

Summary
High-throughput cytostatic and cell death assays are a critical component of pharmacological screens and mechanism-based interrogations into cellular biology. We developed a method for single-cell and population-level analyses using real-time kinetic labeling (abbreviated “SPARKL”) with non-toxic fluorescent probes and high-content live-cell imagers. The protocols herein detail the steps, specifics, and suggested utilization of the SPARKL method within several “label-and-go” zero-handling workflows.
For complete details on the use and execution of this protocol, please refer to Gelles et al. (2019).
Graphical Abstract
Highlights
-
•
An optimized protocol for capturing the kinetics of cell death in real time
-
•
Multi-parametric analyses of cell death kinetics for comparative studies
-
•
Label-and-go multiplex workflows to characterize cell proliferation and death
We present a set of protocols for high-throughput studies quantifying cytostatic and cytotoxic effects on cultured cells in response to perturbagens. Utilizing live-cell imagers, we detail label-and-go workflows with variants for simultaneously quantifying cell proliferation and death, characterizing cell death kinetics and pathways, and tracking sequential labeling of individual cells. These protocols for single-cell and population-level analyses using real-time kinetic labeling (abbreviated “SPARKL”) were adapted from Gelles et al. (2019).
BEFORE YOU BEGIN
Determine the Appropriate Variant of SPARKL
Timing: 10 min
SPARKL is a versatile method built upon live-cell microscopy and detection of fluorescent reporters. Investigators can characterize different aspects of cell death biology by selecting specific reporters and methods of analysis.
-
1.
Variant A: Simultaneous measurement of cell proliferation and death
Note: This Variant quantifies the number of total cells in addition to the dying cells. Use this Variant to normalize cell death data for total cell number or measure treatment-induced cytostatic effects.
-
2.
Variant B: Single- or dual-labeling method for analysis of cell death kinetics
Note: This Variant quantifies dying cells by measuring distinct cell death phenotypes. Individually, or in tandem, reporters in this Variant characterize cell death kinetics for comparative analyses.
-
3.
Variant C: Sequential labeling studies to study penetrance of a cell death response
Note: This Variant uses external software to track sequential labeling of individual cells for population analysis and statistics.
Determine Scan Parameters for the Experiment
-
4.
Objective magnification: a 10× objective is suggested as it will capture a large surface area while retaining resolution to capture cell morphology.
-
5.
Length of experiment: determined by cellular responses and treatments. 12−48 hours are suggested for cell death studies; 48−96 hours are suggested for proliferation studies.
-
6.
Scan frequency: determined by experimental question and anticipated results. Scan intervals are suggested to be 1−2 hours for cell death studies and 4−6 hours for proliferation studies.
-
7.
Plate format: we strongly recommend designing your experiments for a 96-well plate. Smaller wells prevent excessive consumption of reagents, reduce uneven cell distribution within the well, and increase throughput.
-
8.
Frames per well: we strongly recommend acquiring 1 frame per well. Additional frames will not significantly alter the data or statistical power if cells are seeded at appropriate densities. Utilizing technical replicates is a preferred design.
Optimize Cell Seeding Density
SPARKL using an IncuCyte® imager is optimized and validated for immortalized and primary adherent cell lines. Due to the autofocus mechanism, cultures growing in 3D will require additional steps to optimize and may not be suitable with this protocol. Suspension cells which have been adhered to the bottom of the tissue culture plate can be analyzed using SPARKL with varying success. Refer to the limitations section for details regarding which cell types are suitable for this protocol.
-
9.
Cell death assays are best accomplished when wells are 50−85% confluent. When cells are too confluent, it becomes difficult to segment fluorescent events and can result in miscounts; too few cells can affect statistical power of the data.
-
10.
Set up a preliminary experiment with cells at varying densities and monitor their confluency for the length of time of your intended experiment. Select the density which keeps your cellular population from becoming over-confluent but does not require sparse seeding.
-
11.
Optional: For experiments using suspension cell lines, coat the bottom of the tissue culture plate with a binding agent (e.g., collagen, poly-lysine) to adhere the cells. This will prevent them from leaving the focal plane or migrating during scans.
CRITICAL: Attempting to scan cells which are too sparse can affect the autofocus mechanism of an IncuCyte® imager. Ensure that cells are seeded at appropriate densities and evenly distributed within the well.
Fluorescent Reporters and Channels
-
12.
Select reporters for each fluorescent channel
-
a.
(Variant A) Green channel: SYTO21; Red channel: Annexin V-594 or YOYO3
-
b.
(Variant B/C) Green channel: Annexin V-488; Red channel: YOYO3
-
13.
Set up a preliminary experiment using a titration of fluorescent reporters to determine the lowest concentration necessary to obtain robust and sustained signal. Refer to the “Create Processing Definition” section below.
Note: Suggested concentrations of fluorescent reporters are usually optimized for flow cytometry where labeling occurs in larger volumes and is subsequently washed out. It is common that you will use reporters at much lower concentrations since they will be incubated in the culture media.
Optional: In lieu of commercially available Annexin V, recombinant Annexin V can be expressed, purified, and labeled in-house (detailed protocols can be found in Logue et al. 2009 and in the Materials and Methods of Gelles et al. 2019).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| ABT-737 | Selleck Chemicals | Cat#: S1002 |
| Annexin V-488/594 | Thermo Fisher Scientific | Cat#: A13201, A13203 |
| Cycloheximide | N/A | N/A |
| mTNFα | Peprotech | Cat#: 315-01A |
| Necrostatin-1 | Selleck Chemicals | Cat#: S8037 |
| SYTO™ 21 | Thermo Fisher Scientific | Cat#: S7556 |
| YOYO™-3 Iodide | Thermo Fisher Scientific | Cat#: Y3606 |
| zVAD-fmk | Selleck Chemicals | Cat#: S7023 |
| Recombinant DNA | ||
| pProEx.Htb.annexinV | Seamus Martin (Trinity College, Dublin, Ireland) | NM_001154; Logue et al., 2009 |
| Software and Algorithms | ||
| Excel v16.16.9 | Microsoft | N/A |
| ImageJ/Fiji v2.0.0-rc-69/1.52n | National Institutes of Health | Schindelin et al., 2012, Schneider et al., 2012 |
| IncuCyte® ZOOM v2018A | Essen Biosciences | N/A |
| Prism v8.1.1 | Graphpad Software | N/A |
| SPARKL_pipeline | Jerry Chipuk (Icahn School of Medicine at Mount Sinai) | https://doi.org/10.5281/zenodo.3458574; Gelles et al., 2019 |
| Other | ||
| The code utilized in Variant C | Jerry Chipuk (Icahn School of Medicine at Mount Sinai) | https://github.com/ChipukLab/SPARKL_pipeline.git (https://doi.org/10.5281/zenodo.3458574) under the GNU General Public License v3.0. |
MATERIALS AND EQUIPMENT
-
•
IncuCyte® ZOOM or S3, equipped with 10× or 20× objective
-
•
(Variant A) Annexin V conjugated to Alexa Fluor 594 (purchased or self-generated)
-
•
(Variant A) SYTO21 green fluorescent nucleic acid stain
-
•
(Variant B/C) Annexin V conjugated to Alexa Fluor 488 (purchased or self-generated)
-
•
(Variant B/C) YOYO3
-
•
Microsoft Excel and/or Graphpad Prism
-
•
(Variant C) ImageJ/Fiji
-
•
(Variant C) Python 3.x
-
•
(Variant C) SPARKL_pipeline.zip (Gelles and Chipuk, 2019)
Alternatives:
-
•
The IncuCyte® brand imager can be replaced by a similar high-content live-cell imager, such as the IN Cell Analyzer (GE) or Cytation Multi-Mode Reader (BioTek/Agilent). However, the sections describing image analysis and object detection will no longer be applicable and users will have to use either ImageJ or the built-in software of that imager.
-
•
YOYO3 can be replaced by a suitable, non-toxic cell-impermeant viability dye (e.g., SYTOX®)
-
•
Low riboflavin culture media (<0.2 mg/L) can reduce green background fluorescence
Note: This method has been validated in several immortalized cell lines and primary cells. Most culturable adherent cell types should be compatible with this method provided they grown in a monolayer. While not validated herein, adherent cells requiring specific extracellular matrices should be compatible provided the cells seed in a single focal plane. Suspension cells and cells growing in 3D cultures have not been validated and may not be compatible with this method.
Note: In addition to phase contrast, an IncuCyte® imager has a green LED (Ex: 460 [440,480] nm; Em: 524 [504,544] nm) and a red LED (Ex: 585 [565,605] nm; Em: 635 [625,705] nm). Therefore, choose fluorescent reporters which are optimized for these excitation and emission ranges. Avoid using common dyes which fluoresce in the deep-red range (e.g., Draq7) or which may have adverse effects on cells with prolonged incubation (e.g., 7-AAD or propidium iodide).
STEP-BY-STEP METHOD DETAILS
Seed Cells
This section describes how cells should be prepared.
-
1.
Trypsinize, or otherwise detach, the cells from the culture vessel and determine the concentration of cells.
-
2.
Dilute cells to an appropriate concentration for optimal seeding density. Ideally, plate cells at an approximate density of 30−50%, based on the proliferation rate of the cells and the duration of your planned experiment.
Note: Assuming a 96-well plate format and 100 μL cultured per well, the seeding stock is generally in the range of 1−5×104 cells/mL (1000−5000 cells/well).
-
3.
Optional: Aliquot, incubate, and aspirate a binding agent to the plate (e.g., collagen, poly-lysine, laminin) for cell types which do not readily adhere to tissue culture plates.
-
4.
Use a multichannel pipet and reagent reservoir to aliquot the seeding stock into the plate wells. We have found that a multichannel pipet is reliable for seeding an equivalent number of cells in each well and reducing variation.
Figure 1.

Pipet-Induced Vortices Will Result in Uneven Seeding of Cells
(A) Avoid ejecting against the corner of a well as this will cause cells to concentrate into the center of the well.
(B) Avoid ejecting at an angle as the wall curvature will generate a circular vortex concentrating cells into the center of the well.
(C) Place the pipet tip perpendicular to the wall of the well and gently eject the cell suspension to achieve an even distribution of cells.
-
5.
Permit the cells to reattach and reach homeostasis for 16−24 hours.
Pause Point: The following steps will be conducted the next day once the cells have reattached.
Treat Cells
This section describes preparing experimental conditions for the SPARKL workflow.
-
6.
Prepare 2× labeling media stock. Measure a volume of complete media sufficient to aliquot 50 μL for each well containing cells (plus 5−10% excess for pipetting errors). Add fluorescent reporters to this stock to create a 2× solution.
-
7.
Prepare 2× treatment media stock. For each treatment condition, add perturbagens to a volume of culture media sufficient to aliquot 50 μL for sample wells (plus 5−10% excess for pipetting errors).
Note: Every experiment should include a "rapid death" condition; this condition will report maximal death kinetics and signal for analysis. For apoptotic studies, co-treatment of either TNFα with cycloheximide or cycloheximide with ABT-737 (an inhibitor to anti-apoptotic BCL-2 family proteins) is well suited.
-
8.
Aspirate culture media off of cells in small batches. We recommend diluting the vacuum force by attaching pipet tips to the aspirator, such as a P10 stacked on a P200 stacked on a P1000, which will minimize loss of cells during aspiration (Figure 2A). Cells do not need to be washed prior to adding labeling or treatment media.
Figure 2.

Replace Culture Media with Media Containing Labels and Perturbagens
(A) In small batches, aspirate the culture media from wells. Stacking pipet tips will dilute the vacuum force and prevent cell loss.
(B) Prepare phenol red-free labeling media with fluorescent labels and use a multichannel to aliquot 50% of the final volume into aspirated wells. Do this step rapidly to avoid cell desiccation.
(C) Prepare phenol red-free treatment media containing the treatments for your experiment. Aliquot 50% of the final volume to the appropriate wells on top of labeling media.
-
9.
Immediately following aspiration, aliquot the labeling media onto the cells to prevent them from desiccating. Using a multichannel pipet can reduce the time that cells are not in media (Figure 2B).
-
10.
Add treatment media to the appropriate wells. Avoid bubbles on the surface of the media (Figure 2C).
-
11.
Place the plate in the IncuCyte® imager and allow the plate to return to temperature prior to scanning (approximately 30 minutes). When the plate is colder than the temperature of the incubator, condensation will form on the plastic and will hinder autofocusing.
-
12.
Add the plate to the IncuCyte® software.
-
a.
Add appropriate vessel to the corresponding tray.
-
b.
Define scanning pattern for plate.
-
c.
Select channels: these protocols make use of phase, green, and red.
-
d.
Define spectral unmixing: this parameter is variant specific.
-
e.
Define scan schedule and frequency.
-
f.
Optional: Apply real-time analysis job. Requires a processing definition (see next step).
Note: We suggest separating the labeling and treatment media stocks and aliquoting the labeling media first after removing the culture media. Since every well will use the same labels, it is faster to aliquot the labeling media, which will help prevent the cells from desiccating. Once the cells are covered in media, there is less of a rush to add the various treatments to the appropriate wells.
Note: If labeling the plate lid, avoid writing directly over the wells. Markings above the well may affect autofocusing and cause cells to appear blurry.
Note: Background fluorescence increases with media depth in the well. For a 96-well plate, we recommend always having a final volume of 100 μL per well, which should limit evaporation during the experiment while minimizing contributions to background signal.
Create Processing Definition
This part of the protocol is specific to an IncuCyte® brand live-cell imager; analogous real-time imagers may or may not have their own built-in software for analysis. Generating a "processing definition" (the specific parameters for automated object detection within collected images) is required the first time you conduct an experiment. Once generated, this processing definition can be applied to future experiments in real-time (or previous experiments, retroactively). For more comprehensive instructions, consult the IncuCyte® Technical Note documents.
Note: Processing definitions are specific to the cell type, fluorescent reporters, and magnification. Future experiments which change one of these components will require generating a new processing definition.
-
13.
Make an Image Stack of representative frames.
-
a.
Select frames from first and last timepoints as well as regularly spaced timepoints along the duration of your experiment.
-
b.
Include frames which represent the range of label strength and number of cells being labeled. Including a "rapid death" condition is highly recommended to gauge the range of signal intensity and set mask parameters appropriately.
-
14.
Define analysis parameters for image correction and object detection (Figure 3). Do this for each channel being analyzed.
Figure 3.

Processing Definitions Normalize Images and Detect Fluorescent Objects
(A) Top-hat processing determines background fluorescence and subtracts it across the image. This method for image processing is particularly useful for uneven background within the collected images.
(B) Pixels above a threshold fluorescence will be considered a positive object. Adjusting the edge sensitivity and area filter parameters will improve the quantification of objects while excluding artifacts.
Graphs and masks are illustrative and not quantitative.
-
a.
Select Top Hat.
-
i.
This setting considers the background trend across the image and subtracts it appropriately before determining fluorescent objects. This method results in more robust image normalization and object detection.
-
b.
Adjust the Radius.
-
i.
This determines the size of the sampling disk in Top Hat correction.
-
ii.
Smaller radii are better for background elimination and normalization (∼30 μm).
-
iii.
Avoid using a radius which is smaller than an object, which result in decreased sensitivity to detect positive fluorescent objects.
-
c.
Adjust the Threshold.
-
i.
This value determines the sensitivity for what is considered a positive object.
-
ii.
Find a value which eliminates masking background but does not exclude objects.
-
d.
Optimize Edge Segmentation.
-
i.
This setting controls the sensitivity for segmenting masks for adjacent objects.
-
ii.
We recommend setting this value at approximately -30 − -40.
-
e.
Define mask filters to optimize object detection and quantification.
-
i.
Area: set a minimum to gate out cell debris and non-specific labeling.
-
ii.
Mean Intensity: set a minimum to gate out objects with heterogeneous labeling. This may be useful if large areas of the cell body label, but typically is not necessary to include.
Optional: Include the "Overlap" metric to quantify the number of objects displaying double positivity. This metric can be a useful quality control step to assess how proficiently objects will be detected in each channel. Additionally, this metric can be used in downstream analyses.
EXPECTED OUTCOMES
This section describes methods to analyze, visualize, interpret, and compare data generated with the SPARKL workflow. The steps included in this section are not specific to an IncuCyte® brand imager and can be adopted downstream of any live-cell imaging system.
Images of cell labeling will be analyzed and object masks will be generated (Figure 4A). For each channel, export the quantification of objects. Import your data into graphing software and graph fluorescent events over time (Figure 4B). Use data from the "rapid death" control treatment to determine y-axis.
Figure 4.

Expected Outcomes and Analyses for Kinetic Data Collected with SPARKL
(A) SPARKL images cells at regular intervals and autonomously detects fluorescent objects within the images. (Top) Sample frames from MEFs instigated to die by TNFα and cycloheximide co-treatment labeling with Annexin V (green) followed by YOYO3 (red). (Bottom) Generated detection masks for Annexin V (magenta) and YOYO3 (cyan) fluorescent events. Scale bar, 50 μm.
(B) Quantification of detection masks from data as in (A) are visualized as continuous graphs expressing cell death over time.
(C) Area under the curve (AUC) calculated from (B) is a convenient method to illustrate differences between conditions.
(D) The first derivative of data from (B) plots the rate of cell death at each time point. Treatments can be compared by the timepoint of maximal rate of cell death.
(E) Non-linear fit analysis of data from (B) calculates parameters such as time-to-initial death (t′) and initial rate of death (RD).
Data and graphs reproduced from Gelles et al. (2019).
Parameterize Kinetic Data for Comparative Analyses (Optional)
SPARKL generates rich datasets by capturing the kinetics of cell death. Data points are not limited to reporting static cell death values at a single timepoint. Here, we summarize several methods for comparing conditions in your experiment (further detailed in Gelles et al., 2019).
-
•
Area under the curve (AUC).
This is the simplest parameterization method. Several metrics of the kinetic data are compressed into a single parameter for comparison. AUC can be calculated using Prism or a similar program (Figure 4C).
Note: AUC calculations are influenced by the amplitude of the curve, and therefore differential cell numbers in samples will affect this method of analysis. Only use this method in cells that have equivalent numbers (minimal proliferation differences) or that have been normalized as detailed in Variant A below.
-
•
Derivatization.
This parameterization method calculates the slope of kinetic data and provides the rate of death within the population. The derivative can be calculated using Prism or a similar program.
Derivatized kinetic data can be shown over time and the maximal value depicts the time when the largest number of cells labeled with a cell death marker (Figure 4D). Similarly, quartiles of population death can be identified. For comparisons, data can be show as bar graphs depicting the time to quartile or maximal death.
Data can also be shown as a histogram of cells dying over time. Data viewed this way is particularly useful for observing heterogeneity of cellular responses (depicted as "shelves" within the histogram).
-
•
Non-linear fit analysis.
This method fits your kinetic data and calculates parameters which can be compared across conditions. We have had the most success using a Lag One-Phase Exponential (LOPE) function for fit analyses. Fitting your data can be accomplished using Prism or a similar program.
Data fit with a LOPE function generate two parameters: lag phase time (t′) and initial rate of death (RD). These parameters can be compared individually or together across conditions (Figure 4E).
Note: LOPE functions assume that the time immediately following the lag phase is the greatest rate of death. This is not completely accurate and therefore comparison of the RD metric may not be informative in certain conditions.
Note: LOPE functions will not be able to fit data in conditions with little, or no, cell death.
Alternatives: Rates of cell death may be more accurately modeled by a sigmoidal fit analysis if there is sufficient data to establish the plateaus. Conditions with rapid cell death typically cannot be fit with sigmoidal fits.
QUANTIFICATION AND STATISTICAL ANALYSIS
This section will detail methods for multiplex analysis utilizing data from both fluorescent channels. These methods are not specific to an IncuCyte® brand imager and can be applied to data gathered and exported from any similar live-cell imaging system.
Variant A: Simultaneous Measurement of Cell Proliferation and Death
-
1.
Seed and treat cells as described above.
-
•
Green label: SYTO21
-
•
Red label: Annexin V-594 or YOYO3
-
2.
Apply your processing definition (either at run-time or retroactively) and analyze fluorescent events over time as detailed above.
-
3.
Import your data into graphing software and graph.
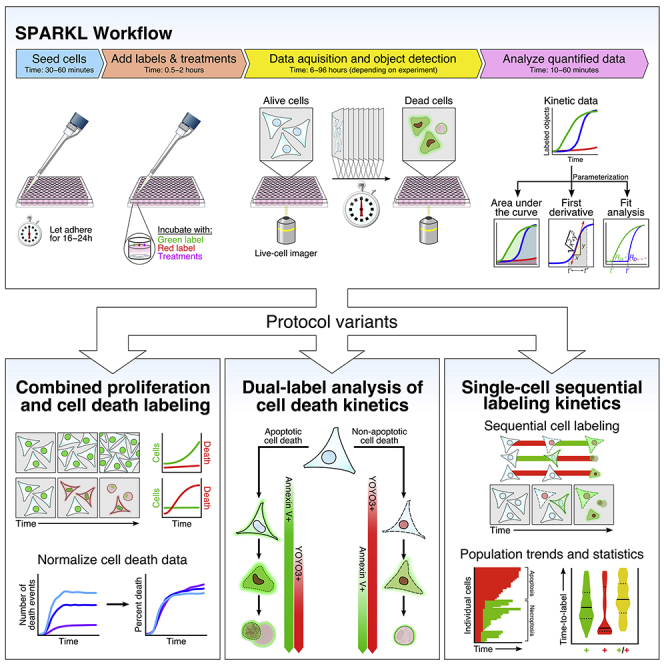
Note: Green channel data (SYTO21) will quantify cell number. If cells are proliferating, this value will increase over time and generate a proliferation curve (Figure 5A).
Figure 5.
Expected Outcomes of Simultaneously Quantifying Cell Proliferation and Death (Variant A)
(A) SYTO21 labels the cellular population for quantification of cell number and generation of proliferation curves.
(B) The number of cell death events varies in wells with dissimilar numbers of cells.
(C) Cell number quantified by SYTO21 can normalize cell death data from (B) into "percent death."
Data and graphs reproduced from Gelles et al. (2019).
Note: Red channel data (Annexin V or YOYO3) will quantify dying cells.
-
4.
Optional: Normalize cell death data to account for cell number variability.
Note: Number of events is dependent on cell number. Therefore, variation in cell number between replicates, conditions, or cell types can obfuscate data and make comparative analysis difficult (Hafner et al., 2016). This step can normalize cell death values to account for cell number variation due to differential seeding, proliferation, or treatment-specific cytostatic effects (Figure 5B).
Note: Depending on cells, treatment, imager sensitivity, and processing definitions, object detection and quantification may not be consistent across fluorescent channels. Therefore, we provide three different strategies to normalize cell death data and obtain a "percent death" metric (Figure 5C).
-
a.
Method 1: Divide red data by green data.
For a given well, divide the cell death data by the corresponding total cell data for each timepoint. Do this for each replicate before determining the mean or deviation.
Note: This is the simplest method and suited if the green and red channels detect and segment objects similarly.
Alternatives: If this method results in values much greater than 100% cell death, then the channels are not detecting equally and you should attempt Method 2 below.
-
b.
Method 2: Divide overlap data by green data.
Instead of using data from the red channel, use overlap data. Calculate as described in Method 1. This method will only normalize SYTO21-positive cells.
Note: This method is useful if there are more cell death objects than total cell objects. Disparate counts between channels can occur when object segmentation is complicated due cell clumping or changes in cellular morphology.
Alternatives: Loss of SYTO21 data over time will result in varying and inconsistent percentages. If cell number decreases over time, then attempt Method 3 below.
-
c.
Method 3: Divide red data by normalized green data.
Calculate the "percent death" values as described in Method 1 or Method 2. Calculate the normalization factor for the experiment: subtract the lowest SYTO21 value from the largest SYTO21 value and divide by 100. Consider values from the entire experiment. Divide each "percent death" by the normalization factor (these mathematical adjustments are detailed in Gelles and Chipuk, 2016).
Note: This method will scale cell death data by the relative minimums and maximums.
Variant B: Single- or Dual-Labeling Method for Analysis of Cell Death Kinetics
-
1.
Seed and treat cells as described above.
-
•
Green label: Annexin V-488
-
•
Red label: YOYO3
-
2.
Apply your processing definition (either at run-time or retroactively) and analyze fluorescent events over time as detailed above.
-
3.
Import your data into graphing software and graph.
Note: Green channel data (Annexin V) will quantify exposure of phosphatidylserine on the outer leaflet of the plasma membrane (Tait et al., 1989). Exposure of phosphatidylserine is a hallmark of apoptosis and demonstrates a cellular commitment to death (Koopman et al., 1994). Annexin V-positive cells can be considered "dying" cells (but biological processes are still being carried out).
Note: Red channel data (YOYO3) will quantify cells which have lost plasma membrane integrity by entering the cell and binding DNA. Plasma membrane permeability occurs in necroptosis and as a late-stage event in apoptosis. YOYO3-positive cells can be considered "dead" since the cell is no longer viable following plasma membrane permeabilization.
-
4.
Optional: Compare Annexin V and YOYO3 labeling to characterize cell death kinetics.
The SPARKL dual-label workflow utilizes two cell death reporters that quantify distinct phenotypes. Comparing Annexin V and YOYO3 kinetic data may suggest which cell death pathway activates following treatment of your perturbagen (Figure 6). Coupled with pathway-specific inhibitors or genetic cell models, investigators can use SPARKL to determine relevant cell death machinery as exampled below (and detailed in Gelles et el., 2019).
-
a.
Death receptor-mediated apoptosis
-
•
Annexin V: cells will exhibit rapid positivity prior to YOYO3 and contraction.
-
•
YOYO3: cells will exhibit positivity shortly after Annexin V.
-
•
Phenotype: cells will contract and form apoptotic bodies ("airbags").
-
•
ABT-737: may increase cell death kinetics if cells have sufficient Type II signaling.
-
•
zVAD-fmk: will decrease apoptotic cell death; if increased cell death is observed, cells may have engaged the necroptosis pathway.
-
b.
Mitochondrial pathway of apoptosis
-
•
Annexin V: cells will exhibit positivity prior to YOYO3 and contraction.
-
•
YOYO3: cells will exhibit positivity after Annexin V following a lag phase.
-
•
Phenotype: cells will contract and form apoptotic bodies ("airbags").
-
•
ABT-737: will increase cell death kinetics.
-
•
zVAD-fmk: will decrease apoptotic cell death.
Note: The length of time between Annexin V-labeling and YOYO3-labeling varies greatly between treatments and is indicative of underlying signal transduction. Characterizing this differential labeling can be quite informative and is detailed below in Variant C.
-
c.
Necroptosis
-
•
Annexin V: cells will exhibit slow positivity after YOYO3.
-
•
YOYO3: cells will exhibit positivity before Annexin V or phenotypic changes.
-
•
Phenotype: cells will not contract and will exhibit label positivity before any phenotypic changes.
-
•
zVAD-fmk: will induce necroptosis.
-
•
Necrostatin-1: will inhibit necroptosis by inhibiting RIP1.
-
d.
Ferroptosis
-
•
Annexin V: cells will exhibit positivity with similar kinetics as YOYO3.
-
•
YOYO3: cells will exhibit positivity with similar kinetics as Annexin V.
-
•
Phenotype: cells will contract.
-
•
Erastin: will induce ferroptosis.
-
•
ABT-737: will have no effect on cell death kinetics.
-
•
zVAD-fmk: will have no effect on cell death kinetics.
Figure 6.

Expected Outcomes for Differential Labeling of Dying Cells with Annexin V and YOYO3 (Variant B)
(A) Cells undergoing apoptosis will label with Annexin V prior to labeling with YOYO3. For single-label experiments, Annexin V is the preferred label as it more accurately reflects the biology of apoptosis.
(B) Cells undergoing non-apoptotic death (e.g., necroptosis) will label with YOYO3 prior to labeling with Annexin V. For single-label experiments, YOYO3 is the preferred label as it more accurately reflects the biology of necroptosis.
Variant C: Sequential Labeling Studies to Study Penetrance of a Cell Death Response
This Variant utilizes object masks generated from the IncuCyte® (or similar live-cell imager) and processes them with ImageJ/Fiji to cross reference objects between channels. The resulting data tracks the time to single- and double-positive signal for individual objects and can capture heterogeneous responses to perturbagen within isogenic cell populations (Roux et al., 2015). This Variant requires the "SPARKL_pipeline" script (see reference Gelles and Chipuk, 2019).
-
1.
Seed and treat cells as described above.
-
•
Green label: Annexin V-488
-
•
Red label: YOYO3
-
2.
Apply your processing definition (either at run-time or retroactively) and analyze fluorescent events over time as detailed above.
-
3.
Export the detection masks for the green, red, and overlap channels.
-
a.
For your conditions of interest, select a representative well from which to export the object masks. TIFF files are the preferred image type.
-
b.
Give the exported images a prefix that denotes the channel (e.g., "Green", "Red", or "Yellow"). Save these in a folder marked "Images".
-
4.
Run the "SPARKL_pipeline" script from the command line on your computer.
-
•
Mac: Open Terminal. Type "python3 " (space included). Either drag the script "main.py" into the window or type the path to the script. Hit enter to execute.
-
•
PC: Open cmd.exe. Type "py " (space included). Either drag the script "main.py" into the window or type the path to the script. Hit enter to execute.
-
5.
Provide the files when prompted and hit enter:
-
a.
The ImageJ/Fiji macro is part of the pipeline package and is called "IJmacro.ijm".
-
b.
Identify the folder ("directory") containing image files from Step 3, which will be called "Images".
-
c.
Type the prefix given to the files from the channel representing the first signal. For example, in an apoptosis experiment using Annexin V and YOYO3, Annexin V is expected to be the first signal and would be from the green channel.
-
d.
Type the prefix given to the files from the overlap channel.
-
e.
Identify the file which will be used to identify regions of interest (ROI). This file is the overlap ("Yellow") file from the last timepoint being analyzed.
-
6.
The script will open ImageJ/Fiji, convert the masks to binary, create image stacks, realign the images, compare with identified ROI, and record the frame at which the ROI became signal positive. Several files will be generated in the same folder containing the Images folder:
-
a.
"outputGY.csv" – this file contains the data for graphing. It contains the time to single- and double-positive signal for each ROI.
Note: the "GY" is taken from the prefixes you provided; if running a red analysis then the file will contain "RY".
-
b.
"resultsGREEN.csv", "resultsYELLOW.csv" – these files contain the status of each ROI at each slice of the image stack. The "mean" refers to the number of pixels within the ROI that are labeled. A "mean" that is greater than zero indicates labeling of the ROI.
Note: the file names will match the prefixes provided.
-
c.
"ROI.tif" – an image of ROI masks taken from the file provided in Step 5e.
-
d.
"ROIset.zip" – ROI Manager data which can be uploaded into ImageJ if needed.
-
e.
"stackGREEN.tif", "stackYELLOW.tif" – these files are the image stacks created by ImageJ which can be used for additional ImageJ workflows.
Note: the file names will match the prefixes provided.
-
7.
Graph the data from the "output" file.
-
a.
Use data from "t1" and "Δt" to make a graph in Prism (or equivalent software).
-
b.
Plot the "Δt" data set as a "stacked" dataset with "t1". This will generate bar graphs displaying the timespans of each signal in sequence (Figure 7A).
-
c.
The data may be more easily visualized as a violin plot. Graphing "t1", "t2", and "Δt" as violin plots will report distribution quartiles and be more informative for comparisons between labeling kinetics and treatment conditions (Figure 7B).
-
8.
Optional: The data represents the population of cells and therefore can be subjected to population statistics. Using a Mann-Whitney U test between labels or treatments can demonstrate statistical significance between population trends.
Note: It may be informative to run this Variant once using the data from the green channel and then again with data from the red channel. These analyses can reveal the sequential labeling kinetics on a population scale and may inform on underlying biology.
Figure 7.
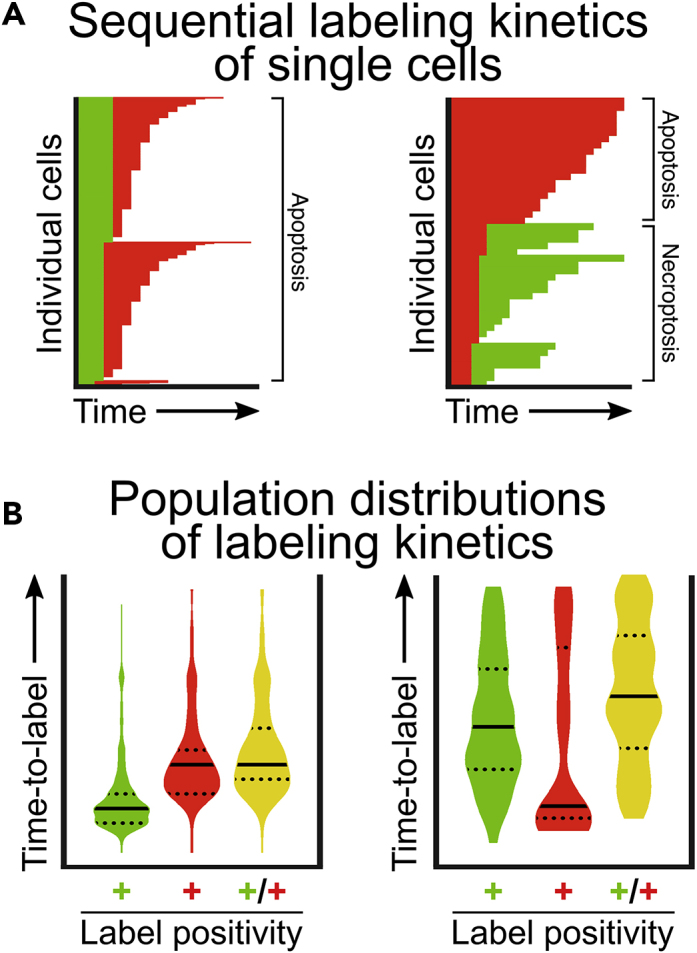
Expected Outcomes for Single-Cell Sequential Labeling and Population Analysis (Variant C)
(A) Tracking labeling kinetics for individual cells can characterize the mode of cell death and the heterogeneity of responses to a perturbagen.
(B) Individual cell data from (A) can be visualized as population distributions for the time-to-label with each label. Green plot: time to Annexin V positivity; red plot: time to YOYO3 positivity; yellow plot: time to double positivity; lines denote quartiles.
Illustrative data reproduced from Gelles et al. (2019).
LIMITATIONS
This method has a few limitations.
Microscopy used in SPARKL is optimized for adherent cells growing in a monolayer. Cells that grow in colonies or in a "cobble-stone" pattern may require more stringent optimization of the processing definitions or may not be suited for this workflow.
Suspension cells are not optimized for the SPARKL workflow. One option is to coat the tissue culture plate with a binding agent (e.g., collagen, poly-lysine) and adhere the cells prior to imaging. However, clustering of suspension cells will make segmentation during detection more complicated.
Quantification of dying cells requires apoptotic bodies to remain attached to the plate. Cell lines which detach from the plate surface during apoptosis will result in a loss of counts over time.
The "SPARKL_pipeline" script utilized in Variant C is limited to detecting cells that exhibit double positivity at the final time point. Cells that only exhibit single positivity are not detected by this script and not quantified in the analysis.
High-content live-cell imagers require robust fluorescent signal for detection. As a consequence, certain applications (e.g., transient transfection of fluorescent reporters) may not be detectable within these workflows.
TROUBLESHOOTING
Problem
My cells are all crowded in the center or the periphery of the well.
Potential Solution
This most likely happened due to pipet-generated vortices. Avoid ejecting into the corner of a well (Figure 1A) or at an angle as these will generate a vortex and pool your cells (Figure 1B). When seeding your cells, eject perpendicularly against the well wall and avoid excessive ejector force (Figure 1C).
Problem
Cells detach when I aspirate culture media and add labeling/treatment media.
Potential Solution
If your cell line detaches easily, you can add your treatment media to the culture media in lieu of aspirating and replacing the media. Seed your cells in a 90 μL volume (adjusting the culture dilution to plate an equivalent number of cells), make your treatment media as a 10× solution, and add 10 μL to the culture media for a final volume of 100 μL in each well. Remember to culture your cells in phenol red-free media.
Problem
In my first time point, my cells are dead and labeled with a cell death marker.
Potential Solution
Rapid and indiscriminate cell death labeling usually occurs due to the preparation of the cells. The most common reason is that cells desiccated after aspirating culture media. Treat wells in small batches to limit the amount of time a well is dry. Another reason could be that your cells were unable to condition the treatment media and became stressed. If your cells are sensitive to media changes, consider adding concentrated treatment media to the wells without aspirating the culture media.
Problem
My cells are not in focus.
Potential Solution
If your cells are out of focus for only the first time point, then the issue is condensation forming on the plate. Give the plate sufficient time to return to temperature before the first scan is scheduled to occur. If the same wells are out of focus throughout the experiment, there may be something on the lid. Remember to avoid writing on the top of the well If you label the lid.
Problem
My scanned images have very high fluorescent background or a halo effect.
Potential Solution
There will always be some degree of halo due to the plastic and media. Top-Hat analysis will correct for this and generate images with a more even background (Figure 3A). Background can be further diminished by doing the following: use phenol-red free media; use media without riboflavin; reduce the volume of treatment media.
Problem
My non-dying cells label with Annexin V.
Potential Solution
Some cell lines accumulate puncta of Annexin V-positivity in a death-independent manner. We believe this occurs when phosphatidylserine stochastically equilibrates to the outer leaflet of the plasma membrane and interacts with Annexin V in the media (Devaux, 1991). Normally, phosphatidylserine would be transferred back to the inner leaflet by resident flippases (Segawa et al., 2014), but the binding of Annexin V prevents this from occurring. These small puncta can easily be excluded during analysis by adding an area filter to your processing definition.
Problem
My cells are not labeling with Annexin V (including the "rapid death" control).
Potential Solution
There are a few reasons why cells may not label with Annexin V. 1) Confirm that treatment media has sufficient free Ca2+ (1.5−2 mM) or supplement as needed (Meers and Mealy, 1993). 2) The cell type may not sufficiently expose phosphatidylserine during cell death – use a cell-impermeable dye instead (e.g., YOYO3). 3) The method of cell death may not result in robust phosphatidylserine exposure (e.g., necroptosis) – use a cell-impermeable dye instead.
Problem
The fluorescent intensity of YOYO3 decreases over time and I lose positive objects.
Potential Solution
DNA-binding viability dyes lose signal as DNA is cleaved and fragmented (particularly in apoptosis) (van Engeland et al., 1998). For experiments lasting several days, a decrease in YOYO3+ events is expected. If loss of signal over time confounds downstream analysis, consider a cell death label which is not dependent on DNA content.
Problem
Both my green and red data decrease over time.
Potential Solution
If both channels lose objects over time, then dead cells may be lifting off the plate and leaving the focal plane of the microscope. Most adherent cell lines remain on the plate after dying unless the vessel is agitated. Adhesion of dead cells may be increased by coating the plate (e.g., collagen, poly-ornithine, poly-lysine). Alternatively, try Variant A to normalize cell death data by total number of cells, which will account for any lost cells during the experiment.
Problem
The morphology of my dead cells causes them to be detected as several objects, so my dead cell counts are higher than the total cell counts (Variant A).
Potential Solution
First, try adjusting the segmentation parameter in the channel detecting cell death events as this may help reduce the number of extra counts for dying cells (Figure 3B, middle panel). Additionally, try using Method 3 to normalize your data. If this does not work, you may have to do a manual assessment of the cells in order to normalize your data. Alternatively, you could keep your data as events instead of normalizing.
Problem
Downstream data analysis is very noisy.
Potential Solution
Scan your plate more frequently to acquire more data and smooth the curve. You can also use a smoothing function (e.g., in Prism) for certain applications without compromising the data interpretation.
Acknowledgments
This work was supported by NIH grants R01 CA157740 (J.E.C.), R01 CA206005 (J.E.C.), R01 CA253139 (J.E.C.), and F31 AA024681 (J.D.G.); the JJR Foundation; the William A. Spivak Fund; the Fridolin Charitable Trust; an American Cancer Society Research Scholar Award; a Leukemia & Lymphoma Society Career Development Award; and an Irma T. Hirschl/Monique Weill-Caulier Trust Research Award. This work was also supported in part by two research grants (5FY1174 and 1FY13416) from the March of Dimes Foundation, a Collaborative Pilot Award from the Melanoma Research Alliance, a Developmental Research Pilot Project Program within the Department of Oncological Sciences at the Icahn School of Medicine at Mount Sinai, and the Tisch Cancer Institute Cancer Center Support grant (P30 CA196521).
Author Contributions
Conceptualization and Methodology: J.D.G.; Funding Acquisition and Writing: J.D.G. and J.E.C.
Declaration of Interests
The authors declare no competing interests.
Contributor Information
Jesse D. Gelles, Email: jesse.gelles@icahn.mssm.edu.
Jerry E. Chipuk, Email: jerry.chipuk@mssm.edu.
References
- Devaux P.F. Static and dynamic lipid asymmetry in cell membranes. Biochemistry. 1991;30:1163–1173. doi: 10.1021/bi00219a001. [DOI] [PubMed] [Google Scholar]
- Gelles J.D., Chipuk J.E. Robust high-throughput kinetic analysis of apoptosis with real-time high-content live-cell imaging. Cell Death Dis. 2016;8:e2758. doi: 10.1038/cddis.2017.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelles J.D., Chipuk J.E. Github.com/ChipukLab/SPARKL_pipeline. Zenodo 2019. 2019. [DOI]
- Gelles J.D., Mohammed J.N., Santos L.C., Legarda D., Ting A.T., Chipuk J.E. Single-cell and population-level analyses using real-time kinetic labeling couples proliferation and cell death mechanisms. Dev. Cell. 2019;51:277–291. doi: 10.1016/j.devcel.2019.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M., Niepel M., Chung M., Sorger P.K. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat. Methods. 2016;13:521–527. doi: 10.1038/nmeth.3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman G., Reutelingsperger C.P., Kuijten G.A., Keehnen R.M., Pals S.T., van Oers M.H. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood. 1994;84:1415–1420. [PubMed] [Google Scholar]
- Logue S., Elgendy M., Martin S. Expression, purification and use of recombinant AV for the detection of apoptotic cells. Nat. Protoc. 2009;4:1383–1395. doi: 10.1038/nprot.2009.143. [DOI] [PubMed] [Google Scholar]
- Meers P., Mealy T. Calcium-dependent annexin V binding to phospholipids: stoichiometry, specificity, and the role of negative charge. Biochemistry. 1993;32:11711–11721. doi: 10.1021/bi00094a030. [DOI] [PubMed] [Google Scholar]
- Roux J., Hafner M., Bandara S., Sims J.J., Hudson H., Chai D., Sorger P.K. Fractional killing arises from cell-to-cell variability in overcoming a caspase activity threshold. Mol. Syst. Biol. 2015;11:803. doi: 10.15252/msb.20145584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segawa K., Kurata S., Yanagihashi Y., Brummelkamp T.R., Matsuda F., Nagata S. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science. 2014;344:1164–1168. doi: 10.1126/science.1252809. [DOI] [PubMed] [Google Scholar]
- Tait J.F., Gibson D., Fujikawa K. Phospholipid binding properties of human placental anticoagulant protein-I, a member of the lipocortin family. J. Biol. Chem. 1989;264:7944–7949. [PubMed] [Google Scholar]
- van Engeland M., Nieland L.J., Ramaekers F.C., Schutte B., Reutelingsperger C.P. Annexin V-affinity assay: a review on an apoptosis detection system based on phosphatidylserine exposure. Cytometry. 1998;31:1–9. doi: 10.1002/(sici)1097-0320(19980101)31:1<1::aid-cyto1>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]