

SUMMARY
Synaptic plasticity is a key mechanism of learning and memory. Synaptic plasticity mechanisms within the nucleus accumbens (NAc) mediate differential behavioral adaptations. Feedforward inhibition in the NAc occurs when glutamatergic afferents onto medium spiny neurons (MSNs) collateralize onto fast-spiking parvalbumin (PV)-expressing interneurons (PV-INs), which exert GABAergic control over MSN action potential generation. Here, we find that feedforward glutamatergic synapses onto PV-INs in the NAc core selectively express Ca2+-permeable AMPA receptors (CP-AMPARs). Ca2+ influx by CP-AMPARs on PV-INs triggers long-term depression (LTD) mediated by endocannabinoid (eCB) signaling at presynaptic cannabinoid type-1 (CB1) receptors (CB1Rs). Moreover, CP-AMPARs authorize tonic eCB signaling to negatively regulate glutamate release probability. Blockade of CP-AMPARs in the NAc core in vivo is sufficient to disinhibit locomotor output. These findings elucidate mechanisms by which PV-IN-embedded microcircuits in the NAc undergo activity-dependent shifts in synaptic strength.
Graphical Abstract
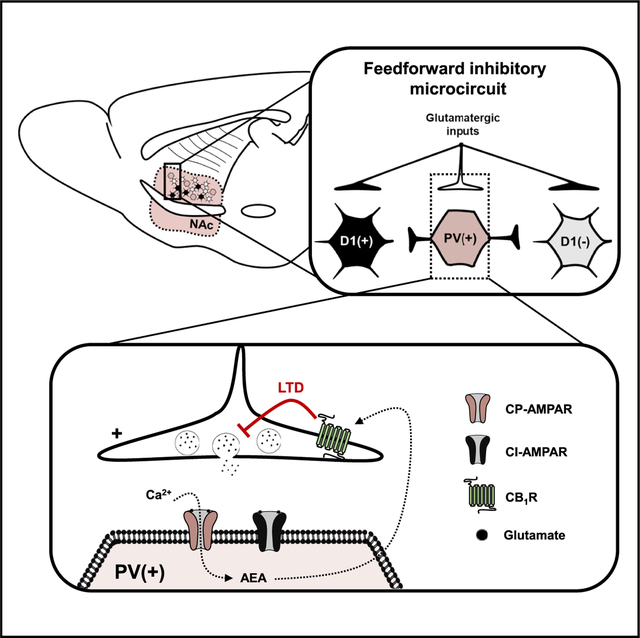
In Brief
Manz et al. show that CP-AMPARs are expressed at glutamatergic synapses onto PV-INs but not D1- or D2-expressing MSNs in the NAc core. Ca2+ influx through CP-AMPARs triggers endocannabinoid-dependent tone and synaptic plasticity. Intra-NAc blockade of CP-AMPARs in vivo increases basal locomotion.
INTRODUCTION
Excitatory synaptic plasticity within the nucleus accumbens (NAc) is a substrate for adaptive motivational behavior implicated in various neuropsychiatric disease states, including addiction and depression (Malenka and Bear, 2004). The NAc gates motivational behavior by scaling downstream access to limbic-motor centers (Turner et al., 2018a). A component of this gate is formed by interneuron (IN)-embedded microcircuits, such as disynaptic feedforward inhibition, that regulate synaptic events mediating NAc output (Winters et al., 2012; Qi et al., 2016; Burke et al., 2017). Long-range glutamatergic inputs from corticolimbic structures collateralize onto GABAergic fast-spiking parvalbumin (PV)-expressing INs (PV-IN) to synchronize the activity of D1 and D2 dopamine (DA) receptor-expressing medium spiny neurons (MSNs) (Qi et al., 2016; Scudder et al., 2018; Trouche et al., 2019). Although GABAergic transmission at PV-IN-to-MSN synapses is a recognized regulatory element within striatal microcircuits, mechanisms regulating glutamatergic transmission onto PV-INs in the NAc remain relatively unexplored.
PV-INs in the NAc display hodological similarities to D1 and D2 MSNs, receiving glutamatergic afferents from the hippocampus, prefrontal cortex, mediodorsal thalamus, basolateral amygdala (BLA), and ventral tegmental area (VTA) (Qi et al., 2016; Yu et al., 2017; Trouche et al., 2019). An important distinction between PV-INs and other striatal cell types is the selective expression of GluA2-lacking Ca2+-permeable AMPA receptors (CP-AMPARs) on PV-INs in drug-naive states (Hu et al., 2014; Yu et al., 2017). CP-AMPARs exhibit greater single-channel conductance, faster deactivation kinetics, and an inwardly rectifying biophysical profile (Liu and Cull-Candy, 2000; Nissen et al., 2010). The fast-spiking, electrotonic, and synaptic properties of PV-INs allow these cells to rapidly transduce shifts in corticolimbic circuit activity into a GABAergic signal coordinating MSN output (O’Hare et al., 2017; Tepper et al., 2018). Therefore, CPAMPAR-predominant synapses onto PV-INs in the NAc may have broad regulatory consequences on NAc circuit function (Soler-Llavina and Sabatini, 2006). Indeed, potentiating synaptic strength at BLA inputs onto PV-INs in the NAc shell expedites cocaine self-administration, supporting the notion that a critical locus within NAc feedforward microcircuits is excitatory drive onto PV-INs (Yu et al., 2017).
In the present study, we investigated synaptic plasticity mechanisms at glutamatergic synapses onto PV-INs in the NAc core. We used in vivo behavioral pharmacology and whole-cell patch-clamp electrophysiology in cell-type-specific reporter mice. We report that NAc-specific blockade of CP-AMPARs, the basal expression of which is restricted to PV-positive cells (PV(+)-INs) and not D1 DA receptor [D1(+)] or D2 DA receptor [D1([−])] MSNs, elicits a hyperlocomotor phenotype. Adapting a low-frequency stimulation (LFS) protocol that elicits long-term depression (LTD) at synapses onto D2 MSNs, we find that LFS triggers LTD of glutamatergic transmission onto PV(+)-INs by CP-AMPARs. LTD at these synapses requires Ca2+ influx by CP-AMPARs that evokes endocannabinoid (eCB) signaling at presynaptic cannabinoid type-1 receptors (CB1Rs). In addition, CP-AMPARs gate tonic CB1R signaling by regulating the production of anandamide (AEA). These findings elucidate a behaviorally relevant circuit element regulating the synaptic strength of PV-IN-embedded microcircuits in the NAc core.
RESULTS
CP-AMPARs Are Functionally Restricted to Glutamatergic Synapses onto PV-INs in the NAc Core
Fast-spiking INs, of which most contain the Ca2+-binding protein PV, express GluA2-lacking CP-AMPARs throughout the fore-brain (Hu et al., 2014). CP-AMPARs are glutamate-gated ion channels lacking the post-transcriptionally edited GluA2 subunit (Lalanne et al., 2018). Although developmentally regulated, GluA2-lacking CP-AMPARs mediate fast excitatory transmission at privileged synaptic loci within IN-embedded microcircuits (Hu et al., 2014; Schall et al., 2020). CP-AMPARs exhibit greater single-channel conductance, inward rectification, and sharp activation-deactivation kinetics that confer specialized synaptic properties (Twomey et al., 2017, 2018). To determine if PV-INs [PV(+)-INs] in the NAc core differ from D1(+)- and D1(−)-expressing MSNs in functional CP-AMPAR content, we prepared acute ex vivo brain slices from PVCre-tdTomato(tdT)f/-STOP-fl (PVtdT) and D1tdTomato transgenic reporter mice (Figure 1A). This strategy allows PV(+) and D1(+) cells in the NAc to be visualized ex vivo, as described previously (Scudder et al., 2018; Manz et al., 2019). To confirm that tdT(+) cells in PVtdT mice were PV-INs, we performed current-clamp recordings in tdT(+) cells to determine if tdT(+) cells displayed a fast-spiking electrophysiological profile. Depolarizing current injection exceeding the action potential (AP) threshold in tdT(+) cells elicited high-frequency AP firing with short-duration waveforms and steep afterhyperpolarizations (AHPs), consistent with fast-spiking PV-INs in the NAc. In contrast, D1(+) and D1(−) MSNs in D1tdTomato mice exhibited a regular-spiking electrophysiological profile, hyperpolarized resting membrane potential (VRMP), and prolonged AHPs, consistent with MSN properties described previously (Figure 1B; data shown: AP frequency at 350 pA, PV(+): 157.5 ± 8.7 Hz, n = 10; D1(+): 22.8 ± 1.4 Hz, n = 12; D1(−): 26.2 ± 3.5 Hz, n = 9; 1-way ANOVA, p < 0.001).
Figure 1. CP-AMPARs Are Expressed Basally at Glutamatergic Synapses onto PV(+)-INs in the NAc Core.
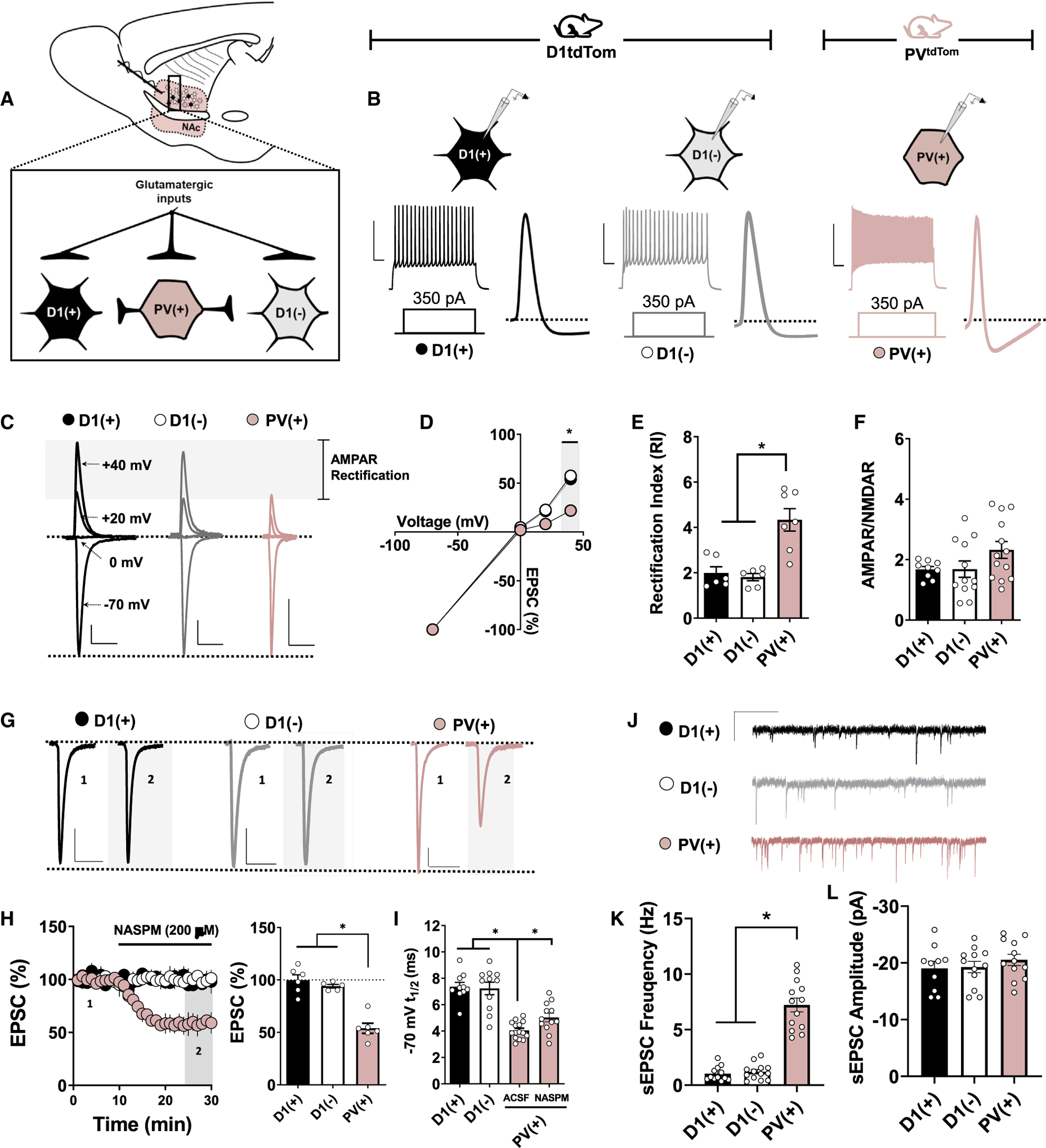
(A) Schematic depicting transgenic reporter strategy and electrophysiological configuration within feedforward microcircuitry. D1(+) MSNs, black-filled circles; D1(−) MSNs, open circles; PV(+)-INs, pink-filled circles.
(B) Representative traces of APs elicited in PV(+), D1(+), and D1(−) cells after a 350-pA somatic current injection. Scale bar: 50 mV/100 ms.
(C) Representative AMPAR-mediated EPSCs in PV(+), D1(+), and D1(−) cells when voltage clamped at −70, 0, +20, and +40 mV. Scale bars: 100 pA/50 ms.
(D and E) AMPAR I-V relationship (D) and RI quantified (E) in PV(+), D1(+), and D1(−) cells.
(F) Average AMPAR:NMDAR ratios obtained from PV(+), D1(+), and D1(−) cells showing no difference between cell types.
(G) Representative traces of EPSCs obtained from PV(+), D1(+), and D1(−) cells at baseline and in the presence of NASPM. Scale bars: 50 pA/50 ms.
(H) Time course summary of normalized EPSC amplitude and Quantification of average EPSC amplitude at PV(+), D1(+), and D1(−) synapses during NASPM bath application.
(I) Decay t1/2 of EPSCs at −70 mV in PV(+), D1(+), and D1(−) cells showing differences in AMPAR decay kinetics and the contribution of CP-AMPARs.
(J) Representative traces of sEPSCs in PV(+), D1(+), and D1(−) cells.
(K) Average sEPSC frequency in PV(+), D1(+), and D1(−) cells over a 5-min recording period. Scale bar: 20 pA/60 s.
(L) Average sEPSC amplitude in PV(+), D1(+), and D1(−) cells over a 5-min recording period. Error bars indicate SEM. *p < 0.05.
We next obtained electrically evoked excitatory postsynaptic currents (EPSCs) in PV(+)-INs of PVtdT mice and D1(+) and D1(−) MSNs of male D1tdTomato mice. AMPAR-mediated EPSCs were pharmacologically isolated by incorporating a GABAA receptor (GABAAR) antagonist, picrotoxin (50 μM), and a NMDAR antagonist, APV (50 μM), into the artificial cerebral spinal fluid (ACSF) bath. To assess stoichiometric differences in AMPAR content at glutamatergic synapses onto PV(+)-INs relative to D1(+) and D1(−) MSNs, we examined the current-voltage (I-V) relationship of AMPAR-mediated EPSCs in each cell type. The rectification index (RI), calculated as the amplitude ratio of EPSCs obtained at −70 mV relative to +40 mV, was significantly higher in PV(+)-INs than both MSN subtypes, indicating the presence of inwardly rectifying GluA2-lacking CP-AMPARs in PV(+)-INs (Figures 1C–1E; RI, PV(+): 4.32 ± 0.57, n = 7; D1(+): 1.83 ± 0.30, n = 6; D1(−): 1.79 ± 0.17, n = 6; 1-way ANOVA, p = 0.002). Despite an increased RI in PV(+)-INs, we did not detect a difference in the AMPAR/NMDAR ratio between cell types (Figure 1F; D1(+) A/N: 1.67 ± 0.098, n = 9; D1(−) A/N: 1.69 ± 0.269, n = 12; PV(+) A/N: 2.32 ± 0.288, n = 13; 1-way ANOVA: F(2, 31) = 2.27, p = 0.119). Bath application of a CP-AMPAR-selective AMPAR antagonist, 1-naphthylacetyl spermine (NASPM, 200 μM), also significantly decreased EPSC amplitude in PV(+)-INs without altering EPSC amplitude in D1(+) or D1(−) MSNs (Figures 1H and 1I; NASPM, PV(+): 54.45% ± 6.57%, n = 5; D1(+): 98.68% ± 6.74%, n = 5; D1(−): 94.04% ±1.91%, n = 5; 1-way ANOVA, p < 0.001). Furthermore, AMPAR-mediated EPSCs in PV(+)-INs exhibited significantly faster decay kinetics (t1/2) than D1(+) and D1(−) MSNs (Figures 1F–1J; t1/2, PV(+): 4.04 ± 0.18 ms, n = 15; D1(+): 7.35 ± 0.35 ms, n = 10; D1(−): 7.23 ± 0.50, n = 11; 1-way ANOVA p < 0.001). The t1/2 of EPSCs in PV(+)-INs was increased in the presence of NASPM but was still less than the t1/2 of EPSCs in D1(+) and D1(−) MSNs, pointing to potential electrotonic differences in dendritic charge transfer in PV(+)-INs (Figure 1I; NASPM t1/2, ACSF PV(+): 4.04 ± 0.18 ms, n = 15; NASPM PV(+): 5.12 ± 0.33, n = 12; p = 0.004). Consistent with our AMPAR/NMDAR measurements, spontaneous EPSC (sEPSC) amplitude did not differ between cell types, but sEPSC frequency was significantly increased in PV(+)-INs relative to D1(+) and D1(−) MSNs (Figures 1J–1L) (sEPSC frequency, D1(+): 1.03 ± 0.18 Hz, n = 12; D1(−): 1.20 ± 0.20 Hz, n = 13; PV(+): 7.22 ± 0.61 Hz; n = 13, 1-way ANOVA; cell effect: F(2, 35) = 79.32, p < 0.001) (sEPSC amplitude, D1(+): −19.05 ± 1.24 pA, n = 10; D1(−): 19.26 ± 0.99 pA, n = 12; PV(+): −20.53 ± 0.95 pA, n = 12; 1-way ANOVA; cell effect: F(2, 31) = 0.571, p = 0.963). Together, these data indicate that glutamatergic synapses onto PV-INs are uniquely enriched in CP-AMPARs and exhibit distinct synaptic properties from MSNs.
Blockade of Ca2+-Permeable AMPARs in the NAc Core Increases Locomotor Output
The functional relevance of CP-AMPAR-invigorated synapses in the NAc following drug cessation is well-documented; yet, little is known about how CP-AMPARs expressed basally regulate behavioral output (Wright et al., 2020). To determine how CP-AMPARs in the NAc control basal locomotor activity, we cannulated the dorsomedial NAc core of 8- to 12-week-old mice and microinfused selective CP-AMPAR antagonist, NASPM (5 μg/μL), or saline (SAL) prior to a 30-min open field assay (OFA) (Figure 2A). Relative to SAL-infused control mice, mice infused with NASPM in the NAc core exhibited a progressive increase in locomotor activity that was greatest at the 30-min time point (Figure 2B; 2-way repeated measures ANOVA [RM-ANOVA], p < 0.05 at t = 20–30 min, F(1,24) = 34.34). NASPM-infused mice displayed greater total locomotor activity throughout the OFA task, indicating that CP-AMPAR-containing synapses in the NAc functionally constrain baseline locomotor output (Figures 2B and 2C; 30-min total, SAL: 4153 ± 940.7 cm, n = 8; NASPM: 8858 ± 1503 cm, n = 9; p = 0.021).
Figure 2. CP-AMPAR Blockade in the NAc Core Increases Locomotor Output.
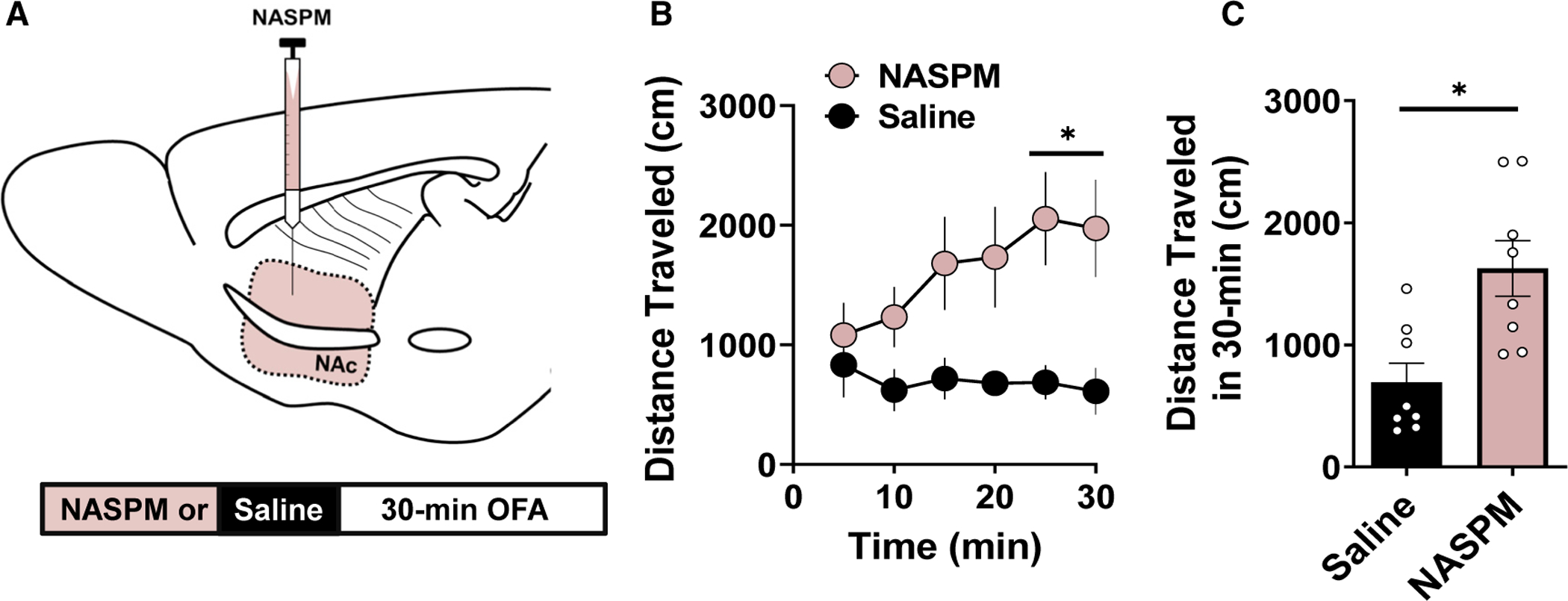
(A) Schematic depicting cannulated delivery of saline (SAL; black) or NASPM into the NAc core prior to a 30-min open-field task.
(B) Locomotor activity in NASPM (pink-filled circles)- and SAL (black-filled circles)-infused mice binned every 5 min showing that NASPM increases locomotor output.
(C) Total locomotor activity averaged over the 30-min OFA with NASPM (pink-filled bars)-infused mice exhibiting increased locomotor output than SAL (black-filled bar)-infused mice. Error bars indicate SEM. *p < 0.05.
LFS Elicits CP-AMPAR-Dependent LTD of Glutamatergic Transmission onto PV(+)-INs
PV(+)-INs receive collateralizing glutamatergic input from corticolimbic afferents that target D1(+) and D1(−) MSNs. At synapses onto D2-GFP(+) [D1(−)] MSNs in the NAc core, LFS (10 Hz) triggers robust LTD (Grueter et al., 2010; Turner et al., 2018b). To determine if LFS modulates glutamatergic synaptic strength onto PV(+)-INs, LFS was delivered locally for 5 min following a 10-min EPSC baseline obtained from PV(+)-INs. LFS elicited high-fidelity EPSCs in PV(+)-INs throughout the induction protocol (Figures 3A and 3B). LFS resulted in a persistent decrease in EPSC amplitude throughout the recording period, indicating the induction of LTD of glutamatergic synapses onto PV(+)-INs. (Figures 3A and 3D; LTD, PV(+): 54.67% ±6.79%, n = 9, p < 0.001). We next asked whether intracellular Ca2+ signaling in PV(+)-INs is required for the induction of this LTD, as activity-dependent shifts in Ca2+ signaling underlie various forms of plasticity in the NAc (Grueter et al., 2010; Francis et al., 2019). To test this possibility, we included a fast-acting Ca2+ chelator, BAPTA (30 mM), in the intracellular solution of the patch pipette. BAPTA completely blocked LFS-induced LTD, suggesting that a rise in intracellular Ca2+ in PV(+)-INs is required for LTD (Figures 3D and 3I; BAPTA,76.37% ± 9.86%, n = 6, p = 0.032).
Figure 3. LFS Triggers Ca2+ and CP-AMPAR-Dependent LTD of Glutamatergic Transmission onto PV(+)-INs.
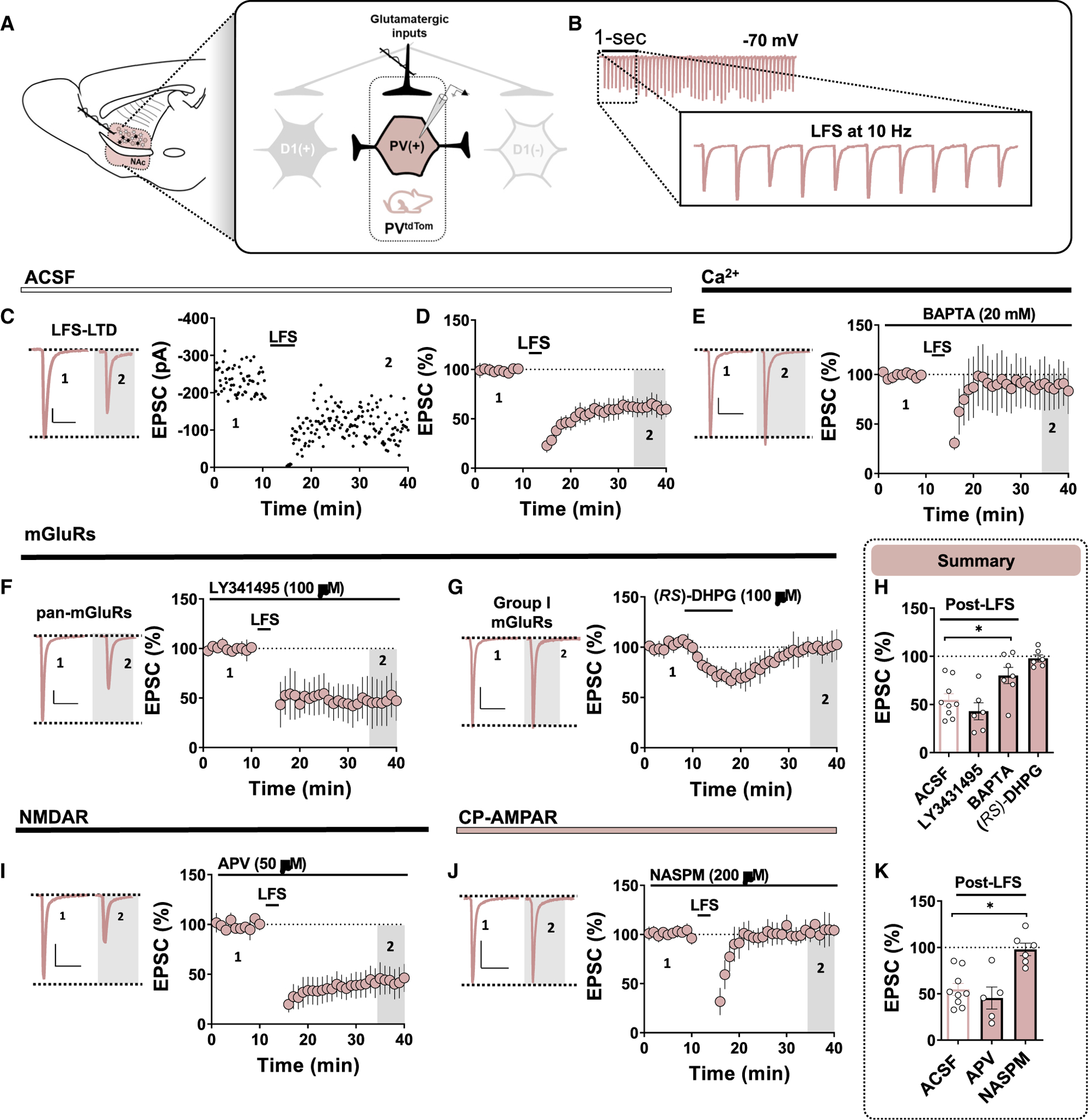
(A) Schematic depicting PVtdTom transgenic reporter strategy and electrophysiological configuration within feedforward microcircuitry. Note that all experiments were performed in the dorsomedial NAc core near the corticoaccumbens interface. Scale bar for all panels: 50 pA/50 ms.
(B) Representative traces of high-fidelity EPSCs recorded from PV(+) during the 10-Hz LFS induction protocol.
(C) Representative traces and experiment in PV(+)-INs at baseline and post-LFS showing that LFS induces robust LTD of glutamatergic transmission.
(D) Time course summary of normalized EPSCs in PV(+)-INs during LFS recording period.
(E) Representative EPSCs and normalized time-course summary of LFS-induced LTD assessed with a Ca2+ chelator, BAPTA, included in the internal solution.
(F) Representative EPSCs and normalized time course of LFS-induced LTD assessed in the presence of a pan-mGluR antagonist, LY341495.
(G) Representative EPSCs and normalized time course summary showing that group I mGluR agonist, (RS)-DHPG, fails to elicit LTD at synapses onto PV(+)-INs.
(H) Quantification of normalized EPSC amplitude post-LFS t(gray) (35–40) min in ACSF, LY341495, and BAPTA and average EPSC amplitude post-DHPG.
(I) Representative EPSCs and normalized time course summary of LFS-induced LTD assessed in the presence of an NMDAR antagonist, APV.
(J) Representative EPSCs and normalized time course of LFS-induced LTD assessed in the presence of NASPM.
(K) Quantification of normalized EPSC amplitude post-LFS t(gray) (35–40) min in APV and NASPM. ACSF average depicts the same data in (H). Error bars indicate SEM. *p < 0.05.
To begin to identify the synaptic source of Ca2+ triggering LTD at these synapses, we first tested the contribution of metabo-tropic glutamate receptors (mGluRs), as group I mGluRs are required for the induction of LFS-LTD at MSN synapses in the NAc core and shell. Incorporation of a pan-mGluR antagonist, LY341495 (100 μM), into the ACSF bath prior to the induction protocol failed to prevent LTD at glutamatergic synapses onto PV(+)-INs (Figures 3F–3H; mGluRs, 42.96% ± 8.88%, n = 6, p = 0.163). Moreover, bath application of a selective group I mGluR agonist, (RS)-dihydroxyphenylglycine (RS-DHPG, 100 μM), elicited a transient depression in EPSC amplitude at PV(+)-IN synapses that returned to baseline following drug washout, indicating a lack of group I mGluR-induced LTD at these synapses (Figures 3G and 3H; DHPG, 100.37% ± 5.75%, n = 6, p = 0.454). These findings indicate that LFS-induced LTD of glutamatergic transmission onto PV(+)-INs is mGluR independent. To determine if LFS instead recruits postsynaptic NMDAR function, we repeated these experiments in the presence of an NMDAR antagonist, APV (50 μM). APV also failed to block LFS-induced LTD at PV(+)-IN synapses (Figures 3I and 3K; APV, 45.34% ± 13.3%, n = 5, p = 0.232). Given that NASPM-sensitive CP-AMPARs are selectively expressed at PV(+)-IN synapses, we hypothesized that Ca2+ influx through CP-AMPARs contributes to the rise in intracellular Ca2+ necessary for LTD. To test this hypothesis, we incorporated CP-AMPAR antagonist NASPM (200 μM) into the ACSF bath for 30 min prior to establishing an EPSC baseline. Interestingly, we were unable to elicit LFS-LTD in the presence of NASPM, indicating that LTD triggered at these synapses requires CP-AMPAR-mediated Ca2+ entry (Figures 3J and 3K; NASPM, 97.72% ± 7.40%, n = 6, p = 0.003).
CP-AMPARs Gate Tonic eCB Signaling through CB1Rs at Glutamatergic Synapses onto PV(+)-INs
NASPM superfusion was accompanied by a surprising reduction in the coefficient of variance (CV) and paired pulse ratio (PPR) of EPSCs recorded from PV(+)-INs, metrics which inversely correlate with presynaptic release probability (Figures 4A–4C; PPR ACSF: 1.44 ± 0.05, n = 22; PPR NASPM: 1.16 ± 0.07; n = 20, p = 0.001) (CV ACSF: 1.18 ± 0.16, n = 12; CV NASPM: 0.58 ±0.10; n = 9, p = 0.01). Although shifts in CV also correspond to the number of functional release sites, blockade of CP-AMPAR-predominant synapses would theoretically increase CV. Thus, we hypothesized that CP-AMPARs control a tonically active, Ca2+-sensitive retrograde messenger at these synapses, a probable candidate being eCBs. To determine if eCBs tonically regulate glutamatergic transmission onto PV(+)-INs by presynaptic CB1Rs, we first measured 50-ms PPR in the presence of a CB1R inverse agonist, AM251 (5 μM). Preincubation in AM251 significantly reduced PPR relative to ACSF control slices and occluded the NASPM-induced decrease in PPR, suggesting that both pharmacological manipulations involve presynaptic CB1R signaling (Figures 4D and 4E; PPR AM251: 1.14 ± 0.10, n = 8, p = 0.025; PPR AM251 + NASPM: 1.07 ± 0.06, n = 9, p = 0.003; 1-way ANOVA, AM251 versus AM251+NASPM, p = 0.985). We next assessed whether AEA or 2-arachidonylglycerol (2-AG), two canonical arachidonic-acid-derived eCBs regulating synaptic strength in the NAc, mediate tonic CB1R signaling at this synapse (Kreitzer and Malenka, 2005; Grueter et al., 2010; Lee et al., 2015). In slices incubated in URB597 (1 μM), an inhibitor of the AEA degradative enzyme fatty acid amide hydrolase (FAAH), PPR was significantly increased relative to ACSF control slices, whereas preincubation in DO34 (1 μM), a selective inhibitor of a 2-AG synthetic enzyme, diacylglycerol lipase (DAGL), failed to evoke a significant change in PPR (Figures 4D and 4E; PPR URB597: 1.77 ± 0.08, n = 16, p = 0.002; PPR DO34: 1.48 ± 0.05, p = 0.998). These data provide evidence that CP-AMPARs at PV-IN synapses promote tonic CB1R activity mediated primarily by AEA signaling.
Figure 4. CP-AMPARs Gate Tonic eCB Signaling through CB1Rs at Glutamatergic Synapses onto PV(+)-INs.
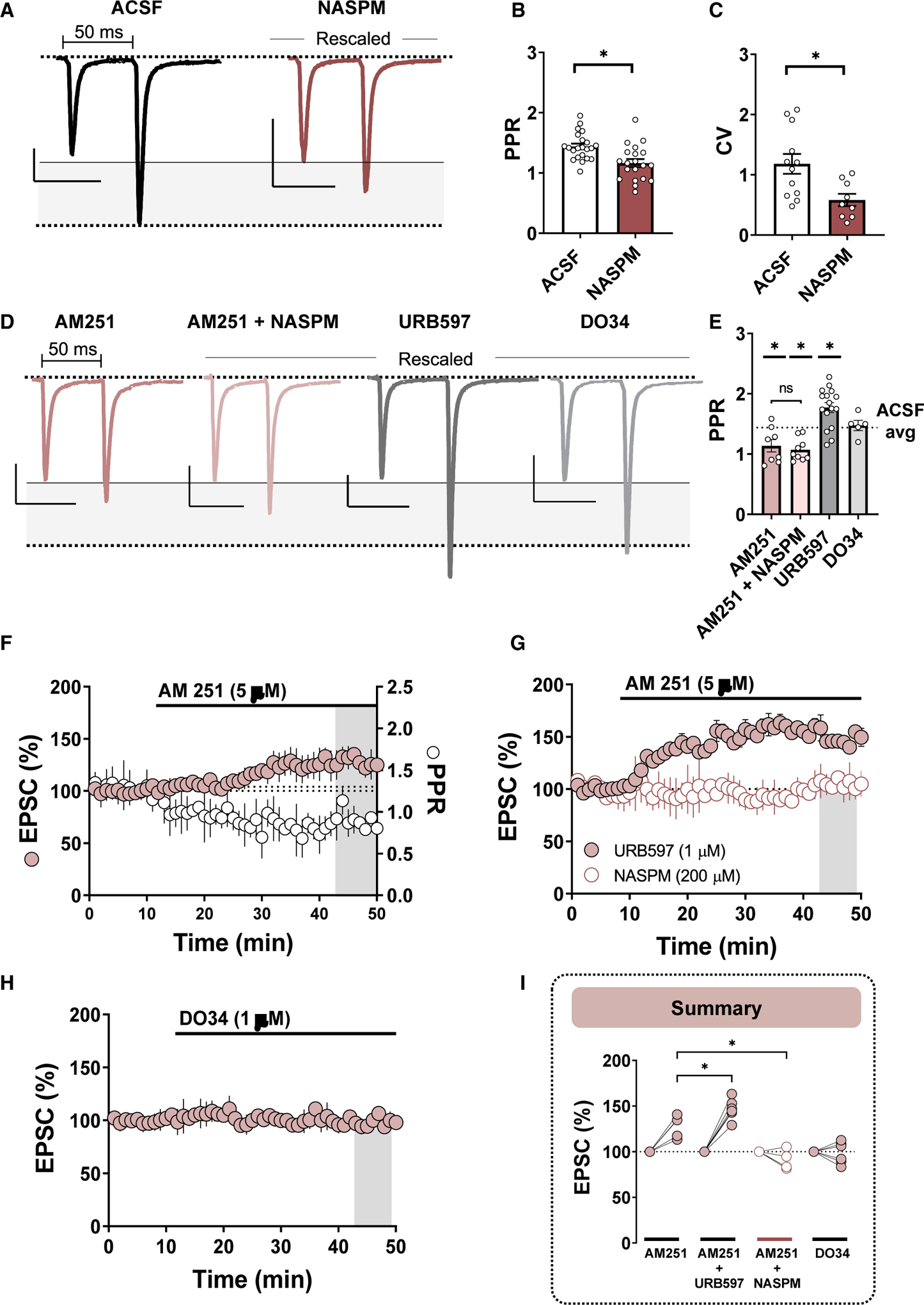
Gray-shaded region highlights the amplitude difference between EPSC1 and EPSC2 of the paired pulses during 50-ms paired-pulse stimulation. All traces scaled to have comparable EPSC1 amplitudes.
(A) Representative traces of 50-ms paired-pulse EPSCs obtained from PV(+)-INs in ACSF (black)- and NASPM (maroon)-incubated slices. Scale bars: 50 pA/50 ms.
(B and C) Quantification of average PPR (B) and CV (C) obtained from PV(+)-INs in ACSF versus NASPM.
(D) Representative traces of 50-ms paired-pulse EPSCs obtained from PV(+)-INs in slices incubated in AM251 (pink), AM251 + NASPM (light pink), URB597 (gray), and DO34 (light gray). Scale bars for all traces: 50 pA/50 ms.
(E) Average PPR following each pharmacological manipulation. Dotted line corresponds to average EPSC amplitude plotted for visual and statistical purposes.
(F) Left y axis: normalized time course summary of average EPSC amplitude in PV(+)-INs during bath application of AM251. Right y axis: raw PPR time course during AM251 superfusion (open circles).
(G) Normalized time course summary of average EPSC amplitude during AM251 bath application in slices incubated and continuously perfused with URB597 (pink-filled circles) or NASPM (open circles).
(H) Normalized time course summary of EPSC amplitude during acute bath application of DO34.
(I) Quantification of average EPSC amplitude following each pharmacological manipulation. Error bars indicate SEM. *p < 0.05.
To assess this mechanism more thoroughly, we examined the effects of AM251 on EPSC amplitude when acutely incorporated into the ACSF bath. Following a 10-min EPSC baseline, AM251 was bath applied for a prolonged exposure period (40 min) to capture changes in EPSC amplitude. Bath application of AM251 resulted in an increase in EPSC amplitude that was accompanied by a decrease in the PPR, consistent with a presynaptic enhancement of glutamate release probability observed above (Figures 4F and 4I; EPSCs, 128.27% ± 6.06%, n = 6, p < 0.001; PPR baseline = 1.31 ± 0.180; PPR post-AM251 = 0.93 ± 0.17, n = 6; paired t test, p < 0.001). If AEA signaling mediates eCB tone, preincubating slices in a FAAH inhibitor should enhance the AM251-evoked increase in EPSC amplitude. Consistent with this hypothesis, bath application of AM251 in URB597-incubated slices unmasked a significant increase in EPSC amplitude relative to baseline and control conditions (Figures 4G and 4I; AM251 + URB: 146.63% ±5.01%, n = 6, p < 0.001; 1-way ANOVA, AM251 versus AM251+URB, F(3,16) = 28.16, p = 0.017). Furthermore, prolonged bath application of DO34 had no effect on EPSC amplitude (Figures 4H and 4I; DO34: 98.65% ± 5.36%, n = 6, p = 0.396). To ascertain whether CP-AMPARs contribute to the AM251-evoked increase in EPSC amplitude, we incorporated NASPM into the ACSF bath prior to AM251. NASPM completely abolished the AM251-induced increase in EPSC amplitude, corroborating our initial pharmacological findings that CP-AMPARs promote tonic eCB signaling at glutamatergic synapses onto PV(+)-INs (Figures 4H and 4I; 91.29% ± 6.37%, n = 4, p = 0.107).
CB1R Signaling Mediates the Expression of LTD Triggered by CP-AMPARs at Glutamatergic Synapses onto PV(+)-INs
Our data indicate that glutamatergic synapses onto PV(+)-INs in the NAc core are tonically regulated by eCBs. To determine if these synapses undergo phasic modes of CB1R-dependent plasticity, we first examined depolarization-induced suppression of excitation (DSE) (Figure 5A). Following a 60-s EPSC baseline sampled at 0.2 Hz, PV(+)-INs underwent a 10-s depolarization step to +40 mV, resulting in a transient reduction in EPSC amplitude consistent with the presence of DSE (Figures 5B and 5C; 61.92% ± 4.05%, n = 8, p < 0.001). To ensure that DSE at these synapses reflects a Ca2+-dependent mobilization of eCBs, we repeated these experiments in EGTA-loaded intracellular solution to chelate depolarization-induced elevations in intracellular Ca2+. EGTA completely abolished the expression of DSE relative to control conditions (Figures 5D and 5E; DSE in EGTA: 95.88% ± 4.24%, n = 6, 1-way ANOVA, ACSF versus EGTA, F(2,18) = 19.21, p < 0.001). Additionally, prior application of AM251 significantly reduced the magnitude of DSE, indicating that synapses onto PV(+)-INs undergo CB1R-dependent short-term plasticity facilitated by intracellular Ca2+ signaling (Figures 5D and 5E; 86.14% ± 3.78%, n = 7, p = 0.007).
Figure 5. CB1R Signaling Mediates the Expression of LTD Triggered by CP-AMPARs at Glutamatergic Synapses onto PV(+)-INs.
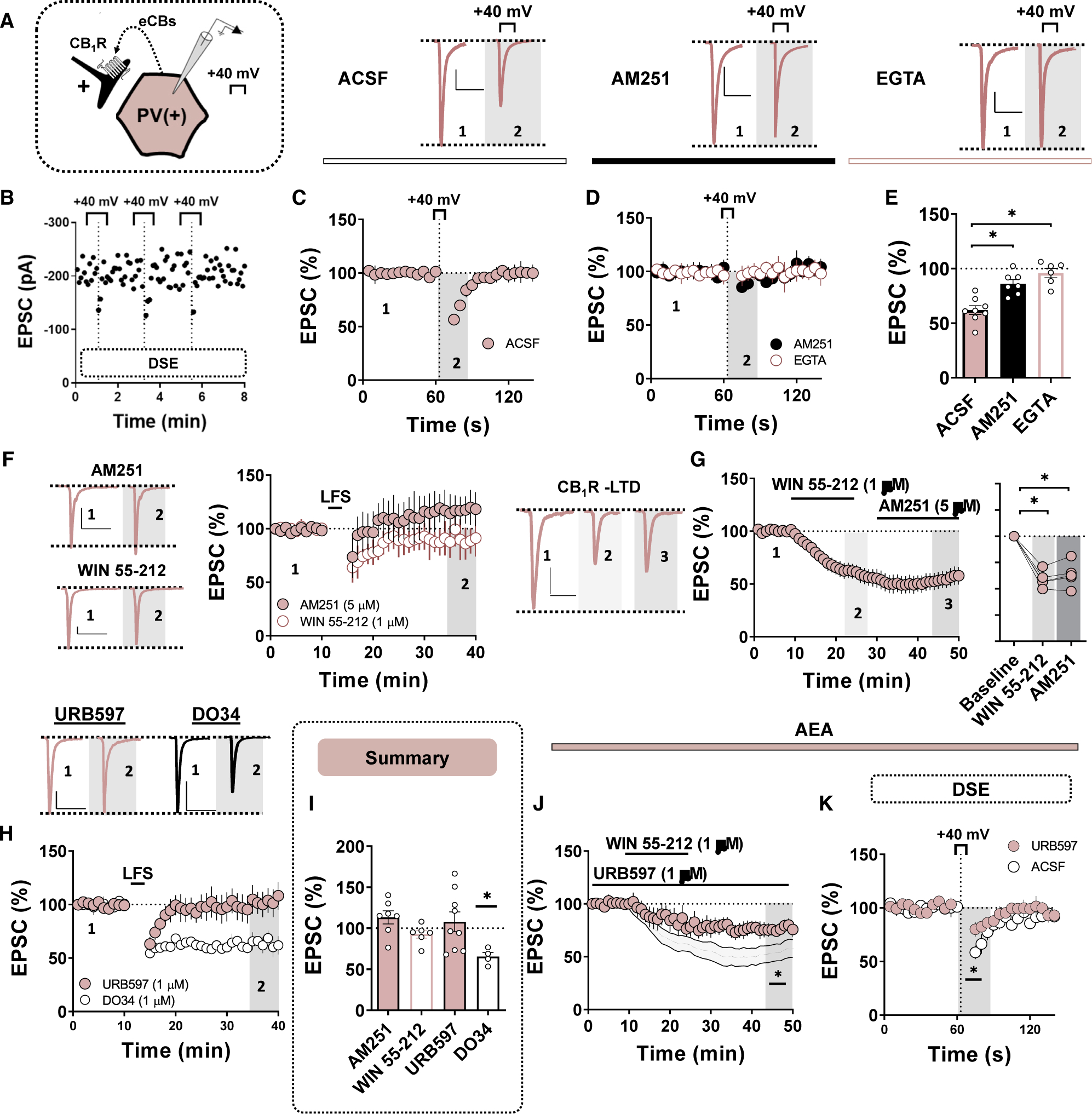
All scale bars: 50 pA/50 ms.
(A) Schematic depicting retrograde eCB signaling at glutamatergic synapses onto PV-INs.
(B) Representative DSE experiment performed in triplicate for each PV(+)-IN with EPSC sampling rate increased to 0.2 Hz.
(C) Top: representative traces of EPSCs obtained pre- and post-DSE in ACSF. Bottom: time course summary of normalized EPSCs obtained from PV(+)-INs during DSE experiment performed in ACSF.
(D) Top: representative traces of EPSCs obtained pre- and post-DSE in AM251 (black circle) and EGTA-containing internal solution (pink open circle). Bottom: time course summary of normalized EPSCs obtained from PV(+)-INs during DSE experiment performed in AM251 (black circles) and EGTA (pink open circles).
(E) Quantification of average EPSC amplitude post-DSE following each pharmacological manipulation.
(F) Representative traces and normalized time course summary of EPSCs obtained from PV(+)-INs pre- and post-LFS in AM251 (top, pink-filled circles) and WIN 55–12 (bottom, pink open circles).
(G) Representative traces and normalized time course summary of EPSCs obtained from PV(+)-INs at baseline, following WIN 55–212 application and in AM251.
(H) Quantification of EPSCs during WIN 55–212 and AM251 bath application.
(I) Representative traces and normalized time course summary of EPSCs obtained from PV(+)-INs pre- and post-LFS in URB597 (pink-filled circles) and DO34 (open circles).
(J) Average EPSC amplitude post-LFS following each pharmacological treatment.
(K) Normalized time-course summary of EPSCs obtained from PV(+)-INs during WIN 55–212 superfusion in slices incubated in URB497. Gray-shaded summary depicts control WIN 55–212 experiments performed in (G).
(L) Time course summary of normalized EPSCs obtained from PV(+)-INs during DSE experiments repeated in ACSF- and in URB597-incubated slices. Error bars indicate SEM. *p < 0.05.
Increased intracellular Ca2+ signaling contributes to the induction of signaling events required for the expression of LTD (Winder and Sweatt, 2001; Grueter et al., 2010; Fitzjohn and Collingridge, 2002). Given that eCBs regulate glutamatergic transmission onto PV(+)-INs in a CP-AMPAR-dependent manner, we hypothesized that CB1R activity underlies the expression of LFS-induced LTD at glutamatergic synapses onto PV(+)-INs. To investigate this mechanism, we examined the integrity of LTD in the presence of a CB1R inverse agonist, AM251 (5 μM). Preincubation of slices in AM251 blocked LFS-induced LTD and unmasked a small potentiation in EPSC amplitude (Figures 5F and 5I; AM251, 113.26% ± 11.89%, n = 5, p = 0.002). If LTD at these synapses requires CB1R activity, then prior activation of CB1R with a CB1/2R agonist, WIN 55–212 (1 μM), should also occlude the expression of LTD. Indeed, continuous application of WIN 55–212 into the ACSF bath prevented the expression of LFS-LTD, strongly suggesting that CB1R activity mediates the expression of CP-AMPAR-dependent LTD at PV(+)-IN synapses (Figures 5F and 5I; WIN, 92.70% ± 5.27%, n = 6, p = 0.002). To determine if CB1R activation alone is sufficient to induce LTD at PV(+)-IN synapses, we superfused WIN 55–212 into the ACSF bath followed by AM251. WIN 55–212 significantly decreased EPSC amplitude in PV(+)-INs that persisted in the presence of AM251, indicating that pharmacological activation of CB1R is sufficient to trigger LTD of glutamatergic transmission onto PV(+)-INs (Figure 4G; WIN-AM251, 63.49% ± 5.49%, n = 5, p < 0.001). Collectively, these data indicate that eCB signaling by CB1R mediates the expression of LTD triggered by CP AMPARs.
To investigate which eCB mediates LFS-LTD, we first examined the contribution of 2-AG by delivering LFS in slices treated with DO34. LFS-induced LTD remained completely intact in the presence of DO34, indicating that this plasticity does not require DAGL-synthesized 2-AG (Figures 5H and 5I; LFS in DO34: 65.45% ± 5.69%, n = 4, p = 0.002). Interestingly, LFS delivered in slices preincubated in a FAAH inhibitor, URB597, failed to elicit LTD and often unmasked an enhancement in EPSC amplitude (Figures 5H and 5I; 107.87% ± 14.17%, n = 9, p = 0.373). Given that AEA signaling mediates tonic CB1R function at this synapse, we speculated that inhibiting the degradation of AEA functionally occludes the expression of LFS-induced LTD. To test this hypothesis, we examined the sensitivity of glutamatergic transmission onto PV(+)-INs to DSE and WIN 55–212 in URB597-treated slices. URB597 blunted the expression of DSE and the magnitude with which WIN 55–212 decreased EPSC amplitude, indicating that preventing the degradation of AEA by FAAH likely increases functional CB1R occupancy, thereby preventing subsequent LTD induction with LFS (Figures 4H and 4I) (DSE ACSF: 69.81% ±4.15%, n = 9; DSE URB: 83.75% ± 3.81%, n = 13, p = 0.028) (WIN URB: 77.44% ± 5.71%, n = 6, p[versus ACSF] = 0.020). Altogether, these data elucidate a mechanism wherein CB1R/CP-AMPAR LTD at synapses onto PV(+)-INs proceeds independently of 2-AG signaling and is heavily regulated by tonic AEA signaling.
DISCUSSION
We offer functional evidence that CP-AMPARs on PV(+)-INs within feedforward microcircuits dynamically regulate glutamatergic transmission in the NAc core through eCB signaling at CB1Rs. Using behavioral pharmacology and whole-cell patch-clamp electrophysiology in PV-IN and D1 MSN-specific transgenic reporter mice, we report that pharmacological blockade of CP-AMPARs in the NAc core increases locomotor output. Under basal conditions, CP-AMPARs are functionally restricted to glutamatergic synapses onto PV(+)-INs but not D1(+) or D1(−) MSNs. Ca2+ influx through CP-AMPARs at PV(+)-IN synapses is recruited during LFS to trigger eCB-dependent LTD. Additionally, we find that tonic eCB signaling at this synaptic locus is mediated by AEA and constitutively regulated by Ca2+ entry through CP-AMPARs. This study directly examines physiological processes governed by CP-AMPARs within PV-IN-embedded microcircuits in the NAc core.
CP-AMPARs in the NAc Constrain Locomotor Output and Are Functionally Restricted to PV-INs within Feedforward Microcircuits
CP-AMPARs on MSNs have been implicated in the pathogenesis of both rewarding- and depressive-like behavioral pheno-types. However, a significant gap remains as to how CP-AM-PARs actually influence synaptic physiology in the NAc (Wolf, 2016; Lim et al., 2012). We find that CP-AMPARs on PV(+)-INs contribute to the fast kinetics of AMPAR-mediated EPSCs detected at these synapses. Rapid detection of glutamatergic input, alongside the electrotonic transfer of charge along PV-IN dendrites, supports the unique role of PV-INs in disynaptic feedforward inhibition (Scudder et al., 2018). As a synaptic intermediate between afferent-directed excitation of MSNs, PV-INs transduce shifts in corticolimbic circuit activity into feedforward GABAergic output. Thus, the biophysical properties of CP-AM-PARs may permit rapid integration of the same corticolimbic inputs driving NAc output.
The stoichiometric profile of AMPARs at glutamatergic synapses in the NAc has important implications for reward-related behavior (Ferrario et al., 2011; Graziane et al., 2016; Wolf, 2016; Conrad et al., 2008). Withdrawal from repeated cocaine exposure leads to the progressive incorporation of GluA2-lacking CP-AMPARs on MSNs in a cell-type- and input-specific manner (Lee et al., 2013; Pascoli et al., 2014; Wright et al., 2020). Time-contingent adaptations at these synapses contribute to the “incubation of cocaine craving” that drives the reinstatement of reward-seeking behavior. In the present study, we provide additional functional evidence that CP-AMPARs are expressed basally at glutamatergic synapses onto PV(+)-INs but not D1(+) or D1(−) MSNs, consistent with prior assess ments of excitatory transmission in the NAc shell (Yu et al., 2017). Interestingly, we find that selectively targeting PV-IN activity by intra-NAc blockade of CP-AMPARs increases locomotor output in drug-naive mice. This finding invites the interpretation that excitatory inputs onto PV-INs constrains overall behavioral output, consistent with recent studies showing that (1) disabling PV-IN output decreases behavioral output during drug reward tasks and (2) CP-AMPAR blockade compromises feedforward inhibition of NAc shell MSNs (Wright et al., 2017; Yu et al., 2017; Wang et al., 2018). However, PV-INs may instead organize purposeful behavioral responses to salient environmental stimuli. For example, chemo- and opto-genetically silencing PV-IN activity in the NAc increases impulsivity in a 5-choice reaction time test (Pisansky et al., 2019). By reducing CP-AMPAR-mediated feedforward input onto PV-INs through CP-AMPARs, afferent inputs to the NAc may drive asynchronous MSN output, leading to enhanced locomotor activity. Future studies are needed to understand how excitatory transmission onto PV-INs programs functional MSN output to downstream limbic-motor centers.
CP-AMPARs Promote Tonic and Phasic eCB Signaling at PV-IN Synapses
In the NAc core, stimulating glutamatergic inputs onto D1(−) MSNs for 5 min at 10 Hz results in LTD mediated by presynaptic CB1R and postsynaptic TRPV1 receptor function. AEA-induced TRPV1 activation results in Ca2+ and dynamin-dependent AMPAR endocytosis, whereas presynaptic CB1R activation results in a shift in the phosphorylation state of vesicular release proteins, such as RIM1α (Grueter et al., 2010). Given that the same glutamatergic inputs onto MSNs collateralize onto synaptically connected PV-INs, we asked whether glutamatergic synapses onto PV-INs also undergo LFS-induced LTD. We initially hypothesized that LTD is unlikely to occur at these synapses, as the electrotonic properties of PV-IN dendrites rapidly transfer membrane potential shifts along the somatodendritic axis and the lack of a dendritic spines fails to confine intracellular effectors recruited during the induction and/or expression of LTD (Eggermann and Jonas, 2011; Hu et al., 2014).
To our surprise, LFS recruits CP-AMPARs to induce robust LTD of glutamatergic transmission onto PV(+)-INs in the NAc core. CP-AMPARs expressed at discrete synapses throughout the limbic network have been associated with the expression of homeostatic and Hebbian plasticity mechanisms (Liu and Cull-Candy, 2000; Soler-Llavina and Sabatini, 2006). A relatively unexplored question, however, is how CP-AMPARs define the molecular requirements for activity-dependent shifts in synaptic strength. We provide evidence that CP-AMPARs on PV(+)-INs trigger a form of LTD mediated by eCB signaling and authorize the release of tonic eCBs that act on presynaptic CB1R to regulate glutamate release probability. Indeed, LFS-induced LTD of glutamatergic transmission onto PV(+)-INs was absent following both CB1R blockade and activation. Several pieces of data point to AEA signaling over 2-AG as the principal eCB governing glutamatergic transmission at this synapse. First, disrupting 2-AG synthesis by inhibiting the Ca2+-sensitive enzyme DAGL had no significant effect on the magnitude of LTD or tonic CB1R activity. In contrast, increasing ambient AEA by blocking the activity of FAAH increased eCB tone, functionally occluded LTD, and significantly attenuated the effects of DSE and WIN 55–212 on glutamatergic transmission. Interestingly, blocking CB1R or inhibiting FAAH during the LFS induction protocol often potentiated EPSC amplitude. Although dissecting this observation is beyond the scope of this study, an intriguing hypothesis is that LFS-facilitated AEA release acts on TRPV1 receptors expressed at a subset of inputs onto PV-INs, leading to increased glutamate release probability (Deroche et al., 2020). Collectively, our data support a mechanism wherein Ca2+ influx by CP-AMPARs is coupled to AEA signaling, which negatively regulates glutamatergic transmission onto PV(+)-INs.
Recent reports using an afferent-specific optogenetic approach indicate that fast-spiking, putative PV-INs in the NAc shell are more strongly innervated by corticolimbic inputs than MSNs, a finding that explains, in part, the high frequency of sEPSCs detected at PV(+)-IN synapses. The extensive innervation pattern of PV-INs necessitates greater regulatory control over the timing, strength, and synchronicity with which glutamatergic transmission drives PV-IN output. Accordingly, tonic AEA signaling by CB1R may serve as a gain-sorting mechanism ensuring appropriate input-output characteristics of NAc feedforward microcircuits. eCB release coupled to Ca2+ influx through CP-AMPARs may serve as an ideal gain-sensing element calibrating eCB tone to corticolimbic input. Although not tested directly, our findings also suggest that LFS increases eCB production beyond tonic levels, as AEA signaling contributes to both tonic and phasic CB1R activity. An activity-dependent rise in local eCB production has vast functional consequences on which glutamatergic inputs to the NAc are constrained by feedforward inhibition. Moreover, eCB-dependent neuromodulation at this synaptic locus, rather than at GABAergic PV-IN-to-MSN synapses, may serve as a substrate for adaptive reward learning (Winters et al., 2012).
A critical question posed by our data is whether the somatodendritic properties of glutamatergic synapses onto PV-INs differ across inputs. Indeed, CP-AMPAR blockade with NASPM blocked excitatory transmission by ~50%. Although a probable hypothesis is that CP-AMPARs are expressed on PV-INs alongside GluA2-containing Ca2+-impermeable AMPARs, an alternative possibility is that CP-AMPARs are enriched at distinct inputs. Inputs with greater innervation ratios relative to MSNs, for example, may possess distinct synaptic properties that differentially drive feedforward inhibition. Thus, superfusion of NASPM may serve as a pharmacological means to isolate CP-AMPAR-poor synapses onto PV-INs. Despite the prospect of inputs with distinct stoichiometric profiles of AMPARs, our data support a cohesive model of plasticity in which CPAMPAR- and CB1R-dependent LTD are elicited by LFS at overlapping synapses. Ongoing efforts are required to characterize afferent-specific differences in feedforward microcircuit function and how these properties regulate reward-related behavior.
Conclusions
PV(+)-INs in the NAc are fast-spiking GABAergic neurons embedded within a feedforward inhibitory network that coordinates functional NAc circuit output. We report that PV(+)-INs, unlike D1(+) an D1(−) MSNs, are enriched in GluA2-lacking CP-AMPARs that confer specialized synaptic properties to feedforward glutamatergic synapses. Importantly, we find that Ca2+ influx through CP-AMPARs triggers (1) eCB-dependent LTD by presynaptic CB1R and (2) tonic eCB signaling by retrograde AEA signaling. These findings provide functional evidence that CP-AMPARs are linked to the release of eCBs that gate tonic-and phasic-dependent shifts in glutamatergic synaptic strength. Understanding how CP-AMPARs contribute to synaptic function may lead to the development of therapeutic tools targeting IN microcircuits for the treatment of maladaptive motivational states, such as addiction and depression.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Brad Grueter (brad.grueter@vumc.org).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Raw electrophysiological traces and behavior videos have not been deposited in a public repository but are available upon request. No custom code was developed.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Mice were bred and housed at Vanderbilt University Medical Center in accordance with IACUC. Male mice 8–16 weeks of age were used for all electrophysiological and in vivo experiments. Experimental mice were housed in groups of 3–5/cage on a 12-hr light-dark cycle with ad lib access to standard food and water. Breeding cages were given 5LOD chow (PicoLab ®, 28.7% protein, 13.4% fat, 57.9% carbohydrate) to improve litter viability. For all electrophysiological experiments examining PV(+)-INs, Cre-induced STOPfl/fl-tdTomato mice (Ai9, Gt(ROSA)26Sortm9(CAG-tdTomato)Hze) obtained from Jackson Laboratory (Stock No.: 007909) were crossed with PV-IRES-Cre (PVCre, Pvalbtm1(cre)Arbr/J, Stock No.: 008069), generating PVCre-tdTomatofl/fl (PVtdT) mice. For all experiments examining D1 and D2 MSN physiology, C57BL/6J mice were bred to harbor a bacterial artificial chromosome (BAC) carrying the tdTomato fluorophore under control of the Drd1a (D1 receptor) promoter. In a subset of experiments, PVtdT mice received either a single home cage injection of saline or cocaine HCl (15 mg/kg) administered intraperitoneal (IP).
METHOD DETAILS
Electrophysiology
Whole-cell patch-clamp electrophysiological recordings were obtained in acute brain slice preparations from PVtdT and D1tdTomato BAC transgenic mice. Mice were euthanized under isoflurane anesthesia after which parasagittal slices (250 μM) containing the NAc core were prepared from whole brain tissue using a Leica Vibratome in oxygenated (95% O2; 5%CO2) ice-cold N-methyl-D-glucamine (NMDG)-based solution (in mM: 2.5 KCl, 20 HEPES, 1.2 NaH2PO4, 25 Glucose, 93 NMDG, 30 NaHCO3, 5.0 sodium ascorbate, 3.0 sodium pyruvate, 10 MgCl2, and 0.5cCaCl2-2H2O). Slices were then recovered in NMDG-based recovery solution for 10–15-min at 30–32c°C before being transferred to a chamber containing artificial cerebral spinal fluid (ACSF, in mM: 119 NaCl, 2.5 KCl, 1.3 MgCl2-6H2O, 2.5 CaCl2-2H2O, 1.0 NaH2PO4-H2O, 26.2 NaHCO3, and 11cglucose; 287–295 mOsm). All experiments were performed using a Scientifica Slicescope Pro System with continuously perfused 28–32°C ACSF at 2cmL/min. PV-INs or MSNs in the NAc core were visualized using Scientifica PatchVision software and patched with 3–6 MΩ recording pipettes. (P1000 Micropipette Puller). For current-clamp recordings, experiments were performed in K+-based intracellular solution: (in mM: 135 K+-gluconate, 5 NaCl, 2 MgCl2, 10 HEPES, 0.6 EGTA, 3 Na2ATP, 0.4 Na2GTP; 290 mOsm). For voltage-clamp recordings, a Cs+-based intracellular solution was used (in mM: 120 CsMeSO3, 15 CsCl, 8 NaCl, 10 HEPES, 0.2 EGTA, 10 TEA-Cl, 4.0 Mg-ATP, 0.3 Na-GTP, 0.1 spermine, and 5.0c QX 314 bromide). In PVtdT or D1tdTomato mice, PV(+)-INs and D1(+) and D1(−) (putative D2) MSNs were differentiated according to the expression of the red tdTomato fluorophore via 530 nm LED light. D1(−) MSNs were distinguished from interneuron cell types based on morphological (size, shape) and biophysical properties (e.g., capacitance, membrane resistance, and AMPAR decay kinetics).
For voltage-clamp recordings, electrically evoked excitatory postsynaptic currents (eEPSCs) were obtained at a command voltage of −70 mV and isolated by incorporating GABAAR antagonist, picrotoxin (PTX, 50cμM), into the ACSF bath. To obtain the current-voltage (I-V) function of AMPAR-mediated EPSCs, AMPAR-mediated EPSCs were isolated by also including NMDAR antagonist, APV (50cμM), into the ACSF bath, though NMDAR-mediated EPSCs contribute minimally to EPSC amplitude at −70 mV. EPSC decay kinetics were obtain from t1/2 obtained time, T, following peak EPSC amplitude. In experiments examining local glutamatergic transmission, a bipolar electrode was placed at the corticoaccumbens interface and stimulated at 0.1 Hz. Paired pulse ratios (PPR) were obtained within-experiment by delivering two 0.3-ms duration pulses with a 50-ms interstimulus interval and calculating the amplitude ratio of the second eEPSC to the first eEPSC (eEPSC2/eEPSC1) at the indicated time-point. sEPSC analysis was performed with Clampfit 10.4 using a stringent best-fit template obtained from preliminary 10-min recording bouts in D1(+) and D1(−) MSNs. Each recording bout yielded a rise/day time (≤3-ms) and amplitude (≥5 pA) selection criteria that was reflected in the overall template score. For current-clamp recordings, cells were permitted 5-min after entering whole-cell configuration to equilibrate to the intracellular dialysate, after which a depolarizing plateau potential was established to maintain cells at approximately −70 mV. To assess intrinsic membrane excitability, action potentials (APs) were elicited in PV(+)-INs or MSNs following −50 pA current steps increasing from 400 to 400 pA with an 800-ms step duration. Membrane resistance and series resistance (RS) were monitored continuously during all experiments, with >20% change in RS resulting in the omission of that experiment.
Behavior
For microinfusion studies, bilateral guide cannulas (26 gauge, cut to 3mm length, 2mm center to center distance, C235GS-5–2.0/ SPC- Plastics One, 22 Roanoke VA) were implanted above the NAc core (AP: 1.45, ML: ± 1.00, DV: 3.00). Infusion guides were secured into place using dental cement and then fitted with a dummy cannula (C235DCS-5/SPC, Plastics One, Roanoke VA) and dust cap (303DC/1 Plastics One, Roanoke VA). Animals were allowed to recover for at least 5 days before habituation for behavioral assay.
Animals were habituated to handling for 3 consecutive days in which they were restrained for increasing amounts of time (30 s, 60 s, 120 s). The following 2 consecutive days, animals were restrained for 120 s to mimic time required to perform infusion and then habituated to activity chambers for 1h (ENV-510; Med Associates). On test day, a bilateral infusion cannula (4mm cut length; C235IS-5/SPC, Plastics One, Roanoke VA) was connected to a 1 uL syringe (#7001, Hamilton Company, Reno, NV) by polyethylene tubing (0.46 mm in diameter; Plastics One). NASPM was microinfused at a dose of 0 (saline) or 8 μg/ml at a rate of 0.25 μL per hemisphere over 60 s and was allowed to absorb for 60 s before cannula was slowly removed (PMID: 31874107). Animals were immediately placed into activity chambers for assessment of basal locomotor activity.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data analysis
Electrophysiological experiments were analyzed using Clampfit 10.4 and GraphPad Prism v7.0. Changes in baseline EPSC amplitude, coefficient of variance (CV), and PPR were calculated by comparing mean values during 5cmin intervals specified in each time-course to baseline PPR and CV values. A depression was defined as a significant difference in eEPSC amplitude from baseline calculated during the time interval specified in the recording. After obtaining each dataset, Shapiro-Wilk tests were performed to assess normality. Data depicted in Figures were determined to be normally distributed. Thus, paired or unpaired t tests were used to analyze statistical differences between datasets. Sidak’s post hoc analyses were used for analyses requiring multiple comparisons. Power analyses were performed with preliminary data during the acquisition of each new dataset. The sample size obtained from each power analysis calculation was then compared to sample sizes reported in the literature for similar experiments. Errors bars depicted in figures represent SEM. For all analyses, α was set as 0.05, with p values < α indicating a statistically significant difference.
ADDITIONAL RESOURCES
No unpublished custom code, software, or algorithm was generated in this study.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant ProteinsPubchem CID | ||
| 1-Naphthyl acetyl spermine (NASPM) trihydrochloride | Sigma-Aldrich, St. Louis, MO | 16219727 |
| WIN 55,212–2 mesylate | Tocris/Bio-Techne, Minneapolis, MN | 6604176 |
| AM251 | Tocris/Bio-Techne, Minneapolis, MN | 2125 |
| Picrotoxin | Sigma-Aldrich, St. Louis, MO | 31304 |
| NBQX disodium salt | Tocris/Bio-Techne, Minneapolis, MN | 6098006 |
| LY341495 | Tocris/Bio-Techne, Minneapolis, MN | 9819927 |
| (RS)-DHPG | Tocris/Bio-Techne, Minneapolis, MN | 108001 |
| BAPTA | Tocris/Bio-Techne, Minneapolis, MN | 104751 |
| EGTA | Tocris/Bio-Techne, Minneapolis, MN | 6207 |
| DO-34 | Glixx Laboratories | GLXC-09757 |
| D-AP5 | Tocris/Bio-Techne, Minneapolis, MN | 135342 |
| Experimental Models: Organisms/Strains | ||
| B6.Cg-Pvalb-T2A-Cre-D | The Jackson Laboratory, Sacramento, CA | Stock No: 022863 |
| Mouse: B6.Cg-Tg(Drd1a-tdTomato) 6Calak/J | The Jackson Laboratory, Sacramento, CA | Stock No: 016204 |
| Mouse: B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J | The Jackson Laboratory, Sacramento, CA | Stock No: 007909 |
| Software and Algorithms | ||
| pClamp 10 | Molecular Devices, 1311 Orleans Drive, Sunnyvale, CA | http://www.moleculardevices.com |
| EthoVision XT | Noldus Information Technology, Leesburg, VA | http://www.noldus.com |
| Prism | GraphPad Software, Inc. 7825 Fay Avenue, Suite 230 La Jolla, CA | https://www.graphpad.com:443/scientific-software/prism/ |
Highlights.
Synapses onto PV-INs in the NAc core express Ca2+-permeable AMPA receptors (CP-AMPARs)
Ca2+ influx through CP-AMPARs on PV-INs triggers long-term depression mediated by CB1 receptors
CP-AMPARs promote tonic endocannabinoid signaling, inhibiting glutamate release
Blockade of CP-AMPARs in the NAc core disinhibits locomotor output
ACKNOWLEDGMENTS
This study was supported by National Institute on Drug Abuse (NIDA) grant R01DA040630 (to B.A.G.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Burke DA, Rotstein HG, and Alvarez VA (2017). Striatal Local Circuitry: A New Framework for Lateral Inhibition. Neuron 96, 267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng L-J, Shaham Y, et al. (2008). Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature 454, 118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deroche MA, Lassalle O, Castell L, Valjent E, and Manzoni OJ (2020). Cell-Type- and Endocannabinoid-Specific Synapse Connectivity in the Adult Nucleus Accumbens Core. J. Neurosci 40, 1028–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermann E, and Jonas P (2011). How the “slow” Ca(2+) buffer parvalbumin affects transmitter release in nanodomain-coupling regimes. Nat. Neurosci 15, 20–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrario CR, Loweth JA, Milovanovic M, Ford KA, Galiñanes GL, Heng L-J, Tseng KY, and Wolf ME (2011). Alterations in AMPA receptor subunits and TARPs in the rat nucleus accumbens related to the formation of Ca2+-permeable AMPA receptors during the incubation of cocaine craving. Neuropharmacology 61, 1141–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzjohn SM, and Collingridge GL (2002). Calcium stores and synaptic plasticity. Cell Calcium 32, 405–411. [DOI] [PubMed] [Google Scholar]
- Francis TC, Yano H, Demarest TG, Shen H, and Bonci A (2019). High-Frequency Activation of Nucleus Accumbens D1-MSNs Drives Excitatory Potentiation on D2-MSNs. Neuron 103, 432–444.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziane NM, Sun S, Wright WJ, Jang D, Liu Z, Huang YH, Nestler EJ, Wang YT, Schlüter OM, and Dong Y (2016). Opposing mechanisms mediate morphine- and cocaine-induced generation of silent synapses. Nat. Neurosci 19, 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueter BA, Brasnjo G, and Malenka RC (2010). Postsynaptic TRPV1 triggers cell type-specific long-term depression in the nucleus accumbens. Nat. Neurosci 13, 1519–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Gan J, and Jonas P (2014). Interneurons. Fast-spiking, parvalbumin+ GABAergic interneurons: from cellular design to microcircuit function. Science 345, 1255263. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, and Malenka RC (2005). Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J. Neurosci 25, 10537–10545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalanne T, Oyrer J, Farrant M, and Sjöström PJ (2018). Synapse Type-Dependent Expression of Calcium-Permeable AMPA Receptors. Front. Synaptic Neurosci 10, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BR, Ma Y-Y, Huang YH, Wang X, Otaka M, Ishikawa M, Neumann PA, Graziane NM, Brown TE, Suska A, et al. (2013). Maturation of silent synapses in amygdala-accumbens projection contributes to incubation of cocaine craving. Nat. Neurosci 16, 1644–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-H, Ledri M, Tóth B, Marchionni I, Henstridge CM, Dudok B, Kenesei K, Barna L, Szabó SI, Renkecz T, et al. (2015). Multiple Forms of Endocannabinoid and Endovanilloid Signaling Regulate the Tonic Control of GABA Release. J. Neurosci 35, 10039–10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim BK, Huang KW, Grueter BA, Rothwell PE, and Malenka RC (2012). Anhedonia requires MC4R-mediated synaptic adaptations in nucleus accumbens. Nature 487, 183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SQ, and Cull-Candy SG (2000). Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature 405, 454–458. [DOI] [PubMed] [Google Scholar]
- Malenka RC, and Bear MF (2004). LTP and LTD: an embarrassment of riches. Neuron 44, 5–21. [DOI] [PubMed] [Google Scholar]
- Manz KM, Baxley AG, Zurawski Z, Hamm HE, and Grueter BA (2019). Heterosynaptic GABAB receptor function within feedforward microcircuits gates glutamatergic transmission in the nucleus accumbens core. J. Neurosci 39, 9277–9293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen W, Szabo A, Somogyi J, Somogyi P, and Lamsa KP (2010). Cell type-specific long-term plasticity at glutamatergic synapses onto hippocampal interneurons expressing either parvalbumin or CB1 cannabinoid receptor. J. Neurosci 30, 1337–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hare JK, Li H, Kim N, Gaidis E, Ade K, Beck J, Yin H, and Calakos N (2017). Striatal fast-spiking interneurons selectively modulate circuit output and are required for habitual behavior. eLife 6, e26231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascoli V, Terrier J, Espallergues J, Valjent E, O’Connor EC, and Lüscher C (2014). Contrasting forms of cocaine-evoked plasticity control components of relapse. Nature 509, 459–464. [DOI] [PubMed] [Google Scholar]
- Pisansky MT, Lefevre EM, Retzlaff CL, Trieu BH, Leipold DW, and Rothwell PE (2019). Nucleus Accumbens Fast-Spiking Interneurons Constrain Impulsive Action. Biol. Psychiatry 86, 836–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi J, Zhang S, Wang H-L, Barker DJ, Miranda-Barrientos J, and Morales M (2016). VTA glutamatergic inputs to nucleus accumbens drive aversion by acting on GABAergic interneurons. Nat. Neurosci 19, 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schall TA, Wright WJ, and Dong Y (2020). Nucleus accumbens fast-spiking interneurons in motivational and addictive behaviors. Mol. Psychiatry, Published online February 18, 2020 10.1038/s41380-020-0683-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scudder SL, Baimel C, Macdonald EE, and Carter AG (2018). Hippo-campal-Evoked Feedforward Inhibition in the Nucleus Accumbens. J. Neurosci 38, 9091–9104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler-Llavina GJ, and Sabatini BL (2006). Synapse-specific plasticity and compartmentalized signaling in cerebellar stellate cells. Nat. Neurosci 9, 798–806. [DOI] [PubMed] [Google Scholar]
- Tepper JM, Koós T, Ibanez-Sandoval O, Tecuapetla F, Faust TW, and Assous M (2018). Heterogeneity and Diversity of Striatal GABAergic Interneurons: Update 2018. Front. Neuroanat 12, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trouche S, Koren V, Doig NM, Ellender TJ, El-Gaby M, Lopes-Dos-Santos V, Reeve HM, Perestenko PV, Garas FN, Magill PJ, et al. (2019). A Hippocampus-Accumbens Tripartite Neuronal Motif Guides Appetitive Memory in Space. Cell 176, 1393–1406.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner BD, Kashima DT, Manz KM, Grueter CA, and Grueter BA (2018a). Synaptic Plasticity in the Nucleus Accumbens: Lessons Learned from Experience. ACS Chem. Neurosci 9, 2114–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner BD, Rook JM, Lindsley CW, Conn PJ, and Grueter BA (2018b). mGlu1 and mGlu5 modulate distinct excitatory inputs to the nucleus accumbens shell. Neuropsychopharmacology 43, 2075–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twomey EC, Yelshanskaya MV, Grassucci RA, Frank J, and Sobolevsky AI (2017). Channel opening and gating mechanism in AMPA-subtype glutamate receptors. Nature 549, 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twomey EC, Yelshanskaya MV, Vassilevski AA, and Sobolevsky AI (2018). Mechanisms of Channel Block in Calcium-Permeable AMPA Receptors. Neuron 99, 956–968.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Gallegos DA, Pogorelov VM, O’Hare JK, Calakos N, Wetsel WC, and West AE (2018). Parvalbumin Interneurons of the Mouse Nucleus Accumbens are Required For Amphetamine-Induced Locomotor Sensitization and Conditioned Place Preference. Neuropsychopharmacology 43, 953–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder DG, and Sweatt JD (2001). Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat. Rev. Neurosci 2, 461–474. [DOI] [PubMed] [Google Scholar]
- Winters BD, Krüger JM, Huang X, Gallaher ZR, Ishikawa M, Czaja K, Krueger JM, Huang YH, Schlüter OM, and Dong Y (2012). Cannabinoid receptor 1-expressing neurons in the nucleus accumbens. Proc. Natl. Acad. Sci. USA 109, E2717–E2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME (2016). Synaptic mechanisms underlying persistent cocaine craving. Nat. Rev. Neurosci 17, 351–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright WJ, Schlüter OM, and Dong Y (2017). A Feedforward Inhibitory Circuit Mediated by CB1-Expressing Fast-Spiking Interneurons in the Nucleus Accumbens. Neuropsychopharmacology 42, 1146–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright WJ, Graziane NM, Neumann PA, Hamilton PJ, Cates HM, Fuerst L, Spenceley A, MacKinnon-Booth N, Iyer K, Huang YH, et al. (2020). Silent synapses dictate cocaine memory destabilization and reconsolidation. Nat. Neurosci 31792465 23, 32–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Yan Y, Li K-L, Wang Y, Huang YH, Urban NN, Nestler EJ, Schlüter OM, and Dong Y (2017). Nucleus accumbens feedforward inhibition circuit promotes cocaine self-administration. Proc. Natl. Acad. Sci. USA 114, E8750–E8759. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw electrophysiological traces and behavior videos have not been deposited in a public repository but are available upon request. No custom code was developed.