

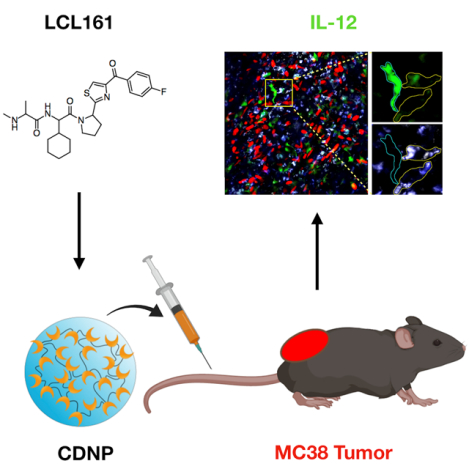
Graphical Abstract
Keywords: Cancer, Immunotherapy, High-Content Drug Screening, Dendritic Cells, Interleukin-12, Nanoparticle, Drug Delivery
SUMMARY
Immune checkpoint blockers can promote sustained clinical responses in a subset of cancer patients. Recent research has shown that a subpopulation of tumor-infiltrating dendritic cells function as gatekeepers, sensitizing tumors to anti-PD-1 treatment via production of interleukin-12 (IL-12). Hypothesizing that myeloid cell-targeted nanomaterials could be used to deliver small molecule IL-12 inducers, we performed high-content image-based screening to identify the most efficacious small molecule compounds. Using one lead candidate, LCL161, we created a myeloid-targeted nanoformulation that induced IL-12 production in intratumoral myeloid cells in vivo, slowed tumor growth as a monotherapy and had no significant systemic toxicity. These results pave the way for developing combination immunotherapeutics by harnessing IL-12 production for immunostimulation.
INTRODUCTION
The past few years have witnessed accelerating development of numerous immunotherapy strategies for cancer treatment(Tang et al., 2018). Immune checkpoint blockade has now demonstrated clinical efficacy against several types of cancer. Despite these promising results, significant hurdles remain. Side effects are not uncommon(Pauken et al., 2019), and immunotherapy is only effective in a fraction of patients(Fares et al., 2019). In an effort to further improve checkpoint therapies, a series of recent studies have focused on elucidating cellular mechanisms of action in vivo in order to better understand limited therapeutic efficacy and resistance(Arlauckas et al., 2017; Garris et al., 2018; Moynihan et al., 2016; Ruffell et al., 2014; Spranger et al., 2015). Through these and other studies, a clearer picture of myeloid cells’ previously underappreciated role is emerging(Engblom et al., 2016). As a result, we now know that effective anti-PD-1 therapy requires intratumoral production of interleukin-12 (IL-12) by myeloid cells and, in particular, dendritic cells (DCs)(Garris et al., 2018).
Several therapeutic strategies may enhance IL-12 production in the tumor environment(Lasek et al., 2014). Directly, systemically administering the cytokine has had limited success due to broad immunotoxicity(Lasek et al., 2014; Wang et al., 2017). Alternative strategies for more selective tumoral delivery have included intratumoral injection, delivery via viral vectors and vaccination with IL-12-positive tumor cells(Cody et al., 2012; Lasek et al., 2004; Rodolfo et al., 1996; Song et al., 2000). IL-12 production in DCs and other myeloid cells can also be increased by stimulating TNF receptor superfamily members (e.g., CD40, OX40 or LTBR) with agonistic antibodies(Hassan et al., 2014; Jahan et al., 2018; Lukashev et al., 2006; Ma et al., 2019; Sun, 2017; Vonderheide and Glennie, 2013; Vonderheide, 2018). Finally, another strategy would be to increase IL-12 production via small molecule inhibitors of certain myeloid pathways(Dougan and Dougan, 2018). Small molecules can access intracellular targets, can be repurposed rapidly and are comparatively inexpensive. Unfortunately, many existing small molecule pharmaceutical classes have unknown effects on IL-12 production, are not targeted to myeloid cells, have unfavorable pharmacokinetics and show off-target toxicities when administered systemically.
We hypothesized that pharmacological candidates could be identified and rank ordered through high-content screening of IL-12 production in reporter cells. Furthermore, we expected that a nanoformulation could be used to deliver inhibitors to tumoral myeloid cells to enhance IL-12 production locally within the tumor microenvironment. Prior research has shown that small molecule biomaterial carriers are an effective strategy to deliver drugs more selectively to phagocytic cells, including both macrophages and dendritic cells, in the tumor microenvironment(Weissleder et al., 2005). Nanocarriers can also solubilize drugs that otherwise have poor phase solubility, thereby enhancing immunomodulation by modifying drug pharmacokinetics(Weissleder et al., 2014; Rodell et al., 2018). To date, however, little work has been done to identify how such strategies could activate tumoral myeloid cells toward an immunotherapeutically critical IL-12-producing state in vivo.
Herein, we developed and used a small molecule high-content screen in primary bone marrow- derived DCs to identify compounds that enhance IL-12 production. Of the compounds screened, multiple inhibitors of cIAP (cellular inhibitor of apoptosis) promoted IL-12 production by activating the non-canonical NFkB pathway. We subsequently focused on the particular cIAP inhibitor, LCL161, which had excellent drug activity but limited pharmacological utility due to poor phase solubility. We successfully complexed LCL161 to cyclodextrin nanoparticles (CDNP-LCL161), thereby allowing for delivery to abundant intratumoral myeloid cells. Monotherapy with CDNP-LCL161 attenuated tumor growth, and realtime intravital imaging confirmed a remarkable increase in the IL-12-producing immune cell infiltrate.
RESULTS
High-content screening of agents that induce IL-12 in dendritic cells.
We developed a high-content screening approach to identify IL-12-inducing agents (summarized Fig. 1). We used commercially available IL-12 reporter mice. These mice have an internal ribosomal entry site-enhanced yellow fluorescent protein (IRES-eYFP) sequence inserted downstream of the endogenous IL12B gene, encoding for the p40 subunit of IL-12. We generated bone marrow-derived dendritic cells (BMDCs) by treating bone marrow cells with recombinant murine FLT3 ligand. Cells were seeded into 384-well plates; 9 days later, cells were treated for 27 hours with compounds. We screened 42 compounds at 7 doses, ranging from 31.6 nM to 31.6 μM, at 1/2-log titration. Following treatment, cells were fixed and then stained with Hoechst 33342 and wheat germ agglutinin to mark the nuclei and cell boundaries, respectively. Upon preliminary examination of our data, we observed considerable single cell heterogeneity in YFP signal. As a result, to score for YFP induction, we generated a single cell distribution of YFP intensities and extracted the intensity at the 95th percentile. We found that this method provided a robust signal that effectively discriminated negative (DMSO) and positive (LPS and IFNγ) controls.
Figure 1. Developing a high-content screen for interleukin-12 soliciting agents.
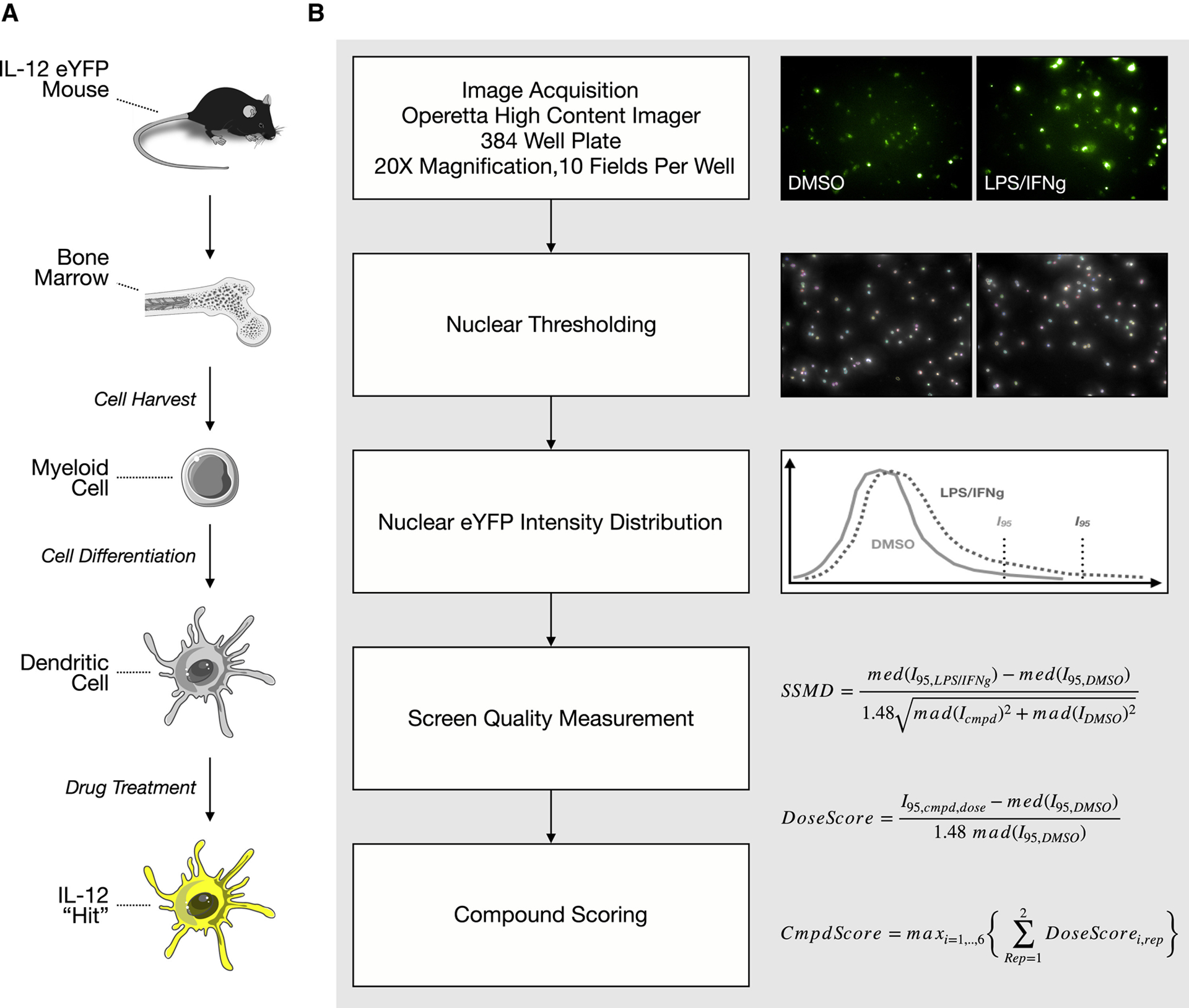
a, Graphic abstract of screen. Bone marrow cells were isolated from an IL-12 reporter mouse and differentiated into dendritic cells, which were subsequently used for a high-content screen. b, Image analysis pipeline. After nuclear thresholding, YFP scores were computed and used to rank order compounds.
We performed the screen twice and generated a cumulative score by averaging scores from the two independent replicates. Each compound was ultimately scored by the maximum effect captured from the dose (Fig. 1). Rank-ordering the compound scores showed that dual TLR and innate immune agonists were among the most effective, consistent with our expectations (Fig. 2). Scoring just below the TLR agonists was a panel of inhibitors of apoptosis protein (cIAP) cellular inhibitor and X-linked inhibitor of apoptosis protein (XIAP). These drugs are second mitochondria-derived activators of caspases (SMAC) mimetics and were originally designed to sensitize cancer cells for apoptosis. More recently, it has become clear that these drugs activate the non-canonical NFkB pathway by inhibiting of cIAP1/2(Chesi et al., 2016). That all small molecule cIAP inhibitors scored supports cIAP inhibition as these drugs’ relevant mechanism of action. We ultimately decided to focus on these drugs over the TLR agonists, as their potential as immunomodulatory agents has been less thoroughly explored. cIAP inhibitors’ immunostimulatory effects have been examined in T cells and NK cells(Clancy-Thompson et al., 2018; Dougan et al., 2010), but focused work on their mechanism in dendritic cells is only beginning to emerge(Garris et al., 2018). We additionally expected these drugs to be more compatible with nanomaterials and thought that enhanced efficacy in vivo could be achieved by dual immunomodulation in the tumor microenviroment as well as direct pro-apoptotic effects on tumor cells via inhibiting XIAP.
Figure 2. High-content screening identifies agents that reliably induce interleukin-12 expression.
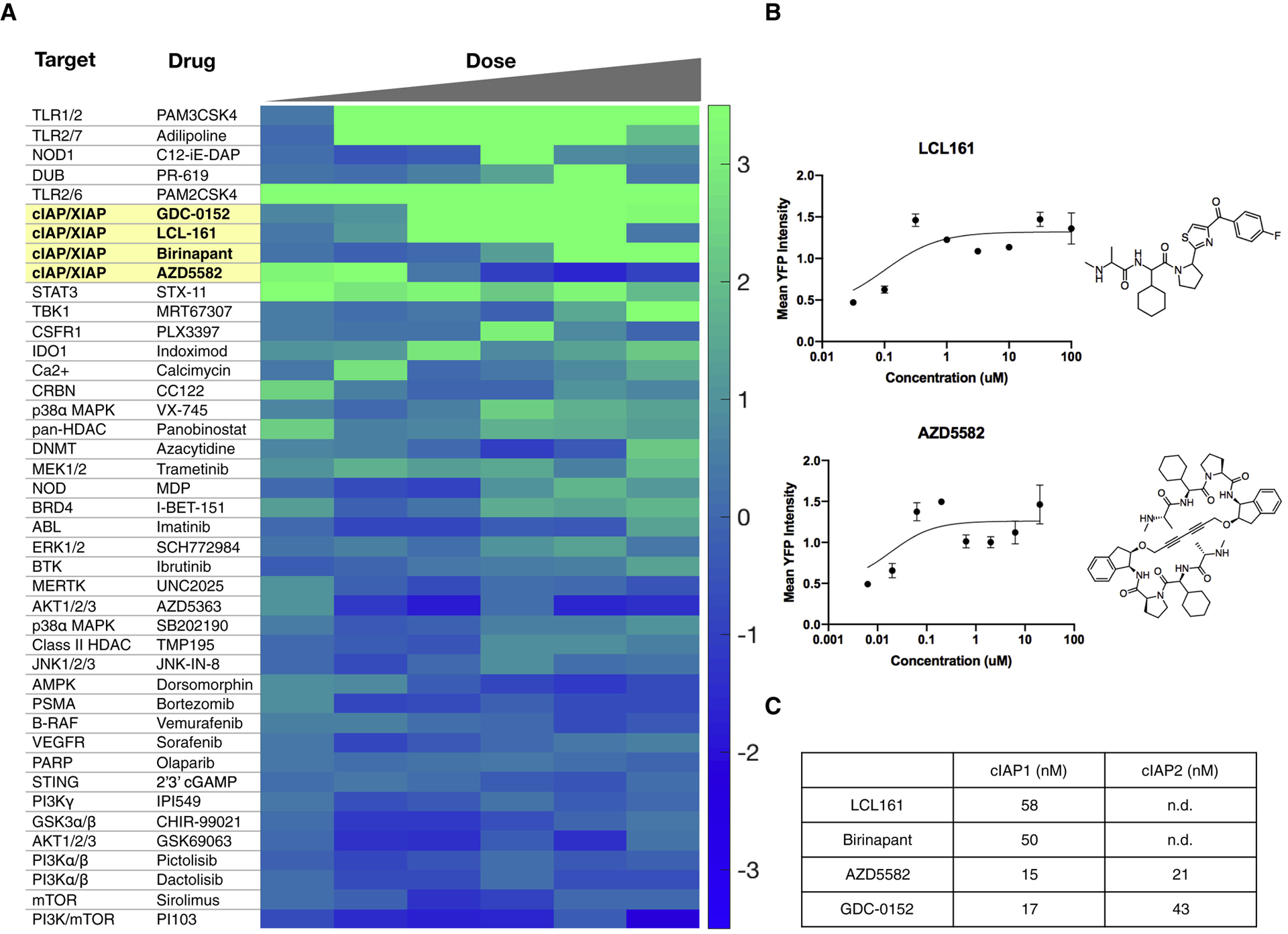
a, Heatmap of compound bioactivities for IL-12 YFP induction. Compounds were screened from 10 μM to 31.6 nM at 1/2 log titration. The scores from the first five doses (10 μM - 100 nM) are averages from two separate, independent screens while the score at the sixth dose (31.6 nM) was obtained from only one screen. b, Dose response curves and structures of cIAP1/2 inhibitors LCL161 and AZD5582. Data plotted as mean ± s.d.; n=2. c, Biochemical IC50 values for cIAP inhibitors. Data were collected from PubChem and SelleckChem.
Validating LCL161 in dendritic cells.
We focused on LCL161 for follow up studies because there are several ongoing and completed clinical trials (e.g. NCT01955434, NCT01968915, NCT02649673) using it for a number of solid tumors as well as blood malignancies (Fulda, 2015; Infante et al., 2014; Pemmaraju et al., 2016) and it is compatible with nanoparticle delivery. We generated BMDCs as before, using FLT3 ligand to differentiate bone marrow progenitor cells into DCs (Fig. 3a). Re-testing the cIAP1/2 inhibitors LCL161 and AZD5582 confirmed they instigate the eYFP reporter (Fig. 2b). We next ascertained that promoting eYFP correlated with increased IL-12 levels (Fig. 3b) and was not a false positive due to a spurious effect (e.g. intrinsic compound fluorescence). In addition to up-regulating IL-12b mRNA levels in bone marrow-derived dendritic cells, LCL161 also promoted IL-12 induction in bone marrow-derived macrophages, though to a lesser extent than in DCs, potentially due to endogenous differences in pathway activation (Fig. 3c).
Figure 3. The cIAP inhibitor LCL161 solicits interleukin-12 expression through the non-canonical NFkB pathway.
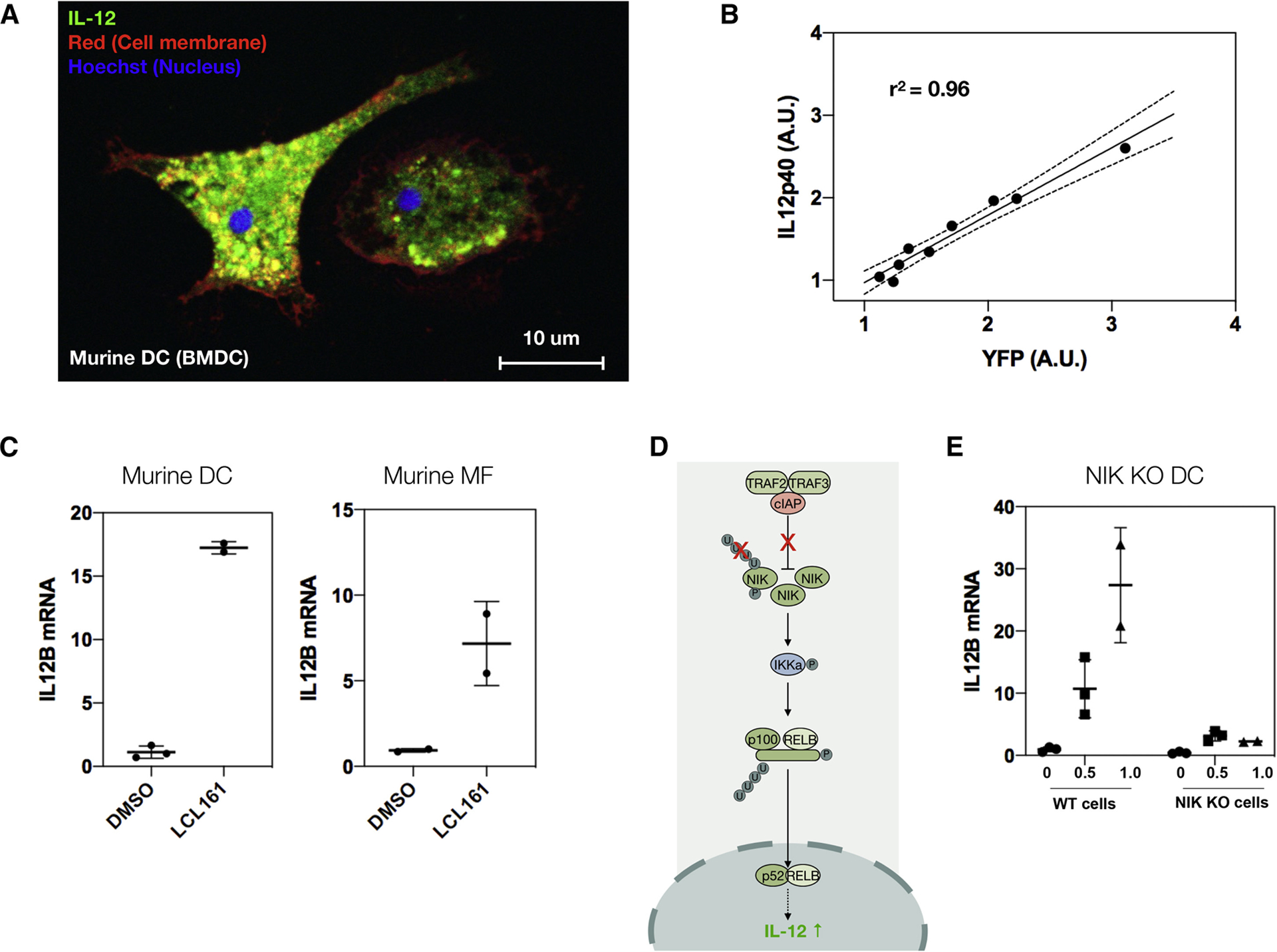
a, Representative image of IL-12-eYFP BMDC. b, Correlation of YFP levels with IL-12p40 production. Stimulating BMDCs with increasing doses of LCL161 (100 nM to 10 μM at 1/4 log titration) upregulates the eYFP reporter, which correlates with endogenous IL-12p40 levels, as measured by indirect immunofluorescence for IL-12p40. Black line: linear regression ± 95% CI (dotted line). c, LCL161 (0.316 μM, 1 day) elevates/raises IL-12p40 in both bone marrow-derived dendritic cells and macrophages, though more so in dendritic cells. Data reported as mean ± s.d.; n=2 or 3, as indicated on plot. d, Cartoon schematic of non-canonical NFkB pathway. LCL161 inhibits the cIAP E3 ligase complex, preventing ubiquitination of NIK, thereby blocking proteasomal degradation. This increases NIK, which leads to nuclear translocation of the active p52 and RelB NFkB subunits. This in turn causes IL-12 production. e, The response to LCL161 (doses indicated, 1 day) in NIK KO BMDCs is markedly reduced compared to WT BMDCs.
We subsequently confirmed that LCL161 induces IL-12 production via the non-canonical NFkB pathway. cIAP1/2 form a complex with the E3 ligases TRAF2 and TRAF3. This complex inhibits expression of NF-kB inducing kinase (NIK), by marking it for proteasomal degradation (Fig. 3d). Blocking cIAP1/2 by LCL-161 allows NIK to be expressed, causing IKKa phosphorylation, which then leads to p100 processing. This in turn results in NFkB’s p52 and c-Rel subunits translocating into the nucleus where they elevate levels of pro-inflammatory cytokines. To test this mechanism of action, we treated BMDCs from a NIK KO mouse and saw minimal IL-12 induction compared to in WT mice (Fig. 3e), confirming that LCL161 elicits its effects via the non-canonical NFkB pathway.
Synthesizing drug-laden supramolecular nanocarriers.
Cyclodextrin nanoparticles (CDNPs) can be used to solubilize small molecule therapeutics and deliver them selectively to phagocytic immune cells in the tumor microenvironment(Rodell et al., 2018). CDNPs thus avoid the poor pharmacokinetic properties common to many small molecule immune modulators while also delivering drugs selectively to phagocytic cells in the tumor microenvironment, thereby mitigating off-target toxicity. For these reasons, CDNPs are a particularly attractive delivery modality for immunotherapeutics.
To demonstrate that LCL161 can be complexed to CDNPs to form the desired drug-laden nanoformulation (LCL161-CDNP, Fig. 4a), we first confirmed interaction between cyclodextrin and LCL161. Adding LCL161 to a phenolphthalein-cyclodextrin complex raised phenolphthalein absorbance (Fig. 4b), indicating that LCL161 competes with phenolphthalein to occupy the hydrophobic cavity within the cyclodextrin macrocycle, and that drug solubilization by guest-host complexation should therefore be possible.
Figure 4. Cyclodextrin nanoacrriers solubilize LCL161 for systemic delivery through guest-host complexation.
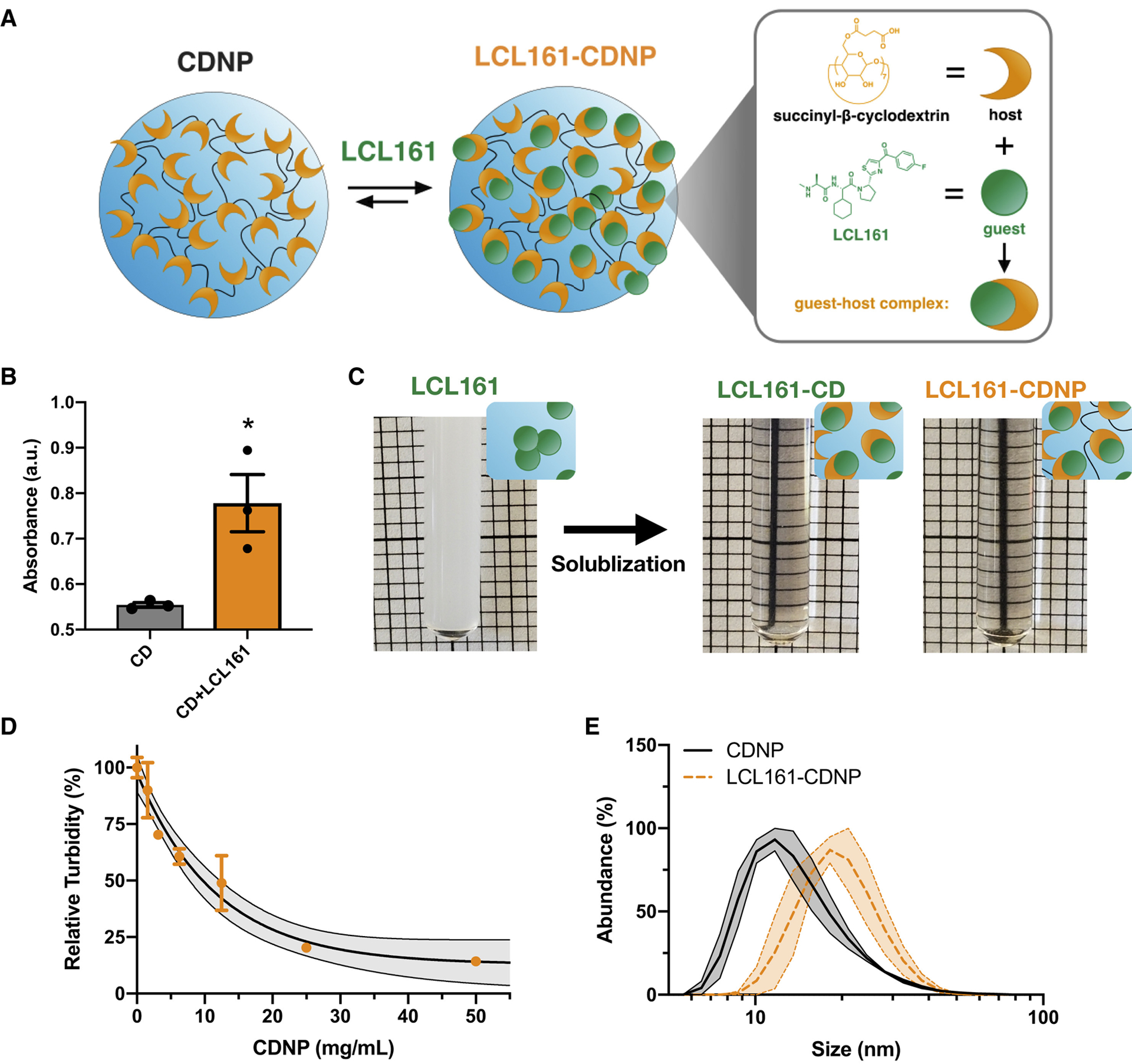
a, Schematic of cyclodextrin nanoparticles (CDNPs) prepared by L-lysine crosslinking of cyclodextrin succinate (orange). LCL161 (green) was subsequently complexed with the nanoparticle through supramolecular interaction (expanded, right) between the host (cyclodextrin) and guest (LCL161) to form a guest-host complex. b, Phenolphthalein absorbance (200 μM, λ=550 nm) in the presence of cyclodextrin (0.2 mM) before and after addition of LCL161 (1.0 mM). Mean ± s.d.; n=3; *P < 0.05, Student’s t-test. c, Macroscopic images of LCL161 insolubility in PBS (5 mM, turbid due to drug aggregation) and solublization by 2-hydroxypropyl-β-cyclodextrin (LCL161-CD, middle) or the supramolecular nanocarrier (LCL161-CDNP, right). d, Phase solubility assessment of LCL161 by turbidity measurement (5 mM, λ=500 nm) in CDNP solutions. Mean ± s.d.; n=3. Black line: exponential decay ± 95% CI (shaded). e, Dynamic light scattering measurement of hydrodynamic diameter for blank and drug-laden nanoparticle preparations. Black line: mean ± s.e.m. (shaded); n=3.
Indeed, LCL161 was highly insoluble in aqueous conditions, as indicated by visually apparent sample turbidity. The drug was readily solubilized upon addition of either 2-hydroxypropyl-β-cyclodextrin (HPβCD; for in vivo administration, vide infra) or CDNPs themselves (Fig. 4c). To quantify solubilization by CDNPs specifically, we examined LCL161’s phase solubility at increasing CDNP concentrations. Turbidity decreased with increasing CDNP concentration, and the drug was fully solubilized at 50 mg/mL nanoparticle concentrations used for formulations administered in subsequent studies. Lastly, we characterized nanoparticle size (DLS, Fig. 4e) and found that a moderate increase in CDNP diameter (19.1±1.6 nm) occurred following drug loading (22.8 ± 3.4 nm), as expected based on prior studies. Large drug aggregates were not observed in the sample preparations.
Therapeutic efficacy against MC38 tumors.
Having demonstrated LCL161’s capacity to induce IL-12 in vitro and the CDNP carrier’s ability to complex the drug for delivery, we went on to examine therapeutic efficacy against the growth of established MC38 tumors (Fig. 5a). Mice were treated by intravenous injections every other day, commencing 8 days after tumor inoculation to allow formation of established (~100 mm3), vascularized tumors. For free drug controls, LCL161 was necessarily solubilized by HPβCD (LCL161-CD). Drug administrations moderately attenuated tumor growth relative to control mice receiving the blank nanoparticle (not significant, Fig. 5b). LCL161-CDNP halted tumor growth without causing undesirable loss of body weight during the course of study (Fig. 5d), in contrast with immune agonists which induce body weight loss. The LCL161-CDNP treatment cohort had a homogenous response to therapy, while individual tumor growth curves (Fig. 5c) highlight only a partial response in LCL161-CD free drug controls.
Figure 5. LCL161 monotherapy attenuates tumor growth and is enhanced by nanoformulation.
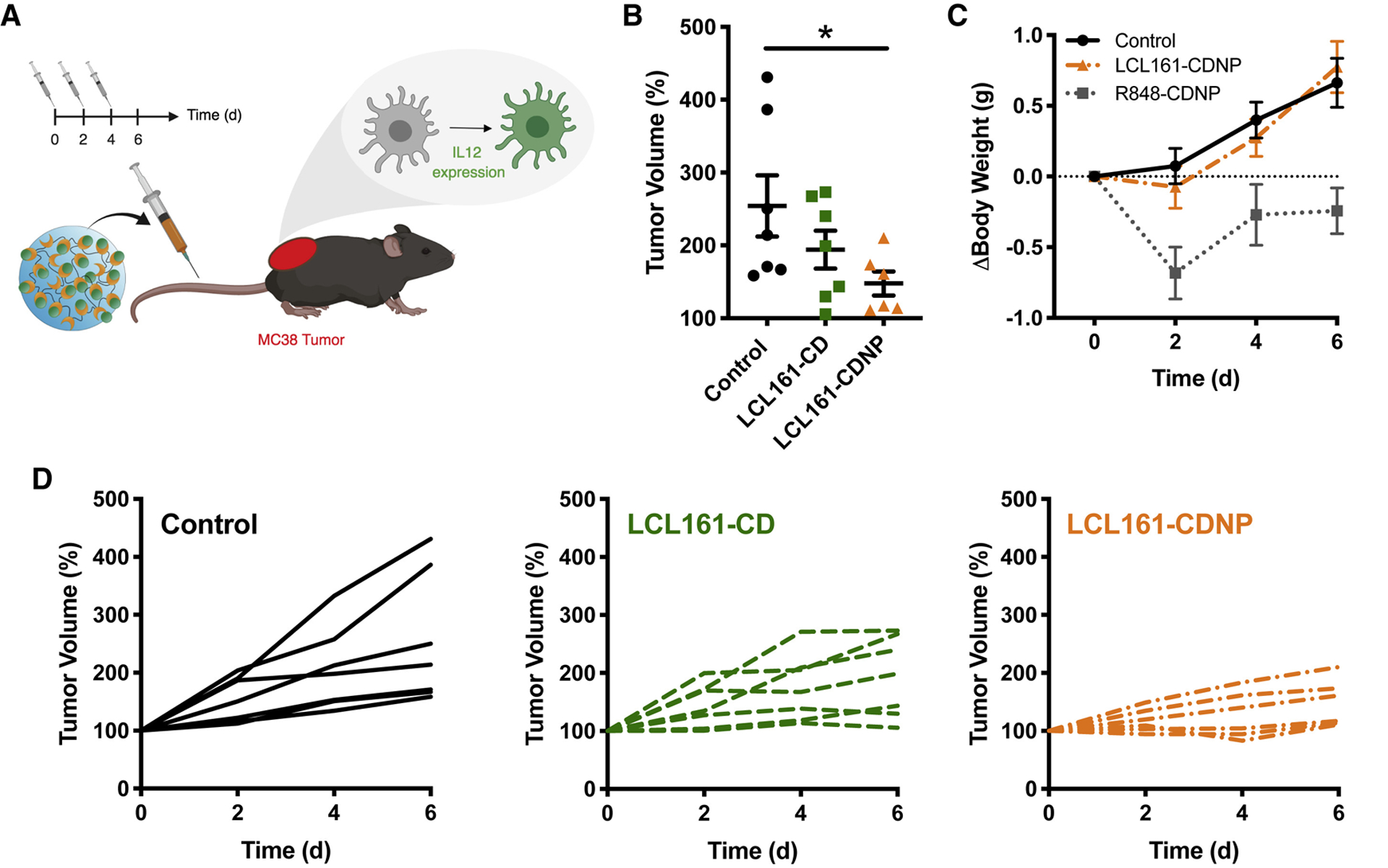
a, Schematic overview of study. Treatments (control: blank nanoparticle, LCL161-CD: drug solublized by HPβCD and LCL161-CDNP: drug-nanoparticle complex) were administered every other day in mice with a single established MC38 tumor. b, Change in tumor volume at day six, relative to animal baseline. Mean ± s.e.m.; n=7; *P < 0.05, Dunn’s multiple comparison. c, Individual tumor growth in response to treatment by control (left, back), LCL161 (middle, green) or LCL161-CDNP (right, orange). d, Change in body weight in response to treatment by control (black), LCL161-CDNP (orange) or R848-CDNP (gray; 10 mg/kg, data from previous study). Mean ± s.e.m.; n=7. See also Figures S1, S4, S5, S6.
LCL161 is pro-apoptotic and can induce cell death by sensitizing the apoptosis threshold of cancer cells(Chen et al., 2012). Thus, it is possible that the anti-tumor effects seen above are not due solely to immunostimulation, but also to direct anti-cancer effect of the drug. While the premise of our nanoparticle formulation is to deliver the drug predominately to tumoral myeloid cells, tumor cells may yet be inadvertently exposed to LCL161. To consider the possibility that LCL161 exerts some anti-tumor effect via direct action, we treated MC38 cells, in vitro, with 10 doses of LCL161 for 1 day and for 1 week. In both cases, we found no effect on cellular viability (Fig. S1), indicating that the anti-tumor effects in the MC38 model are primarily due to immunostimulation.
Intravital imaging of LCL161-CDNP treatment.
Increased IL-12 induction was shown in vitro following LCL161 treatments. However, these results may not fully recapitulate the complex tumor microenvironment, where competing signals may neutralize efficacy and drug delivery to the cell populations of interest is a critical factor. In order to directly examine the therapeutic distribution and effect on IL-12 production in vivo (Fig. 6a), we employed a p40-IRES-eYFP reporter mouse fitted with a dorsal skinfold window chamber for intravital confocal fluorescence microscopy. Pacific Blue-labeled dextran was administered before imaging to identify myeloid cells. Immediately preceding treatment by LCL161-CDNP, myeloid cells expressed only low levels of eYFP. Within minutes after therapeutic injection, LCL161-CDNP distributed throughout the vasculature and began to accumulate in myeloid cells. At these early time points, within 1 hour of administration, IL-12 induction, as measured by the eYFP proxy, was not apparent. At 24 hours after treatment, however, myeloid cells throughout the tumor accumulated LCL161-CDNP and eYFP-positive cells starkly increased. A nearly tenfold increase in the number of eYFP positive cells occurred as a result of LCL161-CDNP treatment (Fig. 6b,c), and IL-12 high expressers were highly motile within the tumor (Mov. S1).
Figure 6. Cyclodextrin nanocarriers loaded with LCL161 distribute rapidly to tumor-associated myeloid cells, eliciting interleukin-12 production in vivo.
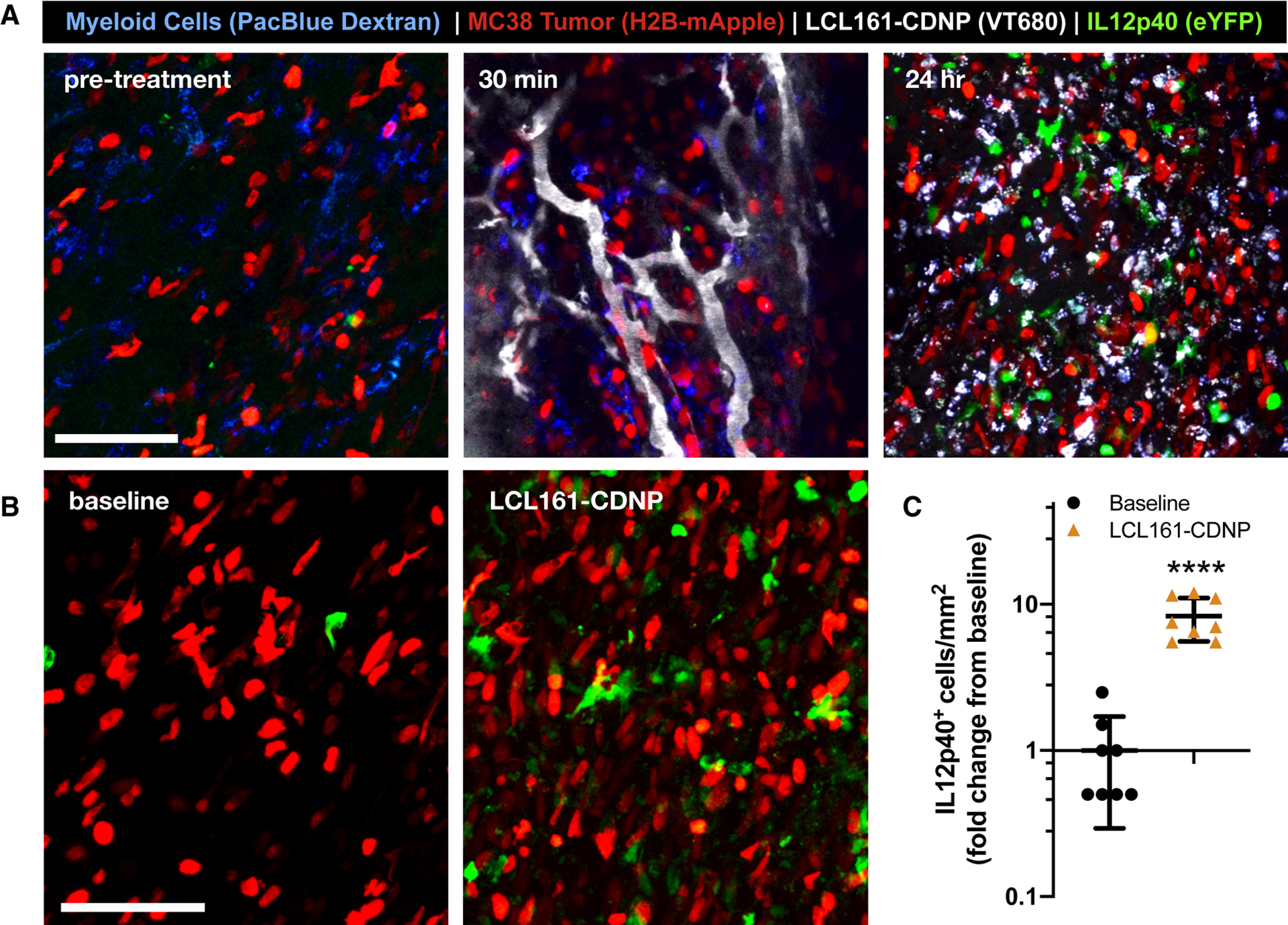
The cellular distribution of the LCL161-CDNP complex was examined by confocal fluorescence microscopy through a dorsal window chamber in an IL-12p40-eYFP mouse bearing an MC38-H2B-mApple tumor. a, The CDNP complex (VT680, gray) rapidly distributed throughout the tumor and accumulated in tumor-associated myeloid cells (blue). At 24 hours, both myeloid-associated nanoparticle uptake and IL-12p40-eYFP expression were evident. Scale bars: 100 μm. b, Expression of IL-12p40-eYPF was assessed before treatment (baseline, left) and at 24 hr post-treatment (LCL161-CDNP, right) c, Corresponding quantification of IL-12hi cells. Mean ± s.d., n=8 fields of view per condition; ****P < 0.0001, Student’s t-test. See also Figures S2, S3 and Movie S1.
PK/PD of LCL161-CDNPs
Pharmacokinetic analysis of the distribution of CDNPs in myeloid cells via intravital microscopy is complicated by the absence of mouse models to readily distinguish myeloid cell subsets at a resolution currently possible by scRNAseq(Pittet et al., 2018). Using fluorescently labeled CDNPs, however, we have previously shown that CDNPs primarily localize to tumor associated macrophages and to a lesser extent, dendritic cells and neutrophils(Rodell et al., 2018). Additional analysis of our intravital imaging data shows that tumor cells did not take up appreciable amounts of CDNPs (fig. S2).
A subtype of dendritic cells are known to be the primary producers of IL-12 at basal level(Garris et al., 2018; Zilionis et al., 2019). As shown in Fig. 3, LCL161 can induce IL-12 in both dendritic cells and macrophages, with higher induction in dendritic cells. To study IL-12 production in vivo, we further analyzed time-lapse intravital microscopy recordings. We identified two populations of IL-12-expressing immune cells upon LCL161 treatment. The first population included motile, elongated cells with minimal internalization of Pacific Blue-dextran that were strongly positive for the eYFP reporter (Mov. S1). The second population were sessile, round cells that had high levels of Pacific Blue-dextran, and a comparatively lower level of eYFP reporter (Mov. S1). Based on morphology and Pacific Blue-dextran levels, these groups likely correspond to dendritic cells and macrophages respectively. The possibility that IL-12 is more strongly induced in dendritic cells than in macrophages is consistent with in vitro data referenced above.
To further study pharmacodynamics, we considered the effect of LCL161-CDNP on IL-12 production at the organismal level. Tumors, tumor draining lymph nodes, and axillary lymph nodes from a LCL161-CDNP treated mouse were harvested, and cellular IL12-eYFP intensity was quantified. We found that the tumors and tumor draining lymph node were strongly positive for Il12-eYFP cells, while the axillary lymph nodes were not (Fig. S3). Confocal microscopy confirmed these findings (Fig. S3). These results suggest that the anti-tumor effect of IL-12 largely arises from myeloid cells proximal to the tumor. Note that this is consistent with previous biodistribution studies indicating that nanoparticles are heavily concentrated in the tumor.
Toxicity analysis of LCL161-CDNP
Existing small molecule cIAP inhibitors often have poor solubility and potentially systemic side effects through their inhibition of apoptosis in tissues that experience peak concentrations and cellular uptake of the small molecule. Reported clinical side effects for systemically administered LCL161 include cytokine release syndrome, nausea, neutropenia, diarrhea, pneumonia, and pyrexia(Bardia et al., 2018; Infante et al., 2014). The overall goal of the nanoparticle formulation in this work was to drive the cIAP inhibitor more selectively into phagocytes in the tumor microenvironment while a secondary intention was to reduce high systemic peak concentrations as is commonly observed with nanoformulations(Ventola, 2017).
Measurements of whole body weight over time indicated that the LCL161-CDNPs were well tolerated (Fig. 5d). To further analyze toxicity, we performed additional measurements in tissues of interest. CDNPs by themselves have an excellent safety profile; indeed, cyclodextrin itself is already used in a number of commercial products(Davis and Brewster, 2004; Rodell et al., 2015; Rodell et al., 2018; Szejtli, 1998; Ventola, 2017; Zhang and Ma, 2013). Several other studies to date using CDNPs have not documented any significant toxicities(Machelart et al., 2019; Rodell et al., 2018). Previous studies using fluorescently labeled CDNPs indicate that they also strongly localize to the tumor through phagocytic uptake, so the LCL161 should be highly concentrated there(Kim et al., 2018; Rodell et al., 2018; Rodell et al., 2019). However, CDNPs are cleared through the reticular endothelial system, which includes cells of the liver and spleen. As both organs contain numerous, regenerative cells as well as white blood cells, potential toxicity may first manifest at these sites.
To address this, we treated mice with LCL161-CDNPs, and examined for acute hepatic and splenocyte toxicity via hematotoxylin and eosin (H&E) staining. The livers and spleen did not show any toxicity compared to CDNP and saline treatment, and likewise had no excessive infiltrate of immune cells (Fig. S4). To further consider toxicity, we also measured weights of organs from mice treated with either saline, CDNPs, LCL161-CD, or LCL161-CDNPs (Fig. S5). No significant decreases were evident. There was an increase in weight of small intestine, though this effect is not suggestive of toxicity.
In regards to immunotoxicity, we treated tumor-bearing mice with saline, CDNPs, or LCL161-CDNPs, and measured serum concentrations of IL-12. We found that mice treated with LCL161-CDNPs had mildly elevated serum IL-12 concentrations (Fig. S6), as would be expected for an efficient immunotherapeutic. However, these concentrations were below those indicative of toxicity(Abdi et al., 2018).
DISCUSSION
Stimulating IL-12 production, predominantly produced in dendritic cells and macrophages(Lasek et al., 2014), has emerged as a therapeutic strategy for cancer immunotherapy. IL-12 elicits an anti-tumor response through a variety of mechanisms in multiple cell types. Further, IL-12 has been shown to promote IFNγ production, thereby activating T and NK cells, enhancing their cytotoxicity and triggering a Th1 type response(Lasek et al., 2014; Tait Wojno et al., 2019). Various other mechanisms, such as anti-angiogenic effects of tumor vasculature, in non-immune cell types may also be relevant and possibly complementary to tumor immune infiltration(Angiolillo et al., 1996; Lasek et al., 2014). Finally, it has been shown that IL-12 production in a specific subset of tumor-associated dendritic cells is critical for mounting a successful anti-tumor immune response(Garris et al., 2018).
A number of strategies to augment tumor IL-12 production are in clinical trials. Perhaps most advanced is the technology behind the ImmunoPulse tavokinogene telseplasmid, an electroporation method that delivers IL-12 plasmid directly to tumors(Daud et al., 2008). Additionally, agonistic antibodies against CD40 (TNF receptor superfamily 5: TNFRSF5), a 48 kDa type I transmembrane protein expressed by antigen-presenting cells, have been shown to increase IL-12 and suppress tumor growth(Hassan et al., 2014; Johnson et al., 2015; Vonderheide and Glennie, 2013). Various antibodies, such as CP-870,893 and ADC-1013, are likewise in clinical trials (e.g. NCT01103635, NCT02379741). Here, we explored a different strategy, namely blocking a non-canonical NFkB pathway inhibitor. Small molecule inhibitors of cIAP have previously been described(Chesi et al., 2016; Cong et al., 2019; Garris et al., 2018) but have delivery challenges and considerable side effects(Fulda, 2015; Infante et al., 2014). We circumvented these issues by complexing LCL161 to a hydrophilic cyclodextrin nanoparticle. The latter has affinity for phagocytic myeloid cells(Ahmed et al., 2019; Rodell et al., 2018). We show a 10-fold increase in tumoral IL-12 production, lack of systemic toxicity and modest efficacy when used as monotherapy.
The current landscape of immunotherapeutics covers many approaches including small molecules, biologics, cellular therapies, and gene therapies(Cody et al., 2012; Daud et al., 2008; Engeland and Bell, 2020; Osipov et al., 2019; Sanmamed and Chen, 2018; Tang et al., 2018; Wang and Mooney, 2018). The approach described in the current manuscript is complementary and offers potential advantages. By using a small molecule, we avoid some of the challenges pertinent to biologics and protein therapeutics. Small molecules are also more likely to access intracellular targets. By targeting an intracellular, downstream node, cIAP1/2, we theoretically minimize the chance of resistance that often occurs with biologics targeting immunological synapses on the cell surface(Sharma et al., 2017). Within the space of small molecules, the choice of target is somewhat unique. To date, most small molecule immunoactivators target pattern recognition receptors(Helms et al., 2019; Ramanjulu et al., 2018), whereas we target a downstream node, in a comparatively less explored pathway. Additionally, while LCL161 was not toxic against the MC38 cell line used in this study, it is reported to have pro-apoptotic effects against other cell lines. Thus, it is possible in a clinical setting that it may exert both pro-apoptotic and immunostimulatory effects. In fact, we would expect these effects to synergies if they indeed co-existed. Lastly, LCL161 is already in clinical trials, so it could potentially be rapidly repurposed, as discussed earlier.
We combine a small molecule with a nanoparticle formulation for enhanced delivery. Systemic delivery of a drug-nanoparticle formulation, may increase dosage and costs required for this type of therapy, but with the advent of intratumoral dosing, lower doses may ultimately become sufficient. Nanoparticles also have longer retention time in the tumor which leads to accumulation of large molecules in the tumor. This may also limit dosage required, and thereby limit systemic exposure to the drug. Overall, our approach combines existing small molecule drugs with a novel and versatile delivery system to maximize therapeutic efficacy.
In the future, we envision combining IL-12 stimulation with checkpoint blockade. The rationale for this approach is clear(Garris et al., 2018): effective anti-tumor responses to anti-PD-1 therapy requires tumor-infiltrating dendritic cells to produce interleukin 12 (IL-12) to fuel the anti-tumor reaction. Given that LCL161 induces substantial IL-12 production in dendritic cells, therapies combining LCL161 nanoformulations with checkpoint blockade could prove effective. Finally, in addition to its immunostimulatory effects, LCL161 also has direct pro-apoptotic effects by limiting XIAP. While not explored in this research, complexing LCL161 to a CDNP should result in some delivery to tumor cells, as through local release from immune cells(Miller et al., 2015) leading to apoptosis of cancer cells. Ensuing cell death could even further enhance immunogenicity, fueling an even stronger anti-tumor response. LCL161, as well as other cIAP inhibitors, represent attractive therapeutics to be used in the interface of cancer immunotherapy and nanomaterials.
STAR Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and request for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ralph Weissleder (rweissleder@mgh.harvard.edu). There are restrictions on the availability of the cyclodextrin nanoparticles generated in this study due to our need to maintain the supply. We may require a completed Materials Transfer Agreement if there is potential for commercial application.
EXPERIMENTAL MODELS
Mice:
The following mouse strains and reporters were used for cell isolation or tumor implantation: i) C57BL/6J mice (Jackson; n=1 for individual in vitro assays, n=6–8 for tumor implantation); ii) p40-IRES-eYFP reporter mice (Jackson; n=3 for primary screens, n=1 for subsequent in vitro assays, n=1 for intravital); iii) Map3k14 (NIK)-KO (Jackson; n=1 for in vitro assays). All animal studies were conducted in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Protocols were approved by the Institutional Animal Care and Use Committees at Massachusetts General Hospital.
For the in vivo studies, MC38 tumors were initiated in female C57BL/6 mice (Jackson, 6–8 weeks of age) by intradermal injection (2×106 cells in 50 μl of PBS). At 8 days post-injection, treatment cohorts were assigned such that tumor size and body weight were normalized across groups. Mice were treated every other day by intravenous injection of CDNP (5.0 mg/mouse), LCL161 (25 mg/kg; 0.5 mg/mouse) or LCL161-CDNP (0.5 mg/mouse LCL161, 5 mg/mouse CDNP). Tumor growth was assessed by caliper measurement and is reported as tumor volume according to ellipsoidal approximation (V=(L*W2)/2).
For intravital microscopy, examination was performed in dorsal skinfold window chambers implanted in female, p40-IRES-eYFP IL-12 reporter mice(Reinhardt et al., 2006). Tumors were initiated 8 days prior to treatment by inoculation with 2×106 MC38–H2B-mApple cells in 20 μl of PBS.
Cell Models.
Bone marrow-derived dendritic cells and macrophages were obtained using standard procedures(Arlauckas et al., 2017; Garris et al., 2018). Wild type C57BL/6J or reporter mice (see below) were sacrificed and femur and tibia bones were flushed with PBS to remove the bone marrow. Cells were then filtered through a 40 μm nylon mesh and centrifuged at 300xg for 10 min. To lyse red blood cells, the pellet was resuspended in 1 mL of ACK lysing buffer (Lonza) and incubated for 2 min on ice. Cells were then washed in PBS and resuspended in media. A single mouse typically yielded 20–30 million cells.
Cells were cultured in a base medium of Iscove’s Modified Dulbecco’s Medium (IMDM, Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (Gibco) and 1% penicillin-streptomycin (Corning). Cells were maintained at 37° C and 5% CO2. For dendritic cell differentiation, bone marrow cells were cultured in IMDM supplemented with 300 ng/mL recombinant murine FLT3 ligand (BioLegend) for 9 days. For macrophage differentiation, bone marrow cells were treated with 50 ng/mL of recombinant murine M-CSF (PeproTech) for 7 days, with media exchanged every other day. In all assays, growth factor containing medium was replaced with base IMDM prior to drug treatment.
The MC38 mouse colon adenocarcinoma cell line was provided courtesy of M. Smyth (QIMR Berghofer Medical Research Institute) and maintained in IMDM supplemented with 10% fetal calf serum (Gibco), 100 IU penicillin (Invitrogen) and 100 μg/mL streptomycin (Invitrogen). All cell lines were regularly screened for mycoplasma contamination.
METHOD DETAILS
Materials.
Unless otherwise indicated, reagents were obtained from Sigma-Aldrich and used as received, water used was of MilliQ grade and reagents were maintained under sterile conditions. LCL161 was obtained from MedChem Express, prepared as 100 mM stock solution in DMSO and stored at −20° C until use.
Drug Screening.
p40-IRES-eYFP mouse-derived BMDCs were seeded into Corning 3764 384-well plates, with 100,000 cells/well in 50 μL of medium. After 9 days of differentiation, medium was washed out with IMDM, and cells were treated with experimental compounds.
Cells were treated with 7 doses from 31.6 μM to 31.6 nM at 1/2 log titration, in replicate, for 1 day. Cells were then fixed with 4% paraformaldehyde for 30 min at room temperature and stained with a cocktail of Hoechst 333412 (Invitrogen, 10 μg/mL) and wheat germ agglutinin Alexa Fluor 647 conjugate (Invitrogen, 5 μg/mL). After 15 min, the plates were washed with PBS and then imaged in the YFP, Hoechst 33342 and Alexa Fluor 647 channels using an Operetta High-Content Imaging system.
Compounds Used for Screening.
Screening compounds were obtained from MedChemExpress, Selleckchem and Tocris as well as the Institute for Chemical and Cell Biology (ICCB) at Harvard Medical School. We screened a set of 42 small molecules that were diverse in both chemical and biological space. The molecules could be divided into roughly 5 classes: i) kinase inhibitors against oncogenic pathways, ii) PI3K/mTOR/Akt pathway inhibitors, iii) innate immune ligands, iv) apoptosis (IAP) inhibitors and v) epigenetic and other IL-12 focused drugs. Class (i) included very common kinase inhibitors, which interact with several kinases involved in oncogenic pathways. Many of these drugs are in current clinical use. Class (ii) specifically focused on the PI3K/mTOR/Akt pathways, due to evidence of this pathway’s role in IL-12 regulation. Class (iii) included agonists of innate immune receptors. These drugs are generally pro-inflammatory. Class (iv) was a set of cIAP/XIAP inhibitors originally designed to sensitize cancer cells to apoptosis. Class (v) included epigenetic drugs as well as a collection of other drugs that, based on literature searches and mechanistic studies, we hypothesized may affect IL-12 production.
PCR.
BMDCs or BMDMs were plated into 24-well plates in 1 mL of appropriate medium. After 1 day of treatment, cells were lysed, and mRNA was extracted using RNeasy kit (Qiagen). cDNA was generated using a High Capacity Reverse Transcriptase kit (Thermo). For qPCR, the Taqman probes (Applied Biosystems) used were Mm01288989_m1 (murine IL12B), and Mm01545399_m1 (murine HPRT). Samples were run on a 7500 Real-Time PCR System using Fast Advanced Master Mix (Applied Biosystems). Ct values were computed using instrument software, and IL-12p40 mRNA fold changes were calculated using the ΔΔCt method.
IL-12 Immunofluorescence.
BMDCs were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) for 20 min and were then washed with PBS. Cells were blocked and permeabilized in a buffer of 2% BSA and 0.2% Triton X-100 for 30 min, then incubated with an anti-mouse IL-12 p40 antibody (R&D, AF-419) at 1:200 dilution in the same buffer overnight at 4° C. The next day, the cells were washed with PBS and incubated in the aforementioned blocking/permeabilization buffer with an Alexa Fluor 647 anti-goat secondary antibody (10 μg/mL) and Hoechst 33342 (10 μg/mL) for 1 hour. Cells were washed in PBS and then imaged on an Operetta High-Content Imaging System.
Preparation of Cyclodextrin Nanoparticles (CDNP).
Cyclodextrin nanoparticles (CDNPs) were prepared as previously described(Rodell et al., 2018). Briefly, succinyl-β-cyclodextrin (250 mg, 1.0 eq. succinylate), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (Fisher; 1482 mg, 10.0 eq. to succinylate) and N-hydroxysuccinimide (550 mg, 5.0 eq. to succinylate) were dissolved in 6.0 mL MES buffer (50 mM, pH 6) and L-lysine (Sigma; 70 mg, 0.5 eq. to succinylate, in 1.5 mL MES buffer) was added after 30 minutes. After overnight crosslinking, nanoparticles were recovered by the addition of brine (200 μL) and precipitation from ice cold ethanol. Nanoparticles were re-dissolved in water and purified by size exclusion chromatography (GE Healthcare; Sephadex G-25 desalting column). Glucose-positive fractions were concentrated in centrifugal filters (Amicon; 10 kDa MWCO) and washed with water prior to lyophilization. Resulting CDNPs were dissolved at 50 mg/mL in water and stored at −20° C. Nanoparticles drug loading was performed by dissolving LCL161 (25 mg/kg; 0.5 mg/mouse) in 100 μL of 2-hydroxypropyl-β-cyclodextrin (20 %wt/vol) or CDNP (50 mg/mL) in PBS for LCL161 and LCL161-CDNP treatments, respectively.
CDNP Characterization.
The guest-host interaction of LCL161 with cyclodextrin was examined using an established assay for recovering phenolphthalein absorbance at 550 nm (1.0 mM LCL161; 0.2 mM β-cyclodextrin, 200 μM phenolphthalein in 125 mM carbonate buffer, pH 10.5)(Higuti et al., 2004; Rodell et al., 2018). CDNP formulation drug solubilization was examined by turbidity measurement. LCL161 was prepared at concentrations identical to formulations used for subsequent animal administration (5 mg/mL in PBS) at CDNP concentrations up to 50 mg/mL. Absorbance at 500 nm was measured (Tecan, Spark) in 384-well plates (Corning) and was normalized to CDNP free controls. For both CDNP and LCL161-CDNP, particle size was calculated by dynamic light scattering (Malvern, Zetasizer APS) in PBS at a concentration of 5 mg/mL; n=3 independent samples.
Intravital Microscopy.
Images were acquired on an FV1000MPE confocal imaging system (Olympus)(Pittet et al., 2018). Pacific Blue, GFP/YFP, mApple and VivoTag 680 (VT680) were excited sequentially using 405, 473, 559 and 635 nm diode lasers and BA430– 455, BA490–540, BA575–620 and BA655–755 emission filters with SDM473, SDM560 and SDM640 beam splitters. Examination was performed in dorsal skinfold window chambers implanted in p40-IRES-eYFP IL-12 reporter mice(Reinhardt et al., 2006). IL-12 expression was examined at baseline (preceding treatment) and at 24 hr following treatment. Pacific Blue-dextran was used to label myeloid cells. This dextran conjugate was prepared by reacting Pacific Blue succinimidyl ester dye (Thermo) with amino dextran 10,000 MW (Thermo), as done previously(Rodell et. al., 2018).
Toxicity Studies
Healthy C57BL/6 mice were treated with saline, empty CDNP, LCL161-CD, or LCL161-CDNP as described in the main text. Mice were sacrificed 24 hours after treatment, and organs (liver, spleen, heart, lungs, kidney, pancreas, small and large intestines, brain, femur) were harvested and weighed. Organs were subsequently fixed in 10% formalin solution (Fisher Chemical). The fixed organs were paraffin-embedded and sectioned for Hematoxylin and Eosin (H&E) staining (Millipore Sigma). The sections were scanned by using NanoZoomer 2.0RS (Hamamatsu, Japan) and the images were processed and analyzed by using FIJI.
For immunotoxicity studies, MC38 tumors were subcutaneously implanted into C57BL/6 mice (2 million cells/mouse), as described in the main text. Mice were treated with either saline, empty CDNPs, or LCL161-CDNPs. Twenty four hours after treatment, blood was collected via cardiac puncture. Blood was allowed to clot for 2 hours, and serum was then collected by centrifugation (2000 x g for 20 min). IL-12 levels were detected by ELISA Quantikine kit (R&D Systems, M1270).
Activation of Dendritic Cells in Lymph Node
Bilateral hindleg MC38 tumors were established subcutaneously in C75BL/6J mice on the left and right sides in female mice, 6–8 weeks of age. Mice were treated with LCL161-CDNP for 24 hours, and then harvested both tumors, as well as tumor draining lymph nodes on both sides, as well as non-draining distal, axillary lymph nodes. Total eYFP signal was determined using a Sapphire Biomolecular Imager (Azure Biosciences). Quantification was done in ImageJ. Confocal microscopy was performed as described in the main text.
Cell viability
Viability assay to measure effect of LCL161 on MC38 cells was done using the Cell Titer Glo Viabiliity Assay (Promega) in accord with manufacturer protocol.
QUANTIFICATION AND STATISTICAL ANALYSIS
Image Processing.
For drug screening, analysis was performed on the Harvard Medical School Columbus server (Perkin Elmer). In brief, nuclei were first segmented using the Hoechst channel, and then surrounding cellular areas were marked with Alexa Fluor 647 channel. YFP fluorescent intensities were extracted for each cell. For each well, a distribution of YFP intensities was constructed and the 95th percentile value was calculated. Compound scores at each dose were calculated according to the formula in Fig. 1 (shown below), and scores were averaged across the replicates. For IL-12 fluorescence staining (Fig. 3), image analysis was performed similarly as in the screen (above), except cellular areas were determined by the IL-12 stain. IL-12 and YFP intensities were quantified, with local background correction. Final data in figure 2 was prepared in MATLAB (MathWorks).
For intravital microscopy, images were pseudo-colored and processed in FIJI (ImageJ, NIH) by adjusting brightness/contrast, creating z-projections of image stacks and performing a rolling ball background subtraction. To quantify IL-12hi cell populations, the mApple channel was subtracted from YFP, automated thresholding was applied by the RenyiEntropy method and ROIs were automatically generated for the corresponding masked image. Cell intensities were normalized to pre-treatment conditions.
Statistical Analysis.
Statistical analyses were performed in GraphPad Prism 8. Data are presented as mean ± standard deviation (s.d.) for in vitro studies or mean ± standard error of the mean (s.e.m.) for in vivo studies. Two tailed Student’s t-test was used to compare two groups. One-way ANOVA with post hoc Tukey’s HSD test was used to compare multiple groups.
For tumor growth models, comparison was performed by Friedman’s Test with post hoc Dunn’s test after excluding outliers by Grubb’s test. Significance was assigned at P<0.05.
Quantification of Nanoparticle Uptake
Image analysis was done in Cell Profiler using the images from intravital microscopy, described in the main text. Whole image correlation values were determined using the Colocalization tool. For measuring nanoparticle uptake, macrophages were segmented using Pacific Blue-Dextran (Alexa Fluor 405), and tumors were segmented using the MC38 H2B Apple (Alexa Fluor 546 channel). Nanoparticle intensity was then calculated using the Alexa Fluor 650 channel.
DATA AND CODE AVAILABILITY
The data supporting the findings of this study are available within the paper. Image analysis pipelines were constructed on a proprietary, commercial server and cannot be freely shared; however, alternative analysis pipelines on other software are available upon request from the Corresponding Author.
Supplementary Material
KEY RESOURCES
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse IL-12 p40 Antibody | R&D | Cat # AF-419 |
| Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Invitrogen | Cat # A-21447 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant murine FLT3 Ligand | BioLegend | Cat # 550704 |
| Recombinant murine M-CSF | Peprotech | Cat # 315–02 |
| Succinyl-β-cyclodextrin | Sigma | Cat # 85990–5G |
| 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide | Fisher | Cat # 22980 |
| N-hydroxysuccinimide | Sigma | Cat # 130672–5G |
| L-lysine | Sigma | Cat # L5501–5G |
| 2-hydroxypropyl-β-cyclodextrin | Sigma | Cat # 332593–25G |
| β--cyclodextrin | TCI America | Cat # C0900 |
| Phenolphthalein | Sigma | Cat # 319236–100ML |
| Hoechst 333412 | Thermo Fisher | Cat # H3570 |
| Wheat germ agglutinin Alexa Fluor 647 conjugate | Invitrogen | Cat # W32466 |
| LCL-161 | MedChemExpress | Cat # HY-15518 |
| Hematoxylin | Millipore Sigma | Cat # 234–12 |
| Eosin Solution Alcoholic | Sigma | Cat # HT110132–1L |
| Dextran, Amino 10,000 MW | Thermo Fisher | Cat # D1860 |
| Pacific Blue Succinimidyl Ester | Thermo Fisher | Cat # P10163 |
| Pacific Blue Labeled Dextran | This manuscript | N/A |
| Cyclodextrin Nanoparticles | This manuscript | N/A |
| Critical Commercial Assays | ||
| RNeasy Mini Kit | Qiagen | Cat # 74104 |
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher | Cat # 4368814 |
| Applied Biosystems TaqMan Fast Advanced Master Mix | Thermo Fisher | Cat # 4444557 |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat # G7571 |
| Mouse IL-12 p70 Quantikine ELISA kit | R&D | Cat # M1270 |
| Experimental Models: Cell Lines | ||
| MC38 Mouse Colon Adenocarcinoma Line | M. Smyth, QIMR Berghover Medical Research Institute | N/A |
| Bone marrow derived dendritic cells (BMDCs) from mouse | Jackson Laboratories | See mouse model |
| Bone marrow derived macrophages (BMDMs) from mouse | Jackson Laboratories | See mouse model |
| Experimental Models: Organisms/Strains | ||
| Mouse: Wild type C57BL/6J | Jackson Laboratories | Stock # 000644 |
| Mouse: p40-IRES-eYFP, B6.129-Il12btm1.1Lky/J | Jackson Laboratories | Stock # 006412 |
| Mouse: Map3k14 (NIK) KO, B6N.129-Map3k14tm1Rds/J | Jackson Laboratories | Stock # 025557 |
| Oligonucleotides | ||
| Taqman probe – murine IL12B | Applied Biosystems | Cat # Mm01288989_m1 |
| Taqman probe – murine HPRT | Applied Biosystems | Cat # Mm01545399_m1 |
| Software and Algorithms | ||
| FiJi (ImageJ) | NIH | N/A |
| Columbus Image Data Storage and Analysis System | Perkin Elmer | N/A |
| MATLAB | Mathworks | N/A |
| Prism 8 | GraphPad | N/A |
| CellProfiler-3.1.8 | Broad Institute | N/A |
| Other | ||
| Sephadex G-25 in PD-10 Desalting Column | GE Healthcare | Cat # 17085101 |
| Amicon Ultra-0.5 Centrifugal Filter Unit 10 kDa MWCO | Millipore Sigma | Cat # UFC5010BK |
SIGNIFICANCE.
Efficient PD1 therapy requires that there are sufficient levels of intratumoral IL-12, generally produced by DC3 cells in the tumor microenvironment. Motivated by this finding, we conducted a small molecule screen to identify IL-12 inducing agents, and found that the cellular Inhibitor of Apoptosis (cIAP) inhibitor, LCL161, promoted IL-12 production in dendritic cells via the non-canonical NFkB pathway. Moreover, to improve pharmacokinetic properties and delivery of LCL161 to dendritic cells, we complexed the drug to cyclodextrin nanoparticles. The resultant LCL161-nanoparticle formulation regressed tumors, had minimal toxicity, and outperformed the free drug control. Together, this work suggests that nanoparticle formulations of cIAP inhibitors may have clinical utility in cancer immunotherapy.
ACKNOWLEDGMENTS
Part of this work was supported in part by grants from the US National Institutes of Health (NCI 5T32CA079443, NCI 5R01CA204019 NCI 5R01CA206890). We thank members of the Institute of Cell and Chemical Biology (ICCB) as well as the Laboratory of Systems Pharmacology (LSP) at Harvard Medical School for assistance with the screen. We also thank Sean Arlauckas, Christopher Garris, and Marie Siwicki for help with the p40-IRES-eYFP reporter mouse and Alexandra Dibrindisi, Yoshi Iwamoto and Greg Wojtkiewicz for assistance with toxicity studies
Footnotes
DECLARATION OF INTERESTS
CBR and RW are listed on a patent filed by Partners Healthcare. The authors declare no competing financial interests.
REFERENCES
- Abdi K, Laky K, Padhan K, Petrovas C, Skinner J, Kabat J, Dorward DW, Brzostowski J, Long EO, Trinchieri G et al. (2018). Cutting Edge: Quantitative Determination of CD40L Threshold for IL-12 and IL-23 Production from Dendritic Cells. J Immunol 201, 2879–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed MS, Rodell CB, Hulsmans M, Kohler RH, Aguirre A, Nahrendorf M, and Weissleder R (2019). A Supramolecular Nanocarrier for Delivery of Amiodarone Anti-Arrhythmic Therapy to the Heart. Bioconjug Chem 30, 733–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angiolillo AL, Sgadari C, and Tosato G (1996). A role for the interferon-inducible protein 10 in inhibition of angiogenesis by interleukin-12. Ann N Y Acad Sci 795, 158–167. [DOI] [PubMed] [Google Scholar]
- Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, Miller MA, Carlson JC, Freeman GJ, Anthony RM et al. (2017). In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci Transl Med 9, eaal3604 DOI: 10.1126/scitranslmed.aal3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardia A, Parton M, Kümmel S, Estévez LG, Huang CS, Cortés J, Ruiz-Borrego M, Telli ML, Martin-Martorell P, López R et al. (2018). Paclitaxel With Inhibitor of Apoptosis Antagonist, LCL161, for Localized Triple-Negative Breast Cancer, Prospectively Stratified by Gene Signature in a Biomarker-Driven Neoadjuvant Trial. J Clin Oncol JCO2017748392. [DOI] [PubMed] [Google Scholar]
- Chen KF, Lin JP, Shiau CW, Tai WT, Liu CY, Yu HC, Chen PJ, and Cheng AL (2012). Inhibition of Bcl-2 improves effect of LCL161, a SMAC mimetic, in hepatocellular carcinoma cells. Biochem Pharmacol 84, 268–277. [DOI] [PubMed] [Google Scholar]
- Chesi M, Mirza NN, Garbitt VM, Sharik ME, Dueck AC, Asmann YW, Akhmetzyanova I, Kosiorek HE, Calcinotto A, Riggs DL et al. (2016). IAP antagonists induce anti-tumor immunity in multiple myeloma. Nat Med 22, 1411–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy-Thompson E, Ali L, Bruck PT, Exley MA, Blumberg RS, Dranoff G, Dougan M, and Dougan SK (2018). IAP Antagonists Enhance Cytokine Production from Mouse and Human iNKT Cells. Cancer Immunol Res 6, 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cody JJ, Scaturro P, Cantor AB, Yancey Gillespie G, Parker JN, and Markert JM (2012). Preclinical evaluation of oncolytic δγ(1)34.5 herpes simplex virus expressing interleukin-12 for therapy of breast cancer brain metastases. Int J Breast Cancer 2012, 628697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong H, Xu L, Wu Y, Qu Z, Bian T, Zhang W, Xing C, and Zhuang C (2019). Inhibitor of Apoptosis Protein (IAP) Antagonists in Anticancer Agent Discovery: Current Status and Perspectives. J Med Chem 62, 5750–5772. [DOI] [PubMed] [Google Scholar]
- Daud AI, DeConti RC, Andrews S, Urbas P, Riker AI, Sondak VK, Munster PN, Sullivan DM, Ugen KE, Messina JL et al. (2008). Phase I trial of interleukin-12 plasmid electroporation in patients with metastatic melanoma. J Clin Oncol 26, 5896–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis ME, and Brewster ME (2004). Cyclodextrin-based pharmaceutics: past, present and future. Nat Rev Drug Discov 3, 1023–1035. [DOI] [PubMed] [Google Scholar]
- Dougan M, Dougan S, Slisz J, Firestone B, Vanneman M, Draganov D, Goyal G, Li W, Neuberg D, Blumberg R et al. (2010). IAP inhibitors enhance co-stimulation to promote tumor immunity. J Exp Med 207, 2195–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougan SK, and Dougan M (2018). Regulation of innate and adaptive antitumor immunity by IAP antagonists. Immunotherapy 10, 787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engblom C, Pfirschke C, and Pittet MJ (2016). The role of myeloid cells in cancer therapies. Nat Rev Cancer 16, 447–462. [DOI] [PubMed] [Google Scholar]
- Engeland CE, and Bell JC (2020). Introduction to Oncolytic Virotherapy. Methods Mol Biol 2058, 1–6. [DOI] [PubMed] [Google Scholar]
- Fares CM, Van Allen EM, Drake CG, Allison JP, and Hu-Lieskovan S (2019). Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients. Am Soc Clin Oncol Educ Book 39, 147–164. [DOI] [PubMed] [Google Scholar]
- Fulda S (2015). Promises and Challenges of Smac Mimetics as Cancer Therapeutics. Clin Cancer Res 21, 5030–5036. [DOI] [PubMed] [Google Scholar]
- Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, Engblom C, Pfirschke C, Siwicki M, Gungabeesoon J et al. (2018). Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-γ and IL-12. Immunity 49, 1148–1161.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan SB, Sørensen JF, Olsen BN, and Pedersen AE (2014). Anti-CD40-mediated cancer immunotherapy: an update of recent and ongoing clinical trials. Immunopharmacol Immunotoxicol 36, 96–104. [DOI] [PubMed] [Google Scholar]
- Helms MW, Jahn-Hofmann K, Gnerlich F, Metz-Weidmann C, Braun M, Dietert G, Scherer P, Grandien K, Theilhaber J, Cao H et al. (2019). Utility of the RIG-I agonist triphosphate RNA for melanoma therapy. Mol Cancer Therapeutics. In Press. DOI: 10.1158/1535-7163.MCT-18-1262. [DOI] [PubMed] [Google Scholar]
- Higuti IH, Silva PAD, Papp J, Okiyama VMDE, Andrade EAD, Marcondes ADA, and Nascimento AJD (2004). Colorimetric determination of alpha and beta-cyclodextrins and studies on optimization of CGTase production from B. firmus using factorial designs. Brazilian Archives of Biology and Technology 47, 837–841. [Google Scholar]
- Infante JR, Dees EC, Olszanski AJ, Dhuria SV, Sen S, Cameron S, and Cohen RB (2014). Phase I dose-escalation study of LCL161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors. J Clin Oncol 32, 3103–3110. [DOI] [PubMed] [Google Scholar]
- Jahan N, Talat H, and Curry WT (2018). Agonist OX40 immunotherapy improves survival in glioma-bearing mice and is complementary with vaccination with irradiated GM-CSF-expressing tumor cells. Neuro Oncol 20, 44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson P, Challis R, Chowdhury F, Gao Y, Harvey M, Geldart T, Kerr P, Chan C, Smith A, Steven N et al. (2015). Clinical and biological effects of an agonist anti-CD40 antibody: a Cancer Research UK phase I study. Clin Cancer Res 21, 1321–1328. [DOI] [PubMed] [Google Scholar]
- Kim HY, Li R, Ng TSC, Courties G, Rodell CB, Prytyskach M, Kohler RH, Pittet MJ, Nahrendorf M, Weissleder R et al. (2018). Quantitative Imaging of Tumor-Associated Macrophages and Their Response to Therapy Using 64Cu-Labeled Macrin. ACS Nano 12, 12015–12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasek W, Basak G, Switaj T, Jakubowska AB, Wysocki PJ, Mackiewicz A, Drela N, Jalili A, Kamiński R, Kozar K et al. (2004). Complete tumour regressions induced by vaccination with IL-12 gene-transduced tumour cells in combination with IL-15 in a melanoma model in mice. Cancer Immunol Immunother 53, 363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasek W, Zagożdżon R, and Jakobisiak M (2014). Interleukin 12: still a promising candidate for tumor immunotherapy. Cancer Immunol Immunother 63, 419–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashev M, LePage D, Wilson C, Bailly V, Garber E, Lukashin A, Ngam-ek A, Zeng W, Allaire N, Perrin S et al. (2006). Targeting the lymphotoxin-beta receptor with agonist antibodies as a potential cancer therapy. Cancer Res 66, 9617–9624. [DOI] [PubMed] [Google Scholar]
- Ma HS, Poudel B, Torres ER, Sidhom JW, Robinson TM, Christmas B, Scott B, Cruz K, Woolman S, Wall VZ et al. (2019). A CD40 Agonist and PD-1 Antagonist Antibody Reprogram the Microenvironment of Nonimmunogenic Tumors to Allow T-cell-Mediated Anticancer Activity. Cancer Immunol Res 7, 428–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machelart A, Salzano G, Li X, Demars A, Debrie AS, Menendez-Miranda M, Pancani E, Jouny S, Hoffmann E, Deboosere N et al. (2019). Intrinsic Antibacterial Activity of Nanoparticles Made of β-Cyclodextrins Potentiates Their Effect as Drug Nanocarriers against Tuberculosis. ACS Nano 13, 3992–4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MA, Zheng YR, Gadde S, Pfirschke C, Zope H, Engblom C, Kohler RH, Iwamoto Y, Yang KS, Askevold B et al. (2015). Tumour-associated macrophages act as a slow-release reservoir of nano-therapeutic Pt(IV) pro-drug. Nat Commun 6, 8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynihan KD, Opel CF, Szeto GL, Tzeng A, Zhu EF, Engreitz JM, Williams RT, Rakhra K, Zhang MH, Rothschilds AM et al. (2016). Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat Med 22, 1402–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipov A, Saung MT, Zheng L, and Murphy AG (2019). Small molecule immunomodulation: the tumor microenvironment and overcoming immune escape. J Immunother Cancer 7, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauken KE, Dougan M, Rose NR, Lichtman AH, and Sharpe AH (2019). Adverse Events Following Cancer Immunotherapy: Obstacles and Opportunities. Trends Immunol 40, 511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pemmaraju N, Carter BZ, Kantarjian HM, Cortes JE, Kadia TM, Garcia-Manero G, DiNardo CD, Bose P, Pierce S, and Zhou L (2016). Results for phase II clinical trial of LCL161, a SMAC mimetic, in patients with primary myelofibrosis (PMF), post-polycythemia vera myelofibrosis (post-PV MF) or post-essential thrombocytosis myelofibrosis (post-ET MF).
- Pittet MJ, Garris CS, Arlauckas SP, and Weissleder R (2018). Recording the wild lives of immune cells. Sci Immunol 3, eaaq0491 DOI: 10.1126/sciimmunol.aaq0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang SY, Tran JL, Moore P, Lehmann S, Eberl HC et al. (2018). Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 564, 439–443. [DOI] [PubMed] [Google Scholar]
- Reinhardt RL, Hong S, Kang SJ, Wang ZE, and Locksley RM (2006). Visualization of IL-12/23p40 in vivo reveals immunostimulatory dendritic cell migrants that promote Th1 differentiation. J Immunol 177, 1618–1627. [DOI] [PubMed] [Google Scholar]
- Rodell CB, Arlauckas SP, Cuccarese MF, Garris CS, Li R, Ahmed MS, Kohler RH, Pittet MJ, and Weissleder R (2018). TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat Biomed Eng 2, 578–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodell CB, Mealy JE, and Burdick JA (2015). Supramolecular Guest-Host Interactions for the Preparation of Biomedical Materials. Bioconjug Chem 26, 2279–2289. [DOI] [PubMed] [Google Scholar]
- Rodell CB, Ahmed MS, Garris CS, Pittet M, and Weissleder R (2019). Development of adamantane-conjugated TLR7/8 agonists for supramolecular delivery and cancer immunotherapy. Theranostics. In Press. DOI: 10.7150/thno.35434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodolfo M, Zilocchi C, Melani C, Cappetti B, Arioli I, Parmiani G, and Colombo MP (1996). Immunotherapy of experimental metastases by vaccination with interleukin gene-transduced adenocarcinoma cells sharing tumor-associated antigens. Comparison between IL-12 and IL-2 gene-transduced tumor cell vaccines. J Immunol 157, 5536–5542. [PubMed] [Google Scholar]
- Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, Daniel D, Hwang ES, Rugo HS, and Coussens LM (2014). Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 26, 623–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanmamed MF, and Chen L (2018). A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. Cell 175, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Hu-Lieskovan S, Wargo JA, and Ribas A (2017). Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 168, 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song K, Chang Y, and Prud’homme GJ (2000). IL-12 plasmid-enhanced DNA vaccination against carcinoembryonic antigen (CEA) studied in immune-gene knockout mice. Gene Ther 7, 1527–1535. [DOI] [PubMed] [Google Scholar]
- Spranger S, Bao R, and Gajewski TF (2015). Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235. [DOI] [PubMed] [Google Scholar]
- Sun SC (2017). The non-canonical NF-κB pathway in immunity and inflammation. Nat Rev Immunol 17, 545–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szejtli J (1998). Introduction and General Overview of Cyclodextrin Chemistry. Chem Rev 98, 1743–1754. [DOI] [PubMed] [Google Scholar]
- Tait Wojno ED, Hunter CA, and Stumhofer JS (2019). The Immunobiology of the Interleukin-12 Family: Room for Discovery. Immunity 50, 851–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Shalabi A, and Hubbard-Lucey VM (2018). Comprehensive analysis of the clinical immuno-oncology landscape. Ann Oncol 29, 84–91. [DOI] [PubMed] [Google Scholar]
- Ventola CL (2017). Progress in Nanomedicine: Approved and Investigational Nanodrugs. P T 42, 742–755. [PMC free article] [PubMed] [Google Scholar]
- Vonderheide RH (2018). The Immune Revolution: A Case for Priming, Not Checkpoint. Cancer Cell 33, 563–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonderheide RH, and Glennie MJ (2013). Agonistic CD40 antibodies and cancer therapy. Clin Cancer Res 19, 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, and Mooney DJ (2018). Biomaterial-assisted targeted modulation of immune cells in cancer treatment. Nat Mater 17, 761–772. [DOI] [PubMed] [Google Scholar]
- Wang P, Li X, Wang J, Gao D, Li Y, Li H, Chu Y, Zhang Z, Liu H, Jiang G et al. (2017). Re-designing Interleukin-12 to enhance its safety and potential as an anti-tumor immunotherapeutic agent. Nat Commun 8, 1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissleder R, Kelly K, Sun EY, Shtatland T, and Josephson L (2005). Cell-specific targeting of nanoparticles by multivalent attachment of small molecules. Nat Biotechnol 23, 1418–1423. [DOI] [PubMed] [Google Scholar]
- Weissleder R, Nahrendorf M, and Pittet MJ (2014). Imaging macrophages with nanoparticles. Nat Mater 13, 125–138. [DOI] [PubMed] [Google Scholar]
- Zhang J, and Ma PX (2013). Cyclodextrin-based supramolecular systems for drug delivery: recent progress and future perspective. Adv Drug Deliv Rev 65, 1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, Krishnan I, Maroni G, Meyerovitz CV, Kerwin CM et al. (2019). Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 50, 1317–1334.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are available within the paper. Image analysis pipelines were constructed on a proprietary, commercial server and cannot be freely shared; however, alternative analysis pipelines on other software are available upon request from the Corresponding Author.