

Abstract
Diabetic neuropathy is one of the clinical syndromes characterized by pain and substantial morbidity primarily due to a lesion of the somatosensory nervous system. The burden of diabetic neuropathy is related not only to the complexity of diabetes but also to the poor outcomes and difficult treatment options. There is no specific treatment for diabetic neuropathy other than glycemic control and diligent foot care. Although various metabolic pathways are impaired in diabetic neuropathy, enhanced cellular oxidative stress is proposed as a common initiator. A mechanism-based treatment of diabetic neuropathy is challenging; a better understanding of the pathophysiology of diabetic neuropathy will help to develop strategies for the new and correct diagnostic procedures and personalized interventions. Thus, we review the current knowledge of the pathophysiology in diabetic neuropathy. We focus on discussing how the defects in metabolic and vascular pathways converge to enhance oxidative stress and how they produce the onset and progression of nerve injury present in diabetic neuropathy. We discuss if the mechanisms underlying neuropathy are similarly operated in type I and type II diabetes and the progression of antioxidants in treating diabetic neuropathy.
1. Introduction
Diabetic late complications are described as macrovascular complications comprising cardiovascular diseases and microvascular complications, including retinopathy, nephropathy, and neuropathy [1]. Diabetic neuropathy, one of the clinical syndromes, is characterized by pain and substantial morbidity primarily due to a lesion of the somatosensory nervous system [2, 3]. The most common clinical pattern is the neuropathy of the feet and the hands with a distal-to-proximal gradient of severity [1–3]. Currently, the only treatment for diabetic neuropathy is glucose control and foot care [2–5].
There are two major predictors of diabetic neuropathy: the duration of diabetes and the levels of haemoglobin A1c [6]. The latter is commonly associated with metabolic factors, genetic risk factors, environmental factors, common cardiovascular risk factors, and poor glycemic control [7–9]. The question, as to by what molecular mechanisms that diabetes mellitus could target sensory neurons, remains unclear [10]. However, a novel concept involving oxidative stress as a potent causative factor of diabetic neuropathy has been put forward [9, 10].
Oxidative stress is caused by an imbalance between production of reactive oxygen species (ROS) and antioxidant systems. It modulates functions of nerve cells through many molecular signalling pathways [10, 11]. Impaired glucose metabolism in diabetes is a critical mechanism to induce oxidative stress as a result of shunting excess glucose to other metabolic or nonmetabolic pathways [7, 8]. This causes accumulation of the toxic metabolites and overconsumption of nicotinic acid adenine dinucleotide phosphate (NADPH). These converge to increase intracellular redox stress and abnormal modifications on protein [12], lipid [13], and DNA [14], thereby adding mitochondrial injury and causing overproduction of ROS. This damages the peripheral nervous system, as indicated by loss of Schwann cells, myelinated axons, and sensory neurons located in the dorsal root ganglia [10, 15, 16]. Meanwhile, insufficient mitochondrial energy production loses the ability to traffic down axons, thereby further promoting axonal injury [17], causing many chronic degenerative diseases, including diabetic neuropathy [7, 8, 18–20].
In this review, we discuss the following points. Firstly, how impaired pathway(s) of glucose metabolism in diabetes lead to oxidative stress, thereby producing the onset and progression of nerve injury present in diabetic neuropathy. The altered pathways include polyol pathway, the hexosamine pathway, the advanced glycation end products (AGE), and protein kinase C (PKC) pathway. Secondly, are the mechanisms underlying neuropathy in type I and type II diabetes distinct? Thirdly, is any antioxidative drug specific and effective for relieving painful diabetic neuropathy? Finally, to highlight areas of research needed for improving the fate of patients with painful diabetic neuropathy.
2. General Concept of Oxidative Stress
The term of oxidative stress describes a condition (Figure 1(a)) when the balance between the generation of free radical and antioxidant system is unfavorable [21, 22]. A free radical can be defined as any molecular species capable of either donating or accepting an electron therefore behaving as oxidants [22], while antioxidants are the molecules stable enough to neutralize the free radicals, thereby maintaining a balance. Despite the chemical structural differences, free radicals share similar mechanisms for damage at the level of biomolecules [23].
Figure 1.
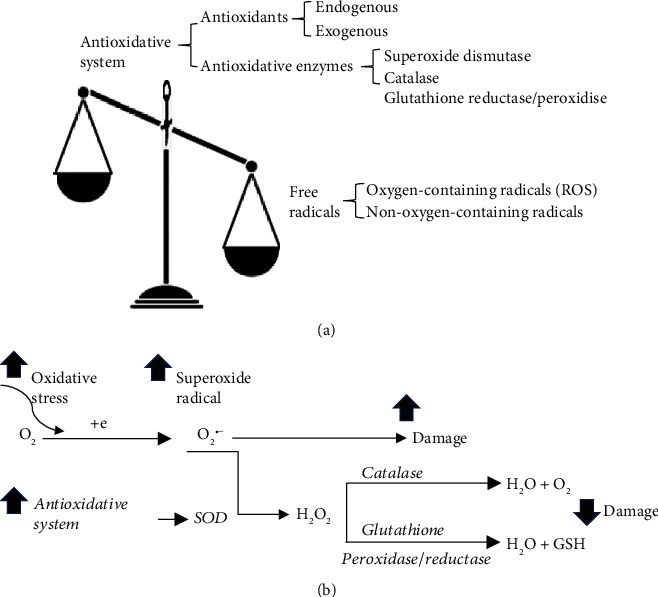
Components of oxidative stress and working mechanism of antioxidative system. (a) Oxidative stress consists of two components as the free radicals and the antioxidative system. The term of oxidative stress describes a condition when the generation of free radicals and the antioxidative system is imbalanced. Free radicals can be the radicals with or without reactivity of oxygen. Antioxidative system consist of antioxidants, that are derived either endogenous or exogenous, and antioxidative enzymes, such as superoxide dismutase, glutathione reductase/peroxidise, and catalase. (b) Antioxidative system works through mechanisms suppressing and scavenging not only free radicals but also de novo antioxidant. Superoxide (O2•−) is the major ROS produced in the mitochondria with increased oxidative stress. However, conversion of O2•− to H2O by coupled reactions of superoxide dismutase (SOD), catalase, and glutathione reductase/peroxidise is accompanied by the formation of GSH and elimination of the detrimental effect of O2•− on damage the cells.
Among the free radicals, ROS are the oxygen-containing free radicals as the natural by-products of the metabolism of oxygen, including hydroxyl radical, superoxide anion radical, hydrogen peroxide, oxygen singlet, hypochlorite, nitric oxide radical, and peroxynitrite radical. They are derived within organelles including peroxisomes, endoplasmic reticulum, and mitochondria, the major site of ROS production.
Antioxidant system consists of antioxidants and antioxidative enzymes (Figure 1(a)). The resources of antioxidants can be endogenous and exogenous. Glutathione is the most abundant endogenous antioxidant in most cell types with the reduced form (GSH) as biologically active [24]. The exogenous antioxidants are derived from either diet or supplement, such as vitamin A, C, and E [21, 25] and antioxidant minerals (copper, zinc, manganese, selenium). The major antioxidative enzymes, including superoxide dismutase, glutathione reductase/peroxidise, and catalase, metabolize free radicals to nontoxic intermediates. They work in synergy (Figure 1(b)) through mechanisms not only suppressing and scavenging free radicals before they can damage cells, but also repairing de novo antioxidants [24, 26].
3. Diabetic Neuropathy
3.1. General Aspects
Diabetic neuropathy, a damage occurred in sensory neurons, causes neuropathic pain with either central or peripheral syndromes in different patterns (e.g., pain and numbness) [7, 9]. Clinically, the most common pattern is the distal symmetric polyneuropathy of the feet and hands, with a distal-to-proximal gradient of severity [1, 9, 10, 27]. Of note, pain is reported by approximately one third of patients with diabetes, regardless of associated neurological deficits [27].
The cause of diabetic neuropathy has been attributed to diabetes [8, 9], which is a metabolic disorder characterized by impaired glucose metabolism with chronic hyperglycaemia and dysfunction of endogenous insulin (insufficiency of secretion as type I or action as type II). Based on the large clinical studies in patients with type I or type II diabetes, a strong correlation between chronic hyperglycaemia and diabetic microvascular complications has been established [28–33]. At least 50% of individuals with diabetes develop diabetic neuropathy with time [34].
Increased glucose levels in diabetes affect primarily those cells that have a limited capacity to regulate their glucose intake, including vascular cells, Schwann cells, and neurons of the peripheral and central nervous systems [35, 36]. It is unclear whether high glucose triggers axonal degeneration by promoting intrinsic programmes within axons [36–38], nor is whether peripheral axons or their associated Schwann cells, the first target. Clinical findings have demonstrated that Schwann cells are targeted in patients with diabetic neuropathy [39]. Experimental studies in diabetic rodents have associated endoplasmic reticulum stress with diabetes-mediated peripheral nerve damage [40]. At the early stage of diabetes, hyperglycaemia causes abnormalities in blood flow and in vascular permeability [7]. With time, impaired glucose metabolism reduces intracellular levels of NADPH and decreases the synthesis of myo-inositol that is particularly required for the normal function of nerves [41, 42]. Collectively, diabetic neuropathy might be caused by a direct effect of hyperglycaemia on damaging cells [43, 44] and an indirect effect on affecting cellular functions [7, 45]. Loss of microvascular cell occurs, in parallel with reduced production of endothelial and neuronal cells, which leads to degeneration of peripheral nerves [46].
3.2. Diabetic Neuropathy in Type I and Type II Diabetes
The major predictors of diabetic neuropathy are the duration of diabetes and the blood levels of haemoglobin A1c (HbA1c) [6]. The severity of diabetic complications correlates with the severity of hyperglycaemia, suggesting that the complications are triggered by the elevation in glucose levels [28]. However, rapid glucose control significantly increased the risk of severe hypoglycemic episodes [47] and resulted in treatment-induced neuropathy in both type I and type II diabetes [48–50].
Diabetic neuropathy can be found late in type I diabetes but early in type II diabetes, and the cause of this occurrence remains unclear. As the consistent feature between type I and type II is hyperglycaemia, one would assume that controlling hyperglycaemia would be the best preventive treatment for diabetic neuropathy regardless the diabetic type. Of interest, the incidence of neuropathy is higher in diabetic patients with type II than those with type I [51–54], whereas the prevalence of diabetic neuropathy was similar in type II diabetic patients [55–57] as seen in type I [6, 58]. Clinically, efficient glucose control significantly reduced or delayed the incidence of developing neuropathy in type I diabetic patients [59, 60], whereas it remains elusive with type II diabetes [29, 47, 61–63]. In addition, lowing haemoglobin A1c in type II diabetic patients has little effect on diabetic neuropathy [64], whereas a greater improvement has been observed in type I diabetic patients after 18 months of glycemic control [48]. Collectively, it suggests that hyperglycaemia is not the prime driving cause of all complications [35, 65], and mechanisms underlying diabetic neuropathy in type I and type II diabetes could be fundamentally different [47].
The most recent findings with alterations in DNA methylation have been suggested as a contributor to diabetic neuropathy in type I and type II diabetic patients [14, 66]. Of interest, spliceosome dysregulation has been proposed as a key neurodegenerative mechanism underlying diabetic neuropathy in type I diabetic patients [67]. Spliceosome is a complex assembled from small nuclear RNA and proteins in nucleus and required to catalyze pre-mRNA splicing in nuclear speckles [68]. Whether splicing abnormalities are identifiable in type II diabetes remains unexplored.
The current approaches to managing diabetic neuropathy focus on improving glycaemic control, mainly in type I diabetic patients, and lifestyle modifications, mainly in type II diabetic patients [35, 69]. Although diabetic neuropathy is the strongest predictor of mortality in type II diabetes, it remains the only microvascular complication of diabetes without a specific treatment owing to our lack of basic understanding of this disease.
4. Oxidative Stress in Diabetic Neuropathy
4.1. Main Sources of ROS Production in Diabetic Neuropathy
The main proof of oxidative stress involvement in diabetic neuropathy was the accumulation of free radicals and reduced activity of antioxidant enzymes in the diabetic animals with diabetic neuropathy, and the effect was ameliorated, in parallel with the alleviation of symptoms, upon antioxidant treatment [70]. Ample evidence strongly support that hyperglycaemia leads to increased oxidative stress that plays a pivotal role in the development of diabetic neuropathy [7] by damaging the cells including endothelial, retinal, mesangial, and neural cells [2, 8].
Impaired mitochondrial glucose oxidation is believed as a main source of ROS production in diabetes [7, 8]. To understand how hyperglycaemia increases ROS production, a brief overview of glucose metabolism is helpful (Figure 2(a)). Under a physiological condition, after being taken-up, intracellular glucose is converted to glucose-6-phosphate, then followed by glycolysis and oxidation to produce NADH and acetyl CoA. Electrons carried by NADH are transferred to oxygen following mitochondrial electron transport chain (ETC), along with pumping protons out of the mitochondrial matrix thereby creating a proton gradient that is used by ATP synthase to produce ATP (Figure 2(a)). Under normal condition, there are only 0.2-2% of the electrons in the ETC leaking out to produce ROS [71], and neurons have sufficient capacity to remove ROS by innate cellular antioxidants (Figure 1(b)) [72]. Under a diabetic condition, however, impaired glucose metabolism shunts glucose or intermediates of glycolysis to other metabolic and nonmetabolic pathways (Figure 2(b)), which causes mitochondrial injury with higher rates of protons returning to the mitochondria without generating ATP and an overwhelming production of ROS in a neuron [11]. Axons are rich of mitochondria, having a direct access to nerve blood supply. The inability of the neuron to detoxify the excess ROS together with insufficient ATP production leave axons being more susceptible to ROS-mediated damage in hyperglycaemia [11], in part because of their dependence on local mitochondria for energy, which in turn precipitates axonal degeneration [11].
Figure 2.
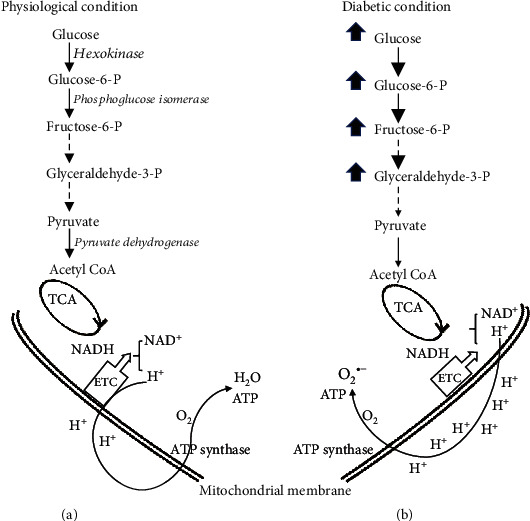
Major glucose metabolism pathway under physiological condition and diabetic condition. (a) Under a physiological condition, intracellular glucose is converted to glucose-6-phosphate, via hexokinase, then following isomerization to fructose-6-phosphate. Along glycolysis pathway, glyceraldehyde-3-phosphate travels down to pyruvate and acetyl CoA via pyruvate dehydrogenase, which then enters tricarboxylic acid (TCA) cycle. NADH, as electron carrier and generated during the process of glycolysis and glucose oxidation, can donate reducing equivalents to the mitochondrial electron transport chain (ETC), thereby creating a proton gradient that is used by ATP synthase to produce ATP and converting O2 to H2O. (b) Under diabetic condition, glucose metabolism is impaired, causing accumulation of glucose and the glycolytic intermediates, resulting mitochondrial injury, thereby converting O2 to superoxide radical (O2•−) instead of H2O. As a result, ATP production is reduced.
4.2. Molecular Mechanisms of ROS Production in Diabetic Neuropathy
There are four damaging pathways (Figure 3) that can explain the detrimental effects of ROS in hyperglycaemia-induced diabetic neuropathy, including polyol pathway and hexosamine pathway that have been consistently observed in patients with diabetic neuropathy [73–76]. The AGE and PKC pathway modify proteins, lipids, and DNAs via a direct [77–80] or an indirect effect of glucose [81, 82]. All of which are linked to diabetic neuropathy by a single event: overproduction of ROS [7], which is a consistent differentiating feature common to all cell types that are damaged by hyperglycaemia [8].
Figure 3.
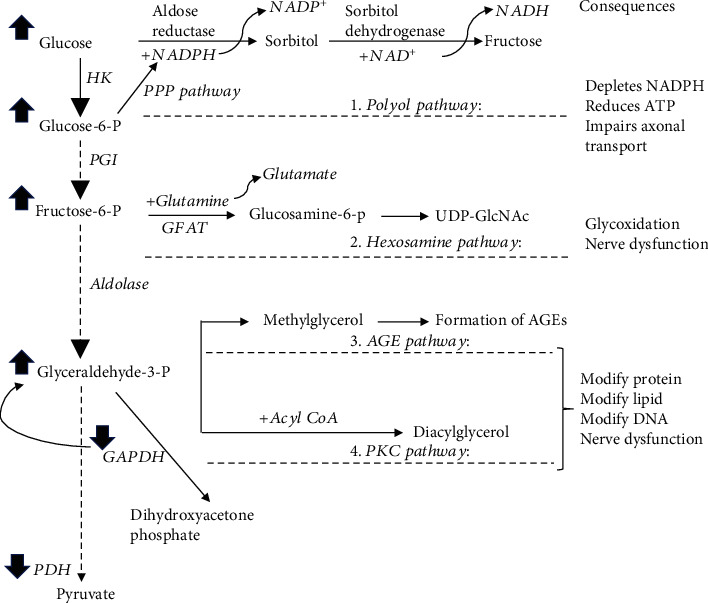
Four damaging pathways that can explain the detrimental effects of ROS in hyperglycaemia-induced diabetic neuropathy. The impaired glucose metabolism in diabetic condition causes an accumulation of glucose and glycolytic intermediates, which, instead of travel along glycolysis pathway, shunts to other metabolic or nonmetabolic pathways, resulting activation of the polyol pathway, hexosamine pathway, and AGE and PKC pathway. Superoxide inhibits glyceraldehyde-3-phosphate dehydrogenase (GAPDH) activity, which is proposed to be a reason causing accumulation of all the glycolytic intermediates. Pentose phosphate pathway (PPP pathway) is to generate NADPH, which is used in polyol pathway. GFAT: glutamine-fructose-6-phosphate aminotransferase; GAPDH: glyceraldehyde-3 phosphate dehydrogenase; PDH: pyruvate dehydrogenase.
4.2.1. Activated Polyol Pathway
The polyol pathway is a two-step metabolic process (Figure 3), promoted by a mass action of excess glucose to activate aldose reductase. Aldose reductase, a cytosolic protein, normally has low affinity to glucose, and function for reducing toxic aldehydes in tissues such as nerve, retina, lens, glomerulus, and vascular cells [83, 84]. In many of which, glucose moves freely across the cell membrane independent of insulin, and intracellular levels of glucose rise with hyperglycaemia in parallel with an increased affinity of aldose reductase for glucose. This favors the excess glucose to generate sorbitol, instead of down to glycolysis, with consumption of nicotinamide adenine dinucleotide phosphate (NADPH) to NADP+ (Figure 3). Sorbitol is subsequently oxidized to fructose through sorbitol dehydrogenase, with NAD+ as a cofactor. To note, compared to glucose, glyceraldehyde 3-phosphate, a glycolytic intermediate, has been suggested as a relevant substrate for aldose reductase, owing to its higher affinity to aldose reductase under pathological conditions [85].
The well accepted mechanism for hyperglycaemia-induced polyol pathway is the increased redox stress due to the consumption of NADPH, which is derived from pentose phosphate pathway for generating GSH from glutathione (Figure 3). This notion is further supported by the observation that overexpression of human aldose reductase in diabetic mice reduced the expression of regulatory genes for regenerating glutathione [86]. Meanwhile, excess fructose, as a product, promotes glycation and further depletion of NADPH thereby causing and exacerbating intracellular oxidative stress.
Relevant to diabetic neuropathy, accumulation of sorbitol and fructose were observed in the peripheral nerves of diabetic rats [87], while shunting glycolytic intermediates to polyol pathway also promotes glycation and formation of diacylglycerol in dorsal root ganglia of the diabetic mice [88]. Inhibiting aldose reductase prevented accumulation of sorbitol and fructose in peripheral nerves of the diabetic rats [89, 90] and restored diabetes-induced defect in nerve conduction velocity in diabetic dogs [91]. In addition, patients with a high aldose reductase expression are commonly having an early diabetic neuropathy relative to the patients with a low aldose reductase expression [92, 93]. Thus, inhibiting the polyol pathway continues to be a drug target in the treatment of diabetic neuropathy.
4.2.2. Activated Hexosamine Pathway
The well accepted mechanism that hexosamine pathway contributes to diabetic neuropathy is the effect of intracellular UDP-GlcNAc on modification of proteins [94, 95]. Under physiological condition, hexosamine pathway is a minor branch of the glycolytic pathway [96, 97] with only 2-5% [97] of fructose-6-phosphate being converted to glucosamine-6-phosphate by glutamine-fructose-6-phosphate aminotransferase (GFAT), the rate limiting enzyme. Under hyperglycaemia condition, however, the increased production of mitochondrial ROS inhibits glyceraldehyde-3 phosphate dehydrogenase activity, a glycolytic enzyme (Figure 3), thereby blocking fructose-6-phosphate flow through glycolysis [98]. Subsequently, glucosamine-6-phosphate along with acetyl-CoA and uridine-5′-triphosphate are used to produce the amino sugar uridine-5′-diphosphat-N-acetylglucosamine (UDP-GlcNAc) [97], which controls activity of O-linked N-acetylglucosamine transferase [99]. The latter is a cytosolic and nuclear enzyme, catalyzing a reversible posttranslational protein modification by transferring GlcNAc to specific serine and threonine residues on proteins [99, 100]. Of particular interest at proteins modified by O-GlcNAcylation are insulin receptor substrates-1 and 2 [101, 102] as well as glucose transporter 4 [103].
Relevant to diabetic neuropathy, an increase in GFAT activity and UDP-GlcNAc concentrations was evident in muscle of ob/ob mice [104]. Conversely, a reduced UDP-GlcNAc concentration was observed, in parallel with improved insulin sensitivity in muscles of the rats with chronic caloric restriction [104]. Clinically, GFAT activity is increased in muscle biopsies obtained from insulin resistant patients with type II diabetes [105], while insulin resistance is improved markedly by insulin treatment in patients with severely insulin resistant, uncontrolled, obese, type II diabetes, concomitant with 40% increase in the levels of UDP-GlcNAc in muscle [106]. In contrary, a positive correlation among UDP-GlcNAc circulating levels of FFA and leptin was found in adipocytes, but not in muscle of type II diabetic patients relative to nondiabetic control [107]. Hyperglycaemia-induced overmodification of proteins by glucosamine results in pathological changes in gene expression, especially transcription factors, which contribute to the pathogenesis of diabetic complications with the strongest evidence for the role in diabetic complications [102, 108]. However, it is not yet clear what kind of peripheral nerve proteins can be modified by activated hexosamine pathway following diabetes, nor is the causal connection. Thus, the contribution of hexosamine pathway to diabetic neuropathy remains to be further explored.
4.2.3. Activated AGE and PKC Pathway
The common feature of both pathways is the modification of proteins thereby modulating diabetic complications via activating transcription factors. Activation of hexosamine pathway causes an elevated level of glyceraldehyde-3 phosphate, which upon conjugation with fatty acid, produces diacylglycerol [109, 110] to activate PKC. Thus, both activated polyol pathway and hexosamine pathway in diabetes can activate AGE and PKC pathway.
(1) Activation of AGE Pathway. AGEs are intracellular and extracellular adducts formed by covalent linking reducing sugars or its metabolites to lysine or arginine on proteins [111–113]. Hyperglycaemia is recognized as the primary initiating event in the formation of AGEs [78]. There are two forms of AGEs precursors, glyoxal and methylglyoxal, which can be generated through three major pathways: auto-oxidation of glucose to form glyoxal, a smallest dialdehyde [80]; abnormal metabolism of glyceraldehyde-3 phosphate from glycolysis to form methylglyoxal; and degradation of glyceraldehyde and glycolaldehyde (fructose-lysine adducts) to form both glyoxal and methylglyoxal. Compared to glyoxal, methylglyoxal is highly reactive and causing vascular endothelial cells to be more sensitive to damage [114].
Relevant to diabetic neuropathy, the well accepted mechanism is that extracellular AGEs interact with a specific AGE receptor: known as RAGE on the cell surface [115], causing overproduction of ROS, thereby activating nuclear factor kappa B (NF-κB) to initiate multiple pathological changes in gene expression [116]. Activation of AGE-RAGE-NF-κB axis appears to be sustained in both clinical [117] and laboratory settings [112, 117]. In the streptozotocin-induced diabetic mice, knockdown of RAGE gene significantly improved electrophysiological and anatomical markers of diabetic neuropathy [112] as well as restored pain perception in sciatic nerves [117], in parallel with a decreased expression of NF-κB in peripheral nerves [117], and particularly in Schwann cells [112]. Clinically, activated NF-κB was colocalized with RAGE within the vasa nervorum in the sural nerve biopsies from the patients with diabetic neuropathy [117]. In relevant, diabetic patients have increased expression of endothelia RAGE and less collateral vessels, compared with nondiabetic controls, which contributes to increased rates of lower limb amputation [118]. Thus, AGE-RAGE-NF-κB axis might promote diabetic neuropathy through its impact on microvessels within the sensory neurons. Up to date, effective treatment modalities of AGE-induced nerve injury is not available clinically [67].
(2) Activation of Protein Kinase C. Activation of classic PKC is dependent on both Ca2+ ions and phosphatidylserine and is greatly enhanced by diacylglycerol [119]. The primary function of PKC is phosphorylating targeted proteins, which in turn operating on gene expression in diabetes, thereby mediating abnormalities of blood flow and permeability via inhibiting NO production [120] or activating NF-κB [121] and microvascular matrix proteins [122] in both diabetic patients [123] and animal models [124].
Of relevance, the PKC β-isoform in particular has been linked to the development of diabetic nephropathy in the diabetic animal models, such as in the streptozotocin-induced diabetic rats; PKCβ inhibitors actually improve motor nerve conduction velocity and endoneurial blood flow [125, 126]. However, clinical studies using ruboxistaurin, a selective PKCβ inhibitor, for treatment of painful diabetic neuropathy did not achieve significance [127]. Based on six randomized controlled trials, it was concluded that PKCβ inhibitor offered no benefit in the treatment of diabetic neuropathy [128]. Up to date, the role for PKC activation in diabetic neuropathy remains unclear.
5. Management of Treating Diabetic Neuropathy in Relation to Oxidative Stress
5.1. Assessment
There are two newer techniques, besides a number of other tools [65], for clinician to assess diabetic neuropathy, including the visual quantification of intraepidermal nerve fibers through skin biopsy for peripheral vs. MR imaging for central neuropathy, which allows noninvasive in vivo imaging of corneal nerves [129, 130].
5.2. Newest Approach in Pain Medicine
The newest approach was released on May 2020 by Vienna University of Technology that chronic pain can be reduced by stimulating the vagus nerve in the ear with a tiny electrodes, in which a 3D computer model is created to calculate the optimal stimulation of nerve branches [131, 132]. The approach has now been successfully tested on patients [132]. Although this is not specifically for treatment of diabetic neuropathy, it is an important step forward.
In clinical practice, the assessment with combination pharmacotherapy is often applied for managing diabetic neuropathy. Of interest, pharmacological approaches aiming at targeting antioxidative stress are the few strategies that reduce pain in diabetic neuropathy patients in clinical trials [133–138].
5.3. Targeting Oxidative Stress
Several strategies aiming at antioxidative stress have been employed (Figure 4) to combat nerve dysfunction in diabetes, including directly against ROS, against individual oxidative stress pathways, or targeted at mitochondria.
Figure 4.
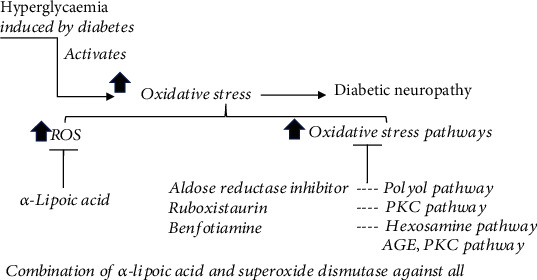
Strategies of antioxidative drugs in treating diabetic neuropathy include directly against ROS and against individual oxidative stress pathways. The listed drugs showed effect on improving symptoms of diabetic neuropathy. However, none of them have been FDA-approved due to the lack of clinical significance.
5.4. Targeting ROS
Including α-lipoic acid (ALA), vitamins A, C, and E, acetyl L-carnitine, taurine, and melatonin, taurine, acetyl L-carnitine, and N-acetylcysteine have been demonstrated to reduce the progression of diabetic neuropathy [139], whereas the effect of vitamins A, C, and E in diabetic neuropathy needs more research to ascertain [9, 21]. ALA is thought to be a valuable therapeutic option for diabetic neuropathy as the treatment ameliorated the symptoms of diabetic neuropathy in the clinical trials [135, 137, 138]. ALA is a water- and fat-soluble compound known to reduce oxidative stress by inhibiting hexosamine and AGEs pathways [140]. The combination of ALA and superoxide dismutase has improved symptoms and electroneurographic parameters in the patients with diabetic neuropathy [141]. Currently, ALA has been licensed in Germany to treat symptomatic diabetic neuropathy with 600 mg daily dosage [142].
5.5. Targeting Individual Oxidative Stress Pathways
Including aldose reductase inhibitors, anti-AGE agents, and PKC inhibitors, in contrast to ruboxistaurin, a specific PKCβ inhibitor failed to achieve its clinical significance; aldose reductase inhibitors and anti-AGE agents have shown the therapeutic effect.
Aldose reductase inhibitors are used to reduce the flux of glucose into the polyol pathway. The positive effect of inhibiting aldose reductase on diabetic neuropathy includes enhancing sural motor and sensory nerve conduction velocities, improving wrist and ankle F-wave latency together with and alleviating neuropathic pain [143], which have been observed in diabetic mice [143] and in diabetic patients [144]. However, its efficacy needs further investigation [145].
Anti-AGE agents are used to prevent the formation and accumulation of AGEs. Benfotiamine, a lipid-soluble analogue of vitamin B1, has the effect on preventing the activation of the hexosamine pathway and the AGE and PKC pathway induced by diabetes [7, 8]. Importantly, benfotiamine has shown the therapeutic efficacy in the patients with diabetic neuropathy [134, 146, 147] and in diabetic rats [8, 133], which suggests that benfotiamine may extend the treatment option for diabetic neuropathy based on causal influence on impaired glucose metabolism [134].
The clinical translation is challenging, as none of these drugs are currently FDA-approved yet, in part, due to the failure in clinical trials [148, 149]. One of the common contributors to the failure in the clinical trials is the irreproducibility of the improvement in the treated animal models vs. in humans, for example, sorbinil (an aldose reductase inhibitor), which was shown to inhibit diabetes-induced nerve conduction deficit in streptozotocin-induced diabetic rats [150]; however, it did not produce similar results in humans [151]. Another concern is the side effect, such as photosensitive skin rash, discouraging the further usage [152]. Extensive preclinical research is still on going to investigate further mechanisms and new targets with improved efficacy and safety for treating diabetic neuropathy.
6. Conclusion
Although considerable research has been devoted to understanding mechanisms of diabetic neuropathy in general, treatment options to eliminate the initial causes are still “lacking. There have been disparities between the results obtained from animals and human studies. Currently, genetic- or diet-induced animal models are commonly used to investigate neuropathy in type II diabetes, while high-dose streptozotocin-induced diabetic animal models are used to mimic the metabolic phenotype of type I diabetes. However, no single rodent model accurately mimics human diabetic neuropathy [153] with the major problems not only in pain assessment that is hard to measure in rodents [154] but also in the knowledge about the molecular mechanisms. Most knowledge about mechanisms of diabetic neuropathy was gained in genetically homogenous male rodents, while patients vary in sex, ethnicity and genetic background, age, and duration of diabetes. Thus, these factors should be considered in designing the individualized treatment plans for patients with diabetic neuropathy.
Hyperglycaemia-induced oxidative stress remains the most accepted mechanism for the progression of diabetes to diabetic neuropathy (Figure 5). The impaired glucose metabolism in diabetes leads to hypoxia and acidosis, which trigger other abnormalities responsible for mitochondrial and bioenergetic dysfunction by increasing ROS production to cause membrane hyperexcitability and reduction of ATP production. Treatment options with antioxidants have been investigated; none are satisfactory. Treatment which repairs nerves, in patients with diabetic neuropathy, has yet to be translated into clinical trials. Thus, future research must establish the most efficacious drug combinations on combating hyperglycaemia and oxidative stress for the prevention/treatment of diabetic neuropathy, in addition to explore the new mechanisms. Clinically, predictors and biomarkers need to be validated for both clinical trials and clinical practice.
Figure 5.
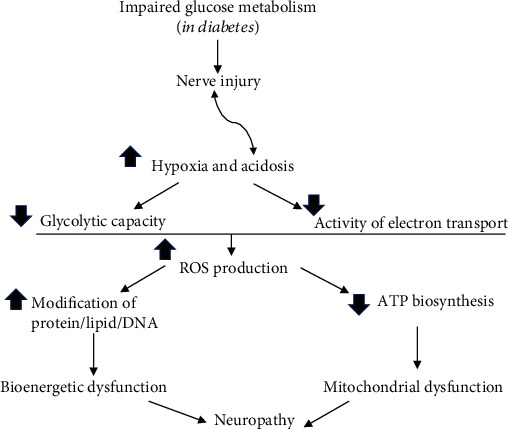
Summary of the major contributors to diabetic neuropathy. Diabetes-induced impairment of glucose metabolism causes hypoxia and acidosis, which contributes to and exacerbates the nerve injury. As a result, both glycolytic capacity and activity of electron transport chain are reduced, leading to overproduction of ROS, which, not only reducing ATP production but also initiating various modifications on protein/lipid and DNA. Thus, mitochondrial and bioenergetic dysfunction leads to neuropathy.
Acknowledgments
The authors' research was supported by the National Natural Science Foundation of Jilin Province (nos. 20190201061JC and 20200201321JC), the National Natural Science Foundation (no. 81970228), and the Educational Commission of Jilin Province of China (JJKH20201085KJ and JJKH20201047KJ).
Contributor Information
Haichun Ma, Email: mahaichun2003@163.com.
Xin Yu, Email: yoson911@yeah.net.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Authors' Contributions
Lei Pang and Xin Lian contributed equally to this work.
References
- 1.Callaghan B. C., Cheng H. T., Stables C. L., Smith A. L., Feldman E. L. Diabetic neuropathy: clinical manifestations and current treatments. Lancet Neurology. 2012;11(6):521–534. doi: 10.1016/S1474-4422(12)70065-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boulton A. J., Vinik A. I., Arezzo J. C., et al. Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes Care. 2005;28(4):956–962. doi: 10.2337/diacare.28.4.956. [DOI] [PubMed] [Google Scholar]
- 3.Little A. A., Edwards J. L., Feldman E. L. Diabetic neuropathies. Practical Neurology. 2007;7(2):82–92. [PubMed] [Google Scholar]
- 4.Vinik A. I., Maser R. E., Mitchell B. D., Freeman R. Diabetic autonomic neuropathy. Diabetes Care. 2003;26(5):1553–1579. doi: 10.2337/diacare.26.5.1553. [DOI] [PubMed] [Google Scholar]
- 5.Vinik A. I., Mehrabyan A. Diabetic neuropathies. The Medical Clinics of North America. 2004;88(4):947–999. doi: 10.1016/j.mcna.2004.04.009. xi. [DOI] [PubMed] [Google Scholar]
- 6.Tesfaye S., Chaturvedi N., Eaton S. E., et al. Vascular risk factors and diabetic neuropathy. The New England Journal of Medicine. 2005;352(4):341–350. doi: 10.1056/NEJMoa032782. [DOI] [PubMed] [Google Scholar]
- 7.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(6865):813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 8.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54(6):1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 9.Feldman E. L., Callaghan B. C., Pop-Busui R., et al. Diabetic neuropathy. Nature Reviews Disease Primers. 2019;5(1) doi: 10.1038/s41572-019-0092-1. [DOI] [PubMed] [Google Scholar]
- 10.Feldman E. L., Nave K. A., Jensen T. S., Bennett D. L. H. New horizons in diabetic neuropathy: mechanisms, bioenergetics, and pain. Neuron. 2017;93(6):1296–1313. doi: 10.1016/j.neuron.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fernyhough P., McGavock J. Mechanisms of disease: mitochondrial dysfunction in sensory neuropathy and other complications in diabetes. Handbook of Clinical Neurology. 2014;126:353–377. doi: 10.1016/B978-0-444-53480-4.00027-8. [DOI] [PubMed] [Google Scholar]
- 12.Ott M., Gogvadze V., Orrenius S., Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007;12(5):913–922. doi: 10.1007/s10495-007-0756-2. [DOI] [PubMed] [Google Scholar]
- 13.Vincent A. M., Hayes J. M., McLean L. L., Vivekanandan-Giri A., Pennathur S., Feldman E. L. Dyslipidemia-induced neuropathy in mice: the role of oxLDL/LOX-1. Diabetes. 2009;58(10):2376–2385. doi: 10.2337/db09-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo K., Elzinga S., Eid S., et al. Genome-wide DNA methylation profiling of human diabetic peripheral neuropathy in subjects with type 2 diabetes mellitus. Epigenetics. 2019;14(8):766–779. doi: 10.1080/15592294.2019.1615352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chowdhury S. K. R., Smith D. R., Fernyhough P. The role of aberrant mitochondrial bioenergetics in diabetic neuropathy. Neurobiology of Disease. 2013;51:56–65. doi: 10.1016/j.nbd.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 16.Fernyhough P. Mitochondrial dysfunction in diabetic neuropathy: a series of unfortunate metabolic events. Current Diabetes Reports. 2015;15(11) doi: 10.1007/s11892-015-0671-9. [DOI] [PubMed] [Google Scholar]
- 17.Rumora A. E., Lentz S. I., Hinder L. M., et al. Dyslipidemia impairs mitochondrial trafficking and function in sensory neurons. The FASEB Journal. 2017;32(1):195–207. doi: 10.1096/fj.201700206r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vincent A. M., Edwards J. L., Sadidi M., Feldman E. L. The antioxidant response as a drug target in diabetic neuropathy. Current Drug Targets. 2008;9(1):94–100. doi: 10.2174/138945008783431754. [DOI] [PubMed] [Google Scholar]
- 19.Vincent A. M., Feldman E. L. New insights into the mechanisms of diabetic neuropathy. Reviews in Endocrine & Metabolic Disorders. 2004;5(3):227–236. doi: 10.1023/B:REMD.0000032411.11422.e0. [DOI] [PubMed] [Google Scholar]
- 20.Vincent A. M., Russell J. W., Low P., Feldman E. L. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocrine Reviews. 2004;25(4):612–628. doi: 10.1210/er.2003-0019. [DOI] [PubMed] [Google Scholar]
- 21.Rock C. L., Jacob R. A., Bowen P. E. Update on the biological characteristics of the antioxidant Micronutrients. Journal of the American Dietetic Association. 1996;96(7):693–702. doi: 10.1016/S0002-8223(96)00190-3. quiz 703-704. [DOI] [PubMed] [Google Scholar]
- 22.Cheeseman K. H., Slater T. F. An introduction to free radical biochemistry. British Medical Bulletin. 1993;49(3):481–493. doi: 10.1093/oxfordjournals.bmb.a072625. [DOI] [PubMed] [Google Scholar]
- 23.Halliwell B., Whiteman M. Measuring reactive species and oxidative damage in vivo and in cell culture: how should you do it and what do the results mean? British Journal of Pharmacology. 2004;142(2):231–255. doi: 10.1038/sj.bjp.0705776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi H., Noguchi N., Niki E. Comparative study on dynamics of antioxidative action of alpha-tocopheryl hydroquinone, ubiquinol, and alpha-tocopherol against lipid peroxidation. Free Radical Biology & Medicine. 1999;27(3-4):334–346. doi: 10.1016/S0891-5849(99)00053-2. [DOI] [PubMed] [Google Scholar]
- 25.Levine M., Rumsey S. C., Daruwala R., Park J. B., Wang Y. Criteria and recommendations for vitamin C intake. JAMA. 1999;281(15):1415–1423. doi: 10.1001/jama.281.15.1415. [DOI] [PubMed] [Google Scholar]
- 26.Maritim A. C., Sanders R. A., Watkins J. B. Diabetes, oxidative stress, and antioxidants: a review. Journal of Biochemical and Molecular Toxicology. 2003;17(1):24–38. doi: 10.1002/jbt.10058. [DOI] [PubMed] [Google Scholar]
- 27.Sloan G., Shillo P., Selvarajah D., et al. A new look at painful diabetic neuropathy. Diabetes Research and Clinical Practice. 2018;144:177–191. doi: 10.1016/j.diabres.2018.08.020. [DOI] [PubMed] [Google Scholar]
- 28.Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The New England Journal of Medicine. 1993;329(14):977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 29.UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33) Lancet. 1998;352(9131):837–853. doi: 10.1016/S0140-6736(98)07019-6. [DOI] [PubMed] [Google Scholar]
- 30.Nathan D. M. Long-term complications of diabetes mellitus. The New England Journal of Medicine. 1993;328(23):1676–1685. doi: 10.1056/NEJM199306103282306. [DOI] [PubMed] [Google Scholar]
- 31.Ruderman N. B., Williamson J. R., Brownlee M. Glucose and diabetic vascular disease1. The FASEB Journal. 1992;6(11):2905–2914. doi: 10.1096/fasebj.6.11.1644256. [DOI] [PubMed] [Google Scholar]
- 32.Clark C. M., Jr., Lee D. A. Prevention and treatment of the complications of diabetes mellitus. The New England Journal of Medicine. 1995;332(18):1210–1217. doi: 10.1056/NEJM199505043321807. [DOI] [PubMed] [Google Scholar]
- 33.Ziyadeh F. N. The extracellular matrix in diabetic nephropathy. American Journal of Kidney Diseases. 1993;22(5):736–744. doi: 10.1016/S0272-6386(12)80440-9. [DOI] [PubMed] [Google Scholar]
- 34.Callaghan B. C., Kerber K. A., Lisabeth L. L., et al. Role of neurologists and diagnostic tests on the management of distal symmetric polyneuropathy. JAMA Neurology. 2014;71(9):1143–1149. doi: 10.1001/jamaneurol.2014.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosenberger D. C., Blechschmidt V., Timmerman H., Wolff A., Treede R. D. Challenges of neuropathic pain: focus on diabetic neuropathy. Journal of Neural Transmission (Vienna) 2020;127(4):589–624. doi: 10.1007/s00702-020-02145-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scholz J., Broom D. C., Youn D. H., et al. Blocking caspase activity prevents transsynaptic neuronal apoptosis and the loss of inhibition in lamina II of the dorsal horn after peripheral nerve injury. The Journal of Neuroscience. 2005;25(32):7317–7323. doi: 10.1523/JNEUROSCI.1526-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Campbell J. N., Meyer R. A. Mechanisms of neuropathic pain. Neuron. 2006;52(1):77–92. doi: 10.1016/j.neuron.2006.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.May A. Chronic pain may change the structure of the brain. Pain. 2008;137(1):7–15. doi: 10.1016/j.pain.2008.02.034. [DOI] [PubMed] [Google Scholar]
- 39.Dunnigan S. K., Ebadi H., Breiner A., et al. Conduction slowing in diabetic sensorimotor polyneuropathy. Diabetes Care. 2013;36(11):3684–3690. doi: 10.2337/dc13-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lupachyk S., Watcho P., Stavniichuk R., Shevalye H., Obrosova I. G. Endoplasmic reticulum stress plays a key role in the pathogenesis of diabetic peripheral neuropathy. Diabetes. 2013;62(3):944–952. doi: 10.2337/db12-0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clements R. S., Jr., Vourganti B., Kuba T., Oh S. J., Darnell B. Dietary myo-inositol intake and peripheral nerve function in diabetic neuropathy. Metabolism. 1979;28(4):477–483. doi: 10.1016/0026-0495(79)90060-X. [DOI] [PubMed] [Google Scholar]
- 42.Salway J. G., Whitehead L., Finnegan J. A., Karunanayaka A., Barnett D., Payne R. B. Effect of myo-inositol on peripheral-nerve function in diabetes. Lancet. 1978;2(8103):1282–1284. doi: 10.1016/s0140-6736(78)92043-3. [DOI] [PubMed] [Google Scholar]
- 43.Trachootham D., Lu W., Ogasawara M. A., Valle N. R. D., Huang P. Redox regulation of cell survival. Antioxidants & Redox Signaling. 2008;10(8):1343–1374. doi: 10.1089/ars.2007.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poulsen H. E., Specht E., Broedbaek K., et al. RNA modifications by oxidation: a novel disease mechanism? Free Radical Biology & Medicine. 2012;52(8):1353–1361. doi: 10.1016/j.freeradbiomed.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 45.Giacco F., Brownlee M. Oxidative stress and diabetic complications. Circulation Research. 2010;107(9):1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williams B. Angiotensin II, VEGF, and diabetic retinopathy. Lancet. 1998;351(9105):837–838. doi: 10.1016/S0140-6736(05)78974-1. [DOI] [PubMed] [Google Scholar]
- 47.Callaghan B. C., Little A. A., Feldman E. L., Hughes R. A. Enhanced glucose control for preventing and treating diabetic neuropathy. Cochrane Database of Systematic Reviews. 2012;6(6, article CD007543) doi: 10.1002/14651858.cd007543.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gibbons C. H., Freeman R. Treatment-induced diabetic neuropathy: a reversible painful autonomic neuropathy. Annals of Neurology. 2010;67(4):534–541. doi: 10.1002/ana.21952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gibbons C. H. Treatment-induced neuropathy of diabetes. Current Diabetes Reports. 2017;17(12):p. 127. doi: 10.1007/s11892-017-0960-6. [DOI] [PubMed] [Google Scholar]
- 50.Hwang Y. T., Davies G. 'Insulin neuritis' to 'treatment-induced neuropathy of diabetes': new name, same mystery. Practical Neurology. 2016;16(1):53–55. doi: 10.1136/practneurol-2015-001215. [DOI] [PubMed] [Google Scholar]
- 51.Pop-Busui R., Boulton A. J. M., Feldman E. L., et al. Diabetic neuropathy: a position statement by the American Diabetes Association. Diabetes Care. 2016;40(1):136–154. doi: 10.2337/dc16-2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ang L., Jaiswal M., Martin C., Pop-Busui R. Glucose control and diabetic neuropathy: lessons from recent large clinical trials. Current Diabetes Reports. 2014;14(9):p. 528. doi: 10.1007/s11892-014-0528-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martin C. L., Albers J. W., Pop-Busui R., for the DCCT/EDIC Research Group Neuropathy and related findings in the diabetes control and complications trial/epidemiology of diabetes interventions and complications study. Diabetes Care. 2013;37(1):31–38. doi: 10.2337/dc13-2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pop-Busui R., Lu J., Brooks M. M., et al. Impact of glycemic control strategies on the progression of diabetic peripheral neuropathy in the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) Cohort. Diabetes Care. 2013;36(10):3208–3215. doi: 10.2337/dc13-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Franklin G. M., Kahn L. B., Baxter J., Marshall J. A., Hamman R. F. Sensory neuropathy in non-insulin-dependent diabetes mellitus. The San Luis Valley Diabetes Study. American Journal of Epidemiology. 1990;131(4):633–643. doi: 10.1093/oxfordjournals.aje.a115547. [DOI] [PubMed] [Google Scholar]
- 56.Partanen J., Niskanen L., Lehtinen J., Mervaala E., Siitonen O., Uusitupa M. Natural history of peripheral neuropathy in patients with non-insulin-dependent diabetes mellitus. The New England Journal of Medicine. 1995;333(2):89–94. doi: 10.1056/NEJM199507133330203. [DOI] [PubMed] [Google Scholar]
- 57.Dyck P. J., Kratz K. M., Karnes J. L., et al. The prevalence by staged severity of various types of diabetic neuropathy, retinopathy, and nephropathy in a population-based cohort: the Rochester Diabetic Neuropathy Study. Neurology. 1993;43(4):817–824. doi: 10.1212/WNL.43.4.817. [DOI] [PubMed] [Google Scholar]
- 58.Boulton A. J. M., Knight G., Drury J., Ward J. D. The prevalence of symptomatic, diabetic neuropathy in an insulin-treated population. Diabetes Care. 1985;8(2):125–128. doi: 10.2337/diacare.8.2.125. [DOI] [PubMed] [Google Scholar]
- 59.Fullerton B., Jeitler K., Seitz M., Horvath K., Berghold A., Siebenhofer A. Intensive glucose control versus conventional glucose control for type 1 diabetes mellitus. Cochrane Database of Systematic Reviews. 2014;2014(2, article CD009122) doi: 10.1002/14651858.CD009122.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Albers J. W., Herman W. H., Pop-Busui R., et al. Effect of prior intensive insulin treatment during the Diabetes Control and Complications Trial (DCCT) on peripheral neuropathy in type 1 diabetes during the Epidemiology of Diabetes Interventions and Complications (EDIC) Study. Diabetes Care. 2010;33(5):1090–1096. doi: 10.2337/dc09-1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pantalone K. M., Misra-Hebert A. D., Hobbs T. M., et al. Effect of glycemic control on the Diabetes Complications Severity Index score and development of complications in people with newly diagnosed type 2 diabetes. Journal of Diabetes. 2018;10(3):192–199. doi: 10.1111/1753-0407.12613. [DOI] [PubMed] [Google Scholar]
- 62.Ismail-Beigi F., Craven T., Banerji M. A., et al. Effect of intensive treatment of hyperglycaemia on microvascular outcomes in type 2 diabetes: an analysis of the ACCORD randomised trial. Lancet. 2010;376(9739):419–430. doi: 10.1016/S0140-6736(10)60576-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Calles-Escandon J., Lovato L. C., Simons-Morton D. G., et al. Effect of intensive compared with standard glycemia treatment strategies on mortality by baseline subgroup characteristics: the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Diabetes Care. 2010;33(4):721–727. doi: 10.2337/dc09-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.The Action to Control Cardiovascular Risk in Diabetes Study Group. Effects of intensive glucose lowering in type 2 diabetes. The New England Journal of Medicine. 2008;358(24):2545–2559. doi: 10.1056/NEJMoa0802743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tesfaye S., Boulton A. J. M., Dyck P. J., et al. Diabetic neuropathies: update on definitions, diagnostic criteria, estimation of severity, and treatments. Diabetes Care. 2010;33(10):2285–2293. doi: 10.2337/dc10-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Roshandel D., DCCT/EDIC Research Group, Chen Z., et al. DNA methylation age calculators reveal association with diabetic neuropathy in type 1 diabetes. Clinical Epigenetics. 2020;12(1):p. 52. doi: 10.1186/s13148-020-00840-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kobayashi M., Zochodne D. W. Diabetic neuropathy and the sensory neuron: new aspects of pathogenesis and their treatment implications. Journal of Diabetes Investigation. 2018;9(6):1239–1254. doi: 10.1111/jdi.12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Girard C., Will C. L., Peng J., et al. Post-transcriptional spliceosomes are retained in nuclear speckles until splicing completion. Nature Communications. 2012;3(1, article 994) doi: 10.1038/ncomms1998. [DOI] [PubMed] [Google Scholar]
- 69.Qaseem A., Wilt T. J., Kansagara D., et al. Hemoglobin A1cTargets for glycemic control with pharmacologic therapy for nonpregnant adults with type 2 diabetes mellitus: a guidance statement update from the American College of Physicians. Annals of Internal Medicine. 2018;168(8):569–576. doi: 10.7326/M17-0939. [DOI] [PubMed] [Google Scholar]
- 70.Cameron N. E., Cotter M. A., Archibald V., Dines K. C., Maxfield E. K. Anti-oxidant and pro-oxidant effects on nerve conduction velocity, endoneurial blood flow and oxygen tension in non-diabetic and streptozotocin-diabetic rats. Diabetologia. 1994;37(5):449–459. doi: 10.1007/s001250050131. [DOI] [PubMed] [Google Scholar]
- 71.Turrens J. F. Mitochondrial formation of reactive oxygen species. The Journal of Physiology. 2003;552(2):335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nishikawa T., Edelstein D., du X. L., et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404(6779):787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 73.Greene D. A., Arezzo J. C., Brown M. B. Effect of aldose reductase inhibition on nerve conduction and morphometry in diabetic neuropathy. Neurology. 1999;53(3):580–591. doi: 10.1212/WNL.53.3.580. [DOI] [PubMed] [Google Scholar]
- 74.Behl T., Kaur I., Kotwani A. Implication of oxidative stress in progression of diabetic retinopathy. Survey of Ophthalmology. 2016;61(2):187–196. doi: 10.1016/j.survophthal.2015.06.001. [DOI] [PubMed] [Google Scholar]
- 75.Forbes J. M., Coughlan M. T., Cooper M. E. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes. 2008;57(6):1446–1454. doi: 10.2337/db08-0057. [DOI] [PubMed] [Google Scholar]
- 76.Javed S., Petropoulos I. N., Alam U., Malik R. A. Treatment of painful diabetic neuropathy. Therapeutic Advances in Chronic Disease. 2014;6(1):15–28. doi: 10.1177/2040622314552071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wolff S. P., Dean R. T. Glucose autoxidation and protein modification. The potential role of 'autoxidative glycosylation' in diabetes. The Biochemical Journal. 1987;245(1):243–250. doi: 10.1042/bj2450243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wells-Knecht K. J., Zyzak D. V., Litchfield J. E., Thorpe S. R., Baynes J. W. Mechanism of autoxidative glycosylation: identification of glyoxal and arabinose as intermediates in the autoxidative modification of proteins by glucose. Biochemistry. 2002;34:3702–3709. doi: 10.1021/bi00011a027. [DOI] [PubMed] [Google Scholar]
- 79.Fu M. X., Wells-Knecht K. J., Blackledge J. A., Lyons T. J., Thorpe S. R., Baynes J. W. Glycation, glycoxidation, and cross-linking of collagen by glucose. Kinetics, mechanisms, and inhibition of late stages of the Maillard reaction. Diabetes. 1994;43(5):676–683. doi: 10.2337/diab.43.5.676. [DOI] [PubMed] [Google Scholar]
- 80.Chetyrkin S., Mathis M., Pedchenko V., et al. Glucose autoxidation induces functional damage to proteins via modification of critical arginine residues. Biochemistry. 2011;50(27):6102–6112. doi: 10.1021/bi200757d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stitt A. W., Li Y. M., Gardiner T. A., Bucala R., Archer D. B., Vlassara H. Advanced glycation end products (AGEs) co-localize with AGE receptors in the retinal vasculature of diabetic and of AGE-infused rats. The American Journal of Pathology. 1997;150(2):523–531. [PMC free article] [PubMed] [Google Scholar]
- 82.Horie K., Miyata T., Maeda K., et al. Immunohistochemical colocalization of glycoxidation products and lipid peroxidation products in diabetic renal glomerular lesions. Implication for glycoxidative stress in the pathogenesis of diabetic nephropathy. The Journal of Clinical Investigation. 1997;100(12):2995–3004. doi: 10.1172/JCI119853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ramasamy R., Goldberg I. J. Aldose reductase and cardiovascular diseases, creating human-like diabetic complications in an experimental model. Circulation Research. 2010;106(9):1449–1458. doi: 10.1161/CIRCRESAHA.109.213447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hers H. G. The mechanism of the transformation of glucose in fructose in the seminal vesicles. Biochimica et Biophysica Acta. 1956;22(1):202–203. doi: 10.1016/0006-3002(56)90247-5. [DOI] [PubMed] [Google Scholar]
- 85.Bohren K. M., Grimshaw C. E., Gabbay K. H. Catalytic effectiveness of human aldose reductase. Critical role of C-terminal domain. The Journal of Biological Chemistry. 1992;267(29):20965–20970. [PubMed] [Google Scholar]
- 86.Vikramadithyan R. K., Hu Y., Noh H. L., et al. Human aldose reductase expression accelerates diabetic atherosclerosis in transgenic mice. The Journal of Clinical Investigation. 2005;115(9):2434–2443. doi: 10.1172/JCI24819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stewart M. A., Sherman W. R., Kurien M. M., Moonsammy G. I., Wisgerhof M. Polyol accumulations in nervous tissue of rats with experimental diabetes and galactosaemia. Journal of Neurochemistry. 1967;14(11):1057–1066. doi: 10.1111/j.1471-4159.1967.tb09516.x. [DOI] [PubMed] [Google Scholar]
- 88.Uehara K., Yamagishi S. I., Otsuki S., Chin S., Yagihashi S. Effects of polyol pathway hyperactivity on protein kinase C activity, nociceptive peptide expression, and neuronal structure in dorsal root ganglia in diabetic mice. Diabetes. 2004;53(12):3239–3247. doi: 10.2337/diabetes.53.12.3239. [DOI] [PubMed] [Google Scholar]
- 89.Mayer J. H., Tomlinson D. R. Prevention of defects of axonal transport and nerve conduction velocity by oral administration of myo-inositol or an aldose reductase inhibitor in streptozotocin-diabetic rats. Diabetologia. 1983;25(5):433–438. doi: 10.1007/BF00282524. [DOI] [PubMed] [Google Scholar]
- 90.Finegold D., Lattimer S. A., Nolle S., Bernstein M., Greene D. A. Polyol pathway activity and myo-inositol metabolism. A suggested relationship in the pathogenesis of diabetic neuropathy. Diabetes. 1983;32(11):988–992. doi: 10.2337/diab.32.11.988. [DOI] [PubMed] [Google Scholar]
- 91.Engerman R. L., Kern T. S., Larson M. E. Nerve conduction and aldose reductase inhibition during 5 years of diabetes or galactosaemia in dogs. Diabetologia. 1994;37(2):141–144. doi: 10.1007/s001250050084. [DOI] [PubMed] [Google Scholar]
- 92.Demaine A. G. Polymorphisms of the aldose reductase gene and susceptibility to diabetic microvascular complications. Current Medicinal Chemistry. 2003;10(15):1389–1398. doi: 10.2174/0929867033457359. [DOI] [PubMed] [Google Scholar]
- 93.Thamotharampillai K., Chan A. K. F., Bennetts B., et al. Decline in neurophysiological function after 7 years in an adolescent diabetic cohort and the role of aldose reductase gene polymorphisms. Diabetes Care. 2006;29(9):2053–2057. doi: 10.2337/dc06-0678. [DOI] [PubMed] [Google Scholar]
- 94.McClain D. A. Hexosamines as mediators of nutrient sensing and regulation in diabetes. Journal of Diabetes and its Complications. 2002;16(1):72–80. doi: 10.1016/S1056-8727(01)00188-X. [DOI] [PubMed] [Google Scholar]
- 95.McClain D. A., Lubas W. A., Cooksey R. C., et al. Altered glycan-dependent signaling induces insulin resistance and hyperleptinemia. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(16):10695–10699. doi: 10.1073/pnas.152346899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Buse M. G. Hexosamines, insulin resistance, and the complications of diabetes: current status. American Journal of Physiology. Endocrinology and Metabolism. 2006;290(1):E1–E8. doi: 10.1152/ajpendo.00329.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Marshall S., Bacote V., Traxinger R. R. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. The Journal of Biological Chemistry. 1991;266:4706–4712. [PubMed] [Google Scholar]
- 98.Du X.-L., Edelstein D., Rossetti L., et al. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(22):12222–12226. doi: 10.1073/pnas.97.22.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kreppel L. K., Blomberg M. A., Hart G. W. Dynamic glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. The Journal of Biological Chemistry. 1997;272(14):9308–9315. doi: 10.1074/jbc.272.14.9308. [DOI] [PubMed] [Google Scholar]
- 100.Lubas W. A., Frank D. W., Krause M., Hanover J. A. O-linked GlcNAc transferase is a conserved nucleocytoplasmic protein containing tetratricopeptide repeats. The Journal of Biological Chemistry. 1997;272(14):9316–9324. doi: 10.1074/jbc.272.14.9316. [DOI] [PubMed] [Google Scholar]
- 101.Andreozzi F., D’Alessandris C., Federici M., et al. Activation of the hexosamine pathway leads to phosphorylation of insulin receptor substrate-1 on Ser307 and Ser612 and impairs the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin insulin biosynthetic pathway in RIN pancreatic beta-cells. Endocrinology. 2004;145(6):2845–2857. doi: 10.1210/en.2003-0939. [DOI] [PubMed] [Google Scholar]
- 102.Federici M., Menghini R., Mauriello A., et al. Insulin-dependent activation of endothelial nitric oxide synthase is impaired by O-linked glycosylation modification of signaling proteins in human coronary endothelial cells. Circulation. 2002;106(4):466–472. doi: 10.1161/01.CIR.0000023043.02648.51. [DOI] [PubMed] [Google Scholar]
- 103.Buse M. G., Robinson K. A., Marshall B. A., Hresko R. C., Mueckler M. M. Enhanced O-GlcNAc protein modification is associated with insulin resistance in GLUT1-overexpressing muscles. American Journal of Physiology. Endocrinology and Metabolism. 2002;283(2):E241–E250. doi: 10.1152/ajpendo.00060.2002. [DOI] [PubMed] [Google Scholar]
- 104.Buse M. G., Robinson K. A., Gettys T. W., McMahon E. G., Gulve E. A. Increased activity of the hexosamine synthesis pathway in muscles of insulin-resistant ob/ob mice. The American Journal of Physiology. 1997;272, 6 Part 1:E1080–E1088. doi: 10.1152/ajpendo.1997.272.6.E1080. [DOI] [PubMed] [Google Scholar]
- 105.Yki-Jarvinen H., Daniels M. C., Virkamaki A., Makimattila S., DeFronzo R. A., McClain D. Increased glutamine:fructose-6-phosphate amidotransferase activity in skeletal muscle of patients with NIDDM. Diabetes. 1996;45(3):302–307. doi: 10.2337/diab.45.3.302. [DOI] [PubMed] [Google Scholar]
- 106.Pouwels M. J. J., Span P. N., Tack C. J., et al. Muscle uridine diphosphate-hexosamines do not decrease despite correction of hyperglycemia-induced insulin resistance in type 2 diabetes. The Journal of Clinical Endocrinology and Metabolism. 2002;87(11):5179–5184. doi: 10.1210/jc.2002-020440. [DOI] [PubMed] [Google Scholar]
- 107.Pouwels M. J. J., Tack C. J., Span P. N., et al. Role of hexosamines in insulin resistance and nutrient sensing in human adipose and muscle tissue. The Journal of Clinical Endocrinology and Metabolism. 2004;89(10):5132–5137. doi: 10.1210/jc.2004-0291. [DOI] [PubMed] [Google Scholar]
- 108.Kolm-Litty V., Sauer U., Nerlich A., Lehmann R., Schleicher E. D. High glucose-induced transforming growth factor beta1 production is mediated by the hexosamine pathway in porcine glomerular mesangial cells. The Journal of Clinical Investigation. 1998;101(1):160–169. doi: 10.1172/JCI119875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Inoguchi T., Battan R., Handler E., Sportsman J. R., Heath W., King G. L. Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(22):11059–11063. doi: 10.1073/pnas.89.22.11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shiba T., Inoguchi T., Sportsman J. R., Heath W. F., Bursell S., King G. L. Correlation of diacylglycerol level and protein kinase C activity in rat retina to retinal circulation. American Journal of Physiology-Endocrinology and Metabolism. 1993;265(5):E783–E793. doi: 10.1152/ajpendo.1993.265.5.e783. [DOI] [PubMed] [Google Scholar]
- 111.Ahmed N. Advanced glycation endproducts--role in pathology of diabetic complications. Diabetes Research and Clinical Practice. 2005;67(1):3–21. doi: 10.1016/j.diabres.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 112.Toth C., Rong L. L., Yang C., et al. Receptor for advanced glycation end products (RAGEs) and experimental diabetic neuropathy. Diabetes. 2008;57(4):1002–1017. doi: 10.2337/db07-0339. [DOI] [PubMed] [Google Scholar]
- 113.Gallet X., Charloteaux B., Thomas A., Brasseur R. A fast method to predict protein interaction sites from sequences. Journal of Molecular Biology. 2000;302(4):917–926. doi: 10.1006/jmbi.2000.4092. [DOI] [PubMed] [Google Scholar]
- 114.Yao D., Taguchi T., Matsumura T., et al. High glucose increases angiopoietin-2 transcription in microvascular endothelial cells through methylglyoxal modification of mSin3A. The Journal of Biological Chemistry. 2007;282(42):31038–31045. doi: 10.1074/jbc.M704703200. [DOI] [PubMed] [Google Scholar]
- 115.Ramasamy R., Yan S. F., Schmidt A. M. Arguing for the motion: yes, RAGE is a receptor for advanced glycation endproducts. Molecular Nutrition & Food Research. 2007;51(9):1111–1115. doi: 10.1002/mnfr.200700008. [DOI] [PubMed] [Google Scholar]
- 116.Goldin A., Beckman J. A., Schmidt A. M., Creager M. A. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation. 2006;114(6):597–605. doi: 10.1161/CIRCULATIONAHA.106.621854. [DOI] [PubMed] [Google Scholar]
- 117.Bierhaus A., Haslbeck K. M., Humpert P. M., et al. Loss of pain perception in diabetes is dependent on a receptor of the immunoglobulin superfamily. The Journal of Clinical Investigation. 2004;114(12):1741–1751. doi: 10.1172/JCI18058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Abaci A., Oguzhan A., Kahraman S., et al. Effect of diabetes mellitus on formation of coronary collateral vessels. Circulation. 1999;99(17):2239–2242. doi: 10.1161/01.CIR.99.17.2239. [DOI] [PubMed] [Google Scholar]
- 119.Geraldes P., King G. L. Activation of protein kinase C isoforms and its impact on diabetic complications. Circulation Research. 2010;106(8):1319–1331. doi: 10.1161/CIRCRESAHA.110.217117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pugliese G., Pricci F., Pugliese F., et al. Mechanisms of glucose-enhanced extracellular matrix accumulation in rat glomerular mesangial cells. Diabetes. 1994;43(3):478–490. doi: 10.2337/diab.43.3.478. [DOI] [PubMed] [Google Scholar]
- 121.Yerneni K. K., Bai W., Khan B. V., Medford R. M., Natarajan R. Hyperglycemia-induced activation of nuclear transcription factor kappaB in vascular smooth muscle cells. Diabetes. 1999;48(4):855–864. doi: 10.2337/diabetes.48.4.855. [DOI] [PubMed] [Google Scholar]
- 122.Craven P. A., Studer R. K., Felder J., Phillips S., DeRubertis F. R. Nitric oxide inhibition of transforming growth factor-beta and collagen synthesis in mesangial cells. Diabetes. 1997;46(4):671–681. doi: 10.2337/diab.46.4.671. [DOI] [PubMed] [Google Scholar]
- 123.Christiansen J. S., Gammelgaard J., Frandsen M., Parving H. H. Increased kidney size, glomerular filtration rate and renal plasma flow in short-term insulin-dependent diabetics. Diabetologia. 1981;20(4):451–456. doi: 10.1007/BF00253406. [DOI] [PubMed] [Google Scholar]
- 124.Hostetter T. H., Troy J. L., Brenner B. M. Glomerular hemodynamics in experimental diabetes mellitus. Kidney International. 1981;19(3):410–415. doi: 10.1038/ki.1981.33. [DOI] [PubMed] [Google Scholar]
- 125.Nakamura J., Kato K., Hamada Y., et al. A protein kinase C-beta-selective inhibitor ameliorates neural dysfunction in streptozotocin-induced diabetic rats. Diabetes. 1999;48(10):2090–2095. doi: 10.2337/diabetes.48.10.2090. [DOI] [PubMed] [Google Scholar]
- 126.Cameron N. E., Cotter M. A., Jack A. M., Basso M. D., Hohman T. C. Protein kinase C effects on nerve function, perfusion, Na(+), K(+)-ATPase activity and glutathione content in diabetic rats. Diabetologia. 1999;42(9):1120–1130. doi: 10.1007/s001250051280. [DOI] [PubMed] [Google Scholar]
- 127.Vinik A. I., Bril V., Kempler P., et al. Treatment of symptomatic diabetic peripheral neuropathy with the protein kinase C β-inhibitor ruboxistaurin mesylate during a 1-year, randomized, placebo-controlled, double-blind clinical trial. Clinical Therapeutics. 2005;27(8):1164–1180. doi: 10.1016/j.clinthera.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 128.Bansal D., Badhan Y., Gudala K., Schifano F. Ruboxistaurin for the treatment of diabetic peripheral neuropathy: a systematic review of randomized clinical trials. Diabetes and Metabolism Journal. 2013;37(5):375–384. doi: 10.4093/dmj.2013.37.5.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tavakoli M., Petropoulos I. N., Malik R. A. Corneal confocal microscopy to assess diabetic neuropathy: an eye on the foot. Journal of Diabetes Science and Technology. 2013;7(5):1179–1189. doi: 10.1177/193229681300700509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lauria G., Cornblath D. R., Johansson O., et al. EFNS guidelines on the use of skin biopsy in the diagnosis of peripheral neuropathy. European Journal of Neurology. 2005;12(10):747–758. doi: 10.1111/j.1468-1331.2005.01260.x. [DOI] [PubMed] [Google Scholar]
- 131.Dabiri B., Kampusch S., Geyer S. H., et al. High-resolution episcopic imaging for visualization of dermal arteries and nerves of the auricular cymba conchae in humans. Frontiers in Neuroanatomy. 2020;14 doi: 10.3389/fnana.2020.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Technology VUo. Novel electric impulses relieve the pain. ScienceDaily; 2020. [Google Scholar]
- 133.Sanchez-Ramirez G. M., Caram-Salas N. L., Rocha-Gonzalez H. I., et al. Benfotiamine relieves inflammatory and neuropathic pain in rats. European Journal of Pharmacology. 2006;530(1-2):48–53. doi: 10.1016/j.ejphar.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 134.Stracke H., Gaus W., Achenbach U., Federlin K., Bretzel R. G. Benfotiamine in diabetic polyneuropathy (BENDIP): results of a randomised, double blind, placebo-controlled clinical study. Experimental and Clinical Endocrinology & Diabetes. 2008;116(10):600–605. doi: 10.1055/s-2008-1065351. [DOI] [PubMed] [Google Scholar]
- 135.Ziegler D., Ametov A., Barinov A., et al. Oral treatment With -Lipoic Acid Improves Symptomatic Diabetic Polyneuropathy: The SYDNEY 2 trial. Diabetes Care. 2006;29(11):2365–2370. doi: 10.2337/dc06-1216. [DOI] [PubMed] [Google Scholar]
- 136.Ziegler D., Movsesyan L., Mankovsky B., Gurieva I., Abylaiuly Z., Strokov I. Treatment of symptomatic polyneuropathy with actovegin in type 2 diabetic patients. Diabetes Care. 2009;32(8):1479–1484. doi: 10.2337/dc09-0545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ametov A. S., Barinov A., Dyck P. J., et al. The sensory symptoms of diabetic polyneuropathy are improved with alpha-lipoic acid: the SYDNEY trial. Diabetes Care. 2003;26(3):770–776. doi: 10.2337/diacare.26.3.770. [DOI] [PubMed] [Google Scholar]
- 138.Papanas N., Ziegler D. Efficacy of α-lipoic acid in diabetic neuropathy. Expert Opinion on Pharmacotherapy. 2014;15(18):2721–2731. doi: 10.1517/14656566.2014.972935. [DOI] [PubMed] [Google Scholar]
- 139.Negre-Salvayre A., Coatrieux C., Ingueneau C., Salvayre R. Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. British Journal of Pharmacology. 2008;153(1):6–20. doi: 10.1038/sj.bjp.0707395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Du X., Edelstein D., Brownlee M. Oral benfotiamine plus alpha-lipoic acid normalises complication-causing pathways in type 1 diabetes. Diabetologia. 2008;51(10):1930–1932. doi: 10.1007/s00125-008-1100-2. [DOI] [PubMed] [Google Scholar]
- 141.Bertolotto F., Massone A. Combination of alpha lipoic acid and superoxide dismutase leads to physiological and symptomatic improvements in diabetic neuropathy. Drugs in R&D. 2012;12(1):29–34. doi: 10.2165/11599200-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Shakher J., Stevens M. J. Update on the management of diabetic polyneuropathies. Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy. 2011;4:289–305. doi: 10.2147/DMSO.S11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yagihashi S., Yamagishi S. I., Wada Ri R., et al. Neuropathy in diabetic mice overexpressing human aldose reductase and effects of aldose reductase inhibitor. Brain. 2001;124(12):2448–2458. doi: 10.1093/brain/124.12.2448. [DOI] [PubMed] [Google Scholar]
- 144.Kawai T., Takei I., Tokui M., et al. Effects of epalrestat, an aldose reductase inhibitor, on diabetic peripheral neuropathy in patients with type 2 diabetes, in relation to suppression of Nɛ-carboxymethyl lysine. Journal of Diabetes and its Complications. 2010;24(6):424–432. doi: 10.1016/j.jdiacomp.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 145.Boulton A. J. M., Kempler P., Ametov A., Ziegler D. Whither pathogenetic treatments for diabetic polyneuropathy? Diabetes/Metabolism Research and Reviews. 2013;29(5):327–333. doi: 10.1002/dmrr.2397. [DOI] [PubMed] [Google Scholar]
- 146.Winkler G., Pal B., Nagybeganyi E., Ory I., Porochnavec M., Kempler P. Effectiveness of different benfotiamine dosage regimens in the treatment of painful diabetic neuropathy. Arzneimittel-Forschung. 1999;49(3):220–224. doi: 10.1055/s-0031-1300405. [DOI] [PubMed] [Google Scholar]
- 147.Haupt E., Ledermann H., Kopcke W. Benfotiamine in the treatment of diabetic polyneuropathy--a three-week randomized, controlled pilot study (BEDIP study) International Journal of Clinical Pharmacology and Therapeutics. 2005;43(2):71–77. doi: 10.5414/CPP43071. [DOI] [PubMed] [Google Scholar]
- 148.Dewanjee S., Das S., Das A. K., et al. Molecular mechanism of diabetic neuropathy and its pharmacotherapeutic targets. European Journal of Pharmacology. 2018;833:472–523. doi: 10.1016/j.ejphar.2018.06.034. [DOI] [PubMed] [Google Scholar]
- 149.Grewal A. S., Bhardwaj S., Pandita D., Lather V., Sekhon B. S. Updates on aldose reductase inhibitors for management of diabetic complications and non-diabetic diseases. Mini Reviews in Medicinal Chemistry. 2015;16(2):120–162. doi: 10.2174/1389557515666150909143737. [DOI] [PubMed] [Google Scholar]
- 150.Cameron N. E., Tuck Z., McCabe L., Cotter M. A. Effect of the hydroxyl radical scavenger, dimethylthiourea, on peripheral nerve tissue perfusion, conduction velocity and nociception in experimental diabetes. Diabetologia. 2001;44(9):1161–1169. doi: 10.1007/s001250100626. [DOI] [PubMed] [Google Scholar]
- 151.Edwards J. L., Vincent A. M., Cheng H. T., Feldman E. L. Diabetic neuropathy: mechanisms to management. Pharmacology & Therapeutics. 2008;120(1):1–34. doi: 10.1016/j.pharmthera.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sima A. A. F., Bril V., Nathaniel V., et al. Regeneration and repair of myelinated fibers in sural-nerve biopsy specimens from patients with diabetic neuropathy treated with sorbinil. The New England Journal of Medicine. 1988;319(9):548–555. doi: 10.1056/NEJM198809013190905. [DOI] [PubMed] [Google Scholar]
- 153.O'Brien P. D., Sakowski S. A., Feldman E. L. Mouse models of diabetic neuropathy. ILAR Journal. 2014;54(3):259–272. doi: 10.1093/ilar/ilt052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tappe-Theodor A., Kuner R. Studying ongoing and spontaneous pain in rodents--challenges and opportunities. The European Journal of Neuroscience. 2014;39(11):1881–1890. doi: 10.1111/ejn.12643. [DOI] [PubMed] [Google Scholar]