

Abstract
Changes in chromosome numbers may strongly affect reproductive barriers, because individuals heterozygous for distinct karyotypes are typically expected to be at least partially sterile or to show reduced recombination. Therefore, several classic speciation models are based on chromosomal changes. One import mechanism generating variation in chromosome numbers is fusion and fission of existing chromosomes, which is particularly likely in species with holocentric chromosomes, i.e. chromosomes that lack a single centromere. Holocentric chromosomes evolved repeatedly across the tree of life, including in Lepidoptera. Although changes in chromosome numbers are hypothesized to be an important driver of the spectacular diversification of Lepidoptera, comparative studies across the order are lacking. We performed the first comprehensive literature survey of karyotypes for Lepidoptera species since the 1970s and tested if, and how, chromosomal variation might affect speciation. Even though a meta-analysis of karyological differences between closely related taxa did not reveal an effect on the degree of reproductive isolation, phylogenetic diversification rate analyses across the 16 best-covered genera indicated a strong, positive association of rates of chromosome number evolution and speciation. These findings suggest a macroevolutionary impact of varying chromosome numbers in Lepidoptera and likely apply to other taxonomic groups, especially to those with holocentric chromosomes.
This article is part of the theme issue ‘Towards the completion of speciation: the evolution of reproductive isolation beyond the first barriers’.
Keywords: chromosomal speciation, chromosome number, chromoSSE, diversification rate analysis, macroevolution, holocentric chromosomes
1. Introduction
The order Lepidoptera, which comprises more than 160 000 described species of butterflies and moths, is one of the most speciose branches of the tree of life. Its remarkable diversity is accompanied by a tremendous variation in chromosome numbers, ranging from 5 to 223 chromosomes in the haploid karyotype [1,2]. However, this variation is not randomly distributed among genera, as most show the presumed ancestral haploid karyotype of n=31, while other genera vary widely ([1], figure 1). In several genera, increased diversity in chromosome numbers appears associated with bursts in species numbers, suggesting that chromosomal variation may contribute to speciation [1,3–5]. This view is supported by theory, predicting that chromosomal variation can act as an intrinsic barrier to gene flow, either because hybrids between individuals with different chromosome numbers are at least partially sterile, or because chromosomal rearrangements suppress recombination [6,7]. Nevertheless, empirical evidence for the role of varying chromosome numbers in speciation is mixed, in part contrasting the theoretical predictions. Closely related species with different chromosome numbers can often be crossed [8,9] and hybrid fitness may not necessarily be reduced [10,11]. Moreover, evolutionary modes of diversification within genera in relation to varying chromosome numbers may range from neutral [4,12] to adaptive [5] evolution. However, a comprehensive study across Lepidoptera is lacking. With these inconsistencies at hand, we aim to infer the impact of interspecific chromosomal differentiation on reproductive isolation and rates of speciation across genera. We then discuss potential underlying mechanisms.
Figure 1.

Distribution of chromosome numbers in Lepidoptera based on 2399 taxa (electronic supplementary material, table 1.1) with boxplots summarizing chromosome numbers for the 16 genera used for the phylogenetic analysis. The number under each boxplot indicates the available number of taxa with chromosome counts.
Lepitopteran chromosomes are holocentric, i.e. they lack a centromeric region that concentrates all kinetochores, which allow attachment of the spindle tubules during mitosis and meiosis. Instead, species with holocentric chromosomes evolved mechanisms that allow kinetochore proteins to bind along the entire chromosome, permitting microtubules to attach broadly (reviewed in [13]). Holocentric chromosomes have evolved from monocentric ancestors at least 13 times in groups as diverse as plants and arthropods [13]. In plants, varying chromosome numbers have been shown to promote species diversification, for instance, in sedges (the genus Carex, with about 2000 species among the largest plant genera), by leading to hybrid dysfunction as a result of the formation of meiotic multivalents [14]. For species with holocentric chromosomes, changes in chromosome numbers evolve by either fusing two chromosomes into a single one, or through fission of a chromosome into two smaller chromosomes. As a consequence of holocentricity, fragmented chromosomes are initially more likely to be retained since the fragments maintain kinetochore function, which may make it more likely for chromosomal variation to evolve in the first place [13]. Additionally, in species with holocentric chromosomes, hybrid incompatibility between closely related species may be countered by mechanisms that some of the species have evolved in order to avoid meiotic mistakes. In the wood white butterfly (Pieris sinapis), for instance, the order of meiotic events is inverted, which is presumed to underlie rescued fitness of chromosomal hybrids [10]. Thus, while on the one hand, holocentric chromosomes may facilitate karyotype evolution in some lineages, chromosomal numbers are often conserved in other lineages, suggesting additional genomic control mechanisms that suppress fusion and fission.
Despite the evolutionary relevance across the tree of life, the molecular features that underlie fusion and fission of chromosomes in species with holocentric chromosomes are not resolved, and likely differ between species [15,16]. Among the potential features are repetitive sequences such as ribosomal DNA, GC rich DNA segments and transposable elements, which have been suggested to facilitate chromosomal fusion and fission by creating artificial centromere-like regions [17]. However, comparative genomic studies are rare and based on few species [16,18,19], relying often on short-read sequencing technologies that limit the study of repetitive parts of the genome. While some of these studies suggested a higher number of retrotransposons than expected by chance at fusion sites [18] others do not show such an enrichment [16]. Independent of karyotypic changes, rearranged Lepidoptera genomes show evidence for conserved synteny blocks that are maintained across even very distantly related species [16,20,21].
Chromosomal speciation implies that chromosomal rearrangements cause reproductive isolation between populations and, therefore, promote speciation [22,23]. Yet, causal effects of fusion and fission on the rate of speciation remain contentious [7,15]. Classic chromosomal speciation models were based on hybrid sterility, i.e. where individuals that are heterozygous for chromosomal rearrangements are partially or completely sterile [7,22]. Under these scenarios, differentially fixed chromosomal rearrangements between closely related species may in theory themselves quickly generate strong reproductive isolation and act as Dobzhansky–Muller incompatibilities (DMIs), potentially reinforced upon secondary contact, where heterozygotes would suffer from reduced fertility [24]. The problem with these classic models is that they require chromosomal rearrangements to be fixed in order to be of major effect, yet the conditions under which fixation of novel chromosomal rearrangements is likely would result in shallow reproductive barriers. Specifically, newly arising chromosomal rearrangements would typically be underdominant, i.e. they lead to reduced fitness of hybrid individuals. While strong underdominance makes it unlikely that they spread to fixation, weak underdominance may allow for fixation, but would ensure that chromosomal rearrangements represent only shallow barriers, and are therefore unlikely to cause speciation [7,25]. Empirical evidence for such chromosomal speciation comes from mammals that have monocentric chromosomes, including mice [26] and wallabies [27]. Here, monobrachial homology, i.e. multiple chromosomal fusions with one or more common arms in different fusion arrangements, causes reproductive isolation. Differences in chromosome numbers have also been suggested to act as DMIs for plants with holocentric chromosomes [14], however, to which degree this might apply to other systems, including Lepidoptera, is not known
More recent theoretical approaches have attempted to overcome the underdominance paradox by focusing on the changes in recombination associated with chromosomal rearrangements [6,7,25,28]. In essence, under these recent models, rearranged chromosomes can become fixed by drift or selection when two or more adaptive loci become physically coupled, enhancing existing reproductive isolation by reducing recombination [6,25]. Such re-arranged regions of reduced recombination may act as barrier loci and promote further differentiation, which may eventually lead to post-zygotic isolation through the build-up of genic DMIs [6,7,25]. By suppressing recombination, chromosomal rearrangements could help to increase reproductive isolation, which may be further enhanced by sexual selection or reinforcement [29] and may thus promote speciation upon secondary contact. Also, as a consequence of chromosomal speciation, the effective population size (Ne) may initially become reduced, which could, in turn, affect rates of speciation [30] and change the fixation probabilities of new karyotypes in allopatry. Indeed, for mammals, families with large geographical distributions but whose species have restricted geographical ranges showed a greater probability for fixing different karyotypes [31].
To understand the evolution of varying chromosome numbers and their potential implications on the speciation process, we reviewed the karyotypic literature on Lepidoptera and compiled the current knowledge on karyotypic diversity, which has doubled since the last attempt almost half a century ago [1]. We first assessed if published estimates of reproductive isolation [32] would differ between closely related species pairs with the same, or different karyotypes. While karyotypic changes in Lepidoptera evolve through fusion and fission events rather than genome duplications [33], transposable elements that may underlie such fission sites could perhaps lead to an increase in genome size [34]. Consequently, we also tested whether chromosome numbers are correlated with genome size. Combining karyotypic with genetic data, we finally assessed if the rate of chromosome number evolution is positively associated with the rate of speciation across the best-covered genera. In the light of the results, we then discuss the different roles of chromosomal variation in Lepidoptera, with a focus on the best-studied butterfly genera. Chromosomal variation can also include karyotypic changes through sex chromosome evolution (reviewed in [35]). Although information on sex chromosome evolution is limited to relatively few taxa [36], the current data suggest that the Z chromosome is highly conserved in Lepidoptera, while the evolution of neo-W chromosomes through fusion of autosomes may be common [37]. Although sex chromosomes also promote speciation [38], our study focuses more broadly on karyotype evolution through fusion and fission processes.
2. Material and methods
The previous comprehensive compilation of chromosome numbers in Lepidoptera was published by Robinson [1] almost 50 years ago, comprising data for 1183 taxa. After digitizing this list, we used Google Scholar in July 2019 to search for publications containing chromosome numbers that were not covered by [1]. Search terms were [Lepidoptera OR butterfly OR moth] AND karyotype, and [Lepidoptera OR butterfly OR moth] AND chromosome number. Our search yielded another 30 publications (in the electronic supplementary material, table 1.1), several of which are themselves compilations of chromosome numbers from multiple studies, e.g. [18]. We subsequently removed duplicate entries and ambiguous cases where taxa were not fully identified. The 97 cases in which intraspecific chromosomal variation was reported (e.g. Pieris sinapis; n = 28–54 [16]) are included in electronic supplementary material, table 1.1 but excluded from subsequent karyotype specific analyses, because the karyotype of the individuals for which the associated data was collected is unknown.
Genome size estimates for Lepidoptera were taken from the NCBI Genome Database (https://www.ncbi.nlm.nih.gov/genome) on 15 September 2019. From the 66 sequenced genomes, 64 had chromosome numbers available in our database (electronic supplementary material, table 1.2). Flow cytometric estimates of genome sizes were available for another 19 species from the Animal Genome Size Database 2.0 (www.genomesize.com); all also represented in our database. Flow cytometric estimates were converted to base-pair size using the formula from [39]: DNA content (pg) = genome size (bp)/(0.978 × 109). We then used a phylogenetic linear model in R 3.5.1 [40], package phylolm v. 2.6 [41] with genome size as response variable, chromosome number as fixed factor with and without accounting for the method of genome size estimate (sequence or flow cytometry). Non-independence of species data due to shared ancestry was incorporated by including a phylogenetic tree of the sampled species computed from mitochondrial COI sequences downloaded from GenBank (electronic supplementary material, table 1.3) and reconstructed using RAxML v. 8.2.8 [42]. To select an appropriate model for the error term, we fitted all implemented phylogenetic models that allow for measurement error (i.e. Brownian motion, Ohrnstein–Uhlenbeck with fixed or random root, kappa, delta, and Early Burst [41]). Genome size was normalized by log-transformation and the best-fitting model was determined based on AIC values.
We next tested if published estimates of reproductive isolation [32] differ between closely related species that share the same number of chromosomes (N = 49) or not (N = 19; electronic supplementary material, table 1.4). We used two linear mixed effect models with reproductive isolation (total isolation index in [32]) as a response variable and the genus as random effect. In one model, we used as fixed factor a categorical variable, i.e. if chromosome numbers differed or not, and in the other the actual difference in chromosome numbers. It was not possible to account for the phylogenetic error structure, because such an analysis requires the inclusion of a phylogeny with the most recent common ancestor of each species pair. We could not construct such a phylogeny due to the lack of phylogenetic data for, or resolution among, many of these closely related species, e.g. [5]. For the same reason, it was not possible to account for the age (divergence times) of each species pair.
To test if chromosome number evolution has an effect on species diversification rates, we selected all genera that had enough karyotype data, DNA sequence data, and species representation to warrant phylogenetic investigation. Because missing species can critically affect diversification rate analyses [43] and estimated rates of trait evolution [44], we only included the 16 genera for which we had both karyotype information and DNA sequence data for more than 25% of the genus (electronic supplementary material, table 1.5), and no generic para- or polyphyly was indicated (thus genera correspond to clades). This large sampling (representing ca 1055 species—371 of which had karyotypes) will thus likely provide insights about the association of chromosome number evolution and species diversification across Lepidoptera.
For each genus, we reconstructed a phylogenetic hypothesis with branch lengths proportional to divergence times, to be able to compare genera on the same measurement scale. We employed a Bayesian approach, to take full account of uncertainty in phylogeny reconstruction and propagate it in downstream analyses. First, we used the pipeline OneTwoTree [45] to obtain DNA sequence data. This pipeline downloads all sequence data on NCBI GenBank (https://www.ncbi.nlm.nih.gov/genbank/) for a set of input taxon names, clusters the sequences based on OrthoMCL [46] to define groups of homologous sequences, and aligns these using MAFFT [47]. Thus, the approach guides marker selection objectively, based on sequence information rather than sequence headers. To run OneTwoTree, we provided the name of each genus, plus one outgroup taxon based on the global Lepidoptera phylogeny of [48]. Genbank IDs and details on taxon sampling are provided in electronic supplementary material, table 1.6. We then groomed alignments after manual inspection by removing loci available for fewer than 10% of the species, and eliminated sites with greater than 90% missing data using PhyUtility [49].
Phylogenetic inference was based on a MrBayes v. 3.2.7a [50] analysis for each genus. We employed GTR substitution models and gamma-distributed rate variation among sites, performing two runs of four metropolis-coupled Markov chains per analysis, using default proposal mechanisms and temperatures. We made sure that runs converged to the target distribution based on the potential scale reduction factors (approaching 1 for all parameters), average standard deviations of split frequencies (being well below 0.1), effective sample sizes (≫200 for each parameter), and by inspecting the traces. The number of MCMC generations required for convergence differed between genera, from 1 to 10 million (electronic supplementary material, table 1.5). We then combined trees from both runs after excluding 25% as burnin, and thinned it uniformly to a sample from the posterior distribution of 100 trees (hereafter, ‘posterior') per genus using BurnTrees v. 0.3.0 (https://github.com/nylander/Burntrees). We rooted each tree with the outgroup, and performed divergence time estimation using a relaxed, correlated molecular clock fitted based on penalized likelihood [51], implemented in the ape package in R [52]. The split between in- and outgroup was dated based on the median ages reported in [48]. Subsequently, the outgroup and stem lineage were pruned, yielding a posterior sample of 100 dated trees per genus.
To test for an association of chromosomal evolution and species diversification rates, we used a Bayesian approach to fit a phylogenetic model (ChromoSSE) tailored for chromosome evolution [53], implemented in the statistical software RevBayes [54], to each genus. This model jointly describes the evolution of chromosome numbers through fusion and fission (i.e. a change in chromosome number by −1 or +1, respectively), and the origination and extinction of phylogenetic lineages (hereafter, species). Chromosome numbers are thus allowed to evolve along branches (i.e. anagenetic change) or at speciation events (i.e. cladogenetic change). Specifically, we fitted three speciation rate parameters: fission-associated speciation, fusion-associated speciation, and speciation without chromosomal change; and two parameters for anagenetic chromosomal change: one for fission and one for fusion. We estimated a single species turnover rate per genus. As such, we did not allow for dysploidy and polyploidy, because these processes are not documented in Lepidoptera [1,2]. We fitted the model sequentially to each of the 100 trees in the posterior, feeding the final MCMC sample of one tree as the starting values for the next. After computing a generous burnin of 300 generations on the first tree, we computed 20 generations per tree. We then combined MCMC samples across trees, evaluated MCMC performance using Tracer v. 1.7.1 [55], and thinned it uniformly to 100 samples per genus. It was computationally not feasible to fit the ChromoSSE model to the data of Polyommatus, probably because of its very large range of chromosome numbers (range 10–223, figure 1) that made exponentiation of the instantaneous rate matrix computationally prohibitive. Therefore, we also employed a computationally simpler approach where we computed for each genus a single speciation rate using a Yule model of lineage diversification and a single rate of chromosomal evolution based on Brownian motion, using the R-packages diversitree v. 0.9–11 [56] and phytools v. 0.6-99 [57], respectively. Even though, at least in principle, chromosome number evolution is poorly described by a random drift process, the results were overall fully congruent with the ChromoSSE model, irrespective of assumptions on extinction rates, divergence time uncertainty. These results are detailed in electronic supplementary material, S3.
To attempt to reject the hypothesis that the rate of speciation is not related to the rate of chromosome number evolution, we performed a phylogenetic linear regression analysis using the R package phylolm 2.6 [41]. The rate of species diversification was the response variable and defined as the sum of all estimated speciation rate parameters, while the rate of chromosome evolution was the predictor and constructed as the sum of all fusion and fission rate parameters. After computing species-means, checking assumptions and performing the relevant log-transformations, we determined the best evolutionary model for the error term by fitting all implemented models that account for measurement error (i.e. Brownian motion, Ohrnstein–Uhlenbeck with fixed or random root, kappa, delta and early-burst models [41]) and selected the model with the lowest (best) AIC score. This analysis accounts for the non-independence among observations (i.e. genera) by including the phylogeny of [48] pruned to just represent the phylogenetic relations among the included genera. To also account for phylogenetic uncertainty, we repeated the analysis 100 times by randomly sampling values for each genus from their respective posterior distributions, and checking significance and slope.
3. Results
Our literature survey identified 2399 lepidopteran taxa for which a chromosome number was reported (electronic supplementary material, table 1.1), about double from the previous comprehensive survey. However, chromosome numbers were only available for 41 of the 124 Lepidoptera families [58] with a strong bias to some groups of butterflies (e.g. almost half of the observations came from two families: Nymphalidae, N = 869; Lycaenidae, N = 239). Only 610 (25.4%) taxa with chromosome numbers were moths. The median chromosome number was n = 29 (range 5–223) and the most common karyotype was the putatively ancestral chromosome number of n = 31 (N = 630; figure 1). The effect of chromosome number on genome size was best described using an OU-model for the error term (AIC = 56.2, irrespective of root assumptions; AIC for other models ranged 59.3–61.3), and included a weak (r2 = 0.01) yet significant, positive effect of chromosome number (t = 3.53, p = 0.0006, figure 2), also when including method (sequence or flow cytometry) as an additional factor (t = 2.91, p = 0.045). In particular species with few chromosomes had smaller genomes. However, the available data covered only a small range of known chromosomal variation (sampled range 12–60, median: 29.5; known range 5–223; electronic supplementary material, table 1.1 and 1.2).
Figure 2.

Weakly positive relationship between genome size and chromosome numbers (phylogenetic linear model, t = 3.53, p = 0.0006, r2 = 0.01). Data on genome size is either based on genome sequences (open circles) or estimates from flow cytometry (filled circles).
In our species-pairs analysis, we could not detect a significant effect of the presence of chromosome number difference on reproductive isolation (figure 3). This was independent of whether the difference was coded as a categorical variable ( p = 0.305), or the actual differences in chromosome numbers were used ( p = 0.836).
Figure 3.
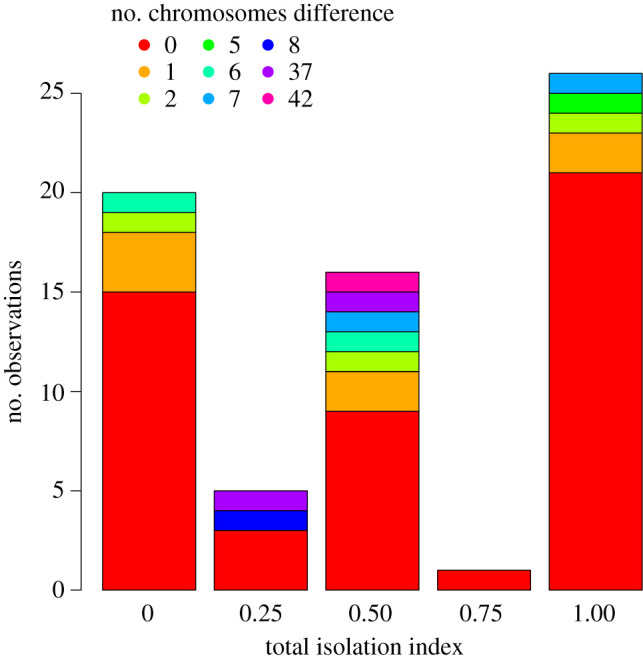
Estimates of reproductive isolation (total isolation index from [32]) between closely related species pairs that either differ in their karyotype or not.
The groomed sequence matrices contained on average 50 taxa per genus, representing 70% of the species (range 11–166 taxa, representing 59% and 90% of the species respectively) with an average of 8095 bps of sequence data (range 3790–23 905 bps; electronic supplementary material, table 1.5). Dated phylogenies with tip states are provided in electronic supplementary material, S2.
Analyses based on ChromoSSE models strongly supported the hypothesis that rates of speciation and chromosome evolution are related. All rate parameters differed strongly across the 15 genera for which we could fit the ChromoSSE model (electronic supplementary material, figure 4.1). The posterior mean net diversification rate (i.e. the difference between speciation and extinction rates) per genus ranged from 0.047 (Lycaena) to 0.305 (Lysandra) species per species per million years, with the posterior mean species turnover fraction ranging from 0.13 (Erebia) to 0.98 (Lysandra; electronic supplementary material, S3). The relation between total speciation rates and total chromosome evolution rates across posterior-mean values was strongly positive (figure 4; effect size 0.630 ± 0.193 in log-log space, t = 3.26, p = 0.006, using a BM model for the error term, AIC = 53.5; AIC for other models ranged 55.3–55.5). To further confirm this result, we replaced each species-mean value by a random draw from the respective posterior distribution and checked significance of the relation. Repeating this process 100 times yielded a significant, positive relationship in each.
Figure 4.

Joint phylogenetic analyses of chromosomal evolution and speciation rates based on the ChromoSSE model across 15 Lepidoptera genera. (a) Total speciation (the sum of all speciation rate parameters) is positively associated with total chromosomal variation (the sum of all chromosomal change parameters—phylogenetic linear model, t = 3.26, p = 0.006, black line). Dots indicate posterior mean rates estimated for each genus, with error bars, extending 1 s.d. in either direction. Names of genera for each observation are indicated. (b) The cladogenetic component of chromosomal change (in % of total chromosomal evolution) differs strongly among genera, but is not significantly associated with total chromosomal evolution (phylogenetic linear model, logit-transformation, t = 1.52 p = 0.150). Annotation as in (a). (c) Rates of fission exceed rates of fusion (summing cladogenetic and anagenetic components) in most genera, indicated by their position above the dashed line that indicates y = x. Annotation as in (a).
Comparing the fits of the ChromoSSE models across genera yielded further insights into the role of chromosome evolution in species diversification. Foremost, the overall importance of chromosomal speciation was underlined by the finding that speciation rates with chromosomal change exceeded speciation without chromosomal change in 12 out of 15 genera (the exceptions being Colias, Heliconius and Papilio; electronic supplementary material, S4, figure 4.2). In most genera, chromosomal change was more frequently anagenetic (along a branch) than cladogenetic (at speciation; figure 4b; electronic supplementary material, S4, figure 4.3). Although the three exceptions Lysandra, Oleria and Pteronymia were those with also the highest rates of chromosomal change, there was no general association between the absolute rate of chromosomal evolution and the importance of its cladogenetic component (figure 4b). Overall, the absolute importance of the cladogenetic component of chromosomal change ranged from 3.6% in Lycaena to 98% in Lysandra.
The ChromoSSE models also allowed us to infer whether fission or fusion events are more common and more commonly implicated in speciation. Overall, fission events occurred at higher rates than fusion events in all but two genera (Papilio and Memphis; electronic supplementary material, S4, figure 4.4). However, the cladogenetic component did not consistently differ between fission and fusion events, where it was significantly higher for three and seven genera for fission and fusion events, respectively (electronic supplementary material, S4, figure 4.5).
Our complementary approach, based on Brownian motion for chromosomal evolution rates and Yule diversification rates allowed us to include the chromosomally most diverse genus Polyommatus and yielded fully congruent results, irrespective of how we accounted for extinction and dating uncertainty (electronic supplementary material, S3).
4. Discussion
The karyological variation in Lepidoptera has attracted much interest over the past decades, yet many aspects underlying its spectacular diversity remain enigmatic. Lepidoptera show the highest known range in chromosome numbers among non-polyploid eukaryotes [2]. Among hexapods only hemipterans, that also have holocentric chromosomes, are known to have up to a hundred chromosomes [59]. In plants, polyploidization often generates tremendous variation in chromosome numbers [60], but the highest known range that is not attributed to polyploidization occurs in genera with holocentric chromosomes, such as Carex (n = 6–62, [61]). Performing the most comprehensive literature survey on chromosome numbers to date, we found that most lepidopteran species show the putative ancestral chromosome number of n = 31, or a number close to this (figure 1). This is consistent with the previous systematic review [1] that covered half as many taxa. However, variation in chromosome numbers differs strikingly among genera (figure 1). Interestingly, while our analyses based on the ChromoSSE model suggests that rates of chromosomal fission are generally higher than those of chromosomal fusions (figure 4c), a reduction in chromosome numbers from the ancestral number seems to be more common among the extant species (figure 1). This could be because fission events are predicted to more likely result in deleterious meiotic products, and may therefore be more often be selected against, though this effect is debated in Lepidoptera [62].
While karyotypic variation has been extensively studied in some Lepidoptera genera (e.g. [1,3,16]), the macroevolutionary impact of varying chromosome numbers on the dynamics of clade diversification had not been assessed. By employing a phylogenetic diversification rate analysis for the best-covered genera, we show that, overall, increased rates of chromosomal evolution are associated with increased rates of speciation (figure 4a). Similar positive relationships between rates of speciation and karyotypic variation were reported for Sceloporus lizards [63], several plant genera, including Carex and Helianthus (reviewed in [64]), and mammals [65].
In principle, it is possible that factors covarying with chromosome number exert effects on speciation, rather than chromosomal evolution per se. Changes in genome size have been suggested to affect rates of speciation themselves [66], and indeed, we observed that genome size significantly increases with the number of chromosomes (figure 2). It is, however, unlikely that the effects we ascribe to karyotypical variation (figure 4) are primarily due to genome size differences. This is because the effect of genome size differs among taxonomic groups. Whereas increased genome size correlates positively with speciation rates in mammals, the opposite is true for insects including Lepidoptera [66]. For plants, the rate of genome size evolution rather than genome size itself is positively correlated with speciation [67]. Also, chromosome number is only loosely associated with genome size (r2 = 0.01; figure 2), while the association of chromosome number with speciation rates was tight (figure 4). The paucity of broadly sampled species level phylogenies with associated genome size estimates for Lepidoptera (electronic supplementary material, table 1.2) precludes testing for associations between genome size and speciation directly. However, we cannot rule out other unaccounted factors. For example, genetic diversity was previously found to be positively correlated with chromosome numbers rather than genome size in Lepidoptera [68], though this association has to be considered with care, given the small sample size (N = 34) and the limited range of chromosome numbers covered (range n: 13–34). Among the factors that could explain some of the variation in genome size that we observed are the genetic features suggested to underlie fusion and fission sites. These include transposable elements and may lead to increased genome size, as has been found in Pieris [34]. However, the currently available taxonomic breadth and sample sizes are limited (electronic supplementary material, figure 1.2) and comparative studies of the presence and abundance of transposable elements are missing.
The reliability of our results also critically depends on the robustness of our analytical approach. The ChromoSSE model belongs to a family of state-dependent speciation and extinction models (SSE; [69]) that evaluate the effect of a focal character state on the rate of lineage origination and loss. Though widely used, their statistical performance remains debated, and much potentially undesired behaviour has been evaluated, including unbalanced prevalence of the focal character [70], assumptions about its root state [71], and effects of covarying, unevaluated characters [72,73]. The effects of these issues are mixed, i.e. they can lead to inflated Type I [74] or Type II error [70] rates. They are addressed in more recent implementations (e.g. [73]) and alternatives approaches (e.g. [75]), that themselves have also received criticism. Overall, SSE models require careful interpretation and adequate accounting for relevant sources of error [76]. The recently developed ChromoSSE has been evaluated under a wide range of simulated conditions [53], that demonstrate its reliability for our study: although parameter estimates were typically accurate and precise, cladogenetic components of chromosomal change tended to be underestimated relative to anagenetic change [53], making the approach in our context rather conservative. Most importantly, even when as little as 10% of extant species are sampled, the accuracy of ChromoSSE model estimates was only marginally compromised [53]. Given that we accounted for various sources of error in a Bayesian framework and only included the most densely sampled genera, these findings suggest that our estimated parameters are robust. Our implementation of the phylogenetic linear model analysis [41] further accounted for inaccuracy of parameter estimates for individual clade. Importantly, our result was also robust regarding phylogenetic uncertainty and different analytical approaches, as the analyses based on the ChromoSSE model (figure 4) yielded results fully congruent with those based on Brownian motion, while accounting for extinction rates and dating uncertainty (electronic supplementary material, S3). Our statistically significant results are therefore unlikely to be artefactual.
The contribution of chromosomal speciation to all cladogenetic events differed among genera (figure 4b), where the rate of anagenetic chromosomal change, i.e. along branches, exceeded that of cladogenetic chromosomal changes in nine genera (electronic supplementary material, S4, figure 4.3). This suggests that only some fusion and fission events may directly lead to speciation and that the probability of chromosomal speciation differs considerably among genera. The difference between anagenetic and cladogenetic changes furthermore suggest that the evolutionary mechanisms underlying the role of chromosomal change in speciation may differ among genera: for genera where cladogenetic changes predominate, chromosomal changes may act as DMIs as has been found in plants with holocentric chromosomes [14]. Conversely, when chromosomal changes are predominantly anagenetic, novel chromosomes may suppress recombination, leading eventually to the build-up of genic DMIs, suggesting indirect, gradual effects of anagenetic chromosomal change on speciation. These are hypotheses and need thorough investigation (see §5).
Interestingly, species turnover, measured as the ratio of extinction over speciation rates, was highest in the genera Lysandra, Oleria and Pteronymia, (figure 4b) that also had highest rates of chromosomal change, suggesting that new species form frequently through chromosomal change but may not persist [77]. If selection against new karyotypes is weak, novel karyotypes may form new species and proliferate before going extinct also reducing effective population sizes, with suspected effects promoting extinction rate [78]. Conversely, if selection is stronger, new karyotypes may be selected against immediately without ever giving rise to new species. While the former scenario is congruent with the pattern in the aforementioned three genera, the latter scenario would result in an apparent macroevolutionary stasis as seen in the genera with lowest rates of chromosomal change (figure 4; see electronic supplementary material, S2 for the phylogenetic distribution of chromosome numbers per genus). Understanding why the effect of chromosomal change differs among genera might thus be achieved by comparing the potentially different selective forces acting on newly arising karyotypes.
Whereas our phylogenetic analyses suggest that increased chromosomal variation is associated with increased rates of speciation, we did not detect a significant effect of difference in chromosome number on reproductive isolation between closely related species pairs (figure 3). This observation could reflect that pre-zygotic barriers may be more likely to drive reproductive isolation in some genera [3,79]. However, we note that the available number of estimates for reproductive isolation was limited and that the data were strongly phylogenetically structured, yet the lack of relevant phylogenetic information precluded a formal phylogenetic analysis (electronic supplementary material, table 1.4). As a consequence, we could also not account for differences in reproductive isolation due to different evolutionary ages. In the following, we discuss the evidence for chromosomal variation driving diversification among the best-studied genera of Lepidoptera.
With a range of 10–223 chromosomes in the haploid karyotype, Polyommatus is karyotypically the most diverse known Lepidoptera genus (figure 1; [2,3]). Together with its sister genus Lysandra, Polyommatus showed the highest speciation rates in our analyses based on Brownian motion (1.80 sp−1my; electronic supplementary material, S3), which is consistent with former genus-specific inferences [4,80,81]. Species of both genera occur across the Palaearctic region and have diversified recently, i.e. over the last 1–3 Myr [4,12,80]. Comparative phylogenetic analyses suggested that chromosomal variation may gradually accumulate in a random walk manner, consistent with neutral evolution [12], where the fixation of a particular karyotype has been suggested to occur through bottleneck events [4]. While hybrids between Polyommatus species with distinct karyotypes can suffer from reduced fertility due to segregation problems during meiotic division, promoting reproductive barriers [3,81], in some cases, hybridization can lead to homoploid hybrid speciation [82], further boosting species diversification. Karyotypic changes in Polyommatus are thought to primarily accumulate in allopatry and speciation to become complete through reinforcement upon secondary contact [3]. Closely related Polyommatus species indeed exhibit a higher karyotypic difference in sympatry than closely related allopatric populations where reinforcement leads to increased phenotypic differentiation in zones of secondary contact [3,81]. The genomic features underlying fusion and fission sites in both Polyommatus and Lysandra are not resolved and genomic data are lacking. Jointly, these data suggest that chromosomal change likely has an important role in driving speciation in these genera potentially as intrinsic post-zygotic barrier, however, causality remains to be shown.
In contrast to Polyommatus, species of the family Pieridae often show the putatively ancestral karyotype of n = 31, with comparatively little interspecific karyotypic variation (electronic supplementary material, table 1.1). Consistent with this observation, we documented both low rates of chromosomal evolution and low rates of species diversification for the genera Colias, Eurema and Pieris (figure 4). This result is in line with the idea that genera that remained close to the ancestral chromosome number of 31 diversify at lower rates than those in which chromosomal change has been substantial. However, while in Pieridae karyotypes rarely differ between species, intraspecific and even intra-population chromosomal variation can occur, e.g. in wood whites – Leptidea [10,83]. Leptidea sinapis shows the highest non-polyploid intraspecific chromosomal variation documented to date in Lepidoptera (n = 28–54) [10]. The polymorphic Leptidea karyotypes are thought to result from rapid accumulation of fusion and fission events, as well as other complex rearrangements, followed by extinction of intermediate forms [83]. Notably, heterozygotes between chromosomal races of Leptidea are abundant and do not appear to be selected against. The lack of fitness disadvantages of such chromosomal hybrids may be a result of inverted meiosis, in which the order of the meiotic steps is switched in order to facilitate the proper segregation of chromosomes [10]. Despite the lack of hybrid dysfunction, chromosomal rearrangements are still expected to promote the evolution of reproductive isolation by reducing gene flow and recombination among chromosomal races of Leptidea [84] or through the evolution of novel sex chromosomes [83]. Smaller chromosomal rearrangements may furthermore be abundant within genera that show little karyotypic variation. For example, in a recent comparative study on Pieris napi and P. rapae, the genomes of both species were shown to be reorganized into collinear blocks mainly through translocations, with a minor role for fusion and fission. The rearranged genomic sections were locally enriched with functional gene clusters, highlighting the potential selective advantage of chromosomal rearrangements [16]. In the case of Pieris, diversification is mainly driven by an arms race with their Brassicaceae host plants, though the potential role of chromosomal rearrangements for speciation has not been assessed [85]. Our results suggest these effects are rather weak in this genus (figure 4).
The well-studied radiation of Heliconius butterflies has emerged over the last 10–13 Myr in the Neotropics, where speciation has been shown to be predominantly driven by strong natural selection on wing patterns, resulting in different mimicry rings. Interspecific gene flow and adaptive introgression occurs among distantly related species that have the same karyotype [20,86], where different co-adapted loci are in some cases maintained by small-scale chromosomal rearrangements such as inversions [87]. While most Heliconius species have only 21 chromosomes, higher chromosome numbers have evolved at least twice, i.e. in the doris group and more recently in the sapho group (electronic supplementary material, table 1.1, [88]). In contrast to the rest of the radiation, very little is known about these species and none of their genomes have so far been sequenced (electronic supplementary material, table 1.2). If differences in chromosome numbers restrict interspecific gene flow, the otherwise abundant adaptive introgression is expected to be significantly reduced or absent and may thus limit the evolutionary potential in these groups. While mimicry is similarly prevalent in many other Neotropical butterfly groups, these often also show karyotypic variation that is thought to have evolved through non-adaptive processes such as drift or genetic bottlenecks, and which may further reinforce speciation [89].
Taken together, the evolution of chromosomal variation may be a significant factor for speciation, but its effect and magnitude seems to differ among (figure 4) and potentially within genera. The latter is indicated by the observation that the strength of reproductive isolation caused by differences in chromosome numbers can be limited when species have only recently diverged [10,11]. While some large-scale chromosomal rearrangements may act as DMIs, suggested by our inferred cases of cladogenetic chromosomal change (electronic supplementary material, S4.3), changes are more often anagenetic and may suggest that chromosomal rearrangements could, if at all, promote speciation by suppressing recombination in genomic regions underlying adaptation [6,7]. Combined with other evolutionary forces such as reinforcement or sexual selection, they may then lead towards complete reproductive isolation and overall accelerate speciation as is indicated by our macroevolutionary inferences.
5. Knowledge gaps and future directions
The evolutionary mechanisms that may lead towards the completion of speciation are still not fully understood [90]. Chromosomal rearrangements resulting in karyotypic variation have been suggested to promote reproductive isolation, either by promoting hybrid sterility [7,22,24] or by suppressing recombination promoting the accumulation of reproductive isolation over time [6,7,25]. Importantly, the theory underlying the aforementioned predictions was developed for species with monocentric chromosomes. To which degree they also apply for species with holocentric chromosomes needs further investigation. By executing 16 parallel case studies of the karyologically best-covered genera, our analyses suggest that chromosomal variability in Lepidoptera is overall associated with increased rates of speciation (figure 4). The underlying evolutionary mechanisms seem to differ with the timing of chromosomal change relative to speciation: while they are primarily cladogenetic in some, they are anagenetic in most genera (electronic supplementary material, figure 4.3). Further in-depth studies are thus needed to understand if cladogenetic events represent cases where karyological differences result in DMIs or if speciation is rapidly completed by other factors such as sexual selection or reinforcement, with a minor role of chromosomal change. Similarly, genomic investigations are needed to assess if and to which degree novel chromosomes may suppress recombination particularly in clades that show primarily anagenetic speciation events such as Erebia. Given that karyotypes were only available for a third of all Lepidoptera families (electronic supplementary material, table 1.1), further investigations comprising genera from many more families are needed to assess the generality of our observed pattern across the order of Lepidoptera, ideally including a very high fraction of extant species sampled for more speciose genera, which would also allow for accurate extinction rate estimates.
As for the evolutionary processes, our understanding of the genomic architecture of fusion and fission sites is limited. Only few genomes are currently sequenced, with a bias towards a few model species such as the genus Heliconius (electronic supplementary material, table 1.2), where we document exceptionally low chromosome-associated speciation (figure 4). The sequenced species primarily cover taxonomic groups that show little karyotypic variation, and have karyotypes that evolved mainly through chromosomal fusions from the putative ancestral karyotype. The few genomic studies suggest genus-specific mechanisms and genomic features that could underlie chromosomal rearrangements [16,18,91]. However, the genomic features responsible for increased rates of chromosomal fission, as e.g. seen in Lysandra and Polyommatus, are unresolved. Also, it remains unknown whether fusion and fission processes always involve the same chromosomes, and whether species groups that show conservatism in terms of chromosome numbers may have degenerated fusion and fission sites, and are thus genetically constrained [15]. Given that similar genomic architectures are likely to be at play across very distinct taxonomic groups [14,15,17], resolving the aforementioned issues—by using e.g. novel long-read sequencing methodologies and a broader taxonomic scope—will help to resolve the evolution of one of the most speciose taxonomic orders and provide insights for evolution in species with holocentric chromosomes in general.
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Acknowledgements
Phylogenetic analyses were performed at sciCORE (http://scicore.unibas.ch/) scientific computing centre at the University of Basel.
Data accessibility
Data supporting this study have been deposited in the Zenodo repository at http://doi.org/10.5281/zenodo.3735954 [92]
Authors' contributions
K.L. conceived of the study; J.M.d.V. and K.L. designed analyses; K.L. and H.A. collected the data, J.M.d.V. and L.B. analysed the data, K.L. and J.M.d.V. wrote the manuscript with input from H.A. and L.B.
Competing interests
We declare we have no competing interests.
Funding
This research was supported by the Swiss National Science Foundation grant nos. 310030_184934 to K.L. and 310030_185251 to J.M.d.V. We thank the Editor and two anonymous reviewers for their insightful comments that helped us improve the manuscript.
References
- 1.Robinson R. 1971. Lepidoptera genetics. Oxford, UK: Pergamon Press Inc. [Google Scholar]
- 2.Lukhtanov V. 2015. The blue butterfly Polyommatus (Plebicula) atlanticus (Lepidoptera, Lycaenidae) holds the record of the highest number of chromosomes in the non-polyploid eukaryotic organisms. Comp. Cytogenet. 9, 683–690. ( 10.3897/CompCytogen.v9i4.5760) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lukhtanov VA, Kandul NP, Plotkin JB, Dantchenko AV, Haig D, Pierce NE. 2005. Reinforcement of pre-zygotic isolation and karyotype evolution in Agrodiaetus butterflies. Nature 436, 385–389. ( 10.1038/nature03704) [DOI] [PubMed] [Google Scholar]
- 4.Talavera G, Lukhtanov VA, Rieppel L, Pierce NE, Vila R. 2013. In the shadow of phylogenetic uncertainty: the recent diversification of Lysandra butterflies through chromosomal change. Mol. Phylogenet. Evol. 69, 469–478. ( 10.1016/j.ympev.2013.08.004) [DOI] [PubMed] [Google Scholar]
- 5.Lucek K. 2018. Evolutionary mechanisms of varying chromosome numbers in the radiation of Erebia butterflies. Genes 9, 1–9. ( 10.3390/genes9030166) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guerrero RF, Kirkpatrick M. 2014. Local adaptation and the evolution of chromosome fusions. Evolution 68, 2747–2756. ( 10.1111/evo.12481) [DOI] [PubMed] [Google Scholar]
- 7.Faria R, Navarro A. 2010. Chromosomal speciation revisited: rearranging theory with pieces of evidence. Trends Ecol. Evol. 25, 660–669. ( 10.1016/j.tree.2010.07.008) [DOI] [PubMed] [Google Scholar]
- 8.Lorkovic Z. 1958. Some peculiarities of spatially and sexually restricted gene exchange in the Erebia tyndarus group. Cold Spring Harb. Symp. Quant. Biol. 23, 319–325. ( 10.1101/SQB.1958.023.01.032) [DOI] [PubMed] [Google Scholar]
- 9.Descimon H, Mallet J. 2009. Bad species. In Ecology of butterflies in Europe. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 10.Lukhtanov VA, Dinca V, Friberg M, Šíchová J, Olofsson M, Vila R, Marec F, Wiklund C. 2018. Versatility of multivalent orientation, inverted meiosis, and rescued fitness in holocentric chromosomal hybrids. Proc. Natl Acad. Sci. USA 115, E9610–E9619. ( 10.1073/pnas.1802610115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hora KH, Marec F, Roessingh P, Menken SBJ. 2019. Limited intrinsic postzygotic reproductive isolation despite chromosomal rearrangements between closely related sympatric species of small ermine moths (Lepidoptera: Yponomeutidae). Biol. J. Linn. Soc. 128, 44–58. ( 10.1093/biolinnean/blz090) [DOI] [Google Scholar]
- 12.Vershinina AO, Lukhtanov VA. 2017. Evolutionary mechanisms of runaway chromosome number change in Agrodiaetus butterflies. Sci. Rep. 7, 8199 ( 10.1038/s41598-017-08525-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Melters DP, Paliulis LV, Korf IF, Chan SWL. 2012. Holocentric chromosomes: convergent evolution, meiotic adaptations, and genomic analysis. Chrom. Res. 20, 579–593. ( 10.1007/s10577-012-9292-1) [DOI] [PubMed] [Google Scholar]
- 14.Escudero M, Hahn M, Brown BH, Lueders K, Hipp AL. 2016. Chromosomal rearrangements in holocentric organisms lead to reproductive isolation by hybrid dysfunction: the correlation between karyotype rearrangements and germination rates in sedges. Am. J. Bot. 103, 1529–1536. ( 10.3732/ajb.1600051) [DOI] [PubMed] [Google Scholar]
- 15.Dion-Côté A.-M, Barbash DA. 2017. Beyond speciation genes: an overview of genome stability in evolution and speciation. Curr. Op. Genet. Dev. 47, 17–23. ( 10.1016/j.gde.2017.07.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hill JA, et al. 2019. A butterfly chromonome reveals selection dynamics during extensive and cryptic chromosomal reshuffling. Sci. Adv. 5, eaau3648 ( 10.1126/sciadv.aau3648) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li S-F, Su T, Cheng G-Q, Wang B-X, Li X, Deng C-L, Gao W-J. 2017. Chromosome evolution in connection with repetitive sequences and epigenetics in plants. Genes 8, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahola V, et al. 2014. The Glanville fritillary genome retains an ancient karyotype and reveals selective chromosomal fusions in Lepidoptera. Nat. Commun. 5, 4737 ( 10.1038/ncomms5737) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanost MR, et al. 2016. Multifaceted biological insights from a draft genome sequence of the tobacco hornworm moth, Manduca sexta. Insect Biochem. Mol. Biol. 76, 118–147. ( 10.1016/j.ibmb.2016.07.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nadeau NJ, et al. 2016. The gene cortex controls mimicry and crypsis in butterflies and moths. Nature 534, 106–110. ( 10.1038/nature17961) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.d'Alencon E, et al. 2010. Extensive synteny conservation of holocentric chromosomes in Lepidoptera despite high rates of local genome rearrangements. Proc. Natl Acad. Sci. USA 107, 7680–7685. ( 10.1073/pnas.0910413107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.White MJD. 1978. Modes of speciation. San Francisco, CA: W. H. Freeman. [Google Scholar]
- 23.King M. 1995. Species evolution. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 24.Coyne JA, Orr HA. 2004. Speciation. Sunderland, MA: Sinauer Associates. [Google Scholar]
- 25.Navarro A, Barton NH. 2003. Accumulating postzygotic isolation genes in parapatry: a new twist on chromosomal speciation. Evolution 57, 447–459. ( 10.1111/j.0014-3820.2003.tb01537.x) [DOI] [PubMed] [Google Scholar]
- 26.Garagna S, Page J, Fernandez-Donoso R, Zuccotti M, Searle JB. 2014. The Robertsonian phenomenon in the house mouse: mutation, meiosis and speciation. Chromosoma 123, 529–544. ( 10.1007/s00412-014-0477-6) [DOI] [PubMed] [Google Scholar]
- 27.Potter S, Bragg JG, Blom MP. K., Deakin JE, Kirkpatrick M, Eldridge MD. B, Moritz C. 2017. Chromosomal speciation in the genomics era: disentangling phylogenetic evolution of rock-wallabies. Front. Genet. 8, 10 ( 10.3389/fgene.2017.00010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rieseberg LH. 2001. Chromosomal rearrangements and speciation. Trends Ecol. Evol. 16, 351–358. ( 10.1016/S0169-5347(01)02187-5) [DOI] [PubMed] [Google Scholar]
- 29.Trickett AJ, Butlin RK. 1994. Recombination suppressors and the evolution of new species. Heredity 73, 339–345. ( 10.1038/hdy.1994.180) [DOI] [PubMed] [Google Scholar]
- 30.Lanfear R, Kokko H, Eyre-Walker A. 2014. Population size and the rate of evolution. Trends Ecol. Evol. 29, 33–41. ( 10.1016/j.tree.2013.09.009) [DOI] [PubMed] [Google Scholar]
- 31.Martinez PA, Jacobina UP, Fernandes RV, Brito C, Penone C, Amado TF, Fonseca CR, Bidau CJ. 2017. A comparative study on karyotypic diversification rate in mammals. Heredity, 118, 366–373. ( 10.1038/hdy.2016.110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Presgraves DC. 2002. Patterns of postzygotic isolation in Lepidoptera. Evolution 56, 1168–1183. ( 10.1111/j.0014-3820.2002.tb01430.x) [DOI] [PubMed] [Google Scholar]
- 33.Nakatani Y, McLysaght A. 2019. Macrosynteny analysis shows the absence of ancient whole-genome duplication in lepidopteran insects. Proc. Natl Acad. Sci. USA 116, 1816–1818. ( 10.1073/pnas.1817937116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Talla V, Suh A, Kalsoom F, Dinca V, Vila R, Friberg M, Wiklund C, Backström N. 2017. Rapid increase in genome size as a consequence of transposable element hyperactivity in wood-white (Leptidea) butterflies. Genome Biol. Evol. 9, 2491–2505. ( 10.1093/gbe/evx163) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nguyen P, Carabajal Paladino L. 2016. On the neo-sex chromosomes of Lepidoptera. In Evolutionary biology, 171–185. Cham, Switzerland: Springer International Publishing. [Google Scholar]
- 36.Sahara K, Yoshido A, Traut W. 2012. Sex chromosome evolution in moths and butterflies. Chromosome Res. 20, 83–94. ( 10.1007/s10577-011-9262-z) [DOI] [PubMed] [Google Scholar]
- 37.Fraïsse C, Picard MAL, Vicoso B. 2017. The deep conservation of the Lepidoptera Z chromosome suggests a non-canonical origin of the W. Nat. Commun. 8, 1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carabajal PLZ, Provazníková I., Berger M, Bass C, Aratchige NS, López SN, Marec F, Nguyen P. 2019. Sex chromosome turnover in moths of the diverse superfamily Gelechioidea. Genome Biol. Evol. 11, 1307–1319. ( 10.1093/gbe/evz075) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dole el J, Barto J, Voglmayr H, Greilhuber J. 2003. Nuclear DNA content and genome size of trout and human. Cytometry 51A, 127–128. [DOI] [PubMed] [Google Scholar]
- 40.R Core Team. 2018. R: a language and environment for statistical computing, v 3.5.1. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- 41.Tung HLS, Ané C. 2014. A Linear-time algorithm for Gaussian and non-Gaussian trait evolution models. Syst. Biol. 63, 397–408. ( 10.1093/sysbio/syu005) [DOI] [PubMed] [Google Scholar]
- 42.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. ( 10.1093/bioinformatics/btu033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang J, Rabosky DL, Alfaro ME. 2020. Estimating diversification rates on incompletely-sampled phylogenies: theoretical concerns and practical solutions. Systematic Biol. 69, 602–611. ( 10.1093/sysbio/syz081) [DOI] [PubMed] [Google Scholar]
- 44.Rabosky DL. 2015. No substitute for real data: a cautionary note on the use of phylogenies from birth–death polytomy resolvers for downstream comparative analyses. Evolution 69, 3207–3216. ( 10.1111/evo.12817) [DOI] [PubMed] [Google Scholar]
- 45.Drori M, Rice A, Einhorn M, Chay O, Glick L, Mayrose I. 2018. OneTwoTree: an online tool for phylogeny reconstruction. Mol. Ecol. Resour. 18, 1492–1499. ( 10.1111/1755-0998.12927) [DOI] [PubMed] [Google Scholar]
- 46.Li L, Stoeckert CJ, Roos DS. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. ( 10.1101/gr.1224503) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Katoh K, Standley DM. 2013. mafft multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. ( 10.1093/molbev/mst010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chazot N., et al. 2019. Priors and posteriors in Bayesian timing of divergence analyses: the age of butterflies revisited. Syst. Biol. 0, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith SA, Dunn CW. 2008. Phyutility: a phyloinformatics tool for trees, alignments and molecular data. Bioinformatics 24, 715–716. ( 10.1093/bioinformatics/btm619) [DOI] [PubMed] [Google Scholar]
- 50.Ronquist F., et al. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. ( 10.1093/sysbio/sys029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paradis E. 2013. Molecular dating of phylogenies by likelihood methods: a comparison of models and a new information criterion. Mol. Phylo. Evol. 67, 436–444. ( 10.1016/j.ympev.2013.02.008) [DOI] [PubMed] [Google Scholar]
- 52.Paradis E, Schliep K. 2019. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528. ( 10.1093/bioinformatics/bty633) [DOI] [PubMed] [Google Scholar]
- 53.Freyman WA, Höhna S. 2017. Cladogenetic and anagenetic models of chromosome number evolution: a Bayesian model averaging approach. Syst. Biol. 67, 195–215. ( 10.1093/sysbio/syx065) [DOI] [PubMed] [Google Scholar]
- 54.Höhna S, Landis MJ, Heath TA, Boussau B, Lartillot N, Moore BR, Huelsenbeck JP, Ronquist F. 2016. RevBayes: Bayesian phylogenetic inference using graphical models and an interactive model-specification language. Syst. Biol. 65, 726–736. ( 10.1093/sysbio/syw021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. 2018. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904. ( 10.1093/sysbio/syy032) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.FitzJohn RG. 2012. Diversitree: comparative phylogenetic analyses of diversification in R. Methods Ecol. Evol. 3, 1084–1092. ( 10.1111/j.2041-210X.2012.00234.x) [DOI] [Google Scholar]
- 57.Revell LJ. 2011. phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 3, 217–223. ( 10.1111/j.2041-210X.2011.00169.x) [DOI] [Google Scholar]
- 58.Kristensen NP, Scoble MJ, Karsholt O. 2007. Lepidoptera phylogeny and systematics: the state of inventorying moth and butterfly diversity. Zootaxa 1668, 699–747. ( 10.11646/zootaxa.1668.1.30) [DOI] [Google Scholar]
- 59.Blackmon H, Ross L, Bachtrog D. 2016. Sex determination, sex chromosomes, and karyotype evolution in insects. J. Hered. 108, 78–93. ( 10.1093/jhered/esw047) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rice A, Glick L, Abadi S, Einhorn M, Kopelman NM, Salman-Minkov A, Mayzel J, Chay O, Mayrose I. 2015. The Chromosome Counts Database (CCDB)—a community resource of plant chromosome numbers. New Phytol. 206, 19–26. ( 10.1111/nph.13191) [DOI] [PubMed] [Google Scholar]
- 61.Cuacos M, Franklin FC H, Heckmann S. 2015. Atypical centromeres in plants—what they can tell us. Front. Plant. Sci. 6, 1247 ( 10.3389/fpls.2015.00913) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pringle EG, Baxter SW, Webster CL, Papanicolaou A, Lee SF, Jiggins CD. 2007. Synteny and chromosome evolution in the Lepidoptera: evidence from mapping in Heliconius melpomene. Genetics 177, 417–426. ( 10.1534/genetics.107.073122) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leaché A. D., Banbury BL, Linkem CW, de Oca AN.-M.. 2016. Phylogenomics of a rapid radiation: is chromosomal evolution linked to increased diversification in North American spiny lizards (genus Sceloporus)? BMC Evol. Biol. 16, 63 ( 10.1186/s12862-016-0628-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.De Storme N, Mason A.. 2014. Plant speciation through chromosome instability and ploidy change: cellular mechanisms, molecular factors and evolutionary relevance. Curr. Plant Biol. 1, 10–33. ( 10.1016/j.cpb.2014.09.002) [DOI] [Google Scholar]
- 65.Bush GL, Case SM, Wilson AC, Patton JL. 1977. Rapid speciation and chromosomal evolution in mammals. Proc. Natl Acad. Sci. USA 74, 3942–3946. ( 10.1073/pnas.74.9.3942) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kraaijeveld K. 2010. Genome size and species diversification. Evol. Biol. 37, 227–233. ( 10.1007/s11692-010-9093-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Puttick MN, Clark J, Donoghue PC. J. 2015. Size is not everything: rates of genome size evolution, not C-value, correlate with speciation in angiosperms. Proc. R. Soc. B. 282, 20152289 ( 10.1098/rspb.2015.2289) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mackintosh A, Laetsch DR, Hayward A, Charlesworth B, Waterfall M, Vila R, Lohse K. 2019. The determinants of genetic diversity in butterflies. Nat. Commun. 10, 3466 ( 10.1038/s41467-019-11308-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maddison WP, Midford PE, Otto SP. 2007. Estimating a binary character's effect on speciation and extinction. Syst. Biol. 56, 701–710. ( 10.1080/10635150701607033) [DOI] [PubMed] [Google Scholar]
- 70.Davis MP, Midford PE, Maddison W. 2013. Exploring power and parameter estimation of the BiSSE method for analyzing species diversification. BMC Evol. Biol. 13, 38 ( 10.1186/1471-2148-13-38) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gamisch A. 2016. Notes on the statistical power of the binary state speciation and extinction (BiSSE) Model. Evol. Bioinform. 12, 164–174. ( 10.4137/EBO.S39732) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maddison WP, FitzJohn RG. 2014. The unsolved challenge to phylogenetic correlation tests for categorical characters. Syst. Biol. 64, 127–136. ( 10.1093/sysbio/syu070) [DOI] [PubMed] [Google Scholar]
- 73.Beaulieu JM, O'Meara BC. 2016. Detecting hidden diversification shifts in models of trait-dependent speciation and extinction. Syst. Biol. 65, 583–601. ( 10.1093/sysbio/syw022) [DOI] [PubMed] [Google Scholar]
- 74.Rabosky DL, Goldberg EE. 2015. Model inadequacy and mistaken inferences of trait-dependent speciation. Syst. Biol. 64, 340–355. ( 10.1093/sysbio/syu131) [DOI] [PubMed] [Google Scholar]
- 75.Rabosky DL. 2014. Automatic detection of key innovations, rate shifts, and diversity-dependence on phylogenetic trees. PLoS ONE 9, e89543 ( 10.1371/journal.pone.0089543) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.O'Meara BC, Beaulieu JM. 2016. Past, future, and present of state-dependent models of diversification. Am. J. Bot. 103, 792–795. ( 10.3732/ajb.1600012) [DOI] [PubMed] [Google Scholar]
- 77.Rosenblum EB, Sarver BA. J., Brown JW, Roches Des S, Hardwick KM, Hether TD, Eastman JM, Pennell MW, Harmon LJ. 2012. Goldilocks meets Santa Rosalia: an ephemeral speciation model explains patterns of diversification across time scales. Evol. Biol. 39, 255–261. ( 10.1007/s11692-012-9171-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Frankham R. 2005. Genetics and extinction. Biol. Cons. 126, 131–140. ( 10.1016/j.biocon.2005.05.002) [DOI] [Google Scholar]
- 79.Mérot C, Salazar C, Merrill RM, Jiggins CD, Joron M. 2017. What shapes the continuum of reproductive isolation? Lessons from Heliconius butterflies. Proc. R. Soc. B. 284, 20170335 ( 10.1098/rspb.2017.0335) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kandul NP, Lukhtanov VA, Dantchenko AV, Coleman JWS, Sekercioglu CH, Haig D, Pierce NE. 2004. Phylogeny of Agrodiaetus Hübner 1822 (Lepidoptera: Lycaenidae) inferred from mtDNA sequences of COI and COII and nuclear sequences of EF1-alpha: karyotype diversification and species radiation. Syst. Biol. 53, 278–298. ( 10.1080/10635150490423692) [DOI] [PubMed] [Google Scholar]
- 81.Kandul NP, Lukhtanov VA, Pierce NE. 2007. Karyotypic diversity and speciation in Agrodiaetus butterflies. Evolution 61, 546–559. ( 10.1111/j.1558-5646.2007.00046.x) [DOI] [PubMed] [Google Scholar]
- 82.Lukhtanov VA, Shapoval NA, Anokhin BA, Saifitdinova AF, Kuznetsova VG. 2015. Homoploid hybrid speciation and genome evolution via chromosome sorting. P. R. Soc. B. 282, 20150157 ( 10.1098/rspb.2015.0157) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Šíchová J, Ohno M, Dinca V, Watanabe M, Sahara K, Marec F. 2016. Fissions, fusions, and translocations shaped the karyotype and multiple sex chromosome constitution of the northeast-Asian wood white butterfly, Leptidea amurensis. Biol. J. Linn. Soc. 118, 457–471. ( 10.1111/bij.12756) [DOI] [Google Scholar]
- 84.Lukhtanov VA, Dinca V, Talavera G, Vila R. 2011. Unprecedented within-species chromosome number cline in the wood white butterfly Leptidea sinapis and its significance for karyotype evolution and speciation. BMC Evol. Biol. 11, 109 ( 10.1186/1471-2148-11-109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Edger PP, et al. 2015. The butterfly plant arms-race escalated by gene and genome duplications. Proc. Natl Acad. Sci. USA 112, 8362–8366. ( 10.1073/pnas.1503926112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pardo-Diaz C, Salazar C, Baxter SW, Merot C, Figueiredo-Ready W, Joron M, Mcmillan WO, Jiggins CD. 2012. Adaptive introgression across species boundaries in Heliconius butterflies. PLoS Genet. 8, e1002752 ( 10.1371/journal.pgen.1002752) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Joron M, et al. 2011. Chromosomal rearrangements maintain a polymorphic supergene controlling butterfly mimicry. Nature 477, 203–206. ( 10.1038/nature10341) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kozak KM, Wahlberg N, Neild AFE, Dasmahapatra KK, Mallet J, Jiggins CD. 2015. Multilocus species trees show the recent adaptive radiation of the mimetic Heliconius butterflies. Syst. Biol. 64, 505–524. ( 10.1093/sysbio/syv007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Saura A, Schoultz Von B, Saura AO, Brown KSJ. 2013. Chromosome evolution in Neotropical butterflies. Hereditas 150, 26–37. ( 10.1111/j.1601-5223.2013.00008.x) [DOI] [PubMed] [Google Scholar]
- 90.Kulmuni J, Butlin RK, Lucek K, Savolainen V, Westram AM. 2020. Towards the completion of speciation: the evolution of further reproductive isolation beyond the first barriers. Phil. Trans. R. Soc. B 375, 20190528 ( 10.1098/rstb.2019.0528) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ahola V, Wahlberg N, Frilander MJ. 2017. Butterfly genomics: insights from the genome of Melitaea cinxia. Ann. Zool. Fennici 54, 275–291. ( 10.5735/086.054.0123) [DOI] [Google Scholar]
- 92.de Vos JM, Augustijnen H, Bätscher L, Lucek K. 2020. Data from: Speciation through chromosomal fusion and fission in Lepidoptera Zenodo. ( 10.5281/zenodo.3735954) [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- de Vos JM, Augustijnen H, Bätscher L, Lucek K. 2020. Data from: Speciation through chromosomal fusion and fission in Lepidoptera Zenodo. ( 10.5281/zenodo.3735954) [DOI] [PMC free article] [PubMed]
Supplementary Materials
Data Availability Statement
Data supporting this study have been deposited in the Zenodo repository at http://doi.org/10.5281/zenodo.3735954 [92]