

Abstract
Substoichiometric iron mediates the thioetherification of unactivated aliphatic C–H bonds directed by resident silylperoxides. Upon exposure to a catalytic amount of iron(II) triflate, TIPS-protected peroxides bearing primary, secondary, and tertiary C–H sites undergo chemoselective thioetherification of remote C–H bonds with diaryl disulfides. The reaction demonstrates a broad substrate scope and functional group tolerance without the use of any noble metal additives. Mechanistic experiments suggest that the reaction proceeds through 1,5-H atom abstraction by a hydroxyl radical generated with iron.
Graphical Abstract

INTRODUCTION
Thioethers are versatile synthetic intermediates, as they are readily oxidized to either the sulfoxide or the sulfone.1 Sulfoxides are well-known for enabling syn eliminations,2 Mislow–Evans rearrangements,3,4 Pummerer reactions,5,6 and sulfoxide–metal exchanges7,8 while also acting as a useful ligand class for transition metal catalysis.9 The higher-oxidation state sulfones also provide ample opportunities in organic synthesis, most notably as alkene precursors in the Julia–Lythgoe olefination,1,10 Kocienski-modified Julia olefination,11,12 and Ramberg–Bäcklund rearrangement.13,14 A recent report has also highlighted the utility of sulfones as radical cross-coupling partners.15 The preparation of alkyl thioethers generally proceeds through nucleophilic displacement of a (pseudo)halide with a thiol or the trapping of an alkyl radical or organometallic nucleophile with a disulfide.16 In addition to the synthetic versatility of thioethers, thioethers themselves are found in a variety of bioactive molecules such as Zofenopril, an FDA-approved angiotensin converting enzyme (ACE) inhibitor with antihypertensive and cardioprotective properties (Figure 1).17Čeković has reported that phenylsulfenyl ethers promote the selective thiophenylation of unactivated C–H bonds under photolytic conditions.18 With this limited precedent, a robust, catalytic method for the selective thioetherification of C(sp3) –H bonds remains desirable. While directed C–H functionalization has developed quickly over the past 20 years,19 strict steric and electronic requirements imposed by the transition metal-mediated C–H activation step have limited extension of this strategy to C(sp3) –H bonds. Here, we offer a directed, radical C–H thioetherification reaction that can overcome such limitations and enable facile C–H oxidations.
Figure 1.
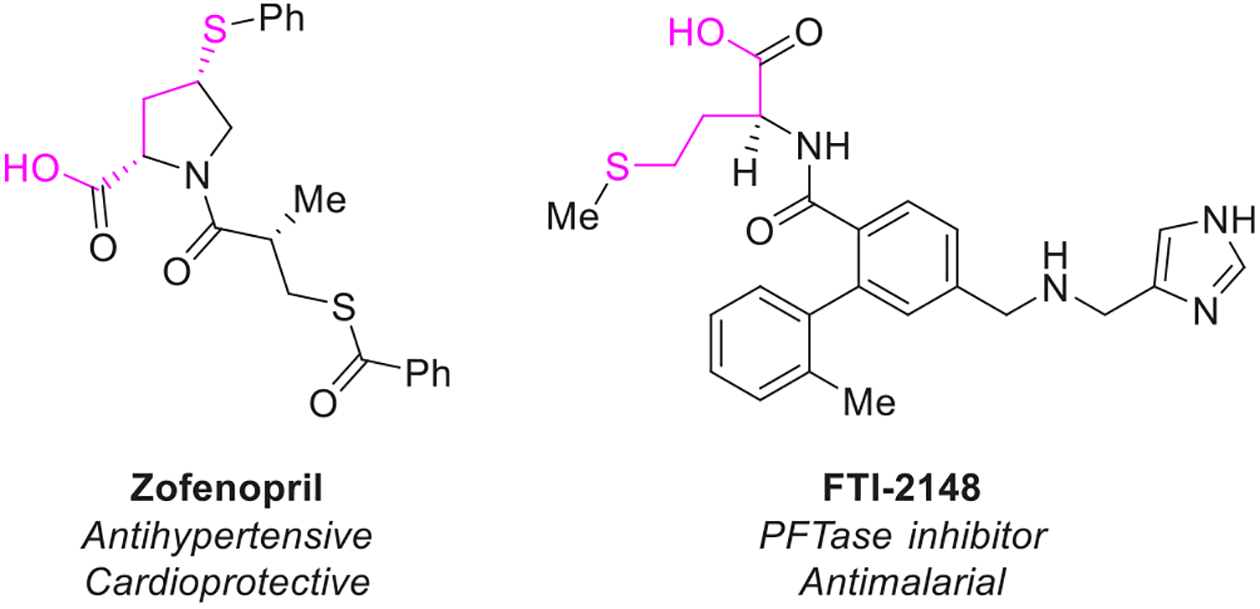
Bioactive molecules bearing a 1,4-oxy-thio motif.
The Fenton reaction (Figure 2a) represents the most widely implemented C–H oxidation in history.20–23 Inspired by this precedent, we sought to investigate an intramolecular variant of Fenton’s initial discovery. We hypothesized that by harnessing the facile 1,5-H-atom abstraction observed in other heteroatom-based radical systems, we could employ easily obtained peroxides in the presence of iron to generate the requisite H-atom abstractor.
Figure 2.

Directed radical chemistry as a route to C(sp3)–H thioetherification.
RESULTS AND DISCUSSION
To test the hypothesis, a series of peroxides were prepared from the oxidation of the primary alcohol (see the Supporting Information for full details). We have previously demonstrated that Fe(OTf)2 is an effective catalyst for the generation of N-centered radicals (Figure 2b).24 Interestingly, exposure of symmetrical peroxide 1 to Fe(OTf)2 in the presence of excess disulfide trap provided the desired product in 50% NMR yield (Scheme 1). Encouraged by this preliminary result, we sought to improve the reaction with the goal of developing a general method for selective C–H thioetherification.
Scheme 1.

Thioetherification of an Unactivated C–H Bond
The use of an unsymmetric peroxide improves the atom economy of the transformation but also introduces a challenge of selectivity for the desired alkoxy radical during O–O bond cleavage. We selected dodecyl-methyl peroxide 3a as a model substrate for optimization of the reaction parameters (Table 1). Although the room-temperature reaction did not proceed at a low concentration, we found that at 0.5 M, the reaction with 5 mol % Fe(OTf)2 and 1.5 equiv of disulfide provided 4a in 41% yield (entries 1 and 2, Table 1). Although higher reaction temperatures, catalyst loadings, or disulfide loadings did not substantially improve the reaction (entries 3, 4, and 6–8, Table 1), we were pleased to find that the disulfide loading could be decreased to 1.1 equiv with no loss of yield (entry 5, Table 1). As with the iron-catalyzed peroxide reductions that Bao has reported,25,26 all other precatalysts that wee evaluated were inferior to Fe(OTf)2 (see the Supporting Information). At this stage, we noticed that a number of reactions with Fe(OTf)2 provided yields between 40% and 45% with a nearly 1:1 4a:1-dodecanol ratio. Both of these outcomes suggested that all steps of the putative catalytic cycle (O–O cleavage, HAT, radical quench, and catalyst turnover) were proceeding efficiently, but the O–O cleavage simply lacked selectivity. Therefore, we reasoned that replacing the methyl with a variety of substituents would be the most likely way to improve the reaction beyond its current state.
Table 1.
Optimization of Pertinent Reaction Parameters
 | |||||
|---|---|---|---|---|---|
| entry | Ph2S2 (equiv) | temp (°C) | % Fe | product (%)a | 1-dodecanol (%) |
| 1b | 1.5 | rt | 5 | 0 | 10 |
| 2 | 1.5 | rt | 5 | 41 | 43 |
| 3 | 1.5 | rt | 20 | 23 | 52 |
| 4 | 1.5 | 40 | 5 | 41 | 45 |
| 5 | 1.1 | 40 | 5 | 41 | 43 |
| 6 | 1.5 | 80 | 5 | 45 | 35 |
| 7 | 1.5 | 80 | 20 | 35 | 42 |
| 8 | 4.0 | rt | 5 | 43 | 42 |
NMR yield using BHT as the internal standard. Full conversion unless otherwise noted.
Reaction run at a 0.05 M concentration and 24% conversion.
A series of sacrificial substituents with varying electronic and steric properties were evaluated in the C–H thioetherification reaction (Table 2). The hydroperoxide provided a very similar 39% yield (entry 2, Table 2), perhaps due to the similar pKa values of a primary alcohol and water.27 Surprisingly, acyl peroxides performed poorly in the reaction (entry 3, Table 2).28 Considering the mechanism of the Fenton reaction, the acyl peroxide should exhibit high selectivity for the alkoxy radical and carboxylate with negligible formation of the opposite pair. The 6% yield from trifluoroacetyl peroxide 3c, however, was the highest of all acyl peroxides examined (see the Supporting Information). Although most silyl groups were comparable to the methyl- and hydroperoxides (entries 4–9, Table 2), triisopropylsilyl (TIPS)-protected peroxide 3j stood out because of its incomplete conversion and 2:1 4a:1-dodecanol ratio (entry 10, Table 2), the first sign of selective O–O bond cleavage. Further experiments revealed that the reaction proceeded most efficiently at higher temperatures and lower concentrations, with an optimal 71% yield and 3.5:1 selectivity at 80 °C and 0.1 M (entry 11, Table 2). Further experiments revealed that the reaction proceeded to full conversion within 5 h. Slight modifications to the reaction conditions, such as addition of Fe(OTf)2 as a solution in methanol (3 is insoluble in methanol), an increased disulfide loading, or an increased catalyst loading, had a minimal impact on the reaction. The TIPS group remained superior after an extensive screening of other silyl groups (see the Supporting Information for additional details).
Table 2.
Investigation of Nonsymmetrical Peroxides
 | |||||
|---|---|---|---|---|---|
| entry | R group | yield (%)a | RSM (%) | 1-dodecanol (%) | |
| 1 | Me | 3a | 41 | 0 | 43 |
| 2 | H | 3b | 39 | 0 | 37 |
| 3 | C(O)CF3 | 3c | 6 | 21 | 28 |
| 4 | Si(OEt)3 | 3d | 43 | 0 | nd |
| 5 | SiMe3 | 3e | 34 | 4 | 36 |
| 6 | SiEt3 | 3f | 37 | 0 | 36 |
| 7 | Si(C2H3)Me2 | 3g | 50 | 0 | 37 |
| 8 | SiMePh2 | 3h | 44 | 0 | 30 |
| 9 | SiMe2tBu | 3i | 44 | 0 | 27 |
| 10 | SiiPr3 | 3j | 23 | 55 | 11 |
| 11b | SiiPr3 | 71 | 0 | 20 | |
NMR yield using BHT as an internal standard.
At 80 °C and 0.1 M.
With optimized conditions for the directed C(sp3)–H thioetherification, we evaluated the performance of the reaction across a number of substrates. Pleasingly, the reaction is effective for the functionalization of primary, secondary, and tertiary C–H sites (Table 3), with the highest yields obtained for secondary systems (Table 3, 4a, 8, 9, and 11–23). Interestingly, the 2-hexyl peroxide provided a 5:1 mixture of [1,5] and [1,6] products, but 8 was isolated as a single isomer. The improved yield of primary thioether 6 in comparison to 5 is likely the result of a Thorpe–Ingold effect imposed by the methyl group. As expected, a secondary C–H undergoes preferential (1:0.8) thioarylation over primary C–H bonds (Table 3, 11). Moreover, alcohols bearing chloro, ester, amide, and sulfonic ester functional groups (14–20) function well under the reaction conditions. Usefully, silyl-protected alcohols undergo desilylation during the reaction to produce thioarylated diols (22 and 23).
Table 3.
Substrate Scope for Thioetherificationa

|
Isolated yield.
Reaction run for 10 h.
Isolated as a mixture with another regioisomer with a 1:0.08 ratio.
Reaction run for 8 h.
Total thioarylation yield, as deacylation occurred in the reaction.
TIPS deportection of alcohol observed during the reaction.
TBDS deprotection of alcohol observed during the reaction, reaction run for 12 h.
Next, we examined the scope of various disulfide traps (Table 4). Although naphthyl disulfide 4b proved to be less effective (perhaps due to its poor solubility in methanol), we found that both electron-poor and electron-rich aryl disulfides 24–31 performed similarly to Ph2S2. In contrast, 4-nitrodisulfide 31 provided very low yields, and no product was observed with 2-carboxyphenyl disulfide 32. Attempts to extend the methodology to dialkyl disulfides proved difficult as alkyl disulfides trap alkyl radicals more slowly than aryl disulfides.29,30 For example, dimethyl disulfide 33 and dioctyl disulfide fail to produce any product.
Table 4.
Disulfide Scope for Thioetherification











Isolated yield. Full conversion in all cases.
To examine the electronic preference during C–S bond formation, we tested the reaction with electronically differentiated disulfides (Table 5). Little difference was observed in a competition between phenyl and naphthyl (product ratio of 1.03). Upon comparison of electron-poor (4-nitrophenyl) or electron-rich (4-methoxyphenyl) to phenyl disulfides, however, the reaction clearly favored the product that incorporated the more electron-rich sulfur system.
Table 5.
Thioetherification with an Unsymmetrical Disulfide

|
The presumption of a Fenton mechanism guided the conceptual development of this thioetherification reaction. To test this mechanistic hypothesis, experiments were conducted to uncover evidence of the presence of radical intermediates. Providing support for an oxy-radical intermediate, 1-pentenyl peroxide 34 undergoes 5-exo-trig cyclization and subsequent trapping to form the substituted tetrahydrofuran 35. Additionally, cyclopentyl peroxide 36 provides α-cleavage product 37 in 40% isolated yield as the dimethyl acetal (Scheme 2a). Interestingly, substrate 38 could undergo 5-exo-dig cyclization to form tetrasubstituted enol 39 in a remarkable 41% yield. Furthermore, cyclopropylmethyl substrate 40 undergoes complete ring opening to afford primary sulfide 41 in 51% isolated yield (Scheme 2b). The sum of these results suggests that the reaction occurs via a radical intermediate with a lifetime of approximately 10−3–10−4 s.31
Scheme 2.

Radical-Clock Experiments
If the iron catalyst acted as an initiator and the reaction proceeded thereafter through a radical chain propagation, then PhS• should be capable of initiating the reaction. When PhSH and AIBN were used to generate PhS•, the peroxide underwent full conversion with no sign of thioetherification (entry 1, Table 6). In contrast, with Ph2S2 (2.1 or 1.1 equiv) and AIBN, we observed low yields of the product (entries 2 and 3, Table 6). Additional experimentation revealed that the reaction did not require an exogenous initiator as the reaction with 1.1 equiv of Ph2S2 and no AIBN provided a 54% yield (entry 4, Table 6). Interestingly, the thermal, iron-free reactions produced 7–15% 1-dodecanal as a side product while the iron-catalyzed variants produced, at best, trace quantities of 1-dodecanal.
Table 6.
Disulfide Scope for Thioetherification
 | |||||
|---|---|---|---|---|---|
| entry | precursor | yield (%)a | RSM (%) | 1-dodecanol (%) | 1-dodecanal (%) |
| 1 | PhSH | 0 | 0 | 75 | 8 |
| 2b | Ph2S2 | 9 | 69 | 7 | 7 |
| 3 | Ph2S2 | 26 | 36 | 17 | 12 |
| 4c | Ph2S2 | 54 | 0 | 21 | 15 |
NMR yield using BHT as the internal standard.
With 2.1 equiv of Ph2S2.
No AIBN.
Intrigued by this result, we examined the thermal profile of the background reaction in greater detail. When run to 18 h, thermal decomposition of the O–O bond begins between 50 and 60 °C. At temperatures above 80 °C, the maximum yield is 59%. Importantly, we found that at 80 °C, thermal decomposition only begins to set in at the 5 h mark, and the half-life of the peroxide is approximately 10 h (see the Supporting Information for full details), thereby providing strong evidence of an iron-mediated mechanism.
In conclusion, we developed a mild silylperoxide-directed C–H thioetherification reaction catalyzed by a simple iron salt, Fe(OTf)2. The reaction proceeds in good yield with broad functional group tolerance under simple reaction conditions. Importantly, the use of a TIPS protecting group biases the scission of the O–O bond and enables a selective alkoxy-radical formation. The reaction proceeds through short-lived radical intermediates that provide high selectivity for 1,5-HAT.
EXPERIMENTAL PROCEDURES AND CHARACTERIZATIONS
General Methods.
Analytical grade solvents and commercially available reagents were purchased from commercial sources and used directly without further purification unless otherwise stated. THF and DME were purified according to the Grubbs procedure.32 Thin layer chromatography (TLC) was carried out on Merck 60 F254 precoated, glass silica plates that were visualized with ultraviolet light or stained with KMnO4. 1H NMR and 13C{1H} NMR spectra were recorded at room temperature using a Varian I400 or VXR400 (1H NMR at 400 MHz and 13C{1H} NMR at 100 MHz), Varian I500 (1H NMR at 500 MHz and 13C{1H} NMR at 125 MHz), and Varian I600 (1H NMR at 600 MHz and 13C{1H} NMR at 150 MHz) instruments. 19F NMR spectra were recorded at room temperature using a Varian I400 or VXR400 instrument (19F NMR at 376 MHz). Chemical shifts are reported in parts per million with reference to solvent signals [1H NMR, CDCl3 (7.26 ppm) and acetone-d6 (2.05 ppm); 13C NMR, CDCl3 (77.2 ppm)]. 19F NMR was externally referenced to a trifluoroacetic acid standard (−77.55 ppm). Signal patterns are indicated as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Infrared spectra (IR) were recorded on a Bruker Tensor II FTIR spectrometer analyzed as a thin film and recorded in wavenumbers (cm−1). High-resolution mass spectrometry (HRMS) analysis was performed using either an Agilent 7890B/7250 GCQTOF system for electron-impact ionization (EI) and reported as m/ z (relative intensity) for the molecular ion [M]/suitable fragment ion or a Waters/Micromass LCT Classic system (ESI-TOF) for electrospray ionization (ESI) or atmospheric-pressure chemical ionization (APCI) and reported molecular ion [M + H] or [M + Na] or a suitable fragment ion.
General Procedure A. Synthesis of Alkyl Mesylates.
To a round-bottom flask with a stir bar were added alcohol (1 equiv), DCM (0.4 M), and methanesulfonyl chloride (1.1 equiv). At room temperature, triethylamine (1.2 equiv) was added dropwise. The reaction mixture was stirred at room temperature for 3–15 h, the reaction quenched with 1 M aqueous HCl, and the mixture diluted with water and DCM and transferred to a separatory funnel. The organic layer was removed, and the aqueous layer was then extracted three times with DCM. The combined organic layers were washed with saturated aqueous NaHCO3 and then brine. The organic layer was dried (MgSO4), filtered, and concentrated by rotary evaporation. In general, the crude alkyl mesylate was pure as determined by 1H NMR and did not require further purification. The spectra of 1-butyl, 1-pentyl, 1-hexyl, 2-hexyl, 1-dodecyl, 4-pentenyl, and cyclopentyl mesylates matched the reported spectra.
General Procedure B. Synthesis of Dialkyl Peroxides.33
To a round-bottom flask with a stir bar were added alkyl mesylate (2 equiv), methanol (1.0 M), and 35% aqueous H2O2 (1 equiv). The stirred solution was cooled in an ice bath, and 50% (w/w) aqueous KOH (2 equiv) was added dropwise. The reaction mixture was allowed to warm to room temperature and stirred for 14–24 h. After this time, the reaction mixture was diluted with water and hexanes. The organic layer was removed, and the aqueous layer was extracted three times with hexanes. The combined organic layers were washed once with 1 M aqueous NaOH to remove residual hydrogen peroxide, washed three times with water until the washings were neutral, followed by brine, and then dried (MgSO4), filtered, and concentrated by rotary evaporation (keeping the water bath below 40 °C) to afford a clear, colorless oil. The peroxides were purified by silica flash chromatography to clear, colorless oils. Typical Rf values are 0.65–0.75 in 25% EtOAc/hexanes.
General Procedure C. Synthesis of Hydroperoxides from Alkyl Mesylate.34
To a round-bottom flask with a stir bar were added alkyl mesylate (1 equiv), methanol/water (9:1, 0.8 M), and 35% aqueous H2O2 (6 equiv). The stirred solution was cooled in an ice bath, and 50% (w/w) aqueous KOH (1.5 equiv) was added dropwise. The reaction mixture was allowed to warm to room temperature and stirred for 14–24 h. Then, the reaction mixture was diluted with water and diethyl ether. The organic layer was removed, and the aqueous layer was extracted three times with ether. The combined organic layers were washed three times with water and twice with brine, dried (MgSO4), filtered, and concentrated by rotary evaporation (keeping the water bath below 40 °C) to afford a clear, colorless oil. The hydroperoxides were purified by silica flash chromatography to clear, colorless oils. Hydroperoxides stain brightly with KMnO4, and typical Rf values are 0.3–0.4 in 15% EtOAc/hexanes.
General Procedure D. Synthesis of Hydroperoxides from Alkyl Iodide.35
The hydroperoxides were prepared by adding silver trifluoroacetates (1.2 equiv) in small portions to an ice-cooled solution in the ether of the appropriate alkyl halide and hydrogen peroxide in a ≤1-fold excess. The reactions were run for 1–3 h. The silver halide was filtered off, and the trifluoroacetic acid formed; the excess of hydrogen peroxide was removed by treatment with aqueous sodium bicarbonate, and the solvent was removed to leave the crude hydroperoxide. The hydroperoxides were purified by silica flash chromatography to clear, colorless oils. Hydroperoxides stain brightly with KMnO4, and typical Rf values are 0.3–0.4 in 15% EtOAc/hexanes.
General Procedure E. Silylation of Hydroperoxides.36
To a flame-dried round-bottom flask with a stir bar were added hydroperoxide (1.0 equiv) and pentane (0.5 M). The flask was cooled in an ice bath, and the silyl chloride or triflate (1.0 equiv) was added dropwise, followed by the dropwise addition of distilled 2,6-lutidine (1.0 equiv). The reaction mixture was slowly warmed to room temperature and stirred for 3–24 h. After the specified time, the reaction mixture was diluted with diethyl ether and washed with water, followed by brine. The organic layer was dried (MgSO4), filtered, and concentrated to provide a clear, colorless oil. If necessary, the alkyl silyl peroxide was purified by silica flash chromatography.
General Procedure F. Silyl Hydroperoxide Alkylation.
To a reaction vial with a stir bar were added triisopropyl hydroperoxide (1 equiv) and pentane (0.5 M). The solution was cooled in an ice bath. Ag2O (3 equiv) was added, followed by the dropwise addition of alkyl iodide (3 equiv). The reaction vessel was covered with foil and left in the ice bath to warm to room temperature slowly. After 14 h, the reaction mixture was diluted with diethyl ether, filtered, and concentrated by rotary evaporation. The alkyl silyl peroxide product was purified by silica flash chromatography. The alkyl iodide closely elutes in many cases but can often be removed under high vacuum.
General Procedure G. C–H Thioetherification Reaction.
To a flame-dried 40 mL reaction vial with a septum-lined cap and a stir bar were added TIPS peroxide (1.0 equiv), disulfide (1.1 equiv), and Fe(OTf)2 (0.05 equiv). The flask was evacuated and backfilled three times with N2. Degassed MeOH (0.1 M) was added via syringe, and then the reaction mixture was heated to 80 °C on a preheated oil bath. After 5 h, the reaction mixture was cooled, diluted with ethyl acetate (5 mL), filtered through a silica plug, washing with ~10 mL of EtOAc, and concentrated. Screening efforts were usually run on a 0.05–0.2 mmol scale. Products were isolated by flash chromatography or analyzed by crude NMR using BHT as the internal standard.
Preparation and Characterization of the Starting Material.
1-Hydroperoxydodecane (3b).
1-Dodecyl mesylate (2.652 g, 10.0 mmol) was subjected to general procedure C, using diethyl ether (1.25 M) and 12:1 MeOH/H2O (0.3 M) as the solvents, for 42 h. Silica flash chromatography provided 3b (1.522 g, 75% yield) as a clear, colorless oil that matched the reported spectra:37 1H NMR (400 MHz, CDCl3) δ 7.80 (s, 1H), 4.02 (t, J = 6.6 Hz, 2H), 1.62 (p, J = 7.1 Hz, 2H), 1.42–1.18 (m, 18H), 0.88 (t, J = 6.7 Hz, 3H); TLC Rf = 0.40 (20% EtOAc/hexanes).
(Dodecylperoxy)triisopropylsilane (3j).
1-Dodecyl hydroperoxide 3b (1.089 g, 5.0 mmol, 1 equiv) was subjected to general procedure E using triisopropylsilyl triflate (1.40 mL, 5.0 mmol, 1 equiv) for 23 h. The reaction mixture was diluted with 10 mL of hexanes, filtered through a silica plug, washing with an additional 5 mL of hexanes, and concentrated to provide 3j (1.375 g, 76% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 3.99 (t, J = 6.6 Hz, 2H), 1.58 (p, J = 6.7 Hz, 2H), 1.39–1.14 (m, 21H), 1.10 (d, J = 6.8 Hz, 18H), 0.88 (t, J = 6.8 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 76.9, 32.1, 29.8, 29.8, 29.8, 29.7, 29.6, 29.5, 27.9, 26.3, 22.9, 18.1, 14.3, 11.7; LRMS (GCMS) mass fragmentation 315.3 (100), 273.1 (8), 131.2 (13), 103.1 (13), 77.1 (13), 43.2 (6); IR (neat) 2922, 2865, 1737, 1463, 1377, 997, 882, 677 cm−1; TLC Rf = 0.74 (15% EtOAc/hexanes).
(Butylperoxy)triisopropylsilane.
TIPSOOH (295 mg, 1.5 mmol, 1 equiv) was subjected to general procedure F for 15 h using iodobutane (0.51 mL, 4.5 mmol, 3 equiv). Silica flash chromatography provided (butylperoxy)triisopropylsilane (197.1 mg, 53% yield) as a clear, colorless oil: 1H NMR (600 MHz, CDCl3) δ 4.00 (t, J = 6.6 Hz, 2H), 1.58 (p, J = 6.9 Hz, 2H), 1.38 (sext, J = 7.3 Hz, 2H), 1.20 (sext, J = 7.3 Hz, 2H), 1.10 (d, J = 7.4 Hz, 18H), 0.92 (t, J = 7.4 Hz, 3H); 13C{1H} NMR (125 MHz, CDCl3) δ 76.7, 30.1, 19.6, 18.1, 14.1, 11.7; HRMS (APCI) calcd for C13H31O2Si [M + H]+ 247.2093, found 247.2086; IR (neat) 2943, 2866, 1463, 1382, 1064, 881, 676 cm−1; TLC Rf = 0.63 (5% EtOAc/hexanes).
Triisopropyl(pentan-2-ylperoxy)silane.
TIPSOOH (380 mg, 2.0 mmol, 1 equiv) was subjected to general procedure F for 16 h using 2-iodopentane (0.60 mL, 4.6 mmol, 2.3 equiv). Silica flash chromatography provided triisopropyl(pentan-2-ylperoxy)silane (247 mg, 47% yield) as a clear, colorless oil: 1H NMR (500 MHz, CDCl3) δ 4.04 (sextet, J = 6.0 Hz, 1H), 1.67–1.53 (m, 1H), 1.45–1.28 (m, 3H), 1.25–1.14 (m, 6H), 1.09 (d, J = 7.3 Hz, 18H), 0.94–0.88 (m, 3H); 13C{1H} NMR (125 MHz, CDCl3) δ 81.1, 36.8, 18.9, 18.7, 18.2, 14.4, 11.8; LRMS (GCMS) mass fragmentation 201.3 (3), 173.3 (6), 147.2 (20), 105.2 (43), 103.1 (30), 77.2 (100), 76.1 (35), 61.1 (17), 43.3 (14); IR (neat) 2960, 2866, 1738, 1463, 1370, 997, 881, 676 cm−1; TLC Rf = 0.71 (10% EtOAc/hexanes).
(Isopentylperoxy)triisopropylsilane.
1-Iodo-3-methylbutane (1.98 g, 10 mmol) was subjected to general procedure D for 2 h. Silica flash chromatography provided 1-hydroperoxy-3-methylbutane (392 mg, 38% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.46 (s, 1H), 4.06 (t, J = 6.8 Hz, 2H), 1.69 (dp, J = 13.3, 6.7 Hz, 1H), 1.52 (q, J = 6.9 Hz, 2H), 0.92 (d, J = 6.6 Hz, 6H); 13C{1H} NMR (100 MHz, CDCl3) δ 75.95, 36.46, 25.39, 22.84; TLC Rf = 0.5 (20% EtOAc/hexanes). 1-Hydroperoxy-3-methylbutane (330 mg, 3.17 mmol) was subjected to general procedure E for 3 h. Silica flash chromatography provided (isopentylperoxy)triisopropylsilane (640 mg, 78% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 4.02 (t, J = 6.8 Hz, 2H), 1.69 (dq, J = 13.4, 6.7 Hz, 1H), 1.55–1.40 (m, 2H), 1.29–1.13 (m, 3H), 1.15–0.99 (m, 18H), 0.90 (d, J = 6.6 Hz, 6H); 13C{1H} NMR (126 MHz, CDCl3) δ 75.5, 36.7, 25.5, 22.8, 18.1, 11.7; LRMS (GCMS) mass fragmentation 201.3 (1), 175.2 (1), 168.2 (1), 147.2 (1), 129.2 (8), 119.2 (27), 77.2 (100), 76.1 (35), 69.2 (35), 61.1 (20), 43.3 (10); IR (neat) 2945, 2867, 1464, 1367, 1014, 997, 881, 819, 743 cm−1; TLC Rf = 0.5 (5% EtOAc/hexanes).
1-Hydroperoxyhexane.
1-Hexyl mesylate (1.465 g, 8.0 mmol) was subjected to general procedure C for 20 h. Silica flash chromatography provided 1-hydroperoxyhexane (623 mg, 66% yield) as a clear, colorless oil that matched the reported spectra:38 1H NMR (600 MHz, CDCl3) δ 7.91–7.84 (br s, 1H), 4.02 (t, J = 6.7 Hz, 2H), 1.61 (p, J = 7.0 Hz, 2H), 1.45–1.20 (m, 6H), 0.89 (t, J = 6.8 Hz, 3H); TLC Rf = 0.31 (15% EtOAc/hexanes).
(Hexylperoxy)triisopropylsilane.
1-Hydroperoxyhexane (258 mg,2.2 mmol, 1 equiv) was subjected to general procedure E using triisopropylsilyl triflate (0.59 mL, 2.2 mmol, 1 equiv) for 14 h. The reaction mixture was diluted with 3 mL of pentane, filtered through a silica plug, washing with an additional 5 mL of pentane, and concentrated to provide (hexylperoxy)triisopropylsilane (366 mg, 60% yield) as a clear, colorless oil that did not require further purification: 1H NMR (400 MHz, CDCl3) δ 3.99 (t, J = 6.6 Hz, 2H), 1.59 (p, J = 6.9 Hz, 2H), 1.41–1.25 (m, 6H), 1.21 (hept, J = 7.9, 7.1 Hz, 3H), 1.10 (d, J = 7.4 Hz, 18H), 0.89 (t, J = 6.8 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 76.9, 31.8, 27.9, 26.0, 22.7, 18.1, 14.1, 11.7; HRMS (EI) calcd for C12H27O2Si [M – C3H7]+ 231.1775, found 231.1769; IR (neat) 2942, 2866, 1741, 1463, 1381, 882, 676 cm−1.
2-Hydroperoxyhexane.
2-Hexyl mesylate (3.62 g, 20 mmol, 1 equiv) was subjected to general procedure C at a concentration of 0.3 M (12:1 MeOH/H2O) for 19 h. Silica flash chromatography provided 2-hydroperoxyhexane (879 mg, 37% yield) as a clear, colorless oil:39 1H NMR (600 MHz, CDCl3) δ 7.69–7.54 (m, 1H), 4.06 (h, J = 6.1 Hz, 1H), 1.71–1.55 (m, 1H), 1.49–1.26 (m, 5H), 1.22 (d, J = 6.2 Hz, 3H), 0.90 (t, J = 7.0 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 81.9, 33.8, 27.7, 22.9, 18.3, 14.1; IR (neat) 3374, 2933, 2862, 1737, 1461, 1375, 1114 cm−1; TLC Rf = 0.31 (15% EtOAc/hexanes).
(Hexan-2-ylperoxy)triisopropylsilane.
2-Hexyl hydroperoxide (473 mg, 4.0 mmol, 1 equiv) was subjected to general procedure E using triisopropylsilyl triflate (1.07 mL, 4.0 mmol, 1 equiv) and anhydrous ammonia as the base (sparge for 10 min and then a balloon atmosphere) for 16 h. Silica flash chromatography provided (hexan-2-ylperoxy)triisopropylsilane (796 mg, 72% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 4.03 (sext, J = 6.0 Hz, 1H), 1.70–1.56 (m, 1H), 1.47–1.25 (m, 5H), 1.24–1.15 (m, 6H), 1.09 (d, J =7.3 Hz, 18H), 0.89 (t, J = 6.9 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 81.3, 34.3, 27.8, 23.0, 18.6, 18.2, 14.2, 11.8; HRMS (EI) calcd for C12H27O2Si [M – C3H7]+ 231.1775, found 231.1769; IR (neat) 2942, 2866, 1738, 1463, 1371, 881, 791 cm−1; TLC Rf = 0.75 (10% EtOAc/hexanes).
Triisopropyl[(4-methylpentyl)peroxy]silane.
TIPSOOH (344 mg,1.8 mmol, 1 equiv) was subjected to general procedure F using 1-iodo-4-methylpentane (0.80 mL, 5.4 mmol, 3 equiv). Silica flash chromatography provided triisopropyl[(4-methylpentyl)peroxy]silane (205.5 mg, 41% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 3.97 (t, J = 6.7 Hz, 2H), 1.68–1.46 (m, 3H), 1.29–1.14 (m, 5H), 1.10 (d, J = 7.0 Hz, 18H), 0.88 (d, J = 6.6 Hz, 6H); 13C{1H} NMR (100 MHz, CDCl3) δ 77.2, 35.3, 28.0, 25.8, 22.6, 18.1, 11.7; HRMS (EI) calcd for C12H27O2Si [M – C3H7]+ 231.1775, found 231.1769; IR (neat) 2945, 2867, 1463, 1367, 1250, 1015, 882, 676 cm−1; TLC Rf = 0.60 (5% EtOAc/hexanes).
2-Ethylhexyl Methanesulfonate.
2-Ethylhexan-1-ol (4.69 mL, 30.0 mmol) was subjected to general procedure A for 14 h. After the workup procedure, the crude material was isolated with high purity. We moved to the next step without any further purification (5.933 g, 95% yield): 1H NMR (400 MHz, CDCl3) δ 4.27–4.01 (m, 2H), 3.00 (s, 3H), 1.65 (p, J = 6.0 Hz, 1H), 1.48–1.20 (m, 8H), 1.02–0.82 (m, 6H); 13C{1H} NMR (100 MHz, CDCl3) δ 72.2, 39.2, 37.1, 29.9, 28.8, 23.3, 22.9, 14.0, 10.8; HRMS (ESI) calcd for C9H20O3NaS [M + Na]+ 231.1025, found 231.1026; IR (neat) 2959, 2931, 2861, 1463, 1350, 1172, 939, 845, 816, 748, 527 cm−1; TLC Rf = 0.69 (30% EtOAc/hexanes).
[(2-Ethylhexyl)peroxy]triisopropylsilane.
2-Ethylhexyl methanesulfonate (5.208 g, 25.0 mmol) was subjected to general procedure C, using diethyl ether (1.25 M) and 12:1 MeOH/H2O (0.3 M) as the solvents, for 40 h. Silica flash chromatography provided 3-(hydroperoxymethyl)heptane (1.501 g, 42% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 8.07–7.87 (m, 1H), 3.91 (d, J = 6.1 Hz, 2H), 1.61 (q, J = 5.9 Hz, 1H), 1.45–1.12 (m, 8H), 0.87 (t, J =7.4 Hz, 6H); 13C{1H} NMR (100 MHz, CDCl3) δ 80.4, 38.1, 30.6, 29.1, 24.0, 23.1, 14.2, 11.1; IR (neat) 3423 (b), 2559, 2928, 1540, 1462, 1379, 1172, 941, 816, 412 cm−1; TLC Rf = 0.43 (30% EtOAc/hexanes). Next, 3-(hydroperoxymethyl)heptane (426 mg,2.91 mmol) was subjected to general procedure E for 3 h. Silica flash chromatography provided [(2-ethylhexyl)peroxy]-triisopropylsilane (588 mg, 67% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ δ 3.91 (d, J = 5.9 Hz, 2H), 1.57 (td, J =11.7, 5.7 Hz, 1H), 1.43–1.24 (m, 8H), 1.24–1.14 (m, 3H), 1.14–1.03 (m, 18H), 0.88 (t, J = 7.3 Hz, 6H); 13C{1H} NMR (126 MHz, CDCl3) δ 79.8, 38.7, 30.9, 29.2, 24.3, 23.1, 18.2, 14.2, 11.7, 11.3; HRMS (EI) calcd for C17H38O2Si [M]+ 302.2636, found 302.2632; IR (neat) 2927, 2866, 1463, 1381, 1250, 997, 881, 677 cm−1; TLC Rf = 0.64 (5% EtOAc/hexanes).
(Cyclooctylperoxy)triisopropylsilane.
TIPSOOH (380 mg, 2.0 mmol, 1 equiv) was subjected to general procedure F using iodocyclooctane (1.42 g, 6 mmol, 3 equiv). Silica flash chromatography provided (cyclooctylperoxy)triisopropylsilane (288 mg, 48% yield) as a clear, colorless oil: 1H NMR (400 MHz, chloroform-d) δ 4.06 (tt, J = 8.4, 3.5 Hz, 1H), 1.92 (ddt, J = 14.0, 8.1, 2.8 Hz, 2H), 1.70 (dtd, J = 12.6, 8.0, 3.5 Hz, 2H), 1.62–1.30 (m, 10H), 1.30–1.12 (m, 3H), 1.09 (d, J = 6.7 Hz, 18H); 13C{1H} NMR (101 MHz, chloroform-d) δ 85.8, 30.1, 27.4, 25.9, 23.7, 18.3, 11.8; LRMS (GCMS) mass fragmentation 257.3 (3), 173.2 (3), 147.2 (33), 105.2 (66), 77.2 (100), 69.2 (20), 61.2 (20), 41.3 (13); IR (neat) 2922, 2865, 1463, 1447, 1382, 1366, 1250, 1052, 1014, 997, 881, 776, 676 cm−1; TLC Rf = 0.7 (5% EtOAc/hexanes).
3-Phenylbutyl Methanesulfonate.
3-Phenylbutan-1-ol (1.5 g, 10 mmol) was subjected to general procedure A for 14 h. After the workup procedure, the crude material was isolated with high purity. We moved to the next step without any further purification (2.3 g, 98% yield): 1H NMR (400 MHz, CDCl3) δ 7.34 (dd, J = 8.2, 6.9 Hz, 2H), 7.27–7.19 (m, 3H), 4.34–3.89 (m, 2H), 3.01–2.80 (m, 4H), 2.17–1.94 (m, 2H), 1.33 (d, J = 7.0 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 145.4, 128.8, 127.1, 126.7, 68.6, 37.3, 37.3, 36.3, 27.2, 22.4; HRMS (ESI) calcd for C11H16O3SNa [M + Na]+ 251.0712, found 251.0714; IR (neat) 2962, 2935, 1494, 1348, 1170, 935, 700, 525 cm−1; TLC Rf = 0.7 (30% EtOAc/hexanes).
(4-Hydroperoxybutan-2-yl)benzene.
3-Phenylbutyl methanesulfonate (1.2 g, 5.26 mmol) was subjected to general procedure C, using diethyl ether (1.25 M) and 12:1 MeOH/H2O (0.3 M) as the solvents, for 48 h. Silica flash chromatography provided (4-hydroperoxybutan-2-yl)benzene (340 mg, 39% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.96–7.76 (m, 1H), 7.31 (t, J = 7.7 Hz, 2H), 7.24–7.16 (m, 3H), 3.97–3.86 (m, 2H), 3.14–2.74 (m, 1H), 2.01–1.86 (m, 2H), 1.29 (d, J = 6.9 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 146.6, 128.6, 127.1, 126.3, 75.5, 36.7, 35.9, 22.6; HRMS (ESI) calcd for C10H14O4Na [M + Na + O2]+ 221.0784, found 221.0784; IR (neat) 3372 (b), 2959, 1493, 1451, 1374, 1051, 761, 698, 546 cm−1; TLC Rf = 0.38 (20% EtOAc/hexanes).
Triisopropyl[(3-phenylbutyl)peroxy]silane.
(4-Hydroperoxybutan-2-yl)benzene (238 mg, 2.64 mmol) was subjected to general procedure E for 3 h. Silica flash chromatography provided triisopropyl[(3-phenylbutyl)peroxy]silane (611 mg, 72% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.29 (dd, J = 8.1, 6.9 Hz, 2H), 7.22–7.14 (m, 3H), 3.91 (t, J = 6.7 Hz, 2H), 2.84 (h, J =7.1 Hz, 1H), 1.98–1.81 (m, 2H), 1.27 (d, J = 7.0 Hz, 3H), 1.24–1.12 (m, 3H), 1.12–1.01 (m, 18H); 13C{1H} NMR (126 MHz, CDCl3) δ 146.9, 128.5, 127.1, 126.2, 75.1, 36.8, 36.2, 22.6, 18.1, 11.7; HRMS (EI) calcd for C16H27O2Si [M – C3H7]+ 279.1775, found 279.1776; IR (neat) 2943, 2866, 1493, 1367, 1016, 881, 759, 698, 676 cm−1; TLC Rf = 0.64 (5% EtOAc/hexanes).
6-Chlorohexyl Methanesulfonate.
6-Chlorohexan-1-ol (2.732 g, 20.0 mmol) was subjected to general procedure A for 14 h. After the workup procedure, the crude material was isolated with high purity. We moved to the next step without any further purification (4.1 g, 96% yield)L 1H NMR (400 MHz, CDCl3) δ 4.23 (t, J = 6.5 Hz, 2H), 3.54 (t, J = 6.6 Hz, 2H), 3.01 (s, 3H), 1.90–1.70 (m, 4H), 1.47 (dtt, J = 14.9, 9.1, 5.8 Hz, 4H); 13C{1H} NMR (100 MHz, CDCl3) δ 69.8, 44.8, 37.4, 32.3, 29.0, 26.2, 24.8; HRMS (ESI) calcd for C7H15O3ClNaS [M + Na]+ 237.0323, found 237.0324; IR (neat) 2939, 2863, 1348, 1170, 943, 915, 817, 717, 526 cm−1; TLC Rf = 0.36 (30% EtOAc/hexanes).
[(6-Chlorohexyl)peroxy]triisopropylsilane.
6-Chlorohexyl methanesulfonate (3.64 g, 17.0 mmol) was subjected to general procedure C, using 12:1 MeOH/H2O (0.3 M) as the solvents, for 18 h. Silica flash chromatography provided 1-chloro-6-hydroperoxyhexane (1.317 g, 51% yield) as a clear, colorless oil: 1H NMR (500 MHz, CDCl3) δ 8.16 (s, 1H), 4.02 (t, J = 6.5 Hz, 2H), 3.53 (t, J = 6.7 Hz, 2H), 1.84–1.73 (m, 2H), 1.71–1.60 (m, 2H), 1.51–1.34 (m, 4H); 13C{1H} NMR (126 MHz, CDCl3) δ 77.0, 45.1, 32.6, 27.5, 26.8, 25.3; IR (neat) 3381 (b), 2937, 2861, 1461, 1367, 1309, 109, 763, 648 cm−1; TLC Rf = 0.52 (30% EtOAc/hexanes). Next, 1-chloro-6-hydro-peroxyhexane (763 mg, 5 mmol) was subjected to general procedure E for 3 h. Silica flash chromatography provided [(6-chlorohexyl)-peroxy]triisopropylsilane (1.278 g, 83% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 3.97 (t, J = 6.5 Hz, 2H), 3.51 (t, J =6.7 Hz, 2H), 1.88–1.70 (m, 2H), 1.65–1.55 (m, 2H), 1.50–1.32 (m, 4H), 1.30–1.11 (m, 3H), 1.07 (d, J = 7.0 Hz, 18H); 13C{1H} NMR (126 MHz, CDCl3) δ 75.6, 44.2, 31.6, 26.8, 26.2, 25.9, 24.6, 17.1, 10.7; LRMS (ESI) mass fragmentation 336.1 (11), 308.1 (55), 276.0 (16), 267.1 (29), 250.0 (10), 217.9 (14), 190.4 (8), 178.0 (41), 147.0 (100); IR (neat) 2942, 2865, 1463, 1366, 1015, 882, 781, 676, 661 cm−1; TLC Rf = 0.66 (5% EtOAc/hexanes).
Methyl 8-Bromohexanoate.
A mixture of 8-bromohexanoic acid (2.2 g, 10 mmol) and concentrated H2SO4 (0.3 mL) in methanol (20 mL) was refluxed for 5 h. Then, the volatile was evaporated. The mixture was poured into H2O (30 mL) and extracted with EtOAc (3 × 30 mL). The combined organic layer was washed with a saturated NaHCO3 solution (30 mL) and a brine solution (30 mL) and dried over anhydrous MgSO4. After the solvent had evaporated, methyl 8-bromohexanoate was isolated (1.94 g, 82%) with high purity as a colorless oil that matched the reported spectra:40 1H NMR (500 MHz, CDCl3) δ 3.65 (s, 3H), 3.38 (t, J = 6.8 Hz, 2H), 2.29 (t, J = 7.5 Hz, 2H), 1.89–1.78 (m, 2H), 1.69–1.56 (m, 2H), 1.52–1.37 (m, 2H), 1.35–1.29 (m, 4H); TLC Rf = 0.4 (20% EtOAc/hexanes).
Methyl 8-Iodomohexanoate.
A mixture of methyl 8-bromohexanoate (1.91 g, 8.06 mmol) was added to a solution of NaI (3.6 g,24.2 mmol) in acetone (15 mL), and the mixture was stirred for 2 h at 65 °C. After 2 h, the reaction mixture was cooled to room temperature, the precipitate filtered, and the filtrate evaporated. Then, Et2O (50 mL) was added to the residue and washed with saturated aqueous Na2S2O3 (1 × 50 mL), H2O (1 × 50 mL), and brine (1 × 50 mL). The organic solution was dried over anhydrous MgSO4. After evaporation of the solvent, methyl 8-iodohexanoate was isolated (2.15 g, 94%) with high purity as a colorless oil that matched the reported spectra:41 1H NMR (500 MHz, CDCl3) δ 3.65 (s, 3H), 3.17 (t, J = 7.0 Hz, 2H), 2.29 (t, J = 7.5 Hz, 2H), 1.80 (p, J = 7.1 Hz, 2H), 1.68–1.55 (m, 2H), 1.43–1.35 (m, 2H), 1.31 (p, J = 3.6 Hz, 4H); TLC Rf = 0.41 (20% EtOAc/hexanes).
Methyl 8-Hydroperoxyoctanoate.
Methyl 8-iodohexanoate (1.586 g, 6.19 mmol) was subjected to general procedure D for 2 h. Silica flash chromatography provided methyl 8-hydroperoxyoctanoate hexanoate (810 mg, 69% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 8.20 (s, 1H), 3.99 (ddd, J = 8.4, 5.4, 1.8 Hz, 2H), 3.66 (s, 3H), 2.42–2.10 (m, 2H), 1.61 (dd, J = 9.5, 4.8 Hz, 4H), 1.36–1.25 (m, 6H); 13C{1H} NMR (101 MHz, CDCl3) δ 175.0, 77.1, 51.8, 34.2, 29.1, 29.1, 27.6, 25.8, 24.9; TLC Rf = 0.34 (30% EtOAc/hexanes); HRMS (ESI) calcd for C9H18O4Na [M + Ma]+ 213.1097, found 213.1097; IR (neat) 3400, 2933, 2857, 1736, 1715, 1437, 1363, 1252, 1203, 1171, 1104, 855, 726 cm−1.
Methyl 8-[(Triisopropylsilyl)peroxy]hexanoate.
Methyl 8-hydro-peroxyoctanoate (0.790 g, 4.157 mmol) was subjected to general procedure E for 3 h. Silica flash chromatography provided methyl 8-[(triisopropylsilyl)peroxy]hexanoate (978 mg, 68% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 3.96 (t, J = 6.6 Hz, 2H), 3.65 (s, 3H), 2.29 (t, J = 7.5 Hz, 2H), 1.59 (dt, J = 13.5, 6.9 Hz, 4H), 1.31 (d, J = 4.1 Hz, 6H), 1.25–1.11 (m, 3H), 1.08 (d, J = 7.0 Hz, 18H); 13C{1H} NMR (101 MHz, CDCl3) δ 174.4, 76.8, 51.6, 34.2, 29.2, 29.2, 27.9, 26.1, 25.0, 18.2, 11.7; HRMS (ESI) calcd for C18H38O4NaSi [M + Na]+ 369.2432, found 369.2434; IR (neat) 2941, 2865, 1741, 1463, 1365, 1250, 1198, 1168, 1104, 997, 919, 882, 854, 779, 677, 663, 503 cm−1; TLC Rf = 0.3 (5% EtOAc/hexanes).
Methyl 6-Bromohexanoate.
A mixture of 6-bromohexanoic acid (1.95 g, 10 mmol) and concentrated H2SO4 (0.3 mL) in methanol (20 mL) was refluxed for 5 h, Then, the volatile was evaporated. The mixture was poured into H2O (30 mL) and extracted with EtOAc (3 × 30 mL). The combined organic layer was washed with a saturated NaHCO3 solution (30 mL) and a brine solution (30 mL) and dried over anhydrous MgSO4. After the solvent had evaporated, methyl 6-bromohexanoate was isolated (1.585 g, 76%) with high purity as colorless oil that matched the reported spectra:42 1H NMR (400 MHz, CDCl3) δ 3.67 (s, 3H), 3.41 (t, J = 6.8 Hz, 2H), 2.33 (t, J = 7.4 Hz, 2H), 1.97–1.83 (m, 2H), 1.73–1.60 (m, 2H), 1.53–1.41 (m, 2H); TLC Rf = 0.4 (10% EtOAc/hexanes).
Methyl 6-Iodomohexanoate.
A mixture of methyl 6-bromohexanoate (1.59 g, 7.6 mmol) was added to a solution of NaI (1.245 g, 8.3 mmol) in acetone (10 mL), and the mixture was stirred for 2 h at 65 °C. After 2 h, the reaction mixture was cooled to room temperature, the precipitate filtered, and the filtrate evaporated. Then, Et2O (50 mL) was added to the residue, and the mixture was washed with saturated aqueous Na2S2O3 (1 × 50 mL), H2O (1 × 50 mL), and brine (1 × 50 mL). The organic solution was dried over anhydrous MgSO4. After the solvent had been evaporated, methyl 6-iodohexanoate was isolated (1.710 g, 88%) with high purity as colorless oil that matched the reported spectra:42 1H NMR (400 MHz, CDCl3) δ 3.67 (s, 3H), 3.18 (t, J = 7.0 Hz, 2H), 2.32 (t, J = 7.5 Hz, 2H), 2.04–1.77 (m, 2H), 1.73–1.60 (m, 2H), 1.53–1.34 (m, 2H); TLC Rf = 0.42 (10% EtOAc/hexanes).
Methyl 6-[(Triisopropylsilyl)peroxy]hexanoate.
Methyl 6-iodohexanoate (1.586 g, 6.19 mmol) was subjected to general procedure F for 14 h. Silica flash chromatography provided methyl 6-[(triisopropylsilyl)peroxy]hexanoate (270 mg, 14% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 3.96 (t, J = 6.5 Hz, 2H), 3.64 (s, 3H), 2.29 (t, J = 7.5 Hz, 2H), 1.61 (dq, J = 11.2, 7.1 Hz, 4H), 1.37 (q, J = 8.4 Hz, 2H), 1.26–1.11 (m, 3H), 1.07 (d, J = 7.1 Hz, 18H); 13C{1H} NMR (101 MHz, CDCl3) δ 174.1, 76.5, 51.5, 34.1, 27.6, 25.8, 24.9, 18.1, 11.7; HRMS (EI) calcd for C13H27O4Si [M – C3H7]+ 275.1673, found 275.1676; IR (neat) 2944, 2866, 1741, 1463, 1435, 1366, 1197, 1015, 882, 854, 677 cm−1; TLC Rf = 0.33 (5% EtOAc/hexanes).
N,N-Diethyl-6-iodohexanamide.
To a round-bottom flask with a stir bar were added 6-bromohexanoic acid (3.9 g, 20 mmol) and DCM (60 mL). DMF (0.05 equiv, 73 μL) was added at rt. Oxalyl chloride (30 mmol, 2.5 mL) was added dropwise. The reaction mixture was stirred at room temperature until bubbling stopped (1 h). Without being transferred from the reaction flask, volatile components were removed by rotary evaporation and high vacuum. The crude reaction product was dissolved in DCM (60 mL) and stirred. N,N-Diethylamine (20 mmol, 3.1 mL), followed by triethylamine (30 mmol, 4.2 mL), was added at room temperature, and the reaction mixture was stirred overnight. The reaction was quenched with 1 M aqueous HCl, and the mixture transferred to a separatory funnel. The crude mixture was diluted with DCM (0.1 M) and water (0.1 M). The organic layer was removed, and the aqueous layer was then extracted with DCM (3 × 0.1 M). The combined organic layers were washed with saturated aqueous NaHCO3 and then brine. The organic layer was dried with MgSO4, filtered, and concentrated by rotary evaporation. Without any further purification, we isolated 6-bromo-N,N-diethylhexanamide as an oily liquid (4.5 g, 90%) that matched previously reported spectra:43 1H NMR (400 MHz, CDCl3) δ 3.47–3.33 (m, 4H), 3.29 (q, J = 7.1 Hz, 2H), 2.30 (t, J = 7.5 Hz, 2H), 1.88 (p, J = 7.0 Hz, 2H), 1.67 (p, J = 7.5 Hz, 2H), 1.57–1.37 (m, 2H), 1.16 (t, J = 7.2 Hz, 3H), 1.10 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.8, 42.0, 40.2, 33.9, 33.0, 32.8, 28.2, 24.6, 14.5, 13.3; TLC Rf = 0.17 (30% EtOAc/hexanes). Next, a mixture of 6-bromo-N,N-diethylhexanamide (3 g, 12 mmol) was added to a solution of NaI (2.16 g, 14.4 mmol) in acetone (20 mL), and the mixture was stirred overnight at 65 °C. Then, the reaction mixture was cooled to room temperature, the precipitate filtered, and the filtrate evaporated. Then, Et2O (50 mL) was added to the residue and washed with saturated aqueous Na2S2O3 (1 × 50 mL), H2O (1 × 50 mL), and brine (1 × 50 mL). The organic solution was dried over anhydrous MgSO4. After the solvent had evaporated, N,N-diethyl-6-iodohexanamide was isolated (2.2 g, 62%) as a brown liquid, which matched previously reported spectra.44 Without further purification, we proceeded to the next step: 1H NMR (400 MHz, CDCl3) δ 3.37 (q, J = 7.1 Hz, 2H), 3.30 (q, J = 7.1 Hz, 2H), 3.20 (t, J = 7.0 Hz, 2H), 2.30 (t, J = 7.5 Hz, 2H), 1.85 (p, J = 7.1 Hz, 2H), 1.76–1.57 (m, 2H), 1.52–1.38 (m, 2H), 1.17 (t, J = 7.1 Hz, 3H), 1.10 (t, J = 7.1 Hz, 3H); TLC Rf = 0.21 (30% EtOAc/hexanes).
N,N-Diethyl-6-[(triisopropylsilyl)peroxy]hexanamide.
N,N-Diethyl-6-iodohexanamide (1.1 g, 3.7 mmol) was subjected to general procedure D for 2 h. Silica flash chromatography provided N,N-diethyl-6-hydroperoxyhexanamide with another mixture. We proceeded to the next step with the impure N,N-diethyl-6-hydro-peroxyhexanamide. The mixture with N,N-diethyl-6 hydroperoxyhexanamide was subjected to general procedure E with TIPSOTf (0.71 mL, 2.64 mmol) and lutidine (0.506 mL, 2.64 mL) for 2 h. Silica flash chromatography provided N,N-diethyl-6-[(triisopropylsilyl)peroxy]-hexanamide (278 mg, 21%, after two steps) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 3.96 (t, J = 6.5 Hz, 2H), 3.34 (q, J = 7.1 Hz, 2H), 3.27 (q, J = 7.1 Hz, 2H), 2.37–2.14 (m, 2H), 1.74–1.52 (m, 4H), 1.44–1.31 (m, 2H), 1.24–1.10 (m, 6H), 1.11–1.02 (m, 21H); 13C{1H} NMR (100 MHz, CDCl3) δ 172.1, 76.6, 42.0, 40.1, 33.0, 27.8, 26.1, 25.4, 18.1, 14.5, 13.2, 11.6; LRMS (GC) decomposed in GCMS; IR (neat) 3395, 2970, 2932, 2863, 1616, 1459, 1431, 1379, 1260, 1220, 1140, 1071, 1055, 605 cm−1; TLC Rf = 0.32 (20% EtOAc/hexanes).
6-Hydroperoxyhexyl 3-(Trifluoromethyl)benzoate.
To a flame-dried round-bottom flax equipped with a stir bar was added 6-bromohexan-1-ol (2.6 mL, 20 mmol); 50 mL of dry DCM was then added via syringe. Then, 3-(trifluoromethyl)benzoyl chloride (3.01 mL, 20 mmol) was added dropwise. Then, triethylamine (3.1 mL, 22 mmol) was added to the mixture dropwise (15 min) with constant stirring. The total reaction mixture was stirred at room temperature overnight. After the reaction had reached completion, 10 mL of water was added to it. The DCM layer was separated with a separatory funnel. The DCM layer was washed with saturated NaHCO3 (20 mL) and a saturated NaCl solution. Then, the final organic portion was passed through anhydrous MgSO4 and dried in rotavap. With the concentrated organic portion as 6-bromohexyl 3-(trifluoromethyl)-benzoate (6.4 g, 91%, ~90% pure), we proceeded to the next step without any further purification: 1H NMR (400 MHz, CDCl3) δ 8.29 (s, 1H), 8.22 (d, J = 7.8 Hz, 1H), 7.81 (d, J = 7.8 Hz, 1H), 7.58 (t, J =7.9 Hz, 1H), 4.36 (t, J = 6.6 Hz, 2H), 3.42 (t, J = 6.7 Hz, 2H), 1.98–1.75 (m, 4H), 1.62–1.42 (m, 4H); 13C{1H} NMR (100 MHz, CDCl3) δ 165.4, 132.9, 131.3, 131.1 (q, J = 32.0 Hz), 129.5 (q, J = 4.0 Hz), 129.1, 126.5 (q, J = 4.0 Hz), 123.8 (q, J = 272.5 Hz), 65.6, 33.8, 32.7, 28.6, 27.9, 25.4; 19F NMR (376 MHz, CDCl3) δ −62.84; LRMS (GCMS) mass fragmentation 235.0 (3), 233.0 (3), 191.1 (40), 173.1 (100), 162.1 (40), 164.1 (40), 145.1 (75), 125.1 (10), 95.1 (10), 83.2 (45), 69.2 (10), 55.2 (25), 41.2 (10); IR (neat) 2937, 2860, 1721, 1617, 1440, 1333, 1300, 1248, 1166, 1123, 1086, 1071, 921, 820, 756, 693, 649, 561 cm−1; TLC Rf = 0.5 (20% EtOAc/hexanes). In the next step, a mixture of 6-bromohexyl 3-(trifluoromethyl)benzoate (5.36 g, 15.2 mmol) was added to a solution of NaI (2.5 g, 16.7 mmol) in acetone (45 mL) and the mixture was stirred overnight at 65 °C. Then, the reaction mixture was cooled to room temperature, the precipitate filtered, and the filtrate evaporated. Then, Et2O (50 mL) was added to the residue and washed with saturated aqueous Na2S2O3 (1 × 50 mL), H2O (1 × 50 mL), and brine (1 × 50 mL). The organic solution was dried over anhydrous MgSO4. After the solvent had evaporated, 6-iodohexyl 3-(trifluoromethyl)benzoate was isolated (4.2 g, 68%, ~90% pure) as a brown liquid. Without any further purification, we proceeded to the next step: 1H NMR (400 MHz, CDCl3) δ 8.28 (s, 1H), 8.21 (d, J = 7.8 Hz, 1H), 7.79 (d, J = 7.8 Hz, 1H), 7.57 (t, J = 7.8 Hz, 1H), 4.35 (t, J = 6.6 Hz, 2H), 3.18 (t, J = 6.9 Hz, 2H), 1.82 (dt, J = 19.1, 6.8 Hz, 4H), 1.47 (p, J = 3.5 Hz, 4H); 13C{1H} NMR (100 MHz, CDCl3) δ 165.3, 132.8, 131.3, 131.0 (q, J = 32.3 Hz), 129.3 (q, J = 4.0 Hz), 129.1, 126.5 (q, J = 3.9 Hz), 123.7 (q, J = 273.5 Hz), 65.5, 33.3, 30.2, 28.6, 25.1, 6.7; 19F NMR (376 MHz, CDCl3) δ −62.83; LRMS (GCMS) mass fragmentation 273.1(34), 210.1 (10), 191.1 (37), 173.1 (100), 145.1 (75), 125.1 (10), 83.2 (63), 55.2 (30), 41.1 (10); IR (neat) 2934, 2858, 1721, 1617, 1441, 1333, 1247, 1166, 1123, 1086, 1071, 920, 819, 756, 693, 650, 602 cm−1; TLC Rf = 0.52 (20% EtOAc/hexanes). Finally, 6-iodohexyl 3-(trifluoromethyl)benzoate (2 g, 5 mmol) was subjected to general procedure D for 2 h. Silica flash chromatography provided 6-hydroperoxyhexyl 3-(trifluoromethyl)benzoate (550 mg, 36% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 8.29 (s, 1H), 8.22 (d, J = 7.8 Hz, 1H), 8.02 (s, 1H), 7.81 (d, J = 7.7 Hz, 1H), 7.58 (t, J = 7.8 Hz, 1H), 4.36 (t, J = 6.6 Hz, 2H), 4.03 (t, J = 6.4 Hz, 2H), 1.80 (p, J = 6.7 Hz, 2H), 1.68 (p, J = 6.7 Hz, 2H), 1.51–1.41 (m, 4H); 13C{1H} NMR (126 MHz, chloroform-d) δ 165.6, 132.9, 131.4, 131.2 (q, J = 32.8 Hz), 129.6 (q, J = 3.7 Hz), 129.2, 126.6 (q, J = 3.9 Hz), 123.8 (q, J = 173.9 Hz), 77.02, 65.70, 28.71, 27.63, 25.97, 25.74; 19F NMR (376 MHz, CDCl3) δ −62.85; HRMS (ESI) calcd for C14H17F3O4Na [M + Na]+ 329.0971, found 329.0974; IR (neat) 3414, 2939, 2863, 1721, 1617, 1334, 1248, 1166, 1123, 1087, 1071, 979, 920, 821, 757, 693, 650 cm−1; TLC Rf = 0.35 (20% EtOAc/hexanes).
6-[(Triisopropylsilyl)peroxy]hexyl 3-(Trifluoromethyl)benzoate.
6-Hydroperoxyhexyl 3-(trifluoromethyl)benzoate (496 mg, 1.62 mmol) was subjected to general procedure E for 2 h. Silica flash chromatography provided 6-[(triisopropylsilyl)peroxy]hexyl 3-(trifluoromethyl)benzoate (480 mg, 65% isolated as pure) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) 8.29 (d, J = 2.2 Hz, 1H), 8.22 (d, J = 7.8 Hz, 1H), 7.81 (d, J = 7.7 Hz, 1H), 7.58 (t, J = 7.8 Hz, 1H), 4.35 (t, J = 6.7 Hz, 2H), 4.00 (t, J = 6.5 Hz, 2H), 1.80 (p, J =6.8 Hz, 2H), 1.64 (p, J = 6.7 Hz, 2H), 1.49–1.40 (m, 4H), 1.29–1.13 (m, 3H), 1.14–0.93 (m, 18H); 13C{1H} NMR (126 MHz, chloroform-d) δ 165.4, 132.9, 131.5, 131.2 (q, J = 34.0 Hz), 129.5 (q, J = 3.7 Hz), 129.2, 126.6 (q, J = 3.9 Hz), 123.9 (q, J = 272.2 Hz), 76.64, 65.71, 28.77, 27.88, 26.05, 26.02, 18.13, 11.72; 19F NMR (376 MHz, CDCl3) δ −62.87; HRMS (ESI) calcd for C23H37F3O4SiNa [M + Na]+ 485.2305, found 485.2307; IR (neat) 2943, 2867, 1726, 1464, 1335, 1250, 1169, 1132, 1086, 1072, 920, 883, 757, 694 cm−1; TLC Rf = 0.38 (5% EtOAc/hexanes).
6-Hydroperoxyhexyl Acetate.
6-Iodohexyl acetate (1.37 g, 5.10 mmol) was subjected to general procedure D for 2 h. Silica flash chromatography provided 6-hydroperoxyhexyl acetate (267 mg, 30% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 8.32 (s, 1H), 4.05 (t, J = 6.7 Hz, 2H), 4.01 (t, J = 6.5 Hz, 2H), 2.04 (s, 3H), 1.70–1.58 (m, 4H), 1.46–1.32 (m, 4H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.6, 77.0, 64.6, 28.6, 27.5, 25.8, 25.6, 21.1; LRMS (GCMS) mass fragmentation 115.1 (13), 98.1 (12), 80.1 (10), 69 (20), 61.1 (50), 55.1 (40), 43.1 (100); IR (neat) 3401, 2939, 2862, 1737, 1718, 1389, 1367, 1240, 1036 cm−1; TLC Rf = 0.2 (20% EtOAc/hexanes).
6-[(Triisopropylsilyl)peroxy]hexyl Acetate.
6-Hydroperoxyhexyl acetate (260 mg, 1.47 mmol) was subjected to general procedure E for 2 h. Silica flash chromatography provided 6-[(triisopropylsilyl)-peroxy]hexyl acetate (330 mg, 68% isolated as pure) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 4.02 (t, J = 6.7 Hz, 2H), 3.96 (t, J = 6.5 Hz, 2H), 2.01 (s, 3H), 1.63–1.55 (m, 4H), 1.35 (p, J =3.6 Hz, 4H), 1.24–1.11 (m, 3H), 1.10–1.03 (m, 18H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.3, 76.7, 64.6, 28.6, 27.8, 26.0, 21.1, 18.1, 11.7; HRMS (EI) calcd for C14H29O4Si [M – C3H7]+ 289.1830, found 289.1827; IR (neat) 2942, 2866, 1741, 1463, 1365, 1233, 1037, 919, 882, 781, 677, 606, 462 cm−1; TLC Rf = 0.41 (10% EtOAc/hexanes).
6-Iodohexyl Methanesulfonate.
6-Iodohexan-1-ol (1.71 g, 7.5 mmol) was subjected to general procedure A for 14 h. After the workup procedure, the crude material was isolated with high purity. We moved to the next step without any further purification (1.94 g, 85% yield): 1H NMR (400 MHz, CDCl3) δ 4.21 (t, J = 6.5 Hz, 2H), 3.18 (t, J = 6.9 Hz, 2H), 2.99 (d, J = 1.1 Hz, 3H), 1.90–1.67 (m, 4H), 1.52–1.36 (m, 4H); HRMS (ESI) calcd for C7H15IO3SNa [M + Na]+ 328.9679, found 328.9679; IR (neat) 2935, 2858, 1347, 1331, 1206, 1169, 971, 938, 908, 827, 718, 526, 459 cm−1; TLC Rf = 0.4 (30% EtOAc/hexanes).
6-Hydroperoxyhexyl Methanesulfonate.
6-Iodohexyl 4-methylbenzenesulfonate (0.915 g, 3 mmol) was subjected to general procedure D for 2 h. Silica flash chromatography provided 6-hydroperoxyhexyl methanesulfonate (265 mg, 42% yield) as a clear, colorless oil: 1H NMR (400 MHz, CD3OD) δ 4.87 (s, 1H), 4.38 (t, J = 6.4 Hz, 2H), 4.07 (t, J = 6.4 Hz, 2H), 3.20 (s, 3H), 1.94–1.86 (m, 2H), 1.78 (p, J = 6.7 Hz, 2H), 1.67–1.45 (m, 4H); 13C{1H} NMR (100 MHz, CD3OD) δ 76.0, 70.4, 35.7, 28.7, 27.2, 25.1, 24.9; HRMS (ESI) calcd for C7H16O5SNa [M + Na]+ 235.0611, found 235.0609; IR (neat) 3428, 2940, 2864, 1344, 1168, 973, 952, 926, 820, 528 cm−1; TLC Rf = 0.5 (50% EtOAc/hexanes).
6-[(Triisopropylsilyl)peroxy]hexyl Methanesulfonate.
6-Hydro-peroxyhexyl methanesulfonate (248 mg, 1.17 mmol) was subjected to general procedure E for 2 h. Silica flash chromatography provided 6-[(triisopropylsilyl)peroxy]hexyl methanesulfonate (295 mg, 68%) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 4.18 (t, J = 6.5 Hz, 2H), 3.95 (t, J = 6.4 Hz, 2H), 2.96 (s, 3H), 1.80–1.67 (m, 2H), 1.63–1.53 (m, 2H), 1.45–1.33 (m, 4H), 1.25–1.10 (m, 3H), 1.10–0.98 (m, 18H); 13C{1H} NMR (100 MHz, CDCl3) δ 76.5, 70.1, 37.4, 29.1, 27.7, 25.7, 25.4, 18.1, 11.6; HRMS (ESI) calcd for C16H36O5SSiNa [M + Na]+ 391.1945, found 391.1949; IR (neat) 2942, 2866, 1464, 1354, 1174, 972, 955, 924, 882, 855, 815, 781, 678, 664, 528 cm−1; TLC Rf = 0.63 (30% EtOAc/hexanes).
(5-Iodopentyl)benzene.
A mixture of (5-bromopentyl)benzene (909 mg, 4 mmol) was added to a solution of NaI (2 g, 12 mmol) in acetone (10 mL), and the mixture was stirred overnight at 65 °C. Then, the reaction mixture was cooled to room temperature, the precipitate filtered, and the filtrate evaporated. Then, Et2O (30 mL) was added to the residue and washed with saturated aqueous Na2S2O3 (1 × 20 mL), H2O (1 × 20 mL), and brine (1 × 50 mL). The organic solution was dried over anhydrous MgSO4. After the solvent had evaporated, (5-iodopentyl)benzene was isolated (1.01 g, 93%) as a brown liquid. Without any further purification, we proceeded to the next step:45 1H NMR (400 MHz, CDCl3) δ 7.29 (dd, J = 8.6, 6.6 Hz, 2H), 7.24–7.13 (m, 3H), 3.19 (t, J = 7.0 Hz, 2H), 2.63 (t, J = 7.7 Hz, 2H), 1.87 (p, J = 7.2 Hz, 2H), 1.75–1.61 (m, 2H), 1.51–1.35 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 142.5, 128.6, 128.5, 126.0, 35.9, 33.7, 30.6, 30.4, 7.2; TLC Rf = 0.5 (5% EtOAc/hexanes).
(5-Hydroperoxypentyl)benzene.
(5-Iodopentyl)benzene (1 g, 3.6 mmol) was subjected to general procedure D for 1.5 h. Silica flash chromatography provided (5-hydroperoxypentyl)benzene (279 mg, 43% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.88 (s, 1H), 7.33–7.25 (m, 2H), 7.22–7.15 (m, 3H), 4.02 (t, J = 6.6 Hz, 2H), 2.63 (t, J = 7.7 Hz, 2H), 1.78–1.54 (m, 4H), 1.52–1.35 (m, 2H); 13C{1H} NMR (101 MHz, CDCl3) δ 142.6, 128.6, 128.5, 125.9, 77.2, 36.0, 31.4, 27.6, 27.3, 25.7; TLC Rf = 0.33 (20% EtOAc/hexanes); LRMS (GCMS, mass fragmentation) 162.1 (100), 144 (83), 133.1 (31), 129.1 (54), 120.0 (42), 118.1 (37), 117.1 (25), 105.1 (12), 92.1 (37), 91.0 (4); IR (neat) 3376, 3025, 2933, 2856, 1602, 1495, 1452, 1367, 1030, 745, 697, 493 cm−1.
Triisopropyl[(5-phenylpentyl)peroxy]silane.
(5-Hydroperoxypentyl)benzene (0.180 g, 1.5 mmol) was subjected to general procedure E for 3 h. Silica flash chromatography provided triisopropyl[(5-phenylpentyl)peroxy]silane (201 mg, 40% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.36–7.27 (m, 2H), 7.24–7.17 (m, 3H), 4.03 (t, J = 6.6 Hz, 2H), 2.65 (t, J = 7.7 Hz, 2H), 1.80–1.59 (m, 4H), 1.44 (dddd, J = 14.8, 9.3, 6.6, 3.4 Hz, 2H), 1.32–1.18 (m, 3H), 1.14 (d, J = 6.9 Hz, 18H); 13C{1H} NMR (101 MHz, CDCl3) δ 142.7, 128.6, 128.4, 125.8, 76.8, 36.0, 31.5, 27.9, 26.0, 18.3, 18.2, 18.2, 11.7; HRMS (EI) calcd for C17H29O2Si [M – C3H7]+ 293.1931, found 293.1933; IR (neat) 2940, 2865, 1495, 1462, 1382, 1366, 1250, 1050, 1014, 997, 919, 882, 849, 783, 744, 696, 676, 569, 462 cm−1; TLC Rf = 0.6 (5% EtOAc/hexanes).
7-[(Triisopropylsilyl)oxy]heptan-1-ol.
To a round-bottom flask equipped with a stir bar was added sodium hydride (60% dispersion in mineral oil, 1.2 g, 30 mmol). The sodium hydride was washed three times with hexanes and dried under an argon atmosphere. Thirty milliliters of THF was then added, and the resulting heterogeneous solution was cooled to 0 °C. 1, 7-Heptanediol (4.2 mL, 30 mmol) in 15 mL of THF was added via syringe. The reaction mixture was warmed to room temperature for 30 min and then cooled to 0 °C. Triisopropylsilyl chloride (6.4 mL, 30 mmol) in 15 mL of THF was added via syringe, and the reaction mixture was allowed to stir for 40 min at 0 °C and then overnight at rt. The reaction was quenched by the addition of water, and the mixture was extracted three times with diethyl ether. The combined organic layers were dried over MgSO4 and concentrated in vacuo. The desired product was isolated by silica flash chromatography (2.93 g, 34% yield) as a colorless oil that matched previously reported spectra:46 1H NMR (400 MHz, CDCl3) δ 3.76–3.53 (m, 4H), 1.56 (m, 5H), 1.36 (m, 6H), 1.13–0.99 (m, 21H); HRMS (ESI) calcd for C16H37O2Si [M – H]+ 289.2557, found 289.2559; IR (neat) 3327, 2933, 2891, 2864, 1462, 1382, 1366, 1247, 1101, 1059, 1012, 994, 918, 881, 840, 791, 716, 677, 657 cm−1; TLC Rf = 0.47 (20% EtOAc/hexanes).
[(7-Iodoheptyl)oxy]triisopropylsilane.
A flame-dried flask equipped with a stir bar was charged with PPh3 (4.7 g, 7.2 mmol), imidazole (1.2 g, 7.2 mmol), and I2 (4.5 g, 7.2 mmol) under N2. Anhydrous DCM (0.2 M) was added; the flask was cooled in an ice bath, and the mixture was stirred for 10 min. Alcohol (1.728 g, 6 mmol), diluted in a 5:1 ratio in DCM, was added dropwise at 0 °C, and the reaction mixture was stirred at 0 °C for 1 h. After 1 h, the reaction mixture was allowed to warm to room temperature and stirred for an additional 12 h. The reaction mixture was concentrated under reduced pressure, diluted with 25% EtOAc in hexanes, and filtered through a plug of silica. The filtrate was concentrated under reduced pressure, and silica flash chromatography provided [(7-iodoheptyl)oxy]triisopropylsilane (1.48 g, 62% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 3.67 (t, J = 6.5 Hz, 2H), 3.18 (t, J = 7.0 Hz, 2H), 1.82 (p, J = 7.1 Hz, 2H), 1.53 (p, J = 6.7 Hz, 2H), 1.44–1.27 (m, 6H), 1.05 (d, J = 4.3 Hz, 21H); 13C{1H} NMR (101 MHz, CDCl3) δ 63.5, 33.7, 33.0, 30.6, 28.5, 25.8, 18.2, 12.1, 7.3; HRMS (ESI) calcd for C16H36OISi [M + H]+ 399.1575, found 399.1576; IR (neat) 2931, 2891, 2863, 1461, 1382, 1245, 1103, 994, 918, 881, 789, 717, 678, 654 cm−1; TLC Rf = 0.49 (5% EtOAc/hexanes).
[(7-Hydroperoxyheptyl)oxy]triisopropylsilane.
[(7-Iodoheptyl)-oxy]triisopropylsilane (1.3 g, 3.2 mmol) was subjected to general procedure D for 2 h. Silica flash chromatography provided [(7-hydroperoxyheptyl)oxy]triisopropylsilane (340 mg, 35% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.97 (s, 1H), 4.02 (t, J = 6.6 Hz, 2H), 3.67 (t, J = 6.6 Hz, 2H), 1.67–1.59 (m, 2H), 1.54 (p, J = 6.6 Hz, 2H), 1.44–1.30 (m, 6H), 1.15–0.99 (m, 21H); 13C{1H} NMR (126 MHz, CDCl3) δ 77.3, 63.6, 33.0, 29.0, 27.6, 26.1, 25.9, 18.2, 12.2; HRMS (ESI) calcd for C16H37O3Si [M + H]+ 305.2506, found 305.2508; IR (neat) 3357, 2938, 2864, 1462, 1382, 1366, 1248, 1101, 1067, 1013, 995, 918, 881, 792, 717, 679, 657, 460 cm−1; TLC Rf = 0.11 (5% EtOAc/hexanes).
3,3,14,14-Tetraisopropyl-2,15-dimethyl-4,5,13-trioxa-3,14-disilahexadecane.
[(7-Iodoheptyl)oxy]triisopropylsilane (330 mg, 1.08 mmol) was subjected to general procedure E for 2 h. Silica flash chromatography provided 3,3,14,14-tetraisopropyl-2,15-dimethyl-4,5,13-trioxa-3,14-disilahexadecane (355 mg, 72% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 3.98 (t, J = 6.6 Hz, 2H), 3.66 (t, J = 6.6 Hz, 2H), 1.56 (ddd, J = 22.2, 10.4, 4.3 Hz, 4H), 1.39–1.28 (m, 6H), 1.28–1.14 (m, 3H), 1.15–1.02 (m, 39H); 13C{1H} NMR (126 MHz, CDCl3) δ 76.9, 63.6, 33.1, 29.5, 27.9, 26.3, 25.9, 18.2, 18.1, 12.2, 11.7; HRMS (EI) calcd for C25H57O3Si2 [M]+ 459.3684, found 459.3686; IR (neat) 2940, 2865, 1462, 1382, 1366, 1248, 1101, 1069, 1013, 996, 919, 881, 783, 677, 459 cm−1; TLC Rf = 0.7 (5% EtOAc/hexanes).
5-[(Triisopropylsilyl)oxy]pentan-1-ol.
To a round-bottom flask equipped with a stir bar was added sodium hydride (60% dispersion in mineral oil, 1 g, 25 mmol). The sodium hydride was washed three times with hexanes and dried under an argon atmosphere. Thirty millilitersof THF was then added, and the resulting heterogeneous solution was cooled to 0 °C. 1, 5-Pentanediol (2.6 mL, 25 mmol) in 15 mL of THF was added via syringe. The reaction mixture was warmed to room temperature for 30 min and then cooled to 0 °C. Triisopropylsilyl chloride (5.3 mL, 50 mmol) in 15 mL of THF was added via syringe, and the reaction mixture was allowed to stir for 40 min at 0 °C and then overnight at rt. The reaction was quenched by the addition of water, and the mixture was extracted three times with diethyl ether. The combined organic layers were dried over MgSO4 and concentrated in vacuo. The desired product was isolated by silica flash chromatography (2.73 g, 42% pure isolated) as a colorless oil that matched the reported spectra:47 1H NMR (400 MHz, CDCl3) δ 3.67 (t, J = 6.3 Hz, 2H), 3.63 (t, J = 7.1 Hz, 2H), 1.64–1.51 (m, 4H), 1.46–1.37 (m, 2H), 1.03 (m, 21H); IR (neat) 3330, 2939, 2864, 1462, 1382, 1248, 1102, 1069, 994, 918, 881, 790, 718, 678, 656 cm−1; TLC Rf = 0.45 (20% EtOAc/hexanes).
5-[(Triisopropylsilyl)oxy]pentyl Methanesulfonate.
5-[(Triisopropylsilyl)oxy]pentan-1-ol (2.33 g, 8.87 mmol) was subjected to general procedure A for 14 h. After the workup procedure, the crude material was isolated with high purity. We moved to the next step without any further purification (2.86 g, 95% yield): 1H NMR (400 MHz, CDCl3) δ 4.24 (t, J = 6.6 Hz, 2H), 3.69 (t, J = 6.1 Hz, 2H), 3.00 (s, 3H), 1.79 (p, J = 6.8 Hz, 2H), 1.63–1.45 (m, 4H), 1.18–0.90 (m, 21H); 13C{1H} NMR (100 MHz, CDCl3) δ 70.0, 62.9, 37.3, 32.2, 28.9, 27.1, 21.9, 18.0, 11.9; HRMS (ESI) calcd for C15H35O4SSi [M + H]+ 339.2020, found 339.2020; IR (neat) 2941, 2864, 1463, 1351, 1172, 1110, 949, 881, 828, 719, 677, 657, 527, 459 cm−1; TLC Rf = 0.17 (10% EtOAc/hexanes).
[(5-Hydr op er oxypentyl)oxy]tr iisopropylsi lane.
5-[(Triisopropylsilyl)oxy]pentyl methanesulfonate (2.85 g, 8.46 mmol) was subjected to general procedure C, using 12:1 MeOH/ H2O (0.3 M) and Et2O (0.8 M) as the solvents, for 44 h. Silica flash chromatography provided [(5-hydroperoxypentyl)oxy]-triisopropylsilane (1.19 g, 51% yield) as a clear, colorless oil: 1H NMR (500 MHz, CDCl3) δ 8.12–7.66 (m, 1H), 4.03 (t, J = 6.6 Hz, 2H), 3.69 (t, J = 6.4 Hz, 2H), 1.74–1.63 (m, 2H), 1.62–1.54 (m, 2H), 1.50–1.40 (m, 2H), 1.14–0.91 (m, 21H); 13C{1H} NMR (126 MHz, CDCl3) δ 77.3, 63.3, 32.8, 27.4, 22.4, 18.2, 12.1; IR (neat) 3333, 2938, 2864, 1462, 1382, 1248, 1102, 1069, 994, 918, 881, 791, 718, 678, 656, 454 cm−1; TLC Rf = 0.1 (5% EtOAc/hexanes).
3,3,12,12-Tetraisopropyl-2,13-dimethyl-4,5,11-trioxa-3,12-disilatetradecane.
[(5-Hydroperoxypentyl)oxy]triisopropylsilane (1.19 g,4.31 mmol) was subjected to general procedure E for 2 h. Silica flash chromatography provided 3,3,12,12-tetraisopropyl-2,13-dimethyl-4,5,11-trioxa-3,12-disilatetradecane (780 mg, 42% isolated as pure) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 4.00 (t, J = 6.6 Hz, 2H), 3.68 (t, J = 6.4 Hz, 2H), 1.71–1.51 (m, 4H), 1.48–1.38 (m, 2H), 1.28–1.14 (m, 3H), 1.14–1.01 (m, 39H); 13C{1H} NMR (126 MHz, CDCl3) δ 76.9, 63.3, 33.0, 27.8, 22.7, 18.2, 18.1, 12.2, 11.7; HRMS (ESI) calcd for C23H53O3Si2 [M + H]+ 433.3528, found 433.3530; IR (neat) 2941, 2865, 1462, 1382, 1248, 1102, 1068, 996, 919, 881, 783, 718, 677, 659, 458 cm−1; TLC Rf = 0.7 (5% EtOAc/hexanes).
[(6-Bromohexyl)oxy](tert-butyl)diphenylsilane.
To a round-bottom flask equipped with a stir bar were added 6-bromohexanol (2.6 mL, 30 mmol) and TBDCl (33 mmol, 5.2 mL) in DCM (60 mL). Then, triethylamine (4.6 mL, 45 mol) was added dropwise. The reaction mixture was stirred overnight at room temperature. Then, the reaction was quenched with a 10% NaHCO3 solution (40 mL), and the solution was diluted with ethyl acetate (40 mL). The organic layer was extracted; the aqueous layer was washed with ethyl acetate (3 × 20 mL), and the combined organic layers were dried with MgSO4 and concentrated in vacuo. The pure desired product was isolated by silica gel column chromatography using hexane as the eluent. The isolated pure product (35%, 2.9 g) was a colorless oil that matched previously reported spectra:48 1H NMR (400 MHz, CDCl3) δ 7.81–7.59 (m, 4H), 7.50–7.34 (m, 6H), 3.66 (t, J = 6.4 Hz, 2H), 3.39 (t, J = 6.8 Hz, 2H), 1.83 (td, J = 8.6, 7.8, 5.9 Hz, 2H), 1.70–1.51 (m, 2H), 1.44–1.36 (m, 4H), 1.05 (s, 9H); TLC Rf = 0.6 (2% EtOAc/hexanes).
[(6-Iodoohexyl)oxy](tert-butyl)diphenylsilane.
A mixture of [(6-bromohexyl)oxy](tert-butyl)diphenylsilane (900 mg, 2.15 mmol) was added to a solution of NaI (894 mg, 6 mmol) in acetone (10 mL), and the mixture was stirred overnight at 65 °C. Then, the reaction mixture was cooled to room temperature, the precipitate filtered, and the filtrate evaporated. Then, Et2O (30 mL) was added to the residue and washed with saturated aqueous Na2S2O3 (1 × 20 mL), H2O (1 × 20 mL), and brine (1 × 50 mL). The organic solution was dried over anhydrous MgSO4. After the solvent had evaporated, [(6-iodoohexyl)oxy](tert-butyl)diphenylsilane was isolated (871 mg, 87%) as a brown liquid. Without any further purification, we proceeded to the next step:49 1H NMR (400 MHz, CDCl3) δ 7.75–7.55 (m, 4H), 7.39 (m, 6H), 3.66 (t, J = 6.4 Hz, 2H), 3.39 (q, J = 6.7 Hz, 2H), 1.83 (q, J = 7.0 Hz, 2H), 1.54 (m, 2H), 1.47–1.33 (m, 4H), 1.04 (s, 9H); TLC Rf = 0.6 (5% EtOAc/hexanes).
tert-Butyl[(6-hydroperoxyhexyl)oxy]diphenylsilane.
[(6-Iodoohexyl)oxy](tert-butyl)diphenylsilane (800 mg, 1.7 mmol) was subjected to general procedure D for 2 h. Silica flash chromatography provided tert-butyl[(6-hydroperoxyhexyl)oxy]diphenylsilane (505 mg, 47% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 8.04 (s, 1H), 7.65–7.53 (m, 4H), 7.44–7.23 (m, 6H), 3.90 (t, J = 6.6 Hz, 2H), 3.58 (t, J = 6.5 Hz, 2H), 1.58–1.42 (m, 4H), 1.27 (ddt, J =14.5, 12.7, 5.0 Hz, 4H), 0.97 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 135.7, 134.2, 129.6, 127.7, 77.2, 64.0, 32.5, 27.7, 27.0, 25.8, 25.7, 19.4; HRMS (ESI) calcd for C22H33O3Si [M + H]+ 373.2193, found 373.2195; IR (neat) 3363, 2931, 2857, 1472, 1427, 1389, 1187, 1108, 1093, 822, 739, 700, 688, 613, 503, 488 cm−1; TLC Rf = 0.26 (10% EtOAc/hexanes).
3,3-Diisopropyl-2,14,14-trimethyl-13,13-diphenyl-4,5,12-trioxa-3,13-disilapentadecane.
tert-Butyl[(6-hydroperoxyhexyl)oxy]-diphenylsilane (270 mg, 0.72 mmol) was subjected to general procedure E for 2 h. Silica flash chromatography provided 3,3-diisopropyl-2,14,14-trimethyl-13,13-diphenyl-4,5,12-trioxa-3,13-disilapentadecane (273 mg, 72% isolated as pure) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.89–7.57 (m, 4H), 7.55–7.29 (m, 6H), 4.10–3.89 (m, 2H), 3.73–3.54 (m, 2H), 1.72–1.47 (m, 4H), 1.35 (m, 4H), 1.29–1.16 (m, 3H), 1.10 (m, 18H), 1.06 (s, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 135.7, 134.3, 129.7, 127.8, 76.9, 64.1, 32.6, 28.0, 27.1, 2618, 25.9, 19.4, 18.2, 18.2, 12.2, 11.7; HRMS (ESI) calcd for C31H53O3Si2 [M + H]+ 529.3528, found 529.3528; IR (neat) 2939, 2864, 1462, 1427, 1385, 1362, 1257, 1106, 997, 882, 822, 781, 738, 700, 684, 613, 503, 487 cm−1; TLC Rf = 0.63 (10% EtOAc/hexanes).
Triisopropyl(pent-4-en-1-ylperoxy)silane (34).
TIPSOOH (284 mg, 1.5 mmol, 1 equiv) was subjected to general procedure F using 5-iodo-1-pentene (0.55 mL, 4.5 mmol, 3 equiv). Silica flash chromatography provided 34 (164.9 mg, 42% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 5.81 (ddt, J = 16.9, 10.2, 6.6 Hz, 1H), 5.03 (dd, J = 17.1, 1.8 Hz, 1H), 4.97 (dd, J = 10.1, 1.8 Hz, 1H), 4.00 (t, J = 6.5 Hz, 2H), 2.12 (q, J = 7.1 Hz, 2H), 1.71 (p, J = 7.6, 6.8, 6.8 Hz, 2H), 1.29–1.14 (m, 3H), 1.10 (d, J = 7.1 Hz, 18H); 13C{1H} NMR (100 MHz, CDCl3) δ 138.2, 115.0, 76.1, 30.4, 27.2, 18.1, 11.7; LRMS (GCMS) mass fragmentation 171.2 (15), 169.2 (15), 141.2 (15), 129.2 (25), 105.1 (30), 103.1 (30), 77.1 (100), 76.1 (40), 61.1 (20), 41.2 (10); IR (neat) 3079, 2943, 2866, 1642, 1463, 995, 911, 882, 676 cm−1; TLC Rf = 0.58 (5% EtOAc/hexanes).
Hydroperoxycyclopentane.
Cylopentyl mesylate (1.96 g, 12 mmol, 1 equiv) was subjected to general procedure C at a concentration of 0.3 M (12:1 MeOH/H2O) for 22 h. Silica flash chromatography provided hydroperoxycyclopentane (539 mg, 44% yield) as a clear, colorless oil that matched the reported spectra:50 1H NMR (600 MHz, CDCl3) δ 7.99 (br s, 1H), 4.60 (tt, J = 5.9, 3.1 Hz, 1H), 1.89–1.61 (m, 6H), 1.63–1.48 (m, 2H); TLC Rf = 0.36 (20% EtOAc/hexanes).
(Cyclopentylperoxy)triisopropylsilane (36).
Hydroperoxycyclopentane (322 mg, 3.0 mmol, 1 equiv) was subjected to general procedure E using triisopropylsilyl chloride (0.65 mL, 3.0 mmol, 1 equiv), Ag2O (686 mg, 3.0 mmol, 1 equiv), and anhydrous ammonia as the base (sparge for 10 min and then a balloon atmosphere) for 15 h. The crude mixture was filtered through a silica plug, eluting with 10% EtOAc/hexanes to provide 36 (395 mg, 51% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 4.54 (dt, J = 5.6, 2.9 Hz, 1H), 1.86–1.73 (m, 2H), 1.72–1.57 (m, 5H), 1.58–1.44 (m, 2H), 1.28–1.13 (m, 2H), 1.08 (d, J = 6.8 Hz, 18H); 13C{1H} NMR (100 MHz, CDCl3) δ 87.1, 31.1, 24.2, 18.2, 11.8; LRMS (GCMS) mass fragmentation 199.3 (3), 173.2 (3), 147.2 (15), 105.2 (43), 77.2 (100), 76.1 (40), 61.1 (15), 45.2 (10); IR (neat) 2943, 2866, 1463, 1346, 1071, 881, 675 cm−1; TLC Rf = 0.6 (5% EtOAc/hexanes).
(5-Bromopent-1-yn-1-yl)benzene.
To phenylacetylene (1.7 mL, 15 mmol) in anhydrous THF (28 mL) was added n-BuLi (2.2 M solution in hexanes, 22 mmol) dropwise at room temperature over 5 min before the mixture was heated to reflux for 2 h. The reaction mixture was cooled to room temperature before 1,4-diiodobutane (2.0 equiv) was added as a THF solution (0.4 M). The reaction mixture was refluxed overnight, returned to room temperature, quenched with H2O (10 mL), and extracted with Et2O (3 × 25 mL). The combined organic extracts were dried (MgSO4), filtered, and concentrated to give a residue. Silica flash chromatography provided (5-bromopent-1-yn-1-yl)benzene (1.66 g, 50%) as a clear, colorless oil, which matched previously reported spectra:51 1H NMR (400 MHz, CDCl3) δ 7.46–7.33 (m, 2H), 7.32–7.14 (m, 3H), 3.57 (t, J = 6.5 Hz, 2H), 2.59 (td, J = 6.9, 4.8 Hz, 3H), 2.12 (p, J = 6.7 Hz, 2H).
(5- Iodopent-1-yn-1-yl)benzene.
A mixture of (5-bromopent-1-yn-1-yl)benzene (1.332 g, 6 mmol) was added to a solution of NaI (3 g, 18 mmol) in acetone (20 mL), and the mixture was stirred overnight at 65 °C. Then, the reaction mixture was cooled to room temperature, the precipitate filtered, and the filtrate evaporated. Then, Et2O (40 mL) was added to the residue and washed with saturated aqueous Na2S2O3 (1 × 20 mL), H2O (1 × 30 mL), and brine (1 × 50 mL). The organic solution was dried over anhydrous MgSO4. After the solvent had evaporated, (5-iodopent-1-yn-1-yl)benzene was isolated (1.474 g, 91%) as a brown liquid, which matched previously reported spectra. Without any further purification, we proceeded to the next step:52 1H NMR (400 MHz, CDCl3) δ 7.41 (ddd, J = 6.2, 4.5, 2.7 Hz, 2H), 7.34–7.09 (m, 3H), 3.37 (t, J = 6.8 Hz, 2H), 2.56 (t, J = 6.7 Hz, 2H), 1.22 (t, J = 7.0 Hz, 2H).
Triisopropyl[(5-phenylpent-4-yn-1-yl)peroxy]silane (38).
(5-Iodo-pent-1-yn-1-yl)benzene (1.458 g, 5.4 mmol) was subjected to general procedure D for 2 h. Silica flash chromatography provided (5-hydroperoxypent-1-yn-1-yl)benzene (522 mg, 55% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 8.50 (s, 1H), 7.45–7.37 (m, 2H), 7.27 (dt, J = 4.7, 2.9 Hz, 3H), 4.16 (t, J = 6.2 Hz, 2H), 2.51 (t, J = 7.0 Hz, 2H), 2.10–1.67 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 131.7, 128.3, 127.8, 123.7, 89.3, 81.3, 75.7, 26.9, 16.1; IR (neat) 3369, 3059, 2936, 2232, 1709, 1598, 1489, 1441, 1275, 1176, 1069, 1027, 755, 713, 690, 526 cm−1; TLC Rf = 0.27 (20% EtOAc/hexanes). Next, (5-hydroperoxypent-1-yn-1-yl)benzene (475 mg, 2.7 mmol) was subjected to general procedure E for 2 h. Silica flash chromatography provided triisopropyl[(5-phenylpent-4-yn-1-yl)-peroxy]silane (609 mg, 68% isolated as pure) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.37 (tt, J = 5.7, 2.3 Hz, 2H), 7.30–7.21 (m, 3H), 4.13 (t, J = 6.1 Hz, 2H), 2.49 (t, J = 7.2 Hz, 2H), 1.98–1.89 (m, 2H), 1.26–1.15 (m, 3H), 1.08 (d, J = 7.1 Hz, 18H); 13C{1H} NMR (126 MHz, CDCl3) δ 131.7, 131.7, 128.4, 127.8, 89.5, 75.3, 27.4, 27.3, 18.2, 18.2, 16.5, 11.7; LRMS (GCMS) mass fragmentation 273.1 (27), 231.2 (100), 203.2 (27), 155.2 (7), 115.2 (17), 75.2 (3); IR (neat) 2943, 2865, 1598, 1490, 1463, 1366, 1069, 997, 918, 881, 753, 687 cm−1; TLC Rf = 0.7 (5% EtOAc/hexanes).
(4-Iodobutyl)cyclopropane.
To a 50 mL two-neck flask with a stir bar and a reflux condenser were added 5-hexen-1-ol (0.840 mL, 7.0 mmol, 1 equiv), diiodomethane (0.710 mL, 8.75 mmol, 1.25 equiv), and anhydrous DCM (1.0 M).53 While the mixture was being stirred at room temperature, trimethylaluminum (8.0 mL, 15.4 mmol, 2.2 equiv, 2 M in hexanes) was added dropwise over ~5 min. The reaction mixture was stirred at room temperature for 10 min and then heated to 40 °C overnight. After 14 h, the reaction mixture was cooled to room temperature and diluted with 10 mL of DCM, and the reaction carefully quenched with water (dropwise addition of 1 mL of water, ~1 drop/10 s). After vigorous bubbling had stopped, the mixture was transferred to a separatory funnel with DCM and an additional 3 mL of water was slowly added. Once all bubbling had stopped, an additional ~25 mL of water was added. The mixture was diluted with DCM and 1 M aqueous NaOH (~50 mL each). The organic layer was removed and extracted with 3 × 50 mL of aqueous DCM. The combined organic layers were washed with brine, dried (MgSO4), filtered, and concentrated by rotary evaporation to provide 4-cyclopropylbutan-1-ol as a clear yellow oil. The product was pure as determined by 1H NMR analysis and directly subjected to Appel conditions. To a 50 mL flask with a stir bar were added triphenylphosphine (1.835 g, 7.0 mmol, 1 equiv), imidazole (488 mg, 7.0 mmol, 1 equiv), and iodine (1.769 g, 7.0 mmol, 1 equiv). The flask was placed in an ice bath, and DCM (0.5 M) was added. The resulting opaque yellow solution was stirred at 0 °C for 50 min. A solution of the crude alcohol in 2 mL of DCM was added dropwise, and the reaction mixture was slowly warmed to room temperature overnight. After 14 h, solvents were removed by rotary evaporation, providing a viscous yellow oil. This oil was filtered through a pad of silica, washing with ~100 mL of hexanes, and then concentrated to provide (4-iodobutyl)cyclopropane (1.295 g, 82% yield over two steps) as a clear, colorless oil that did not require further purification: 1H NMR (400 MHz, CDCl3) δ 3.19 (t, J = 7.1 Hz, 1H), 1.86 (p, J =7.2 Hz, 1H), 1.50 (ddd, J = 12.5, 8.2, 6.2 Hz, 1H), 1.21 (q, J = 7.2 Hz, 1H), 0.64 (ddt, J = 9.7, 7.2, 3.6 Hz, 1H), 0.50–0.34 (m, 1H), −0.00 (t, J = 4.8 Hz, 1H); 13C{1H} NMR (101 MHz, chloroform-d) δ 33.8, 33.5, 30.7, 10.8, 7.4, 4.6; HRMS (EI) calcd for C7H13I [M]+ 224.0056, found 224.0053; IR (neat) 3073, 2998, 2923, 2850, 1455, 1426, 1221, 1179, 1166, 1013, 820, 720, 597, 504 cm−1; TLC Rf = 0.68 (10% EtOAc/hexanes).
[(4-Cyclopropylbutyl)peroxy]triisopropylsilane (40).
TIPSOOH (288 mg, 1.5 mmol, 1 equiv) was subjected to general procedure F using (4-iodobutyl)cyclopropane (1.010 g, 4.5 mmol, 3 equiv). Silica flash chromatography provided 40 (118.0 mg, 27% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 4.02 (t, J = 6.6 Hz, 2H), 1.64 (dt, J = 8.7, 6.6 Hz, 2H), 1.57–1.40 (m, 2H), 1.35–1.18 (m, 5H), 1.12 (d, J = 6.9 Hz, 18H), 0.81–0.59 (m, 1H), 0.49–0.35 (m, 2H), 0.07 to −0.05 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 77.0, 34.7, 27.8, 26.3, 18.2, 11.8, 10.9, 4.6; LRMS (GCMS) mass fragmentation 227.3 (1), 185.2 (10), 183.2 (10), 155.2 (20), 131.2 (20), 105.2 (42), 103.2 (30), 77.2 (100), 76.1 (38), 61.1 (20), 41.2 (10); IR (neat) 2942, 2866, 1463, 1382, 1366, 1256, 1014, 997, 881, 859, 789, 676, 663 cm−1; TLC Rf = 0.69 (5% EtOAc/hexanes).
5-Hydroperoxypent-1-ene.
4-Pentenyl mesylate (1.310 g, 8 mmol, 1 equiv) was subjected to general procedure C at a concentration (of 0.3 M 12:1 MeOH/H2O) for 20 h. Silica flash chromatography provided 5-hydroperoxypent-1-ene (380 mg, 46% yield) as a clear, colorless oil that matched the reported spectra:54 1H NMR (400 MHz, CDCl3) δ 7.88 (s, 1H), 5.82 (ddt, J = 16.9, 10.2, 6.6 Hz, 1H), 5.05 (dd, J = 17.2, 1.8 Hz, 1H), 4.99 (dd, J = 10.2, 1.7 Hz, 1H), 4.04 (t, J = 6.5 Hz, 2H), 2.14 (q, J = 7.1 Hz, 2H), 1.75 (p, J = 7.0 Hz, 2H); TLC Rf = 0.38 (20% EtOAc/hexanes).
Hydroperoxytriisopropylsilane (TIPSOOH).
To a 100 mL round-bottom flask with a stir bar were added TIPSCl (4.30 mL, 20 mmol, 1 equiv) and dry Et2O (1.0 M). The solution was cooled in an ice bath, and urea/H2O2 (3.86 g, 40 mmol, 2 equiv) was added in a single portion. The reaction mixture was sparged with anhydrous ammonia for 15 min and then left under an atmosphere of NH3, warming to room temperature overnight. The next morning, the reaction mixture was diluted with hexane and water (~20 mL each). The organic layer was removed, and the aqueous layer extracted with hexanes (2 × 50 mL). The combined organic layers were washed with brine, dried (MgSO4), filtered, and concentrated to provide a clear colorless oil. Silica flash chromatography provided hydroperoxytriisopropylsilane (2.021 g, 53%) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.25–7.14 (br s, 1H), 1.26 (sept, J = 7.3 Hz, 3H), 1.11 (d, J = 7.3 Hz, 18H); 13C{1H} NMR (100 MHz, CDCl3) δ 18.1, 11.6; IR (neat) 3331, 2944, 2867, 1463, 1384, 998, 881, 733, 676 cm−1; TLC Rf =0.40 (15% EtOAc/hexanes) (stains brightly with KMnO4); TLC silanol side product Rf = 0.34 (15% EtOAc/hexanes) (stains faintly with KMnO4)
1-(Methylperoxy)dodecane (3a).
To a 25 mL round-bottom flask with a stir bar were added 1-dodecyl hydroperoxide 391 (404 mg, 2.0 mmol, 1 equiv) and MeOH (0.8 M). The solution was cooled in an ice bath, and 50 wt % aqueous KOH (225 mg, 2.0 mmol, 1 equiv) and iodomethane (0.45 mL, 5.0 mmol, 2.5 equiv) were added successively. The reaction mixture was stirred overnight, warming to room temperature. After 15 h, the mixture was diluted with DCM (~25 mL) and water (~50 mL). The organic layer was removed, and the aqueous layer extracted with 3 × 25 mL of DCM. The combined organic layers were washed sequentially with water, potassium thiosulfate, and brine, dried (MgSO4), filtered, and concentrated by rotary evaporation, keeping the water bath below 40 °C. Silica flash chromatography provided 3a (368 mg, 85% yield) as a clear, colorless oil: 1H NMR (600 MHz, CDCl3) δ 3.97 (t, J = 6.6 Hz, 2H), 3.82 (s, 3H), 1.59 (p, J = 6.8 Hz, 2H), 1.26 (d, J = 4.4 Hz, 18H), 0.88 (t, J =6.7 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 74.2, 62.2, 32.1, 29.8, 29.8, 29.7, 29.7, 29.6, 29.5, 28.0, 26.2, 22.8, 14.3; IR (neat) 2922, 2853, 1742, 1466, 1376, 1017 cm−1; TLC Rf = 0.66 (20% EtOAc/hexanes).
Dodecyl 2,2,2-Trifluoroethaneperoxoate (3c).55
To a screw-cap test tube with a stir bar was added 1-dodecyl hydroperoxide 3b (204.5 mg, 1.0 mmol, 1 equiv). The flask was cooled in an ice bath, and trifluoroacetic anhydride (0.140 mL, 1.0 mmol, 1 equiv) was added dropwise. The reaction mixture was stirred at 0 °C for 2 h, the reaction quenched with 3 mL of water, and the mixture diluted with 10 mL of DCM. The mixture was washed with saturated aqueous NaHCO3, the organic layer removed, and the aqueous layer extracted with DCM (2 × 10 mL). The combined organic layers were washed with brine, dried (MgSO4), filtered, and concentrated by rotary evaporation to provide 3c (241 mg, 81% yield) as a clear, colorless oil in 90% purity (dodecanal as the contaminant). The crude product was used without further purification: 1H NMR (600 MHz, CDCl3) δ 4.38 (t, J = 6.6 Hz, 2H), 1.72 (p, J = 6.8 Hz, 2H), 1.41 (p, J = 7.0 Hz, 2H), 1.36–1.16 (m, 16H), 0.88 (t, J = 6.7 Hz, 3H); 19F NMR (376 MHz, CDCl3) δ −72.43; IR (neat) 2923, 2854, 1785, 1466, 1162, 779 cm−1.
(Dodecylperoxy)triethoxysilane (3d).
1-Dodecyl hydroperoxide 3b (102.4 mg, 0.5 mmol, 1 equiv) was subjected to general procedure E using chlorotriethoxysilane (0.10 mL, 0.5 mmol, 1 equiv) and triethylamine (0.14 mL, 1.0 mmol, 2 equiv) as the base for 15 h. The reaction mixture was diluted with diethyl ether, filtered, and then concentrated by rotary evaporation. NMR analysis showed incomplete conversion; the crude material was dissolved in DCM (3 mL), and triethylamine (0.10 mL, 0.72 mmol, 1.5 equiv) was added. The reaction mixture was stirred for 14 h, filtered, and concentrated to provide 3d (95.3 mg, 52% yield) in ~80% purity by 1H NMR, which was used without further purification: 1H NMR (600 MHz, CDCl3) δ 4.03 (t, J = 6.6 Hz, 2H), 3.87 (q, J = 6.8 Hz, 6H), 1.69–1.51 (m, 4H), 1.39–1.18 (m, 25H), 0.88 (t, J = 7.0 Hz, 3H); TLC Rf = 0.50 (15% EtOAc/hexanes), hydrolyzes on silica gel.
(Dodecylperoxy)triisopropoxysilane (SI-3k).
1-Dodecyl hydroperoxide 3b (103.4 mg, 0.5 mmol, 1 equiv) was subjected to general procedure E using chlorotriisopropoxysilane56 (0.14 mL, 0.5 mmol, 1 equiv) and triethylamine (0.14 mL, 1.0 mmol, 2 equiv) as the base for 15 h. The reaction mixture was diluted with diethyl ether, filtered, and then concentrated by rotary evaporation. NMR analysis showed incomplete conversion; the crude material was dissolved in DCM (3 mL), and triethylamine (0.10 mL, 0.72 mmol, 1.5 equiv) was added. The reaction mixture was stirred for 14 h, filtered, and concentrated to provide SI-3k (138.0 mg, 68% yield) in ~85% purity by 1H NMR, which was used without further purification: 1H NMR (600 MHz, CDCl3) δ 4.24 (hept, J = 6.3 Hz, 3H), 3.99 (t, J = 6.7 Hz, 3H), 1.61 (p, J = 6.9, Hz, 2H), 1.36–1.21 (m, 17H), 1.19 (d, J = 6.1 Hz, 18H), 0.86 (t, J = 6.9 Hz, 3H).
(Dodecylperoxy)trimethylsilane (3e).
1-Dodecyl hydroperoxide 3b (102.5 mg, 0.5 mmol, 1 equiv) was subjected to general procedure E using chlorotrimethylsilane (0.130 mL, 1.0 mmol, 2 equiv) and pyridine (0.080 mL, 1.0 mmol, 2 equiv) as the base at a concentration of 0.2 M for 40 min. The reaction mixture was diluted with pentane, filtered, and then concentrated to provide 3e (74.3 mg, 54% yield) in >95% purity by 1H NMR: 1H NMR (400 MHz, CDCl3) δ 3.97 (t, J = 6.6 Hz, 2H), 1.60 (p, J = 6.9 Hz, 2H), 1.39–1.18 (s, 19H), 0.88 (t, J =6.7 Hz, 3H), 0.20 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 77.3, 32.1, 29.8, 29.8, 29.7, 29.7, 29.6, 29.5, 27.9, 26.2, 22.8, 14.3, −1.3; IR (neat) 2923, 2853, 1250, 879, 732 cm−1; TLC, hydrolyzes on silica gel.
(Dodecylperoxy)triethylsilane (3f).
1-Dodecyl hydroperoxide 3b (40.5 mg, 0.5 mmol, 1 equiv) was subjected to general procedure E using triethylsilyl triflate (0.040 mL, 0.18 mmol, 0.9 equiv). The reaction mixture was diluted with diethyl ether, filtered, and then concentrated to provide 3f (74.3 mg, 54% yield) as a clear, colorless oil in ~85% purity by 1H NMR with contaminating hydroperoxide. 3f decomposed under all purification attempts and was used without further purification: 1H NMR (600 MHz, CDCl3) δ 3.97 (t, J = 6.6 Hz, 2H), 1.61 (p, J = 6.8 Hz, 2H), 1.40–1.20 (m, 18H), 1.00 (t, J =8.0 Hz, 9H), 0.89 (t, J = 7.0 Hz, 3H), 0.70 (q, J = 8.0 Hz, 6H); TLC, hydrolyzes on silica gel.
(Dodecylperoxy)dimethyl(vinyl)silane (3g).
1-Dodecyl hydroperoxide 3b (103.0 mg, 0.5 mmol, 1 equiv) was subjected to general procedure E using chloro(dimethyl)vinylsilane (0.070 mL, 0.5 mmol, 1 equiv) for 14 h. The reaction mixture was diluted with pentane, filtered, and then concentrated to provide 3g (102.5 mg, 71% yield) as a clear, colorless oil in ~80% purity by 1H NMR with contaminating hydroperoxide: 1H NMR (400 MHz, CDCl3) δ 6.26–6.03 (m, 2H), 5.87 (dd, J = 19.8, 4.3 Hz, 1H), 3.97 (t, J = 6.6 Hz, 2H), 1.69–1.55 (m, 2H), 1.44–1.15 (m, 18H), 0.88 (t, J = 6.8 Hz, 3H), 0.28 (s, 6H); TLC, hydrolyzes on silica gel.
(Dodecylperoxy)(methyl)diphenylsilane (3h).
1-Dodecyl hydroperoxide 3b (200.6 mg, 1.0 mmol, 1 equiv) was subjected to general procedure E using chloro(methyl)diphenylsilane (0.070 mL, 0.5 mmol, 1 equiv) and silver triflate (257.0 mg, 1.0 mmol, 1 equiv) for 14 h. To remove residual hydroperoxide, the crude material was diluted with pentane (3 mL) and flushed through a silica plug, washing with an additional ~10 mL of pentane, and concentrated to provide 3h (110.0 mg, 27% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.65 (dt, J = 6.5, 1.6 Hz, 3H), 7.52 (dt, J = 6.7, 1.5 Hz, 1H), 7.47–7.35 (m, 5H), 7.31 (dd, J = 7.9, 6.4 Hz, 1H), 3.93 (t, J = 6.6 Hz, 2H), 1.58–1.47 (m, 2H), 1.40–1.12 (m, 18H), 0.89 (t, J = 6.7 Hz, 3H), 0.76 (s, 3H); TLC Rf = 0.73 (15% EtOAc/hexanes).
tert-Butyl(dodecylperoxy)dimethylsilane (3i).
1-Dodecyl hydroperoxide 3b (120.0 mg, 0.6 mmol, 1 equiv) was subjected to general procedure E using tert-butyldimethylsilyl triflate (0.140 mL, 0.6 mmol, 1 equiv) at a concentration of 0.75 M for 14 h to provide 3i (136.0 mg, 71% yield) as a clear, colorless oil. The crude material was >95% pure as determined by 1H NMR and did not require purification: 1H NMR (400 MHz, CDCl3) δ 3.96 (t, J = 6.6 Hz, 2H), 1.59 (p, J = 6.6 Hz, 2H), 1.40–1.17 (m, 18H), 0.94 (s, 9H), 0.88 (t, J = 6.6 Hz, 3H), 0.16 (s, 6H); 13C{1H} NMR (100 MHz, CDCl3) δ 77.2, 32.1, 29.8, 29.8, 29.7, 29.7, 29.6, 29.5, 27.9, 26.4, 26.3, 22.9, 18.3, 14.3, −5.7; IR (neat) 924, 2854, 1738, 1462, 1250, 835, 733 cm−1.
tert-Butyl(dodecylperoxy)diphenylsilane (SI-3l).
1-Dodecyl hydroperoxide 3b (203.8 mg, 1.0 mmol, 1 equiv) was subjected to general procedure E using tert-butyl(chloro)diphenylsilane (0.26 mL, 1.0 mmol, 1 equiv) and silver triflate (248.4 mg, 0.97 mmol, 1 equiv) for 14 h. The reaction mixture was diluted with diethyl ether, filtered, and then concentrated. Silica flash chromatography provided SI-3l (107.0 mg, 24% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.75 (td, J = 8.7, 7.9, 1.2 Hz, 4H), 7.50–7.31 (m, 6H), 3.92 (t, J = 6.6 Hz, 2H), 1.53–1.44 (m, 2H), 1.37–1.06 (m, 27H), 0.88 (t, J = 6.7 Hz, 3H); IR (neat) 2926, 2855, 1738, 1462, 1112, 906, 730 cm−1; TLC Rf = 0.73 (15% EtOAc/hexanes).
Di-tert-butyl(dodecylperoxy)(isobutyl)silane (SI-3m).
1-Dodecyl hydroperoxide 3b (101.2 mg, 0.5 mmol, 1 equiv) was subjected to general procedure E using di-tert-butyl-isobutylsilyl triflate (0.14 mL, 1.0 mmol, 1 equiv) for 16 h. The reaction mixture was diluted with diethyl ether, filtered, and then concentrated. Elution through a silica plug (hexanes) provided SI-3m (18.7 mg, 9% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 3.98 (t, J = 6.5 Hz, 2H), 2.00 (septet, J = 6.6 Hz, 1H), 1.58 (p, J = 6.9 Hz, 2H), 1.39–1.18 (m, 18H), 1.05 (s, 18H), 0.99 (d, J = 6.5 Hz, 6H), 0.88 (t, J = 6.7 Hz, 3H), 0.68 (d, J = 6.6 Hz, 2H); TLC Rf = 0.79 (5% EtOAc/hexanes).
(2,3-Dimethylbutan-2-yl)(dodecylperoxy)dimethylsilane (SI-3n).
1-Dodecyl hydroperoxide 3b (101.2 mg, 0.5 mmol, 1 equiv) was subjected to general procedure E using chlorodimethylhexylsilane (0.10 mL, 0.5 mmol, 1 equiv) for 16 h. The reaction mixture was diluted with diethyl ether, filtered, and then concentrated. Elution through a silica plug (hexanes) provided SI-3n (24.9 mg, 14% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 3.95 (t, J = 6.6 Hz, 2H), 1.70–1.49 (m, 2H), 1.38–1.18 (m, 18H), 0.94–0.83 (m, 16H), 0.21 (s, 6H); TLC Rf = 0.71 (5% EtOAc/hexanes).
tert-Butoxy(dodecylperoxy)diphenylsilane (SI-3o).
1-Dodecyl hydroperoxide 3b (100.6 mg, 0.5 mmol, 1 equiv) was subjected to general procedure E using tert-butoxy(chloro)diphenylsilane57 (148.1 mg, 0.5 mmol, 1 equiv) for 14 h. The reaction mixture was diluted with diethyl ether, filtered, and then concentrated. Silica flash chromatography provided SI-3o (132.5 mg, 58% yield) as a clear, colorless oil: 1H NMR (600 MHz, CDCl3) δ 7.76–7.63 (m, 4H), 7.47–7.39 (m, 2H), 7.38–7.30 (m, 4H), 3.88 (t, J = 6.6 Hz, 2H), 1.49 (p, J = 6.6 Hz, 2H), 1.36 (s, 9H), 1.34–1.10 (m, 18H), 0.89 (t, J = 6.8 Hz, 3H); TLC Rf = 0.65 (15% EtOAc/hexanes).
(Dodecylperoxy)(isopropoxy)diisopropylsilane (SI-3p).
1-Dodecyl hydroperoxide 3b (198.9 mg, 1.0 mmol, 1 equiv) was subjected to general procedure E using chloro(isopropoxy)diisopropylsilane (216.1 mg, 1.0 mmol, 1 equiv) for 16 h. The reaction mixture was diluted with pentane, filtered, and then concentrated. Silica flash chromatography provided SI-3p (143.0 mg, 38% yield) as a clear, colorless oil: 1H NMR (600 MHz, CDCl3) δ 4.27 (sept, J = 6.0 Hz, 1H), 4.02 (t, J = 6.6 Hz, 2H), 1.60 (p, J = 6.8 Hz, 2H), 1.39–1.23 (m, 18H), 1.20 (d, J = 6.1 Hz, 6H), 1.13–1.01 (m, 14H), 0.88 (t, J = 6.7 Hz, 3H); TLC Rf = 0.73 (15% EtOAc/hexanes).
1,2-Di(naphthalen-2-yl)disulfane.
To a 25 mL flask with a stir bar were added 2-thionaphthol (807 mg, 5 mmol, 1 equiv) and ethyl lactate (0.67 M). The reaction mixture was stirred at 60 °C, open to air.58 After 16 h, the reaction was quenched with water and the mixture transferred to a separatory funnel, diluting with ~50 mL each of ethyl acetate and water. The organic layer was removed, and the aqueous layer was extracted with EtOAc (2 × 50 mL). The combined organic layers were washed with water and then brine, dried (MgSO4), filtered, and concentrated by rotary evaporation. Trituration with diethyl ether provided 1,2-di(naphthalen-2-yl)-disulfane (377 mg, 47% yield) as an off-white solid that matched the reported spectrum:58 1H NMR (400 MHz, CDCl3) δ 7.99 (s, 2H), 7.86–7.68 (m, 6H), 7.62 (dd, J = 8.7, 1.9 Hz, 2H), 7.52–7.38 (m, 4H).
1,2-Bis(3,4-dichlorophenyl)disulfane.
The compound was prepared following the same procedure that was used for 1,2-di(naphthalen-2-yl)disulfane, using 3,4-dichlorothiophenol (0.64 mL, 5 mmol). Trituration with diethyl ether provided 1,2-bis(3,4-dichlorophenyl)disulfane (353 mg, 39% yield) as an off-white solid that matched the reported spectrum:59 1H NMR (400 MHz, CDCl3) δ 7.55 (s, 2H), 7.37 (d, J = 8.5 Hz, 2H), 7.29 (d, J = 8.5 Hz, 2H).
1,2-Bis(4-bromophenyl)disulfane.
The compound was prepared following the same procedure that was used for 1,2-di(naphthalen-2-yl)disulfane, using 4-bromothiophenol (383 mg, 2 mmol). Silica flash chromatography provided 1,2-bis(4-bromophenyl)disulfane (302 mg, 80% yield) as a white solid that matched the reported spectrum:58 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 8.6 Hz, 4H), 7.34 (d, J = 8.5 Hz, 4H); TLC Rf = 0.66 (15% EtOAc/hexanes).
1,2-Bis[4-(trifluoromethyl)phenyl]disulfane.
The compound was prepared following the same procedure that was used for 1,2-di(naphthalen-2-yl)disulfane, using 4-(trifluoromethyl)thiophenol (0.40 mL, 3 mmol). Trituration with diethyl ether provided 1,2-bis[4-(trifluoromethyl)phenyl]disulfane (210 mg, 39% yield) as a white solid that matched the reported spectrum:60 1H NMR (400 MHz, CDCl3) δ 7.58 (s, 8H); 19F NMR (376 MHz, CDCl3) δ −62.64.
1,2-Bis(4-methoxyphenyl)disulfane.
To a 100 mL round-bottom flask with a stir bar were added silica gel (6.4 g) and water (1 M). The mixture was stirred for 5 min to hydrate silica, leaving a free-flowing powder. DCM (0.1 M) and 4-methoxybenzenethiol (0.62 mL, 5 mmol, 1 equiv) were added, and then while the mixture was being stirred at room temperature, a solution of bromine (0.26 mL, 5.05 mmol, 1.01 equiv) in 5 mL of DCM was added dropwise.61 After 20 min, the reaction mixture was filtered, washing with ~75 mL of DCM, and concentrated to provide a clear red-orange oil. Silica flash chromatography provided 1,2-bis(4-methoxyphenyl)disulfane (618 mg, 88% yield) as a clear, viscous yellow oil that matched the reported spectrum:60 1H NMR (600 MHz, CDCl3) δ 7.40 (d, J = 8.7 Hz, 4H), 6.84 (d, J = 8.8 Hz, 4H); TLC Rf = 0.19 (25% EtOAc/hexanes).
1,2-Bis(4-nitrophenyl)disulfane.
The compound was prepared following the same procedure that was used for 1,2-di(naphthalen-2-yl)disulfane, using 4-nitrothiophenol (785 mg, 5 mmol). Trituration with diethyl ether provided 1,2-bis(4-nitrophenyl)disulfane (508 mg, 66% yield) as a light yellow solid that matched the reported spectrum:60 1H NMR (600 MHz, CDCl3) δ 8.19 (d, J = 8.8 Hz, 4H), 7.62 (d, J = 9.0 Hz, 4H); TLC Rf = 0.50 (25% EtOAc/hexanes).
2,2′-Disulfanediyldibenzoic Acid.
The compound was prepared following the same procedure that was used for 1,2-di(naphthalen-2-yl)disulfane, using 2-mercaptobenzoic acid (769 mg, 5 mmol). Trituration with diethyl ether provided 2,2′-disulfanediyldibenzoic acid (194 mg, 25% yield) as a white solid that matched the reported spectrum:62 1H NMR (400 MHz, DMSO-d6) δ 13.55 (br s, 2H), 8.02 (d, J = 7.7 Hz, 2H), 7.69–7.51 (m, 4H), 7.34 (t, J = 7.4 Hz, 2H).
1-(Naphthalen-2-yl)-2-phenyldisulfane.63
To a solution of the 1,2-di(naphthalen-2-yl)disulfane (1.0 mmol) in tetrahydrofuran (THF) (5.0 mL) at 0 °C was added slowly SO2Cl2 (1.1 mmol). The reaction mixture was stirred for 1 h at the same temperature. This solution was used immediately in the next step.
To a solution of the thiophenol (1.0 mmol), triethylamine (2.2 mmol), and water (10 mmol) in THF (4.0 mL) at room temperature was added the solution from the first step (1 mmol of sulfenyl chloride dissolved in THF) while the mixture was being stirred. The reaction mixture was stirred for 6 h at room temperature. The reaction mixture was diluted with DCM (20 mL), washed with a saturated aqueous NaHCO3 solution and water, and then dried over anhydrous MgSO4. The solvent was removed by evaporation, and 1-(naphthalen-2-yl)-2-phenyldisulfane was purified by column chromatography using hexanes as the eluent and isolated as a white solid in 35% yield (94 mg), which matched the reported spectrum:64 1H NMR (400 MHz, chloroform-d) δ 7.95 (s, 1H), 7.85–7.77 (m, 2H), 7.75 (d, J = 8.1 Hz, 1H), 7.60 (dd, J = 8.6, 1.9 Hz, 1H), 7.57–7.50 (m, 2H), 7.50–7.41 (m, 2H), 7.29 (dd, J = 8.4, 6.8 Hz, 2H), 7.25–7.17 (m, 1H).
1-(4-Methoxyphenyl)-2-phenyldisulfane.
To a solution of the 1,2-bis(4-methoxyphenyl)disulfane (1.0 mmol) in tetrahydrofuran (THF) (5.0 mL) at 0 °C was added slowly SO2Cl2 (1.1 mmol). The reaction mixture was stirred for 1 h at the same temperature. This solution was used immediately in the next step.
To a solution of the thiophenol (1.0 mmol), triethylamine (2.2 mmol), and water (10 mmol) in THF (4.0 mL) at room temperature was added the solution from the first step (1 mmol of sulfenyl chloride dissolved in THF) while the mixture was being stirred. The reaction mixture was stirred for 6 h at room temperature. The reaction mixture was diluted with DCM (20 mL), washed with a saturated aqueous NaHCO3 solution and water, and then dried over anhydrous MgSO4. The solvent was removed by evaporation, and 1-(4-methoxyphenyl)-2-phenyldisulfane was purified by column chromatography using a mixture of hexanes and ethyl acetate as the eluent and isolated as an oily liquid in 75% yield (186 mg), which matched the reported spectrum:65 1H NMR (400 MHz, chloroform-d) δ 7.51 (d, J = 7.5 Hz, 2H), 7.43 (d, J = 8.3 Hz, 2H), 7.35–7.27 (m, 2H), 7.27–7.20 (m, 1H), 6.83 (d, J = 8.3 Hz, 2H), 3.77 (s, 2H).
1-(4-Nitrophenyl)-2-phenyldisulfane.
To a solution of 1,2-bis(4-nitrophenyl)disulfane (1.0 mmol) in tetrahydrofuran (THF) (5.0 mL) at 0 °C was added slowly SO2Cl2 (1.1 mmol). The reaction mixture was stirred for 1 h at the same temperature. This solution was used immediately in the next step.
To a solution of the thiophenol (1.0 mmol), triethylamine (2.2 mmol), and water (10 mmol) in THF (4.0 mL) at room temperature was added the solution from the first step (1 mmol of sulfenyl chloride dissolved in THF) while the mixture was being stirred. The reaction mixture was stirred for 6 h at room temperature. The reaction mixture was diluted with DCM (20 mL), washed with a saturated aqueous NaHCO3 solution and water, and then dried over anhydrous MgSO4. The solvent was removed by evaporation, and 1-(4-nitrophenyl)-2-phenyldisulfane was purified by column chromatography using a mixture of hexanes and ethyl acetate as the eluent and isolated as an oily liquid in 55% yield (145 mg), which matched the reported spectrum:65 1H NMR (400 MHz, chloroform-d) δ 8.25–8.10 (m, 2H), 7.71–7.58 (m, 2H), 7.55–7.43 (m, 2H), 7.41–7.25 (m, 3H).
Preparation and Characterization of Thioarylated Products.
4-(Phenylthio)dodecan-1-ol (4a).
(Dodecylperoxy)triisopropylsilane 3j (179.4 mg, 0.5 mmol) was subjected to general procedure G for 5 h. Silica flash chromatography provided 4a (98.4 mg, 66% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.39 (d, J = 7.3 Hz, 2H), 7.28 (t, J = 7.6 Hz, 2H), 7.21 (t, J = 7.2 Hz, 1H), 3.63 (t, J = 6.3 Hz, 2H), 3.10 (p, J = 6.4 Hz, 1H), 1.82–1.36 (m, 10H), 1.26 (s, 11H), 0.88 (t, J = 6.8 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 135.7, 132.0, 128.9, 126.7, 62.9, 49.1, 34.7, 32.0, 30.8, 30.0, 29.6, 29.6, 29.4, 26.9, 22.8, 14.2; HRMS (APCI) calcd for C18H31OS [M + H]+ 295.2096, found 295.2092; FTIR (neat) 3320, 3073, 2923, 2852, 1478, 1090, 738, 691 cm−1; TLC Rf = 0.22 (20% EtOAc/hexanes).
4-(Phenylthio)butan-1-ol (5).
(Butylperoxy)triisopropylsilane (125.7 mg, 0.5 mmol) was subjected to general procedure G for 5 h. Silica flash chromatography provided 5 (15.3 mg, 16% yield) as a clear, colorless oil that matched the reported spectrum:66 1H NMR (400 MHz, CDCl3) δ 7.32 (d, J = 7.7 Hz, 2H), 7.27 (d, J = 15.0 Hz, 2H), 7.16 (t, J = 7.3 Hz, 1H), 3.65 (t, J = 5.9 Hz, 2H), 2.95 (t, J = 6.7 Hz, 2H), 1.80–1.64 (m, 4H), 1.50–1.63 (s, 1H); TLC Rf = 0.11 (25% EtOAc/hexanes).
5-(Phenylthio)pentan-2-ol (6).
Triisopropyl(pentan-2-ylperoxy)-silane (129.8 mg, 0.5 mmol) was subjected to general procedure G for 5 h. Silica flash chromatography provided 6 (32.0 mg, 32% yield) as a clear, colorless oil that matched the reported spectrum:67 1H NMR (400 MHz, CDCl3) δ 7.31 (d, J = 7.4 Hz, 2H), 7.29–7.22 (m, 2H), 7.15 (t, J = 7.3 Hz, 1H), 3.80 (p, J = 6.3 Hz, 1H), 2.93 (t, J = 7.2 Hz, 2H), 1.82–1.72 (m, 2H), 1.44–1.34 (m, 2H), 1.23 (s, 1H), 1.17 (d, J = 6.2 Hz, 3H); TLC Rf = 0.28 (35% EtOAc/hexanes).
3-Methyl-4-(phenylthio)butan-1-ol (7).
(Isopentylperoxy)-triisopropylsilane (130 mg, 0.5 mmol) was subjected to general procedure G for 5 h. Silica flash chromatography provided the pure product (45 mg, 46% yield) as a clear, colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.33 (d, J = 7.5 Hz, 2H), 7.27 (t, J = 7.7 Hz, 2H), 7.21–7.11 (m, 1H), 3.76–3.65 (m, 2H), 2.89 (ddd, J = 66.8, 12.7, 6.6 Hz, 2H), 2.08–1.69 (m, 2H), 1.63–1.46 (m, 1H), 1.44 (s, 0H), 1.06 (d, J = 6.7 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 137.2, 129.1, 129.0, 125.9, 60.9, 41.3, 39.0, 30.1, 19.7; HRMS (EI) calcd for C11H16OS [M]+ 196.0916, found 196.0920; FTIR (neat) 3329, 2955, 2924, 2871, 1583, 1479, 1437, 1088, 735, 689 cm−1; TLC Rf = 0.21 (20% EtOAc/hexanes).
4-(Phenylthio)hexan-1-ol (8).
(Hexylperoxy)triisopropylsilane (137.7 mg, 0.5 mmol) was subjected to general procedure G for 5 h. Silica flash chromatography provided 7 (65.0 mg, 62% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.44–7.35 (m, 2H), 7.33–7.24 (m, 2H), 7.24–7.17 (m, 1H), 3.64 (t, J = 6.2 Hz, 2H), 3.12–3.00 (m, 1H), 1.83–1.55 (m, 6H), 1.51 (s, 1H), 1.02 (t, J = 7.4 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 135.6, 132.0, 128.9, 126.7, 62.9, 50.7, 30.3, 30.1, 27.5, 11.3; HRMS (APCI) calcd for C12H19OS [M + H]+ 211.1157, found 211.1151; FTIR (neat) 3345, 3073, 2961, 2872, 1478, 1057, 1024, 908, 731 cm−1; TLC Rf =0.26 (35% EtOAc/hexanes).
5-(Phenylthio)hexan-2-ol (9).
(Hexan-2-ylperoxy)-triisopropylsilane (137.1 mg, 0.5 mmol) was subjected to general procedure G for 10 h. The crude reaction mixture showed a 5:1 9:9-isomer mixture. Silica flash chromatography provided 9 (67.8 mg, 64% yield, sole isomer) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.39 (d, J = 8.1 Hz, 2H), 7.28 (t, J = 7.7, 7.0 Hz, 2H), 7.21 (t, J = 7.4 Hz, 1H), 3.78 (sextet, J = 5.6 Hz, 1H), 3.22 (heptet, J = 6.5 Hz, 1H), 1.68–1.48 (m, 4H), 1.28 (dd, J = 6.7, 1.5 Hz, 3H), 1.18 (dd, J = 6.2, 2.4 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 135.3, 132.1, 132.1, 128.9, 126.9, 126.8, 68.1, 68.0, 43.6, 43.4, 36.7, 36.5, 32.9, 32.7, 23.7, 23.7, 21.4, 21.3; HRMS (APCI) calcd for C12H19OS [M + H]+ 211.1157, found 211.1150; FTIR (neat) 3374, 3074, 2965, 2925, 1478, 1024, 906, 728 cm−1; TLC Rf = 0.29 (25% EtOAc/ hexanes).
4-Methyl-4-(phenylthio)pentan-1-ol (10).
Triisopropyl[(4-methylpentyl)peroxy]silane (137.9 mg, 0.5 mmol) was subjected to general procedure G for 5 h. Silica flash chromatography provided 10 (15.8 mg, 15% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.54–7.48 (m, 2H), 7.38–7.29 (m, 3H), 3.65 (t, J = 6.5 Hz, 2H), 1.83–1.73 (m, 2H), 1.57–1.49 (m, 2H), 1.34 (s, 1H), 1.25 (s, 6H); 13C{1H} NMR (100 MHz, CDCl3) δ 137.6, 132.3, 128.8, 128.6, 63.4, 49.1, 38.5, 29.0, 28.4; HRMS (APCI) calcd for C12H19OS [M + H]+ 211.1157, found 211.1157; FTIR (neat) 3330, 3072, 2942, 2866, 1472, 1451, 1056, 748, 693 cm−1; TLC Rf = 0.16 (30% EtOAc/hexanes).
2-Ethyl-4-(phenylthio)hexan-1-ol (11).
[(2-Ethylhexyl)peroxy]-triisopropylsilane (150 mg, 0.5 mmol) was subjected to general procedure G for 8 h. The crude reaction mixture showed a 1:0.07 mixture of regioisomers. Silica flash chromatography provided the desired product with 2-ethylhexan-1-ol as an inseparable mixture, and then high vacuum provided the pure product (69 mg, 58% yield, 1:1 dr) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.43–7.34 (m, 2H), 7.32–7.22 (m, 2H), 7.23–7.13 (m, 1H), 3.63–3.52 (m, 2H), 3.18–3.02 (m, 1H), 1.83–1.67 (m, 1H), 1.67–1.44 (m, 4H), 1.44–1.20 (m, 3H), 0.99 (t, J = 7.4 Hz, 3H), 0.87 (td, J = 7.5, 2.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 135.6, 135.5, 132.2, 132.1, 129.1, 129.0, 128.9, 126.8, 126.8, 125.9, 65.3, 64.9, 48.9, 48.7, 39.9, 39.5, 35.9, 35.5, 28.3, 28.2, 24.1, 23.6, 11.2, 11.10, 11.07, 11.06; HRMS (EI) calcd for C14H22OS [M]+ 238.1386, found 238.1386; FTIR (neat) 3331, 2960, 2926, 2873, 1583, 1460, 1437, 1378, 1090, 1036, 1025, 914, 738, 690 cm−1; TLC Rf = 0.37 (20% EtOAc/hexanes).
4-(Phenylthio)cyclooctan-1-ol (12).
(Cyclooctylperoxy)-triisopropylsilane (150 mg, 0.5 mmol) was subjected to general procedure G for 5 h. Silica flash chromatography provided the desired product with cyclooctanol as an inseparable mixture, and then high vacuum provided the pure product (55 mg, 47% yield, 1:1 dr) as a clear, colorless oil: 1H NMR: (400 MHz, chloroform-d) δ 7.31 (dt, J = 8.1, 1.5 Hz, 2H), 7.26–7.18 (m, 2H), 7.18–7.11 (m, 1H), 3.79 (ddq, J = 14.9, 7.5, 4.4 Hz, 1H), 3.45–3.18 (m, 1H), 2.18–1.88 (m, 1H), 1.87–1.58 (m, 7H), 1.58–1.46 (m, 2H), 1.46–1.11 (m, 3H); 13C{1H} NMR (101 MHz, chloroform-d) δ 135.8, 135.7, 132.1, 131.9, 129.0, 126.9, 126.9, 72.0, 71.6, 48.3, 48.2, 34.3, 34.1, 33.0, 32.8, 30.6, 30.5, 27.9, 27.8, 25.5, 25.2, 22.9, 22.3; HRMS (EI) calcd for C14H20OS [M]+ 236.1229, found 236.1229; FTIR (neat) 3345, 2923, 2852, 1583, 1473, 1437, 1089, 1043, 1024, 1001, 977, 737, 690 cm−1; TLC Rf = 0.14 (20% EtOAc/hexanes).
3-Phenyl-4-(phenylthio)butan-1-ol (13).
Triisopropyl[(3-phenylbutyl)peroxy]silane (81 mg, 0.25 mmol) was subjected to general procedure G for 5 h. Silica flash chromatography provided the desired product with 3-phenylbutan-1-ol as an inseparable mixture, and then high vacuum provided the pure product (13 mg, 21% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.31–7.16 (m, 7H), 7.16–7.06 (m, 3H), 3.55–3.34 (m, 2H), 3.20–3.04 (m, 2H), 2.93 (dtd, J = 10.4, 7.2, 4.6 Hz, 1H), 2.15 (dtd, J = 14.1, 7.2, 4.6 Hz, 1H), 1.82 (ddt, J = 13.7, 10.2, 5.8 Hz, 1H), 1.07 (s, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 143.2, 136.6, 129.2, 128.9, 128.7, 127.6, 126.9, 126.0, 60.9, 42.1, 40.8, 38.1; HRMS (EI) calcd for C16H18OS [M]+ 258.1073, found 258.1074; FTIR (neat) 3346, 3026, 2925, 1582, 1479, 1452, 1438, 1087, 1042, 1025, 737, 699 cm−1; TLC Rf =0.22 (20% EtOAc/hexanes).
6-Chloro-4-(phenylthio)hexan-1-ol (14).
[(6-Chlorohexyl)-peroxy]triisopropylsilane (148 mg, 0.5 mmol) was subjected to general procedure G for 8 h. Silica flash chromatography provided the desired product with 6-chlorohexan-1-ol as an inseparable mixture, and then high vacuum provided the pure product (56 mg, 46% yield) as a clear, colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.52–7.39 (m, 2H), 7.39–7.29 (m, 2H), 7.30–7.24 (m, 1H), 3.66 (t, J = 6.2 Hz, 2H), 3.39–3.17 (m, 1H), 2.11–1.94 (m, 2H), 1.92–1.60 (m, 4H), 1.55 (s, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 134.3, 132.7, 129.1, 127.4, 62.6, 46.5, 42.6, 37.7, 31.3, 29.9; HRMS (EI) calcd for C12H17ClOS [M]+ 244.0689, found 244.0683; FTIR (neat) 3319, 2936, 2866, 1582, 1475, 1437, 1274, 1055, 1023, 937, 911, 740, 691, 658 cm−1; TLC Rf = 0.24 (30% EtOAc/hexanes).
1.5 mmol Scale Reaction.
To a flame-dried 100 mL Schlenk flask with a stir bar were added [(6-chlorohexyl)peroxy]triisopropylsilane (1.5 mmol, 459 mg), disulfide (1.65 mmol, 360 mg), and Fe(OTf)2 (0.075 mmol, 26.5 mg). The flask was evacuated and backfilled three times with N2. Degassed MeOH (15 mL) was added via syringe, and then the reaction mixture was heated to 80 °C on a preheated oil bath in a nitrogen atmosphere. After 12 h, the reaction mixture was cooled, diluted with ethyl acetate (15 mL), filtered through a silica plug, washing with ~30 mL of EtOAc, and concentrated. Products were isolated by flash chromatography using a mixture of ethyl acetate and hexanes as the eluent. The product was eluted in a 23% mixture of ethyl acetate and hexanes. The isolated yield was 42% (152 mg).
Methyl 8-Hydroxy-5-[(4-methoxyphenyl)thio]octanoate (15).
Methyl 8-[(triisopropylsilyl)peroxy]octanoate (173 mg, 0.5 mmol) was subjected to general procedure G for 8 h. Silica flash chromatography provided the desired product (82 mg, 53% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.40–7.24 (m, 2H), 6.93–6.75 (m, 2H), 3.78 (s, 3H), 3.64 (s, 3H), 3.61 (t, J = 6.4 Hz, 2H), 2.86 (p, J = 6.4 Hz, 1H), 2.28 (t, J = 7.4 Hz, 2H), 1.91–1.63 (m, 5H), 1.63–1.42 (m, 4H); 13C{1H} NMR (101 MHz, CDCl3) δ 174.1, 159.6, 135.9, 124.7, 114.6, 62.8, 55.4, 51.7, 50.0, 34.0, 33.9, 30.6, 30.0, 22.3; HRMS (ESI) calcd for C16H24O4SNa [M + Na]+ 335.1288, found 335.1289; FTIR (neat) 3400, 2939, 2867, 1732, 1591, 1569, 1492, 1437, 1282, 1241, 1171, 1056, 1028, 827, 798, 640 cm−1; TLC Rf = 0.14 (30% EtOAc).
Methyl 6-Hydroxy-3-(phenylthio)hexanoate (16).
Methyl 6-[(triisopropylsilyl)peroxy]hexanoate (127 mg, 0.4 mmol) was subjected to general procedure G for 8 h. Silica flash chromatography provided the desired product with methyl 6-hydroxyhexanoate as an inseparable mixture, and then high vacuum provided the pure product (35 mg, 35% yield) as a clear, colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.47–7.32 (m, 2H), 7.24 (dd, J = 8.2, 6.4 Hz, 2H), 7.21–7.17 (m, 1H), 3.65–3.54 (m, 5H), 3.44 (tdd, J = 8.1, 6.6, 4.5 Hz, 1H), 2.63–2.39 (m, 2H), 1.83–1.72 (m, 1H), 1.72–1.63 (m, 2H), 1.61–1.51 (m, 1H), 1.47 (s, 2H); 13C{1H} NMR (126 MHz, CDCl3) δ 172.2, 133.8, 133.1, 129.1, 127.7, 62.5, 51.9, 44.9, 40.6, 30.9, 30.0; HRMS (ESI) calcd for C13H18O3SNa [M + Na]+ 277.0869, found 277.0871; FTIR (neat) 3375, 2946, 2865, 1732, 1583, 1475, 1436, 1354, 1231, 1152, 1055, 1023, 754, 692 cm−1; TLC Rf = 0.17 (30% EtOAc/hexanes).
N,N-Diethyl-6-hydroxy-3-(phenylthio)hexanamide (17).
N,N-Diethyl-6-[(triisopropylsilyl)peroxy]hexanamide (72 mg, 0.2 mmol) was subjected to general procedure G for 8 h. Silica flash chromatography provided the desired product (21 mg, 36% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.48–7.38 (m, 2H), 7.32–7.26 (m, 2H), 7.24–7.19 (m, 1H), 4.00–3.61 (m, 3H), 3.46–3.12 (m, 4H), 2.72–2.44 (m, 2H), 1.93–1.52 (m, 4H), 1.25 (s, 1H), 1.16–1.02 (m, 6H); 13C{1H} NMR (126 MHz, CDCl3) δ 170.3, 135.1, 131.8, 129.1, 127.0, 61.8, 43.8, 42.0, 40.6, 39.5, 30.8, 29.8, 29.5, 14.4, 13.2; HRMS (ESI) calcd for C16H25O2NNaS [M + Na]+ 318.1498, found 318.1500; FTIR (neat) 3405, 2930, 2871, 1699, 1437, 1380, 1362, 1259, 1220, 1141, 1068, 1030, 787, 745, 692, 639, 516 cm−1; TLC Rf = 0.25 (60% EtOAc/hexanes).
6-Hydroxy-3-(phenylthio)hexyl 3-(Trifluoromethyl)benzoate (18).
6-[(Triisopropylsilyl)peroxy]hexyl 3-(trifluoromethyl)benzoate (115 mg, 0.25 mmol) was subjected to general procedure G for 8 h. Silica gel chromatography provided the desired product (54 mg, 55% yield) as a clear, colorless oil, using dichloromethane as the eluent: 1H NMR (500 MHz, CDCl3) δ 8.24 (s, 1H), 8.17 (d, J = 7.8 Hz, 1H), 7.82 (d, J = 7.8 Hz, 1H), 7.58 (t, J = 7.8 Hz, 1H), 7.49–7.35 (m, 2H), 7.30–7.23 (m, 2H), 7.23–7.19 (m, 1H), 4.69–4.39 (m, 2H), 3.67 (t, J = 6.0 Hz, 2H), 3.26 (tt, J = 7.9, 5.2 Hz, 1H), 2.22–1.97 (m, 2H), 1.94–1.66 (m, 4H), 1.25 (s, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 165.3, 134.7, 133.0, 132.6, 131.2 (q, J = 32.7 Hz) 131.2, 129.6 (q, J = 3.8 Hz), 129.2, 129.2, 127.4, 126.6 (q, J = 3.8 Hz), 123.8 (q, J = 272.2 Hz), 63.4, 62.7, 46.5, 34.2, 31.4, 30.1; 19F NMR (376 MHz, chloroform-d) δ −62.82; HRMS (ESI) calcd for C20H21F3O3SNa [M + Na]+ 421.1056, found 421.1056; FTIR (neat) 3399, 3074, 2930, 2859, 1721, 1439, 1333, 1247, 1167, 1123, 1070, 755, 691, 650 cm−1; TLC Rf = 0.26 (30% EtOAc/hexanes).
6-Hydroxy-3-(phenylthio)hexyl Acetate (19) and 3-(Phenylthio)-hexane-1,6-diol (19-diol).
6-[(Triisopropylsilyl)peroxy]hexyl acetate (83 mg, 0.25 mmol) was subjected to general procedure G for 8 h. Silica flash chromatography provided the desired product with 6-hydroxyhexyl acetate as an inseparable mixture, and then high vacuum provided the pure product (23 mg, 35% yield) as a clear, colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.49–7.37 (m, 2H), 7.33–7.28 (m, 2H), 7.28–7.24 (m, 1H), 4.27 (t, J = 6.5 Hz, 2H), 3.66 (t, J = 6.2 Hz, 2H), 3.19 (tt, J = 7.7, 5.4 Hz, 1H), 2.03 (s, 3H), 2.00–1.55 (m, 7H); 13C{1H} NMR (126 MHz, CDCl3) δ 171.2, 134.6, 132.6, 129.0, 127.3, 62.6, 62.2, 46.1, 33.9, 31.2, 30.0, 21.0; HRMS (ESI) calcd for C14H20O3SNa [M + Na]+ 291.1025, found 291.1024; FTIR (neat) 3399, 2937, 2865, 1735, 1583, 1475, 1365, 1233, 1024, 743, 691, 605 cm−1; TLC Rf = 0.14 (30% EtOAc/hexanes).
Silica flash chromatography provided 3-(phenylthio)hexane-1,6-diol with hexane-1,6-diol (<10%) as an inseparable mixture (17 mg, 29% yield) as a clear, colorless oil: 1H NMR (500 MHz, chloroform-d) δ 7.45–7.40 (m, 2H), 7.29 (dd, J = 8.3, 6.7 Hz, 2H), 7.25–7.20 (m, 1H), 3.91–3.76 (m, 2H), 3.64 (t, J = 6.1 Hz, 2H), 3.39–3.24 (m, 1H), 2.08–1.47 (m, 8H); 13C{1H} NMR (126 MHz, CDCl3) δ 134.9, 132.4, 129.1, 127.2, 62.8, 60.7, 46.2, 37.5, 31.4, 29.9; HRMS (EI) calcd for C12H18O2S [M]+ 226.1022, found 226.1019; FTIR (neat) 3332, 2933, 2872, 1583, 1477, 1438, 1258, 1168, 1025, 745, 692, 640 cm−1; TLC Rf = 0.2 (50% EtOAc/hexanes).
6-Hydroxy-3-(phenylthio)hexyl Methanesulfonate (20).
6-[(Triisopropylsilyl)peroxy]hexyl methanesulfonate (92 mg, 0.25 mmol) was subjected to general procedure G for 8 h. Silica flash chromatography provided the desired product with methyl 6-hydroxyhexanoate as an inseparable mixture, and then high vacuum provided the pure product (31 mg, 41% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.47–7.37 (m, 2H), 7.36–7.21 (m, 3H), 4.59–4.38 (m, 2H), 3.66 (t, J = 6.0 Hz, 2H), 3.29–3.17 (m, 1H), 2.94 (s, 3H), 2.14–2.01 (m, 1H), 2.00–1.87 (m, 1H), 1.87–1.60 (m, 4H), 1.25 (s, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 134.0, 132.7, 129.2, 127.6, 67.8, 62.5, 45.5, 37.3, 34.4, 31.3, 29.9; HRMS (ESI) calcd for C13H20O4S2Na [M + Na]+ 327.0695, found 327.0698; FTIR (neat) 3381, 2936, 2863, 1582, 1474, 1438, 1346, 1169, 956, 913, 798, 743, 692, 526 cm−1; TLC Rf = 0.18 (50% EtOAc/hexanes).
5-Phenyl-4-(phenylthio)pentan-1-ol (21).
Triisopropyl[(5-phenylpentyl)peroxy]silane (139 mg, 0.41 mmol) was subjected to general procedure G for 5 h. Silica flash chromatography provided the pure product (65 mg, 59% yield) as a clear, colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.43–7.36 (m, 2H), 7.33–7.25 (m, 4H), 7.25–7.20 (m, 2H), 7.19–7.15 (m, 2H), 3.59 (t, J = 6.2 Hz, 2H), 3.36 (tdd, J = 8.2, 5.9, 4.3 Hz, 1H), 3.00 (dd, J = 14.0, 5.9 Hz, 1H), 2.80 (dd, J =13.9, 8.3 Hz, 1H), 1.85 (dddd, J = 12.7, 9.1, 6.6, 2.3 Hz, 1H), 1.72 (dddd, J = 15.0, 12.5, 8.2, 5.1 Hz, 2H), 1.64–1.51 (m, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 139.3, 135.3, 132.2, 129.3, 129.0, 128.5, 127.0, 126.5, 62.8, 50.6, 41.7, 30.0, 29.9; HRMS (APCI) calcd for C17H21OS [M + H]+ 273.1308, found 273.1309; FTIR (neat) 3328, 3059, 3025, 2934, 2853, 1582, 1494, 1477, 1437, 1055, 1024, 739, 692 cm−1; TLC Rf = 0.24 (20% EtOAc/hexanes).
4-[(4-Methoxyphenyl)thio]heptane-1,7-diol (22).
3,3,14,14-Tetraisopropyl-2,15-dimethyl-4,5,13-trioxa-3,14-disilahexadecane (115 mg, 0.25 mmol) was subjected to general procedure G for 8 h. Silica flash chromatography provided the pure product (41 mg, 61% yield) as a clear, colorless oil: 1H NMR (400 MHz, chloroform-d) δ 7.42–7.29 (m, 2H), 6.86–6.71 (m, 2H), 3.77 (s, 3H), 3.60 (t, J = 6.4 Hz, 4H), 2.98–2.83 (m, 1H), 1.73 (ddt, J = 13.1, 9.3, 6.8 Hz, 6H), 1.65–1.49 (m, 4H); 13C{1H} NMR (101 MHz, chloroform-d) δ 159.4, 135.6, 124.6, 114.4, 62.6, 55.3, 49.9, 30.6, 29.8; HRMS (ESI) calcd for C14H23O3S [M + H]+ 271.1362, found 271.1363; FTIR (neat) 3273, 2927, 2854, 1592, 1493, 1445, 1283, 1240, 1173, 1089, 1057, 1026, 816, 636 cm−1; TLC Rf = 0.41 (60% EtOAc/hexanes).
3-[(4-Methoxyphenyl)thio]hexane-1,6-diol (23).
3,3-Diisopropyl-2,14,14-trimethyl-13,13-diphenyl-4,5,12-trioxa-3,13-disilapentadecane (132 mg, 0.25 mmol) was subjected to general procedure G for 12 h. Silica flash chromatography provided the pure product (38 mg, 59% yield) as a clear, colorless oil: 1H NMR (400 MHz, chloroform-d) δ 7.40 (dd, J = 8.5, 1.9 Hz, 2H), 6.93–6.72 (m, 2H), 4.04–3.75 (m, 5H), 3.65 (t, J = 6.3 Hz, 2H), 3.07 (td, J = 8.3, 4.1 Hz, 1H), 1.87–1.70 (m, 4H), 1.70–1.53 (m, 3H), 1.25 (s, 1H); 13C{1H} NMR (126 MHz, chloroform-d) δ 159.7, 136.0, 124.3, 114.7, 62.8, 60.9, 55.5, 47.3, 37.3, 31.2, 30.0; HRMS (ESI) calcd for C13H21O3S [M + H]+ 257.1206, found 257.1208; FTIR (neat) 3265, 2923, 2854, 1592, 1493, 1454, 1296, 1247, 1170, 1097, 1045, 1019, 815, 800, 642 cm−1; TLC Rf = 0.37 (60% EtOAc/hexanes).
4-(Naphthalen-2-ylthio)hexan-1-ol (24).
(Hexylperoxy)triisopropylsilane (67 mg, 0.25 mmol) was subjected to general procedure G for 8 h. Silica flash chromatography provided the pure product (30 mg, 47% yield) as a clear, colorless oil: 1H NMR (500 MHz, CDCl3; 400 MHz, chloroform-d) δ 7.85 (s, 1H), 7.77 (dd, J = 17.9, 8.3 Hz, 3H), 7.46 (h, J = 6.9, 6.0 Hz, 3H), 3.65 (t, J = 6.1 Hz, 2H), 3.20 (p, J = 6.2 Hz, 1H), 1.86–1.60 (m, 6H), 1.35 (s, 1H), 1.05 (t, J = 7.4 Hz, 3H); 13C{1H} NMR (101 MHz, chloroform-d) δ 133.9, 133.3, 132.2, 130.3, 129.7, 128.4, 127.8, 127.4, 126.6, 126.0, 63.0, 50.6, 30.4, 30.2, 27.6, 11.4; HRMS (ESI) calcd for C16H20O2NaS [M + O + Na]+ 299.1077, found 299.1077; FTIR (neat) 3331, 3052, 2928, 2870, 1624, 1586, 1499, 1453, 1377, 1132, 1061, 942, 851, 811, 742, 633, 602 cm−1; TLC Rf = 0.16 (20% EtOAc/hexanes).
4-[(3,4-Dichlorophenyl)thio]hexan-1-ol (25).
(Hexylperoxy)triisopropylsilane (67 mg, 0.25 mmol) was subjected to general procedure G for 8 h. Silica flash chromatography provided the pure product (48 mg, 70% yield) as a clear, colorless oil: 1H NMR (500 MHz, CDCl3; 400 MHz, chloroform-d) δ 7.45 (d, J = 2.1 Hz, 1H), 7.33 (d, J = 8.4 Hz, 1H), 7.19 (dd, J = 8.4, 2.1 Hz, 1H), 3.66 (t, J =6.1 Hz, 2H), 3.07 (p, J = 6.3 Hz, 1H), 1.83–1.51 (m, 6H), 1.31 (s, 1H), 1.01 (t, J = 7.4 Hz, 3H); 13C{1H} NMR (101 MHz, chloroform-d) δ 136.4, 132.9, 132.9, 130.8, 130.6, 62.9, 51.0, 30.3, 30.0, 27.5, 11.3; HRMS (ESI) calcd for C12H16O2Cl2NaS [M + O + Na]+ 317.0140, found 317.0138; FTIR (neat) 3318, 2931, 2871, 1568, 1545, 1457, 1362, 1130, 1095, 1057, 1028, 810, 675, 554 cm−1; TLC Rf = 0.17 (20% EtOAc/hexanes).
4-[(4-Bromophenyl)thio]hexan-1-ol (26).
(Hexylperoxy)triisopropylsilane (67 mg, 0.25 mmol) was subjected to general procedure G for 8 h. Silica flash chromatography provided the pure product (44 mg, 61% yield) as a clear, colorless oil: 1H NMR (500 MHz, chloroform-d) δ 7.44–7.39 (m, 2H), 7.30–7.23 (m, 2H), 3.75–3.57 (m, 2H), 3.11–2.99 (m, 1H), 1.81–1.58 (m, 6H), 1.32 (d, J = 4.1 Hz, 1H), 1.03 (t, J = 7.4 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 134.9, 133.5, 132.0, 120.8, 62.8, 51.0, 30.3, 30.0, 27.5, 11.3; HRMS (ESI) calcd for C12H17BrO2SNa [M + O + Na]+ 329.0003, found 329.0004; FTIR (neat) 3319, 2930, 2871, 1566, 1471, 1383, 1089, 1066, 1006, 921, 809, 728, 480 cm−1; TLC Rf = 0.25 (20% EtOAc/hexanes).
4-{[4-(Trifluoromethyl)phenyl]thio}hexan-1-ol (27).
(Hexylperoxy)triisopropylsilane (67 mg, 0.25 mmol) was subjected to general procedure G for 8 h. Silica flash chromatography provided the pure product (38 mg, 55% yield) as a clear, colorless oil with a minor unknown isomer (6:1 ratio): 1H NMR (400 MHz, chloroform-d) δ 7.51 (d, J = 8.1 Hz, 2H), 7.42 (d, J = 8.1 Hz, 2H), 3.66 (d, J = 6.0 Hz, 2H), 3.21 (p, J = 5.9, 5.4 Hz, 1H), 1.93–1.58 (m, 6H), 1.35–1.27 (m, 1H), 1.03 (t, J = 7.3 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 141.9, 130.0, 128.1 (q, J = 32.8 Hz), 125.8 (q, J = 3.8 Hz), 124.3 (q, J = 272.2 Hz), 62.8, 49.8, 30.4, 30.0, 27.6, 11.3; 19F NMR (376 MHz, CDCl3) δ −62.51; HRMS (ESI) calcd for C13H17O2F3NaS [M + O + Na]+ 317.0794, found 317.0795; FTIR (neat) 3335, 2934, 2874, 1606, 1454, 1401, 1321, 1162, 1120, 1093, 1061, 1012, 825, 701, 592, 494 cm−1; TLC Rf = 0.24 (20% EtOAc/hexanes).
4-[(4-Chlorophenyl)thio]hexan-1-ol (28).
(Hexylperoxy)triisopropylsilane (67 mg, 0.25 mmol) was subjected to general procedure G for 8 h. Silica flash chromatography provided the pure product (43 mg, 70% yield) as a clear, colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.36–7.28 (m, 2H), 7.26–7.22 (m, 2H), 3.64 (t, J = 6.2 Hz, 2H), 3.08–2.97 (m, 1H), 1.89–1.51 (m, 6H), 1.43–1.21 (m, 1H), 1.01 (t, J = 7.4 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 134.2, 133.5, 132.9, 129.1, 62.9, 51.2, 30.3, 30.1, 27.5, 11.3; HRMS (EI) calcd for C12H17ClOS [M]+ 244.0683, found 244.0683; FTIR (neat) 3321, 2931, 2872, 1572, 1474, 1458, 1387, 1093, 1057, 1011, 921, 816, 744 cm−1; TLC Rf = 0.22 (20% EtOAc/hexanes).
4-[(4-Methoxyphenyl)thio]hexan-1-ol (29).
(Hexylperoxy)triisopropylsilane (67 mg, 0.25 mmol) was subjected to general procedure G for 8 h. Silica flash chromatography provided the pure product (36 mg, 60% yield) as a clear, colorless oil: 1H NMR (500 MHz, chloroform-d) δ 7.45–7.31 (m, 2H), 6.96–6.65 (m, 2H), 3.79 (s, 3H), 3.72–3.54 (m, 2H), 2.91–2.76 (m, 1H), 1.82–1.68 (m, 2H), 1.67–1.50 (m, 4H), 1.36 (s, 1H), 1.06–0.94 (m, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 159.4, 135.7, 125.2, 114.5, 62.9, 55.4, 52.0, 30.2, 30.1, 27.4, 11.3; HRMS (ESI) calcd for C13H20O3NaS [M + O + Na]+ 279.1025, found 279.1027; FTIR (neat) 3331, 2932, 2871, 1591, 1492, 1460, 1282, 1241, 1171, 1092, 921, 826, 640, 526 cm−1; TLC Rf = 0.15 (20% EtOAc/hexanes).
4-(p-Tolylthio)hexan-1-ol (30).
(Hexylperoxy)triisopropylsilane (67 mg, 0.25 mmol) was subjected to general procedure G for 8 h. Silica flash chromatography provided the pure product (40 mg, 72% yield) as a clear, colorless oil with a minor unknown isomer (15:1 ratio): 1H NMR (400 MHz, chloroform-d) δ 7.36–7.28 (m, 2H), 7.09 (d, J = 7.8 Hz, 2H), 3.64 (t, J = 6.5 Hz, 2H), 2.96 (dq, J = 7.3, 6.1 Hz, 1H), 2.32 (s, 3H), 1.83–1.68 (m, 2H), 1.68–1.53 (m, 4H), 1.36 (s, 1H), 1.01 (t, J = 7.3 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 137.1, 133.0, 131.6, 129.7, 63.0, 51.2, 30.3, 30.1, 27.5, 21.2, 11.3; HRMS (ESI) calcd for C13H21OS [M + H]+ 225.1308, found 225.1308; FTIR (neat) 3320, 2928, 2871, 1491, 1449, 1377, 1057, 1017, 921, 807, 632 cm−1; TLC Rf = 0.24 (20% EtOAc/hexanes).
4-[(4-Nitrophenyl)thio]hexan-1-ol (31).
(Hexylperoxy)triisopropylsilane (67 mg, 0.25 mmol) was subjected to general procedure G for 8 h. Silica flash chromatography provided the pure product (11 mg, 18% yield) as a clear, colorless oil with a minor unknown isomer (6:1 ratio): 1H NMR (400 MHz, CDCl3) δ 8.18–8.06 (m, 2H), 7.49–7.32 (m, 2H), 3.68 (m, 2H), 3.34 (t, J = 6.2 Hz, 1H), 1.92–1.63 (m, 6H), 1.25 (s, 1H), 1.04 (t, J = 7.4 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 147.6, 145.4, 128.1, 124.1, 62.7, 49.1, 30.3, 30.0, 27.5, 11.3; HRMS (EI) calcd for C12H17NO3S [M]+ 255.0924, found 255.0925; FTIR (neat) 3346, 2982, 2856, 1576, 1507, 1476, 1456, 1332, 1182, 1081, 851, 837, 741, 683 cm−1; TLC Rf = 0.61 (50% EtOAc/hexanes).
Reaction with Unsymmetrical Disulfide: Reaction with 1-(Naphthalen-2-yl)-2-phenyldisulfane.
(Hexylperoxy)triisopropylsilane (55 mg, 0.2 mmol) and 1-(naphthalen-2-yl)-2-phenyldisulfane (60 mg, 0.22 mmol) were subjected to general procedure G. After the reaction mixture was stirred at 80 °C for 8 h, the reaction mixture was filtered through a 1 cm silica pad and washed with an additional 10 mL of ethyl acetate. The organic portion was concentrated, and to it was added 10.5 mg of 1,3,5-trimethoxybenzene as an internal standard. The yield was calculated by 1H NMR analysis of the crude reaction mixture, comparing the previously isolated pure products. The yield of compound 24 was 36%. The yield of compound 8 was 35%.
Reaction with 1-(4-Nitrophenyl)-2-phenyldisulfane.
(Hexylperoxy)triisopropylsilane (28 mg, 0.1 mmol) and 1-(4-nitrophenyl)-2-phenyldisulfane (29 mg, 0.21 mmol) were subjected to general procedure G. After the reaction mixture was stirred at 80 °C for 8 h, the reaction mixture was filtered through a 1 cm silica pad and washed with an additional 10 mL of ethyl acetate. The organic portion was concentrated, and to it was added 8.2 mg of 1,3,5-trimethoxybenzene as internal standard. The yield was calculated by 1H NMR analysis of the crude reaction mixture, comparing the previously isolated pure products. The yield of compound 31 was 19%. The yield of compound 8 was 29%.
Reaction with 1-(4-Methoxyphenyl)-2-phenyldisulfane.
(Hexylperoxy)triisopropylsilane (28 mg, 0.1 mmol) and 1-(4-methoxyphenyl)-2-phenyldisulfane (29 mg, 0.21 mmol) were subjected to general procedure G. After the reaction mixture was stirred at 80 °C for 8 h, reaction mixture was filtered through a 1 cm silica pad and washed with an additional 10 mL of ethyl acetate. The organic portion was concentrated, and to it was added 6.2 mg of 1,3,5-trimethoxybenzene as internal standard. The yield was calculated by 1H NMR analysis of the crude reaction mixture, comparing the previously isolated pure products. The yield of compound 28 was 42%. The yield of compound 8 was 37%.
2-[(Phenylthio)methyl]tetrahydrofuran (35).
Triisopropyl(pent-4-en-1-ylperoxy)silane 34 (129.1 mg, 0.5 mmol) was subjected to general procedure G for 5 h. Silica flash chromatography provided 35 (62.4 mg, 64% yield) as a clear, colorless oil that matched the reported spectrum:68 1H NMR (400 MHz, CDCl3) δ 7.37 (d, J = 7.3 Hz, 2H), 7.27 (t, J = 7.6 Hz, 2H), 7.17 (t, J = 7.3 Hz, 1H), 4.06 (p, J =6.6 Hz, 1H), 3.96–3.86 (m, 1H), 3.81–3.71 (m, 1H), 3.24–3.11 (m, 1H), 2.97 (dd, J = 13.0, 6.8 Hz, 1H), 2.15–2.00 (m, 1H), 1.99–1.80 (m, 2H), 1.74–1.60 (m, 1H); TLC Rf = 0.34 (15% EtOAc/hexanes).
(5,5-Dimethoxypentyl)(phenyl)sulfane(37).
(Cyclopentylperoxy)triisopropylsilane 36 (130.3 mg, 0.5 mmol) was subjected to general procedure G for 5 h. Silica flash chromatography provided 37 (48.1 mg, 40% yield) as a clear, colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.37–7.23 (m, 4H), 7.17 (t, J = 7.2 Hz, 1H), 4.34 (t, J = 5.6 Hz, 1H), 3.31 (s, 6H), 2.92 (t, J = 7.3 Hz, 2H), 1.74–1.56 (m, 4H), 1.56–1.43 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 136.9, 129.1, 129.0, 125.9, 104.5, 52.9, 33.6, 32.2, 29.1, 24.0; HRMS (EI) calcd for C13H20O2S [M] 240.1184, found 240.1171; FTIR (neat) 3073, 2985, 2828, 1583, 1438, 1123, 1024, 908, 734 cm−1; TLC Rf = 0.43 (25% EtOAc/hexanes).
7-(Phenylthio)hept-4-en-1-ol (41).
[(4-Cyclopropylbutyl)peroxy]-triisopropylsilane 40 (72 mg, 0.25 mmol) was subjected to general procedure G for 5 h. Silica flash chromatography provided 42 (28 mg, 51%) as a clear colorless liquid in a 3.5:1 isomeric ratio: 1H NMR (400 MHz, chloroform-d) δ 7.30–7.16 (m, 4H), 7.09 (tt, J = 6.6, 1.5 Hz, 1H), 5.53–5.14 (m, 2H), 3.56 (dt, J = 12.8, 6.4 Hz, 2H), 2.87 (td, J = 7.3, 1.8 Hz, 2H), 2.44–2.16 (m, 2H), 2.13–1.95 (m, 2H), 1.83–1.39 (m, 2H), 1.21 (m, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 136.7, 131.9, 131.2, 129.1, 129.0, 129.0, 128.6, 128.0, 126.0, 126.0, 124.2, 62.6, 33.8, 33.7, 32.6, 32.4, 32.4, 29.9, 29.0, 27.1; HRMS (EI) calcd for C13H18OS [M]+ 222.1073, found 222.1072; FTIR (neat) 3332, 2925, 2848, 1583, 1479, 1437, 1055, 1024, 967, 736, 689 cm−1; TLC Rf = 0.35 (20% EtOAc/hexanes).
2-[Phenyl(phenylthio)methylene]tetrahydrofuran (39).
Triisopropyl[(5-phenylpent-4-yn-1-yl)peroxy]silane 38 (83 mg, 0.25 mmol) was subjected to general procedure G for 8 h. Silica flash chromatography provided 2-[phenyl(phenylthio)methylene]-tetrahydrofuran (27 mg, 41%) as a clear colorless liquid: 1H NMR (500 MHz, chloroform-d) δ 7.73–7.66 (m, 2H), 7.26–7.15 (m, 3H), 7.11 (d, J = 5.0 Hz, 3H), 7.08–7.03 (m, 1H), 7.00–6.93 (m, 1H), 4.33 (t, J = 6.9 Hz, 2H), 2.97 (t, J = 7.7 Hz, 2H), 2.05–1.80 (m, 2H); 13C{1H} NMR (126 MHz, chloroform-d) δ 166.6, 139.0, 138.3, 129.0, 128.4, 128.0, 126.1, 125.3, 124.7, 98.0, 73.9, 32.1, 24.3; HRMS (EI) calcd for C14H20OS [M]+ 236.1228, found 236.1228; FTIR (neat) 295 6, 2922, 2852, 1625, 1578, 1474, 1438, 1368, 1240, 1176, 1074, 1043, 1024, 799, 754, 735, 689 cm−1; TLC Rf = 0.33 (5% EtOAc/hexanes).
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge funds from Indiana University in partial support of this work. The authors also gratefully acknowledge the National Science Foundation CAREER Award (CHE-1254783) and the National Institutes of Health (GM121668). Eli Lilly & Co. and Amgen supported this work through the Lilly Grantee Award and the Amgen Young Investigator Award.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.9b01979.
Reaction optimization and spectroscopic data (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Prilezhaeva EN Sulfones and sulfoxides in the total synthesis of biologically active natural compounds. Russ. Chem. Rev 2000, 69, 367. [Google Scholar]
- (2).Trost BM; Conway WP; Strege PE; Dietsche TJ New Synthetic reactions. Alkylative elimination. J. Am. Chem. Soc 1974, 96, 7165. [Google Scholar]
- (3).Evans DA; Andrews GC Allylic sulfoxides. Useful intermediates in organic synthesis. Acc. Chem. Res 1974, 7, 147. [Google Scholar]
- (4).Engstrom KM; Mendoza MR; Navarro-Villalobos M; Gin DY Total Synthesis of (+)-Pyrenolide D. Angew. Chem., Int. Ed 2001, 40, 1128. [DOI] [PubMed] [Google Scholar]
- (5).de Lucchi O; Miotti U; Modena G Org. React 1991, 157. [Google Scholar]
- (6).Bonjoch J; Catena J; Valls N Total Synthesis of (±)-Deethylibophyllidine: Studies of a Fischer Indolization Route and a Successful Approach via a Pummerer Rearrangement/Thionium Ion-Mediated Indole Cyclization. J. Org. Chem 1996, 61, 7106. [DOI] [PubMed] [Google Scholar]
- (7).Lockard J; Schroeck C; Johnson C Synthesis of Optically Active Dialkyl Sulfoxides from Aryl Alkyl Sulfoxides1. Synthesis 1973, 1973, 485. [Google Scholar]
- (8).Satoh T; Kobayashi S; Nakanishi S; Horiguchi K; Irisa S Generation of oxiranyllithiums and oxiranyl Grignard reagents having a carbanion-destabilizing group from sulfinyloxiranes: Their property and an application to asymmetric synthesis of epoxides and alcohols. Tetrahedron 1999, 55, 2515. [Google Scholar]
- (9).Trost BM; Rao M Development of Chiral Sulfoxide Ligands for Asymmetric Catalysis. Angew. Chem., Int. Ed 2015, 54, 5026. [DOI] [PubMed] [Google Scholar]
- (10).Julia M; Paris J-M Syntheses a l’Aide de Sulfones v(+)-Methode de Synthese Generale de Doubles Liaisons. Tetrahedron Lett. 1973, 14, 4833. [Google Scholar]
- (11).Blakemore PR; Cole WJ; Kocieński PJ; Morley A A Stereoselective Synthesis oftrans-1,2-Disubstituted Alkenes Based on the Condensation of Aldehydes with Metallated 1-Phenyl-1H-tetrazol-5-yl Sulfones. Synlett 1998, 1998, 26. [Google Scholar]
- (12).Trost BM Chemical Chameleons. Organosulfones as Synthetic Building Blocks. Bull. Chem. Soc. Jpn 1988, 61, 107. [Google Scholar]
- (13).Paquette LA Organic Reactions 1977, 1. [Google Scholar]
- (14).MaGee DI; Beck EJ The Use of the Ramberg–Bäcklund Rearrangement for the Formation of aza-Macrocycles: a Total Synthesis of Manzamine C. Can. J. Chem 2000, 78, 1060. [DOI] [PubMed] [Google Scholar]
- (15).Merchant RR; Edwards JT; Qin T; Kruszyk MM; Bi C; Che G; Bao D-H; Qiao W; Sun L; Collins MR; Fadeyi OO; Gallego GM; Mousseau JJ; Nuhant P; Baran PS Modular radical cross-coupling with sulfones enables access to sp3-rich (fluoro)alkylated scaffolds. Science 2018, 360, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Procter DJ The synthesis of thiols, selenols, sulfides, selenides, sulfoxides, selenoxides, sulfones and selenones. J. Chem. Soc., Perkin Trans 1 2000, 835. [Google Scholar]
- (17).Ambrosioni E Defining the Role of Zofenopril in the Management of Hypertension and Ischemic Heart Disorders. Am. J. Cardiovasc. Drugs 2007, 7, 17. [DOI] [PubMed] [Google Scholar]
- (18).Petrović G; Saičić R;Čeković Ž Regioselective Free Radical Phenylsulfenation of a Non-Activated δ-Carbon Atom by the Photolysis of Alkyl Benzenesulfenate. Tetrahedron 2003, 59, 187. [Google Scholar]
- (19).Lyons TW; Sanford MS Palladium-catalyzed ligand-directed C-H functionalization reactions. Chem. Rev 2010, 110, 1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wolff ME Cyclization of N-Halogenated Amines (The Hofmann-Loffler Reaction). Chem. Rev 1963, 63, 55. [Google Scholar]
- (21).Fenton HJH LXXIII.—Oxidation of Tartaric Acid in Presence of Iron. J. Chem. Soc., Trans 1894, 65, 899. [Google Scholar]
- (22).Walling C Fenton’s reagent revisited. Acc. Chem. Res 1975, 8, 125. [Google Scholar]
- (23).Guan H; Sun S; Mao Y; Chen L; Lu R; Huang J; Liu L Iron(II)-Catalyzed Site-Selective Functionalization of Unactivated C(sp3)–H Bonds Guided by Alkoxyl Radicals. Angew. Chem., Int. Ed 2018, 57, 11413. [DOI] [PubMed] [Google Scholar]
- (24).Groendyke BJ; AbuSalim DI; Cook SP Iron-Catalyzed, Fluoroamide-Directed C-H Fluorination. J. Am. Chem. Soc 2016, 138, 12771. [DOI] [PubMed] [Google Scholar]
- (25).Zhu N; Zhao J; Bao H Iron Catalyzed Methylation and Ethylation of Vinyl Arenes. Chem. Sci 2017, 8, 2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Jian W; Ge L; Jiao Y; Qian B; Bao H Iron-Catalyzed Decarboxylative Alkyl Etherification of Vinylarenes with Aliphatic Acids as the Alkyl Source. Angew. Chem., Int. Ed 2017, 56, 3650. [DOI] [PubMed] [Google Scholar]
- (27).Olmstead WN; Margolin Z; Bordwell FG Acidities of water and simple alcohols in dimethyl sulfoxide solution. J. Org. Chem 1980, 45, 3295. [Google Scholar]
- (28).Zard SZ Recent progress in the generation and use of nitrogen-centred radicals. Chem. Soc. Rev 2008, 37, 1603. [DOI] [PubMed] [Google Scholar]
- (29).Curran DP; Martin-Esker AA; Ko SB; Newcomb M Rate constants for chalcogen group transfers in bimolecular substitution reactions with primary alkyl radicals. J. Org. Chem 1993, 58, 4691. [Google Scholar]
- (30).Pryor WA; Platt PK Reactions of Radicals. V. Reaction of Phenyl Radicals with Aliphatic Disulfides. J. Am. Chem. Soc 1963, 85, 1496. [Google Scholar]
- (31).Griller D; Ingold KU Free-radical clocks. Acc. Chem. Res 1980, 13, 317. [Google Scholar]
- (32).Pangborn AB; Giardello MA; Grubbs RH; Rosen RK; Timmers FJ Safe and Convenient Procedure for Solvent Purification. Organometallics 1996, 15, 1518. [Google Scholar]
- (33).Welch F; Williams HR; Mosher HS Organic Peroxides.IV. Higher Dialkyl Peroxides. J. Am. Chem. Soc 1955, 77, 551. [Google Scholar]
- (34).Williams HR; Mosher HS Peroxides. I. n-Alkyl Hydroperoxides. J. Am. Chem. Soc 1954, 76, 2984. [Google Scholar]
- (35).Cookson PG; Davies AG; Roberts BP A convenient and general preparation of alkyl hydroperoxides and dialkyl peroxides. J. Chem. Soc., Chem. Commun 1976, 1022. [Google Scholar]
- (36).Buncel E; Davies AG 308. Peroxides of elements other than carbon. Part I. The preparation and reactions of peroxysilanes. J. Chem. Soc 1958, 1550. [Google Scholar]
- (37).Dussault P; Sahli A 2-Methoxy-2-propyl hydroperoxide: a convenient reagent for the synthesis of hydroperoxides and peracids. J. Org. Chem 1992, 57, 1009. [Google Scholar]
- (38).Kyasa S; Puffer BW; Dussault PH Synthesis of Alkyl Hydroperoxides via Alkylation of gem-Dihydroperoxides. J. Org. Chem 2013, 78, 3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Jinsheng L; Pritzkow W; Voerckel V Intramolecular H-transfer reactions during the Decomposition of Alkylhydroperoxides in hydrocarbons as the solvents. J. Prakt. Chem./Chem.-Ztg 1994, 336,43. [Google Scholar]
- (40).Savariar EN; Aathimanikandan SV; Thayumanavan S Supramolecular Assemblies from Amphiphilic Homopolymers: Testing the Scope. J. Am. Chem. Soc 2006, 128, 16224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Xia Y; Qu F; Maggiani A; Sengupta K; Liu C; Peng L Photoactivatable Phospholipids Bearing Tetrafluorophenylazido Chromophores Exhibit Unprecedented Protonation-State-Dependent 19F NMR Signals. Org. Lett 2011, 13, 4248. [DOI] [PubMed] [Google Scholar]
- (42).Gupta PK; Reid RC; Liu L; Lucke AJ; Broomfield SA; Andrews MR; Sweet MJ; Fairlie DP Inhibitors selective for HDAC6 in enzymes and cells. Bioorg. Med. Chem. Lett 2010, 20, 7067. [DOI] [PubMed] [Google Scholar]
- (43).Zhou J; Fu GC Palladium-Catalyzed Negishi Cross-Coupling Reactions of Unactivated Alkyl Iodides, Bromides, Chlorides, and Tosylates. J. Am. Chem. Soc 2003, 125, 12527. [DOI] [PubMed] [Google Scholar]
- (44).Vechorkin O; Proust V; Hu X Functional Group Tolerant Kumada–Corriu–Tamao Coupling of Nonactivated Alkyl Halides with Aryl and Heteroaryl Nucleophiles: Catalysis by a Nickel Pincer Complex Permits the Coupling of Functionalized Grignard Reagents.J. Am. Chem. Soc 2009, 131, 9756. [DOI] [PubMed] [Google Scholar]
- (45).Pouliot M-F; Mahé O; Hamel J-D; Desroches J; Paquin J-F Halogenation of Primary Alcohols Using a Tetraethylammonium Halide/[Et2NSF2]BF4 Combination. Org. Lett 2012, 14, 5428. [DOI] [PubMed] [Google Scholar]
- (46).Bergueiro J; Montenegro J; Saá C; López S One-step chemoselective conversion of tetrahydropyranyl ethers to silyl-protected alcohols. RSC Adv. 2014, 4, 14475. [Google Scholar]
- (47).Crimmins MT; Vanier GS Enantioselective Total Synthesis of (+)-SCH 351448. Org. Lett 2006, 8, 2887. [DOI] [PubMed] [Google Scholar]
- (48).Liu Y; Ji L; Eno M; Kudalkar S; Li A-L; Schimpgen M; Benchama O; Morales P; Xu S; Hurst D; Wu S; Mohammad KA; Wood JT; Zvonok N; Papahatjis DP; Zhou H; Honrao C; Mackie K; Reggio P; Hohmann AG; Marnett LJ; Makriyannis A; Nikas SP (R)-N-(1-Methyl-2-hydroxyethyl)-13-(S)-methylarachidonamide (AMG315): A Novel Chiral Potent Endocannabinoid Ligand with Stability to Metabolizing Enzymes. J. Med. Chem 2018, 61, 8639. [DOI] [PubMed] [Google Scholar]
- (49).Yamada Y; Okada M.-a.; Tanaka K Repetitive stepwise rotaxane formation toward programmable molecular arrays. Chem. Commun 2013, 49, 11053. [DOI] [PubMed] [Google Scholar]
- (50).Barton DHR; Launay F The selective functionalization of saturated hydrocarbons. Part 40. Aspects of FeII based peroxide fragmentation in pyridine solution. Tetrahedron 1997, 53, 14565. [Google Scholar]
- (51).Asao N; Sato K; Menggenbateer; Yamamoto Y AuBr3- and Cu(OTf)2-Catalyzed Intramolecular [4 + 2] Cycloaddition of Tethered Alkynyl and Alkenyl Enynones and Enynals: A New Synthetic Method for Functionalized Polycyclic Hydrocarbons. J. Org. Chem 2005, 70, 3682. [DOI] [PubMed] [Google Scholar]
- (52).Yalavac I; Lyons SE; Webb MR; Procter DJ SmI2–H2O-mediated 5-exo/6-exo lactone radical cyclisation cascades. Chem. Commun 2014, 50, 12863. [DOI] [PubMed] [Google Scholar]
- (53).Janusz JM; Young PA; Ridgeway JM; Scherz MW; Enzweiler K; Wu LI; Gan L; Darolia R; Matthews RS; Hennes D; Kellstein DE; Green SA; Tulich JL; Rosario-Jansen T; Magrisso IJ; Wehmeyer KR; Kuhlenbeck DL; Eichhold TH; Dobson RLM; Sirko SP; Farmer RW New Cyclooxygenase-2/5-Lipoxygenase Inhibitors. 1. 7-tert-Butyl-2,3-dihydro-3,3-dimethylbenzofuran Derivatives as Gastrointestinal Safe Antiinflammatory and Analgesic Agents: Discovery and Variation of the 5-Keto Substituent. J. Med. Chem 1998, 41, 1112. [DOI] [PubMed] [Google Scholar]
- (54).Taillez B; Bertrand MP; Surzur J-M Intramolecular addition of alkoxyl radicals. Part 4. Reductive cyclisation of olefinic hydroperoxides. J. Chem. Soc., Perkin Trans 2 1983, 547. [Google Scholar]
- (55).Hiatt RR; Glover LC; Mosher HS Concerted mechanism and phase effects in decompositions of alkyl peroxy esters. J. Am. Chem. Soc 1975, 97, 1556. [Google Scholar]
- (56).Chappelow CC; Elliott RL; Goodwin JT The Phenylation and Methylation of Alkoxychlorosilanes1. J. Org. Chem 1960, 25, 435. [Google Scholar]
- (57).Kojima S; Fukuzaki T; Yamakawa A; Murai Y Highly (Z)-Selective Synthesis of β-Monosubstituted α,β-Unsaturated Cyanides Using the Peterson Reaction. Org. Lett 2004, 6, 3917. [DOI] [PubMed] [Google Scholar]
- (58).Liu Y; Wang H; Wang C; Wan J-P; Wen C Bio-based green solvent mediated disulfide synthesis via thiol couplings free of catalyst and additive. RSC Adv. 2013, 3, 21369. [Google Scholar]
- (59).Ramirez NP; Gonzalez-Gomez JC Decarboxylative Giese-Type Reaction of Carboxylic Acids Promoted by Visible Light: A Sustainable and Photoredox-Neutral Protocol. Eur. J. Org. Chem 2017, 2017, 2154. [Google Scholar]
- (60).Zheng Y; Qing F-L; Huang Y; Xu X-H Tunable and Practical Synthesis of Thiosulfonates and Disulfides from Sulfonyl Chlorides in the Presence of Tetrabutylammonium Iodide. Adv. Synth. Catal 2016, 358, 3477. [Google Scholar]
- (61).Ali MH; McDermott M Oxidation of thiols to disulfides with molecular bromine on hydrated silica gel support. Tetrahedron Lett. 2002, 43, 6271. [Google Scholar]
- (62).Chatterjee T; Ranu BC Aerobic oxidation of thiols to disulfides under ball-milling in the absence of any catalyst, solvent, or base. RSC Adv. 2013, 3, 10680. [Google Scholar]
- (63).Han M; Lee JT; Hahn H-G A traceless, one-pot preparation of unsymmetric disulfides from symmetric disulfides through a repeated process involving sulfenic acid and thiosulfinate intermediates. Tetrahedron Lett. 2011, 52, 236. [Google Scholar]
- (64).Demkowicz S; Rachon J; Witt D A Versatile and Convenient Preparation of Unsymmetrical Diaryl Disulfides. Synthesis 2008, 2008, 2033. [Google Scholar]
- (65).Tanaka K; Ajiki K Phosphine-free cationic rhodium(I) complex-catalyzed disulfide exchange reaction: convenient synthesis of unsymmetrical disulfides. Tetrahedron Lett. 2004, 45, 5677. [Google Scholar]
- (66).Petrović G; Saičić RN;Čeković Ž Regioselective free radical phenylsulfenation of a non-activated δ-carbon atom by the photolysis of alkyl benzenesulfenate. Tetrahedron 2003, 59, 187. [Google Scholar]
- (67).Cerè V; Pollicino S; Fava A Formation of cyclic sulfonium salts by Me3SiI-promoted intramolecular displacement of hydroxide or methoxide by sulfide. Ring contraction thiepane → thiolane. Tetrahedron 1996, 52, 5989. [Google Scholar]
- (68).Marset X; Guillena G; Ramón DJ Deep Eutectic Solvents as Reaction Media for the Palladium-Catalysed C–S Bond Formation: Scope and Mechanistic Studies. Chem. - Eur. J 2017, 23, 10522. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.