

Abstract
RNA interference techniques represent a promising strategy for therapeutic applications. In addition to small interfering RNA-based approaches, which have been widely studied and translated into clinical investigations, microRNA-based approaches are attractive owing to their “one hit, multiple targets” concept. To overcome challenges with in vivo delivery of microRNAs related to stability, cellular uptake, and specific delivery, our group has developed and characterized chitosan nanoparticles for nucleotide delivery. This platform allows for robust target modulation and antitumor activity following intravenous administration.
Keywords: RNA interference, miR-34a, microRNA delivery, Chitosan nanoparticles, Prostate cancer
1. Introduction
The discovery of RNA interference (RNAi) has rapidly led to unique opportunities for the treatment of many diseases, including cancer [1, 2]. RNAi techniques represent a continuum of specific approaches for mRNA degradation, resulting in inhibition of gene expression [3]. Small interfering RNAs (siRNAs) and microRNAs (miRNAs) are small RNA molecules most frequently used for RNAi-based therapeutic applications [4].
siRNAs are exogenous double-stranded RNAs of about 20–22 nucleotides that are incorporated into RNA-induced silencing complex (RISC). SiRNA loaded into RISC is cleaved by argonaute-2 protein found in RISC, leading to the degradation of the “passenger strand” and activation of RISC with the “guide strand.” Eventually, this activated RISC with single-stranded guide RNA provides recognition of target mRNA through inter-molecular base pairing followed by degradation of the guide RNA. RISC can be recycled and implement numerous cleavage events [5]. The knockdown of a specific target mRNA is achieved through full complementation of siRNA to its target mRNA.
miRNAs are endogenous RNA molecules of about 22 nucleotides that are formed by cleavage of long primary precursors (pre-miRNAs) by Dicer. miRNAs are incorporated into RISC and bind to the 3′ untranslated regions of target mRNAs. This binding leads to either the destruction of target mRNA or translational repression [6, 7]. Unlike siRNA, miRNA regulates the expression of multiple targets owing to its partial complementation to the untranslated region in target mRNA [8]. Given the fact that the majority of oncogenes are undruggable with present strategies such as small-molecule inhibitors and monoclonal antibodies [9–11], new therapeutic approaches are needed for clinical translation. Therefore, both siRNA and miRNA have tremendous potential as therapeutic agents for multiple diseases through downregulation of the associated genes and their mRNA transcripts.
However, several challenges have to be overcome for successful clinical translation of these therapeutic agents. Susceptibility of RNAs to degradation by nucleases can hinder the RNAs from reaching the cytoplasm of target cells. Additional challenges such as stability, poor cellular uptake, or nonspecific delivery of RNAi molecules also must be addressed. The initial criteria in developing an ideal delivery system is the selection of material that is biocompatible, biodegradable, nonimmunogenic, and nontoxic. Several viral or other transfection vectors and carriers such as liposomes, polymeric nanoparticles, iron oxide nanoparticles, carbon nanotubes, and dendrimers have been developed to overcome these issues [12, 13].
Among the various options, chitosan (CH) is a particularly attractive option owing to its low immunogenicity, low toxicity, and biocompatibility [14]. CH is a natural polysaccharide that consists of repeated glucosamine and N-acetyl-glucosamine residues. It is derived from partial deacetylation of chitin, which is abundant in crustacean shells [15]. The cationic nature of CH enables easy and fast complex formation with negatively charged siRNA or miRNA [16], which increases the incorporation efficiency of the payload. In addition, the hydroxyl and amino groups of CH allow for chemical conjugation of specific ligands tailored for targeted therapy [17]. For instance, our group modified CH particles with an Arg-Gly-Asp (RGD) peptide via a thiolation reaction and showed that these RGD-CH-siRNA nanoparticles significantly enhanced selective intratumoral delivery in orthotopic animal models of ovarian cancer [18].
We have demonstrated several applications of siRNA/miRNA delivery using CH nanoparticles [19–26]. To assess in vivo miRNA delivery of CH nanoparticles into endothelial cells, we incorporated Cy3-labeled control miRNA into CH nanoparticles and delivered them intravenously into nude tumor-bearing mice. Forty-eight hours after delivery, mice were sacrificed and endothelial cells were assessed for colocalization of Cy3 signal. The results demonstrated endothelial and periendothelial miRNA localization in HeyA8 tumors [27].
In a recent study, we showed that CH-mediated delivery of miR-34a, a tumor-suppressive miRNA that downregulates multiple gene products involved in prostate cancer progression and metastasis, inhibited tumor growth and preserved bone integrity in a xenograft model representative of established prostate cancer bone metastasis [28]. To determine whether CH could deliver siRNAs to the tumor tissue preferentially, we incorporated Cy5.5-labeled siRNA into CH nanoparticles and administered the carriers intravenously to the mice that had been injected with PC3MM2-LG cells in the femur. An increase in Cy5.5 siRNA signal intensity was observed in the femur with the tumor compared with the femur without the tumor 3 days after siRNA delivery, suggesting that CH-incorporated siRNA was specifically delivered to the tumor tissue. Taken together, these experiments demonstrated that CH nanoparticles can accumulate in tumor tissues after intravenous administration and that miR-34a-CH nanoparticles significantly decrease prostate tumor growth in the bone.
The present chapter clarifies the experimental approaches and methods required to better understand the studies mentioned above. We detail the preparation of CH nanoparticles that were used for in vivo delivery of non-coding RNA. Cell preparation for in vivo injection, the administration, and the in vivo imaging techniques of nanocarriers are delineated. Next, we describe how the in vivo delivery is validated by in situ hybridization. Finally, immunoblotting and immunofluorescence, the procedures required to confirm the miR-34a functionality, are provided.
2. Materials
2.1. Cell Culture
Dulbecco’s Modified of Eagle’s Medium (DMEM)/Ham’s F12 50/50 Mix (Invitrogen or Sigma-Aldrich).
Fetal bovine serum (FBS; Invitrogen).
Penicillin-streptomycin solution, 100× (Corning).
Trypsin-EDTA solution, 0.25%, sterile, filtered, suitable for cell culture, 2.5 g porcine trypsin, and 0.2 g EDTA. (Sigma-Aldrich).
Hank’s Balanced Salt Solution (HBSS; Thermo Scientific).
2.2. Preparation of CH-miRNA or CH-siRNA Nanoparticles
Chitosan, low molecular weight, deacetylated chitin, poly (d-glucosamine) (Sigma-Aldrich).
Glacial acetic acid (Sigma-Aldrich).
Sodium tripolyphosphate (TPP; Sigma-Aldrich).
Molecular biology grade water (Lonza).
2.3. Immunoblotting
RIPA Lysis and Extraction Buffer (Thermo Scientific).
Protease inhibitor cocktail (Sigma-Aldrich).
Phosphatase inhibitor cocktail (Sigma-Aldrich).
Bicinchoninic acid assay kit (BCA; Thermo Scientific).
Laemmli buffer: 1.2 g SDS, 6 mg bromophenol blue, 4.7 mL glycerol, 1.2 mL Tris 0.5 M, and 2.1 mL distilled water; after everything is dissolved, add 0.93 g DTT, then aliquot and keep frozen at −20 °C.
Running buffer: for 10× stock, dissolve 30 g Tris-base and 144 g glycine in 1 L distilled water; for use, dilute to 1× by adding 100 mL stock and 10 mL 10% SDS to 890 mL distilled water for use.
Ready-to-use TGX gels.
Transfer buffer: for 10× stock, dissolve 30 g Tris-base and 144 g glycine in 1 L distilled water; for use, dilute to 1× by adding 100 mL stock and add 200 mL methanol to 700 mL distilled water.
Polyvinylidene difluoride (PVDF) membrane (Thermo Scientific).
Nonfat dry milk powder (Sigma-Aldrich).
Tris-buffered saline (TBS): for 10× stock, 31.5 g Tris-HCl and 80 g NaCl in 1 L distilled water.
TBS-T: add 100 mL 10× TBS and 2 mL Tween 20 (polyox-yethylene-20-sorbitan monolaurate; Thermo Scientific) to 898 mL distilled water.
Primary antibodies (c-Met and c-Myc from Santa Cruz Biotechnology; Axl from Cell Signaling, and GAPDH from Millipore).
Secondary antibodies (horseradish peroxidase-conjugated secondary goat anti-mouse or goat anti-rabbit antibody (Bio-Rad).
HyGLO™ Quick Spray Chemiluminescent HRP Antibody Detection Reagent (Denville Scientific).
Western Blot stripping buffer (Thermo Scientific).
2.4. In Situ Hybridization
Xylene.
Ethanol.
Hematoxylin (Harris).
Eosin (Harris).
Proteinase K (Sigma).
Predesigned digoxigenin-labeled miRCURY locked nucleic acid probe (Exiqon).
Small nuclear RNA U6 (Exiqon).
Polyclonal anti-digoxigenin antibody (Roche).
Alkaline phosphatase-conjugated secondary antibody (Roche).
4-nitro-blue tetrazolium (Roche).
5-bromo-4-chloro-3′-indolylphosphate (Roche).
Microscope imaging software for quantitative analysis of histological staining and fluorescence (Nikon NIS Elements).
2.5. Immunofluorescence (Promega DeadEnd TUNEL System)
Phosphate-buffered saline (PBS), 1× without calcium and magnesium (Corning).
Xylene.
Ethanol.
1× antigen retrieval buffer (Dako).
Pap pen.
3% hydrogen peroxide in methanol (Thermo Scientific).
Fish gelatin 4% in PBS (Sigma-Aldrich).
Primary antibody rat anti-mouse CD31 (BD Bioscience).
Secondary antibody Alexa 594 goat anti-rat (Jackson ImmunoResearch).
Paraformaldehyde 4% in PBS (1:4 dilution from 16% stock).
Triton X-100 0.2% in PBS.
Equilibration buffer (in Promega DeadEnd TUNEL kit).
Nucleotide mix (in Promega DeadEnd TUNEL kit).
Terminal Deoxynucleotidyl Transferase (TdT) enzyme (in Promega DeadEnd TUNEL kit).
2X saline sodium citrate buffer (SSC, diluted from 20X concentrate in Promega DeadEnd TUNEL kit).
Hoechst 33342 mounting media to stain nuclei (Thermo Scientific).
3. Methods
3.1. Gelation of CH with Sodium Tripolyphosphate and RNA
Dissolve CH in 0.25% acetic acid to make the final concentration of 2 mg/mL.
Mix TPP (0.25% w/v) and RNA (1 μg/μL), then add this mixture to CH solution with continuous stirring to spontaneously generate the nanoparticles.
Incubate the mixture at 4 °C for 40 min.
Centrifuge the mixture at 20,817 × g for 50 min at 4 °C to obtain the RNA/CH particles.
Discard the supernatant.
Wash the pellet three times with distilled water to remove unbound chemicals or miRNA (see Note 1).
After three washes, dissolve the pellet in distilled water (5 μg/100 μL) and store at 4 °C.
3.2. Cell Culture
Maintain prostate tumor PC3MM2 cells in 50% Dulbecco modified Eagle medium/50% Ham F12 supplemented with 10% FBS and 1% penicillin-streptomycin at 37 °C in 5% CO2.
3.3. Preparation of Cells for In Vivo Injections
Trypsinize the cells when they are 60–80% confluent.
Centrifuge the trypsinized cells at 290 × g for 5 min at 4 °C (see Note 2).
Wash the cells three times with PBS.
Count the cells with cell counter.
Take one million cells to a new tube and centrifuge at 290 × g.
Discard the supernatant and resuspend the cells in HBSS for each animal.
Inject one million PC3MM2 cells (100 μL) either subcutaneously for the subcutaneous model or into the femur of nude mice for the metastatic model.
3.4. CH-Non-Coding RNA Treatment In Vivo
3.4.1. CH-miRNA Treatment
One week after cell injection, divide the mice into control group and miR-34a treatment group. Administer control miRNA-encapsulated CH nanoparticles (control group; 5 μg/100 μL nanoparticles) or miR-34a-encapsulated CH nanoparticles (treatment group; 5 μg/100 μL nanoparticles) via tail vein injection every 3 days for both subcutaneous and metastatic models.
After 2 weeks, sacrifice the animals and harvest the tumors.
Fix the tumor either in formalin for paraffin embedding for in situ hybridization or snap-freeze in liquid nitrogen for Western blot analysis.
3.4.2. Delivery of CH-siRNA Nanoparticles into Bone
Ten days after cell injection, administer control siRNA-CH or Cy5.5-labeled siRNA-CH nanoparticles (5 μg/100 μL nanoparticles) via tail vein injection.
Three days after nanoparticle injection, sacrifice the mice.
Harvest the femurs.
Image the femurs using IVIS 200 (see Fig. 1).
Fig. 1.
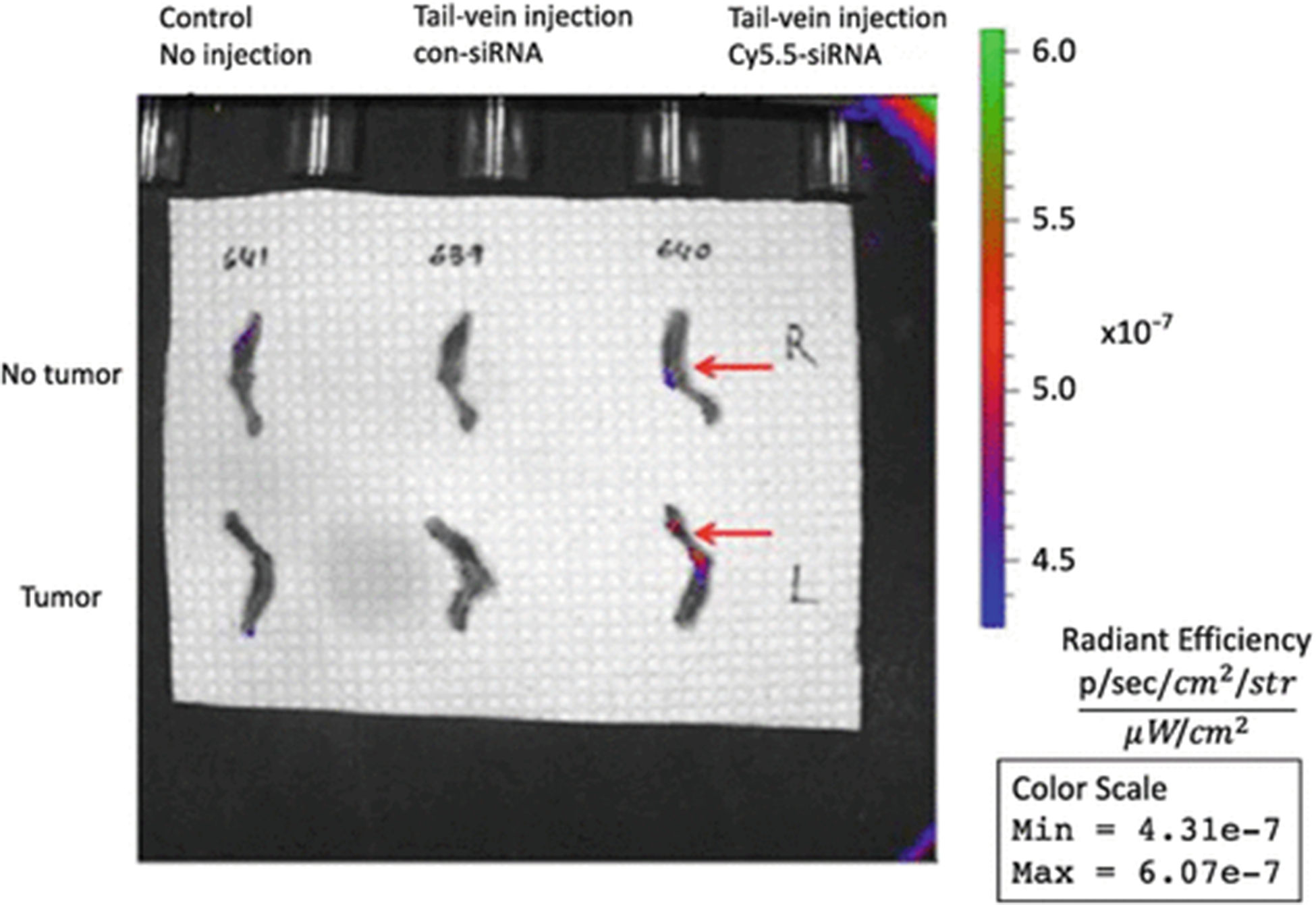
Chitosan nanoparticles deliver Cy5.5-labeled siRNA to the tumor tissue preferentially. Control or Cy5.5-labeled siRNA was administered to the mice with or without a tumor in the femur and Cy5.5 signal was measured by using IVIS 200. An increase in Cy5.5 signal was observed in the femur in the tumor group compared with the nontumor group (reproduced from ref. 28 with permission from Oncotarget)
3.5. Immunoblotting
Homogenize the snap frozen tumor tissue in RIPA Lysis and Extraction Buffer plus protease and phosphatase inhibitor cocktails (10 μL from each for 1000 μL lysis buffer) using homogenizer. Add 200 μL from the mixture onto each tumor tissue.
Incubate lysate on ice for 30 min, by vortexing every 5 min.
Centrifuge the lysate at 15,294 × g for 20 min at 4 °C.
Transfer the supernatant to a new Eppendorf tube.
Perform protein quantification using the BCA assay kit.
Adjust an equal amount of protein (15 or 30 μg) for each sample and mix with 25 μL Laemmli sample buffer.
Boil protein and Laemmli sample buffer mixtures at 100 °C for 5 min.
Load the mixtures (25 μL) onto 8% or 12% polyacrylamide gel and transfer onto a PVDF membrane.
Block the membrane in 5% nonfat dry milk in 1× TBS-T for 1 h at room temperature.
Probe the membrane with a specific primary antibody (e.g., c-Met, c-Myc, Axl, and GAPDH) at 1:1000 dilution overnight at 4 °C.
Remove the primary antibody and wash the membrane with TBS-T for 1 h at room temperature by changing the TBS-T every 15 min.
Re-probe the membrane with horseradish peroxidase-conjugated secondary goat anti-mouse or goat anti-rabbit antibody for 1 h at 1:2000 dilution at room temperature.
Remove the secondary antibody and wash the membrane for 1 h at room temperature, with changing the TBS-T every 15 min.
Remove the TBS-T.
Mix an equal amount (1 mL) of each ECL A and B reagent and add onto the membrane.
Incubate the membrane in this mixture for 1 min (see Note 3).
Remove the ECL reagent and blot the membrane with paper towel to remove extra ECL reagent.
Place the membrane into an X-ray film cassette along with a film for a few minutes.
Strip the membrane for 15 min using stripping buffer and wash with TBS-T for 30 min before probing with another antibody (see Fig. 2).
Fig. 2.
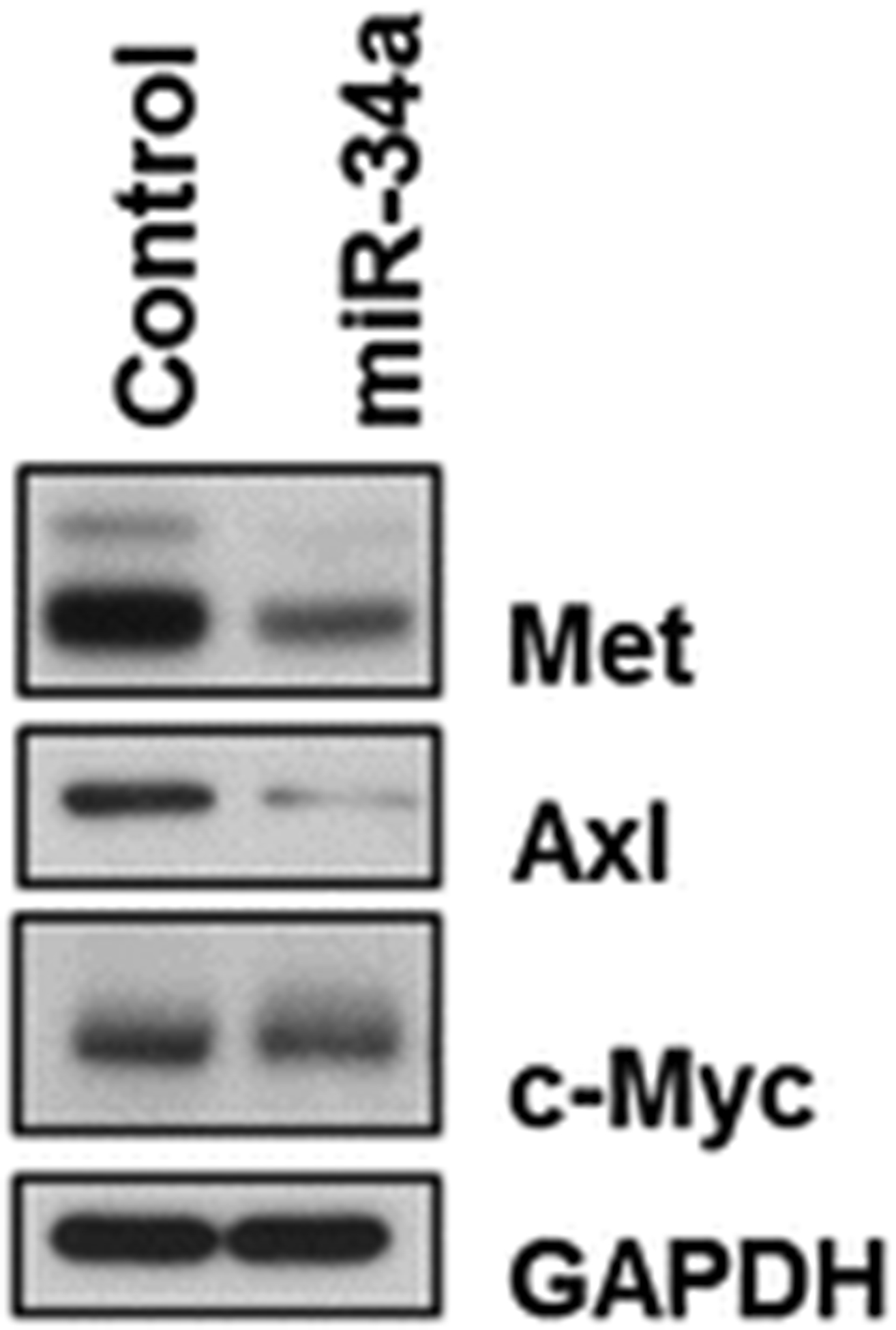
Western blot results for Met, Axl, and c-Myc proteins from harvested tumor tissues treated with control miRNA or miR-34a. Treatment with miR-34a led to downregulation of Met, Axl, and c-Myc (reproduced from ref. 28 with permission from Oncotarget)
3.6. In Situ Hybridization
Immerse the formalin-fixed, paraffin-embedded tissue sections in xylene in a Coplin staining jar for 5 min at room temperature for deparaffinization.
Dehydrate the deparaffinized slides by immersing in 100% ethanol for 5 min and rehydrate in decreasing concentrations of ethanol (95%, 85%, 70%, and 50%) for 3 min each.
Stain one slide from each group (control and miR-34a) with hematoxylin and eosin to validate the integrity of the tissues.
Digest two tissue sections from each group (control and miR-34a) with 10 μg/mL proteinase K for 10 min at room temperature and load onto the Ventana Discovery Ultra system.
Hybridize one of the tissue slides from each group with the double digoxigenin-labeled miRCURY locked nucleic acid probe designed for control miRNA or miR-34a for 2 h at 50 °C.
Use small nuclear RNA U6 as endogenous control.
Detect the digoxigenins with a polyclonal anti-digoxigenin antibody and alkaline phosphatase-conjugated secondary antibody using nitro-blue tetrazolium and 5-bromo-4-chloro-3′-indolyphosphate as the substrate.
Measure the intensities for ten areas from each slide with a microscope imaging software and compute the mean (see Fig. 3).
Fig. 3.
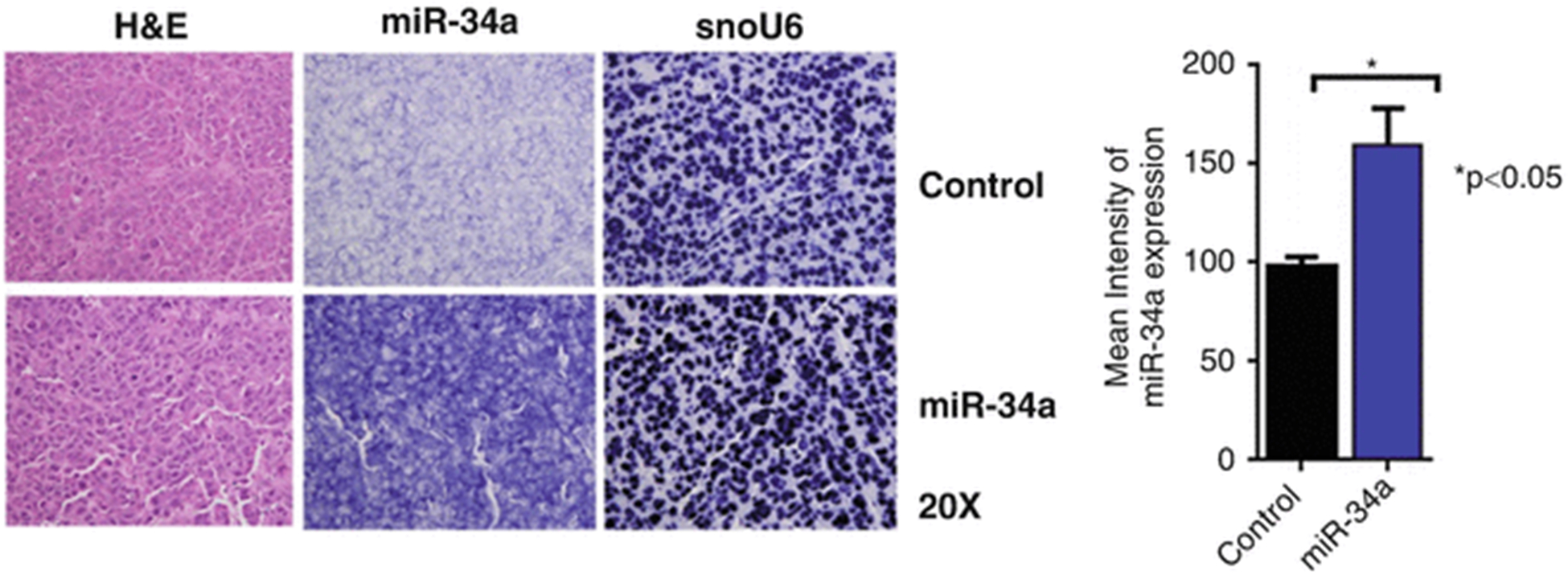
In situ hybridization of miR-34a with small nuclear RNA U6 (snoU6) as endogenous control. Formalin-fixed, paraffin-embedded slides were stained with hematoxylin and eosin (H&E) or in situ hybridization was performed by using a double digoxigenin-labeled miRCURY locked nucleic acid probe designed for control miRNA or miR-34a after a digestion step with proteinase K. The mean (± standard deviation) intensities obtained from ten areas from each slide were measured using the NIS Elements software (reproduced from ref. 28 with permission from Oncotarget)
3.7. Immunofluorescence
Immerse paraffin-embedded sections in xylene in a Coplin staining jar for 5 min at room temperature for deparaffinization. For frozen sections, fix the sections with cold acetone for 10 min and then continue from step 7.
Dehydrate the deparaffinized slides by immersing in 100% ethanol for 5 min and rehydrate in decreasing concentrations of ethanol (95%, 85%, 70%, and 50%) for 3 min each.
Perform antigen retrieval with 1× Dako antigen retrieval buffer.
Wash with PBS three times for 3 min each.
Separate the slides individually, align on a slide holder placed on a humidified chamber. Draw a circle around the tissue with a Pap pen (see Note 4).
Block endogenous peroxidases by incubating the slides in 0.3% hydrogen peroxide in PBS for 3–5 min at room temperature.
Incubate the tissue sections with protein block (4% fish gelatin in PBS) for 20 min at room temperature.
Remove protein block and add rat anti-mouse CD31 antibody (1:500 diluted in 4% fish gelatin), incubate overnight at 4 °C.
Wash with PBS three times for 3 min and incubate with protein block for 5 min.
Remove protein blocking solution and add Alexa 594 goat anti-rat secondary antibody (Jacskon ImmunoResearch, 1:800 diluted in 4% fish gelatin), incubate 1 h at room temperature.
Wash with PBS three times for 3 min each.
Fix the tissue sections by adding 4% paraformaldehyde in PBS on the slides and incubating for 20 min at room temperature.
Incubate the tissues in 0.2% Triton X-100 in PBS for 15 min at room temperature.
Add equilibration buffer (in Promega kit) on tissue sections and incubate for 10 min at room temperature.
Prepare TUNEL incubation buffer by mixing 45 μL equilibration buffer, 5 μL nucleotide mix, and 1 μL TdT enzyme for each slide. Incubate the slides with the buffer for 1 h at 37 °C in the dark.
To terminate the reactions, wash the slides in 2× SSC for 15 min at room temperature.
Do the counterstaining with Hoechst mounting media and visualize under a fluorescence microscope (see Fig. 4).
Fig. 4.
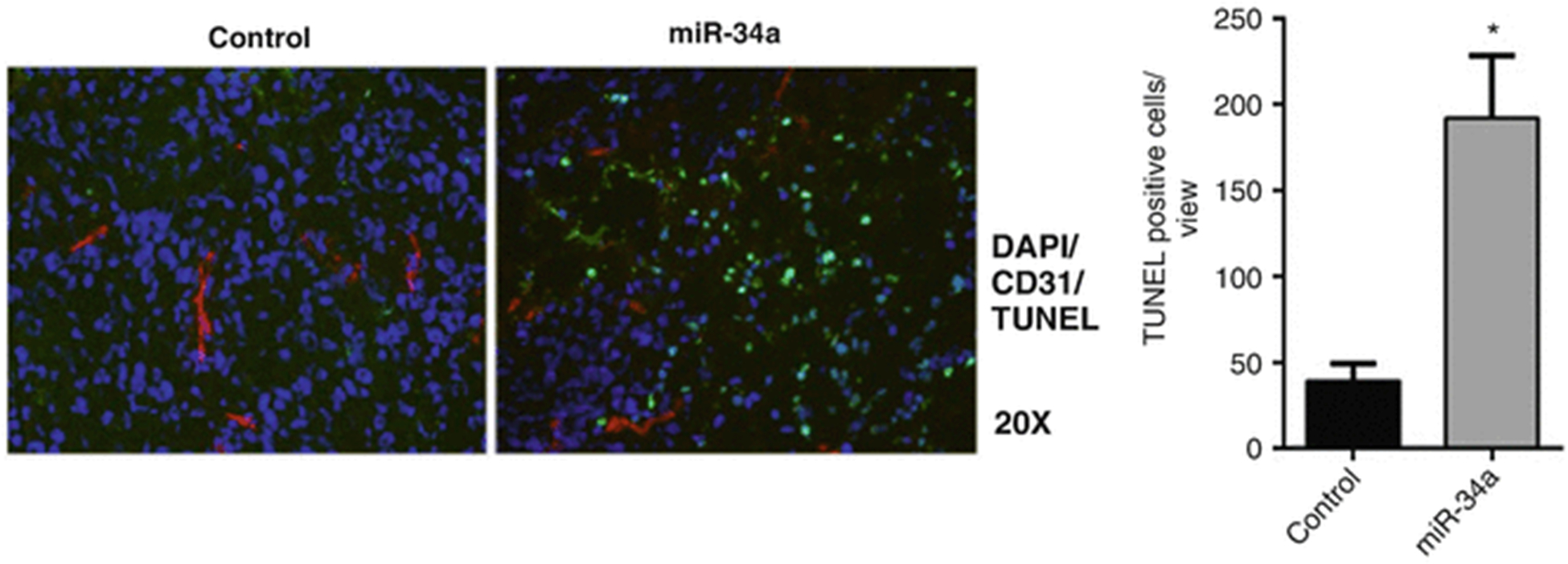
Tissue sections were used for immunofluorescence reactions. Tumors treated with control miRNA and miR-34a were stained by TUNEL (green), CD31 (red), and DAPI (blue). Mean (± standard deviation) TUNEL-positive cells obtained from ten areas from each slide were quantified using the ImageJ software. *p < 0.05 (unpaired two-sided two-sample Student’s t-test) compared with control (reproduced from ref. 28 with permission from Oncotarget)
4. Notes
Once CH nanoparticles are reconstituted in water, do not freeze and thaw the suspension because this process may fracture or rupture the particles. RNA/CH nanoparticles can be stored at 4 °C until they are used.
Use cells at 60–80% confluence for in vivo injections. Injecting overconfluent cells may lead to lower tumor take rates.
Rotate the membrane box by hand to make sure that the ECL mixture covers the membrane.
Try to keep the tissues wet with PBS because letting the tissue dry during the staining procedure may cause high background.
Acknowledgments
Portions of this work were supported by the NIH (CA016672, CA109298, P50 CA083639, P50 CA098258, UH3 TR000943), the Ovarian Cancer Research Fund, Inc. (Program Project Development Grant), the Blanton-Davis Ovarian Cancer Research Program, the RGK Foundation, and the Gilder Foundation.
References
- 1.Bobbin ML, Rossi JJ (2016) RNA interference (RNAi)-based therapeutics: delivering on the promise? Annu Rev Pharmacol Toxicol 56:103–122 [DOI] [PubMed] [Google Scholar]
- 2.SY W, Lopez-Berestein G, Calin GA, Sood AK (2014) RNAi therapies: drugging the undruggable. Sci Transl Med 6 (240):240ps247–240ps247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilson RC, Doudna JA (2013) Molecular mechanisms of RNA interference. Annu Rev Biophys 42:217–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carthew RW, Sontheimer EJ (2009) Origins and mechanisms of miRNAs and siRNAs. Cell 136(4):642–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rana TM (2007) Illuminating the silence: understanding the structure and function of small RNAs. Nat Rev Mol Cell Biol 8 (1):23–36. doi: 10.1038/nrm2085 [DOI] [PubMed] [Google Scholar]
- 6.Meister G, Tuschl T (2004) Mechanisms of gene silencing by double-stranded RNA. Nature 431(7006):343–349 [DOI] [PubMed] [Google Scholar]
- 7.Tijsterman M, Plasterk RH (2004) Dicers at RISC: the mechanism of RNAi. Cell 117 (1):1–3 [DOI] [PubMed] [Google Scholar]
- 8.Lam JK, Chow MY, Zhang Y, Leung SW (2015) siRNA versus miRNA as therapeutics for gene silencing. Mol Ther Nucleic Acids 4 (9):e252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ledford H (2015) Cancer: the Ras renaissance. Nature 520(7547):278–280. doi: 10.1038/520278a [DOI] [PubMed] [Google Scholar]
- 10.Nero TL, Morton CJ, Holien JK, Wielens J, Parker MW (2014) Oncogenic protein interfaces: small molecules, big challenges. Nat Rev Cancer 14(4):248–262. doi: 10.1038/nrc3690 [DOI] [PubMed] [Google Scholar]
- 11.Ramos P, Bentires-Alj M (2015) Mechanism-based cancer therapy: resistance to therapy, therapy for resistance. Oncogene 34 (28):3617–3626. doi: 10.1038/onc.2014.314 [DOI] [PubMed] [Google Scholar]
- 12.Gharpure KM, SY W, Li C, Lopez-Berestein G, Sood AK (2015) Nanotechnology: future of oncotherapy. Clin Cancer Res 21 (14):3121–3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ozcan G, Ozpolat B, Coleman RL, Sood AK, Lopez-Berestein G (2015) Preclinical and clinical development of siRNA-based therapeutics. Adv Drug Deliv Rev 87:108–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Croisier F, Jérôme C (2013) Chitosan-based biomaterials for tissue engineering. Eur Polym J 49(4):780–792 [Google Scholar]
- 15.Shukla SK, Mishra AK, Arotiba OA, Mamba BB (2013) Chitosan-based nanomaterials: a state-of-the-art review. Int J Biol Macromol 59:46–58 [DOI] [PubMed] [Google Scholar]
- 16.Choi C, Nam J-P, Nah J-W (2016) Application of chitosan and chitosan derivatives as biomaterials. J Ind Eng Chem 33:1–10 [Google Scholar]
- 17.Ragelle H, Vandermeulen G, Préat V (2013) Chitosan-based siRNA delivery systems. J Control Release 172(1):207–218 [DOI] [PubMed] [Google Scholar]
- 18.Han HD, Mangala LS, Lee JW, Shahzad MM, Kim HS, Shen D, Nam EJ, Mora EM, Stone RL, Lu C (2010) Targeted gene silencing using RGD-labeled chitosan nanoparticles. Clin Cancer Res 16(15):3910–3922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu C, Han HD, Mangala LS, Ali-Fehmi R, Newton CS, Ozbun L, Armaiz-Pena GN, Hu W, Stone RL, Munkarah A (2010) Regulation of tumor angiogenesis by EZH2. Cancer Cell 18(2):185–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim H-S, Han HD, Armaiz-Pena GN, Stone RL, Nam EJ, Lee J-W, Shahzad MM, Nick AM, Lee SJ, Roh J-W (2011) Functional roles of Src and Fgr in ovarian carcinoma. Clin Cancer Res 17(7):1713–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steg AD, Katre AA, Goodman B, Han H-D, Nick AM, Stone RL, Coleman RL, Alvarez RD, Lopez-Berestein G, Sood AK (2011) Targeting the notch ligand JAGGED1 in both tumor cells and stroma in ovarian cancer. Clin Cancer Res 17(17):5674–5685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu W, Lu C, Dong HH, Huang J, D-y S, Stone RL, Nick AM, Shahzad MM, Mora E, Jennings NB (2011) Biological roles of the delta family notch ligand Dll4 in tumor and endothelial cells in ovarian cancer. Cancer Res 71(18):6030–6039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hussein YR, Sood AK, Bandyopadhyay S, Albashiti B, Semaan A, Nahleh Z, Roh J, Han HD, Lopez-Berestein G, Ali-Fehmi R (2012) Clinical and biological relevance of enhancer of zeste homolog 2 in triple-negative breast cancer. Hum Pathol 43(10):1638–1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ziebarth AJ, Nowsheen S, Steg AD, Shah MM, Katre AA, Dobbin ZC, Han H-D, Lopez-Berestein G, Sood AK, Conner M (2013) Endoglin (CD105) contributes to platinum resistance and is a target for tumor-specific therapy in epithelial ovarian cancer. Clin Cancer Res 19(1):170–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krzeszinski JY, Wei W, Huynh H, Jin Z, Wang X, Chang T-C, Xie X-J, He L, Mangala LS, Lopez-Berestein G (2014) miR-34a blocks osteoporosis and bone metastasis by inhibiting osteoclastogenesis and Tgif2. Nature 512 (7515):431–435 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Aslan B, Monroig P, Hsu MC, Pena GA, Rodriguez-Aguayo C, Gonzalez-Villasana V, Rupaimoole R, Nagaraja AS, Mangala S, Han HD, Yuca E, SY W, Ivan C, Moss TJ, Ram PT, Wang H, Gol-Chambers A, Ozkayar O, Kanlikilicer P, Fuentes-Mattei E, Kahraman N, Pradeep S, Ozpolat B, Tucker S, Hung MC, Baggerly K, Bartholomeusz G, Calin G, Sood AK, Lopez-Berestein G (2015) The ZNF304-integrin axis protects against anoikis in cancer. Nat Commun 6:7351. doi: 10.1038/ncomms8351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pecot CV, Rupaimoole R, Yang D, Akbani R, Ivan C, Lu C, Wu S, Han H-D, Shah MY, Rodriguez-Aguayo C (2013) Tumour angiogenesis regulation by the miR-200 family. Nat Commun 4:2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaur S, Wen Y, Song JH, Parikh NU, Mangala LS, Blessing AM, Ivan C, Wu SY, Varkaris A, Shi Y (2015) Chitosan nanoparticle-mediated delivery of miRNA-34a decreases prostate tumor growth in the bone and its expression induces non-canonical autophagy. Oncotarget 6(30):29161. [DOI] [PMC free article] [PubMed] [Google Scholar]