

Key Points
Loss of Pten during the neonatal period causes sustained fetal hematopoiesis, resulting in pediatric death.
Loss of Nf1 is necessary for cells to be hypersensitive to granulocyte-macrophage colony-stimulating factor and for monocytosis, but it is not a lethal event in juvenile leukemia.
Abstract
It is not clear whether disrupted age-specific hematopoiesis contributes to the complex manifestations in leukemia patients who carry identical mutations, particularly in pediatric and adult patients with similar clinical characteristics. By studying a dual-age–specific mouse model, we demonstrate that (1) loss of Pten during the fetal-to-adult hematopoiesis switch (hematopoiesis switch) causes sustained fetal hematopoiesis, resulting in death in juvenile leukemia; (2) myeloid-biased hematopoiesis in juvenile mice is associated with the sustained fetal properties of hematopoietic stem cells (HSCs); (3) the age specificity of juvenile myelomonocytic leukemia depends on the copy number of Pten and Nf1; (4) single-allelic Pten deletion during the hematopoiesis switch causes constitutive activation of MAPK in juvenile mice with Nf1 loss of heterozygosity (LOH); and (5) Nf1 LOH causes monocytosis in juvenile mice with Pten haploinsufficiency but does not cause lethality until adulthood. Our data suggest that 1 copy of Pten is sufficient to maintain an intact negative-feedback loop of the Akt pathway and HSC function in reconstitution, despite MAPK being constitutively activated in juvenile Pten+/Δ Nf1LOH mice. However, 2 copies of Pten are required to maintain the integrity of the MAPK pathway in juvenile mice with Nf1 haploinsufficiency. Our data indicate that previous investigations of Pten function in wild-type mice may not reflect the impact of Pten loss in mice with Nf1 mutations or other genetic defects. We provide a proof of concept that disassociated age-specific hematopoiesis contributes to leukemogenesis and pediatric demise.
Visual Abstract

Introduction
Leukemia without cell maturational arrest has not been studied in the context of disrupted age-specific hematopoiesis during physiological transitions, such as the fetal-to-adult hematopoiesis switch (hematopoiesis switch), puberty, or menopause. It remains unknown whether an aberrant hematopoiesis switch contributes to the complex manifestations in leukemia patients who carry identical mutations, particularly in pediatric and adult patients with similar clinical characteristics, such as juvenile myelomonocytic leukemia (JMML) and chronic myelomonocytic leukemia (CMML). Both diseases exhibit features of mixed myelodysplastic syndrome (MDS)/myeloproliferative neoplasm (MPN), and JMML is clearly an age-restricted disease. At diagnosis, the median age of JMML patients is 1.8 years,1 and it very rarely develops after the age of 6 years. In contrast, the mean age of CMML patients is 69 years.2 JMML and CMML are regarded as RASopathy diseases. JMML is almost exclusively caused by mutations in the RAS pathway, whereas RASopathy genes were detected in 40% of CMML patients.3 JMML and CMML are in the category of diseases with the least number of mutations per sample among pediatric and adult cancers, respectively.4 Emerging data suggest that epigenetics plays a significant role in the leukemogenesis of JMML and CMML.5-9 Previously, we reported that mice with germline interruptions of Nf1 exon 31 (Nf1+/−) developed a juvenile MPN when Pten was deleted during the neonatal period, whereas T-cell acute lymphoid leukemia (T-ALL) was induced when Pten was deleted in adulthood, suggesting that the timing of the genetic disturbance is critical for the phenotype.10 Here, we report that loss of Pten and Nf1 during the neonatal period, while the hematopoiesis switch is ongoing, causes sustained fetal hematopoiesis in juvenile mice, suggesting a novel mechanism underlying the timing of genetic disturbance in leukemia development. Our data provide a proof of concept that a disassociated age-specific hematopoiesis and sustained fetal hematopoiesis contribute to pediatric leukemia and demise.
Materials and methods
Study design and mice
PtenfloxP/floxP (Ptenfl/fl, B6.129S4-Ptentm1Hwu/J), Nf1Fcr/+ (B6.129S6-Nf1tm1Frc/J), Nf1floxP/floxP (Nf1fl/fl, B6.129 [Cg]-Nf1tm1Par/J), and Mx1-Cre (B6.Cg-Tg [Mx1-Cre]1Cgn/J) founder mice were purchased from The Jackson Laboratory. Mice with experimental genotypes were produced by crossbreeding, as previously reported.10 Pten deletion and Nf1 loss of heterozygosity (LOH) were induced by intraperitoneal injection of 30 µL of polyinosinic-polycytidylic acid (pIpC;InvivoGen) at a concentration of 1 μg/μL on postnatal day 8 (PND8) and PND10 (supplemental Figure 1). Genotyping was performed on PND15. The experimental procedures were approved by the Institutional Animal Care and Use Committee at the University of Arkansas for Medical Sciences.
Serial single hematopoietic stem cell transplantation
For primary (1°) transplants, single long-term hematopoietic stem cells (LT-HSCs; CD45.2+/LIN−Sca1+cKit+CD48−CD150+CD34−CD135−) were freshly sorted by a BD FACSAria II cell sorter (BD Biosciences), as reported,11 into each well of a 96-well plate, containing 2 × 105 bone marrow (BM) cells from wild-type (WT) mice with CD45.1+ in 120 μL of phosphate-buffered saline–5% mouse serum. Donor cells were injected into the retro-orbital venous sinus of preirradiated recipient mice, as previously reported.10 For secondary (2°) and tertiary (3°) transplants, a donor-derived single LT-HSC with CD45.2+ was sorted from 1° or 2° recipient mice at 16 to 18 weeks posttransplant, respectively. Recipient mice were evaluated at 16 to 18 weeks posttransplant.
Blood reconstitution analysis
Peripheral blood (PB) was collected from the retro-orbital venous sinus of recipient mice. Twenty-microliter aliquots were analyzed for white blood cell (WBC) count, and 100-μL aliquots were analyzed for donor-derived leukocytes by flow cytometry analysis. Mice were considered to be reconstituted by LT-HSCs when donor-derived myeloid cells (CD45.2+CD11b+) and lymphoid cells (sum of CD45.2+B220+ and CD45.2+CD3e+) were >0.1% of the WBCs in recipient mice at 16 weeks posttransplant.
For more information, see supplemental Materials and methods.
Results
Pten and Nf1 loss during neonatal period induces age-specific juvenile or adult myelomonocytic leukemia, depending upon Pten copy number
NF1 LOH and PTEN deficiency are frequently found in JMML patients.12-14 To simulate the molecular dynamics in JMML patients who develop NF1 LOH and lose PTEN protein shortly after birth,12,13,15 we generated experimental Ptenfl/flNf1Fcr/flMx1-Cre+ mice on the C57BL6/129 genetic background. Ptenfl/flNf1Fcr/flMx1-Cre+ mice were born healthy but started to show abdominal fullness and elevated WBC counts after 3 weeks of age, and all died at around 6 weeks of age (n = 7). This is likely due to the spontaneous induction of Mx1-Cre by endogenous cytokines. Therefore, we induced myeloid-specific Pten deletion and Nf1LOH on PND8 (equivalent to a full-term newborn age in humans) when WT mice undergo the fetal-to-adult hematopoiesis switch.16,17 Pten and Nf1 deletions were confirmed in mouse blood, spleens, and livers at 3 days post-pIpC injection (supplemental Figures 2 and 3). Mice with biallelic Pten deletion and Nf1LOH (PtenΔ/ΔNf1LOH) showed signs of disease with distressed fur and abdominal fullness in the second week of life. All died before 3 weeks of age (equivalent to 1-3 years old in humans). PtenΔ/ΔNf1LOH mice had significantly shorter lifespans compared with PtenΔ/ΔNf1+/Δ, Pten+/ΔNf1LOH, or WT littermates (P < .001) (Figure 1A). We evaluated the moribund mice and littermates at PND17-19. Substantial hepatosplenomegaly was observed in PtenΔ/ΔNf1LOH mice, and the severity correlated inversely with the copy number of Pten and Nf1 (Figure 1B; supplemental Figure 4A). Hematoxylin and eosin–stained tissue sections revealed severe infiltration of mature myeloid cells in the BM, spleens, livers, and lungs from PtenΔ/ΔNf1LOH mice (Figure 1C). These mice also had significantly elevated WBCs and monocytes, as well as lower hemoglobin levels, than did littermates with the other genotypes (Figure 1D-I). Flow cytometry analyses confirmed that they had significantly increased monocytes/macrophages and granulocytes, as well as a significant reduction in lymphocytes and erythrocytes in BM, blood, and spleen (Figure 1J-O). All of these characteristics suggest that these mice replicate the clinical features of JMML patients. Surprisingly, mice with a single copy of Pten and Nf1 LOH (Pten+/ΔNf1LOH) survived the juvenile period, although they had monocytosis and lymphocytopenia like their PtenΔ/Δ littermates with JMML since PND17 (Figure 1A,D-N). Most of them survived for >3 months but eventually died of severe infiltration of mature myeloid cells in vital organs, including obstruction of intestine/colon (supplemental Figures 4B and 5-7). Although PTEN mutations are rare in CMML patients, Pten+/ΔNf1LOH mice have clinical features that resemble those of CMML patients with RASopathy genes.3,18,19
Figure 1.
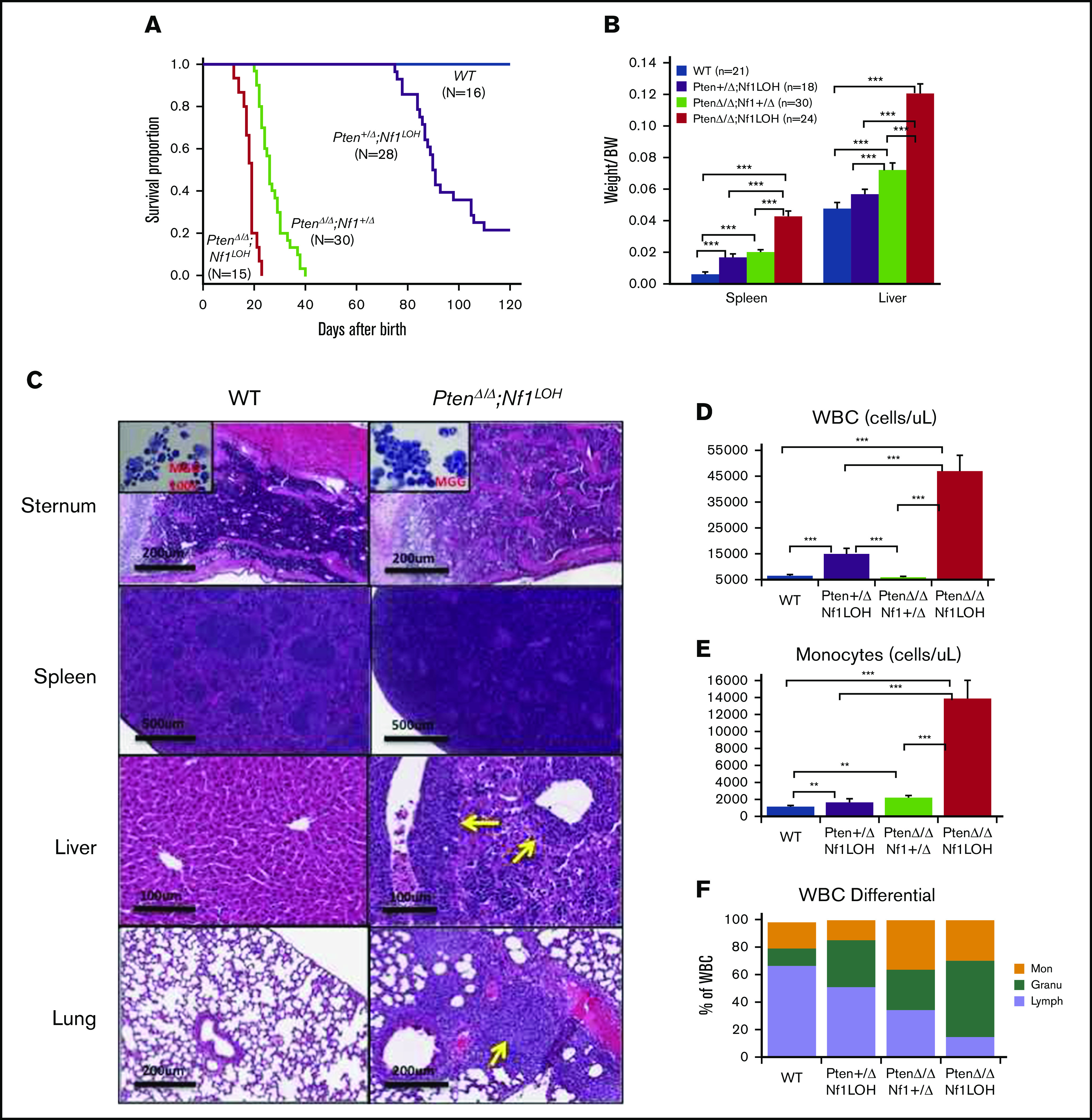
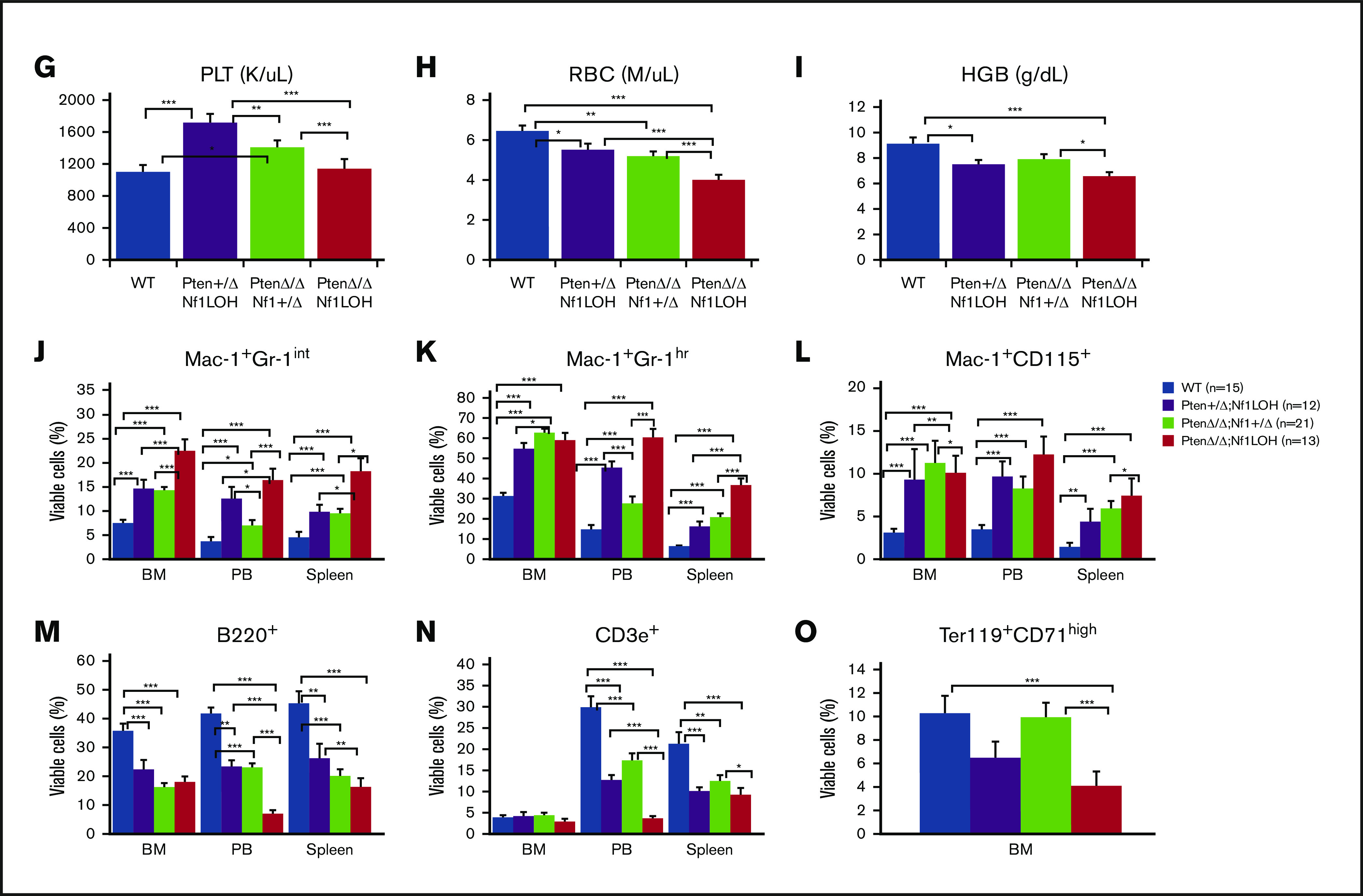
Correlation of phenotype and genotype in the dual-age–specific leukemia mouse model. (A) Kaplan-Meier survival curves show the relationship between survival time and genotype (P < .001). Death was counted in mice without any intervention. Male/female ratios were 2:1 in PtenΔ/ΔNf1LOH mice and Pten+/ΔNf1LOH mice and 1:1 in PtenΔ/ΔNf1+/Δ mice and WT mice. (B) Relationship between the genotype and the spleen and liver weights. Organs from littermates were collected at PND17-19 when PtenΔ/ΔNf1LOH mice with JMML were moribund. Data are presented as median ratios of spleen or liver weight/body weight (BW). (C) Representative photomicrographs of hematoxylin and eosin–stained tissue sections from PtenΔ/ΔNf1LOH mice with JMML (right panels) and WT control littermates (left panels) at PND18. Infiltrates are indicated by yellow arrows. (D-I) Blood profiles from juvenile PtenΔ/ΔNf1LOH mice and littermates at PND17-19 when diseased mice were moribund (n = 12-21). Complete blood counts were performed with a Vet Abc Hematological analyzer. Differentials were manually counted from blood smears stained with May-Grünwald-Giemsa. (J-O) Flow cytometry analysis of cell subpopulations in BM, PB, and spleen. Data are mean ± standard error. *P < .05, ** P <.01, ***P < .001. See additional supportive data in supplemental Figures 1-7. Granu, granulocytes; HGB, hemoglobin; Lymph, lymphocytes; Mon, monocytes; PLT, platelets; RBC, red blood cells.
JMML disease, but not CMML, is transplantable
To investigate whether the age-specific leukemias are transplantable, we performed competitive BM transplants (BMTs) using juvenile (PND17-20) mice with various genotypes and 3-month-old adult Pten+/ΔNf1LOH mice as donors (supplemental Figure 8). All recipient mice transplanted from JMML-diseased donors with PtenΔ/ΔNf1LOHdied before 16 weeks posttransplant (Figure 2A). Recipient mice from PtenΔ/Δ donors exhibited significantly myeloid-predominant engraftment in blood, with a significant reduction in B cells; however, T-cell deficiency was only observed in recipients from PtenΔ/ΔNf1LOH donors (Figure 2B-C). PtenΔ/ΔNf1+/Δ recipients began dying after 10 weeks; 9% (2/22) developed T-ALL as previously reported in recipients from PtenΔ/ΔNf1+/− mice,10 and 64% (14/22) survived through 16 weeks post-BMT (Figure 2A). Our data demonstrate that the JMML-like disease in PtenΔ/ΔNf1LOH mice is transplantable. Surprisingly, recipients transplanted with cells from juvenile and adult Pten+/ΔNf1LOH donors had transient elevated WBC counts at 8 weeks posttransplant, but it returned to the normal range by 12 weeks posttransplant. All of these mice lived through 16-weeks post-BMT (Figure 2A). After 16 weeks posttransplant, flow cytometry analyses showed that Pten+/ΔNf1LOH donor cells engrafted progenies similarly to WT donor cells, regardless of donor age (Figure 2B-C), suggesting that CMML-like disease in Pten+/ΔNf1LOH mice is not transplantable.
Figure 2.
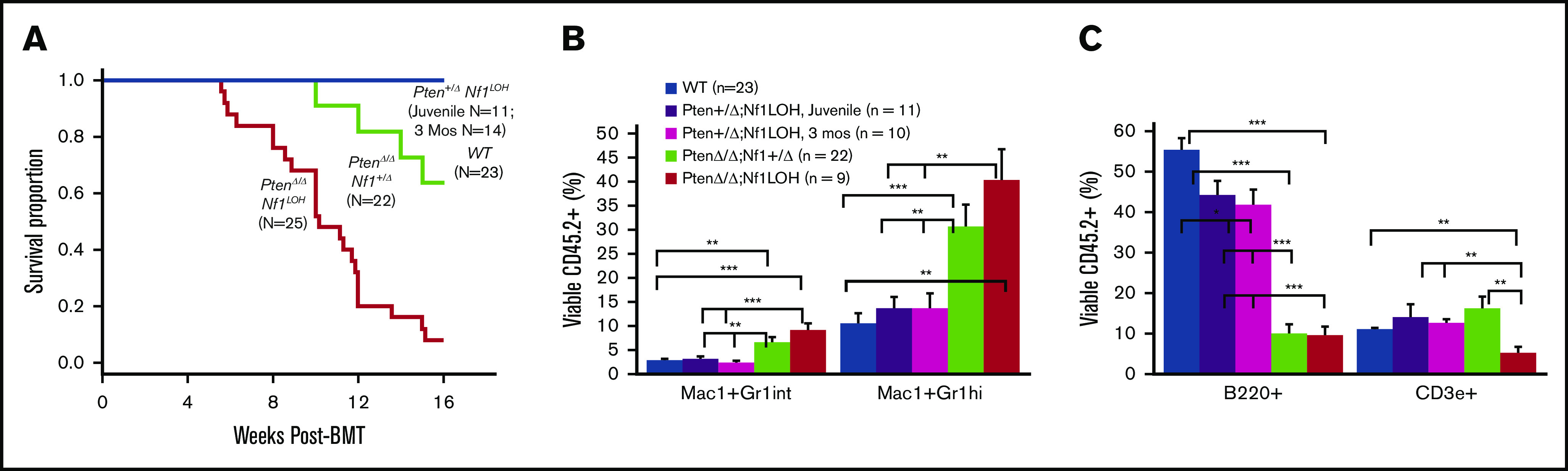
Reconstitution of BM cells from mice with JMML and littermates in competitive BMT. (A) Kaplan-Meier survival curves from recipients transplanted with BM from mice with JMML and juvenile littermates or from Pten+/Δ Nf1LOH mice with CMML at the age of 3 months when they were moribund. (B-C) Blood engraftment data from transplanted mice. Blood was collected from recipient mice at 12 to 16 weeks post-BMT. Data are mean ± standard error. *P < .05, **P < .01, ***P < .001. See additional supportive data in supplemental Figure 8.
Combined with our previous findings,10 we conclude that Pten copy number is critical for determining whether mice die as juveniles or adults. PtenΔ/Δ Nf1LOH mice replicate the clinical features of JMML in terms of age of onset and tissue-specific lesions. Pten+/ΔNf1LOH mice mimic human CMML that is caused by a dysregulated RAS pathway.
Pten deletion contributes to GM-CSF hypersensitivity without depletion of HSCs in juvenile mice with Nf1LOH
The prior notion was that the hypersensitivity of JMML cells to granulocyte-macrophage colony-stimulating factor (GM-CSF) was responsible for the lethal monocytosis that caused vital organ failure. We previously reported that PTEN protein was deficient in 67% of JMML patients, and PTEN deficiency contributed to the increased GM-CSF sensitivity in myeloid leukemia cell lines.12,20 However, GM-CSF hypersensitivity was absent in BM cells from PtenΔ/Δ mice with or without Nf1 haploinsufficiency, although those mice died of a juvenile MPN.10 Other groups reported that mice with WT Pten and somatic Nf1 deletion or Nf1 LOH were hypersensitive to GM-CSF but did not die as juveniles.21,22 To understand what causes GM-CSF hypersensitivity, we investigated the impact of Pten and Nf1 copy number on GM-CSF sensitivity. We found that BM cells from juvenile PtenΔ/ΔNf1LOH mice were hypersensitive to GM-CSF and interleukin-3 (IL-3), in contrast to cells from juvenile littermates with Pten+/ΔNf1LOH or biallelic Pten deletion without Nf1LOH (PtenΔ/ΔNf1+/Δ) (Figure 3A-B). Interestingly, BM cells from adult Pten+/ΔNf1LOH mice displayed GM-CSF hypersensitivity when they developed the full spectrum of CMML features with significantly elevated WBCs (Figure 3C-D; supplemental Figure 9A). Strikingly, we found that Pten protein was lost in adult CMML mice when their WBC counts reached the high levels as observed in mice with JMML (supplemental Figure 9B). Those data support our previous findings in cell lines,20 suggesting that an intact PTEN function is necessary for maintaining GM-CSF sensitivity in blood cells. This further supports the notion that loss of Pten is necessary for GM-CSF hypersensitivity in juvenile Nf1LOH mice, suggesting that NF1 LOH is required for monocytosis in JMML patients with NF1 mutation but is not sufficient to cause juvenile death. Further flow cytometry analyses of the hematopoietic progenitor cells in juvenile littermates showed that PtenΔ/ΔNf1LOH mice with JMML had significantly elevated percentages of LIN−Sca1+cKit+ cells in BM compared with littermates without GM-CSF hypersensitivity (Figure 3E). Our data suggest that Pten protein loss is necessary for cells to be GM-CSF hypersensitive in juvenile Nf1LOH mice without depleting hematopoietic stem cells (HSCs).
Figure 3.
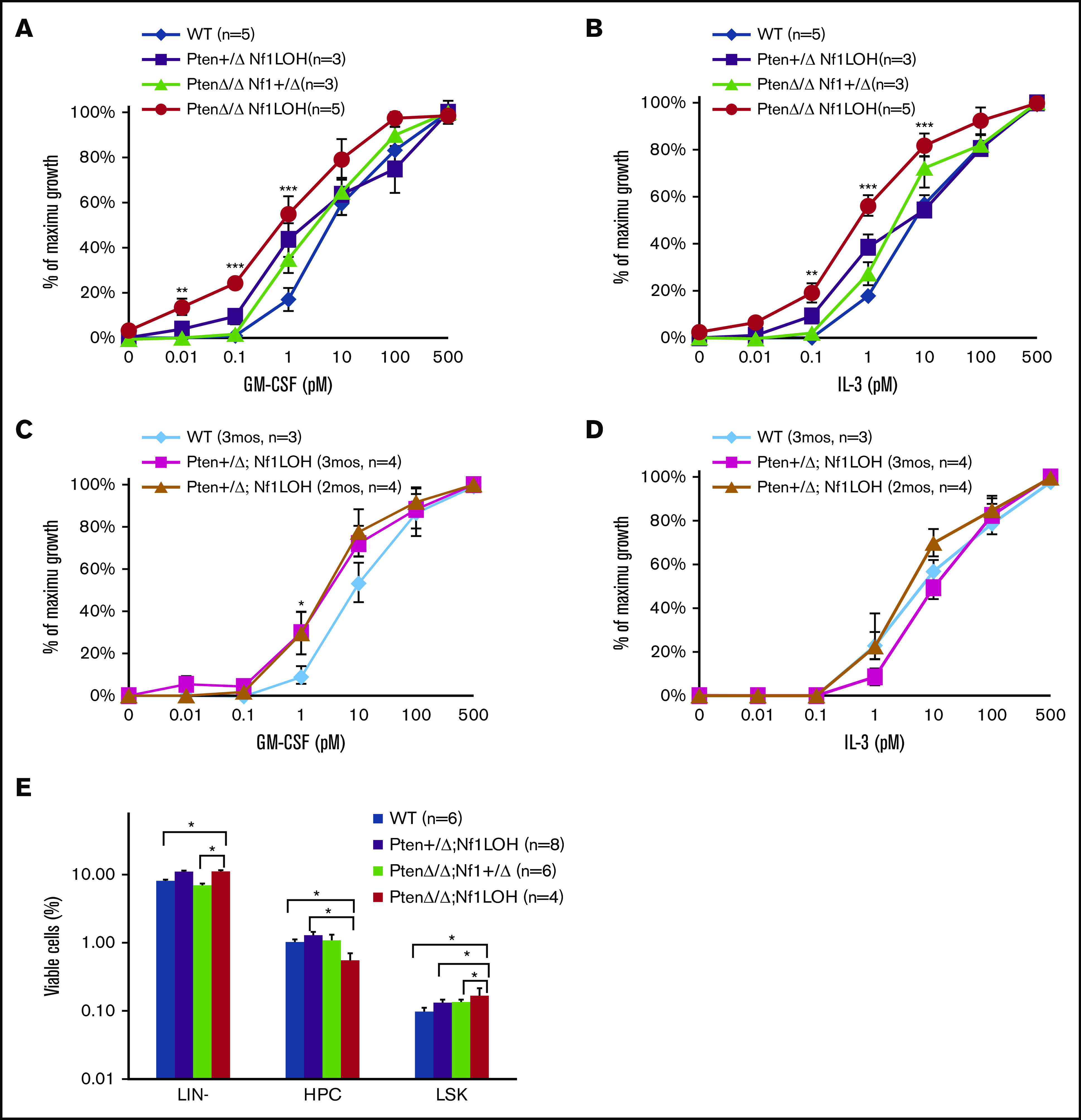
Analysis of BM progenitors in diseased mice and littermates. (A-D) Colony formation assay data. Unfractionated BM nucleated cells (WBM) were collected from PtenΔ/ΔNf1LOH mice with JMML and littermates at PND17-19 or from mice with CMML at age 2 to 3 months when they were moribund. Data from CFU-GM showed that GM-CSF and IL-3 sensitivities were significantly increased in BM cells from PtenΔ/Δ Nf1LOH mice with JMML compared with juvenile littermates, whereas Pten+/ΔNf1LOH mice showed GM-CSF hypersensitivity only as adults. (E) Flow cytometry analysis of hematopoietic progenitors in BM from juvenile mice. WBMs from PtenΔ/ΔNf1LOH mice with JMML and littermates at PND17-19 were analyzed by flow cytometry; data demonstrated that lineage (LIN)-negative cells (negative for Gr1, CD3, B220, and Ter119) and LIN−Sca1+cKit+ cells (LSK) were significantly increased in PtenΔ/ΔNf1LOH mice with JMML, whereas hematopoietic progenitor cells (HPCs; LIN−Sca1−cKit+) were decreased. Data are mean ± standard error. *P < .05, **P < .01, ***P < .001.
Loss of Pten protein disrupts the fetal-to-adult hematopoiesis switch, resulting in juvenile demise
Our data indicated that juvenile death was associated with Pten deletion, instead of monocytosis or lymphocytopenia; juvenile Pten+/ΔNf1LOH mice had worse myeloid-predominant blood counts than did PtenΔ/ΔNf1+/Δ littermates, but they did not die until developing severe CMML in adulthood (Figure 1A,J-L; supplemental Figures 5-7). We also found that BM cells from juvenile mice with the juvenile-lethal genotype (PtenΔ/Δ) reconstituted significantly more myeloid progenies and fewer B cells than did those from Pten+/ΔNf1LOH or WT littermates (Figure 2B-C).
Human newborns have myeloid-predominant blood with high monocytes during the first 48 hours after birth.23,24 The developmental switch from fetal to adult hematopoiesis occurs during the first year in humans and at 1 to 2 weeks after birth in mice.16,17 Patients with NF1 mutation have the highest fetal hemoglobin levels among JMML patients.14 Recent data suggested that a “fetal-like subgroup” of JMML patients was associated with poor prognosis.25 Because JMML targets an age-restricted group (birth to 6 years), and maturational arrest is absent in blood cells from patients and mice, we questioned whether fetal hematopoiesis was sustained in mice with JMML. We first compared the blood profiles of WT mice between ages PND8 and 6 weeks. Flow cytometry analyses showed that PB in WT mice at PND8 was substantially myeloid predominant compared with those aged 6 weeks (Figure 4A). Interestingly, juvenile (PND17) PtenΔ/Δ mice also had myeloid-predominant blood, as observed in WT mice at age PND8, regardless of Nf1 status, whereas juvenile WT mice had a blood profile similar to 6-week-old adult mice (P > .05; Figure 4B). We questioned whether PtenΔ/Δ was associated with fetal-like hematopoiesis in juvenile mice at the age when WT mice normally establish adult hematopoiesis.
Figure 4.
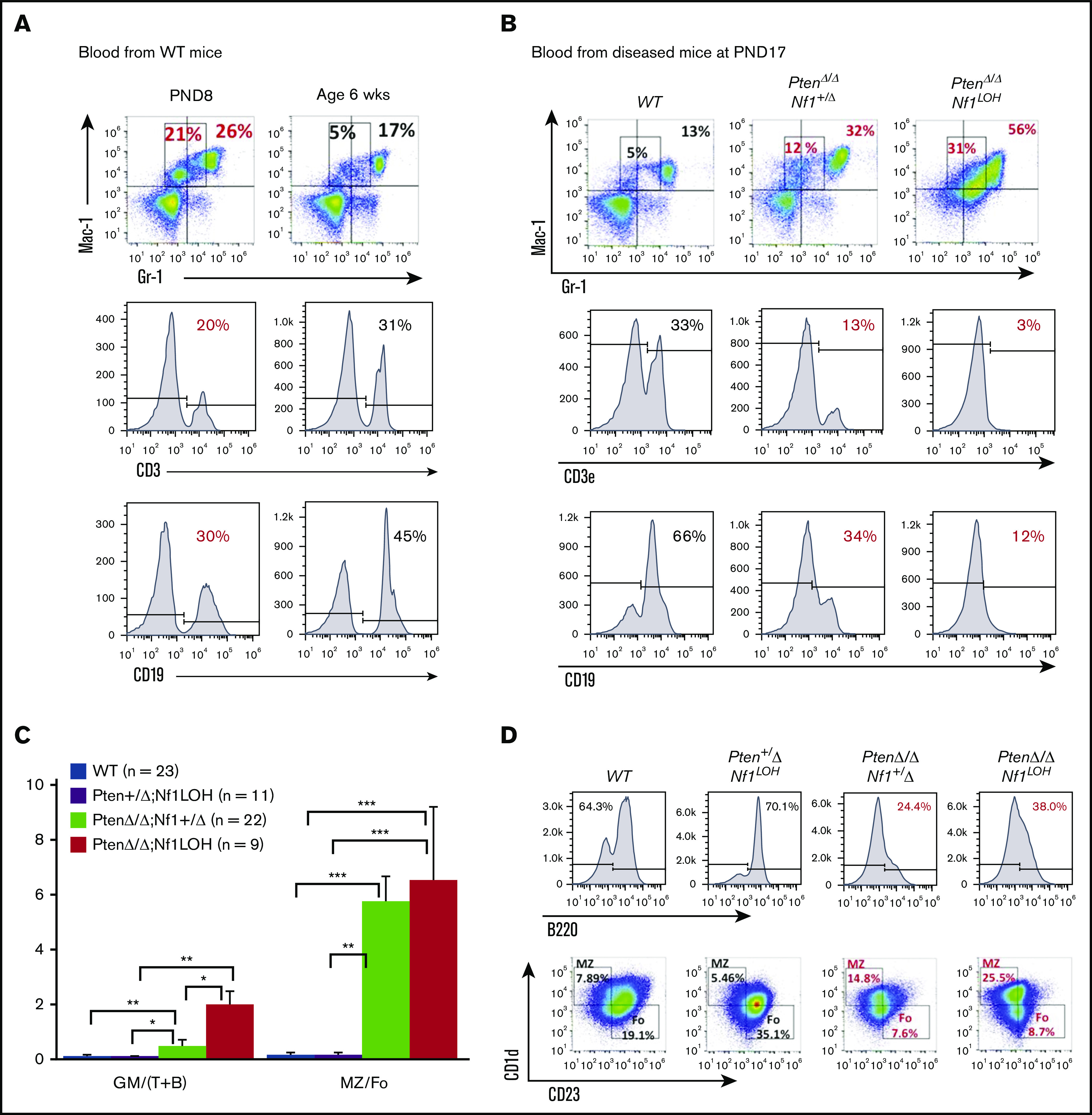
Significant fetal hematopoiesis in juvenile mice with Pten deletion and leukemia. (A) Representative flow cytometry data from WT mouse blood at PND8 and 6 weeks (n = 4 each group). (B) Representative flow cytometry data from blood of WT mice and PtenΔ/Δ mice with leukemia at age 3 weeks (≥10 mice in each group). (C) Relationship between donor-derived (CD45.2+) progenies in PB and MZ/Fo B-cell ratios in spleens of recipients. (D) Representative flow cytometry data from blood and spleens of recipient mice transplanted from donor mice with various copy numbers of Pten and Nf1. GM [blood myeloid; CD45.2+CD11b+], (T+B) [blood lymphoid lineage; sum of CD45.2+B220+ + CD45.2+CD3e+], and blood myeloid/lymphoid ratios (GM/[T+B]) were calculated as ratios of myeloid cells/lymphocytes in total WBCs with CD45.2+ in recipient mice (CD45.1+). Fo, donor-derived follicular B cells are the predominant B cells in adults; MZ, donor-derived splenic marginal zone B cells representing fetal origin HSCs. The ratios of splenic MZ (fetal origin)/Fo (adult origin) B cells represented the fetal properties of donor HSCs. Data are median ± standard error. *P < .05, **P < .01, ***P < .001. See additional supportive data in supplemental Figures 8 and 10.
Fetal hemoglobin cannot be used as a marker for tracking fetal hematopoiesis in mice as it can be in humans. Mouse definitive erythrocytes arise when erythropoiesis shifts to the fetal liver and synthesizes adult hemoglobin, which continues after birth when definitive erythropoiesis shifts to the BM. In tracking mouse fetal hematopoiesis, the difference in the ability of neonatal BM vs adult BM to reconstitute B cells in recipients is considered a distinctive property of mouse fetal and adult HSCs.26-28 Donor-derived splenic marginal zone (MZ) B cells (B220+CD1d+CD23−) are of uniquely fetal origin, and donor-derived follicular (Fo) B cells are the predominant B cells in adults.26,27 To investigate whether PtenΔ/Δ HSCs have significant fetal properties, we evaluated blood profiles and splenic MZ B cells in recipients transplanted from juvenile donors with various copy numbers of Pten and Nf1. Our data show that PtenΔ/Δ BM cells reconstituted a significantly myeloid-predominant blood, but juvenile donor cells from Pten+/ΔNf1LOH mice reconstituted blood similarly to WT cells (Figure 4C). Furthermore, significantly elevated splenic MZ B cells were reconstituted only in recipients transplanted from juvenile PtenΔ/Δ mice. Nf1LOH significantly enhanced myeloid-predominant hematopoiesis with elevated MZ B cells when Pten was absent (Figure 4C-D; supplemental Figure 10). However, Nf1LOH did not have any impact on the reconstitution of splenic MZ B cells in recipients transplanted from Pten+/ΔNf1LOH donors (Figure 4C-D). Our data suggest that, in reconstitution, juvenile donor PtenΔ/Δ HSCs had significantly more fetal properties than did Pten+/ΔNf1LOH or WT HSCs. These data suggest that PTEN loss in early infancy contributes to the sustained fetal hematopoiesis in juveniles, resulting in pediatric death.
Myeloid bias of PtenΔ/Δ HSCs is associated with fetal properties of HSCs
Initially, most recipients transplanted from PtenΔ/ΔNf1LOH donors with JMML died before 16 weeks posttransplant (Figure 2A), which is the time point to properly evaluate the properties of LT-HSCs. Single-cell HSC transplant ensures that all progeny cells detected in recipients arise from the same original cell, and the recipient mice survive longer than 16 weeks posttransplant. To further confirm the fetal properties of leukemic-initiating cells in JMML mice, we performed single-cell transplantations with LT-HSCs from juvenile WT mice and PtenΔ/ΔNf1LOH mice with JMML (supplemental Figure 11). Our data show that HSCs from JMML donors produced various ratios of myeloid to lymphoid blood cells (GM/[T+B]) in recipients, but they constantly escalated after 10 weeks posttransplant, worsening in 2° transplants, whereas WT HSCs derived balanced myeloid/lymphoid progenies (GM/[T+B] ∼ 1) (Figure 5A,C,E). Those progeny ratios for GM/[T+B] represented the properties of each transplanted HSC subtype (clone) in the HSC pool from donors. Interestingly, we found that the elevated ratios of donor-derived GM/[T+B] were consistent with the elevated MZ/Fo B-cell ratios as the result of a significant reduction in adult splenic Fo B cells in recipients (supplemental Figure 12). The same trend continued in the sequential 2° and 3° single LT-HSC transplants (Figure 5). Specifically, we found that the single HSC with myeloid bias from recipient 1°-B (Figure 5C-D) derived a clone in a recipient of a 2° transplant with worse myeloid-biased progenies and a higher MZ/Fo B-cell ratio (representing the property of fetal HSCs) in a recipient of a 2° transplant (recipient 2°-BA; Figure 5E-F), whereas another clone from the same donor (1°-B) derived progenies with less myeloid bias and lower MZ/Fo B-cell ratios (recipient 2°-BC; Figure 5E-F). This trend was replicated in the 3° transplants (recipient 3°-BAK vs 3°-BAB and 3°-BAC; Figure 5G-H). Our data demonstrate that a single LT-HSC from donors with JMML resulted in progenies with diverse myeloid or lymphoid lineages, and myeloid-biased potential was associated with the properties of fetal HSCs, whereas a single HSC from WT donors at the same age reconstituted only balanced myeloid/lymphoid progenies, without the potential for fetal-like HSCs (Figure 5A-B). Our data suggest that the myeloid-biased leukemic-initiating cells from donors with JMML have the potential to reconstitute progenies with more or less myeloid bias in WT recipients, which is related to the properties of fetal HSCs. These data also indicate that the lineage-biased HSCs from mice with JMML have the potential to derive progenies with less myeloid bias, despite the persistent absence of Pten and Nf1. The finding that the engraftment of lineage-balanced HSCs from donors with JMML can be enhanced in WT recipients may have significant clinical applications in autologous HSC transplantation for JMML or other pediatric leukemias.
Figure 5.
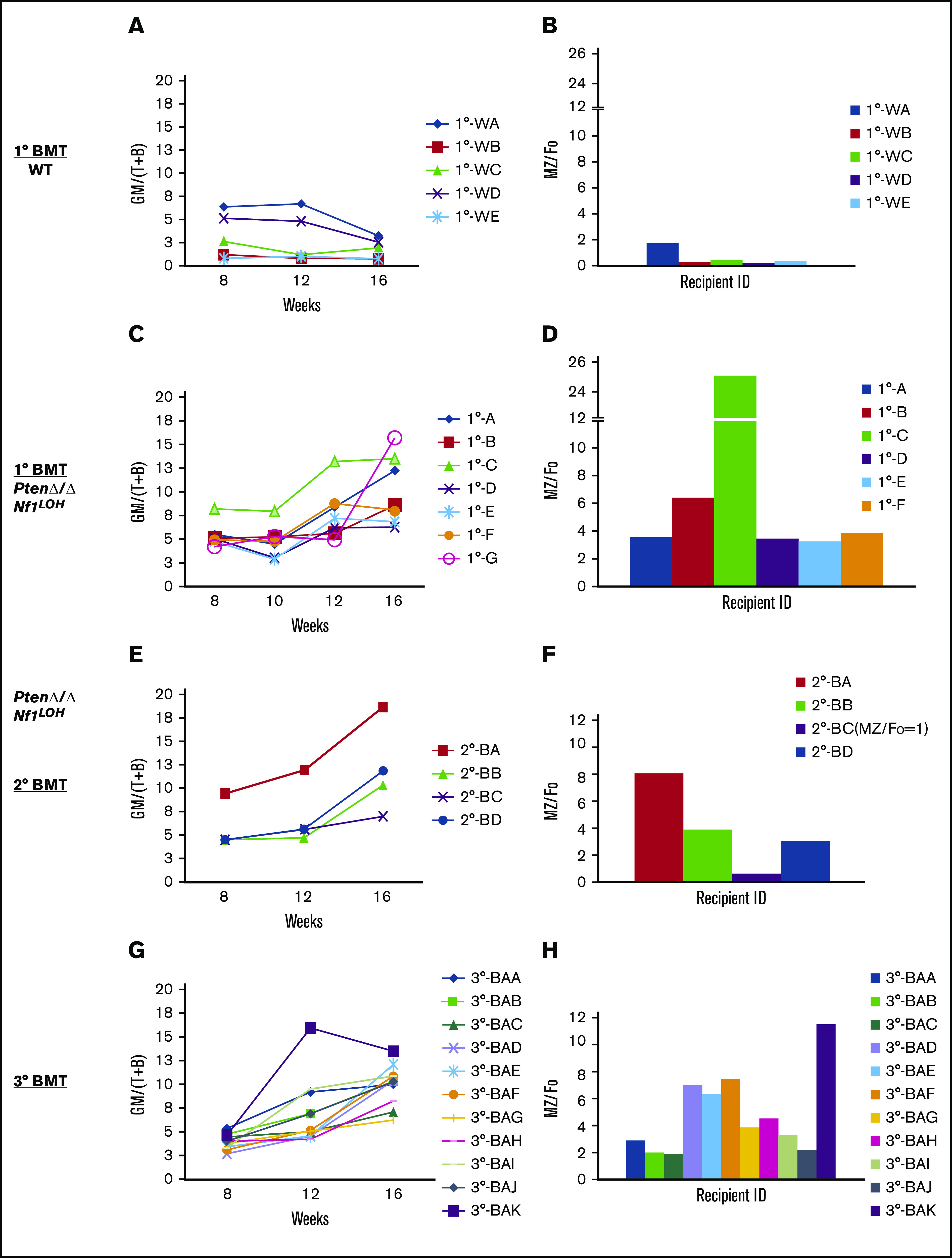
Serial single-HSC transplantation. (A-D) 1° Transplant: single LT-HSC from juvenile WT mice and PtenΔ/ΔNf1LOH mice with JMML was transplanted with 2 × 105 rescue cells into WT recipient mice with CD45.1+, respectively. Sixteen weeks posttransplant, WT donor HSCs reconstituted balanced progenies, whereas HSCs with JMML (PtenΔ/ΔNf1LOH) reconstituted various degrees of myeloid-biased progenies with elevated ratios of MZ/Fo B cells. Recipient 1°-G, which had the worst myeloid-biased blood, died before spleen tissue collection for MZ cell analysis. (E-F) 2° Transplant: single donor-derived HSC (CD45.2+) from primary recipient 1°-B was retransplanted into 2° BMT recipients. 2° Recipient mice continuously reconstituted various degrees of myeloid-biased progenies with elevated MZ/Fo B-cell ratios. Recipient 2°-BC reconstituted balanced progenies with the least splenic MZ cells. (G-H) 3° Transplant: single donor-derived HSC (CD45.2+) from recipient 2°-BA was retransplanted to 3°-BMT recipients. Recipient 3°-BAK had the worst myeloid-biased engraftment with the highest MZ/Fo B-cell ratio. Data represent ≥2 sets of paired experiments. See additional supportive data in supplemental Figures 11 and 12.
Single-allelic Pten deletion caused constitutive activation of MAPK in mice with Nf1 LOH before age 3 weeks
Reportedly, Pten did not have any impact on the Akt pathway in mouse HSCs or multipotential progenitors until 3 to 4 weeks of age, and the MAPK pathway was not altered in neonatal or adult mice with an otherwise WT genome when Pten was deleted on PND2 (equivalent to late fetal age in the third trimester of human pregnancy).29,30 We previously reported that Akt, but not MAPK, was constitutively active in BM cells of juvenile mice with an otherwise WT genome when Pten was deleted on PND8.10 We also reported that Pten deletion in mice with Nf1 haploinsufficiency (PtenΔ/ΔNf1+/−) on PND8 caused preferential MAPK hyperactivity to GM-CSF stimulation over IL-3 in BM cells of mice at age 3 weeks.10 This suggests that the timing of Pten deletion and the hosts’ genetic background have a significant impact on the Akt and MAPK pathways. To understand how Nf1 deficiency affects the role of Pten in regulating the Akt and MAPK pathways, we investigated the pathways’ activities in BM cells from juvenile littermates with various copy numbers of Pten and Nf1 on PND17-19. Consistent with our previous findings,10 Akt was constitutively activated in PtenΔ/Δ mice, regardless of Nf1 status, but it was not activated when 1 copy of Pten was retained in mice with Nf1 LOH (Figure 6A). This suggests that 1 copy of Pten is sufficient to restrain the activation of the Akt pathway, despite the absence of Nf1 function, and an activated Akt pathway is related to sustained fetal hematopoiesis in juvenile PtenΔ/Δ mice. We also found that deletion of Pten in mice with a single-copy somatic deletion of Nf1 (PtenΔ/ΔNf1+/Δ) significantly increased cellular MAPK activity in response to GM-CSF stimulation (Figure 6A), which is consistent with our previous findings in juvenile PtenΔ/Δ mice with germline mutant Nf1 (PtenΔ/ΔNf1+/−).10 Interestingly, single-allelic Pten deletion in juvenile mice with Nf1 LOH (Pten+ΔNf1LOH) caused constitutive activation of the MAPK pathway before age 3 weeks (Figure 6A), which did not occur in mice with Nf1Δ/Δ alone or when Pten deletion was induced on PND2 in mice with an otherwise WT genome.21,29 Our data suggest that 2 copies of Pten are required to maintain the integrity of the MAPK pathway in juvenile mice with Nf1 LOH and confirmed that Nf1 haploinsufficiency impairs the MAPK pathway in juvenile PtenΔ/Δ mice.10 Our data also suggest that what we learned about Pten function in mice with an otherwise WT genome29,30 may not necessarily reflect the impact of Pten loss in mice with Nf1 mutation or other genetic defects. We also observed a significant increase in phosphorylated S6 ribosomal protein (Ser240/244) in juvenile Pten+/ΔNf1LOH mice, which was not associated with hyperactivation of the Akt pathway (Figure 6A), suggesting that an alternative mechanism other than Akt/mTOR regulates the mTOR pathway in Pten+/ΔNf1LOH cells.29
Figure 6.
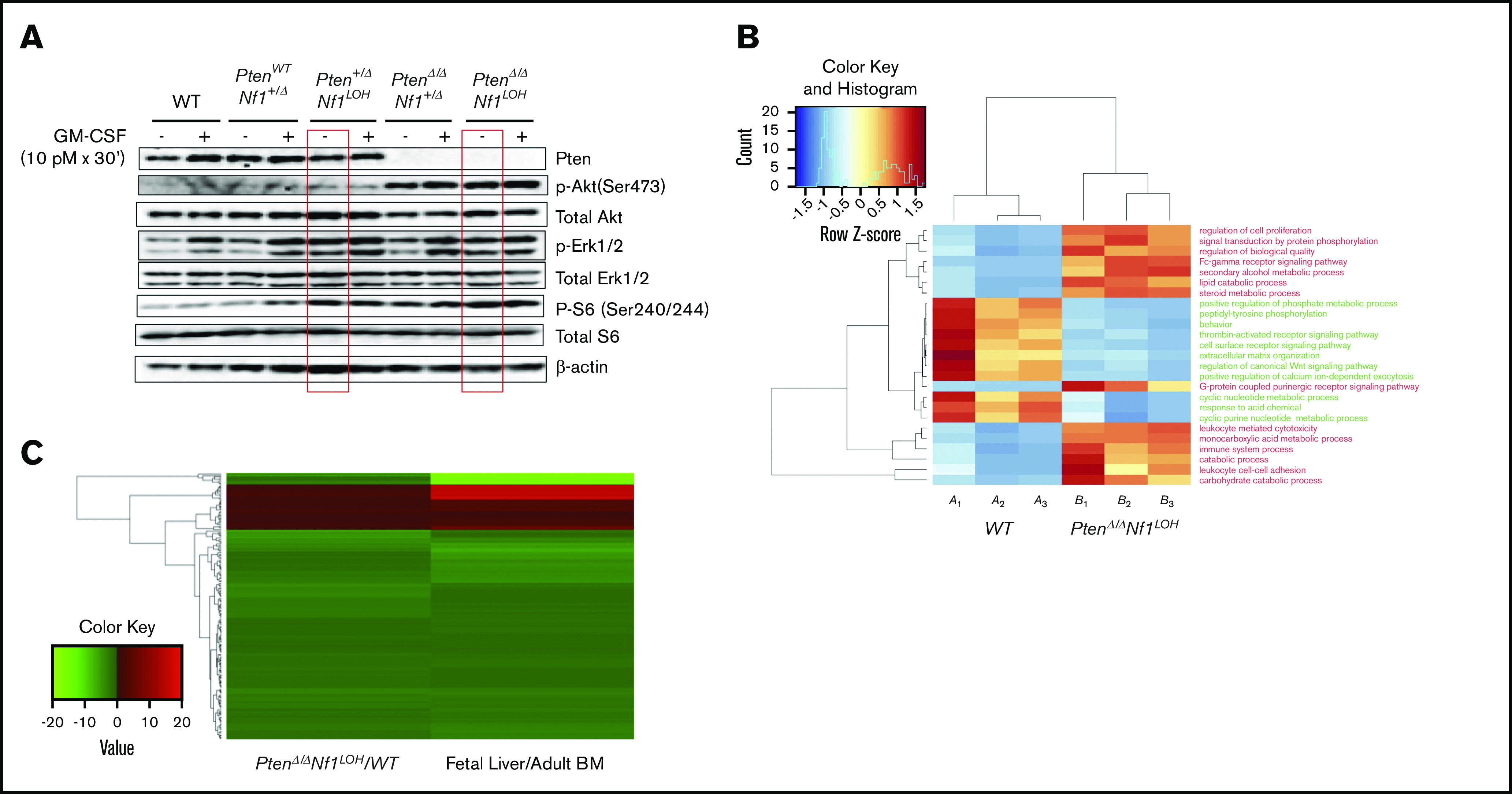
Dysregulated signal transduction pathways and molecular defects in mice with Pten deletion, Nf1 deficiency, and JMML. (A) Representative data from western blot analyses of the elements in GM-CSF signal transduction pathways in unfractionated BM nucleated cells from juvenile mice with various copy numbers of Pten and Nf1. Representative data were from ≥3 experiments from littermate mice at PND17-19. (B) Gene ontology (GO) analysis. Genes that were significantly over- or underexpressed in LIN− BM cells from mice with JMML and WT littermates at age PND17 were used in GO analysis, together with HCA. (C) Heatmap and HCA of 182 genes that were differentially expressed in this study (PtenΔ/Δ Nf1LOH/WT) vs the data (Fetal Liver/Adult BM) reported by Manesia et al.34 We found a log2-fold change in either being positive or negative consistently in both studies (182 genes). See additional supportive data in supplemental Figures 13 and 14 and supplemental Tables 4 and 5 for “GO HeatMap Terms & GeneIDs” and “Combined Analysis DEG Results.”
Multiple dysregulated cellular processes are involved in fetal-like hematopoiesis in mice with JMML
HSC fate decisions in leukemia transformation require a combination of molecules that control HSC survival, proliferation, and self-renewal.31,32 To dissect the molecular mechanism in JMML, we performed RNA sequencing (RNA-seq) on lineage-depleted BM cells from 3 PtenΔ/ΔNf1LOH mice with JMML and 3 WT littermates at age PND17 (supplemental Materials and methods). In addition to the initial data analysis during pipeline processing, RNA-seq exploratory data analysis was performed to summarize the main characteristics of the data from the 3 paired biological replicates using visual methods.33 Overall, exploratory data analysis composite revealed a good separation via heatmap with hierarchical clustering analysis (HCA) and confirmed no systematic bias, which provide evidence of sample separation based on WT vs JMML and allowed for further hypothesis generation (supplemental Figure 13).
DESeq2 standard analysis identified all differentially expressed gene targets meeting the standard statistical criteria (supplemental Table 3). The identified gene targets were further analyzed by gene ontology–based HCA and again showed separation of WT mice vs PtenΔ/ΔNf1LOHmice with JMML, suggesting that the upregulated terms enriched in mice with JMML were related to enhancing cell proliferation, adhesion, and signal transduction, whereas downregulated terms were surprisingly enriched for cell surface receptor and canonical Wnt signaling pathways, positive regulation of phosphate metabolic process, and extracellular matrix organization (Figure 6B; supplemental Table 4). To determine whether mice with JMML have a more fetal-like transcriptional signature than the juvenile WT littermates, we compared our data with the WT mouse fetal liver data published by Manesia et al.34 Heatmap with HCA involving 182 genes shows concordance between the ratio of JMML mice /WT littermates and the ratio of WT mouse fetal liver/adult BM, which was reported by Manesia et al34 (Figure 6C; supplemental Table 5).
Ingenuity pathway analysis utilizing cancer canonical signaling pathways for PI3K/AKT, ERK/MAPK, mTOR, and GM-CSF confirmed that the dysregulated molecules are involved in multiple pathways in regulation of cellular functions, and overexpressed elements in the MAPK pathway were consistent in all 4 analyzed pathways (supplemental Figure 14), suggesting that the MAPK pathway is significantly upregulated in hematopoietic progenitor cells from mice with JMML. Those RNA-seq data provide more leads for further exploring the mechanism underlying the deadly sustained fetal hematopoiesis in JMML.
Discussion
Dramatic changes in HSC functions of newborns occur within a few days after birth.35,36 Many of the changes are coordinated with an abrupt switch in HSC behavior during development.35-37 Adult HSCs make a major contribution to hematopoiesis in the steady-state after fetal-to-adult hematopoiesis switch.38 Beaudin et al reported that developmentally restricted HSCs were incapable of persisting into adulthood in situ.39 Recently, Peng et al reported that luteinizing hormone signaling restricts HSC expansion during puberty.40 It has never been reported that a disassociated age-specific hematopoiesis contributes to leukemogenesis. Here, we demonstrate that the sustained fetal properties of HSCs are associated with myeloid-biased hematopoiesis in juvenile PtenΔ/Δ mice, leading to a lethal juvenile leukemia. Our data suggest that PtenΔ/Δ HSCs have an intrinsic defect to migrate from developmentally restricted HSCs to adult HSCs, resulting in defective adult hematopoiesis in juvenile mice at an age when adult hematopoiesis is normally established in WT mice. This is the first evidence suggesting that Pten loss disrupts the fetal-to-adult hematopoiesis switch and causes aberrant distribution of normal blood cells, without exhausting HSCs. In other words, some leukemias may be a consequence of an unbalanced distribution of lineage-specific HSCs that is caused by disassociated age-specific hematopoiesis. This is particularly critical for pediatric patients with leukemias or MDS/MPNs, because it is time-sensitive and regulated by epigenetics. Our data uncovered a novel mechanism underlying pediatric leukemia. Further studies on the role of age-related hematopoiesis in leukemia development may help to develop novel strategies for treating leukemias and MDS/MPNs.
Recently, 7 key genes were identified that selectively regulate fetal (Sox17, Lin28b, Hmga2, and Ezh2) or adult (Bmi1, Pten, and Cebpa) HSCs in mice.36,41 Pten was reported to negatively regulate signaling pathways exclusively in adult HSCs.21 Pten protein inactivation reduced quiescent (G0) HSCs and compromised the maintenance of LT-HSCs in adult mice.42 The MAPK and phosphatidylinositol 3-kinase (PI3K) pathways are coordinately activated in HSCs to maintain the negative-feedback loop that limits the strength and duration of PI3K/Akt/mTORC1 signaling to allow activated HSCs to return to quiescence.43 When Pten is lost, the MAPK pathway loses the counterpart from PI3K/Akt, resulting in HSC exhaustion.43 Our data suggest that 1 copy of Pten is sufficient for maintaining the integrity of negative feedback in the Akt signaling loop and the HSC function in reconstitution, despite MAPK being constitutively active in Pten+/ΔNf1LOH mice (Figures 2B-C, 3E, and 6A). Our data also suggest that the impact of Pten on the MAPK pathway in early development is Nf1 dependent, and Pten had a different impact on neonatal HSCs in hosts with Nf1 haploinsufficiency compared with those with a WT genome (Figures 3E and 6A).10,29,43
Emerging data demonstrate that upregulated fetal-selective molecules (LIN28B and HMGA2)44 and deficient adult hematopoiesis–required molecules (PTEN) are related to developmental hematopoiesis in JMML patients.12,25 With this knowledge, we propose a leukemogenesis model of JMML, reflecting an aberrant fetal-to-adult hematopoiesis switch that results in the sustained fetal hematopoiesis at an age when healthy juveniles normally establish adult hematopoiesis, leading to pediatric death (Figure 7). Although our data suggest that the MAPK pathway is significantly upregulated in mice with JMML, treating JMML patients with MAPK inhibitors should be performed with caution before confirming whether PTEN is absent, because PTEN is indispensable for cells to respond to MAPK inhibitors in myeloid leukemia.20
Figure 7.

Proposed leukemogenesis model of the aberrant fetal-to-adult hematopoiesis switch in JMML. Fetal and adult hematopoiesis are governed by a developmental/age-specific molecular driving force and executed by fetal-selective genes and adult-necessary genes in hematopoiesis (eg, SOX17, LIN28B, HMGA2, and EZH2 for fetal HSCs, and BMI1, PTEN, and CEBPA for adult HSCs). (A) During the neonatal period when the molecular driving force is balanced in fetal to adult hematopoiesis, healthy newborns can complete the transition from fetal to adult hematopoiesis to meet the needs for growth in normoxia. (B) When the molecular driving force is disrupted in newborns during the fetal-to-adult hematopoiesis switch by mutations or dysregulated epigenetics, fetal hematopoiesis is sustained and causes myeloid/lymphoid distribution errors and insufficient adult hematopoiesis in juveniles, resulting in pediatric demise. This may be the case in patients with JMML who are born with an NF1 mutation and hold an instable molecular driving force from fetal to adult hematopoiesis switch, such as elevated fetal-specific LIN28B and HMGA2 along with PTEN and BMI1 deficiencies. Because JMML patients cannot develop adult hematopoiesis at an age when WT juveniles normally complete their fetal-to-adult hematopoiesis switch, they die of sustained fetal hematopoiesis as juveniles.
In summary, we demonstrate that Pten loss during the fetal-to-adult hematopoiesis switch causes a sustained fetal hematopoiesis in juveniles, resulting in juvenile leukemia and death. A constitutively active MAPK pathway is associated with GM-CSF hypersensitivity and monocytosis but is not responsible for juvenile death. mTOR inhibitors may have the potential for treating patients with JMML or CMML.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Owen W. Stephens for performing A-seq experiments and Brontë Pearson for editing the manuscript.
This work was supported by an Envoys-Seeds of Science Award/Winthrop P. Rockefeller Cancer Institute at University of Arkansas for Medical Sciences (Y.L.L.), Dr. F. E. Joyce Molecular Pathology Laboratory Endowment (P.D.E.), National Institutes of Health, National Cancer Institute (R01 CA122023) (D.Z.), National Institutes of Health, National Institute of General Medical Sciences (P20 GM109005) (D.Z.), University of Arkansas for Medical Sciences Winthrop P. Rockefeller Endowment for Leukemia Research (D.Z.), the Maternal & Child Health Research Institute at Stanford School of Medicine (A.B.), and the Children’s Leukemia Research Association (137046) (A.B.)
Footnotes
Raw data from RNA-seq have been uploaded to the Gene Expression Omnibus (accession number GSE137152) and are available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE137152.
Data sharing requests should be sent to Y. Lucy Liu (ylliu19@stanford.edu) or Peter D. Emanuel (pdemanuel@stvincenthealth.com).
Authorship
Contribution: N.V. designed and performed experiments, analyzed data, and edited the manuscript; Y.L. designed experiments, analyzed data, prepared figures and tables, and wrote the manuscript; Y.Y. designed and performed experiments and prepared figures; S.Y.L. designed experiments, performed statistical analyses, prepared figures, and edited the manuscript; N.C. performed blood profiling experiments and analyzed data; D.R. and J.Z. performed experiments; X.Z. assisted with flow cytometry sorting in single-HSC BMT experiments; E.A.P. performed bioinformatics analysis for RNA-seq experiments, prepared figures and tables, and helped to submit raw data to Gene Expression Omnibus; N.J.B. assisted with blood profiling experiments and edited the manuscript; D.Z. designed and supervised experiments and edited manuscript; A.B. supervised experiments and edited the manuscript; D.J.J. designed and analyzed data for experiments of RNA-seq, prepared figures and tables, and wrote manuscript; P.D.E helped to conceive the project, designed and supervised experiments, analyzed data, and wrote the manuscript; Y.L.L conceived the project, designed/performed/supervised experiments, analyzed data, prepared figures and tables, and wrote the manuscript; and all authors reviewed the final version of the manuscript.
Conflict-of-interest disclosures: The authors declare no competing financial interests.
Correspondence: Y. Lucy Liu, Division of Stem Cell Transplantation and Regenerative Medicine, Department of Pediatrics, Stanford School of Medicine, 240 Pasteur Dr, BMI, Suite 2550, Stanford, CA 94305-5139; e-mail: ylliu19@stanford.edu; and Peter D. Emanuel, Catholic Health Initiatives–St. Vincent, 1 St. Vincent Cir, Suite 450, Little Rock, AR 72205; e-mail: pdemanuel@stvincenthealth.com.
References
- 1.Niemeyer CM, Arico M, Basso G, et al. ; European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS) . Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. Blood. 1997;89(10):3534-3543. [PubMed] [Google Scholar]
- 2.Alfonso A, Montalban-Bravo G, Takahashi K, et al. . Natural history of chronic myelomonocytic leukemia treated with hypomethylating agents. Am J Hematol. 2017;92(7):599-606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geissler K, Jäger E, Barna A, et al. . Chronic myelomonocytic leukemia patients with RAS pathway mutations show high in vitro myeloid colony formation in the absence of exogenous growth factors. Leukemia. 2016;30(11):2280-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sakaguchi H, Okuno Y, Muramatsu H, et al. . Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat Genet. 2013;45(8):937-941. [DOI] [PubMed] [Google Scholar]
- 5.Hofmans M, Lammens T, Helsmoortel HH, et al. . The long non-coding RNA landscape in juvenile myelomonocytic leukemia. Haematologica. 2018;103(11):e501-e504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stieglitz E, Taylor-Weiner AN, Chang TY, et al. . The genomic landscape of juvenile myelomonocytic leukemia [published correction appears in Nat Genet. 2016;48(1):101]. Nat Genet. 2015;47(11):1326-1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lipka DB, Witte T, Toth R, et al. . RAS-pathway mutation patterns define epigenetic subclasses in juvenile myelomonocytic leukemia. Nat Commun. 2017;8(1):2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leoncini PP, Vitullo P, Di Florio F, et al. . Whole genome MBD-seq reveals different CpG methylation patterns in azacytidine-treated juvenile myelomonocytic leukaemia (JMML) patients. Br J Haematol. 2018;182(6):909-912. [DOI] [PubMed] [Google Scholar]
- 9.Leoncini PP, Bertaina A, Papaioannou D, et al. . MicroRNA fingerprints in juvenile myelomonocytic leukemia (JMML) identified miR-150-5p as a tumor suppressor and potential target for treatment. Oncotarget. 2016;7(34):55395-55408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu YL, Yan Y, Webster C, et al. . Timing of the loss of Pten protein determines disease severity in a mouse model of myeloid malignancy. Blood. 2016;127(15):1912-1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang J, Wang Y, Shao L, et al. . Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016;22(1):78-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu YL, Castleberry RP, Emanuel PD. PTEN deficiency is a common defect in juvenile myelomonocytic leukemia. Leuk Res. 2009;33(5):671-677. [DOI] [PubMed] [Google Scholar]
- 13.Steinemann D, Arning L, Praulich I, et al. . Mitotic recombination and compound-heterozygous mutations are predominant NF1-inactivating mechanisms in children with juvenile myelomonocytic leukemia and neurofibromatosis type 1. Haematologica. 2010;95(2):320-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Niemeyer CM. JMML genomics and decisions. Hematology (Am Soc Hematol Educ Program). 2018;2018:307-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bollag G, Clapp DW, Shih S, et al. . Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells [published correction appears in Nat Genet. 1996;12:458]. Nat Genet. 1996;12(2):144-148. [DOI] [PubMed] [Google Scholar]
- 16.Kikuchi K, Kondo M. Developmental switch of mouse hematopoietic stem cells from fetal to adult type occurs in bone marrow after birth. Proc Natl Acad Sci USA. 2006;103(47):17852-17857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Copley MR, Eaves CJ. Developmental changes in hematopoietic stem cell properties. Exp Mol Med. 2013;45(11):e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Itzykson R, Solary E. An evolutionary perspective on chronic myelomonocytic leukemia. Leukemia. 2013;27(7):1441-1450. [DOI] [PubMed] [Google Scholar]
- 19.Tyner JW, Erickson H, Deininger MW, et al. . High-throughput sequencing screen reveals novel, transforming RAS mutations in myeloid leukemia patients. Blood. 2009;113(8):1749-1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang J, Xiang Z, Malaviarachchi PA, et al. . PTEN is indispensable for cells to respond to MAPK inhibitors in myeloid leukemia. Cell Signal. 2018;50:72-79. [DOI] [PubMed] [Google Scholar]
- 21.Le DT, Kong N, Zhu Y, et al. . Somatic inactivation of Nf1 in hematopoietic cells results in a progressive myeloproliferative disorder. Blood. 2004;103(11):4243-4250. [DOI] [PubMed] [Google Scholar]
- 22.Chang T, Krisman K, Theobald EH, et al. . Sustained MEK inhibition abrogates myeloproliferative disease in Nf1 mutant mice. J Clin Invest. 2013;123(1):335-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Christensen RD, Henry E, Jopling J, Wiedmeier SE. The CBC: reference ranges for neonates. Semin Perinatol. 2009;33(1):3-11. [DOI] [PubMed] [Google Scholar]
- 24.Melioli G, Risso FM, Sannia A, et al. . Reference values of blood cell counts in the first days of life. Front Biosci (Elite Ed). 2011;3:871-878. [DOI] [PubMed] [Google Scholar]
- 25.Helsmoortel HH, Bresolin S, Lammens T, et al. . LIN28B overexpression defines a novel fetal-like subgroup of juvenile myelomonocytic leukemia. Blood. 2016;127(9):1163-1172. [DOI] [PubMed] [Google Scholar]
- 26.Carey JB, Moffatt-Blue CS, Watson LC, Gavin AL, Feeney AJ. Repertoire-based selection into the marginal zone compartment during B cell development. J Exp Med. 2008;205(9):2043-2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoshimoto M, Montecino-Rodriguez E, Ferkowicz MJ, et al. . Embryonic day 9 yolk sac and intra-embryonic hemogenic endothelium independently generate a B-1 and marginal zone progenitor lacking B-2 potential. Proc Natl Acad Sci USA. 2011;108(4):1468-1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghosn EE, Yamamoto R, Hamanaka S, et al. . Distinct B-cell lineage commitment distinguishes adult bone marrow hematopoietic stem cells. Proc Natl Acad Sci USA. 2012;109(14):5394-5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Magee JA, Ikenoue T, Nakada D, Lee JY, Guan KL, Morrison SJ. Temporal changes in PTEN and mTORC2 regulation of hematopoietic stem cell self-renewal and leukemia suppression. Cell Stem Cell. 2012;11(3):415-428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.West JR. Fetal alcohol-induced brain damage and the problem of determining temporal vulnerability: a review. Alcohol Drug Res. 1987;7(5-6):423-441. [PubMed] [Google Scholar]
- 31.Lai CK, Moon Y, Kuchenbauer F, et al. . Cell fate decisions in malignant hematopoiesis: leukemia phenotype is determined by distinct functional domains of the MN1 oncogene. PLoS One. 2014;9(11):e112671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wohrer S, Knapp DJ, Copley MR, et al. . Distinct stromal cell factor combinations can separately control hematopoietic stem cell survival, proliferation, and self-renewal. Cell Rep. 2014;7(6):1956-1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tukey JW. Exploratory Data Analysis. Reading, MA: Addison-Wesley; 1977. [Google Scholar]
- 34.Manesia JK, Franch M, Tabas-Madrid D, et al. . Distinct molecular signature of murine fetal liver and adult hematopoietic stem cells identify novel regulators of hematopoietic stem cell function. Stem Cells Dev. 2017;26(8):573-584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bowie MB, Kent DG, Dykstra B, et al. . Identification of a new intrinsically timed developmental checkpoint that reprograms key hematopoietic stem cell properties. Proc Natl Acad Sci USA. 2007;104(14):5878-5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Babovic S, Eaves CJ. Hierarchical organization of fetal and adult hematopoietic stem cells. Exp Cell Res. 2014;329(2):185-191. [DOI] [PubMed] [Google Scholar]
- 37.Dykstra B, Kent D, Bowie M, et al. . Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell Stem Cell. 2007;1(2):218-229. [DOI] [PubMed] [Google Scholar]
- 38.Sawai CM, Babovic S, Upadhaya S, et al. . Hematopoietic stem cells are the major source of multilineage hematopoiesis in adult animals. Immunity. 2016;45(3):597-609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beaudin AE, Boyer SW, Perez-Cunningham J, et al. . A transient developmental hematopoietic stem cell gives rise to innate-like B and T cells. Cell Stem Cell. 2016;19(6):768-783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peng YJ, Yu H, Hao X, et al. . Luteinizing hormone signaling restricts hematopoietic stem cell expansion during puberty. EMBO J. 2018;37(17):e98984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Copley MR, Babovic S, Benz C, et al. . The Lin28b-let-7-Hmga2 axis determines the higher self-renewal potential of fetal haematopoietic stem cells. Nat Cell Biol. 2013;15(8):916-925. [DOI] [PubMed] [Google Scholar]
- 42.Zhang J, Grindley JC, Yin T, et al. . PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441(7092):518-522. [DOI] [PubMed] [Google Scholar]
- 43.Baumgartner C, Toifl S, Farlik M, et al. . An ERK-dependent feedback mechanism prevents hematopoietic stem cell exhaustion. Cell Stem Cell. 2018;22(6):879-892.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Helsmoortel HH, Bresolin S, Lammens T, et al. . LIN28BM overexpression defines a novel fetal-like subgroup of juvenile myelomonocytic leukemia. Blood. 2016;127(9):1163-1172. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.