

Abstract
Background
Invasive lobular carcinoma (ILC) accounts for 10–15% of primary breast cancers and is typically estrogen receptor alpha positive (ER+) and ERBB2 non-amplified. Somatic mutations in ERBB2/3 are emerging as a tractable mechanism underlying enhanced human epidermal growth factor 2 (HER2) activity. We tested the hypothesis that therapeutically targetable ERBB2/3 mutations in primary ILC of the breast associate with poor survival outcome in large public datasets.
Methods
We performed in silico comparison of ERBB2 non-amplified cases of ER+ stage I–III primary ILC (N = 279) and invasive ductal carcinoma (IDC, N = 1301) using METABRIC, TCGA, and MSK-IMPACT information. Activating mutations amenable to HER2-directed therapy with neratinib were identified using existing functional data from in vitro cell line and xenograft experiments. Multivariate analysis of 10-year overall survival (OS) with tumor size, grade, and lymph node status was performed using a Cox regression model. Differential gene expression analyses by ERBB2 mutation and amplification status was performed using weighted average differences and an in silico model of response to neratinib derived from breast cancer cell lines.
Results
ILC tumors comprised 17.7% of all cases in the dataset but accounted for 47.1% of ERBB2-mutated cases. Mutations in ERBB2 were enriched in ILC vs. IDC cases (5.7%, N = 16 vs. 1.4%, N = 18, p < 0.0001) and clustered in the tyrosine kinase domain of HER2. ERBB3 mutations were not enriched in ILC (1.1%, N = 3 vs. 1.8%, N = 23; p = 0.604). Median OS for patients with ERBB2-mutant ILC tumors was 66 months vs. 211 months for ERBB2 wild-type (p = 0.0001), and 159 vs. 166 months (p = 0.733) for IDC tumors. Targetable ERBB2 mutational status was an independent prognostic marker of 10-year OS—but only in ILC (hazard ratio, HR = 3.7, 95% CI 1.2–11.0; p = 0.021). Findings were validated using a novel ERBB2 mutation gene enrichment score (HR for 10-year OS in ILC = 2.3, 95% CI 1.04–5.05; p = 0.040).
Conclusions
Targetable ERBB2 mutations are enriched in primary ILC and their detection represents an actionable strategy with the potential to improve patient outcomes. Biomarker-led clinical trials of adjuvant HER-targeted therapy are warranted for patients with ERBB2-mutated primary ILC.
Keywords: Lobular, Breast cancer, ERBB2, HER2, Mutation, Prognosis, Therapeutic biomarker, Adjuvant
Background
Invasive lobular carcinoma of the breast (ILC) accounts for 10–15% of all breast cancer with an estimated 250,000 cases per year worldwide [1–4]. Nearly all cases of ILC derive from luminal cells that express estrogen receptor alpha but lack E-Cadherin (CDH1) expression or ERBB2 amplification [5].
Clinical evidence suggests that despite favorable prognostic indicators, e.g., ER+ and/or progesterone receptor positive, low Ki67 proliferation index and HER2− status, patients with ILC have similar or worse long-term outcomes compared to those with invasive ductal carcinoma (IDC, otherwise known as invasive carcinoma of no special type) [5, 6]. Adjuvant treatment of ER+ ILC with letrozole, an aromatase inhibitor, may be superior to tamoxifen, and ILC cells demonstrate resistance to tamoxifen in vitro [7, 8]. However, patients with ILC are treated according to identical protocols as those with IDC [9, 10].
HER2-targeted therapy is indicated for patients whose tumors are HER2+ by immunohistochemistry (IHC) or, if IHC is equivocal, where ERBB2 is amplified as detected by in situ hybridization (ISH) [11, 12]. Recent evidence indicates that in ERBB2 non-amplified breast cancer, somatic mutation of ERBB2 (ERBB2mut) and/or ERBB3 (ERBB3mut) may provide an alternative mechanism for upregulation of HER2 activity that is therapeutically tractable using second generation HER2 tyrosine kinase inhibitors such as neratinib [13, 14].
The epidermal growth factor (EGF) family of receptor tyrosine kinases (HER1–4) are activated by ligand-dependent homo-/heterodimerisation and regulate cellular proliferation and tumor progression [15, 16]. In ERBB2-amplified cells, the oncogenic effect of HER2 is mediated by heterodimerisation with HER3 in a ligand-independent manner [17]. Thus HER3 is necessary for HER2 oncogenic activity, and both HER2 and HER3 are therapeutic targets in ERBB2-amplified breast cancer [18]. Mutations in ERBB2 cluster in the tyrosine kinase and extra-cellular domains of HER2 and exert their oncogenic effects by activating tyrosine kinase activity or increasing HER2 dimerization, respectively [13]. In vitro studies of HER2 activity in cell line and xenograft models identified 13 mutations (listed in the “Patients and methods” section) that enhanced proliferation and/or demonstrated growth inhibition with the irreversible HER2/EGFR tyrosine kinase inhibitor, neratinib [13]. Mutations in HER3, a critical binding partner for HER2, have been shown to promote ligand (EGF)-independent transformation of breast epithelial cells only in the presence of kinase-active HER2 [19]. This indicates that known oncogenic mutations in ERBB3, e.g., G284R and E928G, may also be therapeutically targetable via HER2 inhibition [19].
ERBB2 mutations have previously been linked with worse prognosis in CDH1-altered ILC: a study of ILC cases in the TCGA dataset (N = 169) found that ERBB2 mutations (N = 6) occurred exclusively in CDH1-altered tumors (N = 100) [20]. However, prognostic data on the 6 ERBB2mut cases were limited to 2 patient events for both disease-free survival and OS analyses. The overall rate of ERBB2mut in this study of primary ILC was 3.6% (N = 6 out of 169). A study of relapsed CDH1-mutated ILC found ERBB2mut in 18% of cases (N = 4 of 22), suggesting further acquisition of ERBB2 mutations in CDH1-altered ILC due to the selective pressure of treatment [21].
To demonstrate the potential clinical benefit of targeting low frequency somatic mutations, prognostic analyses using large clinical datasets are required. In the MA12 trial comprising 328 premenopausal patients with ER+ primary breast cancer of all histological subtypes, non-silent ERBB2 mutations occurred in 5.2% of patients (N = 17) and were adversely prognostic of OS (p = 0.0114) [22]. A study of 5605 cases of relapsed breast cancer found ERBB2mut in 2.4% of cases (N = 138), of which 20% (N = 27 of 138) were in ILC tumors [23]. However, neither of these larger studies stratified clinical outcome by histological subtype.
An ILC-specific study of 630 cases of primary ILC found ERBB2 and ERBB3 mutations in 5.1 and 3.6% of tumors, respectively [24]. Comparison of cases of ER+, HER2− ILC from the same study (N = 371) with cases of ER+, HER2− IDC from TCGA (N = 338) indicated significant enrichment of both ERBB2 and ERBB3 mutations in ILC (4.3 and 3.5% in ILC vs. 1.5 and 0.6% in IDC) [24]. The study reported limited statistical evidence of a time-dependent effect of ERBB2 mutational status associated with short-term breast cancer-specific survival. However, confirmation in datasets including patients with long-term follow-up and decoupling of activating from silent mutations is needed.
We hypothesized that low-frequency somatic mutations in ERBB2 and ERBB3 are enriched in ER+, ERBB2 non-amplified primary ILC cases and may have a demonstrable prognostic effect. We tested these hypotheses by mining a combined dataset of the three largest primary breast cancer series with data on tumor ERBB2 and ERBB3 mutational status, gene expression, clinicopathological features, and patient survival outcomes. Our overall goal was to determine the association between targetable ERBB2/3 mutations and survival in primary ER+ ILC, and thereby provide evidence for a clinically actionable strategy to improve patient outcomes.
Patients and methods
Patients and outcome measures
Genomic and clinical outcome data associated with tumor samples from patients with primary breast cancer in TCGA 2015 (N = 817), METABRIC 2012 and 2016 (N = 2509), and MSK-IMPACT 2018 (N = 918) were accessed online via CBioportal [25–27]. From these datasets, ER+ and HER2− cases of stage I–III ILC and IDC with both clinical outcome and mutational data called from next-generation sequencing (NGS) analyses were selected (N = 1580). Cases of mixed or non-ILC/IDC histology, ER-negative/undetermined, HER2+/undetermined, carcinoma in situ, and stage 4/undetermined were excluded. For TCGA and MSK datasets, HER2 status was determined by IHC (positive/negative) or, where IHC was indeterminate, by ISH assessment of ERBB2 amplification, in line with standard clinical practice [11]. For METABRIC cases, HER2 status was determined using the Affymetrix SNP6 copy number inference pipeline.
The primary outcome measure, available in all datasets, was OS. Variables included ERBB2 and ERBB3 mutational status. ERBB2mut status was subcategorized as oncogenic or uncharacterized by cross-reference with existing data that identified mutations targetable by HER2-inhibition: G309A/E, S310F, L755S, del755-759, S760A, D769H, D769Y, V777L, P780ins, V842I, and R896C [13]. Cases were denominated oncERBB2mut if tumors harbored at least one oncogenic ERBB2 mutation.
Clinical and NGS mutation data were integrated with clinicopathological features including histological subtype, lymph node (LN) status, and tumor size and grade. Normalized gene expression data were publicly available for METABRIC (Illumina HT12 microarray) and TCGA (RNA-seq) datasets.
Statistical analysis
For analysis of binary somatic mutation status, combined cohort analysis was performed. Enrichment for cases with mutations in candidate genes (ERBB2/3mut) by histological subtype (ILC vs. IDC) was determined by χ2 test for association between categorical variables. For ILC and IDC separately, Kaplan-Meier (KM) survival curves stratified by mutation status were compared using logrank and generalized Wilcoxon tests. Multivariate analysis of OS was performed using a Cox regression model. Covariates included classic prognostic markers tumor grade, size (< 20 mm or ≥ 20 mm), and LN status (positive or negative). Adjustment was made for age at diagnosis (< 50 or ≥ 50 years). Tumor grade was classified as low (grade 1–2) or high (grade 3).
To derive a novel gene expression signature of HER2 activity that accounted for the effect of potentially targetable ERBB2mut in ERBB2 non-amplified tumors, we applied a weighted average difference (WAD) method to gene expression data in cases from the METABRIC 2012 (N = 1980) and TCGA 2015 (N = 817) cohorts [25, 28, 29]. Gene expression in ERBB2mut cases (N = 38, selected by ERBB2 non-amplified status and patient age > 50) was compared with the same order of magnitude of ERBB2 wild-type cases (N = 79, selected by ERBB2 non-amplified status, grade > 1, stage > I, patient age > 50). This was repeated for oncERBB2mut cases (N = 23) using the same comparator and selection criteria. To incorporate the effect of HER2 activity via ERBB2 amplification, the overlap of differentially expressed genes DEGs shared by both comparisons (ERBB2mut and oncERBB2mut vs. ERBB2 wild-type) with DEGs from a further comparison of ERBB2 amplified (N = 247) vs. non-amplified (N = 1733) cases in METABRIC was calculated. Finally, to incorporate the downstream phenotype (HER2 status), the overlap of this list with DEGs from a comparison of clinical HER2+ vs. HER2− cases in TCGA was calculated. In contrast to ERBB2mut cases, matching was not performed for ERBB2 amplified or HER2+ cases because numbers were higher and within an order of magnitude across groups, such that similar variation in gene expression could reasonably be assumed.
Multiple gene expression signatures of HER2 activity have been derived using cell line models and patient tumors [30–33]. We compared our novel gene signature with the HER2 activity signature established by Desmedt et al [31] with respect to its ability to detect potentially targetable ERBB2mut cases in our ILC/IDC dataset. This was achieved by multivariate regression modeling of response to neratinib for each gene signature using breast cancer cell line pharmacogenomic data from the BROAD Institute, accessed online via the CellMinerCDB portal [34, 35]. The Pearson coefficient for each significantly correlated signature gene was used to calculate normalized signature scores for each METABRIC case in the current dataset. To validate the prognostic effect of ERBB2 in ILC, cases were then stratified by gene signature score (upper vs. lower quartiles) and the signatures compared by histological subtype using a Cox regression model of a 10-year OS.
Results
Clinicopathological landscape of the combined cohort
All cases of primary (stage I–III) ER+ and HER2− ILC and IDC from METABRIC, TCGA, and MSK-IMPACT cohorts were selected (total N = 1580, see Table 1). Baseline clinicopathological characteristics by individual cohort are summarized in Table 1.
Table 1.
Baseline clinicopathological characteristics of the combined cohort
| METABRIC (N = 702) | TCGA (N = 330) | MSK-IMPACT (N = 548) | Total (N = 1580) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| N | % | N | % | N | % | N | % | ||
| Histology* | ILC | 76 | 10.8 | 100 | 30.3 | 103 | 18.8 | 279 | 17.7 |
| IDC | 626 | 89.2 | 230 | 69.7 | 445 | 81.2 | 1301 | 82.3 | |
| Age | < 50 years | 125 | 17.8 | 87 | 26.4 | 190 | 34.7 | 402 | 25.4 |
| ≥ 50 years | 577 | 82.2 | 243 | 73.6 | 358 | 65.3 | 1178 | 74.6 | |
| Menopause | Pre- | 125 | 17.8 | 90 | 27.2 | 234 | 42.7 | 449 | 28.5 |
| Post- | 577 | 82.2 | 219 | 66.4 | 309 | 56.4 | 1105 | 69.9 | |
| unknown | 0 | 0 | 21 | 6.4 | 5 | 0.9 | 26 | 1.6 | |
| Stage* | I | 238 | 33.9 | 67 | 49.3 | 270 | 49.3 | 575 | 36.4 |
| II | 418 | 59.5 | 188 | 34.3 | 188 | 34.3 | 794 | 50.3 | |
| III | 46 | 6.6 | 75 | 16.4 | 90 | 16.4 | 211 | 13.4 | |
| Tumor size | < 20 mm | 215 | 30.6 | 96 | 29.1 | 330 | 60.2 | 641 | 40.6 |
| ≥ 20 mm | 487 | 69.4 | 234 | 70.9 | 218 | 39.8 | 939 | 59.4 | |
| Tumor grade | 1 | 61 | 8.7 | 49 | 14.8 | 51 | 9.3 | 161 | 10.2 |
| 2 | 344 | 49.0 | 190 | 57.6 | 191 | 34.9 | 725 | 45.9 | |
| 3 | 268 | 38.2 | 81 | 24.5 | 288 | 52.6 | 637 | 40.3 | |
| Unknown | 29 | 4.1 | 10 | 3.0 | 18 | 3.3 | 57 | 3.6 | |
| Follow-up | < 5 years | 137 | 19.5 | 261 | 82.8 | 454 | 82.8 | 852 | 53.9 |
| 5–10 years | 195 | 27.8 | 61 | 11.7 | 64 | 11.7 | 320 | 20.3 | |
| ≥ 10 years | 370 | 52.7 | 8 | 5.5 | 30 | 5.5 | 408 | 25.8 | |
| Status | Alive** | 310 | 44.2 | 299 | 90.1 | 494 | 90.1 | 1103 | 69.8 |
| Deceased | 392 | 55.8 | 31 | 9.9 | 54 | 9.9 | 477 | 20.3 | |
*Cases of ILC/IDC histology, stage I–III, ER+ and HER2− status with clinical outcome and mutational data were selected via CBioportal
**At last follow-up
To detect prognostic effects of low-frequency mutations and add to the existing body of literature on ILC-specific mutational drivers, a combined cohort of three public datasets was collated. We first evaluated the implications of combining cases of ILC and IDC from potentially disparate datasets.
In the combined cohort, long-term follow-up of patients is dominated by the largest dataset (METABRIC, N = 702: mean follow-up 133 months) while early events are enriched by the two smaller datasets (TCGA and MSK, N = 878: mean follow-up 33 months) (see Supplemental Figure S1(A) in Additional file 1 for KM plot). Compared to the smaller datasets with shorter follow-up (TCGA and MSK), patients in METABRIC were more likely to be over 50 years of age and have T1 tumors (< 20 mm diameter), but no significant difference in grade or LN status was found (see Table 2). As the principle skew in the combined dataset is towards longer follow-up in METABRIC cases, OS analysis was limited to 10 years—thus providing a clinically meaningful endpoint for all patients, irrespective of age. Comparison of METABRIC with combined TCGA and MSK-IMPACT cohorts revealed no significant difference in a 10-year OS (logrank p = 0.225, see Supplemental Figure S1(B) in Additional file 1).
Table 2.
Comparison of long vs. short follow-up cohorts: METABRIC (largest dataset, long follow-up) vs. TCGA and MSK (combined smaller datasets, short follow-up). Significant difference was found with respect to age and tumor size, but not tumor grade or LN status. Table excludes “unknown” cases for each variable
| METABRIC (N = 702) | TCGA AND MSK (N = 878) | χ2 test | ||||
|---|---|---|---|---|---|---|
| N | % | N | % | |||
| Age | < 50 years | 125 | 17.8 | 277 | 31.5 | p < 0.00001 |
| ≥ 50 years | 577 | 82.2 | 601 | 68.5 | ||
| Tumor size | < 20 mm | 215 | 30.6 | 426 | 48.5 | p < 0.00001 |
| ≥ 20 mm | 487 | 69.4 | 452 | 51.5 | ||
| Tumor grade | 1–2 | 405 | 60.2 | 481 | 56.6 | p = 0.158 |
| 3 | 268 | 39.8 | 369 | 43.4 | ||
| LN status | Negative | 370 | 53.9 | 489 | 55.8 | p = 0.457 |
| Positive | 316 | 46.1 | 387 | 44.2 | ||
ERBB2 mutations are enriched in primary ILC and cluster in the HER2 tyrosine kinase domain
We next assessed the prevalence of ERBB2/3mut in our dataset of ILC and IDC (N = 1580). Overall prevalence of ERBB2mut was 2.2% (N = 34). ERBB2mut was enriched in ILC, with prevalence of 5.7% (N = 16) vs. 1.4% in IDC (N = 18) (p < 0.0001). In contrast, prevalence of ERBB3mut was lower (1.6% overall, N = 26), and there was no enrichment of ERBB3mut in ILC vs. IDC (1.1%, N = 3 vs. 1.8%, N = 23; p = 0.604). Due to the small number of cases in ILC, further analysis of ERBB3mut was not performed.
In ILC, ERBB2mut clustered in the tyrosine kinase domain of HER2 (15 out of 16 cases; 93.8%). Of these, the majority have been characterized as oncogenic and potentially targetable using neratinib (oncERBB2mut, 11 out of 15 kinase domain mutations in ILC; 73.3%). In IDC, all kinase domain mutations were known oncogenic (N = 8) and non-characterized mutations were distributed evenly across protein domains (see Fig. 1). The most frequently occurring ERBB3mut coded for E928G, which lies in the tyrosine kinase domain of HER3 (N = 7 out of 26; 27%).
Fig. 1.

Rate of ERBB2 and ERBB3 mutations and their spatial distribution on HER2/3 in ILC and IDC. ERBB2mut were found to be a enriched in ILC (yellow bar) vs. IDC (blue bar) and b clustered in the tyrosine kinase domain of HER2; c ERBB3mut occurred at lower frequency, with the high-frequency outlier in IDC coding for known oncogenic HER3 kinase domain alteration E928G (N = 6). Y-axes show the number of cases harboring at least one ERBB2/3 mutation at a specific amino acid (aa) of HER2/3, shown along the x-axes. Yellow-filled circles indicate oncERBB2mut. Extracellular domains of HER2/3: Receptor L, Furin-like and Growth Factor Receptor IV; intracellular: tyrosine protein kinase. *p < 0.001; n/s = not significant
ERBB2 mutation is an adverse prognostic indicator of survival for patients with primary ILC
Using our meta-cohort of 1580 cases, we next assessed ERBB2mut as a prognostic marker of OS in ILC (N = 279) and IDC (N = 1301). Median duration of patient follow-up was 50 months (range 0–351 months). In patients with ILC, median OS was significantly shorter if tumors were ERBB2mut positive (inclusive of oncogenic and uncharacterized mutations) vs. ERBB2mut negative (66 vs. 211 months, p = 0.0001). In contrast, there was no significant difference in OS for ERBB2mut cases of IDC (159 vs. 166 months, p = 0.733) (see Fig. 2).
Fig. 2.

OS by ERBB2 mutational status in ILC (left) and IDC (right). Gray line indicates ERBB2 wild-type cases; blue line indicates cases with at least one ERBB2mut
ERBB3mut status was not a significant prognostic indicator of OS in cases of IDC (HR = 1.46, 95% CI 0.65–3.28; p = 0.359). There were no events (patient deaths) at 10 years of follow-up in the three cases of ERBB3mut ILC.
Targetable ERBB2 mutation status is an independent adverse prognostic marker of 10-year overall survival in ER+, ERBB2 non-amplified ILC
To test the effect of therapeutically actionable ERBB2 mutations on a clinically relevant endpoint, we stratified 10-year OS by oncERBB2mut status. In ER+, ERBB2 non-amplified ILC, oncERBB2mut status was prognostic of 10-year OS independently of LN status, tumor grade, and size (HR 3.65, 95% CI 1.21–11.00; p = 0.021, see Fig. 3). The independent prognostic value of oncERBBmut status on 10-year OS was retained after further adjusting for age at diagnosis (HR 3.19, 95% CI 1.06–9.62; p = 0.039).
Fig. 3.
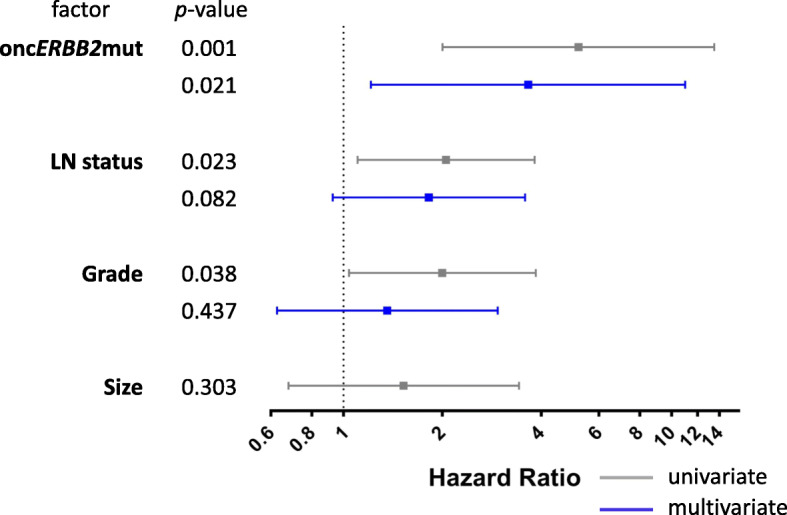
Univariate and multivariate analyses of 10-year OS in N = 279 cases of ER+, ERBB2 non-amplified ILC. Gray square dot indicates hazard ratio (HR) in univariate analysis and the gray bar indicates the 95% confidence interval (CI). Significant prognostic variables in univariate analyses (where 95% CI does not span HR = 1) are included in multivariate analysis, shown in blue. For each variable, cases with unknown values are excluded from the analysis
Unselected ERBB2mut status (including oncogenic and uncharacterized mutations) was also adversely prognostic of 10-year OS in univariate analysis (HR 3.66, 95% CI 1.54–8.73; p = 0.003), but in contrast to characterized oncERBB2mut, this prognostic effect was not independent of LN stage, tumor grade, and/or size. The independent prognostic value of oncERBB2mut was further verified at 5 years of follow-up (HR 3.668, 95% CI 1.096–12.275; p = 0.035).
A novel gene signature of targetable HER2 activity using ERBB2mut status
Since existing gene signatures of HER activity derive from HER2 status (by IHC and/or ISH), we generated a novel gene signature incorporating DEGs in ERBB2mut and oncERBB2mut cases, as described in the “Patients and methods” section and outlined in Fig. 4. Our aim was to establish a novel signature that reflects HER2 pathway activity more completely than existing signatures, whether induced by ERBB2 mutations or amplifications, and to be able to apply it in a wider range of patients. A list of up and downregulated ERBB2 “mutant” DEGs (N = 20) was generated by combining the overlap between DEGs for METABRIC ERBB2 amplified vs. non-amplified, ERBB2 mutated vs. wild-type (ERBB2mut and oncERBB2mut separately), and TCGA HER2+ vs. HER2− (Fig. 4a, and see Additional file 2 for Supplemental Tables S1–3 with all gene lists).
Fig. 4.

A novel gene signature of HER2 activity incorporating ERBB2mut, ERBB2 amplification, and clinical HER2 status. a Generation of a 20-gene signature of HER2 activity. The upper Venn diagrams show the overlap between the top 500 DEGs by WAD score for METABRIC amplified vs. non-amplified, ERBB2 mutant vs. wild-type, and TCGA HER2+ vs. HER2−. Upregulated DEGs are shaded blue and down-regulated DEGs yellow. ERBB2mut and oncERBB2mut vs. wild-type are analyzed separately, and the overlap combined in the lower Venn diagrams. b Comparison with an established 24-gene signature of HER pathway activation [31], using a gene signature (genesig) score derived from multivariate analysis of response to neratinib in breast cancer cell lines. Cases with ERBB2mut (gray lines) and oncERBB2mut (blue lines) clustered in the upper quartile of normalized genesig scores for the novel signature but not the established signature. c 10-year OS analysis of cases in the current study stratified by histological subtype and novel genesig score (upper vs. lower quartiles) indicates that ERBB2mut-associated DEGs are prognostic in ILC but not IDC. GE, geneset enrichment
As shown in Fig. 4b, ERBB2mut and oncERBB2mut clustered in the upper quartile of the novel gene signature score in cases of ER+, HER2− ILC/IDC from METABRIC. In contrast, ERBB2mut and oncERBB2mut cases did not cluster when scored by the established gene signature of HER2 pathway activation but were evenly distributed across the cohort. Both the novel and established HER2 pathway gene signatures were predictive of response to neratinib in multivariate analysis using a pharmacogenomic breast cancer cell line model (Pearson R = 0.9, p < 1e−14 for both; see Supplemental Figure S2 in Additional file 3 for the original plots from the CellMinerCDB online portal). The regression model used incorporates drug response and gene expression data from N = 36 breast cancer cell lines [35, 36]. Our findings using this model verify that the novel signature derived from DEGs in ERBB2mut vs. wild-type cases incorporates oncogenic HER2 activity due to ERBB2mut. This suggests that a gene signature may have value in predicting targetable ERBB2 alterations including ERBB2mut in HER2− breast cancer. Finally, Fig. 4c demonstrates that the novel gene signature score (stratified into upper vs. lower quartiles) was adversely associated with 10-year OS (HR = 2.3, 95% CI 1.04–5.05; p = 0.040), thus providing mRNA level validation of ERBB2mut as an adverse prognostic marker.
Using the same dataset (CellMinerCDB), it was not possible to associate ERBB2mut status with neratinib response as only one breast cancer cell line (MDA-MB-468) was ERBB2mut positive. Response to neratinib for MDA-MB-468 was in the upper tertile of breast cancer cell lines (data in the public domain via CellMinerCDB online portal).
Discussion
In this study, we mined clinical and NGS data from the largest clinical cohorts with data in the public domain to test whether there was an ILC-specific association of ERBB2 mutations with OS. In our meta-cohort of ER+, ERBB2 non-amplified cases of ILC and IDC, we found that ERBB2 mutations are enriched ILC, cluster in the functional kinase domain of HER2, and robustly associate with adverse clinical outcomes—independently of known prognostic clinicopathological features including LN status and tumor grade. In contrast, there was no ILC-specific enrichment of ERBB3mut.
Compared to the largest previous study with data on ERBB2 mutational status in primary ILC (N = 371) [24], the current study found higher frequency of ERBB2mut (5.7 vs. 4.3%) but lower frequency of ERBB3mut (1.1 vs. 3.5%). Hazard proportionality in the study by Desmedt et al 2016 suggested that ERBB2 mutational status had a time-dependent effect associated with short-term risk of breast cancer relapse [24]. The current study adds data on the prognostic effect of specific ERBB2 mutations that were previously shown in cell line, in vitro and xenograft studies to respond to existing clinical compounds such as trastuzumab or neratinib [13]. We demonstrated that the status of these therapeutically targetable ERBB2 mutations is an independent adverse prognostic marker of overall survival in a large cohort of patients with ILC.
Findings from the current study imply that targeted sequencing of ILC tumors for ERBB2 mutations may be an actionable and viable strategy to improve outcomes for patients with primary ILC by providing a biomarker for HER2-targeted therapy in the adjuvant setting. Based on worldwide ILC incidence of 250,000 [1–4], 10% rate of ER− and/or HER2− status in ILC, and 5% rate of potentially targetable ERBB2mut in ER+, ERBB2 non-amplified ILC, we conservatively estimate that 11,250 additional ILC patients per year who have ERBB2-mutated tumors may benefit from HER2-targeting therapy.
Existing clinical studies of HER2-targeting therapy are underway in advanced breast cancer. For example, neratinib (alone or in combination with fulvestrant) was tested in phase II study for patients with advanced, ERBB2 non-amplified, ERBB2mut breast cancer (clinical trial registration number NCT01670877). Of the 16 patients who had evaluable tumor responses, N = 2 had partial response by RECIST criteria (13%). Out of 14 patients with tumors harboring oncERBB2mut, N = 5 (36%) derived clinical benefit, defined as stable disease or partial/complete response by RECIST criteria [37]. Given that the patients in this trial had received multiple prior lines of treatment, it is conceivable that patients with ERBB2 non-amplified primary tumors harboring oncERBB2mut will also derive benefit from (neo) adjuvant HER2-targeted therapy such as neratinib.
In the current cohort of primary ER+, HER2− ILC and IDC, ERBB2 mutations in ILC tumors clustered almost exclusively in the active HER2 tyrosine kinase domain (N = 15 of 16). Our retrospective clinical outcome data indicates an ILC-specific adverse survival association for patients with ERBB2 non-amplified tumors harboring potentially targetable ERBB2 mutations. This primary finding was validated by existing mRNA data via a novel gene signature of HER2 activity, which linked ERBB2mut with response to neratinib. Subject to independent external validation, a gene signature of HER2 activity could have clinical application as a biomarker of response to HER2-targeted small molecules such as neratinib and may be more cost-effective than targeted NGS in the detection of ERBB2mut in primary ILC.
Taken together, these new findings suggest that focusing early phase studies of HER2-inhibition (e.g., with neratinib) in patients with ERBB2 non-amplified, ERBB2mut primary ILC may be an effective strategy to demonstrate feasibility and clinical benefit in the (neo) adjuvant setting.
Limitations of the current study include cohort size, quality of clinical outcome data, and bias in long-term follow-up towards cases from the largest dataset (METABRIC). Statistical power is limited by the small number of patient deaths in cases of ILC and IDC with ERBB2mut (N = 7 and 8, respectively). However, the clear deviation of the KM plot of OS in ILC stratified by ERBB2mut (Fig. 2) is reflected in a low type I error of one in 10,000 (p = 0.0001). In contrast, there was no ILC-specific enrichment of ERBB3mut in our database. However, in IDC there was enrichment of the known oncogenic HER3 kinase domain mutation, E928G. To demonstrate the prognostic effect of mutations affecting HER3, larger datasets will be required.
The effect of incomplete clinical outcome data on the primary endpoint (OS) is difficult to quantify but is accounted for statistically in Cox regression modeling by censoring patients at last follow-up. Patients in METABRIC presented at older age, which is associated with more indolent, less biologically aggressive breast cancer [38]. This implies an underestimation of the adverse impact of ERBB2 mutations on 10-year survival outcomes in the current study. Since the METABRIC dataset did not have complete HER2 IHC or reverse-phase protein array data, it was not possible to associate activating ERBB2mut directly with downstream HER2 protein expression. Instead, we generated a gene signature incorporating clinical HER2 status to validate our findings.
Conclusions
Targetable kinase domain ERBB2 mutations are enriched in primary ILC and their detection by targeted sequencing or validated gene signature surrogate may provide an actionable strategy to improve patient outcomes. Biomarker-led clinical trials of adjuvant HER2-targeted therapy to treat breast tumors with activating ERBB2 mutations are warranted in HER2− primary ILC.
Supplementary information
Additional file 1: Supplemental Figure S1. (A) 10-year OS KM plot stratified by individual study cohort: blue – METABRIC, solid gray – TCGA, dotted gray – MSK-IMPACT; (B) 10-year OS KM plot stratified by study category: blue – long follow-up (METABRIC), gray – short follow-up (TCGA and MSK-IMPACT combined); Logrank p = 0.225.
Additional file 2: Supplemental Tables. (S1) list of genes in the novel (N = 20) and established (Desmedt et al [31], N = 24) gene signatures of HER2 activity; (S2 and S3) top 500 DEGs by WAD score stratified into genes that are up-regulated (S2) and down-regulated (S3) for METABRIC ERBB2 amplified vs. non-amplified (ERBB2amp), METABRIC ERBB2mut vs. wild-type (ERBB2mut) and oncERBB2mut vs. wild-type (oncERBB2mut), and TCGA HER2+ vs. HER2- (TCGA HER2+).
Additional file 3: Supplemental Figure S2. Correlation between the observed response of breast cancer cell lines (N = 36) to neratinib and the response predicted by expression of (A) novel (N = 20) and (B) established (Desmedt et al [31], N = 24) gene panels. Plots were generated using CellMinerCDB online portal and cell line data from the BROAD Institute [34, 35].
Acknowledgements
Not applicable
Abbreviations
- CI
Confidence interval
- DEG
Differentially expressed gene
- EGF
Epidermal growth factor
- ER
Estrogen receptor alpha
- ERBB2/3mut
ERBB2/3 mutant
- HER2/3
Human epidermal growth factor receptor 2/3
- HR
Hazard ratio
- IDC
Invasive ductal carcinoma of the breast
- IHC
Immunohistochemistry
- ILC
Invasive lobular carcinoma of the breast
- ISH
In situ hybridization
- KM
Kaplan-Meier
- LN
Lymph node
- NGS
Next-generation sequencing
- oncERBB2mut
Oncogenic ERBB2 mutant
- OS
Overall survival
- RECIST
Response evaluation criteria in solid tumors
- WAD
Weighted average difference
Authors’ contributions
SK, DMH, WZ, SO, and SJJ conceived, designed, and supervised the study. SK, MA, CM, KB, NPM, and SJJ analyzed and interpreted the data. SK, WZ, SO, and SJJ prepared the manuscript. ARG, IOE, and EAR had supervisory and administrative roles. SJ, TF, and KS had consultative roles in study design. All authors were involved in manuscript preparation and reviewing for submission, gave their final approval, and agreed to be accountable for all aspects of the work.
Funding
The corresponding author (SJJ) is funded by the Wellcome Trust (Royal Academy of Medical Sciences grant number AAM 127669) and the National Institute for Health Research UK. Dr. Oesterreich’s work on ILC is supported by the Breast Cancer Research Foundation and a Komen Scholar awards (SAC160073).
Availability of data and materials
The datasets generated and/or analyzed during the current study are available via the CBioportal online portal (http://www.cbioportal.org) and in the Genome-Phenome Archives EGAS00000000098 and phs000178 (https://ega-archive.org/studies/EGAS00000000098 and https://ega-archive.org/studies/phs000178).
Ethics approval and consent to participate
Ethics approvals and consent for each cohort (METABRIC, TCGA, and MSK-IMPACT) was obtained in the original studies.
Consent for publication
Not applicable
Competing interests
Since the first submission of the article, the corresponding author (SJJ) has moved employment to AstraZeneca, with no competing interests to declare. All other authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s13058-020-01324-4.
References
- 1.Martinez V, Azzopardi JG. Invasive lobular carcinoma of the breast: incidence and variants. Histopathology. 1979;3(6):467–488. doi: 10.1111/j.1365-2559.1979.tb03029.x. [DOI] [PubMed] [Google Scholar]
- 2.Li CI, Anderson BO, Daling JR, Moe RE. Trends in incidence rates of invasive lobular and ductal breast carcinoma. JAMA. 2003;289(11):1421–1424. doi: 10.1001/jama.289.11.1421. [DOI] [PubMed] [Google Scholar]
- 3.Li CI, Daling JR. Changes in breast cancer incidence rates in the United States by histologic subtype and race/ethnicity, 1995 to 2004. Cancer Epidemiol Biomark Prev. 2007;16(12):2773–2780. doi: 10.1158/1055-9965.EPI-07-0546. [DOI] [PubMed] [Google Scholar]
- 4.Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Pineros M, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019;144(8):1941–1953. doi: 10.1002/ijc.31937. [DOI] [PubMed] [Google Scholar]
- 5.Pestalozzi BC, Zahrieh D, Mallon E, Gusterson BA, Price KN, Gelber RD, et al. Distinct clinical and prognostic features of infiltrating lobular carcinoma of the breast: combined results of 15 International Breast Cancer Study Group clinical trials. J Clin Oncol. 2008;26(18):3006–3014. doi: 10.1200/JCO.2007.14.9336. [DOI] [PubMed] [Google Scholar]
- 6.Rakha EA, El-Sayed ME, Powe DG, Green AR, Habashy H, Grainge MJ, et al. Invasive lobular carcinoma of the breast: response to hormonal therapy and outcomes. Eur J Cancer. 2008;44(1):73–83. doi: 10.1016/j.ejca.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 7.Metzger Filho O, Giobbie-Hurder A, Mallon E, Gusterson B, Viale G, Winer EP, et al. Relative effectiveness of letrozole compared with tamoxifen for patients with lobular carcinoma in the BIG 1-98 trial. J Clin Oncol. 2015;33(25):2772–2779. doi: 10.1200/JCO.2015.60.8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sikora MJ, Cooper KL, Bahreini A, Luthra S, Wang G, Chandran UR, et al. Invasive lobular carcinoma cell lines are characterized by unique estrogen-mediated gene expression patterns and altered tamoxifen response. Cancer Res. 2014;74(5):1463–1474. doi: 10.1158/0008-5472.CAN-13-2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.NICE. Early and locally advanced breast cancer: diagnosis and management. Online: National Institute for Health and Care Excellence; 2018 [NICE Pathway]. Available from: https://www.nice.org.uk/guidance/ng101. [PubMed]
- 10.Cardoso F, Kyriakides S, Ohno S, Penault-Llorca F, Poortmans P, Rubio IT, et al. Early breast cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-updagger. Ann Oncol. 2019;30(8):1194–1220. doi: 10.1093/annonc/mdz173. [DOI] [PubMed] [Google Scholar]
- 11.Wolff AC, Hammond MEH, Allison KH, Harvey BE, Mangu PB, Bartlett JMS, et al. Human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. J Clin Oncol. 2018;36(20):2105–2122. doi: 10.1200/JCO.2018.77.8738. [DOI] [PubMed] [Google Scholar]
- 12.Denduluri N, Chavez-MacGregor M, Telli ML, Eisen A, Graff SL, Hassett MJ, et al. Selection of optimal adjuvant chemotherapy and targeted therapy for early breast Cancer: ASCO clinical practice guideline focused update. J Clin Oncol. 2018;36(23):2433–2443. doi: 10.1200/JCO.2018.78.8604. [DOI] [PubMed] [Google Scholar]
- 13.Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013;3(2):224–237. doi: 10.1158/2159-8290.CD-12-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanker A, Koch J, Ye D, Sliwoski G, Sheehan J, Kinch L, et al. Abstract PD3-05: Co-occurring gain-of-function mutations in HER2 and HER3 cooperate to enhance HER2/HER3 binding, HER-dependent signaling, and breast cancer growth. Cancer Res. 2019;79(4 Supplement):PD3–05-PD3. [Google Scholar]
- 15.Eroglu Z, Tagawa T, Somlo G. Human epidermal growth factor receptor family-targeted therapies in the treatment of HER2-overexpressing breast cancer. Oncologist. 2014;19(2):135–150. doi: 10.1634/theoncologist.2013-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009;21(2):177–184. doi: 10.1016/j.ceb.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 17.Junttila TT, Akita RW, Parsons K, Fields C, Lewis Phillips GD, Friedman LS, et al. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell. 2009;15(5):429–440. doi: 10.1016/j.ccr.2009.03.020. [DOI] [PubMed] [Google Scholar]
- 18.Mishra R, Patel H, Alanazi S, Yuan L, Garrett JT. HER3 signaling and targeted therapy in cancer. Oncol Rev. 2018;12(1):355. doi: 10.4081/oncol.2018.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jaiswal BS, Kljavin NM, Stawiski EW, Chan E, Parikh C, Durinck S, et al. Oncogenic ERBB3 mutations in human cancers. Cancer Cell. 2013;23(5):603–617. doi: 10.1016/j.ccr.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 20.Ping Z, Siegal GP, Harada S, Eltoum IE, Youssef M, Shen T, et al. ERBB2 mutation is associated with a worse prognosis in patients with CDH1 altered invasive lobular cancer of the breast. Oncotarget. 2016;7(49):80655–80663. doi: 10.18632/oncotarget.13019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ross JS, Wang K, Sheehan CE, Boguniewicz AB, Otto G, Downing SR, et al. Relapsed classic E-cadherin (CDH1)-mutated invasive lobular breast cancer shows a high frequency of HER2 (ERBB2) gene mutations. Clin Cancer Res. 2013;19(10):2668–2676. doi: 10.1158/1078-0432.CCR-13-0295. [DOI] [PubMed] [Google Scholar]
- 22.Griffith OL, Spies NC, Anurag M, Griffith M, Luo J, Tu D, et al. The prognostic effects of somatic mutations in ER-positive breast cancer. Nat Commun. 2018;9(1):3476. doi: 10.1038/s41467-018-05914-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ross JS, Gay LM, Wang K, Ali SM, Chumsri S, Elvin JA, et al. Nonamplification ERBB2 genomic alterations in 5605 cases of recurrent and metastatic breast cancer: an emerging opportunity for anti-HER2 targeted therapies. Cancer. 2016;122(17):2654–2662. doi: 10.1002/cncr.30102. [DOI] [PubMed] [Google Scholar]
- 24.Desmedt C, Zoppoli G, Gundem G, Pruneri G, Larsimont D, Fornili M, et al. Genomic characterization of primary invasive lobular breast cancer. J Clin Oncol. 2016;34(16):1872–1881. doi: 10.1200/JCO.2015.64.0334. [DOI] [PubMed] [Google Scholar]
- 25.Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell. 2015;163(2):506–519. doi: 10.1016/j.cell.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016;7:11479. doi: 10.1038/ncomms11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell. 2018;34(3):427–438. doi: 10.1016/j.ccell.2018.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kadota K, Nakai Y, Shimizu K. A weighted average difference method for detecting differentially expressed genes from microarray data. Algorithms Mol Biol. 2008;3:8. doi: 10.1186/1748-7188-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferrari A, Vincent-Salomon A, Pivot X, Sertier AS, Thomas E, Tonon L, et al. A whole-genome sequence and transcriptome perspective on HER2-positive breast cancers. Nat Commun. 2016;7:12222. doi: 10.1038/ncomms12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Desmedt C, Haibe-Kains B, Wirapati P, Buyse M, Larsimont D, Bontempi G, et al. Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin Cancer Res. 2008;14(16):5158–5165. doi: 10.1158/1078-0432.CCR-07-4756. [DOI] [PubMed] [Google Scholar]
- 32.Sareyeldin RM, Gupta I, Al-Hashimi I, Al-Thawadi HA, Al Farsi HF, Vranic S, et al. Gene expression and miRNAs profiling: function and regulation in human epidermal growth factor receptor 2 (HER2)-positive breast cancer. Cancers (Basel). 2019;11(5):646. [DOI] [PMC free article] [PubMed]
- 33.Creighton CJ, Hilger AM, Murthy S, Rae JM, Chinnaiyan AM, El-Ashry D. Activation of mitogen-activated protein kinase in estrogen receptor alpha-positive breast cancer cells in vitro induces an in vivo molecular phenotype of estrogen receptor alpha-negative human breast tumors. Cancer Res. 2006;66(7):3903–3911. doi: 10.1158/0008-5472.CAN-05-4363. [DOI] [PubMed] [Google Scholar]
- 34.Rees MG, Seashore-Ludlow B, Cheah JH, Adams DJ, Price EV, Gill S, et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat Chem Biol. 2016;12(2):109–116. doi: 10.1038/nchembio.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rajapakse VN, Luna A, Yamade M, Loman L, Varma S, Sunshine M, et al. CellMinerCDB for integrative cross-database genomics and pharmacogenomics analyses of cancer cell lines. iScience. 2018;10:247–264. doi: 10.1016/j.isci.2018.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Basu A, Bodycombe NE, Cheah JH, Price EV, Liu K, Schaefer GI, et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell. 2013;154(5):1151–1161. doi: 10.1016/j.cell.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma CX, Bose R, Gao F, Freedman RA, Pegram MD, Blackwell K, et al. Phase II trial of neratinib for HER2 mutated, non-amplified metastatic breast cancer (HER2(mut) MBC). J Clin Oncol. 2016;34(15 suppl):516.
- 38.Syed BM, Green AR, Paish EC, Soria D, Garibaldi J, Morgan L, et al. Biology of primary breast cancer in older women treated by surgery: with correlation with long-term clinical outcome and comparison with their younger counterparts. Br J Cancer. 2013;108(5):1042–1051. doi: 10.1038/bjc.2012.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Supplemental Figure S1. (A) 10-year OS KM plot stratified by individual study cohort: blue – METABRIC, solid gray – TCGA, dotted gray – MSK-IMPACT; (B) 10-year OS KM plot stratified by study category: blue – long follow-up (METABRIC), gray – short follow-up (TCGA and MSK-IMPACT combined); Logrank p = 0.225.
Additional file 2: Supplemental Tables. (S1) list of genes in the novel (N = 20) and established (Desmedt et al [31], N = 24) gene signatures of HER2 activity; (S2 and S3) top 500 DEGs by WAD score stratified into genes that are up-regulated (S2) and down-regulated (S3) for METABRIC ERBB2 amplified vs. non-amplified (ERBB2amp), METABRIC ERBB2mut vs. wild-type (ERBB2mut) and oncERBB2mut vs. wild-type (oncERBB2mut), and TCGA HER2+ vs. HER2- (TCGA HER2+).
Additional file 3: Supplemental Figure S2. Correlation between the observed response of breast cancer cell lines (N = 36) to neratinib and the response predicted by expression of (A) novel (N = 20) and (B) established (Desmedt et al [31], N = 24) gene panels. Plots were generated using CellMinerCDB online portal and cell line data from the BROAD Institute [34, 35].
Data Availability Statement
The datasets generated and/or analyzed during the current study are available via the CBioportal online portal (http://www.cbioportal.org) and in the Genome-Phenome Archives EGAS00000000098 and phs000178 (https://ega-archive.org/studies/EGAS00000000098 and https://ega-archive.org/studies/phs000178).