

Abstract
T-type calcium channels, in the central nervous system, are involved in the pathogenesis of many neurodegenerative diseases, including TAF1 intellectual disability syndrome (TAF1 ID syndrome). Here, we evaluated the efficacy of a novel T-type Ca2+ channel enhancer, SAK3 (ethyl 8’-methyl-2’, 4-dioxo-2-(piperidin-1-yl)-2’H-spiro [cyclopentane-1, 3’-imidazo [1, 2-a] pyridine]-2-ene-3-carboxylate) in an animal model of TAF1 ID syndrome. At post-natal day 3, rat pups were subjected to intracerebroventricular (ICV) injection of either gRNA-control or gRNA-TAF1 CRISPR/Cas9 viruses. At post-natal day 21 animals were given SAK3 (0.25 mg/kg, p.o.) or vehicle up to post-natal day 35 (i.e. 14 days). Rats were subjected to behavioral, morphological, electrophysiological, and molecular studies. Oral administration of SAK3 (0.25 mg/kg, p.o.) significantly rescued the behavior abnormalities in beam walking test and open field test caused by TAF1 gene editing. We observed an increase in calbindin-positive Purkinje cells and GFAP-positive astrocytes as well as a decrease in IBA1-positive microglia cells in SAK3-treated animals. In addition, SAK3 protected the Purkinje and granule cells from apoptosis induced by TAF-1 gene editing. SAK3 also restored the excitatory post synaptic current (sEPSCs) in TAF1 edited Purkinje cells. Finally, SAK3 normalized the BDNF/AKT signaling axis in TAF1 edited animals. Altogether, these observations suggest that SAK3 could be a novel therapeutic agent for TAF1 ID syndrome.
Keywords: TAF1, Intellectual disability syndrome, SAK3, Cav3.1, Cerebellum
Introduction
The transcription factor II D (TFIID) complex is an assembly of the TATA-box binding protein (TBP) and of 12–14 TBP-associated factors (TAFs), participating in the preinitiation complex that initiates transcription of RNA polymerase II transcription dependent genes (Bieniossek et al. 2013; Goodrich and Tjian 2010; Warfield et al. 2017). TAF1 is the largest TAF unit of the TFIID complex and plays a key role in the preinitiation complex by facilitating binding to promoter regions (Goodrich and Tjian 2010). The TAF1 gene (GRCh37/hg19, chrX: 70586114–70685855, NM_004606.4 in humans) includes 39 exons and yields more than 20 coding and non-coding transcripts expressed in various tissues, including the central nervous system(Aneichyk et al. 2018). We and others linked variants of TAF1 to neurodevelopmental disorders (Hurst 2018a; Gudmundsson et al. 2019; O’Rawe et al. 2015). TAF1 ID syndrome (also known as mental retardation, X-linked, syndromic-33 disease, MRXS33, OMIM: 300966) occurs mainly in males, leading to abnormalities in global developmental (motor, cognitive, and speech), hypotonia, gait abnormalities, and cerebellar hypoplasia (Hurst 2018a; Gudmundsson et al. 2019). Only missense variants have been reported; the lack of hemi- and homozygous loss-of-function variants in the protein coding part of the canonical TAF1 isoform in human population databases suggests that a complete loss of TAF1 may be embryonic-lethal. A non-coding 2.6 kb insertion of a SINE-VNTR-Alu (SVA)-type retrotransposon in intron 32 of TAF1 causes the neurological disorder X-linked dystonia-parkinsonism (XDP; OMIM: 313650). XDP is a progressive neurodegenerative disorder characterized by involuntary movements (dystonia), most often developing in adult life in combination with Parkinsonism (Makino et al. 2007; Bragg et al. 2017; Herzfeld et al. 2013). Decreased TAF1 levels have also been observed in the striatum of Huntington’s disease patients and transgenic model mice (Hernandez et al. 2020). Taken together, these observations suggest that dysregulation of TAF1 function by different mechanisms can lead to disease in the central nervous system.
Exactly how variants in TAF1 give rise to these neurological deficits remains unclear. TAF1 serves as a scaffold for the assembly of the transcription factor TFIID complex. Furthermore, how the many functions of the TFIID complex are linked to the development of TAF1 ID syndrome is unknown. To gain insight into these questions, we created an animal model of TAF1D syndrome using CRISPR/Cas9 gene editing (Janakiraman et al. 2019). We postulated that deletion of TAF1 by CRISPR/Cas9 editing, into the maturing brain during the post-natal days (using a lentiviral vector) might replicate TAF1 ID symptoms. This allowed for TAF1 gene editing to occur before the last steps of cerebellar maturation involving purkinje and cerebellar neuron differentiation and gliogenesis (Aldrin-Kirk et al. 2014). We found that in neonatal rat pups, removal of TAF1 by CRISPR/Cas9 editing, results in defects in neonatal motor functions(Janakiraman et al. 2019). Furthermore, the motor deficits were associated with loss of Purkinje cells (PC) in the cerebellum; the remaining Purkinje cells displaying abnormal firing frequencies (Janakiraman et al. 2019). In addition, the motor defects persisted in TAF1-edited juvenile pups and were associated with morphological abnormalities within the cerebellum and cerebral cortex (Janakiraman et al. 2019). We also showed that TAF1 regulates the expression of the CaV3.1 T-type Ca2+ channel in vitro and in vivo (Hurst 2018a; Janakiraman et al. 2019). In support of this, transcriptomic analysis of taf1 zebrafish mutants revealed a loss of the cacna1g gene (CaV3.1) orthologue (Gudmundsson et al. 2019). The CaV3.1 T-type channel accounts for the reduction in spontaneous excitatory post-synaptic currents in TAF1-edited animals (Janakiraman et al. 2019). Collectively, these observations suggest that impaired Cav3.1 T-type channel activity plays a functional role in the pathogenesis of TAF1 ID syndrome.
T-type calcium channels are low voltage-activated calcium channels that transiently open to evoke tiny Ca2+ currents (reviewed in (Perez-Reyes 2003)). T-type calcium channels play a crucial role in regulating intracellular calcium homeostasis and maintaining cellular function (Assandri et al. 1999; Chemin et al. 2000; Cazade et al. 2017). Cav3.1 T-type channels are abundant at the cerebellar synapse between parallel fibers and Purkinje cells where they contribute to pre-synaptic depolarization (Ly et al. 2013). Recently, a spirodazopyridine derivative, SAK3, was synthesized and demonstrated to be a potent T-Type channel enhancer (Yabuki et al. 2017). SAK3 has been shown to be effective in improving cognition and promoting neurogenesis in animal models of neurodegenerative disease via activation of CaMKII (Wang et al. 2018; Xu et al. 2018). In this context, we investigated the effects of SAK3 on the behavioral and pathophysiological defects associated with TAF-1 gene editing in our rat model of TAF1 ID syndrome. We also evaluated the molecular mechanism underlying SAK3 effects. Our results provide compelling evidence that T-type Ca2+ channel stimulation can have disease modifying effects in TAF-1 edited animal models.
Materials and Methods
Animals
Pathogen-free, normal E18 pregnant Sprague–Dawley rats (Envigo Laboratories) were housed 1 per cage in temperature- (23 ± 3 °C) and light (12-h light/12-h dark cycle; lights on 07:00–19:00)-controlled rooms with standard rodent chow and water available ad libitum. The neonates are designated as post-natal day 0 (PD0) on the day of birth, the litter size range included in current study was 11–14 pups.
After weaning the rat pups were separated from the dams and maintained 4 per cage. Animals were divided into six groups. SAK3 (Catalog No: SML-2039–5MG, Sigma Aldrich) was dissolved in distilled water and orally administered (0.25 mg/kg, p.o) to the animals from PD21 to PD35 (Figure 1A). We chose this dose of SAK3 because it has been shown to be effective in restoring cognitive function in a mouse model of Alzheimer’s disease (Wang et al. 2018). Rats were sacrificed at PD35. Animals were behaviorally assessed before being euthanized for histological and protein expression analysis. All biochemical, electrophysiology and behavior experiments were performed in a blinded fashion. Animal protocols were approved by the Institutional Animal Care and Use Committee of the College of Medicine at the University of Arizona and conducted in accordance with the Guide for Care and Use of Laboratory Animals published by the National Institutes of Health.
Figure 1. Experimental design for these studies and behavioral assessment of motor functions.

(A) Experimental design for electrophysiology, behavioral, histopathological and molecular studies. The TAF1-edited animals showed behavior deficits compared to naïve and gRNA-control group animals. The TAF1-edited showed increased beam crossing time (B) and increased foot slips errors (C) in the beam walking test. In addition, grooming was increased in the TAF1-edited rats compared to the other groups (D). SAK3 administration to the TAF1 edited animals showed improved the TAF1 edited behavioral abnormalities. Data are shown as mean ± S.E.M., n=24 per experimental condition. *p < 0.05 versus; naïve, #p < 0.05 versus gRNA-control (ANOVA followed by Tukey’s test). The experiments were conducted in an investigator blinded manner.
CRISPR/Cas9-mediated targeting of TAF1 gene
Our strategy to truncate TAF1 focused on targeting exon 1 of the TAF1 gene using a guide RNA (gRNA) has been described previously (Moutal et al. 2017; Moutal, Cai, et al. 2018; Moutal, Sun, et al. 2018; Sandweiss et al. 2018). We targeted this exon to ensure total removal of the TAF1 protein. Using this approach, we expect minimal to none off-target activity of the Cas9 enzyme as we and others verified before (Moutal et al. 2017; Ma et al. 2017). The gRNA sequence (GTGTCTGACATGACGGCGGA, quality score 94) was inserted into the restriction site of the pL-CRISPR.EFS.tRFP lentiplasmid (Cat#57819, Addgene, Cambridge, MA) (Heckl et al. 2014) a plasmid that allows for simultaneous expression of (i) the Cas9 enzyme; (ii) the gude RNA (gRNA); and (iii) a red fluorescent protein (tRFP) – to control for transduction efficiency. All plasmids were verified by Sanger sequencing (Eurofins, Louisville, KY). Lenti-viral plasmids were packaged in lentiviruses by Viracore (USCF, CA) at titers routinely above 107 infectious particles per ml.
Intracerebroventricular injections
Bilateral intracerebroventricular (ICV) injections were performed as previously described in Sprague-Dawley (SD) rat pups on postnatal day 3 (Delenclos et al. 2017; Pang, Cai, and Rhodes 2003). Briefly, newborn SD rat puts were anesthetized by isoflurane. A 10 μl syringe (Hamilton Gastight Syringe, #1701) was used to pierce the skull (coordinates from bregma: −0.6 mm posterior, ± 1.75 mm lateral/medial, and −2.5 mm ventral), and 2.5 μl of CRISPR lentivirus (gRNA-control or gRNA-TAF1) was injected into each cerebral ventricle without opening the scalp. Neonatal rat pups were kept with the dam until weaned.
Cerebellar Slice Preparation
Rat pups were sacrificed between postnatal day10 to 14 for electrophysiological analyses. The animals were decapitated after being deeply anesthetized with isoflurane, and their cerebellums were rapidly removed and placed in an ice-cold dissection ACSF containing (in mM) 220 sucrose, 2.5 KCl, 1.25 Na2HPO4, 3.5 MgCl2, 0.5 CaCl2, 25 NaHCO3, and 20 D-glucose (with pH at 7.4 and osmolarity at 310 mOsm), bubbled with 95% O2 and 5% CO2. Parasagittal slices (320 μm thick) were cut using a VT 1200S vibratome (Leica, Germany). Slices were then incubated for at least 1 hour at 34°C in an oxygenated recording solution containing (in millimolar): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, 25 D-glucose, with pH at 7.4 and osmolarity at 320 mOsm. The slices were then positioned in a recording chamber and continuously perfused with oxygenated recording solution at a rate of 2 to 3 mL/min before electrophysiological recordings at RT.
Whole Cell Patch-clamp Recording
Recordings were made from PCs in lobules IV-VI, which were visually identified based on their location using infrared differential interference contrast video microscopy on an upright microscope (FN1; Nikon, Tokyo, Japan) equipped with a 3 40/0.80 water-immersion objective and a charge-coupled device camera. The pipettes were prepared by pulling glass capillaries on a model p-97 microelectrode puller (Sutter Instrument, Novato, USA). Patch pipettes had resistances of 3–5 MΩ. The internal solution was a K-based solution containing (in mM): 120 potassium gluconate, 20 KCl, 2 MgCl 2, 2 Na 2 -ATP, 0.5 Na-GTP, 20 HEPES, 0.5 EGTA, with pH at 7.28 (with potassium hydroxide [KOH]) and osmolarity at 310 mOsm.
The whole-cell configuration was obtained in voltage-clamp mode. The membrane potential was held at −70 mV using PATCHMASTER software in combination with a patch clamp amplifier (EPC10; HEKA Elektronik, Lambrecht, Germany). To record spontaneous excitatory postsynaptic currents (sEPSCs), bicuculline methiodide (10 μM) was added to the recording solution to block γ-aminobutyric acid-activated currents. SAK3 (0.1 nM) was also added to the perfusion system. Hyperpolarizing step pulses (5 mV in intensity, 50 milliseconds in duration) were periodically delivered to monitor the access resistance (15–25 MΩ), and recordings were discontinued if the access resistance changed by more than 20%. For each PC, sEPSCs were recorded for a total duration of 2 minutes. Currents were filtered at 3 kHz and digitized at 5 kHz. Data were further analyzed by the Mini-Analysis (Synatosoft Inc, NJ) and Clampfit 10.7 Program. The amplitude and frequency of sEPSCs was compared between neurons from three groups.
Open Field Test
The apparatus is 120-cm diameter circular arena, bordered by a 50-cm-high wall made of wood. The floor of this chamber was divided into central and peripheral zone. The rat pups were placed into the peripheral zone of an open field chamber and their behavior was observed for 5 min. Number of grooming; i.e. consisting of licking the fur, washing face or scratching behaviors were noted.
Beam Walking Test
Animals were allowed to walk on a narrow flat stationary wooden beam (L100 cm×W2 cm) placed at a height of 100 cm from the floor to reach an enclosed escape platform. The time taken to cross the beam from one end to the other and the number of foot slip errors were observed as described previously (Rajasankar, Manivasagam, and Surendran 2009).
Hematoxylin & Eosin Staining
After dehydration of the tissue with 30% sucrose, 20 μm sections were cut, stained with hematoxylin and eosin (H&E) dye (Hematoxylin, Catalog No: HHS16–500ML; Eosin, Catalog No: HT110316–500ML, Sigma), and mounted with Richard-Allan scientific mounting medium (Catalog No: 4112, Thermo Scientific) for microscopy.
Nissl Staining
After dehydration of the tissue with 30% sucrose, 20 μm sections were cut, stained with cresyl violet dye (Catalog No: C5042–10G, Sigma), and mounted with Richard-Allan scientific mounting medium (Catalog No: 4112, Thermo Scientific) for microscopy.
Immunohistochemistry
The antibodies used in this study are listed in Table 1. Rats (n = 4 animals per experimental condition) were perfused with saline and 4% formaldehyde in Phosphate buffer saline (PBS) at PD 35, and the brains were extracted and post-fixed in 4% paraformaldehyde for 8 h at 4°C. Cerebellar sections were cut sagittally at 20 μm using a cryostat (Microm HM 505 E). After rinsing the sections in PBS for 5 min, the sections were incubated with a 0.1% H2O2 solution in PBS for 5 min, rinsed in PBS for 5 times for 5 min, and incubated for 30 min with 0.4% Triton X-100, rinsed in PBS and blocked with 8% goat serum, and 1% Triton X-100 in PBS. After blocking, the sections were incubated at 4°C for overnight with the indicated antibodies diluted in 4% goat serum in PBS. The sections were washed in 1% goat serum in PBS, incubated with secondary antibody anti-rabbit Alexa fluor 488 (Life Technologies) or anti-mouse Alexa fluor 488 (Life Technologies), as needed, in 4% goat serum for 2 h, washed with PBS 3 times for 5 mins, and incubated with DAPI (Catalogue No: D1306, Thermofisher Scientific) at concentration of 50 ng/ml for 2 mins. Sections were then washed and further air dried, and cover slipped with glycerol. All procedures were performed at room temperature. stained slides were observed under a fluorescence microscope (LSM510, Carl Zeiss) using a 20x objective and apotome 2.
Table 1.
List of primary and secondary antibodies used in the study.
| Primary antibodies | ||||||
| Anti- | Abbreviation | Host | Company | Catalog number | Application | Dilutiona |
| Bcl-2-associated X protein | Bax | Rabbit | Cell Signaling | 14796S | WB | 1:1000 |
| Brain-derived neurotrophic factor | BDNF | Mouse | Abcam | ab203573 | IHC | 1:500 |
| Calbindin-D28k | Calbindin | Rabbit | Sigma Aldrich | C2724 | IHC | 1:500 |
| Cleaved Caspase 3 | C-Cas-3 | Rabbit | Cell Signaling | 9661S | WB | 1:1000 |
| Calcium/Calmodulin-dependent Protein Kinase II | CaMKII | Rabbit | Cell Signaling | 3362S | WB | 1:500 |
| Phospho-Calcium/Calmodulin-dependent Protein Kinase II | p-CaMKII | Rabbit | Cell Signaling | 12716S | WB | 1:1000 |
| Glial fibrillary acidic protein | GFAP | Rabbit | Dako | Z0334 | IHC | 1:500 |
| Ionizing calcium-binding adaptor molecule 1 | Iba1 | Rabbit | Wako Labs | 019–19,741 | IHC | 1:500 |
| phospho-Protein kinase B | p-AKT | Rabbit | Cell Signaling | 9271S | IHC | 1:500 |
| Tublin | Tublin | Mouse | Promega | G7121 | WB | 1:1000 |
| Secondary antibodies | ||||||
| Anti- | Host | Label | Dilution | Company | ||
| Rabbit IgG | Goat | HRP-linked | 7074S | WB | 1:3000 | Cell Signaling |
| Mouse IgG | Horse | HRP-linked | 7076S | WB | 1:3000 | Cell Signaling |
| Rabbit IgG | Goat | Alexa 488 | A11034 | IHC | 1:500 | Life technologies |
| Mouse IgG | Rabbit | Alexa 488 | A21204 | IHC | 1.500 | Life technologies |
WB = Western blot; IHC = Immunohistochemistry.
TUNEL assay
Cerebellum tissue sections of 20 μm were cut and 5–7 sections chosen according to systematic random sampling scheme from each sample were processed with In Situ Cell Death Detection Kit, Fluorescin (Catalog No: 11684795910) (Roche, Millipore Sigma, USA) according to the manufacturers protocol for tissues. Then the slides were observed under a fluorescence microscope (LSM510, Carl Zeiss) using a 20x objective and apotome 2.
Morphometric analysis
We quantified the number of calbindin+, GFAP+, Iba1a+, and TUNEL+ cells with the granular layer from 12 fields from 4 different animals per experimental condition as previously described(Janakiraman et al. 2019). The immunflourescence for BDNF and pS473-AKT was quantitated in 12 different fields of cerebellar tissue from 4 different animals per experimental a similar fashion.
Western blotting analysis
After decapitation of animals, brain tissue from the cerebellum was dissected, snap frozen in liquid nitrogen prior to storage at −80°C until analysis. Western blot analyses were performed as described(Janakiraman et al. 2019). In brief, frozen samples were homogenized with RIPA buffer and centrifuged at 4°C, 20,000xg for 20 min. Supernatant protein concentrations were determined using BCA method (Catalog No: #23227, Thermo Scientific), and samples were then boiled 5 min in Laemmli’s sample buffer (Catalog No: NP0008, Life Technologies). Equal amounts of protein were loaded onto and run on SDS-polyacrylamide gels (Catalog No: #4568084, Bio-Rad) and then transferred to polyvinylidene difluoride membrane (Catalog No: #1620177, Bio-Rad). After transfer, membranes were blocked with TBST solution (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 0.1% Tween 20) containing 5% non-fat dry milk at 4°C for 1 hour. After blocking membranes were incubated overnight at 4 °C with anti-CaMKII (pan) (1:500; Cell Signaling), anti-phospho-CaMKII (Thr-286) (1:1000; Cell Signaling), anti-cleaved caspase-3 (Asp175) (1:1000; Cell Signaling), anti-Bax (1:1000; Cell Signaling) and β-Tubulin (1:1000; Promega) in 3% BSA. After washing, membranes were incubated with secondary antibody diluted in 3% BSA. Blots were developed using an ECL detection system (Catalog No: 20–300B, Prometheus Protein Biology Products, USA) and signals were quantified using Image Studio Digits software version 5.2 (Li-Cor).
Statistics
All experiments were performed at least twice and in a blinded fashion. All data was first tested for a Gaussian distribution using a D’Agostino-Pearson test (Graphpad Prism 8 Software). The statistical significance of differences between means was determined by a parametric ANOVA followed by Tukey’s post hoc or a non-parametric Kruskal Wallis test followed by Dunn’s post-hoc test depending on whether datasets achieved normality. Differences were considered significant if p≤ 0.05. Error bars in the graphs represent mean ± SEM. All data were plotted in GraphPad Prism 8.
Results
SAK3 improves TAF1 ID-like motor deficits
Motor dysfunction is characteristic of TAF ID syndrome (O’Rawe et al. 2015; Hurst 2018b; Janakiraman et al. 2019). In our previous study, we found concomitant CaV3.1 loss with motor deficits and abnormal grooming behaviors in TAF1-edited animals (Janakiraman et al. 2019). Therefore, we asked whether the T-type Ca2+ channel enhancer, SAK3, could mitigate these behavioral abnormalities in TAF1-edited animals (Fig. 1A). Consistent with our previous findings (Janakiraman et al. 2019), the beam crossing time (Fig. 1B) and number of foot slips errors were higher in the TAF1-edited animals compared to all other experimental groups (Fig. 1C). However, in TAF1-edited animals treated with SAK3, we observed a rescue of the beam crossing time and foot slip errors compared to TAF1-edited animals. TAF1-edited animals displayed an increased grooming frequency which was blunted by SAK3 treatment (Fig. 1D). TAF1-edited animals treated with SAK3 showed similar behavioral patterns as the naïve group. Thus, enhancing T-type Ca2+ channels using SAK3, rescued the behavioral defects associated with TAF1 editing.
SAK3 attenuates the cerebellar abnormalities made by TAF1 deletion
We previously documented that TAF1-editing cause morphological abnormalities in the cells of the cerebellum and cerebral cortex (Janakiraman et al. 2019). Thus, we first evaluated the morphology of the Purkinje cells and cerebral cortex neurons by Hematoxylin and eosin (H&E) and Nissl staining (Supplementary Fig. 1). Our histopathology analysis showed that the architecture of the Purkinje cell layer was abnormal in TAF1-edited animals as we previously described (Supplementary Fig. 1Aii). In addition, the histopathology showed Purkinje cells were markedly hypoplastic in TAF1 treated animals relative to controls (Supplementary Fig. 1B). SAK3 administration improved Purkinje cells morphology and arrangement of the Purkinje cell layer in TAF1 edited animals (Supplementary Fig. 1Avi). No difference of Purkinje cell morphology was found in the Naïve and gRNA-control group independently of the SAK3 treatment.
We next assessed whether SAK3 could prevent the loss of Purkinje cells observed after TAF1 editing (Fig. 2A, B). We stained rat cerebella with the Purkinje cells marker Calbindin and quantified the number of Purkinje cells per linear mm. As before, TAF1 editing reduced the number of Purkinje cells compared to Naïve or gRNA-control rats. SAK3 administration increased the number of calbindin-positive Purkinje cells compared to TAF1-edited animals (Fig. 2B). Treatment with SAK3 had no effect on the Naïve and gRNA-control group compared to the vehicle (water) (Fig. 2B).
Figure 2. SAK3 prevents the loss of Purkinje cell caused by TAF1 gene editing.

(A) Expression of calbindin was decreased in TAF1-edited animals (Av) as compared to naïve and CRISPR-control groups (A i-iv). SAK3 administration to the TAF1 edited animals shows increased the number of Calbindin positive Purkinje cells (Avi). (C) Expression of GFAP was decreased in TAF1-edited animals (Cv) as compared to naïve and CRISPR-control groups (C i-iv). SAK3 administration to the TAF1 edited animals shows increased the number of GFAP positive cells (Cvi). (E) Expression of IBA-1 (microglia marker) was increased in TAF1-edited animals (Ev) as compared to all other experimental groups (Ei-iv). SAK3 administration to the TAF1 edited animals showed a decrease in the number of IBA-1 -positive cells as compared to TAF1 edited group (Evi & v). (B) Summary of the number of Purkinje cells per linear density, (D) number of GFAP positive cells, and (F) the number of IBA-1 positive cells in each of the experimental conditions. Data are shown as mean ± S.E.M., n=12 fields per animal, 4 animals per experimental condition. *p < 0.05 versus; naïve, #p < 0.05 versus gRNA-control (ANOVA followed by Tukey’s test). Scale bars: 200 μm. The experiments were conducted in a blinded fashion.
Glial cells, including astrocytes, oligodendrocytes, and microglia are by far the most abundant cells in the nervous system including in the granular layer of the cerebellum (Domingues et al. 2016). We previously reported a decrease in GFAP-positive astrocytes and increase in Iba1-positive microglia within the granular layer of the cerebellum in TAF1-edited animals (Janakiraman et al. 2019). In the present study, we confirmed these findings (Fig. 2C). We found that SAK3 treatment increased the number of GFAP-positive cells (Fig. 2D) and decreased the number of Iba1-positive cells compared to the TAF1 edited group (Fig. 2E, F). Thus, enhancing T-type Ca2+ channel activity with SAK3 can reverse all morphological alterations induced by TAF1 editing.
SAK3 rescued the decreased frequency of spontaneous excitatory post synaptic current (sEPSCs) in TAF1 edited Purkinje cells
Abnormal motor symptoms in TAF1-edited rats are associated with irregular cerebellar output caused by changes in the intrinsic activity of the Purkinje cells due to loss of pre-synaptic Cav3.1 (Janakiraman et al. 2019). Therefore, we investigated if SAK3 could directly mitigate the defects in presynaptic currents we have noted in TAF1-edited animals. We recorded spontaneous excitatory post synaptic currents (sEPSCs) from gRNA-control and gRNA-TAF1 edited Purkinje cells within cerebellar slices (Fig. 3A) using whole-cell patch clamp electrophysiology (Fig. 3B). As before(Janakiraman et al. 2019), TAF1 editing decreased the frequency of sEPSC compared to gRNA-control (1.27 ± 0.17 Hz vs 2.23 ± 0.26 Hz, *p<0.05). Applying SAK3 (0.1 nM) rescued sEPSC frequency to the level of gRNA control (2.13 ± 0.29 Hz, &p<0.05) (Fig. 3C). We did not observe any changes in the amplitudes of sEPSC for any treatment groups (Fig. 3D). These results show that SAK3 treatment can restore functional neurotransmission in gRNA-TAF1 edited Purkinje cells.
Figure 3. SAK3 rescued the decreased frequency of spontaneous excitatory post synaptic current (sEPSCs) in TAF1 edited Purkinje cells.

(A) Photomicrograph of cerebellar slice preparation with a progressive zoom with the rightmost panel showing positioning of the recording electrode to this region. (B) Representative recording traces of cells from the indicated groups. The cumulative probability of amplitude (C) and inter-event interval (E). Summary of amplitudes (D) and frequencies (F) of sEPSCs for the indicated groups are shown. Data are shown as mean ± S.E.M., n=12 Purkinje cells from at least 2 animals per experimental condition. *p < 0.05 versus; gRNA-control, &p < 0.05 versus gRNA-TAF1 (ANOVA followed by Tukey’s test). The experiments were conducted in an investigator-blinded manner.
SAK3 reduces TAF1 induced neuronal apoptosis
Regulation of apoptosis is critical for cerebellar development (Lossi, Castagna, and Merighi 2018). One of TAF1’s functions is to negatively regulate apoptosis (Curran et al. 2018; Kimura et al. 2008; Wang and Tjian 1994). Because we observed a loss of Purkinje cells in TAF1 edited animals which was rescued by SAK3, we investigated if apoptosis could be enhanced in TAF1 edited animals. We observed an increase in terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) positive cells in TAF1 edited animals (Fig. 4A and Supplementary Fig. 2). SAK3 treatment reduced the number of TUNEL positive cells to control levels in TAF1 edited animals (Fig. 4B). The protective effects of SAK3 against apoptosis was confirmed by SAK3 suppression of activated caspase 3 (Fig. 5A, C) and pro-apoptotic BAX (Fig. 5B, D). We did not observe any changes in Bcl2 levels (data not shown).
Figure 4. The effects of SAK3 on TAF1 induced apoptosis in the cerebellum.

Apoptosis was assessed in cerebellum samples using TUNEL assay. (A) Shown are photomicrographs from gRNA-TAF1 edited animals and gRNA-TAF1 edited animals treated with SAK3. The data from the control groups can be found in the Supplementary data. Note SAK3 reduced the number of TUNEL positive cells in gRNA-TAF1 edited animals. (B) Summary of the number of TUNEL positive cells in each of the experimental conditions. Data are shown as mean ± S.E.M., n=12 fields per animal, 4 animals per experimental condition. *p < 0.05 versus; naïve and gRNA-TAF1 =SAK3 group (ANOVA followed by Tukey’s test). Scale bars: 200 μm. The experiments were conducted in an investigator-blinded manner.
Figure 5. The effects of SAK3 on mediators of apoptosis.

(A) Representative Western analysis is shown from cerebellum samples from animals from each of the experimental conditions. Note increased cleaved caspase 3 in gRNA-TAF1 edited animals and diminished cleaved caspase 3 levels in gRNA-TAF1. Note increased BAX levels in gRNA-TAF1 edited animals, whereas SAK3 reduced BAX levels to control levels (B). (C&D) Quantification of Western analysis form two independent experiments. Data shown are mean + SEM, n = 6 animals per each experimental condition. *p < 0.05 versus gRNA-control water and RNA-control SAK3 group; # p< 0.05 versus gRNA-TAF1 water group (ANOVA followed by Tukey’s test).
SAK3 rescues BDNF/AKT signaling in the cerebellum of TAF1 edited animals
It has been reported that calcium influx via the T-type channel invokes a dynamic interaction between calmodulin and CaV3.1 channels to trigger a signaling cascade that leads to Ca2+/calmodulin-dependent protein kinase II (CaMKII) activation(Asmara et al. 2017). Furthermore, SAK3 has been reported to increase CaMKII activation(Wang et al. 2018; Xu et al. 2018). Therefore, to gain insight into the mechanism by which SAK3 might exert its disease modifying effects in TAF1 edited animals, we investigated the effects of SAK3 on CaMKII activation. TAF1 editing resulted in a reduction of CaMKII activation (measured by phosphorylation at T286) in the cerebellum (Supplementary Fig. 3). However, we did not observe a restoration of CaMKII activation in the cerebellum of SAK3 treated animals (Supplementary Fig. 3). These results imply that SAK3 might exert its neuroprotection in TAF1 edited animals independent of CaMKII signaling.
Brain-derived neurotrophic factor (BDNF) plays a crucial role in promoting survival and differentiation of neurons (Binder 2004). Activation of the PI3K/AKT pathway through BDNF/TrkB interactions can inhibit apoptosis in the developing cerebellum (Schwartz et al. 1997; Yoo et al. 2017). Consequently, we tested if SAK3 could be neuroprotective through this pathway. We found that BDNF levels were markedly lower in TAF1 edited animals compared to Naïve and gRNA-control groups (Fig. 6A, B). However, SAK3 administration restored BDNF expression to the levels in Naïve and gRNA-control groups (Fig. 6A, B). AKT phosphorylation at S473 is associated with its activation (Szymonowicz et al. 2018). We next found that activated AKT (phosphorylated S473) in Purkinje cells was decreased in TAF1 edited animals (Fig. 6C, D) which was prevented by SAK3 treatment (Fig. 6C, D). Taken together, our observations suggest that SAK3 exerts neuroprotective effects by suppressing TAF1-induced neuronal apoptosis (Fig. 4 and 5) and by activating the BDNF-TrkB-PI3K/Akt signaling pathway (Fig. 6).
Figure 6. The effects of SAK3 on BDNF and Akt phosphorylation expression in the cerebellum of TAF1 edited animals.

(A) Expression of BDNF was decreased in TAF1-edited animals (Av) as compared to all other experimental groups (Ai-iv). SAK3 administration to the TAF1 edited animals shows increased number of BDNF-positive cells as compared to TAF1 edited group (Avi & v). (C) Expression of p-AKT was decreased in TAF1-edited animals (Cv) as compared to all other experimental groups (Ci-iv). SAK3 administration to the TAF1 edited animals shows increased expression of p-AKT as compared to TAF1 edited group (Cvi & v). Summary of BDNF expression (B) and expression of p-AKT (D) in each of the experimental conditions. Data are shown as mean ± S.E.M., n=12 fields per animal, 4 animals per experimental condition. *p < 0.05 versus; naïve, #p < 0.05 versus gRNA-control (ANOVA followed by Tukey’s test). Scale bars: 200 μm. The experiments were conducted in an investigator-blinded manner.
Discussion
In the present study, we demonstrated that SAK3 can efficiently reverse behavioral, morphological and biochemical defects induced by TAF1 editing in neonatal rats. We made the following salient observations: (a) SAK3 improves behavioral defects associated with TAF1 gene editing; (b) SAK3 restores the number of Purkinje, astrocytes and microglial cells in the developing cerebellum that were lost following TAF1 editing; (c) SAK3 protects the neurons of the cerebellum from the deleterious effects of TAF1 editing; (d) SAK3 restores excitatory post synaptic current (sEPSCs) in TAF1 edited Purkinje cells; and (e) SAK3 increases BDNF/AKT activation in TAF1 edited animals.
In our previous study, we reported that TAF1 editing produced motor imbalance in beam walking test and increased self-grooming behavior (Janakiraman et al. 2019). These behavioral defects were associated with the loss of Purkinje cells and decrease in Cav3.1 T-type channel expression and function. Deletion of T-type Ca2+ channels impairs motor behaviors, such as walking on a thin elevated beam, which also involves a cerebellar component (Schonewille et al. 2011; De Zeeuw et al. 1998). In the present studies, SAK3 administration improved beam crossing time and decreased foot slip errors while reducing self-grooming. Previous studies reported that, chronic administration of SAK3 to olfactory bulbectomized mice improved depressive-like behavior (Xu et al. 2018) and enhanced cognitive behavior (Yabuki et al. 2017). Collectively, these findings suggest that SAK3 can have beneficial effects on both cognitive and motor deficits in TAF1ID syndromes.
We found that SAK3 administration to the TAF1 edited animals increased the number of GFAP-positive cells and decreased the number of Iba1-positive cells compared to the TAF1 edited group. Microglia are resident immune cells in the brain that continuously monitor the brain microenvironment (Nimmerjahn, Kirchhoff, and Helmchen 2005). They have a very low threshold of activation and rapidly respond within 20–40 min of injury (Davalos et al. 2005). Astrocytes regulate the osmolarity, ionic composition, and pH of the extracellular microenvironment (Kintner et al. 2004; Xu et al. 2001; Sokoloff et al. 1996; Aschner, Mutkus, and Allen 2001). These cells can also clear neurotransmitters from synaptic clefts and provide growth factors and nutrients for neurons (Kintner et al. 2004; Xu et al. 2001; Sokoloff et al. 1996; Aschner, Mutkus, and Allen 2001). Moreover, astrocytes also play a role in regulation of neuronal functions by affecting the strength and number of synapses (Abbott 2002; Rosenberg et al. 2001; Li et al. 2019). Under pathological conditions in the CNS, these two types of glial cells, microglia and astrocytes, become reactive. In the present study, microglial activation appears to occur before reactive astrogliosis which is in an agreement with other brain disorder studies (Li et al. 2019). Our data indicate that the response of these two glial populations to SAK3 ultimately benefit neurons (i.e. Purkinje cells).
Consistent with our earlier studies, we observed that the number of calbindin positive Purkinje cells were decreased in the TAF1 edited animals. To gain insight into the mechanisms underlying the loss of Purkinje cells, we investigated apoptosis as an underlying mechanism for this loss. The mitochondria are central to apoptosis in the central nervous system (Atif, Yousuf, and Agrawal 2009; Lu 2009). A number of genes and proteins can influence or instigate the progression of apoptosis along the mitochondrial pathway (Wang et al. 2013). Key amongst these genes associated with apoptosis are proteins of the Bcl family and caspases (Adams and Cory 2002; Slee, Adrain, and Martin 2001). Bcl-2/Bax family members are key regulatory factors in the mitochondrial apoptotic pathway (Burlacu 2003; Szabo et al. 2011). They are divided into two groups, anti-apoptotic (Bcl-2, Bcl-xL) and pro-apoptotic (Bax, Bad) proteins. Upon stimulation with pro-apoptotic factors, Bax translocates from the cytoplasm to the mitochondrial membrane, which alters the permeability of the mitochondrial membrane and promotes the release of cytochrome c (Cyt c) from the mitochondria into the cytoplasm (Adams and Cory 2002). It has been proven that the activity of the Bcl-2 protein may be regulated through caspase cleavage under various circumstances. The apoptotic cascade is subsequently initiated, eventually leading to apoptosis (Clem et al. 1998). The data from our studies suggests that TAF1 induced apoptosis may be due dysregulation of mitochondrial function in neurons. SAK3 has been shown to attenuate scopolamine-induced apoptosis in SH-SY5Y cells (Suthprasertporn et al. 2020). Our data supports a neuroprotective role for SAK3 against TAF1 induced neuronal apoptosis in vivo.
Stimulation of the BDNF/AKT pathway is well known to inhibit apoptosis (Schwartz et al. 1997; Kim et al. 2017). We also show that SAK3 increased BDNF/AKT activation in TAF1 edited animals. This observation is consistent with previous studies with SAK3 in other animal models of neurologic disease or brain injury (Xu et al. 2018; Izumi et al. 2018; Husain et al. 2018; Yabuki et al. 2017). However, contrary to other studies we did not find activation of CaMKII. This suggests that the mechanism of SAK3 stimulation of the BDNF/AKT pathway in the cerebellum may be different than previous reports. This may include additional targets (besides the Cav3.1 T-type channel) may be involved in its neuroprotective effects. Additional studies directly linking the Cav3.1 T-type channel to the neuroprotective effects of SAK3 in TAF1 edited animals warrant further investigation. Along the same vein, substrates downstream in the BDNF/AKT signaling pathway impacted by SAK3 will require further study. Nevertheless, in the cerebellum, the neuroprotective effects against apoptosis caused by SAK3 appears to involve activation of BDNF/AKT signaling pathway.
In the present study, we focused on investigating the neuroprotective effects of SAK3 against the deleterious effects of TAF1 disruption within the cerebellum. This is because in our previous studies we identified a variant in TAF1 associated with the interference of post-natal cerebellar development (Hurst 2018a) and our neonatal rat pup model replicated these features (Janakiraman et al. 2019). However, the broader set of TAF1 disease variants (i.e. SVA insertion mutation) have been linked to CNS disturbances beyond the cerebellum (Goto et al. 2005; Goto et al. 2013). In our neonatal rat pup model TAF1 deletion, we did not observe any obvious morphologic differences between control and gRNA TAF1 edited animal in other brain regions (i.e. frontal cortex, striatum, hippocampus, substantia nigra, and pons) with the exception of the cerebral cortex (Janakiraman et al. 2019). We are currently evaluating whether SAK3 has any beneficial effects within the cerebral cortex in TAF-1 edited animals
In conclusion, the present study indicates that a novel T-type calcium channel enhancer has disease-modifying effects in animal models of TAF1 editing. Moreover, we provide insights into the molecular mechanism by which SAK3 exerts in pharmacologic effects. Furthermore, our findings imply that the T-VGCCs are novel molecular targets to develop therapeutics to treat TAF1 ID syndrome and that SAK3 is an attractive drug candidate to treat TAF1 associated neurologic disorders.
Supplementary Material
Figure S1. SAK3 improves the morphology of the Purkinje cells in the cerebellum altered by TAF1 gene editing. (A, B) The morphology of the Purkinje cell layer was evaluated by H&E Staining and Nissl Staining. The H&E and Nissl staining showed abnormal architecture of the Purkinje cells layer including hypoplastic Purkinje cells (Av & Bv). SAK3 administration to the TAF1 edited animals showed the morphology of the Purkinje cells were improved as compared to TAF1 edited animals (Avi & Bvi). Scale bars: 500 μm. The experiments were conducted in a blinded fashion.
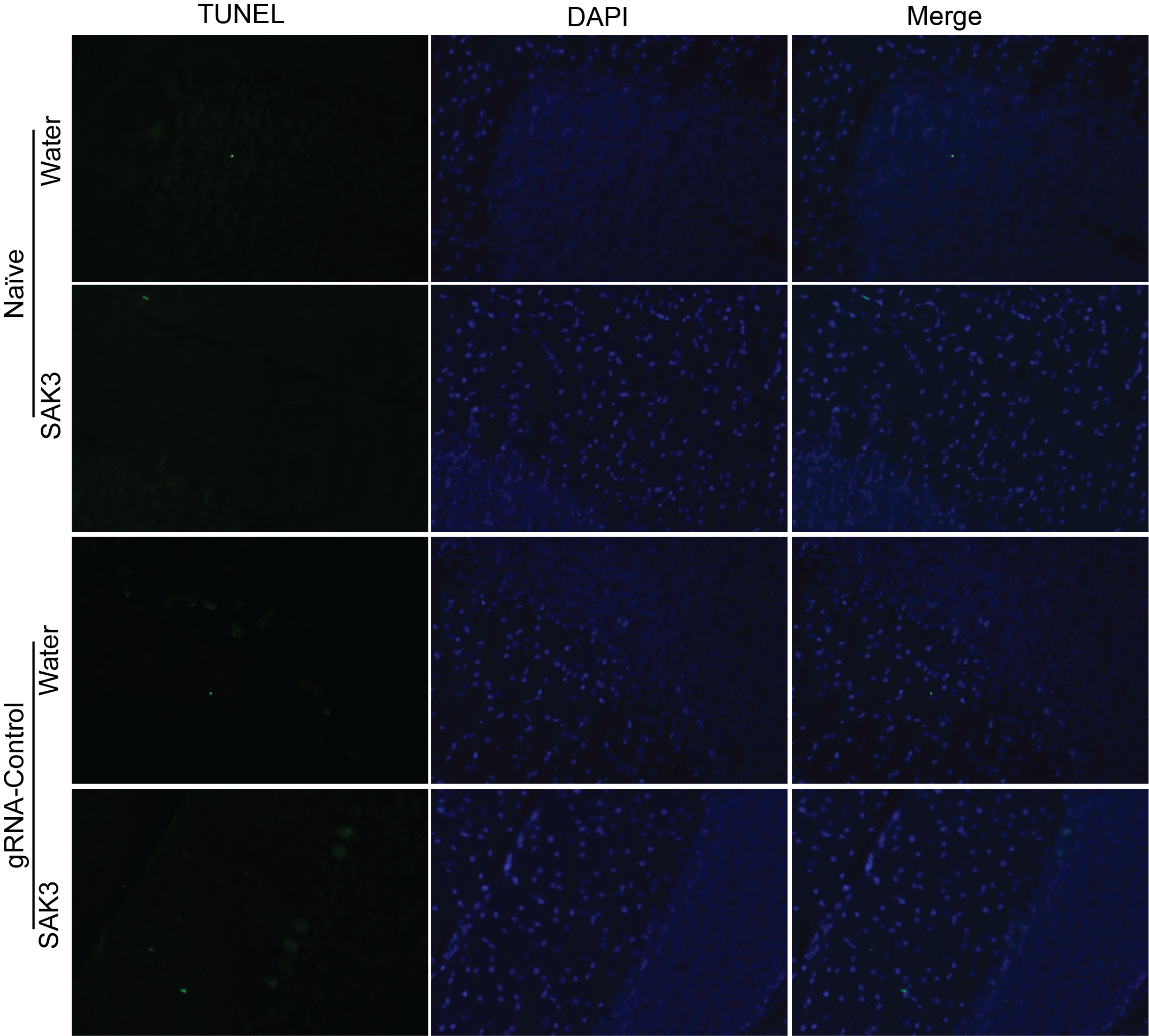
Figure S2. The effects of SAK3 on cerebellum of Naïve and gRNA-control animals. Apoptosis was assessed in cerebellum samples using TUNEL assay. Shown are TUNEL assay photomicrographs from Naive and gRNA-control animals treated with SAK3. Note water and SAK3 treatment to the naïve and gRNA-control animals no significantly changed TUNEL positive cells were observed. Scale bars: 200 μm. The experiments were conducted in an investigator-blinded manner.
{kind=link}
Figure S3. The effects of SAK3 on CaMKII. (A) Representative Western analysis is shown from cerebellum samples from each of the experimental conditions. Note decreased p-CaMKII in gRNA-TAF1 edited animals and increased p-CaMKII levels in gRNA-control. (B) Quantification of p-CaMKII expression on all the experimental condition. Data shown are mean + SEM, n = 4 animals per each experimental condition. No significant changes were observed between the gRNA-TAF1 edited animals and SAK3 treated gRNA-TAF1 animals (Data Analyzed by Kruskal Wallis Test).
{kind=link}
Highlights.
SAK3 improves behavioral defects associated with TAF1 gene editing;
SAK3 reestablishes the number of Purkinje cells and astrocytes within the developing cerebellum that were lost following TAF1 editing
SAK3 protects the neurons of the cerebellum from the deleterious effects of TAF1 editing
SAK3 restores excitatory post synaptic currents (sEPSCs) in TAF1 edited Purkinje cells
SAK3 stimulates the BDNF/AKT pathway in TAF1 edited animals.
Acknowledgements
We acknowledge the funding and support of the Senner Endowment for Precision Health, University of Arizona Health Sciences. This work was also supported by grants from the National Natural Science Foundation of China (81603088) to J.Y., and R01DA042852 from the National Institute on Drug Abuse to R.K.
Footnotes
Conflict of interest
M. Nelson is co-founder of DesertDX, LLC, a molecular diagnostic company. R. Khanna is the co-founder of Regulonix LLC, a company developing non-opioids drugs for chronic pain. The other authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott NJ 2002. ‘Astrocyte-endothelial interactions and blood-brain barrier permeability’, J Anat, 200: 629–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JM, and Cory S 2002. ‘Apoptosomes: engines for caspase activation’, Curr Opin Cell Biol, 14: 715–20. [DOI] [PubMed] [Google Scholar]
- Aldrin-Kirk P, Davidsson M, Holmqvist S, Li JY, and Bjorklund T 2014. ‘Novel AAV-based rat model of forebrain synucleinopathy shows extensive pathologies and progressive loss of cholinergic interneurons’, PLoS One, 9: e100869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aneichyk T, Hendriks WT, Yadav R, Shin D, Gao D, Vaine CA, Collins RL, Domingo A, Currall B, Stortchevoi A, Multhaupt-Buell T, Penney EB, Cruz L, Dhakal J, Brand H, Hanscom C, Antolik C, Dy M, Ragavendran A, Underwood J, Cantsilieris S, Munson KM, Eichler EE, Acuna P, Go C, Jamora RDG, Rosales RL, Church DM, Williams SR, Garcia S, Klein C, Muller U, Wilhelmsen KC, Timmers HTM, Sapir Y, Wainger BJ, Henderson D, Ito N, Weisenfeld N, Jaffe D, Sharma N, Breakefield XO, Ozelius LJ, Bragg DC, and Talkowski ME 2018. ‘Dissecting the Causal Mechanism of X-Linked Dystonia-Parkinsonism by Integrating Genome and Transcriptome Assembly’, Cell, 172: 897–909 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschner M, Mutkus L, and Allen JW 2001. ‘Aspartate and glutamate transport in acutely and chronically ethanol exposed neonatal rat primary astrocyte cultures’, Neurotoxicology, 22: 601–5. [DOI] [PubMed] [Google Scholar]
- Asmara H, Micu I, Rizwan AP, Sahu G, Simms BA, Zhang FX, Engbers JDT, Stys PK, Zamponi GW, and Turner RW 2017. ‘A T-type channel-calmodulin complex triggers alphaCaMKII activation’, Mol Brain, 10: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assandri R, Egger M, Gassmann M, Niggli E, Bauer C, Forster I, and Gorlach A 1999. ‘Erythropoietin modulates intracellular calcium in a human neuroblastoma cell line’, J Physiol, 516 ( Pt 2): 343–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atif F, Yousuf S, and Agrawal SK 2009. ‘S-allyl L-cysteine diminishes cerebral ischemia-induced mitochondrial dysfunctions in hippocampus’, Brain Res, 1265: 128–37. [DOI] [PubMed] [Google Scholar]
- Bieniossek C, Papai G, Schaffitzel C, Garzoni F, Chaillet M, Scheer E, Papadopoulos P, Tora L, Schultz P, and Berger I 2013. ‘The architecture of human general transcription factor TFIID core complex’, Nature, 493: 699–702. [DOI] [PubMed] [Google Scholar]
- Binder DK 2004. ‘The role of BDNF in epilepsy and other diseases of the mature nervous system’, Adv Exp Med Biol, 548: 34–56. [DOI] [PubMed] [Google Scholar]
- Bragg DC, Mangkalaphiban K, Vaine CA, Kulkarni NJ, Shin D, Yadav R, Dhakal J, Ton ML, Cheng A, Russo CT, Ang M, Acuna P, Go C, Franceour TN, Multhaupt-Buell T, Ito N, Muller U, Hendriks WT, Breakefield XO, Sharma N, and Ozelius LJ 2017. ‘Disease onset in X-linked dystonia-parkinsonism correlates with expansion of a hexameric repeat within an SVA retrotransposon in TAF1’, Proc Natl Acad Sci U S A, 114: E11020–E28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burlacu A 2003. ‘Regulation of apoptosis by Bcl-2 family proteins’, J Cell Mol Med, 7: 249–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazade M, Bidaud I, Lory P, and Chemin J 2017. ‘Activity-dependent regulation of T-type calcium channels by submembrane calcium ions’, Elife, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemin J, Monteil A, Briquaire C, Richard S, Perez-Reyes E, Nargeot J, and Lory P 2000. ‘Overexpression of T-type calcium channels in HEK-293 cells increases intracellular calcium without affecting cellular proliferation’, FEBS Lett, 478: 166–72. [DOI] [PubMed] [Google Scholar]
- Clem RJ, Cheng EHY, Karp CL, Kirsch DG, Ueno K, Takahashi A, Kastan MB, Griffin DE, Earnshaw WC, Veliuona MA, and Hardwick JM 1998. ‘Modulation of cell death by Bcl-x(L) through caspase interaction’, Proceedings of the National Academy of Sciences of the United States of America, 95: 554–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran EC, Wang H, Hinds TR, Zheng N, and Wang EH 2018. ‘Zinc knuckle of TAF1 is a DNA binding module critical for TFIID promoter occupancy’, Sci Rep, 8: 4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, and Gan WB 2005. ‘ATP mediates rapid microglial response to local brain injury in vivo’, Nat Neurosci, 8: 752–8. [DOI] [PubMed] [Google Scholar]
- De Zeeuw CI, Hansel C, Bian F, Koekkoek SK, van Alphen AM, Linden DJ, and Oberdick J 1998. ‘Expression of a protein kinase C inhibitor in Purkinje cells blocks cerebellar LTD and adaptation of the vestibulo-ocular reflex’, Neuron, 20: 495–508. [DOI] [PubMed] [Google Scholar]
- Delenclos M, Faroqi AH, Yue M, Kurti A, Castanedes-Casey M, Rousseau L, Phillips V, Dickson DW, Fryer JD, and McLean PJ 2017. ‘Neonatal AAV delivery of alpha-synuclein induces pathology in the adult mouse brain’, Acta Neuropathol Commun, 5: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingues HS, Portugal CC, Socodato R, and Relvas JB 2016. ‘Oligodendrocyte, Astrocyte, and Microglia Crosstalk in Myelin Development, Damage, and Repair’, Front Cell Dev Biol, 4: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich JA, and Tjian R 2010. ‘Unexpected roles for core promoter recognition factors in cell-type-specific transcription and gene regulation’, Nat Rev Genet, 11: 549–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto S, Kawarai T, Morigaki R, Okita S, Koizumi H, Nagahiro S, Munoz EL, Lee LV, and Kaji R 2013. ‘Defects in the striatal neuropeptide Y system in X-linked dystonia-parkinsonism’, Brain, 136: 1555–67. [DOI] [PubMed] [Google Scholar]
- Goto S, Lee LV, Munoz EL, Tooyama I, Tamiya G, Makino S, Ando S, Dantes MB, Yamada K, Matsumoto S, Shimazu H, Kuratsu J, Hirano A, and Kaji R 2005. ‘Functional anatomy of the basal ganglia in X-linked recessive dystonia-parkinsonism’, Ann Neurol, 58: 7–17. [DOI] [PubMed] [Google Scholar]
- Gudmundsson S, Wilbe M, Filipek-Gorniok B, Molin AM, Ekvall S, Johansson J, Allalou A, Gylje H, Kalscheuer VM, Ledin J, Anneren G, and Bondeson ML 2019. ‘TAF1, associated with intellectual disability in humans, is essential for embryogenesis and regulates neurodevelopmental processes in zebrafish’, Sci Rep, 9: 10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckl D, Kowalczyk MS, Yudovich D, Belizaire R, Puram RV, McConkey ME, Thielke A, Aster JC, Regev A, and Ebert BL 2014. ‘Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing’, Nat Biotechnol, 32: 941–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez IH, Cabrera JR, Santos-Galindo M, Sanchez-Martin M, Dominguez V, Garcia-Escudero R, Perez-Alvarez MJ, Pintado B, and Lucas JJ 2020. ‘Pathogenic SREK1 decrease in Huntington’s disease lowers TAF1 mimicking X-linked dystonia parkinsonism’, Brain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzfeld T, Nolte D, Grznarova M, Hofmann A, Schultze JL, and Muller U 2013. ‘X-linked dystonia parkinsonism syndrome (XDP, lubag): disease-specific sequence change DSC3 in TAF1/DYT3 affects genes in vesicular transport and dopamine metabolism’, Hum Mol Genet, 22: 941–51. [DOI] [PubMed] [Google Scholar]
- Hurst SE, Busa-Liktor E, Moutal A, Parker S, Rice S, Szelinger S, Senner G, Hammer MF, Johnstone L, Ramsey K, Naryanan V, Miller-Perez S, Khanna M, Dahlin H, Lewis K, Craig D, Wang EH, Khanna R, and Nelson MA 2018a. ‘A novel variant in TAF1 affects gene expression and is associated with X-linked TAF1 intellectual disability syndrome’, Neuronal Signaling, 2: NS20180141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husain N, Yabuki Y, Shinoda Y, and Fukunaga K 2018. ‘Acute Treatment with T-Type Calcium Channel Enhancer SAK3 Reduces Cognitive Impairments Caused by Methimazole-Induced Hypothyroidism Via Activation of Cholinergic Signaling’, Pharmacology, 101: 309–21. [DOI] [PubMed] [Google Scholar]
- Izumi H, Shinoda Y, Saito T, Saido TC, Sato K, Yabuki Y, Matsumoto Y, Kanemitsu Y, Tomioka Y, Abolhassani N, Nakabeppu Y, and Fukunaga K 2018. ‘The Disease-modifying Drug Candidate, SAK3 Improves Cognitive Impairment and Inhibits Amyloid beta Deposition in App Knock-in Mice’, Neuroscience, 377: 87–97. [DOI] [PubMed] [Google Scholar]
- Janakiraman U, Yu J, Moutal A, Chinnasamy D, Boinon L, Batchelor SN, Anandhan A, Khanna R, and Nelson MA 2019. ‘TAF1-gene editing alters the morphology and function of the cerebellum and cerebral cortex’, Neurobiol Dis, 132: 104539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Lee H, Shin JP, Ahn J, Yoo JM, Song SJ, Kim SJ, Kang SW, and Society Epidemiologic Survey Committee of the Korean Ophthalmologic. 2017. ‘Epiretinal Membrane: Prevalence and Risk Factors from the Korea National Health and Nutrition Examination Survey, 2008 through 2012’, Korean J Ophthalmol, 31: 514–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura J, Nguyen ST, Liu H, Taira N, Miki Y, and Yoshida K 2008. ‘A functional genome-wide RNAi screen identifies TAF1 as a regulator for apoptosis in response to genotoxic stress’, Nucleic Acids Research, 36: 5250–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kintner DB, Su G, Lenart B, Ballard AJ, Meyer JW, Ng LL, Shull GE, and Sun DD 2004. ‘Increased tolerance to oxygen and glucose deprivation in astrocytes from Na+/H+ exchanger isoform 1 null mice’, American Journal of Physiology-Cell Physiology, 287: C12–C21. [DOI] [PubMed] [Google Scholar]
- Li K, Li J, Zheng J, and Qin S 2019. ‘Reactive Astrocytes in Neurodegenerative Diseases’, Aging Dis, 10: 664–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossi L, Castagna C, and Merighi A 2018. ‘Caspase-3 Mediated Cell Death in the Normal Development of the Mammalian Cerebellum’, Int J Mol Sci, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B 2009. ‘Mitochondrial dynamics and neurodegeneration’, Curr Neurol Neurosci Rep, 9: 212–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly R, Bouvier G, Schonewille M, Arabo A, Rondi-Reig L, Lena C, Casado M, De Zeeuw CI, and Feltz A 2013. ‘T-type channel blockade impairs long-term potentiation at the parallel fiber-Purkinje cell synapse and cerebellar learning’, Proc Natl Acad Sci U S A, 110: 20302–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Marti-Gutierrez N, Park SW, Wu J, Lee Y, Suzuki K, Koski A, Ji D, Hayama T, Ahmed R, Darby H, Van Dyken C, Li Y, Kang E, Park AR, Kim D, Kim ST, Gong J, Gu Y, Xu X, Battaglia D, Krieg SA, Lee DM, Wu DH, Wolf DP, Heitner SB, Belmonte JCI, Amato P, Kim JS, Kaul S, and Mitalipov S 2017. ‘Correction of a pathogenic gene mutation in human embryos’, Nature, 548: 413–19. [DOI] [PubMed] [Google Scholar]
- Makino S, Kaji R, Ando S, Tomizawa M, Yasuno K, Goto S, Matsumoto S, Tabuena MD, Maranon E, Dantes M, Lee LV, Ogasawara K, Tooyama I, Akatsu H, Nishimura M, and Tamiya G 2007. ‘Reduced neuron-specific expression of the TAF1 gene is associated with X-linked dystonia-parkinsonism’, Am J Hum Genet, 80: 393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutal A, Cai S, Luo S, Voisin R, and Khanna R 2018. ‘CRMP2 is necessary for Neurofibromatosis type 1 related pain’, Channels (Austin), 12: 47–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutal A, Sun L, Yang X, Li W, Cai S, Luo S, and Khanna R 2018. ‘CRMP2-Neurofibromin Interface Drives NF1-related Pain’, Neuroscience, 381: 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutal A, Yang X, Li W, Gilbraith KB, Luo S, Cai S, Francois-Moutal L, Chew LA, Yeon SK, Bellampalli SS, Qu C, Xie JY, Ibrahim MM, Khanna M, Park KD, Porreca F, and Khanna R 2017. ‘CRISPR/Cas9 editing of Nf1 gene identifies CRMP2 as a therapeutic target in neurofibromatosis type 1-related pain that is reversed by (S)-Lacosamide’, Pain, 158: 2301–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, and Helmchen F 2005. ‘Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo’, Science, 308: 1314–18. [DOI] [PubMed] [Google Scholar]
- O’Rawe JA, Wu Y, Dorfel MJ, Rope AF, Au PY, Parboosingh JS, Moon S, Kousi M, Kosma K, Smith CS, Tzetis M, Schuette JL, Hufnagel RB, Prada CE, Martinez F, Orellana C, Crain J, Caro-Llopis A, Oltra S, Monfort S, Jimenez-Barron LT, Swensen J, Ellingwood S, Smith R, Fang H, Ospina S, Stegmann S, Den Hollander N, Mittelman D, Highnam G, Robison R, Yang E, Faivre L, Roubertie A, Riviere JB, Monaghan KG, Wang K, Davis EE, Katsanis N, Kalscheuer VM, Wang EH, Metcalfe K, Kleefstra T, Innes AM, Kitsiou-Tzeli S, Rosello M, Keegan CE, and Lyon GJ 2015. ‘TAF1 Variants Are Associated with Dysmorphic Features, Intellectual Disability, and Neurological Manifestations’, Am J Hum Genet, 97: 922–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang Y, Cai Z, and Rhodes PG 2003. ‘Disturbance of oligodendrocyte development, hypomyelination and white matter injury in the neonatal rat brain after intracerebral injection of lipopolysaccharide’, Brain Res Dev Brain Res, 140: 205–14. [DOI] [PubMed] [Google Scholar]
- Perez-Reyes E 2003. ‘Molecular physiology of low-voltage-activated t-type calcium channels’, Physiol Rev, 83: 117–61. [DOI] [PubMed] [Google Scholar]
- Rajasankar S, Manivasagam T, and Surendran S 2009. ‘Ashwagandha leaf extract: a potential agent in treating oxidative damage and physiological abnormalities seen in a mouse model of Parkinson’s disease’, Neurosci Lett, 454: 11–5. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Cunningham LA, Wallace J, Alexander S, Estrada EY, Grossetete M, Razhagi A, Miller K, and Gearing A 2001. ‘Immunohistochemistry of matrix metalloproteinases in reperfusion injury to rat brain: activation of MMP-9 linked to stromelysin-1 and microglia in cell cultures’, Brain Res, 893: 104–12. [DOI] [PubMed] [Google Scholar]
- Sandweiss AJ, McIntosh MI, Moutal A, Davidson-Knapp R, Hu J, Giri AK, Yamamoto T, Hruby VJ, Khanna R, Largent-Milnes TM, and Vanderah TW 2018. ‘Genetic and pharmacological antagonism of NK1 receptor prevents opiate abuse potential’, Mol Psychiatry, 23: 1745–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonewille M, Gao Z, Boele HJ, Veloz MF, Amerika WE, Simek AA, De Jeu MT, Steinberg JP, Takamiya K, Hoebeek FE, Linden DJ, Huganir RL, and De Zeeuw CI 2011. ‘Reevaluating the role of LTD in cerebellar motor learning’, Neuron, 70: 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz PM, Borghesani PR, Levy RL, Pomeroy SL, and Segal RA 1997. ‘Abnormal cerebellar development and foliation in BDNF−/− mice reveals a role for neurotrophins in CNS patterning’, Neuron, 19: 269–81. [DOI] [PubMed] [Google Scholar]
- Slee EA, Adrain C, and Martin SJ 2001. ‘Executioner caspase-3, −6, and −7 perform distinct, non-redundant roles during the demolition phase of apoptosis’, J Biol Chem, 276: 7320–6. [DOI] [PubMed] [Google Scholar]
- Sokoloff L, Takahashi S, Gotoh J, Driscoll BF, and Law MJ 1996. ‘Contribution of astroglia to functionally activated energy metabolism’, Dev Neurosci, 18: 344–52. [PubMed] [Google Scholar]
- Suthprasertporn N, Mingchinda N, Fukunaga K, and Thangnipon W 2020. ‘Neuroprotection of SAK3 on scopolamine-induced cholinergic dysfunction in human neuroblastoma SH-SY5Y cells’, Cytotechnology, 72: 155–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo I, Soddemann M, Leanza L, Zoratti M, and Gulbins E 2011. ‘Single-point mutations of a lysine residue change function of Bax and Bcl-xL expressed in Bax- and Bak-less mouse embryonic fibroblasts: novel insights into the molecular mechanisms of Bax-induced apoptosis’, Cell Death Differ, 18: 427–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymonowicz K, Oeck S, Malewicz NM, and Jendrossek V 2018. ‘New Insights into Protein Kinase B/Akt Signaling: Role of Localized Akt Activation and Compartment-Specific Target Proteins for the Cellular Radiation Response’, Cancers (Basel), 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang EH, and Tjian R 1994. ‘Promoter-selective transcriptional defect in cell cycle mutant ts13 rescued by hTAFII250’, Science, 263: 811–4. [DOI] [PubMed] [Google Scholar]
- Wang S, Yabuki Y, Matsuo K, Xu J, Izumi H, Sakimura K, Saito T, Saido TC, and Fukunaga K 2018. ‘T-type calcium channel enhancer SAK3 promotes dopamine and serotonin releases in the hippocampus in naive and amyloid precursor protein knock-in mice’, PLoS One, 13: e0206986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Lu W, Li Y, and Tang B 2013. ‘Alpinetin promotes Bax translocation, induces apoptosis through the mitochondrial pathway and arrests human gastric cancer cells at the G2/M phase’, Mol Med Rep, 7: 915–20. [DOI] [PubMed] [Google Scholar]
- Warfield L, Ramachandran S, Baptista T, Devys D, Tora L, and Hahn S 2017. ‘Transcription of Nearly All Yeast RNA Polymerase II-Transcribed Genes Is Dependent on Transcription Factor TFIID’, Mol Cell, 68: 118–29 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Wang L, Olson JE, and Lu L 2001. ‘Asymmetrical response of p38 kinase activation to volume changes in primary rat astrocytes’, Exp Biol Med (Maywood), 226: 927–33. [DOI] [PubMed] [Google Scholar]
- Xu J, Yabuki Y, Yu M, and Fukunaga K 2018. ‘T-type calcium channel enhancer SAK3 produces anti-depressant-like effects by promoting adult hippocampal neurogenesis in olfactory bulbectomized mice’, J Pharmacol Sci, 137: 333–41. [DOI] [PubMed] [Google Scholar]
- Yabuki Y, Matsuo K, Izumi H, Haga H, Yoshida T, Wakamori M, Kakei A, Sakimura K, Fukuda T, and Fukunaga K 2017. ‘Pharmacological properties of SAK3, a novel T-type voltage-gated Ca(2+) channel enhancer’, Neuropharmacology, 117: 1–13. [DOI] [PubMed] [Google Scholar]
- Yoo JM, Lee BD, Sok DE, Ma JY, and Kim MR 2017. ‘Neuroprotective action of N-acetyl serotonin in oxidative stress-induced apoptosis through the activation of both TrkB/CREB/BDNF pathway and Akt/Nrf2/Antioxidant enzyme in neuronal cells’, Redox Biol, 11: 592–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. SAK3 improves the morphology of the Purkinje cells in the cerebellum altered by TAF1 gene editing. (A, B) The morphology of the Purkinje cell layer was evaluated by H&E Staining and Nissl Staining. The H&E and Nissl staining showed abnormal architecture of the Purkinje cells layer including hypoplastic Purkinje cells (Av & Bv). SAK3 administration to the TAF1 edited animals showed the morphology of the Purkinje cells were improved as compared to TAF1 edited animals (Avi & Bvi). Scale bars: 500 μm. The experiments were conducted in a blinded fashion.
Figure S2. The effects of SAK3 on cerebellum of Naïve and gRNA-control animals. Apoptosis was assessed in cerebellum samples using TUNEL assay. Shown are TUNEL assay photomicrographs from Naive and gRNA-control animals treated with SAK3. Note water and SAK3 treatment to the naïve and gRNA-control animals no significantly changed TUNEL positive cells were observed. Scale bars: 200 μm. The experiments were conducted in an investigator-blinded manner.
Figure S3. The effects of SAK3 on CaMKII. (A) Representative Western analysis is shown from cerebellum samples from each of the experimental conditions. Note decreased p-CaMKII in gRNA-TAF1 edited animals and increased p-CaMKII levels in gRNA-control. (B) Quantification of p-CaMKII expression on all the experimental condition. Data shown are mean + SEM, n = 4 animals per each experimental condition. No significant changes were observed between the gRNA-TAF1 edited animals and SAK3 treated gRNA-TAF1 animals (Data Analyzed by Kruskal Wallis Test).