

Abstract
The Krebs cycle-derived metabolite itaconate is highly upregulated in inflammatory macrophages and exerts immunomodulatory effects through cysteine modifications on target proteins. The NLRP3 inflammasome, which cleaves IL-1β, IL-18, and gasdermin D, must be tightly regulated to avoid excessive inflammation. Here we provide evidence that itaconate modifies NLRP3 and inhibits inflammasome activation. Itaconate and its derivative, 4-octyl itaconate (4-OI), inhibited NLRP3 inflammasome activation, but not AIM2 or NLRC4. Conversely, NLRP3 activation was increased in itaconate-depleted Irg1−/− macrophages. 4-OI inhibited the interaction between NLRP3 and NEK7, a key step in the activation process, and “dicarboxypropylated” C548 on NLRP3. Furthermore, 4-OI inhibited NLRP3-dependent IL-1β release from PBMCs isolated from cryopyrin-associated periodic syndrome (CAPS) patients, and reduced inflammation in an in vivo model of urate-induced peritonitis. Our results identify itaconate as an endogenous metabolic regulator of the NLRP3 inflammasome and describe a process that may be exploited therapeutically to alleviate inflammation in NLRP3-driven disorders.
Keywords: inflammasome, NLRP3, NEK7, pyroptosis, itaconate, metabolite, macrophage, IL-1β, immunometabolism, cysteine modification
Graphical Abstract
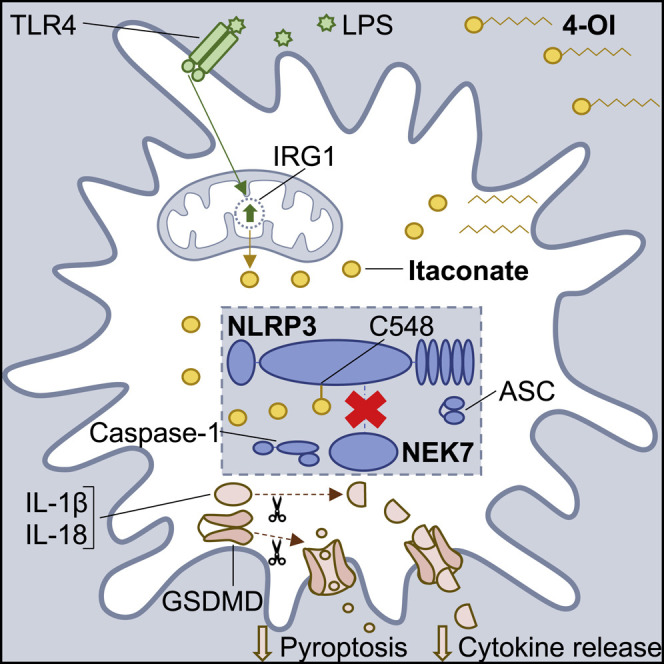
Highlights
-
•
Itaconate and its derivative 4-OI (which generates itaconate) block NLRP3 activation
-
•
Itaconate-depleted Irg1 −/− BMDMs exhibit increased NLRP3 inflammasome activation
-
•
4-OI “dicarboxypropylates” C548 on NLRP3 and blocks the NLRP3-NEK7 interaction
-
•
4-OI reduces peritonitis in vivo and blocks IL-1β release from CAPS patient PBMCs
Hooftman et al. reveal a role for the Krebs cycle-derived metabolite itaconate in regulating the NLRP3 inflammasome. Itaconate specifically blocks NLRP3 inflammasome activation by reducing the NLRP3-NEK7 interaction, likely due to modification of C548 on NLRP3. Furthermore, itaconate inhibits IL-1β release from cells isolated from patients with the NLRP3-mediated disease CAPS.
Introduction
Inflammasomes are intracellular multi-protein complexes that respond to a wide range of stimuli, including invading pathogens, host cell-derived danger signals, and environmental irritants (Lamkanfi and Dixit, 2014; Swanson et al., 2019). Numerous different inflammasomes exist, each responding to its own activating stimuli. The NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasome is one of the best characterized inflammasomes, consisting of its central protein NLRP3, the adaptor protein ASC (also known as PYCARD), the mitotic kinase NIMA-related kinase 7 (NEK7) (He et al., 2016; Schmid-Burgk et al., 2016; Shi et al., 2016b), and the effector protein caspase-1. NLRP3 consists of three domains: an amino-terminal pyrin domain (PYD), a carboxy-terminal leucine-rich repeat (LRR) domain, and a central NACHT domain (Hu et al., 2013). Interactions between NLRP3 and NEK7 are essential for NLRP3 oligomerization and associated inflammasome activation, and these occur at multiple surfaces in the LRR and NACHT domains of NLRP3 (Sharif et al., 2019). Activation of the NLRP3 inflammasome results in caspase-1-mediated cleavage of pro-IL-1β, pro-IL-18, and gasdermin D into their active subunits. Despite advances in our understanding of the interactions that facilitate NLRP3 activation, a unifying molecular pathway from stimulus to NLRP3 activation remains to be elucidated. This is further complicated by the fact that the stimuli that trigger NLRP3 activation are so wide-ranging: potassium efflux (Muñoz-Planillo et al., 2013), chloride efflux (Domingo-Fernández et al., 2017; Tang et al., 2017), lysosomal disruption (Hornung et al., 2008), trans-Golgi network disassembly (Chen and Chen, 2018), and mitochondrial dysfunction (Groß et al., 2016; Shimada et al., 2012) are notable NLRP3-activating stimuli. The NLRP3 inflammasome may also be regulated metabolically. Mitochondrial localization of NLRP3 is thought to precede inflammasome activation (Iyer et al., 2013), and mitochondrial reactive oxygen species (ROS) have been implicated in NLRP3 activation (Groß et al., 2016). Furthermore, the ketone body β-hydroxybutyrate, produced as a result of a metabolic shift to fatty acid oxidation in conditions of low glucose, is an endogenous inhibitor of NLRP3 activation (Youm et al., 2015).
NLRP3 has been implicated in several diseases. Cryopyrin-associated periodic syndrome (CAPS) refers to a group of autoinflammatory disorders characterized by autosomal dominant mutations in Nlrp3, resulting in dysregulated release of IL-1β (Hoffman et al., 2001) and the development of multi-organ systemic inflammation. The role of NLRP3 in disease is not restricted to monogenic NLRP3-driven diseases, however, as NLRP3 has been implicated in a wide range of different autoinflammatory diseases including Alzheimer disease and rheumatoid arthritis (Mangan et al., 2018). The pathological involvement of NLRP3 therefore necessitates that its activation is tightly regulated. A major mechanism involves post-translational modifications. NLRP3 is ubiquitylated under resting conditions (Juliana et al., 2012), whereas its phosphorylation and acetylation promote activation (He et al., 2020; Stutz et al., 2017). Covalent inhibitors of NLRP3, which include parthenolide (Juliana et al., 2010) and oridonin (He et al., 2018), have revealed that NLRP3 is also susceptible to electrophilic modification of reactive cysteines, thus opening up another potential route through which NLRP3 inflammasome activation may be controlled.
Itaconate is an unsaturated dicarboxylic acid that is synthesized from the decarboxylation of the Krebs cycle intermediate cis-aconitate by the enzyme immune-responsive gene 1 (IRG1), also termed ACOD1 (Hooftman and O’Neill, 2019; Michelucci et al., 2013; O’Neill and Artyomov, 2019). Itaconate is a prime example of metabolic reprogramming in macrophages. Although synthesized as a by-product of the Krebs cycle, its production is scaled up upon LPS treatment (Lee et al., 1995) seemingly with the purpose of restraining macrophage immune responses to TLR stimulation. Endogenous itaconate and/or its cell-permeable derivative 4-octyl itaconate (4-OI) have been shown to alkylate cysteine residues on multiple proteins, including kelch-like ECH-associated protein 1 (KEAP1) (Mills et al., 2018), glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Liao et al., 2019), aldolase A (ALDOA) (Qin et al., 2019), and receptor-interacting serine/threonine-protein kinase 3 (RIPK3) (Qin et al., 2020). This form of cysteine alkylation, termed 2,3-dicarboxypropylation or itaconation, has been shown to be an important part of the anti-inflammatory properties of itaconate. As an example, the modification of KEAP1 activates the antioxidant transcription factor nuclear factor erythroid 2-related factor 2 (NRF2), thereby blocking macrophage IL-1β transcription following LPS stimulation, which requires ROS (Mills et al., 2018). However, the effect of itaconate on inflammasome-dependent IL-1β cleavage and release has not been fully explored. Here we provide evidence that itaconate modifies a specific cysteine (C548) on NLRP3 and inhibits NLRP3 activation by interfering with the interaction between NLRP3 and NEK7. Itaconate may therefore be an important negative regulator of NLRP3, which could have utility as a therapy in NLRP3-mediated diseases.
Results
4-OI Specifically Blocks NLRP3 Inflammasome Activation
Treatment of LPS-primed bone marrow-derived macrophages (BMDMs) with 4-OI prior to activation of NLRP3 with ATP or nigericin (Figure 1 A) resulted in a concentration-dependent reduction in IL-1β release (Figures 1B and 1D) as well as a reduction in LDH release (Figures 1C and 1E), which is used as a measure of pyroptosis. Cleavage and release of IL-1β in this assay were NLRP3-dependent (Figures S1A and S1B). The effect of 4-OI was confirmed by western blot as it blocked the cleavage of IL-1β and caspase-1 to their mature p17 and p20 forms, respectively (Figures 1F and 1G, compare lanes 5–7 to lane 4). 4-OI also blocked the cleavage of gasdermin D, the pyroptosis executioner (Shi et al., 2015), to its active N-terminal fragment (Figure S1C, compare lanes 5–7 to lane 4). Dimethyl itaconate (DMI), a different itaconate derivative, similarly blocked IL-1β release and cleavage (Figures S1D and S1E). However, the octyl control compounds 4-octyl succinate (4-OS) and 4-octyl-2-methyl succinate (4-O-2-MS), designed to control for the potentially reactive octyl tail on 4-OI, did not block IL-1β cleavage and release to the same extent (Figures S1F–S1H). The specific NLRP3 inhibitor MCC950 (Coll et al., 2015) was also used as a control in these experiments (Figures S1I and S1J). The cytokine IL-18 is co-secreted with IL-1β in an NLRP3-dependent manner (Ghayur et al., 1997), and its release was completely inhibited by 4-OI (Figure 1H). Upon NLRP3 inflammasome activation, the adaptor protein ASC is recruited by NLRP3 and forms large multimeric complexes (Lu et al., 2014), termed ASC specks. We found that 4-OI inhibited ASC speck formation, as detected by flow cytometry (Figure 1I), and reduced the appearance of large multimeric ASC complexes, as detected by western blot (Figure 1J, compare lane 4 to lane 3).
Figure 1.

4-OI Blocks NLRP3 Inflammasome Activation
(A) Time flow of NLRP3 inflammasome assay with 4-OI in BMDMs.
(B and C) LPS (3 h) and ATP (45 min) induced IL-1β release (B, n = 10) and LDH release (C, n = 4 for ATP alone, n = 7 for others, measured as % LDH release of total lysis control) ± 4-OI.
(D and E) LPS (3 h) and nigericin (45 min) induced IL-1β release (D, n = 7) and LDH release (E, n = 5 for NS alone, n = 6 for others) ± 4-OI.
(F and G) Immunoblot analysis (F) and quantification by densitometry (G, n = 4) of pro- and mature caspase-1 and IL-1β protein in lysates and supernatants of BMDMs treated with LPS (3 h) and nigericin (45 min) ± 4-OI.
(H) LPS (3 h) and nigericin (45 min) induced IL-18 release ± 4-OI (n = 5).
(I) Percent of all cells positive for ASC specks in LPS- (3 h) and nigericin- (45 min) treated BMDMs ± 4-OI (n = 6).
(J) Immunoblot analysis of ASC protein in triton-insoluble pellet and -soluble lysate of LPS- (3 h) and nigericin- (45 min) treated BMDMs ± 4-OI (250 μM).
∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Data are mean ± SEM. Blots are representative of a minimum of 3 independent experiments.
It was important to ensure that any effect was specific to NLRP3 activation rather than priming. We found that 4-OI did not affect tumor necrosis factor α (TNFα) release (Figure S2A) and only inhibited pro-IL-1β expression when added before LPS priming and not after LPS priming (Figure S2B, compare lanes 3–5 to lane 2 and lanes 7–9 to lane 6), in accordance with previous findings (Mills et al., 2018). In order to elucidate whether the effect was specific to the NLRP3 inflammasome, we also tested 4-OI in AIM2, NLRC4, and non-canonical inflammasome assays. 4-OI, when added between LPS and poly(dA:dT) transfection, did not reduce IL-1β release or pyroptosis associated with AIM2 inflammasome activation (Figures 2A and 2B). Furthermore, it had no effect on IL-1β or caspase-1 cleavage (Figures 2C and 2D, compare lanes 5 and 6 to lane 4). We made similar observations when 4-OI was added to cells between LPS and transfection of purified flagellin to activate the NLRC4 inflammasome. IL-1β release, pyroptosis, and cleavage of IL-1β and caspase-1 were unaffected by addition of 4-OI (Figures 2E–2H). The non-canonical inflammasome differs from canonical inflammasome signaling in that it senses cytosolic LPS using the effector protein caspase-11 (caspase-4 and -5 in humans) (Kayagaki et al., 2011; Shi et al., 2014). Although pyroptosis in this model is caspase-11 dependent, IL-1β release remains dependent on canonical NLRP3 activation, as this occurs secondary to K+ efflux resulting from pyroptosis (Rathinam et al., 2019). We added 4-OI in between LPS priming and LPS transfection in a non-canonical inflammasome assay and found that it blocked IL-1β release, but not pyroptosis, in this assay, further indicating the specific targeting of NLRP3 (Figures 2I and 2J).
Figure 2.

4-OI Does Not Block Activation of Other Inflammasomes
(A and B) LPS (3 h) and poly(dA:dT) (6 h) induced IL-1β release (A, n = 5) and LDH release (B, n = 5) ± 4-OI.
(C and D) Immunoblot analysis (C) and quantification by densitometry (D, n = 3) of pro- and mature caspase-1 and IL-1β protein in lysates and supernatants of BMDMs treated with LPS (3 h) and poly(dA:dT) (6 h) ± 4-OI.
(E and F) LPS (3 h) and flagellin (2.5 h) induced IL-1β release (E, n = 8) and LDH release (F, n = 5) ± 4-OI.
(G and H) Immunoblot analysis (G) and quantification by densitometry (H, n = 3) of pro- and mature caspase-1 and IL-1β protein in lysates and supernatants of BMDMs treated with LPS (3 h) and flagellin (2.5 h) ± 4-OI.
(I and J) LPS priming (3 h) and transfection (18 h) induced IL-1β release (I, n = 5) and LDH release (J, n = 5) ± 4-OI.
∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Data are mean ± SEM. Blots are representative of a minimum of 3 independent experiments.
Endogenous Itaconate Regulates NLRP3 Inflammasome Activation
Although 4-OI treatment boosts levels of unmodified itaconate in activated macrophages (Mills et al., 2018), it is unclear whether LPS stimulation is required for this increase and whether the intracellular itaconate arises from de-esterification of 4-OI or from increased production of endogenous itaconate (Swain et al., 2020). We therefore examined whether 4-OI could directly generate itaconate. As shown in Figure S2C, 13C5-labeled itaconate was detected in human macrophages treated with 13C5 octyl itaconate both in the presence and absence of LPS. 13C5 itaconate was detectable as soon as 30 min after treatment with 13C5 octyl itaconate. This confirmed that 4-OI can indeed generate itaconate directly.
It is also unclear as to whether a transporter or cell-surface receptor exists for exogenous itaconate, hence the widespread use of itaconate derivatives in the field of itaconate biology. However, isotope tracing studies have shown that itaconate does accumulate in macrophages exposed to high concentrations of itaconic acid in culture media (Puchalska et al., 2018; Swain et al., 2020). Pre-treatment of BMDMs with itaconic acid (pH 7) prior to LPS and nigericin stimulation resulted in a significant and concentration-dependent reduction in IL-1β release (Figure S2E) and a non-significant reduction in pyroptosis (Figure S2F). Thus, unmodified itaconate, as well as derivatized itaconate, is capable of regulating NLRP3 activation.
The enzyme IRG1 is responsible for the production of itaconate in macrophages, which reaches a concentration of 5 mM in LPS-stimulated BMDMs (Mills et al., 2018). It is therefore possible to deplete or induce the production of endogenous itaconate through manipulation of IRG1. We performed NLRP3 inflammasome assays in Irg1 −/− BMDMs and found that IL-1β release was significantly increased relative to wild-type (WT) BMDMs (Figure 3 A). There was also a non-significant increase in pyroptosis in Irg1 −/− BMDMs (Figure 3B). IL-1β and caspase-1 processing into their mature forms, as detected by western blot, was increased in Irg1 −/− BMDMs (Figures 3C and 3D, compare lane 8 to lane 4). These effects were specific for NLRP3 inflammasome activation, as IL-1β release resulting from AIM2 inflammasome activation was not increased in Irg1 −/− BMDMs (Figure S2G). As NLRP3 expression is LPS-inducible as part of priming, we verified that increased NLRP3 inflammasome activation in Irg1 −/− BMDMs was not due to increased NLRP3 expression (Figure S2H, compare lane 4 to lane 2).
Figure 3.

Endogenous Itaconate Regulates NLRP3 Activation and 4-OI Dicarboxypropylates C548 on NLRP3
(A and B) LPS (3 h) and nigericin (45 min) induced IL-1β release (A, n = 6) and LDH release (B) in wild-type (WT, n = 7) and Irg1−/− (n = 8) BMDMs.
(C and D) Immunoblot analysis (C) and quantification by densitometry (D, n = 4) of pro- and mature caspase-1 and IL-1β protein in lysates and supernatants of wild-type (WT) and Irg1−/− BMDMs treated with LPS (3 h) and nigericin (45 min).
(E) IL-1β release from NLRP3 inflammasome-reconstituted HEK293T cells treated with nigericin (45 min) (n = 3). Immunoblot analysis of pro- and mature IL-1β and IRG1 protein in supernatants and lysates of these cells.
(F) Endogenous co-immunoprecipitation of NLRP3 and NEK7 in BMDMs treated with LPS (3 h) and nigericin (45 min) ± 4-OI (250 μM).
(G) Tandem mass spectrometry spectrum of C548-containing NLRP3 peptide following 4-OI treatment (250 μM, 24 h).
∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Data are mean ± SEM. Blots are representative of a minimum of 3 independent experiments.
It is possible to reconstitute the murine NLRP3 inflammasome in HEK293T cells, as previously described (Shi et al., 2016a). Transfection of the various NLRP3 inflammasome components followed by stimulation with nigericin results in IL-1β cleavage and release from the cell (Figure 3E). We found that overexpression of a plasmid encoding IRG1 in this system resulted in reduced IL-1β cleavage and release relative to transfection of an empty vector plasmid (Figure 3E, compare lane 4 to lane 3). Thus, while the absence of endogenous itaconate boosts NLRP3 inflammasome activation, increased IRG1 expression has the opposite effect, indicating that endogenous itaconate regulates NLRP3.
4-OI Blocks the NLRP3-NEK7 Interaction and Dicarboxypropylates C548 on NLRP3
Itaconate alkylates C151 on KEAP1, in a modification termed 2,3-dicarboxypropylation (Mills et al., 2018), leading to activation of NRF2, a protein that is thought to regulate NLRP3 inflammasome activation (Garstkiewicz et al., 2017; Zhao et al., 2014). However, we found that 4-OI still inhibited NLRP3-dependent caspase-1 and IL-1β processing, as well as IL-1β release in Nfe2l2 −/− (gene encoding NRF2) BMDMs (Figures S3A–S3D), ruling out NRF2 in the inhibitory process. Itaconate also limits inflammation through inhibition of succinate dehydrogenase (SDH) (Cordes et al., 2016; Lampropoulou et al., 2016). The SDH inhibitor dimethyl malonate (DMM), however, did not inhibit IL-1β release in an NLRP3 inflammasome assay (Figure S3E), indicating that SDH inhibition was unlikely to be important here. We also explored whether 4-OI may be interacting with the deglutathionylating enzyme glutathione transferase omega 1 (GSTO1-1), recently described as a regulator of NLRP3 inflammasome activation (Hughes et al., 2019). However, 4-OI still inhibited IL-1β release in an NLRP3 inflammasome assay in Gsto1 −/− BMDMs (Figures S3F and S3G).
We subsequently hypothesized that the acute and specific effect of 4-OI on NLRP3 activation may be the result of targeting NLRP3 and/or NEK7 directly, but not ASC or caspase-1 as these components are shared by other inflammasomes. Upon NLRP3 inflammasome stimulation, NLRP3 and NEK7 interact with each other to promote activation (Sharif et al., 2019). This interaction was blocked by treatment with 4-OI (Figure 3F, compare lane 3 to lane 2). Subsequent tandem mass spectrometry of murine NLRP3, immunoprecipitated from 4-OI-treated NLRP3-overexpressing HEK293T cells, showed that 4-OI dicarboxypropylated (+242.15 Da) C548 on NLRP3 (Figure 3G). C548 is present in the helical domain 2 (HD2) of NLRP3, which is one of the surfaces at which NLRP3 interacts with NEK7 (Sharif et al., 2019). This points to a possible mechanism for inhibition of NLRP3, involving modification of NLRP3 on C548. This might interfere with the interaction between NLRP3 and NEK7, thereby blocking NLRP3 inflammasome activation.
4-OI Reduces NLRP3-Driven Peritonitis In Vivo and Blocks IL-1β Release from Human CAPS PBMCs
Monosodium urate (MSU) crystals, the causative agent of gout, are activators of the NLRP3 inflammasome and cause caspase-1- and ASC-dependent inflammation when injected into mice intraperitoneally (Martinon et al., 2006). Co-injection of 4-OI with MSU crystals reduced IL-1β and IL-6 (which is downstream of IL-1β) concentrations, as well as neutrophil numbers, in the peritoneal lavage fluid (Figures 4A–4C).
Figure 4.

4-OI Reduces Inflammation in a Murine In Vivo Model of Peritonitis and Blocks NLRP3 Inflammasome Activation in Healthy Human and CAPS PBMCs
(A–C) IL-1β concentration (A), IL-6 concentration (B), and neutrophil number (C) in the peritoneal lavage fluid of mice injected for 6 h with MSU crystals (30 mg/kg) ± 4-OI (50 mg/kg) (n = 3 for PBS groups, n = 8 for MSU groups).
(D) LPS or Pam3CSK4 (14 h) and nigericin (2 h) induced IL-1β release (n = 5 for LPS + nigericin, n = 3 for Pam3CSK4 + nigericin) ± 4-OI or 4-O-2-MS (both 250 μM) from healthy human PBMCs.
(E and F) Immunoblot analysis (E) and quantification by densitometry (F, n = 3) of pro- and mature IL-1β protein in lysates and supernatants of human PBMCs treated with LPS (14 h) and nigericin (2 h) ± 4-OI (250 μM).
(G) LPS (1 h) induced IL-1β release (n = 3) ± 4-OI (250 μM) or MCC950 (500 nM) from PBMCs isolated from CAPS patients.
∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Data are mean ± SEM. Blots are representative of a minimum of 3 independent experiments.
Finally, we tested 4-OI on peripheral blood mononuclear cells (PBMCs) from CAPS patients. We first confirmed that 4-OI would block NLRP3 activation in human PBMCs isolated from healthy donors. 4-OI, but not 4-O-2-MS, blocked IL-1β release when added between Pam3CSK4 or LPS and nigericin in human PBMCs (Figure 4D). 4-OI also blocked IL-1β cleavage into its mature form (Figures 4E and 4F, compare lane 5 to lane 4). The efficacy of 4-OI was similar to that of MCC950 and glyburide, albeit at a higher concentration (Figure S4A). PBMCs can also engage an “alternative” inflammasome pathway, which involves caspase-8 and NLRP3, and can be activated by LPS alone (Gaidt et al., 2016). 4-OI blocked IL-1β release from human PBMCs in this assay (Figure S4B). We then tested PBMCs isolated from the whole blood of CAPS patients who have hyperactive NLRP3, which can be stimulated with LPS to release large amounts of IL-1β. We treated CAPS PBMCs with 4-OI after 1 h stimulation with LPS and found that both 4-OI and MCC950 blocked IL-1β release from these cells (Figure 4G).
Discussion
It is now generally accepted that NLRP3 inflammasome signaling plays a critical role in the pathogenesis of several autoimmune disorders, including Alzheimer disease (Heneka et al., 2013), rheumatoid arthritis (Vande Walle et al., 2014), and type 2 diabetes (Masters et al., 2010; Vandanmagsar et al., 2011). This has heightened the need for a greater understanding of how inflammasome activation is regulated endogenously and how it may be inhibited.
We hereby provide evidence of itaconate being a specific endogenous inhibitor of NLRP3 inflammasome activation. Previous studies have pointed toward a role for itaconate in regulating IL-1β cleavage (Lampropoulou et al., 2016; Swain et al., 2020), but by pre-treating cells with itaconate prior to LPS stimulation they were unable to rule out an effect on signal 1. Nor did these studies demonstrate itaconate’s specificity for NLRP3, which we have shown through our AIM2 and NLRC4 experiments.
The mechanism that we propose for this inhibition is itaconate-mediated dicarboxypropylation of C548. This particular modification was also detected by Qin et al. using an itaconate-alkyne (iTALK) probe in Raw264.7 macrophages (Qin et al., 2020). It is possible that modification of NLRP3 at this surface would abolish its ability to interact with NEK7, a process that is necessary for inflammasome activation to take place (Sharif et al., 2019). However, further studies are required to establish (1) whether endogenous itaconate, as well as 4-OI, can cause the same modification—the study by Qin et al. indicates that this might be the case (Qin et al., 2020); (2) whether modification at this surface is functionally relevant with regard to inflammasome activation; and (3) whether there may be other targets for dicarboxypropylation along this pathway. Qin et al. also detected “itaconation sites” on gasdermin D, the pyroptosis executioner (Qin et al., 2020), further emphasizing the inhibitory effect of itaconate on pyroptosis. In addition, the recent observation that NEK7 deglutathionylation of C253 promotes NLRP3 inflammasome activation (Hughes et al., 2019) demonstrates that post-transcriptional modifications of NEK7 are also important in regulating inflammasome function.
Our observation that unmodified itaconic acid can limit NLRP3 activation is important when comparing the compounds widely used to deliver itaconate intracellularly. There has been a focus on how unmodified and derivatized itaconate diverge in their ability to accumulate inside the cell and exert immunomodulatory functions (Swain et al., 2020). However, we demonstrated that 13C5-labeled octyl itaconate was converted into 13C5 itaconate intracellularly and also found consistency in the way 4-OI and itaconic acid inhibited NLRP3 inflammasome activation. In contrast to the view of Swain et al. (2020), we feel that 4-OI can be used as a tool compound in the study of itaconate, since we have shown that it can be taken up and converted to intracellular itaconate by macrophages. We have previously also demonstrated that there is significant overlap in the cysteine residues alkylated by 4-OI and endogenous itaconate (Mills et al., 2018). In spite of this, it must be acknowledged that the relative electrophilicity of 4-OI/DMI, and therefore its ability to impact certain pathways (such as the ATF3-IKBζ axis), is higher than that of unmodified itaconate (Swain et al., 2020). As such, any results obtained with these derivatives must be verified in Irg1 −/− experiments in order to be considered attributable to itaconate.
Our studies performed in Irg1 −/− macrophages have shown that an absence of endogenous itaconate results in dysregulated IL-1β release and increased pyroptosis, establishing the importance of the itaconate-NLRP3 axis in restraining macrophage inflammation. Evidence suggests that, at least in the context of NLRP3 inflammasome activation, the presence of itaconate would limit pathogenic inflammation and resulting damage to the host. Recent studies showed that plasma itaconate levels correlated with improved disease score and reduced severity in patients with rheumatoid arthritis (Daly et al., 2020) and COVID-19 (Song et al., 2020), respectively. These studies add further weight to the concept that itaconate is a critical determinant of innate immune responses, with profound anti-inflammatory effects.
In conclusion, our work provides evidence for the regulation of NLRP3 by itaconate. Furthermore, in 4-OI we present a compound that may be developed further for the treatment of NLRP3-driven disorders, as evidenced by its effect on IL-1β release from CAPS PBMCs. The targeting of NLRP3 by itaconate therefore holds tremendous therapeutic potential and expands the role of itaconate as a key immunometabolite that regulates innate immunity and inflammation.
Limitations of Study
Our study has demonstrated that 4-OI modifies C548 of NLRP3, a mechanism that is likely to be responsible for itaconate-mediated inhibition of inflammasome activation. However, further studies are required to demonstrate the functional effects of this modification, perhaps through site-directed mutagenesis of C548. Furthermore, the identification by Qin et al. of two additional cysteines undergoing modification, C284 and C720 (Qin et al., 2020), should also be functionally investigated. Qin et al. also uncovered modification of gasdermin D on C77 and C192 (Qin et al., 2020), which coupled with our results showing reduced gasdermin D cleavage with 4-OI, means that the effect of itaconate on gasdermin D should be examined. These studies should further emphasize the importance of the targeting of NLRP3-mediated pyroptosis as a key aspect of the anti-inflammatory effects of itaconate.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-β-actin | Sigma | Cat# A5316; RRID: AB_476743 |
| Anti-ASC | Santa Cruz | Cat# Sc-22514-r; RRID: AB_2174874 |
| Anti-Caspase-1 | Adipogen | Cat# AG-20B-0042-C100; RRID: AB_2755041 |
| Anti-CD11b eFluor 660 | Invitrogen | Cat# 50-0112-82; RRID: AB_11218507 |
| Anti-CD16/CD32 | Biolegend | Cat# 101302; RRID: AB_312801 |
| Anti-CD45 APC/Cy7 | Biolegend | Cat# 103116; RRID: AB_312981 |
| Anti-FLAG | Sigma | Cat# F1804; RRID: AB_262044 |
| Anti-GSDMD | Sigma | Cat# G7422; RRID: AB_1850381 |
| Anti-GSTO1-1 | Genetex | Cat# GTX118439; RRID: AB_11174551 |
| Anti-HA | Sigma | Cat# H3663; RRID: AB_262051 |
| Anti-IL-1β | R&D | Cat# AF-401-NA; RRID: AB_416684 |
| Anti-IL-1β (Human) | R&D | Cat# AF-201-NA; RRID: AB_354387 |
| Anti-IRG1 | Abcam | Cat# AB222411 |
| Anti-Ly6G | Biolegend | Cat# 127611; RRID: AB_1877212 |
| Anti-NEK7 | Abcam | Cat# AB133514 |
| Anti-NLRP3 | Cell Signaling | Cat# D4D8T; RRID: AB_2722591 |
| Anti-NRF2 | Cell Signaling | Cat# 12721S; RRID: AB_2715528 |
| Anti-Rabbit IgG Alexa Fluor 488 | Invitrogen | Cat# A-11008; RRID: AB_143165 |
| Rabbit IgG | Merck | Cat# PP64; RRID: AB_97852 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 4-Octyl Itaconate | Professor Richard Hartley, University of Glasgow | Cat# N/A |
| 4-Octyl Succinate | Professor Richard Hartley, University of Glasgow | Cat# N/A |
| 4-Octyl-2-Methyl Succinate | Professor Richard Hartley, University of Glasgow | Cat# N/A |
| A/G PLUS Agarose Beads | Santa Cruz | Cat# sc-2003 |
| ATP | Invivogen | Cat# tlrl-atpl |
| Coomassie Blue R-250 | Cayman | Cat# 14500 |
| Disuccinimidyl Suberate | ThermoFisher | Cat# 21655 |
| Elastase | Promega | Cat# V1891 |
| FuGENE HD Transfection Reagent | Promega | Cat# E2311 |
| Itaconic Acid | Sigma | Cat# I29204 |
| Lipofectamine 2000 | ThermoFisher | Cat# 11668030 |
| LPS from E.coli O127:B8 | Sigma | Cat# L5668 |
| Lymphoprep | StemCell Technologies, Inc. | Cat# 07861 |
| Monosodium Urate Crystals | Invivogen | Cat# tlrl-msu |
| Nigericin | Invivogen | Cat# tlrl-nig |
| Pam3SCK4 | Invivogen | Cat# tlrl-pms |
| Poly (dA:dT) | Invivogen | Cat# tlrl-patn |
| Purified Flagellin from P.aeruginosa | Invivogen | Cat# tlrl-pafla |
| Recombinant Human M-CSF | Peprotech | Cat# 300-25 |
| Strataclean Resin | Agilent Technologies | Cat# 400714 |
| Ultrapure rough LPS from E. coli, serotype EH100 | Enzo | Cat# ALX-581-010-L001 |
| Zombie Green Fixable Viability Kit | Biolegend | Cat# 423111 |
| Critical Commercial Assays | ||
| Cytotox 96 non-radioactive cytotoxicity assay | Promega | Cat# G1780 |
| Human IL-1β DuoSet ELISA | R&D Systems | Cat# DY201 |
| Mouse IL-1β DuoSet ELISA | R&D Systems | Cat# DY401 |
| Mouse IL-1β Quantikine ELISA | R&D Systems | Cat# MLB00C |
| Mouse IL-18 ELISA | Invitrogen | Cat# BMS618-3 |
| Mouse IL-6 DuoSet ELISA | R&D Systems | Cat# DY406 |
| Mouse TNFα DuoSet ELISA | R&D Systems | Cat# DY410 |
| Experimental Models: Cell Lines | ||
| Human: HEK293T cells | Sigma | Cat# 12022001 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | Harlan | N/A |
| Mouse: C57BL/6N-Acod1em1(IMPC)J/J | Jackson | Cat# 029340 |
| Mouse: GSTO1-1−/− 129/SvEv-C57BL/6 | Taconic | Cat# TF1210 |
| Recombinant DNA | ||
| FLAG-NLRP3 | Shi et al., 2016b | Addgene; Cat# 75127 |
| FLAG-Pro-Caspase-1 | Shi et al., 2016b | Addgene; Cat# 75128 |
| FLAG-pro-IL-1β | Shi et al., 2016b | Addgene; Cat# 75131 |
| GFP-IRG1 | Origene | Cat# MG217265 |
| HA-ASC | Hornung et al., 2009 | Addgene; Cat# 41553 |
| HA-NEK7 | Shi et al., 2016b | Addgene; Cat# 75142 |
| Software and Algorithms | ||
| FlowJo v 10.7 | FlowJo | https://www.flowjo.com/solutions/flowjo |
| Graphpad Prism 8.0 | Graphpad | https://www.graphpad.com/ |
| Image Lab 6.1 | Bio-Rad | https://www.bio-rad.com |
| PEAKS Studio 8 | Bioinformatics Solutions | https://www.bioinfor.com/peaks-studio/ |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Luke A.J. O’Neill (laoneill@tcd.ie).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate/analyze datasets/code.
Experimental Model and Subject Details
Animal Details
All mice were on a C57BL/6J background unless stated below. Wild-type mice were purchased from Harlan. Irg1 −/− mice (named C57BL/6N-Acod1em1(IMPC)J/J) were generated by CRISPR-targeted deletion of exon 4 of Acod1, and were purchased from The Jackson Laboratory. Wild-type littermates were used as controls. Gsto1 −/− 129/SvEv-C57BL/6J mice were obtained from Taconic and were originally derived from 129S-5 ES cells and backcrossed to an albino C57BL/6J strain. Bones from Nfe2l2 −/− mice and their wild-type counterparts were kindly provided by Professor Albena Dinkova-Kostova (University of Dundee). Bones from Nlrp3 −/− mice and their wild-type counterparts were kindly provided by Professor Ed Lavelle (Trinity College Dublin). In vivo models were performed with 6-week old female C57BL/6J mice and littermates were randomly assigned to experimental groups. Animals were maintained under specific pathogen-free conditions in line with Irish and European Union regulations. All animal procedures were ethically approved by the Trinity College Dublin Animal Research Ethics Committee prior to experimentation, and conformed with the Directive 2010/63/EU of the European Parliament.
Generation of Murine BMDMs
6-12-week old mice were euthanised in a CO2 chamber, and death was confirmed by cervical dislocation. Bone marrow was subsequently harvested from the tibia and fibula and cells were differentiated in DMEM containing L929 supernatant (10%), fetal calf serum (FCS) (10%), and penicillin/streptomycin (1%) for 6 days, after which cells were counted and plated at 0.5 × 106 cells/mL unless otherwise stated.
CAPS Patient Recruitment
Recruitment was carried out by members of the patient’s own clinical team. Patients were approached to provide a voluntary blood sample for the study. Patients were provided with an information leaflet 24 h in advance of sample donation. All patients or their parents/legal guardians gave informed written consent. Direct travel costs were covered but no incentive or compensation was offered. Patient demographics were not recorded. Patients were receiving standard anti-IL-1 therapy (Anakinra). This treatment was not discontinued prior to blood sampling. The study was conducted in accordance with the Declaration of Helsinki and approved by the Joint Research Ethics Committee of St James Hospital and Tallaght Hospital (REF 2017-11) and the Ethics (Medical Research) Committee of Our Lady’s Children’s Hospital, Crumlin (Now Children’s Health Ireland), REF: GEN/577/17.
Isolation of Human PBMCs
Human blood samples from healthy donors were collected and processed at the School of Biochemistry and Immunology in TBSI (TCD). Blood samples were obtained anonymously and written informed consent for the use of blood for research purposes has been obtained from the donors. All the procedures involving experiments on human samples have been approved by the School of Biochemistry and Immunology Research Ethics Committee (TCD). Experiments were conducted according to the TCD guide on good research practice, which follows the guidelines detailed in the National Institutes of Health Belmont Report (1978) and the Declaration of Helsinki.
30 mL whole blood was layered on 20 mL Lymphoprep (Axis-Shield), followed by centrifugation for 20 min at 400 x g with the brake off, after which the upper plasma layer was removed and discarded. The layer of mononuclear cells at the plasma-density gradient medium interface was retained, and 20 mL PBS was added. Cells were centrifuged for 8 min at 300 x g and the resulting supernatant was removed and discarded. The remaining pellet of mononuclear cells was resuspended, counted and plated at 1 × 106 cells/mL in RPMI supplemented with FCS (10%) and penicillin-streptomycin (1%).
Human Monocyte Differentiation
Frozen human monocytes (Biological Specialty Labs) were thawed and seeded in 10 cm dishes at 1 × 106 cells/mL in macrophage media (RPMI media supplemented with 10% fetal calf serum and penicillin/streptomycin) and 0.1 μg/mL recombinant human M-CSF (Peprotech) and incubated at 37°C and 5% CO2. Cells were cultured for 6 days total to differentiate the monocytes to macrophages, with removal and replacement of media and M-CSF at three days of culture.
Culture of HEK293T Cells
HEK293T cells were obtained from the Centre for Applied Microbiology and Research (Wiltshire, UK) and cultured in DMEM containing FCS (10%) and penicillin-streptomycin (1%).
Method Details
Canonical Inflammasome Assays
BMDMs were plated in 12-well cell culture plates and left overnight to adhere. Cells were treated the following day with LPS from Escherichia coli, serotype EH100 (Enzo Life Sciences, 100 ng/mL) for 3 h. Medium was removed and replaced with serum- and antibiotic-free medium and treated with compounds as required for 45 min. 4-OI, 4-O-2-MS and 4-OS were kindly supplied by Professor Richard Hartley and dissolved in DMSO. Itaconic acid (Sigma Aldrich) was dissolved at 500 mM in PBS and the required volume of NaOH was added in order to achieve a pH of 7. Cells were treated with nigericin (10 μM, Invivogen) or ATP (5 mM, Sigma Aldrich) for 45 min to activate the NLRP3 inflammasome. In order to activate the AIM2 inflammasome, LPS-primed cells were transfected with 1.5 μg poly(dA:dT) (Invivogen) using lipofectamine 2000 (Thermo Fisher Scientific) for 6 h. In order to activate the NLRC4 inflammasome, LPS-primed cells were transfected with 1.6 μg purified flagellin from Pseudomonas aeruginosa (Invivogen) for 2.5 h.
To activate the NLRP3 inflammasome in healthy human PBMCs, cells were treated for 14 h with LPS (200 ng/mL) or Pam3CSK4 (2 μg/mL, Invivogen). Medium was removed and replaced with serum- and antibiotic-free medium and treated with compounds as required for 45 min. Cells were subsequently treated with nigericin (6.5 μM) for 2 h. For alternative inflammasome activation in healthy human PBMCs, cells were pre-treated with compounds for 1 h, after which they were treated with LPS (200 ng/mL) for 14 h, as described previously (Gaidt et al., 2016). Human CAPS PBMCs were treated for 1 h with LPS (100 ng/mL), after which medium was removed and replaced with serum- and antibiotic-free medium and treated with compounds as required for 4 h.
Non-Canonical Inflammasome Assay
Cells were treated with LPS (100 ng/mL) for 3 h, after which the medium was replaced and cells were treated with compounds as required for 45 min. 2 μg LPS was transfected using FuGENE HD (Promega) overnight (18 h) in order to activate the inflammasome.
ASC Speck Assay
Flow cytometry was used to analyze ASC speck formation following inflammasome activation. BMDMs were treated as desired, after which the supernatant was removed and 1 mL of cold PBS was added to the wells. Cells were then detached from the plates using a cell scraper and transferred to round-bottom tubes. 4 mL ethanol (100%) was added to the cells while simultaneously vortexing the tubes, in order to fix the cells and prevent clumping. Cells were incubated at room temperature for 15 min, before being centrifuged at 600 x g for 10 min. Supernatant was removed, and cells were resuspended in FACS buffer (0.1% Sodium azide, 0.1% BSA, 1.5% FCS in PBS) containing FC block (0.4%). Cells were incubated for 20 min at room temperature after which anti-mouse ASC antibody (Santa Cruz, sc-22514-R) was added to a final dilution of 1/1500. Cells were incubated for 90 min at room temperature. 1 mL of FACS buffer was added to the cells and they were centrifuged at 600 x g for 10 min. Supernatant was removed, and the cells were resuspended in Alexa Fluor 488 anti-rabbit IgG antibody (Invitrogen, A27304 1/1500 in FACS buffer), before being incubated in the dark for 45 min at room temperature. Cells were pelleted and resuspended in 200 μL FACS buffer. Samples were acquired on a FACSCanto (BD Biosciences). Samples were gated based on forward light scatter (FSC) versus side scatter (SSC) in order to exclude cell debris. Doublets were also excluded from analysis using FSC-area versus FSC-width analysis. Sorting was conducted by analyzing the pulse height, width and area of the 488nm laser and 530/30nm laser channel. ASC speck positive cells were detected by a decreased width or increased height in the pulse of emitted fluorescence when compared with unstimulated cells.
ASC Oligomerisation
BMDMs were treated as desired. After treatment, cells were washed twice with 200 μL cold PBS before being lysed in crosslinking lysis buffer (50 mM HEPES, 0.5% Triton X-100, 1 mM phenylmethylsulfonyl fluoride (PMSF), 11.5 μg/mL aprotinin, 1 μg/mL leupeptin and 1 mM sodium orthovanadate). Samples were placed on ice for 15 min and benzonase nuclease was added in order to break down DNA in the lysates. Lysates were centrifuged for 15 min at 6000 x g at 4°C and the supernatant was removed and frozen down as the ‘soluble fraction.’ 20 μL of the soluble fraction was mixed with 5 μL of sample lysis buffer (0.125M Tris pH 6.8, 10% glycerol, 0.02% SDS, 5% DTT) and run on a 12% gel. The insoluble pellet was resuspended in HEPES (50 mM) and washed 3 times by centrifuging at 6000 x g at 4°C and removing the supernatant each time. After the final wash, the pellet was resuspended in 500 μL crosslinking buffer (50 mM HEPES, 150 mM NaCl) and disuccinimidyl suberate (DSS, Thermo Fisher, made up in anhydrous DMSO) was added to the final concentration of 2 mM. Immediately following the addition of DSS, the sample was inverted several times and incubated for 45 min at 37°C. The sample was then centrifuged for 15 min at 6000 x g at 4°C, before the supernatant was removed and the pellet was resuspended in 30 μL sample lysis buffer. The resuspended ‘insoluble fraction’ was subsequently boiled for 5 min at 95°C before being run on a gel.
HEK293T Inflammasome Reconstitution
HEK293T cells were plated at a density of 2 × 105 cells/mL in 24-well plates in DMEM containing FCS (10%). The following morning, cells were transfected with plasmids encoding murine GFP-IRG1 (280 ng, Origene, MG217265) and murine NLRP3 inflammasome components: HA-ASC (20 ng, Addgene, 41553), HA-NEK7 (200 ng, Addgene, 75142), FLAG-pro-caspase 1 (100 ng, Addgene, 75128), FLAG-NLRP3 (200 ng, Addgene, 75127), FLAG-Pro-IL-1β (200 ng, Addgene, 75131). Plasmids were transfected using lipofectamine 2000 for 24 h. Following the 24 h transfection, medium was replaced with DMEM containing FCS (10%) and penicillin-streptomycin (1%). 4 h later, nigericin (10 μM) was added to cells in order to activate the inflammasome. Supernatants and cell lysates were harvested 45 min later. This protocol was described previously (Shi et al., 2016a).
Western Blotting
Supernatant was removed from cells following stimulation and lysates were harvested in 30-50 μL lysis buffer (0.125 M Tris pH 6.8, 10% glycerol, 0.02% SDS, 5% DTT) Lysates were subsequently heated to 95°C for 5 min to denature proteins. In order to concentrate supernatants for western blot, 5 μL Strataclean Resin (Agilent) was added to 500 μL of supernatant and vortexed for 1 min. Supernatants were then centrifuged at 210 x g for 2 min at 4°C. Supernatants were removed and discarded, and the remaining pellet was resuspended in 30 μL lysis buffer. SDS-PAGE was used to resolve proteins by molecular weight. Samples were boiled at 95°C for 5 min prior to loading into a 5% stacking gel. The percentage resolving gel depended on the molecular weight of the given protein. The Bio-Rad gel running system was used to resolve proteins and the Bio-Rad wet transfer system was used for the electrophoretic transfer of proteins onto PVDF membrane. Following transfer, the membrane was incubated in milk powder (5% in TBST) for 1 h and subsequently incubated in primary antibody rolling overnight at 4°C. The membrane was incubated for 1 h with secondary antibody (diluted in 5% milk powder) at room temperature. Prior to visualization, the membrane was immersed in WesternBright ECL Spray (Advansta). Protein visualization took place on a ChemiDoc MPTM Imaging System (Bio-Rad), and both chemiluminescent and white light images were taken. Quantification of western blot images was performed using Image Lab Software (Bio-Rad). Adjusted band volume was calculated for each band and for each experimental condition this was presented as target protein/housekeeping protein.
Co-Immunoprecipitation
BMDMs were seeded at 1x106 cells/mL in 10 cm dishes the day prior to treatment. The following day, cells were treated as required. Following treatment, cells were washed with 3 mL PBS before lysis in 500 μL low stringency lysis buffer (50 mM HEPES pH 7.5, 100 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% Nonident P40 (NP-40), 1 mM phenylmethylsulfonyl fluoride (PMSF), 11.5 μg/mL aprotinin, 1 μg/mL leupeptin and 1 mM sodium orthovanadate) on ice. Plates were scraped with a cell scraper and lysate transferred into microcentrifuge tubes. Tubes were agitated at 1100 rpm at 4°C for 15 min before being centrifuged at 21380 x g for 10 min at 4°C. 25 μL of resulting supernatant was mixed with 25 μL sample lysis buffer and treated as the whole cell lysate (WCL) sample. 0.0153 mg/mL anti-NEK7 antibody (Abcam, ab133514) or the equivalent concentration of rabbit IgG antibody (Millipore, pp64), and 40 μL A/G PLUS agarose beads (Santa Cruz) was added to the remaining supernatant, which was subsequently incubated at 4°C in a ferris wheel mixer for 3 h. IP samples were subsequently centrifuged at 400 x g for 2 min at 4°C, supernatant removed, and beads washed three times with 1 mL low stringency lysis buffer. The immune complexes were eluted by addition of 40 μL sample lysis buffer, boiled for 5 min and analyzed by SDS-PAGE.
ELISA
DuoSet ELISA kits for IL-1β, TNFα and IL-6 were purchased from R&D Systems and were carried out according to the manufacturer’s instructions with appropriately diluted cell supernatants added to each plate in duplicate or triplicate. Quantikine ELISA kits for IL-1β (R&D Systems) and IL-18 (Invitrogen) were similarly carried out according to the manufacturer’s instructions. Absorbance at 450 nm was then quantified using a FLUOstar Optima plate reader. Corrected absorbance values were calculated by subtracting the background absorbance, and cytokine concentrations were subsequently obtained by extrapolation from a standard curve plotted on GraphPad Prism 8.0.
LDH Assay
The CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega) was used to quantify lactate dehydrogenase (LDH) release from cells as a measure of cell death in BMDMs following inflammasome stimulation. Freshly harvested supernatants were used in this assay. 50 μL of each supernatant was added to 50 μL Cytotox 96 Reagent and incubated in the dark at room temperature for 30 min. 50 μL acetic acid was added to stop the reaction, and the absorbance at 492 nm was measured using a FLUOstar optima. 100 μL total lysis solution was added to untreated cells 30 min before harvesting and served as a maximum LDH release control. Medium alone was also used to correct for background absorbance.
Generation of 13C5 Octyl Itaconate
All NMR spectra were collected using a Varian 400 MHz NMR. Synthesis was performed by WuXi AppTec. Synthesis of 13C5 octyl itaconate was performed as previously described (Richard et al., 2016) with minor modifications. Uniformly labeled 13C6-citric acid (0.32 mmol) was placed into a distillation apparatus and subjected to pyrolysis by heating to 175°C for 30 min with constant stirring. The reaction was then heated up to 200°C at a pressure of 2 Torr. Heating was maintained for 2 h. The cyclised product was collected as a white solid in the distillation bulb at a temperature of −20°C (3.9% yield). Equimolar quantities of the product and octanol (20 mM final concentration) were mixed in toluene, heated to 100°C and stirred for 12 h. The mixture was concentrated and subjected to preparative HPLC using a Shimadzu LC-8A preparative HPLC with a Waters Xbridge column (150 mm x 25 mm, 5 μM). Mobile phase consisted of 10 mM ammonium bicarbonate in water with a gradient of acetonitrile from 15% to 45% over 20 min with a flow rate of 30 mL/min. Purified yield of 13C5-octylitaconate was 20%. Synthesis of 13C5-octylitaconate was confirmed by 1H NMR and LC/MS (ESI+).
Treatment of Human Macrophages with 13C5 Octyl Itaconate
At the end of the macrophage differentiation, medium was removed and replaced with macrophage medium with or without 100 ng/mL of LPS, from Escherichia coli serotype O127:B8 (Sigma) and 125 μM 13C5 octyl itaconate dissolved in DMSO and incubated for various times at 37°C and 5% CO2. In cultures with 13C5 octyl itaconate alone, supernatants and cells were harvested 0.5, 3, 6, 9 and 27 h after addition of 13C5 octyl itaconate. Cultures with LPS were treated with 13C5 octyl itaconate three h prior or three h following LPS addition. Supernatants and cells were harvested 3, 6 and 24 h post LPS if added after 13C5 octyl itaconate and 6, 9 and 24 h post LPS if added before 13C5 octyl itaconate. Control cultures without 13C5 octyl itaconate included cells alone and 24 h treatment with LPS. Supernatants were transferred to Eppendorf tubes and stored at −80°C until analysis. Cells were washed with D-PBS (GIBCO), trypsinized (GIBCO), scraped with a cell scraper and transferred to conical tubes. Cells were washed three times with 40 mM ammonium formate (Sigma) and transferred to Eppendorf tubes prior to snap freezing on dry ice and stored at −80°C until analysis.
Measurement of 13C5 Itaconate and 13C5 Octyl Itaconate in Human Macrophages by LC-HRMS
Cell pellet samples were thawed on wet ice immediately prior to analysis. 1 mL of ice cold 8:2 methanol-water (Sigma; Milli-Q water) was added to each sample. The tubes were briefly vortexed, sonicated (10 min/ice bath), and briefly vortexed again to lyse cells and extract analytes. Each sample was centrifuged at 6°C for 15 min at 15,000 x g. 450 μL of supernatant was transferred to a 1.5 mL glass autosampler vial, dried down, and derivatized with 3-nitrophenylhydrazine (3-NPH) (Sigma) using a modification of method previously reported method (Han et al., 2013). 100 μL of a 250 mM 3-NPH solution (1:1 acetonitrile-water) and 200 μL of a 143.5 mM EDC.HCl solution (1:1 water:acetonitrile containing 6%(v/v) pyridine) were added to each vial. The samples were briefly vortexed and left at room temperature for 2 h. After adding 500 μL chloroform (Sigma) and 100μL of cold 3N HCl (VWR) each vial was vortexed for about 5 s, and the layers were allowed to separate. 450 μL of the lower organic layer was transferred to a Microsolv Max Recovery 1.2 mL glass vial and dried down. The residue was reconstituted in 100 μL of methanol-water (1:1) for analysis by LC-HRMS.
The LC-HRMS system consisted of an Agilent 1290 HPLC system coupled to an Agilent 6550 Q-TOF mass spectrometer equipped with a Jet Stream ESI source. The chromatographic analysis was performed with a Waters Acquity BEH C18 column (100 × 2.1 mm, 1.7 μm) maintained at 35°C and an elution gradient composed of water (A) and acetonitrile (B). The gradient elution profile was 0.5% B for 2 min, 90% B at 20 min, 90% B to 28 min with column re-equilibration at 0.5% B from 28.05 to 33 min. The flow rate was 0.35 mL/min and the injection volume was 3 μL. The mass spectrometer was operated in negative ion mode with the following source settings: Gas Temp: 290°C, Drying Gas: 12 L/min, Nebulizer: 55 psi, Sheath Gas Temp: 400°C, Sheath Gas Flow: 12, Vcap: 3000V, Nozzle: 1000V. Acquisition rate: 1.5 spectra/s.
Analysis of NLRP3 Modification by 4-OI
HEK293T cells were plated at a density of 3 × 105 cells/mL in 10 cm dishes in DMEM containing FCS (10%). The following morning, cells were transfected with 5 μg of plasmid encoding murine FLAG-NLRP3 (Addgene, 75127) using lipofectamine 2000. 24 h after transfection, cells were treated with 4-OI (250 μM) or vehicle control (DMSO) for a further 24 h. Following treatment, cells were washed with 3 mL PBS before lysis in 500 μL low stringency lysis buffer (50 mM HEPES pH 7.5, 100 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% Nonident P40 (NP-40), 1 mM phenylmethylsulfonyl fluoride (PMSF), 11.5 μg/mL aprotinin, 1 μg/mL leupeptin and 1 mM sodium orthovanadate) on ice. Plates were scraped with a cell scraper and lysate transferred into microcentrifuge tubes. Tubes were agitated at 1100 rpm at 4°C for 15 min before being centrifuged at 21380 x g for 10 min at 4°C. FLAG-tagged NLRP3 was immunoprecipitated using an anti-FLAG antibody (1 mg/mL, Sigma, F1804) and 70 μL A/G PLUS agarose beads (Santa Cruz), placed in a ferris wheel mixer at 4°C for 3 h. IP samples were subsequently centrifuged at 400 x g for 2 min at 4°C, supernatant removed, and beads washed three times with 1 mL low stringency lysis buffer. The immune complexes were eluted by addition of 40 μL sample lysis buffer, boiled for 5 min and run on a 8% gel. The resulting gel was subsequently fixed in Coomassie fixing solution (50% methanol, 10% acetic acid in ddH2O) for 30 min, followed by removal of the fixing solution and incubation in Coomassie blue R-250 buffer (0.125 g Coomassie Blue R-250 (Cayman Chemical), 5 mL acetic acid, 22.5 mL methanol, 22.5 mL ddH2O) for 1 h. Staining solution was then removed from the gel, which was washed repeatedly with de-staining solution (5% methanol, 7.5% acetic acid in ddH2O). The corresponding bands for FLAG-NLRP3 were excised from the gel and subjected to in-gel digest with elastase (Promega). In brief, the gel slices were cut into smaller pieces (1–2 mm3) before reduction with DTT (10 mM) and alkylation with iodoacetamide (50 mM). Gel slices were digested with elastase (1 μg) overnight at 37 °C. Following protease digest, peptides were eluted from the gel pieces and dried down completely in a vacuum centrifuge. Samples were analyzed in an Orbitrap Fusion Lumos coupled to a UPLC ultimate 3000 RSLCnano System (both Thermo Fisher). MS data was analyzed with PEAKS Studio 8 (Bioinformatics Solutions). Precursor mass tolerance was set to 10 ppm, while fragments were detected with a tolerance of 0.5 Da. Oxidation (M), Deamidation (N, Q), Carbamidomethylation (C), dicarboxypropylation of cysteine by 4-OI (C) (+242.15 Da) were defined as variable modifications. Peptide FDR was set to 1%.
MSU-Induced Peritonitis Model
6-week old female C57BL/6J mice were injected intraperitoneally with a mixture of 4-OI (50 mg/kg) in 60% cyclodextrin in PBS and MSU crystals (30mg/kg, Invivogen) suspended in PBS for 6 h. Mice were euthanized in a CO2 chamber and peritoneal lavage was performed using 2.5 mL PBS. The cells in the lavage fluid were pelleted and the supernatant was removed and analyzed by ELISA for IL-1β and IL-6 concentration. Cells were resuspended in 1 mL PBS and passed through a 70 μm filter. 100 μL cells was removed for FACS analysis and the remaining cells were counted using a TC20™ automated cell counter (Biorad) with a minimum size gate of 8 μm and a maximum gate of 20 μm. 100 μL cells were incubated for 20 min at RT with Zombie Green (Biolegend, 1/800). Cells were then washed and incubated for 10 min at RT with anti-mouse CD16/CD32 (BD Biosciences) in 100 μL PBS. After 10 min, 1 μg of the following antibodies was added: APC/Cy7 anti-mouse CD45 (Biolegend), eFluor660 anti-mouse CD11b (ThermoFisher Scientific) and Pacific Blue anti-mouse Ly6G (Biolegend). After 20 min, cells were washed, resuspended in PBS and analyzed on a FACS Canto II Cell Analyzer (BD Biosciences). Neutrophils were identified as Zombie Green negative (live cells), CD45+CD11b+Ly6G+ cells. Analysis of acquired data was performed with the FlowJo software (FlowJo LLC). The total number of neutrophils obtained from each mouse was calculated by multiplying the percentage neutrophils with the total cell count obtained.
Quantification and Statistical Analysis
Details of all statistical analyses performed can be found in the figure legends. Data were expressed as mean ± standard error of the mean (SEM) and p values were calculated using two-tailed Student’s t test for pairwise comparison of variables and one-way ANOVA for multiple comparison of variables. A Sidak’s multiple comparisons test was used as a post-test when performing an ANOVA. A confidence interval of 95% was used for all statistical tests. Statistical tests were not performed on western blot quantification figures due to the semiquantitative nature of these data. Significance was defined as follows: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Sample sizes were determined on the basis of previous experiments using similar methodologies. All depicted data points are biological replicates taken from distinct samples. Each figure consists of a minimum of 3 independent experiments from multiple biological replicates. n = the number of animals or the number of independent experiments with cell lines. For in vivo studies, mice were randomly assigned to treatment groups. For mass spectrometry analyses, samples were processed in random order and experimenters were blinded to experimental conditions.
Acknowledgments
We thank Professor Richard Hartley (University of Glasgow) for synthesizing and supplying 4-OI, 4-OS, and 4-O-2-MS. We thank Professor Albena Dinkova-Kostova (University of Dundee) and Professor Ed Lavelle (Trinity College Dublin) for supplying bones from Nfe2l2 −/− and Nlrp3 −/− mice, respectively. We thank Dylan Ryan (University of Cambridge) for helpful discussions. The O’Neill laboratory acknowledges the following grant support: European Research Council Metabinate (834370), Science Foundation Ireland (12/IA/1531), and The Wellcome Trust (205455).
Author Contributions
A.H. designed and performed experiments and analyzed the data. A.H. and L.A.J.O’N. wrote the manuscript. S.A. performed FACS analysis. S.A. and M.C.R. carried out in vivo experiments. S.H. and R.F. performed tandem mass spectrometry experiments. S.E.C. performed experiments on CAPS PBMCs, isolated from samples provided by A.D.I. C.L., M.C.R., and P.F.S. performed 13C5 octyl itaconate experiments. K.B. generated plasmids for use in experiments. A.F.M. and M.M.H. assisted with experimental design. L.A.J.O’N. conceived ideas and oversaw the research program.
Declaration of Interests
The authors declare no competing interests.
Published: August 12, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cmet.2020.07.016.
Supplemental Information
References
- Chen J., Chen Z.J. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature. 2018;564:71–76. doi: 10.1038/s41586-018-0761-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll R.C., Robertson A.A., Chae J.J., Higgins S.C., Muñoz-Planillo R., Inserra M.C., Vetter I., Dungan L.S., Monks B.G., Stutz A. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015;21:248–255. doi: 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordes T., Wallace M., Michelucci A., Divakaruni A.S., Sapcariu S.C., Sousa C., Koseki H., Cabrales P., Murphy A.N., Hiller K., Metallo C.M. Immunoresponsive gene 1 and itaconate inhibit succinate dehydrogenase to modulate intracellular succinate levels. J. Biol. Chem. 2016;291:14274–14284. doi: 10.1074/jbc.M115.685792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly R., Blackburn G., Best C., Goodyear C.S., Mudaliar M., Burgess K., Stirling A., Porter D., McInnes I.B., Barrett M.P., Dale J. Changes in plasma itaconate elevation in early rheumatoid arthritis patients elucidates disease activity associated macrophage activation. Metabolites. 2020;10:241. doi: 10.3390/metabo10060241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo-Fernández R., Coll R.C., Kearney J., Breit S., O’Neill L.A.J. The intracellular chloride channel proteins CLIC1 and CLIC4 induce IL-1β transcription and activate the NLRP3 inflammasome. J. Biol. Chem. 2017;292:12077–12087. doi: 10.1074/jbc.M117.797126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidt M.M., Ebert T.S., Chauhan D., Schmidt T., Schmid-Burgk J.L., Rapino F., Robertson A.A., Cooper M.A., Graf T., Hornung V. Human monocytes engage an alternative inflammasome pathway. Immunity. 2016;44:833–846. doi: 10.1016/j.immuni.2016.01.012. [DOI] [PubMed] [Google Scholar]
- Garstkiewicz M., Strittmatter G.E., Grossi S., Sand J., Fenini G., Werner S., French L.E., Beer H.D. Opposing effects of Nrf2 and Nrf2-activating compounds on the NLRP3 inflammasome independent of Nrf2-mediated gene expression. Eur. J. Immunol. 2017;47:806–817. doi: 10.1002/eji.201646665. [DOI] [PubMed] [Google Scholar]
- Ghayur T., Banerjee S., Hugunin M., Butler D., Herzog L., Carter A., Quintal L., Sekut L., Talanian R., Paskind M. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature. 1997;386:619–623. doi: 10.1038/386619a0. [DOI] [PubMed] [Google Scholar]
- Groß C.J., Mishra R., Schneider K.S., Médard G., Wettmarshausen J., Dittlein D.C., Shi H., Gorka O., Koenig P.A., Fromm S. K+ efflux-independent NLRP3 inflammasome activation by small molecules targeting mitochondria. Immunity. 2016;45:761–773. doi: 10.1016/j.immuni.2016.08.010. [DOI] [PubMed] [Google Scholar]
- Han J., Gagnon S., Eckle T., Borchers C.H. Metabolomic analysis of key central carbon metabolism carboxylic acids as their 3-nitrophenylhydrazones by UPLC/ESI-MS. Electrophoresis. 2013;34:2891–2900. doi: 10.1002/elps.201200601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y., Zeng M.Y., Yang D., Motro B., Núñez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530:354–357. doi: 10.1038/nature16959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H., Jiang H., Chen Y., Ye J., Wang A., Wang C., Liu Q., Liang G., Deng X., Jiang W., Zhou R. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat. Commun. 2018;9:2550. doi: 10.1038/s41467-018-04947-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M., Chiang H.H., Luo H., Zheng Z., Qiao Q., Wang L., Tan M., Ohkubo R., Mu W.C., Zhao S. An acetylation switch of the NLRP3 inflammasome regulates aging-associated chronic inflammation and insulin resistance. Cell Metab. 2020;31:580–591.e5. doi: 10.1016/j.cmet.2020.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka M.T., Kummer M.P., Stutz A., Delekate A., Schwartz S., Vieira-Saecker A., Griep A., Axt D., Remus A., Tzeng T.C. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–678. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman H.M., Mueller J.L., Broide D.H., Wanderer A.A., Kolodner R.D. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat. Genet. 2001;29:301–305. doi: 10.1038/ng756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooftman A., O’Neill L.A.J. The immunomodulatory potential of the metabolite itaconate. Trends Immunol. 2019;40:687–698. doi: 10.1016/j.it.2019.05.007. [DOI] [PubMed] [Google Scholar]
- Hornung V., Ablasser A., Charrel-Dennis M., Bauernfeind F., Horvath G., Caffrey D.R., Latz E., Fitzgerald K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V., Bauernfeind F., Halle A., Samstad E.O., Kono H., Rock K.L., Fitzgerald K.A., Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z., Yan C., Liu P., Huang Z., Ma R., Zhang C., Wang R., Zhang Y., Martinon F., Miao D. Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science. 2013;341:172–175. doi: 10.1126/science.1236381. [DOI] [PubMed] [Google Scholar]
- Hughes M.M., Hooftman A., Angiari S., Tummala P., Zaslona Z., Runtsch M.C., McGettrick A.F., Sutton C.E., Diskin C., Rooke M. Glutathione transferase omega-1 regulates NLRP3 inflammasome activation through NEK7 deglutathionylation. Cell Rep. 2019;29:151–161.e5. doi: 10.1016/j.celrep.2019.08.072. [DOI] [PubMed] [Google Scholar]
- Iyer S.S., He Q., Janczy J.R., Elliott E.I., Zhong Z., Olivier A.K., Sadler J.J., Knepper-Adrian V., Han R., Qiao L. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39:311–323. doi: 10.1016/j.immuni.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliana C., Fernandes-Alnemri T., Wu J., Datta P., Solorzano L., Yu J.W., Meng R., Quong A.A., Latz E., Scott C.P., Alnemri E.S. Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J. Biol. Chem. 2010;285:9792–9802. doi: 10.1074/jbc.M109.082305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliana C., Fernandes-Alnemri T., Kang S., Farias A., Qin F., Alnemri E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012;287:36617–36622. doi: 10.1074/jbc.M112.407130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N., Warming S., Lamkanfi M., Vande Walle L., Louie S., Dong J., Newton K., Qu Y., Liu J., Heldens S. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M., Dixit V.M. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- Lampropoulou V., Sergushichev A., Bambouskova M., Nair S., Vincent E.E., Loginicheva E., Cervantes-Barragan L., Ma X., Huang S.C., Griss T. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 2016;24:158–166. doi: 10.1016/j.cmet.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.G., Jenkins N.A., Gilbert D.J., Copeland N.G., O’Brien W.E. Cloning and analysis of gene regulation of a novel LPS-inducible cDNA. Immunogenetics. 1995;41:263–270. doi: 10.1007/BF00172150. [DOI] [PubMed] [Google Scholar]
- Liao S.T., Han C., Xu D.Q., Fu X.W., Wang J.S., Kong L.Y. 4-Octyl itaconate inhibits aerobic glycolysis by targeting GAPDH to exert anti-inflammatory effects. Nat. Commun. 2019;10:5091. doi: 10.1038/s41467-019-13078-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu A., Magupalli V.G., Ruan J., Yin Q., Atianand M.K., Vos M.R., Schröder G.F., Fitzgerald K.A., Wu H., Egelman E.H. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 2014;156:1193–1206. doi: 10.1016/j.cell.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan M.S.J., Olhava E.J., Roush W.R., Seidel H.M., Glick G.D., Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018;17:688. doi: 10.1038/nrd.2018.149. [DOI] [PubMed] [Google Scholar]
- Martinon F., Pétrilli V., Mayor A., Tardivel A., Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- Masters S.L., Dunne A., Subramanian S.L., Hull R.L., Tannahill G.M., Sharp F.A., Becker C., Franchi L., Yoshihara E., Chen Z. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat. Immunol. 2010;11:897–904. doi: 10.1038/ni.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelucci A., Cordes T., Ghelfi J., Pailot A., Reiling N., Goldmann O., Binz T., Wegner A., Tallam A., Rausell A. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc. Natl. Acad. Sci. USA. 2013;110:7820–7825. doi: 10.1073/pnas.1218599110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills E.L., Ryan D.G., Prag H.A., Dikovskaya D., Menon D., Zaslona Z., Jedrychowski M.P., Costa A.S.H., Higgins M., Hams E. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. 2018;556:113–117. doi: 10.1038/nature25986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Planillo R., Kuffa P., Martínez-Colón G., Smith B.L., Rajendiran T.M., Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38:1142–1153. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill L.A.J., Artyomov M.N. Itaconate: the poster child of metabolic reprogramming in macrophage function. Nat. Rev. Immunol. 2019;19:273–281. doi: 10.1038/s41577-019-0128-5. [DOI] [PubMed] [Google Scholar]
- Puchalska P., Huang X., Martin S.E., Han X., Patti G.J., Crawford P.A. Isotope tracing untargeted metabolomics reveals macrophage polarization-state-specific metabolic coordination across intracellular compartments. iScience. 2018;9:298–313. doi: 10.1016/j.isci.2018.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W., Qin K., Zhang Y., Jia W., Chen Y., Cheng B., Peng L., Chen N., Liu Y., Zhou W. S-glycosylation-based cysteine profiling reveals regulation of glycolysis by itaconate. Nat. Chem. Biol. 2019;15:983–991. doi: 10.1038/s41589-019-0323-5. [DOI] [PubMed] [Google Scholar]
- Qin W., Zhang Y., Tang H., Liu D., Chen Y., Liu Y., Wang C. Chemoproteomic profiling of itaconation by bioorthogonal probes in inflammatory macrophages. J. Am. Chem. Soc. 2020;142:10894–10898. doi: 10.1021/jacs.9b11962. [DOI] [PubMed] [Google Scholar]
- Rathinam V.A.K., Zhao Y., Shao F. Innate immunity to intracellular LPS. Nat. Immunol. 2019;20:527–533. doi: 10.1038/s41590-019-0368-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J.-V., Delaite C., Riess G., Schuller A.-S. A comparative study of the thermal properties of homologous series of crystallisable n-alkyl maleate and itaconate monoesters. Thermochim. Acta. 2016;623:136–143. [Google Scholar]
- Schmid-Burgk J.L., Chauhan D., Schmidt T., Ebert T.S., Reinhardt J., Endl E., Hornung V. A genome-wide CRISPR (clustered regularly interspaced short palindromic repeats) screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J. Biol. Chem. 2016;291:103–109. doi: 10.1074/jbc.C115.700492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif H., Wang L., Wang W.L., Magupalli V.G., Andreeva L., Qiao Q., Hauenstein A.V., Wu Z., Núñez G., Mao Y., Wu H. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature. 2019;570:338–343. doi: 10.1038/s41586-019-1295-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J., Zhao Y., Wang Y., Gao W., Ding J., Li P., Hu L., Shao F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–192. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- Shi J., Zhao Y., Wang K., Shi X., Wang Y., Huang H., Zhuang Y., Cai T., Wang F., Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- Shi H., Murray A., Beutler B. Reconstruction of the mouse inflammasome system in HEK293T cells. Bio Protoc. 2016;6:1–10. doi: 10.21769/BioProtoc.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H., Wang Y., Li X., Zhan X., Tang M., Fina M., Su L., Pratt D., Bu C.H., Hildebrand S. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 2016;17:250–258. doi: 10.1038/ni.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K., Crother T.R., Karlin J., Dagvadorj J., Chiba N., Chen S., Ramanujan V.K., Wolf A.J., Vergnes L., Ojcius D.M. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J.W., Lam S.M., Fan X., Cao W.J., Wang S.Y., Tian H., Chua G.H., Zhang C., Meng F.P., Xu Z. Omics-driven systems interrogation of metabolic dysregulation in COVID-19 pathogenesis. Cell Metab. 2020;32:188–202. doi: 10.1016/j.cmet.2020.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutz A., Kolbe C.C., Stahl R., Horvath G.L., Franklin B.S., van Ray O., Brinkschulte R., Geyer M., Meissner F., Latz E. NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J. Exp. Med. 2017;214:1725–1736. doi: 10.1084/jem.20160933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain A., Bambouskova M., Kim H., Andhey P.S., Duncan D., Auclair K., Chubukov V., Simons D.M., Roddy T.P., Stewart K.M., Artyomov M.N. Comparative evaluation of itaconate and its derivatives reveals divergent inflammasome and type I interferon regulation in macrophages. Nat Metab. 2020;2:594–602. doi: 10.1038/s42255-020-0210-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson K.V., Deng M., Ting J.P. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019;19:477–489. doi: 10.1038/s41577-019-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang T., Lang X., Xu C., Wang X., Gong T., Yang Y., Cui J., Bai L., Wang J., Jiang W., Zhou R. CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat. Commun. 2017;8:202. doi: 10.1038/s41467-017-00227-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandanmagsar B., Youm Y.H., Ravussin A., Galgani J.E., Stadler K., Mynatt R.L., Ravussin E., Stephens J.M., Dixit V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011;17:179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Walle L., Van Opdenbosch N., Jacques P., Fossoul A., Verheugen E., Vogel P., Beyaert R., Elewaut D., Kanneganti T.D., van Loo G., Lamkanfi M. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature. 2014;512:69–73. doi: 10.1038/nature13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youm Y.H., Nguyen K.Y., Grant R.W., Goldberg E.L., Bodogai M., Kim D., D’Agostino D., Planavsky N., Lupfer C., Kanneganti T.D. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015;21:263–269. doi: 10.1038/nm.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C., Gillette D.D., Li X., Zhang Z., Wen H. Nuclear factor E2-related factor-2 (Nrf2) is required for NLRP3 and AIM2 inflammasome activation. J. Biol. Chem. 2014;289:17020–17029. doi: 10.1074/jbc.M114.563114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets/code.