

Polycomb repressive EED protein orchestrates WNT, BMP, and senescence pathways to ensure proper CNS myelination and repair.
Abstract
Mutations in the polycomb repressive complex 2 (PRC2) can cause Weaver-like syndrome, wherein a patient cohort exhibits abnormal white matter; however, PRC2 functions in CNS myelination and regeneration remain elusive. We show here that H3K27me3, the PRC2 catalytic product, increases during oligodendrocyte maturation. Depletion of embryonic ectoderm development (EED), a core PRC2 subunit, reduces differentiation of oligodendrocyte progenitors (OPCs), and causes an OPC-to-astrocyte fate switch in a region-specific manner. Although dispensable for myelin maintenance, EED is critical for oligodendrocyte remyelination. Genomic occupancy and transcriptomic analyses indicate that EED establishes a chromatin landscape that selectively represses inhibitory WNT and bone morphogenetic protein (BMP) signaling, and senescence-associated programs. Blocking WNT or BMP pathways partially restores differentiation defects in EED-deficient OPCs. Thus, our findings reveal that EED/PRC2 is a crucial epigenetic programmer of CNS myelination and repair, while demonstrating a spatiotemporal-specific role of PRC2-mediated chromatin silencing in shaping oligodendrocyte identity and lineage plasticity.
INTRODUCTION
Chromatin reorganization, regulated by such features as histone modifications, is a major epigenetic event essential for vertebrate development (1, 2). The polycomb repressive complex PRC2 (polycomb repressive complex 2), the sole complex that catalyzes trimethylation of histone H3 lysine K27 (H3K27me3) in mammals (3), has two functional modules, the catalytic EZH1/2 and the methyl group–binding EED, which function as a “writer” and a “reader” of methylated H3K27, respectively (4). PRC2 recruitment facilitates H3K27me3 deposition, resulting in chromatin compaction to maintain a repressive state and inhibit gene expression (3, 5, 6).
PRC2 is critical for cell growth, differentiation, and the maintenance of cell identity. Its dysregulation has been implicated in diseases, including developmental disorders, neurodegenerative diseases, and cancers (7–13). Loss-of-function mutations in individual PRC2 components, EED, enhancer of zeste homolog 2 (EZH2), or suppressor of zeste 12 protein homolog (SUZ12), can cause human Weaver-like or Cohen-Gibson syndrome, a rare congenital disorder characterized by generalized overgrowth, macrocephaly, and intellectual disability (8–10). Recent studies indicate that a cohort of patients with Weaver-like syndrome exhibit white matter defects and delayed myelination in the brain (8–10); however, the functions of PRC2 in the myelination process during central nervous system (CNS) development are poorly defined.
Oligodendrocytes (OLs) are responsible for axonal myelination in the CNS that ensures saltatory nerve conduction (14). Defects in myelinogenesis or failures in remyelination after injury result in various developmental disorders, intellectual disabilities, and autism spectrum disorders (15, 16). Myelin repair defects also underlie neurodegenerative diseases such as multiple sclerosis and leukodystrophies (17, 18).
Subunits of the PRC2 core complex exhibit a spatiotemporal specific pattern of expression, suggesting that individual PRC2 subunits may have distinct functions (19–21). For instance, in contrast to EZH2 function mainly in regulating PRC2 activity, EED has been shown to be an “epigenetic exchange factor” orchestrating the sequential activities of both PRC2 and PRC1 complexes by sensing H3K27 trimethylation (22). At present, it is not clear to what extent the PRC2 core subunit, EED (4), functions in OL development and myelination. In addition, the role of PRC2 during myelin repair after demyelination is unknown.
In this study, we showed that, in contrast to the transient expression of EZH2 observed during the early phase of OL differentiation, EED is persistently expressed during OL maturation. By generating stage-specific conditional knockout mice lacking EED in OL progenitors (OPCs), we demonstrated that EED is necessary for OPC differentiation as well as remyelination after injury. Temporally specific fate-mapping studies indicated that Eed deletion in OPCs results in an OL-to-astrocyte fate switch in a brain region–specific manner. Chromatin accessibility and occupancy profiling revealed that the EED loss alters the chromatin landscape, leading to the loss of expression of promyelination genes and the up-regulation of inhibitory WNT and bone morphogenetic protein (BMP) signaling and cell senescence pathways. Blocking the inhibitory WNT or BMP pathways can partially restore differentiation defects in EED-deficient OPCs. Thus, our findings reveal a previously unidentified role for the EED subunit of PRC2 in epigenetic regulation of OPC differentiation and OL-astrocyte fate choice as well as in myelin repair after injury, suggesting that augmentation of PRC2 activity might enhance remyelination in demyelinating diseases.
RESULTS
Expression patterns of PRC2 components are dynamic and distinct during OL lineage progression
To define the expression patterns of PRC2 components and associated histone modification states during OPC differentiation, OPCs isolated from neonatal rats were induced to differentiate in the presence of triiodothyronine (T3) for 1, 3, or 5 days, representing the progressive stages of OPC maturation. Western blot analysis of PRC2 components indicated that the amounts of EED, SUZ12, and EZH1 were relatively constant throughout the OPC maturation process; in contrast, EZH2 expression was markedly down-regulated as OPCs differentiated (fig. S1A). Similar trends were observed when Eed and Ezh2 mRNA expression was interrogated when OPC differentiated into Mbp+ mature OLs (fig. S1B).
Immunostaining indicated that EED, SUZ12, and EZH2 were mainly expressed in the nuclei of platelet-derived growth factor receptor α–positive (PDGFRα+) OPCs and myelin basic protein–positive (MBP+) mature OLs in culture, whereas EZH1 was detected in both the nucleus and cytoplasm (fig. S1, C and D). Consistent with in vitro observations, in the developing spinal cord, EED was observed in the nuclei of PDGFRα+ OPCs and CC1+ mature OLs (fig. S1, E and F). Similarly, in vivo expression pattern of EZH1 and EZH2 was consistent with that observed in cultured cells (fig. S2A). Expression of EED, SUZ12, and EZH2 was also detected in CC1+ mature OLs in the adult spinal cord (fig. S2B). Together, these observations suggest that individual PRC2 components have dynamic and distinct expression patterns during OPC differentiation.
Since PRC2 activity regulates H3K27me3 levels (6), we examined the H3K27me3 state during OPC maturation. H3K27me3 levels increased as OPCs differentiated, consistent with previous studies (23, 24), while the activating histone mark H3K27ac (H3 acetylated at K27) levels decreased slightly (fig. S2C). In the corpus callosum, H3K27me3 was present at low levels in PDGFRα+ OPCs but at high levels in CC1+ differentiated OLs (fig. S2D). These data suggest that the deposition of histone mark H3K27me3 occurs during the transition of OPCs into myelinating cells.
Mice lacking EED in the OL lineage exhibit severe myelination deficits
To investigate how PRC2 influences CNS myelination, we ablated Eed in OL lineage cells by breeding mice with the Eed floxed allele with an OL lineage expressing Olig1-Cre line that commences in primitive OPCs (Fig. 1A). We compared conditional Eed knockout mice (Eedfl/fl;Olig1-Cre, hereafter referred to as Eed cKO) with their littermate controls, e.g., Eedfl/+;Olig1-Cre or Eedfl/fl mice, which were phenotypically normal in OL development. In Eed cKO mice, immunostaining showed that EED was depleted in PDGFRα+ OPCs and CC1+ mature OLs in the white matter of the spinal cord at postnatal day 7 (P7) (Fig. 1B). Furthermore, H3K27me3 expression was significantly reduced in both PDGFRα+ OPCs and CC1+ OLs in the corpus callosum of the Eed cKO mice (Fig. 1C), suggesting that EED was effectively depleted in the OL lineage.
Fig. 1. Eed cKO mice develop myelination defects in the CNS.
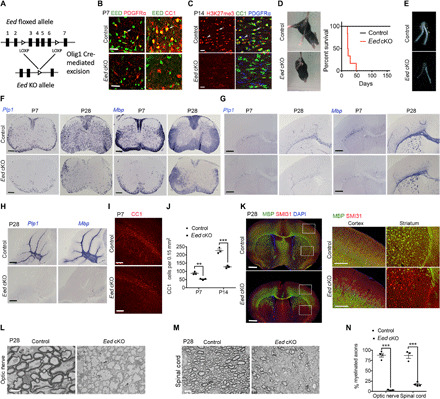
(A) Diagram depicting generation of Eed cKO mice. (B) Immunostaining for EED, PDGFRα, and CC1 in control and Eed cKO spinal cords at P7. (C) Immunostaining for H3K27me3, CC1, and PDGFRα in control and Eed cKO brains at P14. Arrows indicate H3K27me3+CC1+ OLs; arrowheads indicate H3K27me3+ PDGFRα+ OPCs. (D and E) Photographs of control and Eed cKO mice, survival curves, and optic nerves at P28. (F to H) In situ hybridization analyses for Plp1 and Mbp in the spinal cord, brain, and cerebellum from control and Eed cKO mice. (I and J) Immunostaining for CC1 (I) and quantification of CC1+ OLs (J) in control and Eed cKO brains (n = 3 animals per genotype). (K) P28 control and Eed cKO brains immunolabeled for MBP and SMI31 are shown on the left. Boxed regions are magnified to the right. (L to N) Electron micrographs (L and M) and quantification of myelinated axons (N) in P28 control and Eed cKO optic nerves and spinal cords (n > 3 animals per genotype). Scale bars, 20 μm (B and C), 200 μm (F to I), 1 mm (K), 1 μm (L), and 4 μm (M). Data are means ± SEM. **P < 0.01, ***P < 0.001. DAPI, 4′,6-diamidino-2-phenylindole. Photo credit: Jiajia Wang, CCHMC.
Eed cKO mice developed generalized tremors and exhibited hindlimb clasping, a sign of neurological disorder, and died around postnatal week 6 (Fig. 1D). The optic tracts from Eed cKO mice were transparent, indicating a myelin deficiency (Fig. 1E). Furthermore, expression of Mbp and Plp1 was diminished significantly in the spinal cord, cerebral cortex, and cerebellum of mutant mice in comparison to controls from P7 to P28 (Fig. 1, F to H). Consistent with this finding, there were significantly fewer CC1+ mature OLs in the white matter of the mutant brains compared with the controls (Fig. 1, I and J).
We found that expression of myelin protein MBP was markedly decreased in the brain, especially in the superficial cortex and ventral striatum, in Eed cKO mice compared with controls at P28, whereas phosphorylated neurofilaments marked by SMI31 were present at similar levels (Fig. 1K), suggesting that the loss of MBP expression is not due to the gross axonal loss in Eed cKO mice. In light of these observations, we examined the ultrastructure of myelin sheaths in the optic nerves and spinal cords by electron microscopy. We found that axonal ensheathment in the CNS of Eed cKO mice at P28 was severely compromised compared with controls (Fig. 1, L to N). Together, these results indicate that Eed deletion in OL lineage cells results in myelination deficits in the CNS.
EED regulates the transition from OPCs to differentiated OLs
To investigate whether the dysmyelinating phenotype of Eed cKO mice is due to a defect in OPC development, we assessed OPC development by examining an OPC marker Pdgfra. In the Eed cKO animals at P7, Pdgfra+ OPCs were observed in the CNS regions. The OPC number in the deep cortical layers was comparable to the controls (Fig. 2, A and B); however, in the superficial cortical layers and ventral striatum of the forebrain, the number of OPCs was reduced in the Eed cKO animals (Fig. 2, A and B). Similarly, the percentage of Ki67+ PDGFRα+ OPCs was reduced in these regions (Fig. 2, C and D). In contrast, we did not observe substantial alteration of OPC numbers in the spinal cord and cerebellar white matter of control and Eed cKO mice (fig. S3, A and B). The observations suggest a region-specific impact on OPC development in the absence of EED.
Fig. 2. Eed deletion impairs OPC proliferation and differentiation.
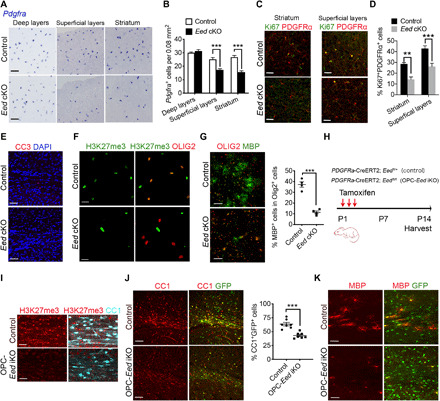
(A and B) In situ hybridization for Pdgfra (A) and quantification of Pdgfra+ OPCs (B) in different brain regions from control and Eed cKO mice at P7 (n = 4 animals per genotype). (C and D) Immunolabeled for Ki67 and PDGFRα (C) and quantification of Ki67+ PDGFRα+ cells (D) in the striatum and superficial layers from P7 control and Eed cKO brains (n = 4 animals per genotype). (E) P14 control and Eed cKO brains labeled for cleaved caspase 3 (CC3) and DAPI. (F) Immunostaining for H3K27me3 and OLIG2 in OPCs isolated from control and Eed cKO cortices. (G) Control and Eed cKO OPCs treated with T3 for 3 days and immunostained for OLIG2, MBP (left). The percentages of MBP+ cells relative to OLIG2+ cells (right, n = 4 independent experiments). (H) Diagram depicting tamoxifen administration strategy on control and OPC-Eed iKO mice. (I) Control and OPC-Eed iKO brains at P14 stained for H3K27me3 and CC1. (J) Immunolabeling for CC1 (left) and quantification of CC1+GFP+ cells (right) in control and OPC-Eed iKO brains at P14 (n = 7 animals per genotype). (K) The white matter of control and OPC-Eed iKO mice at P7 stained for MBP. Scale bars, 50 μm (A), 60 μm (C, I, and K), 100 μm (E, G, and J), and 20 μm (F). Data are means ± SEM. **P < 0.01, ***P < 0.001. Photo credit: Jiajia Wang, CCHMC.
We found a comparable rate of survival (assayed by cleaved caspase 3) between control and Eed cKO cortices (Fig. 2E), suggesting that Eed deletion does not impair OPC survival. To determine whether defects in OL differentiation are a cell-autonomous effect of Eed ablation, we purified OPCs from the neonatal cortices of Eed cKO and control pups, and then treated them with T3 to promote OPC differentiation. Immunostaining showed that H3K27me3 was barely detected in Eed cKO OPCs (Fig. 2F). Control OPCs were able to differentiate into mature MBP+ OLs with a complex morphology; in contrast, Eed cKO OPCs failed to differentiate into MBP+ OLs (Fig. 2G). The percentage of MBP+ cells was approximately 37.3 ± 5.0% in control OPCs, but only 11.3 ± 2.6% in Eed cKO OPCs, suggesting that Eed-depleted OPCs are intrinsically reduced in their differentiation capacity.
To further examine the effects of Eed inactivation on differentiation of OPCs during postnatal development, we generated mice in which Eed can be inducibly deleted in OPCs by breeding Eedfl/fl mice with Pdgfra-CreERT2 mice (25); the latter carry an OPC-specific tamoxifen-inducible cre and a Rosa26:ccGFP reporter (Fig. 2H). The resulting mice (Pdgfra-CreERT2;Eedfl/fl) are hereafter referred to as OPC-Eed iKO mice. Tamoxifen administration from P1 to P3 resulted in depletion of EED as shown by the loss of the PRC2 catalytic product H3K27me3 (Fig. 2I). In accordance with our results of Eed cKO mice, the ablation of Eed in OPCs led to a marked reduction in CC1+ differentiating OLs in the white matter of OPC-Eed iKO mice compared with control littermates (Fig. 2J). In addition, MBP expression was observed at significantly lower levels in OPC-Eed iKO brains at P7 than that of controls (Fig. 2K). Moreover, we also observed reduced OPC proliferation in the superficial cortical layers of the forebrain in OPC-Eed iKO mice compared with the control (fig. S3C). We did not observe any substantial alteration of OPC survival in the cortices of Eed iKO mice (fig. S3D). Together, these data indicate that EED is required for OPC proliferation and differentiation.
Deletion of Eed causes OPC-to-astrocyte fate switch in a brain region–specific manner
Since Eed deletion specifically from OPCs led to a defect in the transition into mature OLs and caused dysmyelination in the CNS, we investigated whether deletion of Eed affects the development of other cell types. By analysis of the astrocyte marker glial fibrillary acidic protein (GFAP) at P28, we found GFAP expression was significantly up-regulated in the superficial cortex and ventral striatum of the forebrain as well as septal regions of the mutant mice compared with controls (Fig. 3A), whereas minimal MBP was observed in the Eed mutants (Fig. 1K). In contrast to Ezh2 cKO mice, GFAP expression was not significantly altered in the brains, despite a delay in OL myelination (fig. S4) as shown previously (24). Similar observations were made upon analysis of the brainstem region: GFAP expression was up-regulated in the center of the brainstem, which exhibited a loss of MBP expression, in the Eed cKO mice (Fig. 3, B and C), indicating a reciprocal relationship between GFAP and MBP expression. Consistent with this, lower MBP expression and higher GFAP expression were observed in Eed cKO brains by Western blotting (Fig. 3D). We did not detect any substantial changes in astrocyte proliferation in the cortex (fig. S3E), suggesting that the astrocytes derived from OPCs in the absence of EED were likely differentiated cells. In addition, there was negligible microglial activation in Eed mutants as shown by labeling with the microglia marker Iba1 (Fig. 3E).
Fig. 3. EED regulates OPC and astrocyte fate switch.
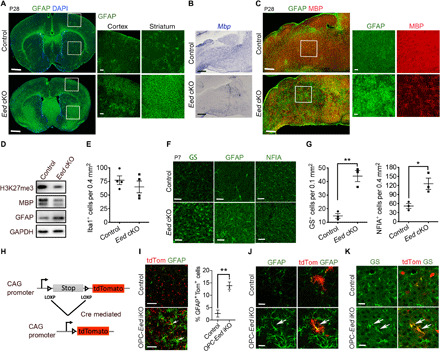
(A) Control and Eed cKO forebrains at P28 stained for GFAP are shown on the left. Boxed areas are magnified to the right. (B) In situ hybridization for Mbp in brainstems from control and Eed cKO mice at P28. (C) The brainstems of control and Eed cKO mice at P28 immunostained for MBP and GFAP are shown on the left. Boxed areas are magnified to the right. (D) Western blot of H3K27me3, MBP, and GFAP from control and Eed cKO brains at P14. (E) Quantification of Iba1+ cells in control and Eed cKO cortices at P28 (n = 4 animals per genotype). (F and G) Immunolabeling for glutamine synthetase (GS), GFAP, and NFIA (F) and quantification of GS+ and NFIA+ astrocytes (G) in control and Eed cKO cortices at P7 (n = 3 animals per genotype). (H) Diagram showing Cre-mediated activation of Tomato after tamoxifen administration. (I) Immunostaining for GFAP (left) and quantification of GFAP+Tomato+ astrocytes (right) in the cortices of control and OPC-Eed iKO mice at P14 (n = 3 animals per genotype). (J and K) Control and OPC-Eed iKO cortices at P14 stained for GFAP (J) or GS (K). Scale bars, 1 mm (A, left; C, left), 100 μm (A, right; B and C, right), 60 μm (F and I), and 20 μm (J and K). Data are means ± SEM. *P < 0.05, **P < 0.01. Photo credit: Jiajia Wang, CCHMC.
To further validate the increase in astrocyte production during development, we immunostained for an astrocyte marker glutamine synthetase (GS), which marks the soma of astrocytes, and for GFAP in the cortices of brains at P7. GS and GFAP levels were up-regulated in Eed cKO cortices compared with control mice (Fig. 3, F and G). Moreover, there was an increase in nuclear factor IA (NFIA), an astrocyte transcriptional regulator, in the Eed cKO brain compared with the controls (Fig. 3, F and G). These data indicate that there are higher frequencies of astrocyte formation in the brain in the absence of EED.
To further trace the fates of Eed-ablated cells, we examined OPC-Eed iKO (Pdgfra-CreERT;Eedfl/fl) mice, which also carried a Rosa26:tdTomato reporter (26) (Fig. 3H). Tamoxifen-induced Cre-mediated LOXP site excision concurrently leads to Eed ablation and activation of Tomato expression. In contrast to the controls, in the cortices of OPC-Eed iKO mice at P14, we observed an increased population of Tomato+ cells expressed GFAP and GS and exhibited characteristic astrocytic morphologies with highly branched bushy processes (Fig. 3, I to K). This indicates that OPCs are converted into astrocytes in the absence of Eed, suggesting that ectopically induced astrocytes are derived from Eed-ablated OPCs.
EED is not essential for myelin maintenance
To determine whether EED plays a role in myelin maintenance in mature OLs, we ablated Eed in adult OLs by crossing Eed-floxed mice with mice that express the Plp-CreERT2 driver in OLs (27). In these Eedfl/fl;Plp-CreERT2 (OL-Eed iKO) mice, tamoxifen treatment from P35 to P40 induced Eed deletion and loss of its catalytic product H3K27me3 in PLP+ mature OLs as shown by analysis at P70 (fig. S5, A and B). Immunostaining showed that the numbers of OLIG2+/CC1+ OLs and MBP expression were comparable between control and OL-Eed iKO mice at P70 (fig. S5, C and D). In addition, ultrastructural analysis showed no signs of demyelination in the optic nerves of tamoxifen-treated OL-Eed iKO adult mice at P70 and P110 (fig. S5, E to H), suggesting that EED is not critical for myelin maintenance at the examined adult stages.
EED is critical for OL remyelination after demyelinating injury
Given the crucial role of EED during developmental myelination, we next investigated whether EED is required for OL regeneration following demyelination induced by lysolecithin (LPC). LPC injection induces a focal demyelinating lesion in the white matter of the spinal cord in mice. OL recruitment is observed at 7 days postlesion (dpl) and a remyelinating phase at dpl 14 (28, 29). In normal adult spinal white matter, the number of cells positive for EED and H3K27me3 was low; at 14 dpl after LPC injection, however, the number increased substantially (Fig. 4A). Notably, EED was detected not only in OLIG2+ OL lineage cells including PDGFRα+ OPCs and CC1+ OLs but also in GFAP+ astrocytes and CD11b+ microglia in the lesion (Fig. 4, B and C).
Fig. 4. EED is required for remyelination after LPC injury.

(A) Immunostaining for EED or H3K27me3 in control and LPC-treated spinal cords. (B and C) Control or LPC-treated spinal cords immunolabeled for EED, H3K27me3, and OLIG2 or PDGFRα or CC1 or GFAP or CD11b. (D) Diagram showing tamoxifen and LPC administration schedule. (E) Immunostaining for H3K27me3 in control and OPC-Eed iKO lesions at dpl 14. (F) In situ hybridization for Mbp and Plp1 (left) and quantification of Plp1+ cells (right) in control and OPC-Eed iKO lesions at dpl 14 (n > 3 animals per genotype). (G) Lesions from control and OPC-Eed iKO spinal cords stained for MBP. (H) Immunostaining for CC1 (left) and quantification of CC1+ cells (right) in lesions from control and OPC-Eed iKO spinal cords (n > 3 animals per genotype). (I) Electron micrographs of LPC lesions (left) and quantification of remyelinated axons (right) in control and OPC-Eed iKO spinal cords at dpl 14 (n > 3 animals per genotype). (J) In situ hybridization for Pdgfra (left) and quantification of Pdgfra+ cells (right) in lesions from control and OPC-Eed iKO mice at dpl 14 (n > 3 animals per genotype). (K) Control and OPC-Eed iKO lesions at dpl 14 stained for GFAP; arrows indicate GFAP+Tomato+ cells. Scale bars, 60 μm (A, E, G, and H), 30 μm (B, C, and K), 100 μm (F and J), and 2 μm (I). Data are means ± SEM. *P < 0.05, ***P < 0.001. Photo credit: Jiajia Wang, CCHMC.
Photo credit: Jiajia Wang, CCHMC
To determine the role of EED in OL regeneration, we administered tamoxifen to adult OPC-Eed iKO mice 5 days before LPC injection to delete Eed in adult OPC-Eed iKO mice, with heterozygous Pdgfra-CreERT:Eedfl/+ mice serving as controls (Fig. 4D). After LPC injection into the ventral white matter of the spinal cord, mice were treated for five additional days with tamoxifen (Fig. 4D), and the lesioned spinal cords were harvested at dpl 14 and dpl 21. Immunostaining showed that expression of H3K27me3 was substantially depleted in the lesions of OPC-Eed iKO mice compared with controls (Fig. 4E), suggesting the loss of the EED/PRC2 function in OPCs.
To determine the extent of remyelination, we examined the expression of myelin genes Mbp and Plp1. At dpl 14 and dpl 21, the number of mature OLs marked by Plp1 and MBP expression was markedly reduced in OPC-Eed iKO animals compared with controls (Fig. 4, F and G). Consistent with this, fewer CC1+ OLs were detected in the lesions of OPC-Eed iKO mice than in controls at dpl 14 and dpl 21 (Fig. 4H). Consistently, the number of remyelinated axons was substantially lower in the lesions of OPC-Eed iKO mice at dpl 14 (Fig. 4I). The loss of Eed did not appear to impair the recruitment of PDGFRα+ OPCs; however, the number of these cells in lesions was reduced in Eed iKO mice (Fig. 4J). In addition, we detected expression of astrocytic GFAP in a population of Eed-ablated OPCs, suggesting an OPC-to-astrocyte conversion occurred in the lesions of Eed-iKO mice (Fig. 4K). Together, these data indicate that Eed deletion leads to a defect in the remyelination process by blocking OPC differentiation and altering their cell fate, suggesting that EED has a crucial role in OPC redifferentiation or remyelination in the context of white matter injury.
EED regulates the transcriptional program necessary for OL differentiation
We next sought to define the mechanism whereby EED regulates OPC differentiation into mature OLs. We isolated OPCs from control and Eed cKO cortices by immunopanning with O4 antibodies (30) and subjected them to transcriptome profiling. Genes significantly up-regulated and down-regulated (fold change >1.5, P < 0.05) in Eed cKO OPCs compared with control OPCs were identified (Fig. 5, A and B). Gene Set Enrichment Analysis (GSEA) revealed that the genes down-regulated in Eed-deficient OPCs were enriched in gene signatures associated with OL differentiation, lipid metabolism, and myelin sheath formation (Fig. 5, C and D), consistent with the dysmyelination phenotype in Eed cKO mice. We found that the target genes of H3K27me3, the EED-PRC2 product, were up-regulated in Eed-deficient OPCs (Fig. 5, C and D); this was expected as H3K27me3 is associated with transcriptional repression (3, 5, 6).
Fig. 5. EED controls a regulatory network necessary for OPC differentiation.
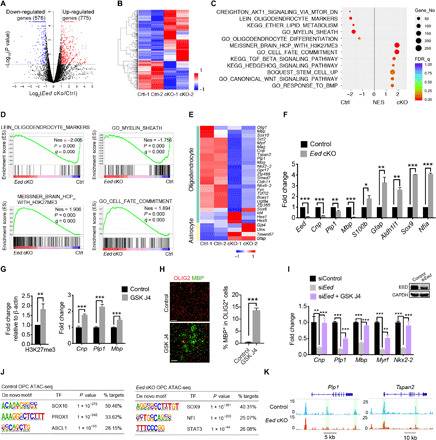
(A) Volcano plot of genes differentially regulated in Eed cKO OPCs (n = 2 experiments; fold change, >1.5; P < 0.05). (B) Heatmap of differentially expressed genes in Eed cKO OPCs from six animals per genotype. (C) GSEA of genes enriched in control or Eed cKO OPCs; NES, net enrichment score. (D) GSEA plots of indicated signature genes. (E and F) Heatmap (E) and quantitative real-time PCR (qRT-PCR) analyses (F, n = 3 experiments) of representative OL and astrocyte genes that are differentially expressed in Eed cKO OPCs. (G) Normalized expression of H3K27me3 relative to β-actin in control and GSK J4–treated Oli-neu cells for 48 hours (left). qRT-PCR analyses of myelination-associated genes (right), n = 3 experiments. (H) Immunostaining for OLIG2 and MBP (left) and quantification of MBP+ cells (right) in mouse OPCs treated with GSK J4 for 48 hours (n = 3 experiments). Scale bars, 100 μm. (I) qRT-PCR analyses of myelination-associated genes in Oli-neu cells treated with siEed or with siEed and GSK-J4 for 48 hours (n = 3 experiments). Western blot of EED to show the knockdown efficiency. (J) De novo motif analysis of ATAC-seq data of control and Eed cKO OPCs. (K) Representative ATAC-seq tracks for OL differentiation genes in control and Eed cKO OPCs. Data are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. Photo credit: Jiajia Wang, CCHMC.
Signatures of stem cell and cell fate commitment were also enriched in Eed-deficient OPCs, consistent with OPC-astrocyte fate conversion in Eed cKO mice (Fig. 5, C and D). Expression of mature OL markers such as Mog, Plp1, Mag, and Mbp was reduced in Eed cKO OPCs compared with controls, whereas astrocyte-associated genes such as Gfap, Gja4, and Tmem67 were up-regulated (Fig. 5E). Quantitative polymerase chain reaction (qPCR) analysis confirmed these observations for the genes evaluated, including astrocyte transcriptional regulators Sox9 and Nfia (Fig. 5F). To identify the genes directly regulated by the PRC2 that are necessary for OPC differentiation, we examined the targets of SUZ12, which is tightly associated with EED in the PRC2 complex, by chromatin immunoprecipitation sequencing (ChIP-seq) analysis (24). We detected SUZ12 binding peaks in the loci of OL differentiation regulators such as Nkx6.2 and Zfp365, while negligible or weak SUZ12-binding signals on the loci of other myelination-associated genes such as Myrf, Plp1, and Mbp (fig. S6A). Thus, EED/PRC2 may differentially regulate expression of OL differentiation regulatory genes through both direct and indirect mechanisms.
To determine whether astrocyte-regulatory factors were repressed by EED in OPCs, we analyzed expression of astrocyte-defining transcription factors such as Nfia and Sox9 and found that Nfia and Sox9 were up-regulated in OPCs isolated from Eed cKO animals (Fig. 5F). In addition, their expression reduced as OPC differentiation proceeded (fig. S6B). However, ChIP-seq analysis indicates negligible SUZ12-binding peaks on the promoter regions of Nfia and Sox9 loci (fig. S6C), suggesting that Nfia and Sox9 might not be direct targets of SUZ12 in OPCs. Nonetheless, given the down-regulation of OPC differentiation and increase in astrocytes in Eed cKO animals, these data suggest that EED regulates a genetic program that controls the OL-astrocyte fate choice in OPCs.
To further investigate whether the depletion of H3K27me3 levels caused by Eed deficiency inhibits the OPC differentiation program, we treated Oli-neu cells, a mouse oligodendroglia cell line (31), or mouse OPCs with the H3K27 demethylase JMJD3 inhibitor GSK-J4 (32). Treatment of GSK-J4 increased H3K27me3 levels and up-regulated expression of differentiation markers Cnp, Plp1, and Mbp in Oli-neu cells (Fig. 5G) and substantially increased OPC differentiation into MBP+ OLs (Fig. 5H), suggesting that increased H3K27me3 levels by pharmacological treatment with GSK-J4 promote OPC maturation. In addition, silencing Eed with siEed in Oli-neu cells significantly decreased expression of myelin genes such as Cnp, Plp1, and Mbp, as well as OL regulatory genes such as Myrf and Nkx2.2 (Fig. 5I); however, cotreatment with GSK-J4 significantly restored expression of these genes (Fig. 5I). These data suggest that EED activity promotes OPC differentiation at least in part through H3K27me3 deposition.
EED establishes an accessible chromatin landscape to activate OL-specific transcriptional programs
Since PRC2 is a histone modifier, we hypothesized that the core subunit EED might be necessary for regulation of the chromatin landscape to facilitate OL lineage progression. To test this hypothesis, we performed an assay for transposase-accessible chromatin (ATAC-seq) (33) to identify regions of open chromatin structure in OPCs from control and Eed cKO animals. The binding motifs of OL-promoting factors SOX10 and ASCL1 were preferentially enriched in the open chromatin regions of control OPCs, whereas there was enrichment for binding motifs of astrocyte lineage regulators SOX9 and NFI within the open regions of Eed-deficient OPCs (Fig. 5J and fig. S7A). Compared with control OPCs, chromatin accessibility in the promoters and proximal enhancers of OL differentiation–related genes such as Plp1 and Tspan2 was decreased in Eed-deficient OPCs (Fig. 5K and fig. S7B). The data suggest that Eed deficiency results in reduced chromatin accessibility at the regulatory elements in a set of genes associated with OPC differentiation program.
Up-regulation of WNT and BMP signaling pathways in Eed-deficient OPCs
The SUZ12-targeted genes were mostly excluded from H3K27ac-enriched gene loci in OPCs (Fig. 6A). We found that 312 genes were targeted by PRC2 and were differentially expressed in Eed-deficient OPCs (Fig. 6B). Among the up-regulated targets, we observed that factors involved in OL differentiation–inhibitory signaling, such as WNT and BMP pathways, were enriched in Eed-deficient OPCs (Fig. 6C).
Fig. 6. Wnt and BMP signaling pathways are up-regulated in Eed-deficient OPCs.
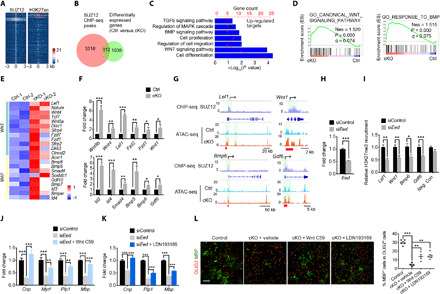
(A) Heatmaps of ±3 kb around SUZ12 and H3K27ac ChIP-seq peak centers from WT OPCs. (B) Venn diagram showing the overlap between SUZ12-bound and differentially expressed genes in Eed cKO OPCs. (C) Enrichment analyses of EED targets that up-regulated in Eed-deleted OPCs. (D) GSEA plots of WNT and BMP pathway signatures. (E and F) Heatmap (E) and qRT-PCR analyses (F, n = 3 experiments) of genes associated with WNT and BMP pathways up-regulated in Eed cKO OPCs. (G) Representative SUZ12 ChIP-seq in rat OPCs and ATAC-seq tracks of indicated genes in control and Eed cKO OPCs. (H) qRT-PCR analyses of Eed in Oli-neu cells at 48 hours after transfection with siControl or siEed (n = 3 experiments). (I) ChIP-qPCR analyses for H3K27me3 enrichment at the promoters of indicated genes in Oli-neu cells treated with siControl or siEed (n = 3 experiments). (J and K) qRT-PCR analyses of myelination-associated genes in Oli-neu cells treated with siControl, siEed, or siEed together with either Wnt C59 or LDN193189 for 48 hours (n = 3 experiments). (L) Immunostaining for OLIG2 and MBP (left) or quantification of MBP+ cells (right) in control and Eed cKO OPCs treated with Wnt C59 or LDN193189 for 3 days (n > 3 experiments). Scale bars, 100 μm. Data are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. Photo credit: Jiajia Wang, CCHMC.
GSEA and transcriptome analyses revealed that WNT and BMP pathway genes were significantly up-regulated in Eed cKO OPCs in contrast to controls (Fig. 6, D and E). Consistent with these data, qPCR analyses showed that expression of WNT/β-catenin effector genes such as Wnt8b, Wnt1, and Lef1, and BMP signaling genes such as Id2/4, BMP6, and Gdf6 were up-regulated in Eed-deficient OPCs (Fig. 6F). Thus, these data suggest that EED deficiency results in an up-regulation of WNT and BMP signaling to inhibit OPC differentiation.
To further examine the targeting of WNT and BMP signaling components by the PRC2 complex, we found that SUZ12 directly bound to the promoter regions of WNT targets Lef1 and Wnt1 and to BMP pathway genes Bmp6 and Gdf6 (Fig. 6G), suggesting that activation of PRC2 inhibits expression of WNT and BMP targets. In addition, chromatin accessibility of the promoters and proximal enhancers of these WNT and BMP signaling genes was significantly increased in Eed-deficient OPCs compared with controls (Fig. 6G and fig. S7C), consistent with their expression change. ChIP-qPCR analysis showed that H3K27me3 enrichment in the WNT target genes, Lef1 and Wnt1, and BMP pathway genes, Bmp6 and Gdf6, was reduced significantly after Eed knockdown, indicating that the up-regulation of these genes is likely a direct consequence of PRC2 loss of function mediated by Eed deletion in OPCs (Fig. 6, H and I). Furthermore, treatment of Eed-deficient OPCs with WNT signaling inhibitor Wnt C59 or with BMP signaling inhibitor LDN193189 resulted in a significant up-regulation of differentiation markers (Fig. 6, J and K) and an increase in MBP+ mature OLs (Fig. 6L), but did not restore substantial H3K27me3 levels (fig. S8). This suggests that blocking WNT or BMP signaling can partially restore OL differentiation without increasing H3K27me3 levels in Eed-deficient OPCs, suggesting that WNT or BMP signaling is one of the downstream pathways regulated by the PRC2 complex. Therefore, these data indicate that the EED-containing PRC2 directly represses expression of differentiation inhibitory WNT and BMP signaling components to facilitate OPC differentiation.
Elevation of cell senescence pathways in Eed-deficient OPCs
Recent studies indicate that senescence of OPCs leads to a defect in remyelination capacity in demyelinating lesions (34–36). p53 and p16INK4a are key regulators of senescence-related pathways to induce cell cycle arrest and aging (37). Notably, we identified an enrichment of genes associated with cellular senescence, such as Cdkn2a, Cdkn2b, Ets1, Igf1r, Igfbp2, and Bmi-1 (34), were up-regulated in Eed-deficient OPCs (Fig. 7, A and B). qPCR analysis indicated that expression of SASP (senescence-associated secretory phenotype) genes, e.g., Fgf5, Il16, and Tgfb1, was up-regulated in Eed-deficient OPCs acutely isolated from P5 cortices (Fig. 7C). Furthermore, the Eed-deficient OPCs exhibited a substantial increase in expression of senescence-associated β-galactosidase (SA-β-Gal), which is indicative of cell senescence and growth arrest (Fig. 7D) (38, 39). Moreover, we found a robust increase in p16INK4a, a marker of cellular senescence (37), and p53, which promotes cell senescence (37), in the Eed cKO OPCs by immunostaining and Western blot analyses (Fig. 7, E and F). These data suggest that the loss of EED promotes the cell senescence program in acutely isolated OPCs. In addition, ChIP-seq analysis showed that the PRC2 core factor, SUZ12, can directly bind to the promoter region of the p16-encoding Cdkn2a gene (Fig. 7G), and chromatin accessibility was increased in the promoter region of Cdkn2a in Eed cKO OPCs compared with controls (Fig. 7G and fig. S9, A and B). Moreover, ChIP-qPCR results showed H3K27me3 recruitment to the promoter region of Cdkn2a, expression of which is reduced upon Eed knockdown in OPCs, suggesting that the EED/PRC2 complex directly regulates Cdkn2a expression (Fig. 7H). Together, these data indicate that EED deficiency may induce OPC senescence due to up-regulation of p16INK4a and p53 expression, thus contributing to the blocking of OL lineage progression.
Fig. 7. Cell senescence pathways are elevated in Eed-deficient OPCs.
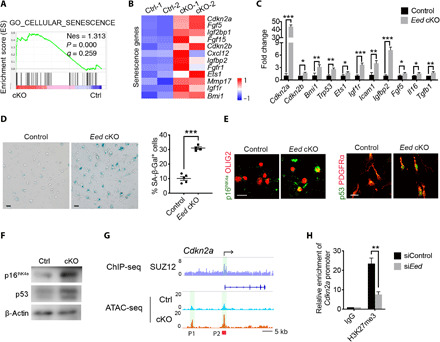
(A) GSEA plots of cellular senescence genes enriched in Eed cKO OPCs. (B) Heatmaps of senescence-associated genes up-regulated in Eed cKO OPCs. (C) qRT-PCR analyses of senescence-associated genes and SASP genes in control and Eed cKO mouse OPCs acutely isolated from cortices at P5 (n = 3 experiments). (D) Representative images of control and Eed cKO OPCs subjected to 5-bromo-4-chloro-3-indolyl-b-D-galactopyranoside (X-Gal) staining. Blue cells are senescence cells (left). Quantification of the percentage of SA-β-Gal+ OPCs (right, n = 5 experiments). (E) Representative images of control and Eed cKO mouse OPCs stained for p16INK4a, OLIG2, p53, and PDGFRα. (F) Western blot of p53, p16INK4a, and β-actin from lysates of OPCs from control and Eed cKO mice. (G) Representative SUZ12 ChIP-seq in rat OPCs and ATAC-seq tracks at the promoter of Cdkn2a in the control and Eed cKO OPCs. P1 and P2, open chromatin sites. (H) ChIP-qPCR analysis of H3K27me3 enrichment at the promoter region of the Cdkn2a gene in control and siEed-treated Oli-neu cells (n = 3 experiments). Scale bars, 20 μm (D and E). Data are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. IgG, immunoglobulin G. Photo credit: Jiajia Wang, CCHMC.
DISCUSSION
Polycomb group proteins play a pivotal role in establishing and maintaining gene expression patterns during development, differentiation, and tumorigenesis (3, 5–8). In this study, we show that EED, a critical mediator of PRC complexes, is required for CNS myelination and myelin repair. Notably, deletion of Eed not only decreases the differentiation of OPCs but also induces an OPC-to-astrocyte fate switch. Although dispensable for myelin maintenance in mature OLs, we find that EED is required for remyelination after demyelinating injury, which recapitulates the critical role of EED in OL lineage development. Our data further indicate that EED-PRC2 represses multiple inhibitory pathways such as BMP and WNT signaling and might prevent the OPC senescence program to safeguard the cascade of events leading to myelination in the CNS (fig. S10).
A stage-specific requirement of PRC2 subunits for OL lineage differentiation
EED was continuously expressed during the OPC maturation process, mainly detected in the nucleus in OPCs and mature OLs. Deletion of Eed from Olig1-expressing lineage cells led to severe dysmyelination. EED depletion in PDGFRα+ OPCs inhibited their differentiation; however, Eed knockout in PLP+ mature OLs did not affect myelin maintenance, suggesting a stage-specific function of EED in OL lineage progression.
Intriguingly, EZH2, a catalytic subunit of PRC2, was enriched in the nucleus of OPCs at the early phase of differentiation but down-regulated in mature OLs; this is in contrast to the persistent expression of EZH1 and EED observed during OPC differentiation. This is in agreement with a previous report that EZH2 is predominantly expressed in proliferating cells (40). Notably, H3K27me3 reduction was less severe in OLs of Ezh2 cKO mice compared with Eed cKO mice (fig. S4). Consistently, deletion of Ezh2 in the OL lineage cells only causes a delay in OL differentiation, and the dysmyelination phenotype is recovered at the late postnatal stages (fig. S4) (24), in contrast to sustained defects in myelination in Eed cKO mice. The difference in the extent and severity of dysmyelination between Ezh2- and Eed-deficient mutants is likely due to transient expression of EZH2 in OPCs over the course of differentiation. It remains to be determined whether EZH1 can compensate for the loss of EZH2 during OL myelination. Nonetheless, our data suggest a temporally specific function of distinct PRC2 subunits during OL differentiation.
Although silencing or pharmacological inhibition of H3K27 methyltransferase EZH2 does not appear to impair OPC differentiation in vitro (23), our in vivo depletion analysis indicated that the activity of PRC2, the sole enzyme for catalyzing H3K27me3, is critical for OPC differentiation during CNS development. Notably, although severe dysmyelination was observed in mice with Eed deletion in the OL lineage, Eed deletion in the Schwann cell lineage does not affect developmental myelination in the peripheral nervous system (41), suggesting distinct roles of EED in central and peripheral myelination. Nonetheless, EED deficiency did result in delayed regeneration of myelinating Schwann cells after peripheral nerve injury (42), similar to remyelination defects in Eed-deficient OLs after spinal cord injury. These observations suggest that PRC2 functions in a cell type–specific manner in developmental myelination and remyelination in the central and peripheral nervous systems.
EED safeguards OL identity and regulates OPC-to-astrocyte fate choice
The capacity of OPCs to adopt an astrocytic fate has been shown in vitro (43). However, there is no evidence that committed OPCs adopt an astrocytic fate during CNS development, suggesting that a mechanism maintains OL lineage identity in OPCs while suppressing alternative cell fates. Our lineage tracing study showed that EED loss can lead to OPC fate conversion to generate astrocytes in specific brain regions, indicating that PRC2-mediated deposition of H3K27me3 maintains commitment to the OPC state and safeguards OL identity. Notably, depletion of EZH2 in the OL lineage did not induce the OPC-astrocyte fate switch (fig. S4), suggesting a unique function of EED in regulating the cell fate choice. The loss of transcriptional regulators such as OLIG2, histone deacetylase 3 (HDAC3), and ZFP36L1 also leads to a switch from an OPC to an astrocytic fate (44–46). Whether and how these factors regulate PRC2 activity and H3K27me3 state remain to be determined. Given that PRC2 can regulate histone deacetylation activity through the interaction with HDACs including HDAC3 (19, 47), it is plausible that EED cooperates with HDAC3 to maintain OL lineage commitment and prevent alternative cell fates. Thus, our findings indicate that PRC2 acts as a key epigenetic modulator to control the cell fate decision between OL and astrocyte lineages in a region-specific manner.
The genomic occupancy analysis indicates that the astrocyte regulatory genes Nfia and Sox9 may not be direct targets of SUZ12 in OPCs. Thus, the increased level of Nfia and Sox9 is likely the consequence of increased astrocyte number in the Eed cKO mice. EED/PRC2 might regulate expression of Nfia and Sox9 through indirect mechanisms to repress astrocyte formation, which remain to be further defined. Our data suggest that Eed deletion alters the genomic landscape and chromatin accessibility, leading to less open chromatin structure on loci differentially expressed in OLs. At present, it remains to be determined how EED achieves cell type–specific functions to coordinate and balance the expression of OL and astrocyte lineage genes. Unexpectedly, global H3K27ac levels were not up-regulated in the absence of EED (fig. S9, C and D), given that the loss of function of PRC2 causes down-regulation of H3K27me3 while reciprocally up-regulating H3K27ac and its associated active transcriptional landscape in other cellular contexts (7, 19). This suggests a cell-specific PRC2 function in regulating the balance levels of chromatin modifications H3K27me3 and H3K27ac.
EED/PRC2 represses WNT and BMP signaling along with cell senescence pathways to promote OL differentiation
The integrated genome-wide mapping of PRC2 occupancy and transcriptomic profiling of mice with deficiencies in EED revealed that PRC2 modulates key regulators of OL lineage development. Notably, we found that PRC2 targets the components of WNT and BMP pathways. These pathways are up-regulated in Eed-deleted OPCs, consistent with previous reports that PRC2 inactivation leads to derepression of the genes associated with loss of H3K27me3 (4, 48). WNT and BMP signaling pathways function as negative regulators of OPC differentiation (49, 50). In addition, up-regulation of BMP signaling promotes astrocyte differentiation (51). Inhibition of WNT and BMP signaling partially restored the OL differentiation program in Eed-deficient OPCs. Thus, the PRC2 may promote and ensure OL lineage differentiation at least in part through silencing WNT and BMP signaling and maintains these genes in a repressed state to prevent adoption of alternate cell fates. It is worth noting that GSEA analysis showed that the AKT–mammalian target of rapamycin (mTOR) signaling, a key signal for OL myelination (52, 53), is down-regulated in Eed-deficient OPCs (Fig. 5C), suggesting that the EED/PRC2 complex may also regulate AKT-mTOR signaling activity for OPC differentiation. This is in keeping with previous study that EZH2/PRC2 activates mTOR signaling during colorectal carcinoma cells (54). It remains to be determined how the PRC2 complex regulates AKT-mTOR signaling activity during OL myelination.
Intriguingly, our analysis of PRC2 genomic binding revealed that the Cdkn2a gene, which encodes the p16INK4a tumor suppressor, is a PRC2 target. Eed ablation led to up-regulation of p53 and its downstream effector p16INK4a in OPCs. p16INK4a expression is a key marker for cell senescence (37). p16INK4a up-regulation causes potent activation of genes associated cellular senescence, is accompanied by cell cycle arrest, and is observed during aging (37, 55, 56). Although the loss of EED elevates p16INK4a and p53 to promote the cell senescence program in acutely isolated OPCs, further study would need to validate the cell senescence in the brain of Eed-cKO mice in vivo. Since our data indicate that EED deficiency impairs OPC proliferation in the superficial cortical layers and ventral striatum, EED depletion in OPCs likely induces p16INK4a-mediated cell senescence program, leading to arrested cell differentiation, which is consistent with previous findings that PRC2 prevents cell senescence in other contexts (40, 56, 57).
PRC2 is relevant in myelinating diseases
Dysregulation of PRC2 components has been implicated in human developmental disorders, neurological diseases, and cancers (7, 8, 13). Here, we demonstrated that EED deficiency in OPCs causes dysmyelination during CNS development. This raises a possibility that the myelination defect caused by EED/PRC2 mutations may contribute to the white matter and myelination defects in a cohort of patients with Weaver-like syndrome (8–10). Furthermore, raising H3K27me3 levels by treatment with H3K27 demethylase JMJD3 inhibitor GSK-J4 partially rescues the OPC differentiation defects caused by EED inactivation and promotes OL differentiation program, suggesting the augmentation of H3K27me3 deposition might have a therapeutic potential to enhance myelination and remyelination in demyelinating diseases. It is worth noting that the OL lineage–specific EED-deficient animals do not recapitulate all the features in the complex disease such as Weaver-like or Cohen-Gibson syndrome due to the systemic loss of EED/PRC2 functions. We show that EED depletion reduces OPC remyelination capacity after injury. Although the exact mechanism for EED regulation of remyelination remains to be defined, it is possible that EED might regulate the developmental myelination and remyelination processes using a similar mechanism. For instance, we observed a similar OL-astrocyte cell fate switch in LPC lesions in the Eed iKO animals.
PRC2 is also dysregulated in several types of human cancers (58). Nonsense and inactivating mutations in Eed or Suz12 are detected in malignant peripheral nerve sheath tumors, and these mutations have been shown to potentiate oncogenesis (7). Moreover, a point mutation of histone H3.3 (H3.3K27M) functions as a dominant negative regulator of PRC2 activity. It promotes the formation of aggressive pediatric gliomas, diffuse intrinsic pontine gliomas, in the brainstem (11, 12) and up-regulates the WNT pathway (59). Similarly, EED depletion in the OL lineage cells led to OPC differentiation defects in the brainstem and elevates WNT signaling, suggesting that EED/PRC2 may play a role in regulating OPC development and gliomagenesis in the brainstem. Together, our present data indicate that the EED-mediated PRC2 function is critical in white matter development and myelin regeneration and suggest that enhancing PRC2 activity or H3K27me3 levels might promote myelin repair in demyelinating diseases.
MATERIALS AND METHODS
Animals
Mice homozygous for floxed alleles of Eed (Eedfl/fl, the Jackson laboratory, stock no. 022727) and Ezh2 (Ezh2fl/fl, the Jackson laboratory, stock no. 022616) were crossed with mice carrying Olig1-Cre to generate OL conditional Eed cKO and Ezh2 cKO mice, respectively. Pdgfra-CreERT2 and green fluorescent protein (GFP)/tdTomato reporter mice were crossed with Eedfl/fl mice to generate the OPC-inducible Eed iKO (OPC-Eed iKO) mice. Plp-CreERT2 mice were mated with Eedfl/fl mice to generate the mature OL-inducible Eed iKO (OL-Eed iKO) mice. Mice of either sex were used in the study, and littermates were used as controls unless otherwise indicated. The mouse strains used in this study were generated and maintained on a mixed C57BL/6; 129Sv background and were housed in a vivarium with a 12-hour light/dark cycle with access to food and water. All studies complied with all relevant animal use guidelines and ethical regulations. All animal use and study protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at the Cincinnati Children’s Hospital Medical Center, Ohio, USA.
The Eedfl/fl line was genotyped using primers 5′-GGGACGTGCTGACATTTTCT-3′ and 5′-GGGACGTGCTGACATTTTCT-3′. The Ezh2fl/fl line was genotyped using primers 5′-CATGTGCAGCTTTCTGTTCA-3′ and 5′-CATGTGCAGCTTTCTGTTCA-3′. The Olig1-Cre line was identified with the primers 5′-CGTTAGTGAAGGGCGCCCCGGGTCG-3′ and 5′-CGTTAGTGAAGGGCGCCCCGGGTCG-3′. Mice positive for the Pdgfra-CreERT2 and Plp-CreERT2 transgenes were identified with the primers 5′-ATCCTGATGATTGGTCTCGTCT-3′ and 5′-TGTTACTCATGTGCCTGATGTG-3′. The GFP reporter line was identified with the primers 5′-GAGCTGGACGGCGACGTAAAC-3′ and 5′-CACCTTGATGCCGTTCTTCTGC-3′. The tdTomato reporter line was genotyped using primers 5′-AAGGGAGCTGCAGTGGAGTA-3′, 5′-CCGAAAATCTGTGGGAAGTC-3′, 5′-CTGTTCCTGTACGGCATGG-3′, and 5′-GGCATTAAAGCAGCGTATCC-3′.
Tamoxifen injections
Tamoxifen (Sigma-Aldrich, T5648) was dissolved to a stock concentration of 5 and 20 mg/ml in a vehicle of ethanol and sunflower seed oil (1:9, v/v). For newborn pups, 20 μl (0.1 mg/20 μl) of tamoxifen was administered by intraperitoneal injection once daily for three consecutive days. For adult mice, tamoxifen (100 mg/kg) was administered by intraperitoneal injection once daily for five consecutive days to indicated mice. Control mice were identically treated.
Tissue processing and in situ hybridization
Mice at indicated developmental stages were anesthetized and perfused with phosphate-buffered saline (PBS) followed by 4% paraformaldehyde (PFA). Brains and spinal cords were dissected, fixed in 4% PFA overnight, washed in PBS for six times, dehydrated in 25% sucrose at 4°C, embedded in OCT, and cryo-sectioned at 16 μm.
In situ hybridization was performed as described previously (16). Cryo-sections were used with digoxigenin (DIG)–labeled antisense riboprobes specific for Mbp, Plp1, and Pdgfra. An anti-DIG antibody (Roche, 16646821) conjugated to alkaline phosphatase was applied to the probe hybridized tissue sections and stained with 5-bromo-4-chloro-3-indolyl phosphate (BCIP)/nitro blue tetrazolium (NBT) chromogenic substrates.
Immunofluorescence staining
For tissues, cryo-sections (16 μm thick) or vibratome sections (50 μm thick) were permeabilized and blocked in blocking buffer (0.2% Triton X-100 and 5% normal donkey serum in PBS) for 1 hour at room temperature and overlaid with primary antibodies overnight at 4°C. For cells, cell culture on coverslip were fixed with 4% PFA for 10 min and then permeabilized and blocked in blocking buffer (0.02% Triton X-100 and 2% normal donkey serum in PBS) for 20 min and incubated with primary antibodies for 45 min at room temperature. Antibodies used in the study were rabbit anti-OLIG2 (Millipore, AB9610), rat anti-PDGFRα (BD Biosciences, 558774), mouse anti–adenomatous polyposis coli (APC) (CC1, Oncogene Research, OP80), goat anti-MBP (Santa Cruz Biotechnology, sc-13914), rabbit anti-GFAP (Cell Signaling Technology, 13-0300), rabbit anti-H3k27ac (Active Motif, 39135), rabbit anti-H3k27me3 (Cell Signaling Technology, 9733), rabbit anti-EZH1 (Thermo Fisher Scientific, PA5-21137), rabbit anti-EZH2 (Cell Signaling Technology, 5246), rabbit anti-SUZ12 (Cell Signaling Technology, 13701S), rabbit anti-EED (Cell Signaling Technology, 85322), rabbit anti-Ki67 (Thermo Fisher Scientific, RM-9106-S0), rabbit anti–cleaved caspase 3 (Cell Signaling Technology, no. 9661), rabbit anti-Iba1 (Wako Pure, 019-19741), mouse anti-GS (Millipore, MAB302), rat anti-CD11b (BD, 561690), mouse anti-p16INK4a (Abcam, ab54210), and mouse anti-p53 (Cell Signaling Technology, 2524S). After washing with 0.2% Triton X-100 in PBS, cells or sections were incubated with secondary antibodies conjugated to Cy2, Cy3, or Cy5 (Jackson ImmunoResearch Laboratories, 715-225-151, 711-165-152, 705-175-147) for 1.5 hours at room temperature, stained in DAPI (4′,6-diamidino-2-phenylindole) for 5 min, washed in PBS, and mounted with Fluoromount-G (SouthernBiotech, 0100-01). Images were quantified in a blinded manner.
Electron microscopy and morphometric analysis
Tissue processing was carried out as previously described (16). In brief, mice were anesthetized and perfused with 0.1 M cacodylate, followed by 4% PFA/2.5% glutaraldehyde in 0.1 M cacodylate. Tissues were dissected and postfixed in 1% OsO4. Ultrathin sections were stained with lead citrate for electron microscopy imaging. The morphometric measurements of axonal sorting defects were performed using electron micrographs of ultrathin sections and analyzed using the National Institutes of Health ImageJ software (http://rsb.info.nih.gov/ij/). The percentage of myelinated axons was analyzed in ultrathin sections using an electron microscope. The g-ratio was calculated as axon diameter/fiber diameter as previously described (16).
Isolation and culture of primary rodent OPCs
Mouse OPCs were isolated from the cortices of indicated genotyped mice at stages of P5 or P6 by immunopanning as previously described (24, 30). Briefly, cell suspension from the cortices was added into panning dish coated with RAN-2, GalC, and O4 antibodies sequentially. OPCs from O4+ plates were collected for analysis. For OPC expansion, isolated mouse OPCs were seeded in poly-d-lysine (PDL)–coated plate and cultured in the proliferation medium with the Sato medium plus 1xB27, NT3, Forskolin (5 μM), and PDGFAA (10 ng/ml) (24, 30). They were differentiated in the differentiation medium with the Sato medium plus ciliary neurotrophic factor (CNTF; 10 ng/ml) and T3 (15 nM).
Rat OPCs were isolated from brains of neonatal pups at P1–P3 as previously described (16). Briefly, mixed glial cells were initially cultured in Dulbecco’s Modified Eagle Medium (DMEM) plus 15% fetal bovine serum and then changed to B104 conditioned medium. OPCs were purified by mechanical detachment in an orbital shaker. Isolated rat OPCs were grown in Sato growth medium plus PDGF-AA (10 ng/ml) and basic fibroblast growth factor (bFGF) (10 ng/ml), and differentiated in Sato medium supplemented with T3 (15 nM) and CNTF (10 ng/ml).
SA-β-Gal staining
For SA-β-Gal staining in OPCs, acutely isolated OPCs were seeded in PDL-coated coverslips with Sato medium for 6 hours to let OPC adhesion to the coverslips, and then fixed with 4% and subjected to analysis. Senescence β-Gal Staining Kit (Cell Signaling Technology, 9860) was applied for senescence-associated SA-β-Gal staining, following the manufacturer’s instructions.
In vitro pharmacological inhibitor treatment
The histone H3 lysine 27 (H3K27) demethylase JMJD3 inhibitor GSK-J4 (Cayman, 12073), WNT inhibitor Wnt C59 (Cayman, 16644), and BMP inhibitor LDN193189 (Cayman, 11802) were dissolved in dimethyl sulfoxide and treated in OPCs and Oli-neu cells.
Western blotting
For Western blotting, acutely isolated mouse OPCs or brain tissues were lysed in radioimmunoprecipitation assay buffer, containing protease and phosphatase inhibitors. Western blot analysis was performed as described previously. Mouse anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Millipore MAB374) was used as an input control. The antibodies used were mouse anti-EZH1 (Sigma-Aldrich, ABE281), rabbit anti-SUZ12 (Cell Signaling Technology, 13701S), rabbit anti-H3k27ac (Active Motif, 39135), rabbit anti-H3k27me3 (Cell Signaling Technology, 9733), rabbit anti-EZH2 (Cell Signaling Technology, 5246), rabbit anti-EED (EMD Millipore, 17-0034), goat anti-MBP (Santa Cruz Biotechnology, sc-13914), mouse anti-GFAP (Sigma-Aldrich, G3893), mouse anti-p53 (Santa Cruz Biotechnology, sc-126), and rabbit anti-p16Ink4a (Santa Cruz Biotechnology, sc-467). Secondary antibodies conjugated to horseradish peroxidase were from Jackson ImmunoResearch Laboratories.
LPC-induced demyelinating injury
LPC-induced demyelination was performed in the ventrolateral spinal white matter of approximately 8-week-old mice as described previously (24). In brief, after exposing the spinal vertebrae at the level of T3–T4, meningeal tissue in the intervertebral space was cleared, and the dura was pierced with a dental needle. One percent LPC (0.5 μl; Sigma-Aldrich, L4129) was injected via a Hamilton syringe attached to a glass micropipette into the ventrolateral white matter via a stereotactic instrument. Spinal cord tissues carrying the lesions were collected at indicated time points. LPC-induced injuries were conducted in a genotype-blinded manner.
Fluorescence activated cell sorting of primary mouse OPCs
Cells were isolated from P5–P6 cortices of control and Eed cKO pups. Single-cell suspension was prepared by papain dissociation. Cells were blocked in 3% bovine serum albumin for 30 min and incubated with APC anti-CD140a (PDGFRα) antibody for 45 min. Isotype and fluorescence-minus-one controls were used to set appropriate gates.
Small interfering RNA transfection
Small interfering RNA (siRNA) transfection in Oli-neu cells was performed by Lipofectamine RNAiMAX (Invitrogen), siRNAs were purchased from Sigma-Aldrich: siEed (SASI_Rn02_00217400).
RNA isolation and quantitative real-time PCR
RNA from purified mouse OPCs from control or Eed cKO mice brain or Oli-neu cells were extracted using TRIzol (Life Technologies, 15-596-018). First-strand cDNA synthesis was performed using iScript Reverse Transcription Supermix (Bio-Rad, 1708841) and was amplified using quantitative SYBR green PCR mix (Bio-Rad, 1725121) according to the manufacturer’s instructions. Quantitative real-time PCR (qRT-PCR) was performed using the StepOnePlus RT-PCR System (Applied Biosystems, 4376600). Expression values for each gene were normalized against Gapdh using the delta-delta CT method. PCR primer sequences are available upon request.
ChIP-qPCR
ChIP-qPCR assays were performed as previously described (24). Oli-neu cells were fixed by 1% formaldehyde to cross-link DNA with endogenous proteins, and chromatin was prepared from shearing with a Covaris S220 sonicator. Immunoprecipitation was overnight at 4°C using H3K27me3 antibody (active motif, catalog no. 61018) or control immunoglobulins [immunoglobulin G (IgG)] in the presence of protein A/G Sepharose CL-4B beads. Primers were designed on the promoter regions, and the amount of DNA was quantified by qRT-PCR using SYBR green PCR mix. The fold enrichment of H3K27me3 was calculated over IgG.
RNA-seq and data analysis
RNA was extracted from OPCs acutely isolated from Eed cKO and control mice using the QIAGEN RNeasy Micro Kit. RNA sequencing (RNA-seq) libraries were prepared using SMART-Seq v4 Ultra Low Input RNA Kit. Massively parallel 75–base pair (bp) PE sequencing was completed on an Illumina HiSeq2500 to acquire ~25 M fragments per sample. All RNA-seq data were aligned to mm10 using TopHat with default settings (http://tophat.cbcb.umd.edu/).
We used Cuff-diff to (i) estimate fragments per kilobase of transcript per million mapped read (FPKM) values for known transcripts and to (ii) analyze differentially expressed transcripts. In all differential expression tests, a difference was considered significant if the P value was less than 0.05. A heatmap of gene expression was generated using the R language (version 3.2.1) and was generated on the basis of log2(FPKM). Gene ontology analysis of gene expression changes was performed using GSEA (www.broadinstitute.org/gsea/index.jsp). Normalized enrichment score reflects the degree to which the gene set is overrepresented at the top or bottom of a ranked list of genes. The GSEA summary plots showing up-regulated and down-regulated pathways were plotted according to www.biostars.org/p/168044/.
ATAC-seq and data analysis
ATAC-seq was performed as described in a previous study (33). Briefly, acutely isolated OPCs were counted and resuspended to obtain 50,000 cells per sample in ice-cold PBS, and then spun down and lysed in cold lysis buffer. After spinning down at 500g for 10 min at 4°C, nuclei were resuspended in transposition mix at 37°C for 30 min. Immediately following transposition, DNA fragments were subsequently amplified using 1× NEBNext PCR master mix to generate single-indexed libraries. A maximum of 12 cycles of PCR was used to prevent saturation biases based on optimization experiments performed using qPCR. Library quality control was carried out using the Bioanalyzer High-Sensitivity DNA Analysis Kit. Libraries were sequenced as single-end 50-bp reads on the Illumina High-Seq 2500 platform.
Reads of ATAC-seq data were aligned to mm10 genome using Bowtie (http://bowtie-bio.sourceforge.net). Peak calling was performed using MACS (Model-based Analysis of ChIP-seq) version 1.4.2 (https://github.com/macs3-project/MACS) with a P value cutoff of 1 × 10−9. Motif discovery was performed by HOMER software. ATAC-seq peak tracking was performed using IGV software. Differential open chromatin regions were assessed with the R package DESeq2 v1.26.0. We used the UCSC liftover tool (https://genome.ucsc.edu/cgi-bin/hgLiftOver) to convert genome position from rat species to mouse species.
Quantification and statistical analysis
All analyses were done using GraphPad Prism 8.00 (San Diego, California, www.graphpad.com). All data are shown as means ± SEM. Data distribution was assumed to be normal, but this was not formally tested. Statistical significance was determined using two-tailed Student’s t tests as indicated. One-way analysis of variance (ANOVA) test was performed by multiple comparisons or pairwise comparisons following Tukey’s ranking tests when comparing multiple groups. Significance was set as *P < 0.05, **P < 0.01, and ***P < 0.001. Quantifications were performed from at least three independent experiments, and data were quantified blindly.
Supplementary Material
Acknowledgments
We would like to thank S. Ogurek for technical support, and K. Berry and E. Hurlock for the comments. Funding: This study was funded in part by a grant from the National Multiple Sclerosis Society (RG-1507) to Q.R.L. Author contributions: Jiajia Wang and Q.R.L. designed the research plan. Jiajia Wang, L.Y., C.D., Jincheng Wang, and Y.Q. performed all the experiments and analyzed the data. Jiajia Wang, L.Y., C.Z., and L.X. performed the RNA-seq, ChIP, ATAC-seq, and data analysis. M.X. and Q.W. provided resources and inputs. Jiajia Wang and Q.R.L wrote the manuscript. Q.R.L supervised the project. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the corresponding authors. All the RNA-seq and ATAC-seq data have been deposited in the NCBI Gene Expression Omnibus (GEO) under accession number GEO: GSE135880. SUZ12 ChIP-seq was from GEO GSE82211.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/33/eaaz6477/DC1
REFERENCES AND NOTES
- 1.Chen T., Dent S. Y. R., Chromatin modifiers and remodellers: Regulators of cellular differentiation. Nat. Rev. Genet. 15, 93–106 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Laugesen A., Helin K., Chromatin repressive complexes in stem cells, development, and cancer. Cell Stem Cell 14, 735–751 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Yu J.-R., Lee C.-H., Oksuz O., Stafford J. M., Reinberg D., PRC2 is high maintenance. Genes Dev. 33, 903–935 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Margueron R., Justin N., Ohno K., Sharpe M. L., Son J., Drury W. J. III, Voigt P., Martin S. R., Taylor W. R., De Marco V., Pirrotta V., Reinberg D., Gamblin S. J., Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461, 762–767 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao R., Wang L., Wang H., Xia L., Erdjument-Bromage H., Tempst P., Jones R. S., Zhang Y., Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298, 1039–1043 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Laugesen A., Højfeldt J. W., Helin K., Molecular mechanisms directing PRC2 recruitment and H3K27 methylation. Mol. Cell 74, 8–18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Raedt T., Beert E., Pasmant E., Luscan A., Brems H., Ortonne N., Helin K., Hornick J. L., Mautner V., Kehrer-Sawatzki H., Clapp W., Bradner J., Vidaud M., Upadhyaya M., Legius E., Cichowski K., PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 514, 247–251 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Spellicy C. J., Peng Y., Olewiler L., Cathey S. S., Rogers R. C., Bartholomew D., Johnson J., Alexov E., Lee J. A., Friez M. J., Jones J. R., Three additional patients with EED-associated overgrowth: Potential mutation hotspots identified? J. Hum. Genet. 64, 561–572 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Gibson W. T., Hood R. L., Zhan S. H., Bulman D. E., Fejes A. P., Moore R., Mungall A. J., Eydoux P., Babul-Hirji R., An J., Marra M. A.; FORGE Canada Consortium, Chitayat D., Boycott K. M., Weaver D. D., Jones S. J. M., Mutations in EZH2 cause Weaver syndrome. Am. J. Hum. Genet. 90, 110–118 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cooney E., Bi W., Schlesinger A. E., Vinson S., Potocki L., Novel EED mutation in patient with Weaver syndrome. Am. J. Med. Genet. A 173, 541–545 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Lewis P. W., Müller M. M., Koletsky M. S., Cordero F., Lin S., Banaszynski L. A., Garcia B. A., Muir T. W., Becher O. J., Allis C. D., Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwartzentruber J., Korshunov A., Liu X.-Y., Jones D. T. W., Pfaff E., Jacob K., Sturm D., Fontebasso A. M., Quang D.-A. K., Tönjes M., Hovestadt V., Albrecht S., Kool M., Nantel A., Konermann C., Lindroth A., Jäger N., Rausch T., Ryzhova M., Korbel J. O., Hielscher T., Hauser P., Garami M., Klekner A., Bognar L., Ebinger M., Schuhmann M. U., Scheurlen W., Pekrun A., Frühwald M. C., Roggendorf W., Kramm C., Dürken M., Atkinson J., Lepage P., Montpetit A., Zakrzewska M., Zakrzewski K., Liberski P. P., Dong Z., Siegel P., Kulozik A. E., Zapatka M., Guha A., Malkin D., Felsberg J., Reifenberger G., von Deimling A., Ichimura K., Collins V. P., Witt H., Milde T., Witt O., Zhang C., Castelo-Branco P., Lichter P., Faury D., Tabori U., Plass C., Majewski J., Pfister S. M., Jabado N., Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Seong I. S., Woda J. M., Song J.-J., Lloret A., Abeyrathne P. D., Woo C. J., Gregory G., Lee J.-M., Wheeler V. C., Walz T., Kingston R. E., Gusella J. F., Conlon R. A., MacDonald M. E., Huntingtin facilitates polycomb repressive complex 2. Hum. Mol. Genet. 19, 573–583 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bercury K. K., Macklin W. B., Dynamics and mechanisms of CNS myelination. Dev. Cell 32, 447–458 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hardan A. Y., Fung L. K., Frazier T., Berquist S. W., Minshew N. J., Keshavan M. S., Stanley J. A., A proton spectroscopy study of white matter in children with autism. Prog. Neuropsychopharmacol. Biol. Psychiatry 66, 48–53 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao C., Dong C., Frah M., Deng Y., Marie C., Zhang F., Xu L., Ma Z., Dong X., Lin Y., Koenig S., Nait-Oumesmar B., Martin D. M., Wu L. N., Xin M., Zhou W., Parras C., Lu Q. R., Dual requirement of CHD8 for chromatin landscape establishment and histone methyltransferase recruitment to promote CNS myelination and repair. Dev. Cell 45, 753–768.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franklin R. J. M., Goldman S. A., Glia disease and repair-remyelination. Cold Spring Harb. Perspect. Biol. 7, a020594 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franklin R. J. M., Gallo V., The translational biology of remyelination: Past, present, and future. Glia 62, 1905–1915 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Ai S., Peng Y., Li C., Gu F., Yu X., Yue Y., Ma Q., Chen J., Lin Z., Zhou P., Xie H., Prendiville T. W., Zheng W., Liu Y., Orkin S. H., Wang D.-Z., Yu J., Pu W. T., He A., EED orchestration of heart maturation through interaction with HDACs is H3K27me3-independent. eLife 6, e24570 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Margueron R., Li G., Sarma K., Blais A., Zavadil J., Woodcock C. L., Dynlacht B. D., Reinberg D., Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 32, 503–518 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang M., Wang Y., Jones S., Sausen M., Mahon K. M., Sharma R., Wang Q., Belzberg A. J., Chaichana K., Gallia G. L., Gokaslan Z. L., Riggins G. J., Wolinksy J.-P., Wood L. D., Montgomery E. A., Hruban R. H., Kinzler K. W., Papadopoulos N., Vogelstein B., Bettegowda C., Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat. Genet. 46, 1170–1172 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao Q., Wang X., Zhao M., Yang R., Malik R., Qiao Y., Poliakov A., Yocum A. K., Li Y., Chen W., Cao X., Jiang X., Dahiya A., Harris C., Feng F. Y., Kalantry S., Qin Z. S., Dhanasekaran S. M., Chinnaiyan A. M., The central role of EED in the orchestration of polycomb group complexes. Nat. Commun. 5, 3127 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu J., Magri L., Zhang F., Marsh N. O., Albrecht S., Huynh J. L., Kaur J., Kuhlmann T., Zhang W., Slesinger P. A., Casaccia P., Chromatin landscape defined by repressive histone methylation during oligodendrocyte differentiation. J. Neurosci. 35, 352–365 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He D., Wang J., Lu Y., Deng Y., Zhao C., Xu L., Chen Y., Hu Y.-C., Zhou W., Lu Q. R., lncRNA functional networks in oligodendrocytes reveal stage-specific myelination control by an lncOL1/Suz12 complex in the CNS. Neuron 93, 362–378 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rivers L. E., Young K. M., Rizzi M., Jamen F., Psachoulia K., Wade A., Kessaris N., Richardson W. D., PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat. Neurosci. 11, 1392–1401 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madisen L., Zwingman T. A., Sunkin S. M., Oh S. W., Zariwala H. A., Gu H., Ng L. L., Palmiter R. D., Hawrylycz M. J., Jones A. R., Lein E. S., Zeng H., A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doerflinger N. H., Macklin W. B., Popko B., Inducible site-specific recombination in myelinating cells. Genesis 35, 63–72 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Franklin R. J. M., Why does remyelination fail in multiple sclerosis? Nat. Rev. Neurosci. 3, 705–714 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Franklin R. J. M., Ffrench-Constant C., Remyelination in the CNS: From biology to therapy. Nat. Rev. Neurosci. 9, 839–855 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Emery B., Dugas J. C., Purification of oligodendrocyte lineage cells from mouse cortices by immunopanning. Cold Spring Harb. Protoc. 2013, 854–868 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Jung M., Krämer E., Grzenkowski M., Tang K., Blakemore W., Aguzzi A., Khazaie K., Chlichlia K., von Blankenfeld G., Kettenmann H., Trotter J., Lines of murine oligodendroglial precursor cells immortalized by an activated neu tyrosine kinase show distinct degrees of interaction with axons in vitro and in vivo. Eur. J. Neurosci. 7, 1245–1265 (1995). [DOI] [PubMed] [Google Scholar]
- 32.Kruidenier L., Chung C.-w., Cheng Z., Liddle J., Che K. H., Joberty G., Bantscheff M., Bountra C., Bridges A., Diallo H., Eberhard D., Hutchinson S., Jones E., Katso R., Leveridge M., Mander P. K., Mosley J., Ramirez-Molina C., Rowland P., Schofield C. J., Sheppard R. J., Smith J. E., Swales C., Tanner R., Thomas P., Tumber A., Drewes G., Oppermann U., Patel D. J., Lee K., Wilson D. M., A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 488, 404–408 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buenrostro J. D., Giresi P. G., Zaba L. C., Chang H. Y., Greenleaf W. J., Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nicaise A. M., Wagstaff L. J., Willis C. M., Paisie C., Chandok H., Robson P., Fossati V., Williams A., Crocker S. J., Cellular senescence in progenitor cells contributes to diminished remyelination potential in progressive multiple sclerosis. Proc. Natl. Acad. Sci. U.S.A. 116, 9030–9039 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oost W., Talma N., Meilof J. F., Laman J. D., Targeting senescence to delay progression of multiple sclerosis. J. Mol. Med. (Berl) 96, 1153–1166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang P., Kishimoto Y., Grammatikakis I., Gottimukkala K., Cutler R. G., Zhang S., Abdelmohsen K., Bohr V. A., Sen J. M., Gorospe M., Mattson M. P., Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer's disease model. Nat. Neurosci. 22, 719–728 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Milanovic M., Fan D. N. Y., Belenki D., Däbritz J. H. M., Zhao Z., Yu Y., Dörr J. R., Dimitrova L., Lenze D., Barbosa I. A. M., Mendoza-Parra M. A., Kanashova T., Metzner M., Pardon K., Reimann M., Trumpp A., Dörken B., Zuber J., Gronemeyer H., Hummel M., Dittmar G., Lee S., Schmitt C. A., Senescence-associated reprogramming promotes cancer stemness. Nature 553, 96–100 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Shay J. W., Roninson I. B., Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene 23, 2919–2933 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Burton D. G., Faragher R. G. A., Cellular senescence: From growth arrest to immunogenic conversion. Age (Dordr.) 37, 27 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hidalgo I., Herrera-Merchan A., Ligos J. M., Carramolino L., Nuñez J., Martinez F., Dominguez O., Torres M., Gonzalez S., Ezh1 is required for hematopoietic stem cell maintenance and prevents senescence-like cell cycle arrest. Cell Stem Cell 11, 649–662 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Ma K. H., Hung H. A., Srinivasan R., Xie H., Orkin S. H., Svaren J., Regulation of peripheral nerve myelin maintenance by gene repression through polycomb repressive complex 2. J. Neurosci. 35, 8640–8652 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma K. H., Duong P., Moran J. J., Junaidi N., Svaren J., Polycomb repression regulates Schwann cell proliferation and axon regeneration after nerve injury. Glia 66, 2487–2502 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raff M. C., Miller R. H., Noble M., A glial progenitor cell that develops in vitro into an astrocyte or an oligodendrocyte depending on culture medium. Nature 303, 390–396 (1983). [DOI] [PubMed] [Google Scholar]
- 44.Lu F., Chen Y., Zhao C., Wang H., He D., Xu L., Wang J., He X., Deng Y., Lu E. E., Liu X., Verma R., Bu H., Drissi R., Fouladi M., Stemmer-Rachamimov A. O., Burns D., Xin M., Rubin J. B., Bahassi E. M., Canoll P., Holland E. C., Lu Q. R., Olig2-dependent reciprocal shift in PDGF and EGF receptor signaling regulates tumor phenotype and mitotic growth in malignant glioma. Cancer Cell 29, 669–683 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weng Q., Wang J., Wang J., He D., Cheng Z., Zhang F., Verma R., Xu L., Dong X., Liao Y., He X., Potter A., Zhang L., Zhao C., Xin M., Zhou Q., Aronow B. J., Blackshear P. J., Rich J. N., He Q., Zhou W., Suvà M. L., Waclaw R. R., Potter S. S., Yu G., Lu Q. R., Single-cell transcriptomics uncovers glial progenitor diversity and cell fate determinants during development and gliomagenesis. Cell Stem Cell 24, 707–723.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang L., He X., Liu L., Jiang M., Zhao C., Wang H., He D., Zheng T., Zhou X., Hassan A., Ma Z., Xin M., Sun Z., Lazar M. A., Goldman S. A., Olson E. N., Lu Q. R., Hdac3 interaction with p300 histone acetyltransferase regulates the oligodendrocyte and astrocyte lineage fate switch. Dev. Cell 36, 316–330 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van der Vlag J., Otte A. P., Transcriptional repression mediated by the human polycomb-group protein EED involves histone deacetylation. Nat. Genet. 23, 474–478 (1999). [DOI] [PubMed] [Google Scholar]
- 48.Shen X., Liu Y., Hsu Y.-J., Fujiwara Y., Kim J., Mao X., Yuan G.-C., Orkin S. H., EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol. Cell 32, 491–502 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weng Q., Chen Y., Wang H., Xu X., Yang B., He Q., Shou W., Chen Y., Higashi Y., van den Berghe V., Seuntjens E., Kernie S. G., Bukshpun P., Sherr E. H., Huylebroeck D., Lu Q. R., Dual-mode modulation of Smad signaling by Smad-interacting protein Sip1 is required for myelination in the central nervous system. Neuron 73, 713–728 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ye F., Chen Y., Hoang T. N., Montgomery R. L., Zhao X.-h., Bu H., Hu T., Taketo M. M., van Es J. H., Clevers H., Hsieh J., Bassel-Duby R., Olson E. N., Lu Q. R., HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the β-catenin-TCF interaction. Nat. Neurosci. 12, 829–838 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schachtrup C., Ryu J. K., Mammadzada K., Khan A. S., Carlton P. M., Perez A., Christian F., Moan N. L., Vagena E., Baeza-Raja B., Rafalski V., Chan J. P., Nitschke R., Houslay M. D., Ellisman M. H., Wyss-Coray T., Palop J. J., Akassoglou K., Nuclear pore complex remodeling by p75NTR cleavage controls TGF-β signaling and astrocyte functions. Nat. Neurosci. 18, 1077–1080 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Figlia G., Gerber D., Suter U., Myelination and mTOR. Glia 66, 693–707 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wood T. L., Bercury K. K., Cifelli S. E., Mursch L. E., Min J., Dai J., Macklin W. B., mTOR: A link from the extracellular milieu to transcriptional regulation of oligodendrocyte development. ASN Neuro 5, e00108 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wei F.-Z., Cao Z., Wang X., Wang H., Cai M.-Y., Li T., Hattori N., Wang D., Du Y., Song B., Cao L.-L., Shen C., Wang L., Wang H., Yang Y., Xie D., Wang F., Ushijima T., Zhao Y., Zhu W.-G., Epigenetic regulation of autophagy by the methyltransferase EZH2 through an MTOR-dependent pathway. Autophagy 11, 2309–2322 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Calcinotto A., Kohli J., Zagato E., Pellegrini L., Demaria M., Alimonti A., Cellular senescence: Aging, cancer, and injury. Physiol. Rev. 99, 1047–1078 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Ito T., Teo Y. V., Evans S. A., Neretti N., Sedivy J. M., Regulation of cellular senescence by polycomb chromatin modifiers through distinct DNA damage- and histone methylation-dependent pathways. Cell Rep. 22, 3480–3492 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bracken A. P., Kleine-Kohlbrecher D., Dietrich N., Pasini D., Gargiulo G., Beekman C., Theilgaard-Mönch K., Minucci S., Porse B. T., Marine J.-C., Hansen K. H., Helin K., The polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 21, 525–530 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Laugesen A., Højfeldt J. W., Helin K., Role of the polycomb repressive complex 2 (PRC2) in transcriptional regulation and cancer. Cold Spring Harb. Perspect. Med. 6, a026575 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nagaraja S., Quezada M. A., Gillespie S. M., Arzt M., Lennon J. J., Woo P. J., Hovestadt V., Kambhampati M., Filbin M. G., Suva M. L., Nazarian J., Monje M., Histone variant and cell context determine H3K27M reprogramming of the enhancer landscape and oncogenic state. Mol Cell 76, 965–980.e12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/33/eaaz6477/DC1