

Abstract
Targeted protein degradation has arisen as a powerful strategy for drug discovery allowing the targeting of undruggable proteins for proteasomal degradation. This approach most often employs heterobifunctional degraders consisting of a protein-targeting ligand linked to an E3 ligase recruiter to ubiquitinate and mark proteins of interest for proteasomal degradation. One challenge with this approach, however, is that only a few E3 ligase recruiters currently exist for targeted protein degradation applications, despite the hundreds of known E3 ligases in the human genome. Here, we utilized activity-based protein profiling (ABPP)-based covalent ligand screening approaches to identify cysteine-reactive small-molecules that react with the E3 ubiquitin ligase RNF4 and provide chemical starting points for the design of RNF4-based degraders. The hit covalent ligand from this screen reacted with either of two zinc-coordinating cysteines in the RING domain, C132 and C135, with no effect on RNF4 activity. We further optimized the potency of this hit and incorporated this potential RNF4 recruiter into a bifunctional degrader linked to JQ1, an inhibitor of the BET family of bromodomain proteins. We demonstrate that the resulting compound CCW 28–3 is capable of degrading BRD4 in a proteasome- and RNF4-dependent manner. In this study, we have shown the feasibility of using chemoproteomics-enabled covalent ligand screening platforms to expand the scope of E3 ligase recruiters that can be exploited for targeted protein degradation applications.
Graphical Abstract
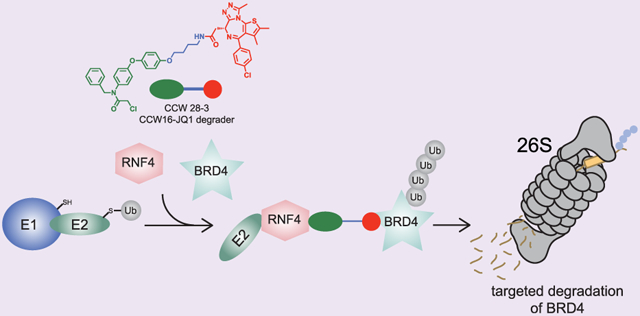
Targeted protein degradation is a groundbreaking drug discovery approach for tackling the undruggable proteome by exploiting the cellular protein degradation machinery to selectively eliminate target proteins.1,2 This technology most often involves the utilization of heterobifunctional degrader molecules consisting of a substrate-targeting ligand linked to an E3 ligase recruiter. These degraders are capable of recruiting E3 ligases to specific protein targets to ubiquitinate and mark targets for degradation in a proteasome-dependent manner. As functional inhibition of the target is not necessary for degrader efficacy, this strategy has the potential to target and degrade any protein in the proteome for which there exists a ligand. However, while there are ~600 different E3 ligases, there are only a few E3 ligases that have been successfully exploited in such a strategy, including small-molecule recruiters for cereblon, VHL, MDM2, and cIAP.2,3 Identifying facile strategies for discovering ligands that bind to E3 ligases remains crucial for expanding the set of E3 ligase recruiters that can be utilized for targeted protein degradation of a given target of interest and ultimately expand the applicability of this modality to any protein that can be subjected to proteasomal degradation regardless of subcellular localization or tissue-specific expression.
Activity-based protein profiling (ABPP) has arisen as a powerful platform for ligand discovery against targets of interest, including proteins commonly considered as undruggable.4–9 ABPP utilizes reactivity-based chemical probes to directly map proteome wide reactive spots and ligand able hotspots in complex biological systems.10,11 When used in a competitive manner, covalently acting small-molecule libraries can be screened for competition against the binding of reactivity-based probes to facilitate covalent ligand discovery against proteins of interest.4–6,8,12,13 In order to discover covalent ligands that may react with E3 ubiquitin ligases, we first investigated whether representative commercially available E3 ligases, MDM2, RNF4, and UBE3A, could be labeled by the cysteine-reactive tetramethylrhodamine-5-iodoacetamide dihydroiodide (IA-rhodamine) reactivity-based probe in vitro. We observed IA-rhodamine labeling of all three E3 ligases in a dose-responsive manner at their expected molecular weights (Figure 1A). While previous studies have already uncovered MDM2 and UBE3A small-molecule modulators,14–16 no chemical tools exist for the E3 ubiquitin ligase RNF4, which recognizes SUMOylated proteins and ubiquitinates these proteins for subsequent proteasomal degradation.17,18 We thus focused our efforts on developing a ligand for RNF4 as an E3 ligase recruitment module for heterobifunctional degraders.
Figure 1.
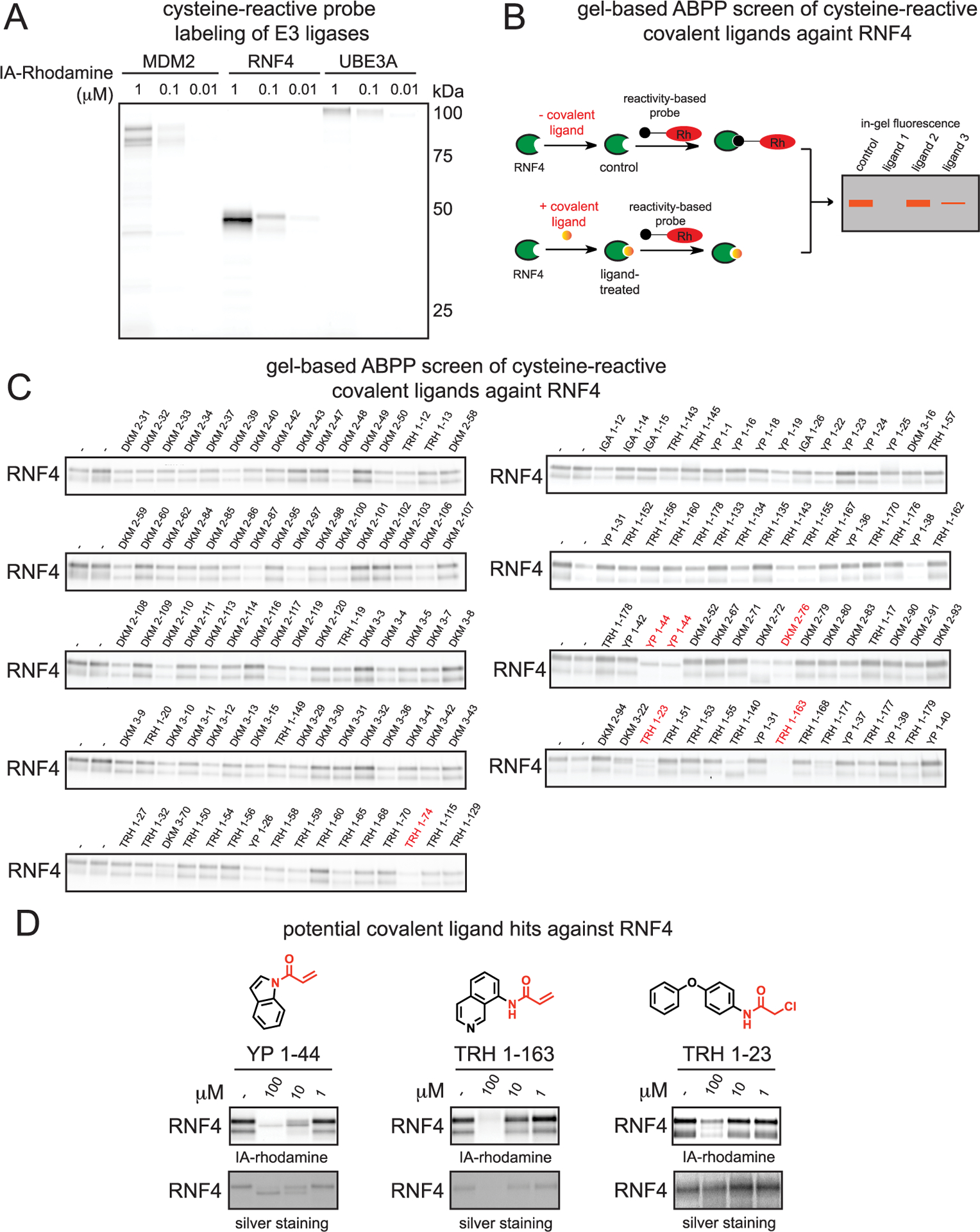
Covalent ligand screen against RNF4 using gel-based ABPP. (A) Gel-based ABPP labeling of E3 ligases MDM2, RNF4, and UBE3A. Purified protein was labeled with IA-rhodamine (IA-Rh) for 30 min at RT, followed by SDS/PAGE and visualization by in-gel fluorescence. (B) Schematic of gel-based ABPP screen of covalent ligands (50 μM) against IA-rhodamine labeling of pure RNF4. (C) Gel-based ABPP screen of cysteine-reactive covalent ligands against IA-rhodamine labeling of RNF4. Covalent ligands were preincubated with pure RNF4 protein for 30 min prior to IA-rhodamine labeling (250 nM) for 1 h. Proteins were subjected to SDS/PAGE and visualized by in-gel fluorescence. Highlighted in red were potential hits from this screen. (D) Chemical structures and gel-based ABPP confirmation of reproducible RNF4 screening hits, performed as described in part C. Gels were also silver stained to identify compounds that induce protein precipitation.
In search of RNF4-targeting ligands, we screened a cysteine-reactive covalent ligand library against IA-rhodamine labeling of pure human RNF4 using gel-based ABPP (Figure 1B,C; Table S1). We note that RNF4 labels with IA-rhodamine as two bands by gel-based ABPP, which may reflect more than one cysteine labeled by the probe. We identified several potential hits from this screen, including TRH 1–74, YP 1–44, DKM 2–76, TRH 1–23, and TRH 1–163. From these, YP 1–44, TRH 1–163, and TRH 1–23 showed reproducible and dose-responsive inhibition of IA-rhodamine labeling of RNF4 (Figure 1D, Figure S1). Based on corresponding silver staining of RNF4 in these experiments, we found that TRH 1–163 may be causing protein precipitation. Based on gel-based ABPP analysis of general cysteine-reactivity in 231MFP lysates, YP 1–44 was much less selective compared to TRH 1–23 (Figure S1). Thus, TRH 1–23 appeared to be the most promising RNF4 hit (Figure 1D).
We next sought to identify the site-of-modification of TRH 1–23 within RNF4. We performed liquid chromatography–tandem mass spectrometry (LC–MS/MS) analysis of tryptic digests from TRH 1–23-treated purified RNF4 protein and found that TRH 1–23 covalently modified either of the two zinc-coordinating cysteines C132 and C135 in the RING domain of RNF4 (Figure 2A,B). While we did not observe modification of both cysteines, we cannot exclude the possibility that TRH 1–23 may engage both C132 and C135 simultaneously. In support of its utility as a functional RNF4 recruiter, we wanted to see if TRH 1–23 had any effect upon RNF4 autoubiquitination activity, since previous studies had shown that mutation of both cysteines to serines inhibited RNF4 function.19–21 Surprisingly, TRH 1–23 treatment did not inhibit RNF4 autoubiquitination activity in an in vitro reconstituted assay (Figure 2C).
Figure 2.
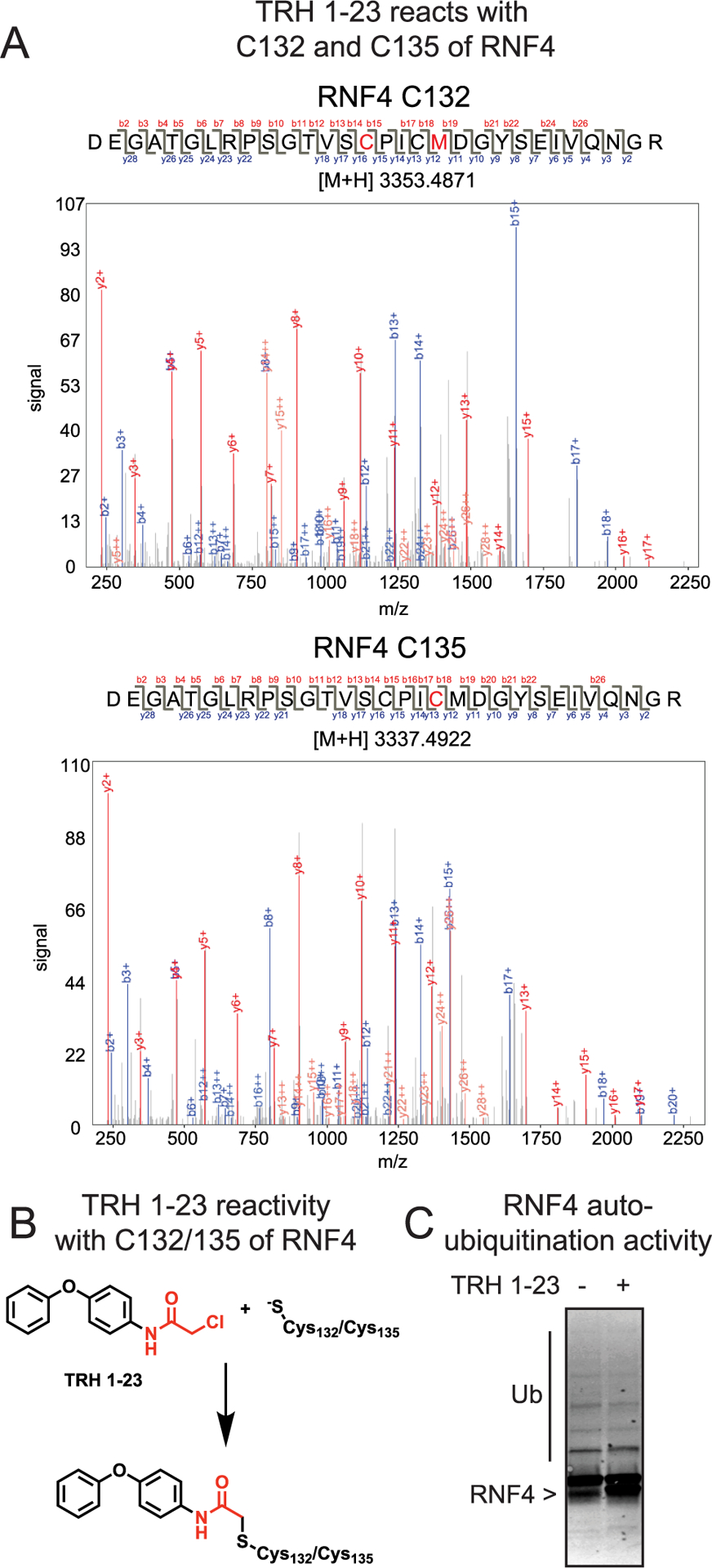
TRH 1–23 reacts nonfunctionally with zinc-coordinating cysteines in RNF4. (A) LC–MS/MS analysis of TRH 1–23 covalent adduct on RNF4. RNF4 was incubated with TRH 1–23 (50 μM) for 30 min at RT. Tryptic digests of RNF4 were analyzed by LC–MS/MS and were searched for the presence of peptides showing the TRH 1–23 adduct. The MS/MS spectra of TRH 1–23-modified C132 and C135 RNF4 tryptic peptides are shown. The confidence level for the peptides shown are 100%. The RNF4 C132 peptide shown also bears an oxidized methionine. Highlighted in red in the peptide sequence is the cysteine that was modified. (B) Schematic of TRH 1–23 reactivity with C132 or C135 of RNF4. (C) TRH 1–23 does not inhibit RNF4 autoubiquitination. RNF4 was preincubated with TRH 1–23 (100 μM) for 30 min at RT followed by addition of UBA1, E2 enzyme, and ATP for 40 min at 37 °C. The reaction was quenched and subjected to SDS/PAGE and Western blotting for RNF4. The gel shown in part C is a representative gel from n = 3.
While a promising nonfunctional ligand against RNF4, we considered the potency of TRH 1–23 with a 50% inhibitory concentration (IC50) in the double digit micromolar range in competitive ABPP suboptimal for usage as an efficient RNF4 recruiter. We thus synthesized several TRH 1–23 analogues and used gel-based ABPP to test their potency and structure–activity relationships against RNF4 (Figure 3A). Among these analogues, we found CCW 16, with an N-benzyl and 4-methoxyphenoxyphenyl substitution on the chloroacetamide scaffold, to be among the most potent of the analogues with inhibition of IA-rhodamine labeling of RNF4 observed down to 1 μM. Further confirmatory studies revealed the IC50 for CCW 16 to be 1.8 μM (Figure 3B).
Figure 3.
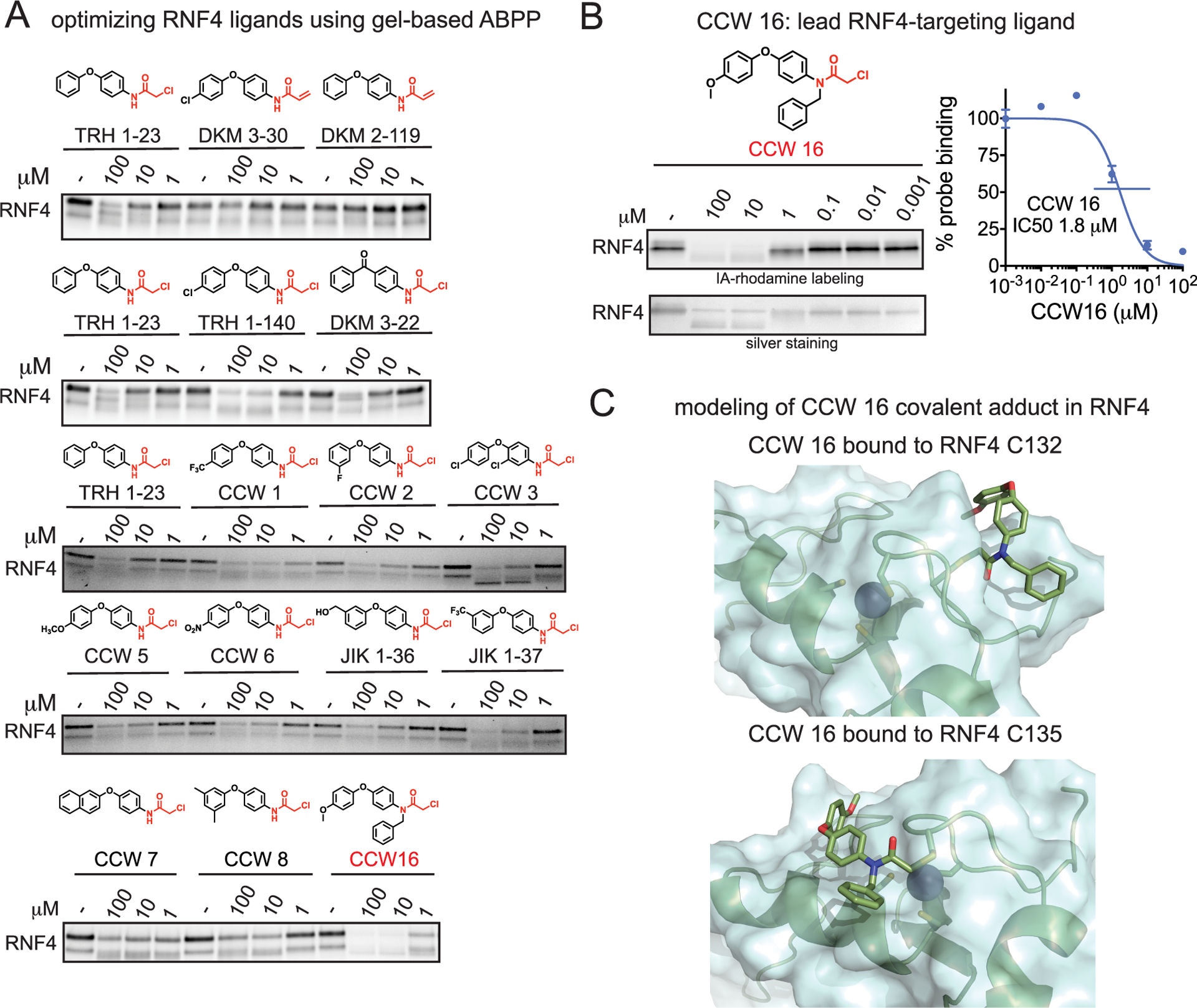
Optimizing RNF4 covalent ligands using gel-based ABPP. (A) Derivatives of TRH 1–23 were tested against IA-rhodamine labeling of RNF4 using gel-based ABPP. Silver stained gels are also shown to visualize total protein content. (B) CCW16 was tested against IA-rhodamine labeling of RNF4 using gel-based ABPP. For parts A and B, covalent ligands were preincubated with pure RNF4 protein for 30 min prior to IA-rhodamine labeling for 1 h. Proteins were subjected to SDS/PAGE and visualized by in-gel fluorescence. In part B, gels were quantified by densitometry to calculate IC50 values. The gel shown in part B is a representative gel from n = 3. (C) Covalent docking of CCW 16 bound to either C132 or C135 in RNF4.
To better understand how CCW 16 was interacting with RNF4, we carried out covalent docking on CCW 16 bound to either C132 or C135 on a publicly available crystal structure of RNF4 (PDB 4PPE) (Figure 3C). In this model, CCW 16 covalent binding to C132 caused a small portion of the backbone to rotate flipping the C132 side chain out into a more surface accessible conformation. The CCW 16 ligand occupied a groove on the RNF4 surface consisting of side-chains R125, T129, and S131, with no obvious polar interactions between the ligands and the protein in this model. Interestingly, CCW 16 did not occupy the zinc-binding site and left the other three zinc-coordinating cysteines (C135, C159, and C162) unperturbed (Figure 3C). In modeling CCW 16 covalently bound to C135, we observed a similar association with a surface groove of RNF4 occupying a region between Q161, S166, and P174. However, to accommodate the ligand, only a dihedral rotation of Cα and Cβ of C132 was predicted as necessary (Figure 3C). Both models indicate that zinc may still be bound by the three remaining cysteines in the site, while CCW 16 binds outside of the zinc-coordinating site. Since CCW 16 and TRH 1–23 are based on the same scaffold, these results may explain why modification by TRH 1–23 did not inhibit RNF4 autoubiquitination activity while the reported mutation of both cysteines 132 and 135 to serine leads to loss of activity.
Based on the structure–activity relationships observed with TRH 1–23 analogues, the 4-methoxy group on CCW16 presented an ideal position for extending a linker to yield a RNF4-based degrader. To demonstrate that we could use CCW 16 as a potential RNF4 recruiter for targeted protein degradation applications, we synthesized CCW 28–3, a bifunctional degrader linking CCW 16 to the BET bromodomain family inhibitor JQ1 (Figure 4A).22 CCW 28–3 was prepared in five steps from commercially available materials. Demethylation of 4-(4-methoxyphenoxy)aniline yielded 4-(4-aminophenoxy)phenol, which underwent reductive amination with benzaldehyde using sodium triacetoxyborohydride as the reducing agent to form CCW 22. Alkylation of phenolic moiety of CCW 22 with 4-(Boc-amino)butyl bromide and subsequent reaction with 2-choloracetyl chloride allowed functionalization of the RNF4 recruiter with a linker containing a “latent” reactive handle, a Boc-protected amino group. Boc deprotection by trifluoroacetic acid restored the functional primary amine which could then be reacted with a hydrolyzed free-acid form of JQ1 through amide coupling, yielding the bifunctional degrader CCW 28–3 as the final product (Supporting Methods). It is noteworthy that the amide coupling reaction should be highly versatile, and thus this synthetic scheme should be applicable for conjugating different protein-targeting ligands with a carboxylic acid moiety onto our RNF4 recruiter.
Figure 4.
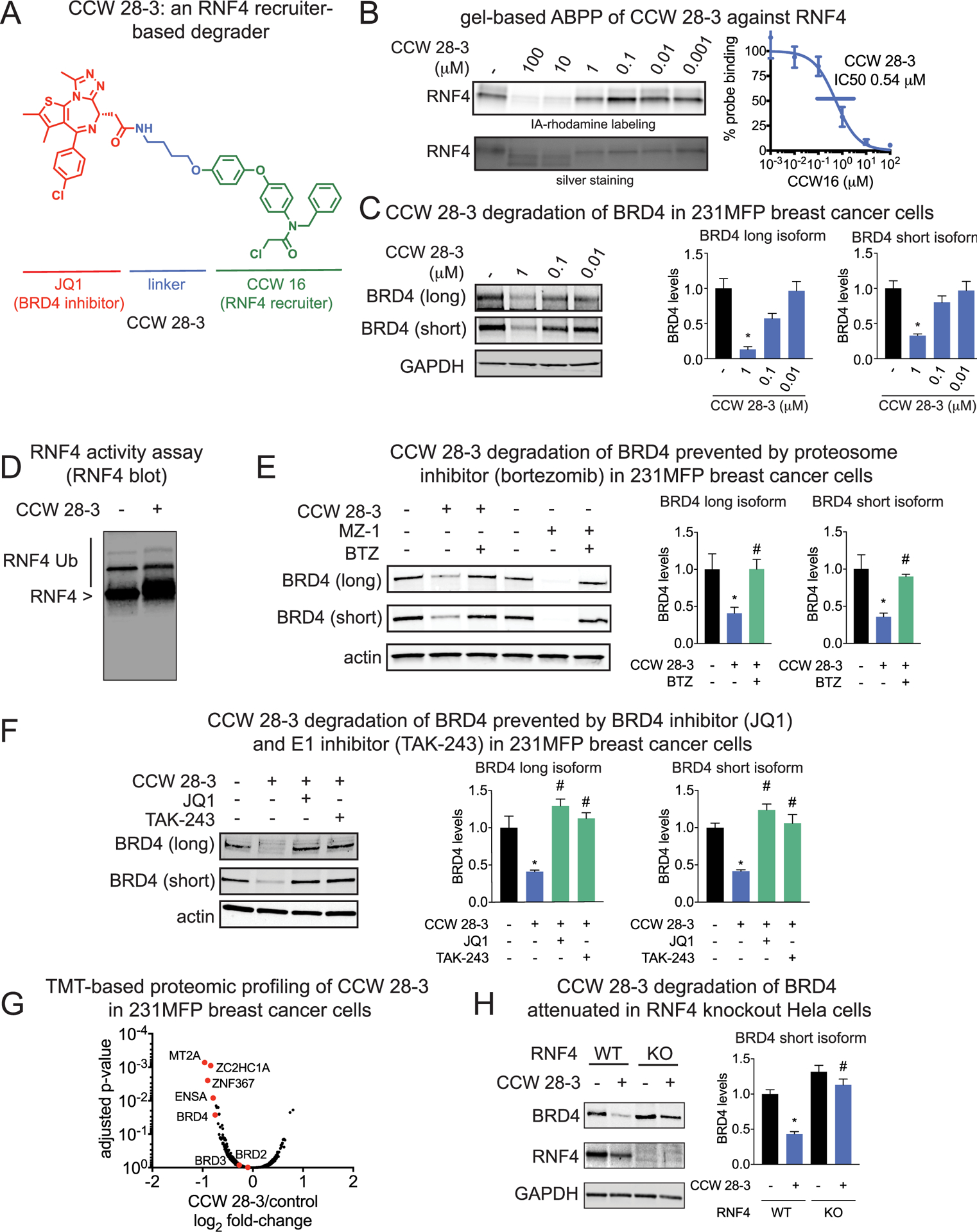
RNF4 recruiter-based BRD4 degrader. (A) Structure of CCW 28–3, an RNF4-recruiter-based degrader linked to BRD4 inhibitor JQ1. (B) Gel-based ABPP analysis of CCW 28–3 against pure human RNF4. CCW 28–3 was preincubated with pure RNF4 protein for 30 min prior to IA-rhodamine labeling for 1 h. Proteins were subjected to SDS/PAGE and visualized by in-gel fluorescence. Gels were quantified by densitometry to calculate IC50 values. (C) CCW 28–3 treatment in 231MFP breast cancer cells leads to BRD4 degradation. 231MFP breast cancer cells were treated with vehicle DMSO or CCW 28–3 for 3 h. Proteins were subjected to SDS/PAGE and Western blotting for BRD4 and GAPDH loading control. (D) CCW 28–3 does not inhibit RNF4 autoubiquitination. RNF4 was preincubated with CCW 28–3 (10 μM) for 30 min at RT followed by addition of UBA1, E2 enzyme, and ATP for 60 min at 37 °C. The reaction was quenched and subjected to SDS/PAGE and Western blotting for RNF4. (E, F) CCW 28–3 treatment in 231MFP breast cancer cells leads to proteasome-, E1 inhibitor-, and BRD4 inhibitor-dependent BRD4 degradation. Vehicle DMSO or proteasome inhibitor bortezomib (BTZ) (10 μM) (E), E1 inhibitor TAK-243 (10 μM), or BRD4 inhibitor JQ1 (10 μM) (F) were preincubated for 30 min prior to treatment with MZ1 (1 μM) or CCW 28–3 (1 μM) for 3 h. Proteins were subjected to SDS/PAGE and Western blotting for BRD4 and actin loading control. (G) TMT-based quantitative proteomic analysis of protein expression changes from CCW 28–3 (1 μM) treatment for 3 h in 231MFP cells. (H) RNF4 wild-type and knockout Hela cells were treated with CCW 28–3 (10 μM) for 5 h and subjected to SDS/PAGE and Western blotting for BRD4, RNF4, and GAPDH. Blots in parts B–F and H were quantified by densitometry. Data in parts B–F and H are from representative gels from n = 3. Bar graphs are average ± standard error of the mean (sem), n = 3–5/group. Significance is expressed as *p < 0.05 compared to vehicle-treated controls and #p < 0.05 compared to CCW 28–3 treated groups in parts E and F and CCW 28–3 treated wild-type cells in part H. Data in part G represent 6 180 protein groups quantified with 2 or more unique peptides in triplicate treatments, see Table S2 for details.
CCW 28–3 showed higher potency for RNF4 than CCW 16 with an IC50 of 0.54 μM in the competitive ABPP assay (Figure 4B). Compellingly, treatment with CCW 28–3 degraded BRD4 in a time and dose-responsive manner in 231MFP breast cancer cells with BRD4 degradation occurring after 1 h (Figure S2; Figure 4C). We showed that this reduced protein expression of BRD4 was not due to transcriptional downregulation of mRNA levels (Figure S2). We also confirmed that CCW 28–3 did not inhibit RNF4 autoubiquitination activity (Figure 4D). While CCW 28–3 did not show as complete or potent degradation compared to previously reported JQ1-based degraders such as MZ1,23,24 CCW 28–3-mediated degradation of BRD4 was still prevented by pretreatment of cells with the proteasome inhibitor bortezomib (BTZ), JQ1 alone, as well as the E1 ubiquitin activating enzyme inhibitor TAK-243 (Figure 4E–4F), indicating the degradation was occurring by induced interaction, ubiquitination, and proteasomal degradation. We next performed tandem mass tagging (TMT)-based quantitative proteomic profiling to determine the selectivity of changes in protein expression from CCW 28–3 treatment. We showed that BRD4 is one of the primary targets degraded by CCW 28–3 in 231MFP breast cancer cells (Figure 4G; Table S2). Notably, RNF4 was not detected in this TMT experiment, indicating that RNF4 has low expression in these cells. We also observed additional downregulated targets such as MT2A, ZC2HC1A, ZNF367, and ENSA, which may represent potential off-targets of JQ1 or transcriptional or post-translationally driven changes in protein expression stemming from on or off-target effects of our RNF4-targeting ligand in 231MFP cells. These targets have not been shown to be downregulated with other JQ1-based degraders.25 Interestingly, CCW 28–3 treatment did not lead to BRD2 and BRD3 degradation. Previously reported JQ1-based degraders have shown varying levels of degradation of BET bromodomain family members with cereblon and VHL-recruiting modules.26–29 This study highlights the utility of having additional E3 ligase recruiters for tuning the efficacy and selectivity of degraders targeting a given protein of interest.
To confirm engagement of RNF4 by CCW 28–3 in situ, we functionalized the optimized RNF4 ligand CCW 16 with an alkyne handle to give the probe CCW 36 (Figure S2). We then overexpressed RNF4 in HEK293T cells, treated these cells with a vehicle or CCW 28–3, enriched for RNF4, and then labeled the enriched RNF4 with CCW 36, followed by a CuAAC-mediated appendage of rhodamine-azide to visualize CCW 36 cysteine-reactivity of RNF4 by in-gel fluorescence. Intriguingly, we only observed ~30% engagement of RNF4 by CCW 28–3 in situ at a concentration 10-times higher than the concentration where we observed significant BRD4 degradation (Figure S2; Figure 4C,E,F). We attribute the low degree of target engagement to poor cell permeability of our yetunoptimized RNF4-targeting ligand. Nonetheless, our results encouragingly suggest that only a modest degree of engagement of RNF4 is sufficient to facilitate the degradation of BRD4 with CCW 28–3. The poor cellular potency observed with BRD4 degradation may also be due to loss of catalytic degradation observed with reversible degraders. However, our lab and the Cravatt lab have independently shown that nanomolar degradation can be observed with covalent E3 ligase recruiters against other E3 ligases such as RNF114 and DCAF16.23,30
We next used two complementary approaches to assess the proteome-wide selectivity of CCW 28–3, isotopic tandem orthogonal proteolysis-enabled ABPP (isoTOP-ABPP) platforms to assess the proteome-wide cysteine-reactivity of CCW 28–34–6,11 and also to perform TMT-based quantitative proteomic profiling of CCW 36-enriched targets (Figure S3; Table S2). We treated 231MFP cells with a vehicle or CCW 28–3 and labeled the resulting proteomes with the cysteine-reactive N-hex-5-ynyl-2-iodo-acetamide (IA-alkyne) probe, followed by an appendage of isotopically light or heavy TEV protease-cleavable biotin-azide tags onto probe-labeled proteins in a vehicle and CCW 28–3-treated groups, respectively. Probe-modified peptides were enriched, eluted, and analyzed using previously described methods for isoTOP-ABPP.4–6,11 Through this analysis, we demonstrated that out of 1114 total quantified probe-modified peptides, only 7 potential off-targets of CCW 28–3 showed isotopically light to heavy ratios greater than 4. This ratio greater than 4 indicated that the covalent ligand displaced IA-alkyne probe labeling at the particular site within the protein by more than 75%. Most notably, none of these off-targets of CCW 28–3 are part of the ubiquitin–proteasome system indicating that the observed BRD4 degradation can likely be attributed to RNF4-based ubiquitination (Figure S2; Table S3). Unfortunately, we were not able to observe the probe-modified peptide for RNF4 in this experiment likely due to its low abundance or labeling efficiency compared to other IA-alkyne labeled proteins. We were thus not able to confirm the degree of RNF4 occupancy by CCW 28–3 using isoTOP-ABPP. We also performed TMT-enabled quantitative proteomic profiling of CCW 36-enriched proteins also competed with CCW 16 in the 231MFP proteome, where we uncovered seven off-targets. Among these seven off-targets, SCP2 was an overlapping target between our isoTOP-ABPP and CCW 36 pulldown TMT proteomics experiments, whereas the six other targets were not overlapping. Again, no RNF4 peptides were observed, likely due to low RNF4 expression (Figure S3; Table S4).
Because CCW 28–3 was not completely selective and targeted zinc-coordinating cysteines on RNF4 that are also conserved across the RING family of E3 ligases, we next sought to confirm the contributions of RNF4 to CCW 28–3-mediated degradation of BRD4. We compared CCW 28–3-mediated BRD4 degradation in wild-type (WT) and RNF4 knockout (KO) HeLa cells. Convincingly, CCW 28–3-mediated degradation of BRD4 observed in HeLa WT cells was not evident in RNF4 KO cells (Figure 4H; Figure S4). These data further support our claim that CCW 28–3 degrades BRD4 specifically through RNF4 recruitment.
In conclusion, our study demonstrates the feasibility of using ABPP-based covalent ligand screening in vitro to rapidly discover chemical entry points for targeting E3 ligases and that these covalent ligand hits can be identified, optimized, and incorporated into degraders for targeted protein degradation applications. While CCW 16 and CCW 28–3 are not yet completely selective for RNF4 and can only achieve fractional target engagement in cells, we demonstrate that we can still degrade BRD4 in a RNF4-dependent manner. We note that CCW 28–3 does not degrade BRD4 as well as other previously reported BRD4 degraders such as MZ1 that utilizes a VHL-recruiter linked to JQ1.26 We also note that RNF4 expression is likely to be quite low in 231MFP breast cancer cells since RNF4 was not detected in TMT-based quantitative proteomic profiling of protein expression, in CCW 36-enriched proteomic profiling, or in isoTOP-ABPP analysis of cysteine-reactivity. This actually highlights the utility of target-based covalent ligand screening against E3 ligases to identify new E3 ligase recruiters for targeted protein degradation applications that cannot necessarily be accessed with proteome-wide chemoproteomic methodologies. Future medicinal chemistry efforts can be employed to optimize the potency, selectivity, and cell permeability of CCW 16 for targeting RNF4 and to improve linker positioning and composition of CCW 28–3 to promote better degradation of protein substrates. Nonetheless, CCW 16 represents a novel, small-molecule E3 ligase recruiter for RNF4, beyond the four other E3 ligase recruiters that have been reported previously, targeting cereblon, VHL, MDM2, and cIAP.2 Beyond RNF4, our study also highlights zinc-coordinating cysteines as a potential ligand able modality that can be targeted with cysteine-targeting covalent ligands. We believe that the approaches described here and insights gained in this study can be utilized for future applications in expanding the scope of E3 ligase recruiters or modulators.
METHODS
Covalent Ligand Library Used in Initial Screen.
The synthesis and characterization of many of the covalent ligands screened against RNF4 have been previously reported.4,5,31,32 Synthesis of TRH 1–156, TRH 1–160, TRH 1–167, YP 1–16, YP 1–22, YP 1–26, YP 1–31, and YP 1–44 have been previously reported.33–40 The synthesis and characterization of covalent ligands that have not been reported are described in the Supporting Information.
Gel-Based ABPP.
Gel-Based ABPP methods were performed as previously described.5,41,42 Pure recombinant human RNF4 was purchased from Boston Biochem (K-220). RNF4 (0.25 μg) was diluted into 50 μL of PBS and 1 μL of either DMSO (vehicle) or covalently acting small molecule to achieve the desired concentration. After 30 min at RT, the samples were treated with 250 nM IARhodamine (Setareh Biotech, 6222, prepared in anhydrous DMSO) for 1 h at room temperature (RT). Samples were then diluted with 20 μL of 4× reducing Laemmli SDS sample loading buffer (Alfa Aesar) and heated at 90 °C for 5 min. The samples were separated on precast 4–20% Criterion TGX gels (Bio-Rad Laboratories, Inc.). Fluorescent imaging was performed on a ChemiDoc MP (Bio-Rad Laboratories, Inc.), and inhibition of target labeling was assessed by densitometry using ImageJ.
LC–MS/MS Analysis of RNF4.
Purified RNF4 (10 μg) was diluted into 80 μL of PBS and treated for 30 min with DMSO or compound (50 μM). The DMSO control was then treated with light iodoacetamide (IA), while the compound treated sample was incubated with heavy IA for 1 h each at RT (100 μM, Sigma-Aldrich, 721328). The samples were precipitated by additional of 20 μL of 100% (w/v) TCA and combined pairwise before cooling to −80 °C for 1 h. The combined sample was then spun at maximum speed for 20 min at 4 °C, the supernatant was carefully removed, and the sample was washed with ice cold 0.01 M HCl/90% acetone solution. The sample was then resuspended in 2.4 M urea containing 0.1% Protease Max (Promega Corp., V2071) in 100 mM ammonium bicarbonate buffer. The samples were reduced with 10 mM TCEP at 60 °C for 30 min. The samples were then diluted 50% with PBS before sequencing grade trypsin (1 μg per sample, Promega Corp., V5111) was added for an overnight incubation at 37 °C. The next day the sample was centrifuged at 13 200 rpm for 30 min. The supernatant was transferred to a new tube and acidified to a final concentration of 5% formic acid and stored at −80 °C until MS analysis.
RNF4 Ubiquitination Assay.
For in vitro autoubiquitination assay, 200 nM RNF4 in 15 μL of ubiquitination assay buffer (50 mM Tris, 150 mM NaCl, 5 mM MgCl2, 5 mM DTT, pH 7.4) was preincubated with DMSO vehicle or the covalently acting compound for 30 min at RT. Subsequently, UBE1 (50 nM, Boston Biochem, E-305), UBE2D1 (400 nM Boston Bichem, E2–615), Flag-ubiquitin (4000 nM, Boston Biochem, U-120), and ATP (200 μM) were added in ubiquitination assay buffer to bring the total volume to 30 μL. The mixture was incubated at 37 °C for the indicated time before quenching with 10 μL of 4× Laemmli’s buffer. Ubiquitination activity was measured by separation on an SDS-PAGE gel and Western blotting as previously described.23
Synthetic Methods and Characterization of Covalent Ligand Analogues and CCW 28–3 Degrader.
Synthetic methods and characterization are detailed in the Supporting Information
Covalent Docking of CCW 16 in RNF4.
For covalent docking, a crystal structure of human RNF4 (PDB code 4PPE) was used.43 This crystal structure was then prepared for docking utilizing Schrödinger’s Maestro (2018–1) protein preparation.44 Missing loops and side chains were added using PRIME, and only the A chain was utilized for docking purposes. Protonation was carried out to optimize H-bond assignments (assuming pH 7.0), and all waters were removed. A restrained minimization (<0.3 Å) was then carried out to optimize the protein. A Zn coordinated by C132, C135, C159, and C162 was removed for docking purposes.
Prior to docking, CCW16 was prepared via LigPrep. To carry out covalent docking using Schrödinger’s covalent docking,45 either C132 or C135 were defined as the center of the binding grid (within 20A). CCW 16 was selected as the ligand, and the appropriate reactive residues were selected. A “nucleophilic substitution” was selected as the reaction type, and calculations were carried out on a Linux workstation with Intel Xeon 2.4 GHz processors running Red Hat 6.8 with 128 GB memory.
Cell Culture.
The 231MFP cells were obtained from Prof. Benjamin Cravatt and were generated from explanted tumor xenografts of MDA-MB-231 cells as previously described.46 RNF4 knockout HeLa cells were purchased from EdiGene USA (CL0033025003A). RNF4 wild-type HeLa cells were provided by EdiGene USA or the UC Berkeley Cell Culture Facility. 231MFP cells were cultured in L-15 media (Corning) containing 10% (v/v) fetal bovine serum (FBS) and maintained at 37 °C with 0% CO2. HeLa cells were cultured in DMEM media (Corning) containing 10% (v/v) fetal bovine serum (FBS) and maintained at 37 °C with 5% CO2.
Cell-Based Degrader Assays.
For assaying degrader activity, cells were seeded (500 000 for 231MFP cells, 300 000 for HeLa cells) into a 6 cm tissue culture dish (Corning) in 2.0–2.5 mL of media and allowed to adhere overnight. The following morning, media was replaced with complete media containing the desired concentration of compound diluted from a 1000× stock in DMSO. At the specified time point, cells were washed once with PBS on ice, before 150 μL of lysis buffer was added to the plate (10 mM sodium phosphate, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X100). The cells were incubated in lysis buffer for 5 min before scraping and transferring to microcentrifuge tubes. The lysates were then frozen at −80 °C or immediately processed for Western blotting. To prepare for Western blotting, the lysates were cleared with a 20 000g spin for 10 min and the resulting supernatants quantified via BCA assay. The lysates were normalized by dilution with PBS to match the lowest concentration lysate, and the appropriate amount of 4× Laemmli’s reducing buffer was added.
Western Blotting.
Antibodies to RNF4 (Proteintech, 17810–1-AP, 1:1000), GAPDH (Proteintech, 60004–1-IG, 1:5000), BRD4 (Abcam, Ab128874, 1:1000), and β-actin (Proteintech Group Inc., 6609–1-IG, 1:7000) were obtained from the specified commercial sources, and dilutions were prepared in 5% BSA/TBST at the specified dilutions. Proteins were resolved by SDS/PAGE and transferred to nitrocellulose membranes using the iBlot system (Invitrogen). Blots were blocked with 5% BSA in Tris-buffered saline containing Tween 20 (TBST) solution for 1 h at RT, washed in TBST, and probed with primary antibody diluted in recommended diluent per the manufacturer overnight at 4 °C. Following washes with TBST, the blots were incubated in the dark with secondary antibodies purchased from Ly-Cor and used at 1:10 000 dilution in 5% BSA in TBST at RT. Blots were visualized using an Odyssey Li-Cor scanner after additional washes. If additional primary antibody incubations were required, the membrane was stripped using ReBlot Plus Strong Antibody Stripping Solution (EMD Millipore, 2504), washed, and blocked again before being reincubated with primary antibody.
IsoTOP-ABPP Chemoproteomic Studies.
IsoTOP-ABPP studies were done as previously reported.4–6,11 Briefly, cells were lysed by probe sonication in PBS and protein concentrations were measured by BCA assay.47 For in situ experiments, cells were treated for 90 min with either DMSO vehicle or a covalently acting small molecule (from 1000× DMSO stock) before cell collection and lysis. For in vitro experiments, proteome samples diluted in PBS (4 mg of proteome per biological replicate) were treated with a DMSO vehicle or covalently acting small molecule for 30 min at RT. Proteomes were subsequently treated with IA-alkyne (100 μM, Chess GmbH, 3187) for 1 h at RT. CuAAC was performed by sequential addition of tris(2-carboxyethyl)-phosphine (TCEP) (1 mM, Sigma), tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) (34 μM, Sigma), copper(II) sulfate (1 mM, Sigma), and biotin-linker-azide, the linker functionalized with a TEV protease recognition sequence as well as an isotopically light or heavy valine for treatment of control or treated proteome, respectively. After CuAAC, proteomes were precipitated by centrifugation at 6500g, washed in ice-cold methanol, combined in a 1:1 control/treated ratio, washed again, then denatured and resolubilized by heating in 1.2% SDS/PBS to 80 °C for 5 min. Insoluble components were precipitated by centrifugation at 6 500g, and soluble proteome was diluted in 5 mL0.2% SDS/PBS. Labeled proteins were bound to avidin-agarose beads (170 μL of resuspended beads/sample, Thermo Pierce) while rotating overnight at 4 °C. Bead-linked proteins were enriched by washing three times each in PBS and water, then resuspended in 6 M urea/PBS (Sigma) and reduced in TCEP (1 mM, Sigma), alkylated with iodoacetamide (IA) (18 mM, Sigma), then washed and resuspended in 2 M urea, and trypsinized overnight with 2 μg/sample sequencing grade trypsin (Promega). Tryptic peptides were eluted off. Beads were washed three times each in PBS and water, washed in TEV buffer solution (water, TEV buffer, 100 μM dithiothreitol), resuspended in buffer with Ac-TEV protease (Invitrogen), and incubated overnight. Peptides were diluted in water and acidified with formic acid (1.2 M, Spectrum) and prepared for analysis.
IsoTOP-ABPP Mass Spectrometry Analysis.
Peptides from all chemoproteomic experiments were pressure-loaded onto 250 μm inner diameter fused silica capillary tubing packed with 4 cm of Aqua C18 reverse-phase resin (Phenomenex no. 04A-4299) which was previously equilibrated on an Agilent 600 series HPLC using a gradient of 100% buffer A to 100% buffer B over 10 min, followed by a 5 min wash with 100% buffer B and a 5 min wash with 100% buffer A. The samples were then attached using a MicroTee PEEK 360 μm fitting (Thermo Fisher Scientific no. p-888) to a 13 cm laser pulled column packed with 10 cm Aqua C18 reverse-phase resin and 3 cm of strong-cation exchange resin for isoTOP-ABPP studies. Samples were analyzed using an Q Exactive Plus mass spectrometer (Thermo Fisher Scientific) using a five-step Multidimensional Protein Identification Technology (MudPIT) program, using 0%, 25%, 50%, 80%, and 100% salt bumps of 500 mM aqueous ammonium acetate and using a gradient of 5–55% buffer B in buffer A (buffer A, 95:5 water/acetonitrile, 0.1% formic acid; buffer B, 80:20 acetonitrile/water, 0.1% formic acid). Data was collected in data-dependent acquisition mode with dynamic exclusion enabled (60 s). One full MS (MS1) scan (400–1800 m/z) was followed by 15 MS2 scans (ITMS) of the nth most abundant ions. The heated capillary temperature was set to 200 °C, and the nanospray voltage was set to 2.75 kV.
Data was extracted in the form of MS1 and MS2 files using Raw Extractor 1.9.9.2 (Scripps Research Institute) and searched against the Uniprot human database using ProLuCID search methodology in IP2 v.3 (Integrated Proteomics Applications, Inc.).48 Cysteine residues were searched with a static modification for carboxyaminomethylation (+ 57.02146) and up to three differential modifications for methionine oxidation and either the light or heavy TEV tags (+ 464.28596 or + 470.29977, respectively). Peptides were required to have at least one tryptic end and to contain the TEV modification. ProLUCID data was filtered through DTASelect to achieve a peptide false-positive rate below 5%. Only those probe-modified peptides that were evident across all two out of three biological replicates were interpreted for their isotopic light to heavy ratios. Those probe-modified peptides that showed ratios >3 were further analyzed as potential targets of the covalently acting small-molecule. For modified peptides with ratios >3, we filtered these hits for peptides were present in all three biological replicates. For those probe-modified peptide ratios >3, only those peptides with 3 ratios >3 were interpreted and otherwise replaced with the lowest ratio. For those probe-modified peptide ratios >4, only those peptides with 3 ratios >4 were interpreted and otherwise replaced with the lowest ratio. MS1 peak shapes of any resulting probe-modified peptides with ratios >3 were then manually confirmed to be of good quality for interpreted peptides across all biological replicates.
Vectors for RNF4 Overexpression.
The pCMV6-Entry-RNF4 (C-term FLAG + Myc tag) vector was purchased from Origene (RC207273). The corresponding pCMV6-Entry-eGFP vector was constructed via Gibson Assembly with primers: GATCTGCCGCCGCGATCGCCatggtgagcaagggcgag, TCGAGCGGCCGCGTACGCGTcttgtacagctcgtccatgcc to amplify the eGFP ORF with desired overlaps, and ACGCGTACGCGGCCG, GGCGATCGCGGCGG to linearize the pCMV6-Entry backbone.
RNF4 Overexpression, Immunoprecipitation, and in Vitro Probe Labeling Experiments.
HEK293T cells were seeded to 30–40% confluency in 10 cm dishes. The day of transfection, media was replaced with DMEM containing 2.5% FBS. For transfection, 500 μL of Opti-MEM (Thermo Fisher) containing 10 μg of pCMV6-Entry-RNF4 or eGFP vector and 30 μg of polyethylenimine was added to the plate. Media was replaced 48 h later with identical media containing DMSO or CCW 28–3 (10 μM). After 1.5 h, cells were washed and harvested in PBS, resuspended in 500 μL of PBS, and lysed by probe tip sonication at 15% amplitude for 2 × 10 s. Lysates were cleared by centrifugation at 21 000g for 20 min, and resulting supernatant was mixed with 30 μL of anti-FLAG resin (Genscript, L00432) and rotated at 4 °C for 1 h. The beads were washed 3× with 500 μL of PBS containing 300 mM sodium chloride, before elution of FLAG-tagged proteins with 100 μL of 250 ng/μL 3 × FLAG peptide (APExBIO, A6001) in PBS.
For labeling of enriched RNF4 with CCW 36 to monitor in situ CCW 28–3 target engagement, 25 μL of eluted protein was treated with 1 μM CCW 36 (alkyne-functionalized probe of CCW 16) for 1h. A rhodamine ligand was appended with copper catalyzed click chemistry by addition of 1 mM copper(II) sulfate, 35 μM Tris(benzyltriazole methylamine) (TBTA), 1 mM TCEP, and 0.1 mM Azide-Fluor 545 (Click Chemistry Tools, AZ109–5). The degree of labeling was assessed by SDS/PAGE and measuring in-gel fluorescence using ImageJ.
TMT-Based Quantitative Proteomic Analysis.
Cell Lysis, Proteolysis, and Isobaric Labeling.
Treated cell-pellets were lysed and digested using the commercially available Pierce Mass Spec Sample Prep Kit for Cultured Cells (Thermo Fisher Scientific, P/N 84840) following manufacturer’s instructions. Briefly, 100 μg of protein from each sample was reduced, alkylated, and digested overnight using a combination of Endoproteinase Lys-C and trypsin proteases. Individual samples were then labeled with isobaric tags using commercially available Tandem Mass Tag 6-plex (TMTsixplex) (Thermo Fisher Scientific, P/N 90061) or TMT11plex (TMT11plex) isobaric labeling reagent (Thermo Fisher Scientific, P/N 23275) kits, in accordance with the manufacturer’s protocols.
High-pH Reversed Phase Separation.
Tandem mass tag labeled (TMT) samples were then consolidated and separated using high-pH reversed phase chromatography (RP-10) with fraction collection as previously described.49 Fractions were speed-vac dried, then reconstituted to produce 24 fractions for subsequent online nanoLC–MS/MS analysis.
Protein Identification and Quantitation by nanoLC–MS/MS.
Reconstituted RP-10 fractions were analyzed on a Thermo Orbitrap Fusion Lumos Mass Spectrometer (Xcalibur 4.1, Tune Application 3.0.2041) coupled to an EasyLC 1200 HPLC system (Thermo Fisher Scientific). The EasyLC 1200 was equipped with a 20 μL loop, setup for 96 well plates. A Kasil-fritted trapping column (75 μm i.d.) packed with ReproSil-Pur 120 C18-AQ, 5 μm material (15 mm bed length) was utilized together with a 160 mm length, 75 μm inner diameter spraying capillary pulled to a tip diameter of approximately 8–10 μm using a P-2000 capillary puller (Sutter Instruments, Novato, CA). The 160 mm separation column was packed with ReproSil-Pur 120 C18-AQ, 3 μm material (Dr. Maisch GmbH, Ammerbuch-Entringen, Germany). Mobile phase A consisted of 0.1% formic acid/2% acetonitrile (v/v), and mobile phase B was 0.1% formic acid/98% acetonitrile (v/v). Samples (18 μL) were injected onto the trapping column using Mobile phase A at a flow rate of 2.5 μL/min. Peptides were then eluted using an 80 min gradient (2% mobile phase B for 5 min, 2–40% B from 5 to 65 min, followed by 70% B from 65 to 70 min, then returning to 2% B from 70 to 80 min) at a flow rate of 300 nL/min on the capillary separation column with direct spraying into the mass spectrometer. Data was acquired on Orbitrap Fusion Lumos Mass Spectrometer in data-dependent mode using synchronous precursor scanning MS3 mode (SPS-MS3), with MS2 triggered for the 12 most intense precursor ions within a mass-to-charge ratio (m/z) range of 300–1500 found in the full MS survey scan event. MS scans were acquired at 60 000 mass resolution (R) at m/z 400, using a target value of 4 × 105 ions, and a maximum fill time of 50 ms. MS2 scans were acquired as CID ion trap (IT) rapid type scans using a target value of 1 × 104 ions, maximum fill time of 50 ms, and an isolation window of 2 Da. Data-dependent MS3 spectra were acquired as Orbitrap (OT) scans, using Top 10 MS2 daughter selection, an automatic gain control (AGC) target of 5 × 104 ions, with a scan range of m/z 100–500. The MS3 maximum injection time was 86 ms, with an HCD collision energy set to 65%. The MS3 mass resolution (R) was set to 15 000 at m/z 400 for TMT6plex experiments and 50 000 at m/z 400 for TMT11-plex experiments. Dynamic exclusion was set to exclude selected precursors for 60 s with a repeat count of 1. The nanospray voltage was set to 2.2 kV, with the heated capillary temperature set to 300 °C and an S-lens rf level equal to 30%. No sheath or auxiliary gas flow is applied.
Data Processing and Analysis.
Acquired MS data was processed using Proteome Discoverer, version 2.2.0.388, software (Thermo) utilizing Mascot version 2.5.1 search engine (Matrix Science, London, U.K.) together with the Percolator validation node for peptide-spectral match filtering.50 Data was searched against the Uniprot protein database (canonical human and mouse sequences, EBI, Cambridge, U.K.) supplemented with sequences of common contaminants. Peptide search tolerances were set to 10 ppm for precursors and 0.8 Da for fragments. Trypsin cleavage specificity (cleavage at K, R except if followed by P) allowed for up to two missed cleavages. Carbamidomethylation of cysteine was set as a fixed modification, and methionine oxidation and TMT-modification of N-termini and lysine residues were set as variable modifications. Data validation of peptide and protein identifications was done at the level of the complete data set consisting of combined Mascot search results for all individual samples per experiment via the Percolator validation node in Proteome Discoverer. Reporter ion ratio calculations were performed using summed abundances with the most confident centroid selected from the 20 ppm window. Only peptide-to-spectrum matches that are unique assignments to a given identified protein within the total data set are considered for protein quantitation. High-confidence protein identifications were reported using a Percolator estimated <1% false discovery rate (FDR) cutoff. Differential abundance significance was estimated using a background-based ANOVA with Benjamini–Hochberg correction to determine adjusted p-values.
Gene Expression by qPCR.
RNA was isolated from cultured cells using TRIzol reagent (Thermo Fisher, 15596026) per the manufacturer’s specification. NEB Protoscript II Reverse Transcriptase (NEB, M0368) was used to synthesize cDNA with oligo dT primers per the manufacturer’s specification. Gene expression was measured from the cDNA using DyNAmo HS SYBR Green qPCR Kit (Thermo Fisher, F410) on a BioRad CFX Connect PCR system. Samples were measured in technical triplicate and BRD4 expression normalized to cyclophilin A levels before comparison. The primers used were:
BRD4 F: ACCTCCAACCCTAACAAGCC
BRD4 R: TTTCCATAGTGTCTTGAGCACC
CycA F: CCCACCGTGTTCTTCGACATT
CycA R: GGACCCGTATGCTTTAGGATGA
CCW 36 Pulldown and Proteomic Analysis.
231 MFP cells were grown to 90% confluency, washed and collected in PBS, and lysed with probe tip sonication. The resulting lysate was and treated with 10 μM CCW 36 alkyne probe or DMSO for 1 h. Downstream click reaction, enrichment, digestion, and isobaric labeling was performed according to the protocol described by Thomas et al.49 Biotin picolyl azide (Sigma, 900912) was used in click conjugation to enhance efficiency. Peptides were labeled with a TMTsixplex kit (Thermo Fisher, 90061). Mass spectrometry and analysis performed as previously described for isoTOP-ABPP mass spectrometry. Identified peptides were quantified, normalized to their respective channel, and CCW 36 treated samples compared against controls. Fold-change enrichment vs control for each protein and p-value (Student’s t test) was reported.
Supplementary Material
ACKNOWLEDGMENTS
We thank the members of the Nomura Research Group and Novartis Institutes for BioMedical Research for critical reading of the manuscript. This work was supported by Novartis Institutes for BioMedical Research and the Novartis-Berkeley Center for Proteomics and Chemistry Technologies (NBCPACT) for all listed authors. This work was also supported by grants from the National Institutes of Health (Grant F31CA225173 for C.C.W.).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.8b01083.
Additional information on methods (PDF)
Table S1, structures of covalent ligands screened against
RNF4 (XLSX)
Table S2, TMT-based quantitative proteomic analysis of CCW 28–3 in 231MFP breast cancer cells in situ (XLSX)
Table S3, isoTOP-ABPP analysis of CCW 28–3 in 231MFP breast cancer cells in situ (XLSX)
Table S4, TMT proteomics data for proteins enriched by CCW 36-labeling and competing with CCW 16 (XLSX)
The authors declare the following competing financial interest(s): J.A.T., J.M.K., and M.S. are employees of Novartis Institutes for BioMedical Research. J.R.T. was an employee of Novartis Institutes for BioMedical Research when this study was submitted but is now an employee of Vertex Pharmaceuticals. This study was funded by the Novartis Institutes for BioMedical Research and the Novartis-Berkeley Center for Proteomics and Chemistry Technologies. D.K.N. is a co-founder, share-holder, and adviser for Artris Therapeutics and Frontier Medicines.
REFERENCES
- (1).Burslem GM, and Crews CM (2017) Small-Molecule Modulation of Protein Homeostasis. Chem. Rev 117, 11269–11301. [DOI] [PubMed] [Google Scholar]
- (2).Lai AC, and Crews CM (2017) Induced Protein Degradation: An Emerging Drug Discovery Paradigm. Nat. Rev. Drug Discovery 16, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Rape M (2017) Ubiquitylation at the Crossroads of Development and Disease. Nat. Rev. Mol. Cell Biol 19, 59–70. [DOI] [PubMed] [Google Scholar]
- (4).Grossman EA, Ward CC, Spradlin JN, Bateman LA, Huffman TR, Miyamoto DK, Kleinman JI, and Nomura DK (2017) Covalent Ligand Discovery against Druggable Hotspots Targeted by Anti-Cancer Natural Products. Cell Chem. Biol 24, 1368–1376.E4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Bateman LA, Nguyen TB, Roberts AM, Miyamoto DK, Ku W-M, Huffman TR, Petri Y, Heslin MJ, Contreras CM, Skibola CF, et al. (2017) Chemoproteomics-Enabled Covalent Ligand Screen Reveals a Cysteine Hotspot in Reticulon 4 That Impairs ER Morphology and Cancer Pathogenicity. Chem. Commun 53, 7234–7237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Backus KM, Correia BE, Lum KM, Forli S, Horning BD, González-Páez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, et al. (2016) Proteome-Wide Covalent Ligand Discovery in Native Biological Systems. Nature 534, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wang C, Weerapana E, Blewett MM, and Cravatt BF (2014) A Chemoproteomic Platform to Quantitatively Map Targets of Lipid-Derived Electrophiles. Nat. Methods 11, 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hacker SM, Backus KM, Lazear MR, Forli S, Correia BE, and Cravatt BF (2017) Global Profiling of Lysine Reactivity and Ligandability in the Human Proteome. Nat. Chem 9, 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Backus KM (2018) Applications of Reactive Cysteine Profiling. Curr. Top. Microbiol. Immunol 420, 375–417. [DOI] [PubMed] [Google Scholar]
- (10).Liu Y, Patricelli MP, and Cravatt BF (1999) Activity-Based Protein Profiling: The Serine Hydrolases. Proc. Natl. Acad. Sci.U. S. A 96, 14694–14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MBD, Bachovchin DA, Mowen K, Baker D, and Cravatt BF (2010) Quantitative Reactivity Profiling Predicts Functional Cysteines in Proteomes. Nature 468, 790–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wang C, Weerapana E, Blewett MM, and Cravatt BF (2014) A Chemoproteomic Platform to Quantitatively Map Targets of Lipid-Derived Electrophiles. Nat. Methods 11, 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Anderson KE, To M, Olzmann JA, and Nomura DK (2017) Chemoproteomics-Enabled Covalent Ligand Screening Reveals a Thioredoxin-Caspase 3 Interaction Disruptor That Impairs Breast Cancer Pathogenicity. ACS Chem. Biol 12, 2522–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. (2004) In Vivo Activation of the P53 Pathway by Small-Molecule Antagonists of MDM2. Science 303, 844–848. [DOI] [PubMed] [Google Scholar]
- (15).Schneekloth AR, Pucheault M, Tae HS, and Crews CM (2008) Targeted Intracellular Protein Degradation Induced by a Small Molecule: En Route to Chemical Proteomics. Bioorg. Med. Chem. Lett 18, 5904–5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Malecka KA, Fera D, Schultz DC, Hodawadekar S, Reichman M, Donover PS, Murphy ME, and Marmorstein R (2014) Identification and Characterization of Small Molecule Human Papillomavirus E6 Inhibitors. ACS Chem. Biol 9, 1603–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Staudinger JL (2017) The Molecular Interface Between the SUMO and Ubiquitin Systems. Adv. Exp. Med. Biol 963, 99–110. [DOI] [PubMed] [Google Scholar]
- (18).Sriramachandran AM, and Dohmen RJ (2014) SUMO-Targeted Ubiquitin Ligases. Biochim. Biophys. Acta, Mol. Cell Res 1843, 75–85. [DOI] [PubMed] [Google Scholar]
- (19).Tan B, Mu R, Chang Y, Wang Y-B, Wu M, Tu H-Q, Zhang Y-C, Guo S-S, Qin X-H, Li T, et al. (2015) RNF4 Negatively Regulates NF-KB Signaling by down-Regulating TAB2. FEBS Lett 589, 2850–2858. [DOI] [PubMed] [Google Scholar]
- (20).Fryrear KA, Guo X, Kerscher O, and Semmes OJ (2012) The Sumo-Targeted Ubiquitin Ligase RNF4 Regulates the Localization and Function of the HTLV-1 Oncoprotein Tax. Blood 119, 1173–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Bilodeau S, Caron V, Gagnon J, Kuftedjian A, and Tremblay A (2017) A CK2-RNF4 Interplay Coordinates Non-Canonical SUMOylation and Degradation of Nuclear Receptor FXR.J. Mol. Cell Biol 9, 195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. (2010) Selective Inhibition of BET Bromodomains. Nature 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Spradlin JN, Hu X, Ward CC, Brittain SM, Ou L, Bussiere DE, Thomas JR, Tallarico JA, McKenna JM, Schirle M, et al. (2018) Harnessing the Anti-Cancer Natural Product Nimbolide for Targeted Protein Degradation. bioRxiv, 436998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zengerle M, Chan K-H, and Ciulli A (2015) Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol 10, 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Savitski MM, Zinn N, Faelth-Savitski M, Poeckel D, Gade S, Becher I, Muelbaier M, Wagner AJ, Strohmer K, Werner T, et al. (2018) Multiplexed Proteome Dynamics Profiling Reveals Mechanisms Controlling Protein Homeostasis. Cell 173, 260–275.E25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zengerle M, Chan K-H, and Ciulli A (2015) Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol 10, 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, and Bradner JE (2015) DRUG DEVELOPMENT. Phthalimide Conjugation as a Strategy for in Vivo Target Protein Degradation. Science 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Gadd MS, Testa A, Lucas X, Chan K-H, Chen W, Lamont DJ, Zengerle M, and Ciulli A (2017) Structural Basis of PROTAC Cooperative Recognition for Selective Protein Degradation. Nat. Chem. Biol 13, 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Nowak RP, DeAngelo SL, Buckley D, He Z, Donovan KA, An J, Safaee N, Jedrychowski MP, Ponthier CM, Ishoey M, et al. (2018) Plasticity in Binding Confers Selectivity in Ligand-Induced Protein Degradation. Nat. Chem. Biol 14, 706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhang X, Crowley VM, Wucherpfennig TG, Dix MM, and Cravatt BF (2018) Electrophilic PROTACs That Degrade Nuclear Proteins by Engaging DCAF16. bioRxiv, 443804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Roberts AM, Miyamoto DK, Huffman TR, Bateman LA, Ives AN, Akopian D, Heslin MJ, Contreras CM, Rape M, Skibola CF, et al. (2017) Chemoproteomic Screening of Covalent Ligands Reveals UBA5 As a Novel Pancreatic Cancer Target. ACS Chem. Biol 12, 899–904. [DOI] [PubMed] [Google Scholar]
- (32).Counihan JL, Wiggenhorn AL, Anderson KE, and Nomura DK (2018) Chemoproteomics-Enabled Covalent Ligand Screening Reveals ALDH3A1 as a Lung Cancer Therapy Target. ACS Chem. Biol 13, 1970–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Kokosza K, Balzarini J, and Piotrowska DG (2014) Novel 5-Arylcarbamoyl-2-Methylisoxazolidin-3-Yl-3-Phosphonates as Nucleotide Analogues. Nucleosides, Nucleotides Nucleic Acids 33, 552–582. [DOI] [PubMed] [Google Scholar]
- (34).Talaty ER, Young SM, Dain RP, and Van Stipdonk MJV (2011) A Study of Fragmentation of Protonated Amides of Some Acylated Amino Acids by Tandem Mass Spectrometry: Observation of an Unusual Nitrilium Ion. Rapid Commun. Mass Spectrom 25, 1119–1129. [DOI] [PubMed] [Google Scholar]
- (35).Timokhin VI, Gastaldi S, Bertrand MP, and Chatgilialoglu C (2003) Rate Constants for the β-Elimination of Tosyl Radical from a Variety of Substituted Carbon-Centered Radicals. J. Org. Chem 68, 3532–3537. [DOI] [PubMed] [Google Scholar]
- (36).Cee VJ, Volak LP, Chen Y, Bartberger MD, Tegley C, Arvedson T, McCarter J, Tasker AS, and Fotsch C (2015) Systematic Study of the Glutathione (GSH) Reactivity of N-Arylacrylamides: 1. Effects of Aryl Substitution. J. Med. Chem 58, 9171–9178. [DOI] [PubMed] [Google Scholar]
- (37).Le Sann C, Huddleston J, and Mann J (2007) Synthesis and Preliminary Evaluation of Novel Analogues of Quindolines as Potential Stabilisers of Telomeric G-Quadruplex DNA. Tetrahedron 63, 12903–12911. [Google Scholar]
- (38).Ikoma M, Oikawa M, and Sasaki M (2008) Synthesis and Domino Metathesis of Functionalized 7-Oxanorbornene Analogs toward Cis-Fused Heterocycles. Tetrahedron 64, 2740–2749. [Google Scholar]
- (39).Cho S-D, Park Y-D, Kim J-J, Lee S-G, Ma C, Song S-Y, Joo W-H, Falck JR, Shiro M, Shin D-S, et al. (2003) A One-Pot Synthesis of Pyrido[2,3-b][1,4]Oxazin-2-Ones. J. Org. Chem 68, 7918–7920. [DOI] [PubMed] [Google Scholar]
- (40).Magolan J, Carson CA, and Kerr MA (2008) Total Synthesis of (±)-Mersicarpine. Org. Lett 10, 1437–1440. [DOI] [PubMed] [Google Scholar]
- (41).Louie SM, Grossman EA, Crawford LA, Ding L, Camarda R, Huffman TR, Miyamoto DK, Goga A, Weerapana E, and Nomura DK (2016) GSTP1 Is a Driver of Triple-Negative Breast Cancer Cell Metabolism and Pathogenicity. Cell Chem. Biol 23, 567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Medina-Cleghorn D, Bateman LA, Ford B, Heslin A, Fisher KJ, Dalvie ED, and Nomura DK (2015) Mapping Proteome-Wide Targets of Environmental Chemicals Using Reactivity-Based Chemoproteomic Platforms. Chem. Biol 22, 1394–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Groocock LM, Nie M, Prudden J, Moiani D, Wang T, Cheltsov A, Rambo RP, Arvai AS, Hitomi C, Tainer JA, et al. (2014) RNF4 Interacts with Both SUMO and Nucleosomes to Promote the DNA Damage Response. EMBO Rep 15, 601–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Schrödinger, LLC (2018) Maestro, release 2018–1, Schrödinger, LLC, New York, NY. [Google Scholar]
- (45).Zhu K, Borrelli KW, Greenwood JR, Day T, Abel R, Farid RS, and Harder E (2014) Docking Covalent Inhibitors: A Parameter Free Approach To Pose Prediction and Scoring. J. Chem. Inf. Model 54, 1932–1940. [DOI] [PubMed] [Google Scholar]
- (46).Jessani N, Humphrey M, McDonald WH, Niessen S, Masuda K, Gangadharan B, Yates JR, Mueller BM, and Cravatt BF (2004) Carcinoma and Stromal Enzyme Activity Profiles Associated with Breast Tumor Growth in Vivo. Proc. Natl. Acad. Sci.U. S. A 101, 13756–13761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, and Klenk DC (1985) Measurement of Protein Using Bicinchoninic Acid. Anal. Biochem 150, 76–85. [DOI] [PubMed] [Google Scholar]
- (48).Xu T, Park SK, Venable JD, Wohlschlegel JA, Diedrich JK, Cociorva D, Lu B, Liao L, Hewel J, Han X, et al. (2015) ProLuCID: An Improved SEQUEST-like Algorithm with Enhanced Sensitivity and Specificity. J. Proteomics 129, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Thomas JR, Brittain SM, Lipps J, Llamas L, Jain RK, and Schirle M (2017) A Photoaffinity Labeling-Based Chemoproteomics Strategy for Unbiased Target Deconvolution of Small Molecule Drug Candidates. Methods Mol. Biol 1647, 1–18. [DOI] [PubMed] [Google Scholar]
- (50).Käll L, Canterbury JD, Weston J, Noble WS, and MacCoss MJ (2007) Semi-Supervised Learning for Peptide Identification from Shotgun Proteomics Datasets. Nat. Methods 4, 923–925. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.