines Rd, La Jolla, CA 92037; e-mail: avondrygalski@ucsd.edu; and Laurent O. Mosnier, Department of Molecular Medicine, IMM-315, The Scripps Research Institute, 10550 N Torrey Pines Rd, La Jolla, CA 92037; e-mail: lmosnier@scripps.edu.
References
- 1.Ruff CT, Giugliano RP, Braunwald E, et al. . Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet. 2014;383(9921):955-962. [DOI] [PubMed] [Google Scholar]
- 2.Camm AJ, Accetta G, Ambrosio G, et al. ; GARFIELD-AF Investigators . Evolving antithrombotic treatment patterns for patients with newly diagnosed atrial fibrillation. Heart. 2017;103(4):307-314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee CH, Fang CC, Tsai LM, et al. . Changing treatment patterns in patients with venous thromboembolism in Taiwan. Circ J. 2020;84(2):283-293. [DOI] [PubMed] [Google Scholar]
- 4.Connolly SJ, Ezekowitz MD, Yusuf S, et al. ; RE-LY Steering Committee and Investigators . Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361(12):1139-1151. [DOI] [PubMed] [Google Scholar]
- 5.Connolly SJ, Eikelboom J, Joyner C, et al. ; AVERROES Steering Committee and Investigators . Apixaban in patients with atrial fibrillation. N Engl J Med. 2011;364(9):806-817. [DOI] [PubMed] [Google Scholar]
- 6.Patel MR, Mahaffey KW, Garg J, et al. ; ROCKET AF Investigators . Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med. 2011;365(10):883-891. [DOI] [PubMed] [Google Scholar]
- 7.Granger CB, Alexander JH, McMurray JJ, et al. ; ARISTOTLE Committees and Investigators . Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365(11):981-992. [DOI] [PubMed] [Google Scholar]
- 8.Prins MH, Lensing AW, Bauersachs R, et al. ; EINSTEIN Investigators . Oral rivaroxaban versus standard therapy for the treatment of symptomatic venous thromboembolism: a pooled analysis of the EINSTEIN-DVT and PE randomized studies. Thromb J. 2013;11(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schulman S, Kearon C, Kakkar AK, et al. ; RE-COVER Study Group . Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med. 2009;361(24):2342-2352. [DOI] [PubMed] [Google Scholar]
- 10.Beyer-Westendorf J, Förster K, Pannach S, et al. . Rates, management, and outcome of rivaroxaban bleeding in daily care: results from the Dresden NOAC registry. Blood. 2014;124(6):955-962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamashita Y, Uozumi R, Hamatani Y, et al. . Current status and outcomes of direct oral anticoagulant use in real-world atrial fibrillation patients - Fushimi AF Registry. Circ J. 2017;81(9):1278-1285. [DOI] [PubMed] [Google Scholar]
- 12.Schiele F, van Ryn J, Canada K, et al. . A specific antidote for dabigatran: functional and structural characterization. Blood. 2013;121(18):3554-3562. [DOI] [PubMed] [Google Scholar]
- 13.Glund S, Moschetti V, Norris S, et al. . A randomised study in healthy volunteers to investigate the safety, tolerability and pharmacokinetics of idarucizumab, a specific antidote to dabigatran. Thromb Haemost. 2015;113(5):943-951. [DOI] [PubMed] [Google Scholar]
- 14.Glund S, Stangier J, Schmohl M, et al. . Safety, tolerability, and efficacy of idarucizumab for the reversal of the anticoagulant effect of dabigatran in healthy male volunteers: a randomised, placebo-controlled, double-blind phase 1 trial. Lancet. 2015;386(9994):680-690. [DOI] [PubMed] [Google Scholar]
- 15.Pollack CV Jr., Reilly PA, Eikelboom J, et al. . Idarucizumab for dabigatran reversal. N Engl J Med. 2015;373(6):511-520. [DOI] [PubMed] [Google Scholar]
- 16.Pollack CV Jr., Reilly PA, van Ryn J, et al. . Idarucizumab for dabigatran reversal - full cohort analysis. N Engl J Med. 2017;377(5):431-441. [DOI] [PubMed] [Google Scholar]
- 17.Connolly SJ, Milling TJ Jr., Eikelboom JW, et al. ; ANNEXA-4 Investigators . Andexanet alfa for acute major bleeding associated with factor Xa inhibitors. N Engl J Med. 2016;375(12):1131-1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Connolly SJ, Gibson CM, Crowther M. Andexanet alfa for factor Xa inhibitor reversal. N Engl J Med. 2016;375(25):2499-2500. [DOI] [PubMed] [Google Scholar]
- 19.Siegal DM, Curnutte JT, Connolly SJ, et al. . Andexanet alfa for the reversal of factor Xa inhibitor activity. N Engl J Med. 2015;373(25):2413-2424. [DOI] [PubMed] [Google Scholar]
- 20.Lu G, DeGuzman FR, Hollenbach SJ, et al. . A specific antidote for reversal of anticoagulation by direct and indirect inhibitors of coagulation factor Xa. Nat Med. 2013;19(4):446-451. [DOI] [PubMed] [Google Scholar]
- 21.Connolly SJ, Crowther M, Eikelboom JW, et al. ; ANNEXA-4 Investigators . Full study report of andexanet alfa for bleeding associated with factor Xa inhibitors. N Engl J Med. 2019;380(14):1326-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sible AM, Nawarskas JJ. Andexanet alfa for reversing factor Xa inhibition. Cardiol Rev. 2019;27(2):108-111. [DOI] [PubMed] [Google Scholar]
- 23.Van der Wall SJ, Lopes RD, Aisenberg J, et al. . Idarucizumab for dabigatran reversal in the management of patients with gastrointestinal bleeding. Circulation. 2019;139(6):748-756. [DOI] [PubMed] [Google Scholar]
- 24.Ansell JE, Bakhru SH, Laulicht BE, et al. . Single-dose ciraparantag safely and completely reverses anticoagulant effects of edoxaban. Thromb Haemost. 2017;117(2):238-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ansell JE, Bakhru SH, Laulicht BE, et al. . Use of PER977 to reverse the anticoagulant effect of edoxaban. N Engl J Med. 2014;371(22):2141-2142. [DOI] [PubMed] [Google Scholar]
- 26.Pollack CV., Jr. Coagulation assessment with the new generation of oral anticoagulants. Emerg Med J. 2016;33(6):423-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.von Drygalski A, Cramer TJ, Bhat V, Griffin JH, Gale AJ, Mosnier LO. Improved hemostasis in hemophilia mice by means of an engineered factor Va mutant. J Thromb Haemost. 2014;12(3):363-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.von Drygalski A, Bhat V, Gale AJ, et al. . An engineered factor Va prevents bleeding induced by anticoagulant wt activated protein C. PLoS One. 2014;9(8):e104304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhat V, von Drygalski A, Gale AJ, Griffin JH, Mosnier LO. Improved coagulation and haemostasis in haemophilia with inhibitors by combinations of superFactor Va and Factor VIIa. Thromb Haemost. 2016;115(3):551-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mann KG, Jenny RJ, Krishnaswamy S. Cofactor proteins in the assembly and expression of blood clotting enzyme complexes. Annu Rev Biochem. 1988;57(1):915-956. [DOI] [PubMed] [Google Scholar]
- 31.Davenport RA, Guerreiro M, Frith D, et al. . Activated protein C drives the hyperfibrinolysis of acute traumatic coagulopathy. Anesthesiology. 2017;126(1):115-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Polderdijk SG, Adams TE, Ivanciu L, Camire RM, Baglin TP, Huntington JA. Design and characterization of an APC-specific serpin for the treatment of hemophilia. Blood. 2017;129(1):105-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao XY, Yegneswaran S, Bauzon M, et al. . Targeted inhibition of activated protein C anticoagulant activity by monoclonal antibody HAPC1573 for treatment of hemophilia [abstract]. Blood. 2016;128(22):80 Abstract 321. [Google Scholar]
- 34.Mesters RM, Houghten RA, Griffin JH. Identification of a sequence of human activated protein C (residues 390-404) essential for its anticoagulant activity. J Biol Chem. 1991;266(36):24514-24519. [PubMed] [Google Scholar]
- 35.Gale AJ, Xu X, Pellequer JL, Getzoff ED, Griffin JH. Interdomain engineered disulfide bond permitting elucidation of mechanisms of inactivation of coagulation factor Va by activated protein C. Protein Sci. 2002;11(9):2091-2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ye X, Fukudome K, Tsuneyoshi N, et al. . The endothelial cell protein C receptor (EPCR) functions as a primary receptor for protein C activation on endothelial cells in arteries, veins, and capillaries. Biochem Biophys Res Commun. 1999;259(3):671-677. [DOI] [PubMed] [Google Scholar]
- 37.Heeb MJ, Schwarz HP, White T, Lämmle B, Berrettini M, Griffin JH. Immunoblotting studies of the molecular forms of protein C in plasma. Thromb Res. 1988;52(1):33-43. [DOI] [PubMed] [Google Scholar]
- 38.Burnier L, Fernández JA, Griffin JH. Antibody SPC-54 provides acute in vivo blockage of the murine protein C system. Blood Cells Mol Dis. 2013;50(4):252-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Radtke KP, Griffin JH, Riceberg J, Gale AJ. Disulfide bond-stabilized factor VIII has prolonged factor VIIIa activity and improved potency in whole blood clotting assays. J Thromb Haemost. 2007;5(1):102-108. [DOI] [PubMed] [Google Scholar]
- 40.Hemker HC, Giesen P, AlDieri R, et al. . The calibrated automated thrombogram (CAT): a universal routine test for hyper- and hypocoagulability. Pathophysiol Haemost Thromb. 2002;32(5-6):249-253. [DOI] [PubMed] [Google Scholar]
- 41.Perzborn E, Strassburger J, Wilmen A, et al. . In vitro and in vivo studies of the novel antithrombotic agent BAY 59-7939–an oral, direct Factor Xa inhibitor. J Thromb Haemost. 2005;3(3):514-521. [DOI] [PubMed] [Google Scholar]
- 42.Wong PC, Pinto DJ, Zhang D. Preclinical discovery of apixaban, a direct and orally bioavailable factor Xa inhibitor. J Thromb Thrombolysis. 2011;31(4):478-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schumacher WA, Bostwick JS, Stewart AB, Steinbacher TE, Xin B, Wong PC. Effect of the direct factor Xa inhibitor apixaban in rat models of thrombosis and hemostasis. J Cardiovasc Pharmacol. 2010;55(6):609-616. [DOI] [PubMed] [Google Scholar]
- 44.Godier A, Miclot A, Le Bonniec B, et al. . Evaluation of prothrombin complex concentrate and recombinant activated factor VII to reverse rivaroxaban in a rabbit model. Anesthesiology. 2012;116(1):94-102. [DOI] [PubMed] [Google Scholar]
- 45.Frost C, Nepal S, Wang J, et al. . Safety, pharmacokinetics and pharmacodynamics of multiple oral doses of apixaban, a factor Xa inhibitor, in healthy subjects. Br J Clin Pharmacol. 2013;76(5):776-786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kubitza D, Becka M, Mueck W, Zuehlsdorf M. Rivaroxaban (BAY 59-7939)–an oral, direct Factor Xa inhibitor–has no clinically relevant interaction with naproxen. Br J Clin Pharmacol. 2007;63(4):469-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jacquemin M, Toelen J, Schoeters J, et al. . The addition of idarucizumab to plasma samples containing dabigatran allows the use of routine coagulation assays for the diagnosis of hemostasis disorders. J Thromb Haemost. 2015;13(11):2087-2092. [DOI] [PubMed] [Google Scholar]
- 48.Quinlan DJ, Eikelboom JW, Weitz JI. Four-factor prothrombin complex concentrate for urgent reversal of vitamin K antagonists in patients with major bleeding. Circulation. 2013;128(11):1179-1181. [DOI] [PubMed] [Google Scholar]
- 49.Frontera JA, Lewin JJ III, Rabinstein AA, et al. . Guideline for reversal of antithrombotics in intracranial hemorrhage: a Statement for Healthcare Professionals from the Neurocritical Care Society and Society of Critical Care Medicine. Neurocrit Care. 2016;24(1):6-46. [DOI] [PubMed] [Google Scholar]
- 50.Gale AJ, Bhat V, Pellequer JL, Griffin JH, Mosnier LO, Von Drygalski A. Safety, stability and pharmacokinetic properties of (super)factor Va, a novel engineered coagulation factor V for treatment of severe bleeding. Pharm Res. 2016;33(6):1517-1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nesheim ME, Taswell JB, Mann KG. The contribution of bovine factor V and factor Va to the activity of prothrombinase. J Biol Chem. 1979;254(21):10952-10962. [PubMed] [Google Scholar]
- 52.Kamisato C, Furugohri T, Morishima Y. A direct thrombin inhibitor suppresses protein C activation and factor Va degradation in human plasma: possible mechanisms of paradoxical enhancement of thrombin generation. Thromb Res. 2016;141:77-83. [DOI] [PubMed] [Google Scholar]
- 53.Incampo F, Carrieri C, Semeraro N, Colucci M. The paradoxical antifibrinolytic effect of dabigatran and argatroban in the presence of soluble thrombomodulin is unrelated to protein C-dependent increase of thrombin generation. Thromb Res. 2014;134(5):1110-1116. [DOI] [PubMed] [Google Scholar]
- 54.Semeraro F, Incampo F, Ammollo CT, et al. . Dabigatran but not rivaroxaban or apixaban treatment decreases fibrinolytic resistance in patients with atrial fibrillation. Thromb Res. 2016;138:22-29. [DOI] [PubMed] [Google Scholar]
- 55.Esmon CT. Targeting factor Xa and thrombin: impact on coagulation and beyond. Thromb Haemost. 2014;111(4):625-633. [DOI] [PubMed] [Google Scholar]
- 56.Sparkenbaugh EM, Chantrathammachart P, Mickelson J, et al. . Differential contribution of FXa and thrombin to vascular inflammation in a mouse model of sickle cell disease. Blood. 2014;123(11):1747-1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Petzold T, Thienel M, Dannenberg L, et al. . Rivaroxaban reduces arterial thrombosis by inhibition of FXa-driven platelet activation via protease activated receptor-1. Circ Res. 2020;126(4):486-500. [DOI] [PubMed] [Google Scholar]
- 58.Cohen MJ, Kutcher M, Redick B, et al. ; PROMMTT Study Group . Clinical and mechanistic drivers of acute traumatic coagulopathy. J Trauma Acute Care Surg. 2013;75(1 suppl 1):S40-S47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Belcheva MM, Coscia CJ. Diversity of G protein-coupled receptor signaling pathways to ERK/MAP kinase. Neurosignals. 2002;11(1):34-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cohen MJ, Christie SA. New understandings of post injury coagulation and resuscitation. Int J Surg. 2016;(33suppl B):242-245. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.