

Abstract
Non-small cell lung cancer (NSCLC) patients with epithermal growth factor receptor (EGFR) mutations can be treated with EGFR-tyrosine kinase inhibitors (EGFR-TKIs), however, development of acquired resistance could significantly limit curative effects of EGFR-TKIs. Different mechanisms of acquired resistance to first-generation and second-generation EGFR TKIs have been widely reported, but there were few reports on the resistant mechanism of third-generation EGFR-TKI such as osimertinib (AZD9291). In the present study, significant upregulation of Bcl-2 was found in AZD9291-resistant H1975 cells (H1975AR) compared with H1975, which may constitute an important resistant mechanism of acquired resistance to AZD9291. More importantly, our study showed that synergism between AZD9291 and Bcl-2 inhibitor ABT263 (0.25 μM) or ABT199 (1 μM) could effectively overcome the acquired resistance of AZD9291 in H1975AR in vitro. Flow cytometry analyses demonstrated that AZD9291+ABT263/ABT199 caused a significantly different cell cycle distribution and produced significantly more apoptosis compared with either AZD9291 or ABT263/ABT199 treatment alone. Further multiscreen/Western blot analyses revealed that NF-κB was significantly downregulated in AZD9291+ABT263/ABT199 treatment groups compared with AZD9291 or ABT263/ABT199 treatment alone, with a more significant reduction of NF-κB in AZD9291+ABT199 compared with AZD9291+ABT263. It is also noticeable that AZD9291+ABT263 specifically caused a significantly reduced expression of p21 compared with AZD9291 or ABT263 treatment alone while AZD9291+ABT199 specifically caused significantly reduced expressions of SQSTM1 and survivin, but increased expression of autophagosome marker LC3-II compared with AZD9291 or ABT199 treatment alone. Furthermore, cytotoxicity of AZD9291+ABT199 could be partially reversed by autophagy inhibitor chloroquine. These results suggest that ABT263 and ABT199 may work through different signaling pathways to achieve synergistic cytotoxicity with AZD9291 in H1975AR. These findings suggest that Bcl-2 inhibitor may provide an effective option in combination therapy with EGFR-TKIs to treat NSCLC with EGFR-TKI acquired resistance.
Keywords: ABT263, ABT199, H1975, EGFR-TKI, NSCLC
Introduction
Non-small-cell lung cancer (NSCLC) patients with epidermal growth factor receptor (EGFR) activating mutations such as EGFR exon 19 deletion (delE746-A750) were sensitive to clinical treatments by first-generation of EGFR tyrosine kinase inhibitors (TKIs) such as gefitinib (Yoneda et al 2019). Osimertinib (AZD9291), as a third-generation EGFR-TKI approved by both Food and Drug Administration (FDA) and the European Commission in 2017 (Santarpia et al 2017), is designed to treat NSCLC positive for the T790M mutation that may be de novo or acquired after treatment with first or second-generation EGFR-TKI such as gefitinib or afatinib (Xu et al 2015). However, acquired resistance to AZD9291 also developed in NSCLC patients within approximately 10-month treatment of the drug (Patel et al 2017) and C797S mutation is regarded as the leading mechanism of resistance (Patel et al 2017). The fourth-generation EGFR-TKI to combat the C797S EGFR resistance problem is still on the discovery and development stage (Patel et al 2017). Besides, our previous study (Liu and Gao 2019) reported that overexpression of Twsit1 was found in AZD9291-resistant H1975 cell line (H1975AR) compared with parental H1975 but knockdown of Twist1 with shRNA was ineffective for overcoming the acquired resistance of AZD9291 in H1975AR. Therefore, other therapeutic strategies to overcome the acquired resistance of third-generation EGFR-TKIs are worthwhile for further investigations.
B-cell lymphoma 2 (Bcl-2), as a member of anti-apoptotic Bcl-2 family proteins (e.g. Bcl-2, Bcl-XL, Bcl-W, Bfl-1, and Mcl-1), plays an important role in the regulation of apoptosis and cell survival (Siddiqui et al 2015). The association between Bcl-2 positive expression and prognosis of NSCLC has been widely reported (Zhang et al 2015). Since Bcl-2 and its family proteins are crucial targets for cancer chemotherapy, it has attracted an increasing interest from medicinal chemists globally for drug discovery using various approaches (Suvarna et al 2019). Small molecules (e.g. ABT263 & ABT199) or natural compounds as Bcl-2 inhibitors have entered into pre-clinical and clinical trials as monotherapy and in combination therapy (Brown and Taygerly 2012; Suvarna et al 2019).
Previous research has revealed that Bcl-2 family proteins acted as central controllers of apoptosis promoting tumor initiation, growth, metastasis, and drug resistance to chemotherapy as well as radiotherapy (Oltersdorf et al 2005; Suvarna et al 2019). Upregulation of Bcl-2 was found to be associated with acquired resistance against EGFR-TKIs in NSCLC (Cheong et al 2018), which developed after 8–12 months of EGFR-TKI treatment. Meanwhile, it was reported knockdown of Bcl-2 could increase sensitivity of first-generation EGFR-TKI gefitinib in NSCLC cell line H1975 harboring T790 mutation with intrinsic resistance to gefitinib (Zou et al 2013).
ABT199 (Venetoclax) is the only Bcl-2 inhibitor approved by FDA in clinical use to treat chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) (Crombie and Davids 2017). ABT263 (Navitoclax), as a potent and orally bioavailable Bcl-2 family inhibitor, inhibits not only Bcl-2, but also Bcl-XL and Bcl-w proteins (Chen et al 2011), which displayed more side effects such as reduced platelet lifespans and thrombocytopenia in monotherapy compared to ABT199 that specifically targets Bcl-2. ABT263 showed more potential in combination therapy against different types of cancers including lung cancer (Suvarna et al 2019). The clinical trial of AZD9291 and ABT263 in treating EGFR-positive patients with previously treated advanced or metastatic NSCLC (ClinicalTrials.gov, 02520778) is still ongoing (Wu and Shih 2018). Also, a most recent study (Xu et al 2019) reported that abivertinib (AC0010), one of the third-generation EGFR-TKIs still in phase II trials in China (Zhou et al 2019), triggered drug resistance in vitro through Bcl-2 overexpression and combination therapy using AC0010 with Bcl-2 inhibitor ABT-263 could effectively overcome the acquired resistance of AC0010 in vitro. However, whether the highly specific Bcl-2 inhibitor ABT199 has the potential to overcome the acquired resistance of the third-generation EGFR-TKI in NSCLC with T790M remains unknown.
The objective of the present study is to investigate the potential of ABT199 in combination with AZD9291 to overcome acquired resistance of AZD9291 in H1975AR with a significant Bcl-2 upregulation compared to parental H1975. Meanwhile, the synergistic effects of ABT199+AZD9291 and ABT263+AZD9291 in H1975AR were compared by MTT assay and flow cytometry analyses. Also, the differences in molecular mechanisms of synergism between ABT199+AZD9291 or ABT263+AZD9291 were further investigated by multiscreen/Western blot analyses.
Materials and Methods
Cell lines and reagents
The NSCLC cell line H1975 was purchased from American Type Culture Collection (ATCC). AZD9291-resistant H1975 (H1975AR) was established in our previous study which was found to have T790M but no C797S mutation (Liu and Gao 2019). H1975 (within five passages) or H1975AR were cultured in RPMI 1640 medium (Thermo Scientific, Logan, UT) containing 5% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA), 100 U/mL penicillin, and 100 μg/mL streptomycin in 5% CO2 incubator at 37 °C.
Osimertinib (AZD9291, 99.87 %), ABT-263 (99.53 %), and ABT-199 (99.70 %) were obtained from selleckchem (Houston, TX, USA). The stocks of AZD9291 (10 mM), ABT263 (10 mM), ABT199 (10 mM) in DMSO was diluted to the required concentrations immediately before used in the growth media. Chloroquine diphosphate (CQ, 98.5%–101.0%) was purchased from Sigma-Aldrich (St. Louis, MI, USA). Primary antibodies including EGFR, phospho-EGFR (Tyr1068), p44/42 MAPK (Erk1/2), phosphop-44/42 MAPK (Erk1/2) (Thr202/Tyr204), Akt, phospho-Akt (Ser473), PARP, cleaved-PARP, NF-κB, caspase-3, cleaved-caspase-3, p21, Bcl-2, Mcl-1, LC3 A/B, survivin, and GAPDH were purchased from Cell Signaling Technology (Danvers, MA, USA) and sequestosome 1 (SQSTM1/p62) was purchased from Santa Cruz (Dallas, TX, USA). Horseradish peroxidase (HRP)-conjugated donkey anti-rabbit IgG or anti-mouse IgG, and chemiluminescence kit were purchased from Santa Cruz (Dallas, TX, USA).
Cell viability assay
Cell viability was evaluated by the MTT assay as described previously (Liu and Gao 2017; Liu and Gao 2019). Briefly, based on the non-significant cytotoxicity of ABT263 or ABT199 observed in H1975AR, 0.25 μM ABT263 or 1.0 μM ABT199 was selected for co-treatment assay. The synergistic effect of AZD9291 and ABT263/ABT199 was evaluated by comparing to vehicle controls for AZD9291 treatments or ABT263 (0.25 μM)/ABT199 (1.0 μM) alone for AZD9291+ABT263/ABT199 treatments, respectively. In addition, 5.0 μM CQ was chosen for co-treatment with AZD9291 and ABT199 for 48 hrs. Experiments were repeated independently three times.
Analysis of apoptosis and cell cycle by flow cytometry
Based on the cell viability assay, a total of nine groups of H1975AR with different treatments for 48 hrs were analyzed for apoptosis and cell cycle, including control, 2.5 μM AZD9291 (AZD2.5), 5 μM AZD9291 (AZD5), 0.25 μM ABT263 (ABT263), 2.5 μM AZD9291+0.25 μM ABT263 (AZD2.5+ABT263), 5.0 μM AZD9291+0.25 μM ABT263 (AZD5+ABT263), 1.0 μM ABT199 (ABT199), 2.5 μM AZD9291+1.0 μM ABT199 (AZD2.5+ABT199), and 5.0 μM AZD9291+1.0 μM ABT199 (AZD5+ABT199). Cells were stained with Nexin or Guava Cell Cycle reagent (Millipore, Billerica, MA) and run on a Guava EasyCyte™ Flow Cytometer (Millipore) as previously described (Liu and Gao 2017). Each sample was run in triplicate and each experiment was repeated three times.
Multiscreen bolt and Western blot analyses
Mini-PROTEAN® II Multiscreen Apparatus (Bio-rad, Hercules, CA, USA) was used to detect potential candidate molecules with significantly different protein expression levels in AZD9291 or AZD9291+ABT263/ABT199 48hr-treated groups. H1975AR exposed to AZD9291 and/or ABT263/ABT199 co-treatments for 48 hrs were further analyzed by Western blot as previously described (Liu and Gao 2019). GAPDH was used as an internal control.
qRT-PCR analyses
Total RNA was isolated from H1975 and H1975AR using the RNeasy Plus Mini Kit (Qiagen) following the manufacturer protocol. One-step RT-PCR Kit with SYBR green was used for amplification of total mRNA (75 ng) following the manufacturer’s protocol (BioRad, Hercules,CA, USA) and our previous study (Liu and Gao 2019). Experiment was performed in triplicate for each group. The PCR primer sequences were as following: Bcl-2 (F): GATGTGATGCCTCTGCGAAG, Bcl-2 (R): CATGCTGATGTCTCTGGAATCT; Bcl-XL (F): TCCTTGTCTACGCTTTCCACG, Bcl-XL(R): GGTCGCATTGTGGCCTTT; GAPDH (F): GGTGGTCTCCTCTGACTTCAACA, and GAPDH (R): GTTGCTGTAGCCAAATTCGTTGT.
Statistical analyses
Factorial ANOVA was performed to test the synergistic effects of AZD9291 and/or ABT263/ABT199 concentrations and incubation times on cell viability. Prism 6.0 was used to calculate the 50% inhibitory concentrations (IC50s). One-way ANOVA was used to determine the difference in the analytical results of flow cytometry among groups followed by a Fisher’s LSD test. Differences with p < 0.05 were considered statistically significant.
Results
Cytotoxicity of co-treatment of AZD9291 and/or ABT199/ABT263 in H1975AR
As shown in Figure 1a and 1b, the protein and gene expressions level of Bcl-2 in H1975AR has significantly increased compared with in H1975, which may account for an important mechanism of acquired resistance to AZD9291 in H1975AR. However, the protein and gene expression levels of Bcl-XL in H1975AR were not significantly changed compared with H1975.
Figure 1. Bcl-2 and Bcl-XL proteins (a) and mRNA (b) expressions in H1975AR and H1975, and cytotoxic effects of AZD9291 (AZD) and/or ABT263/ABT199 in H1975AR at 24 hrs (c) and 48 hrs (d).
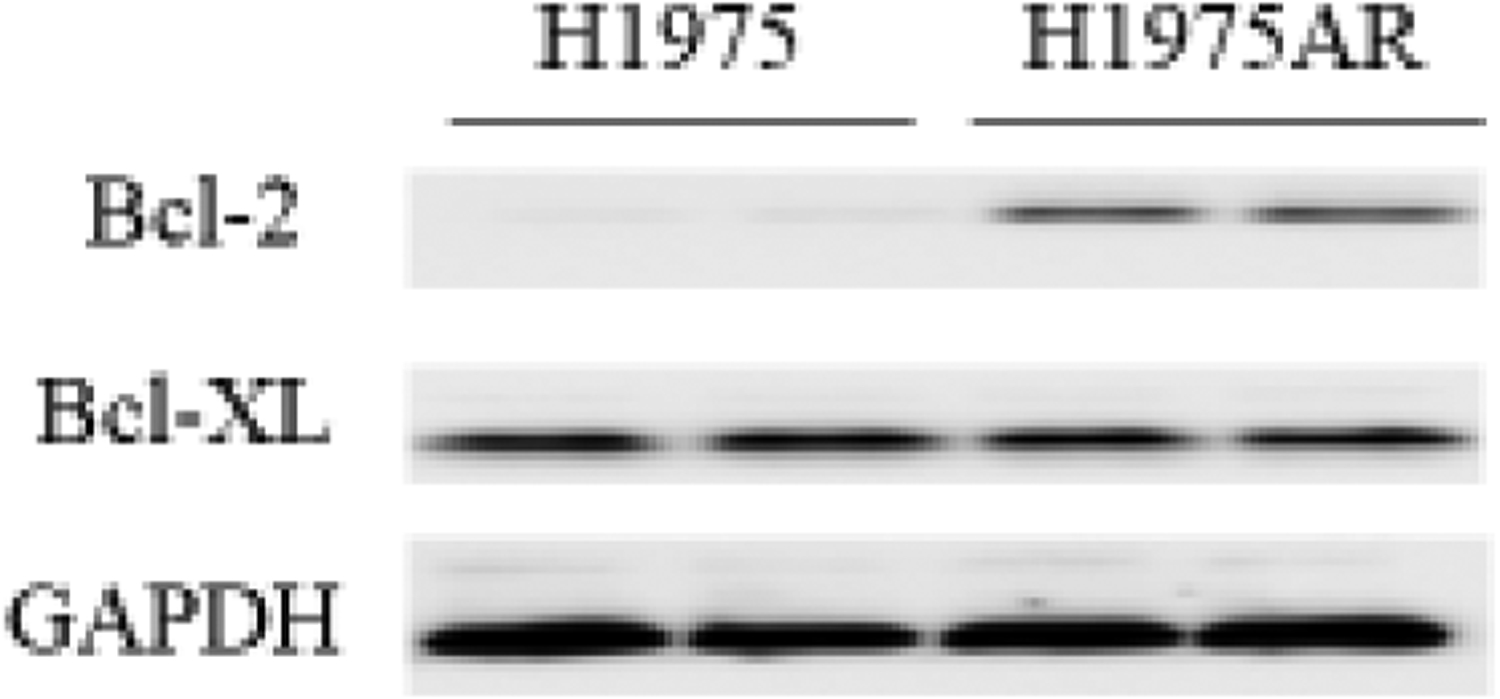
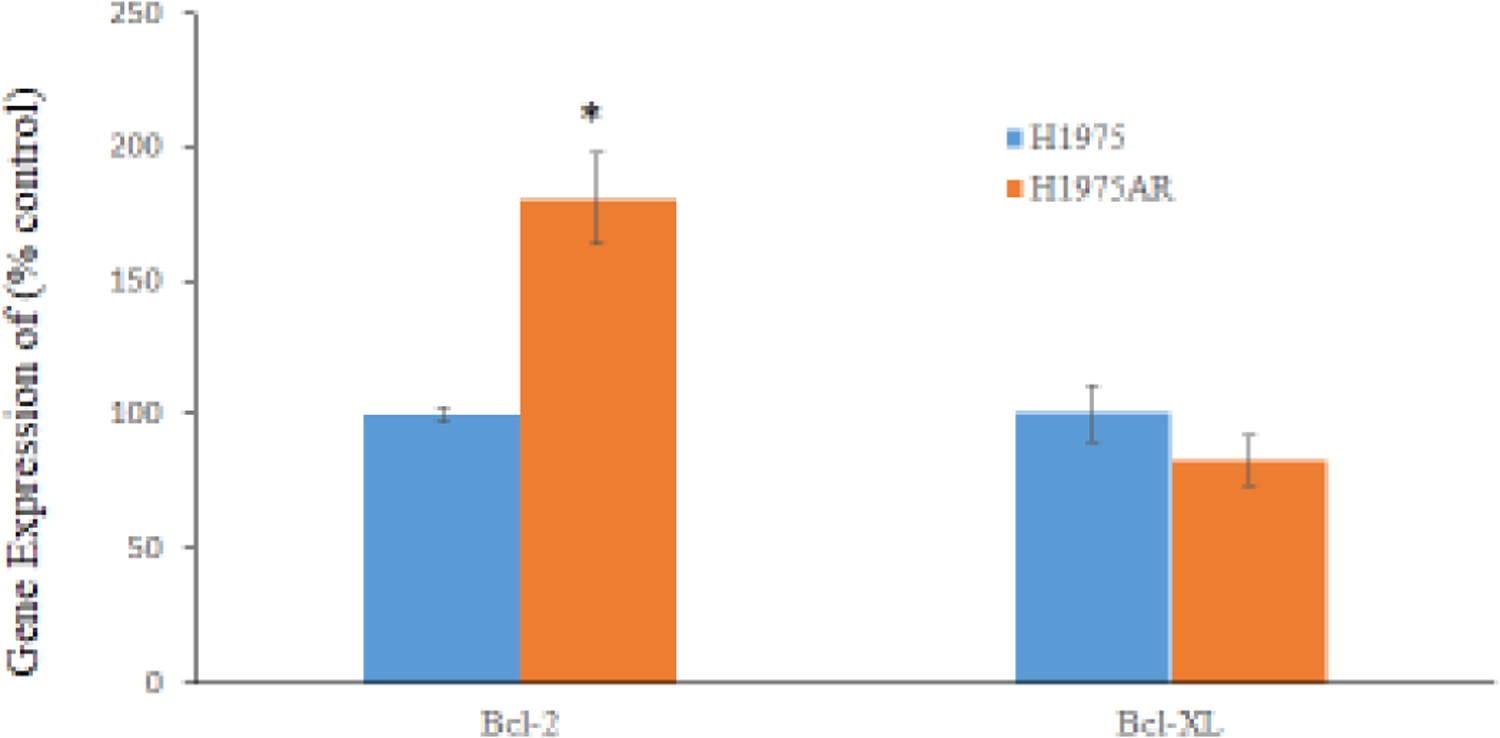
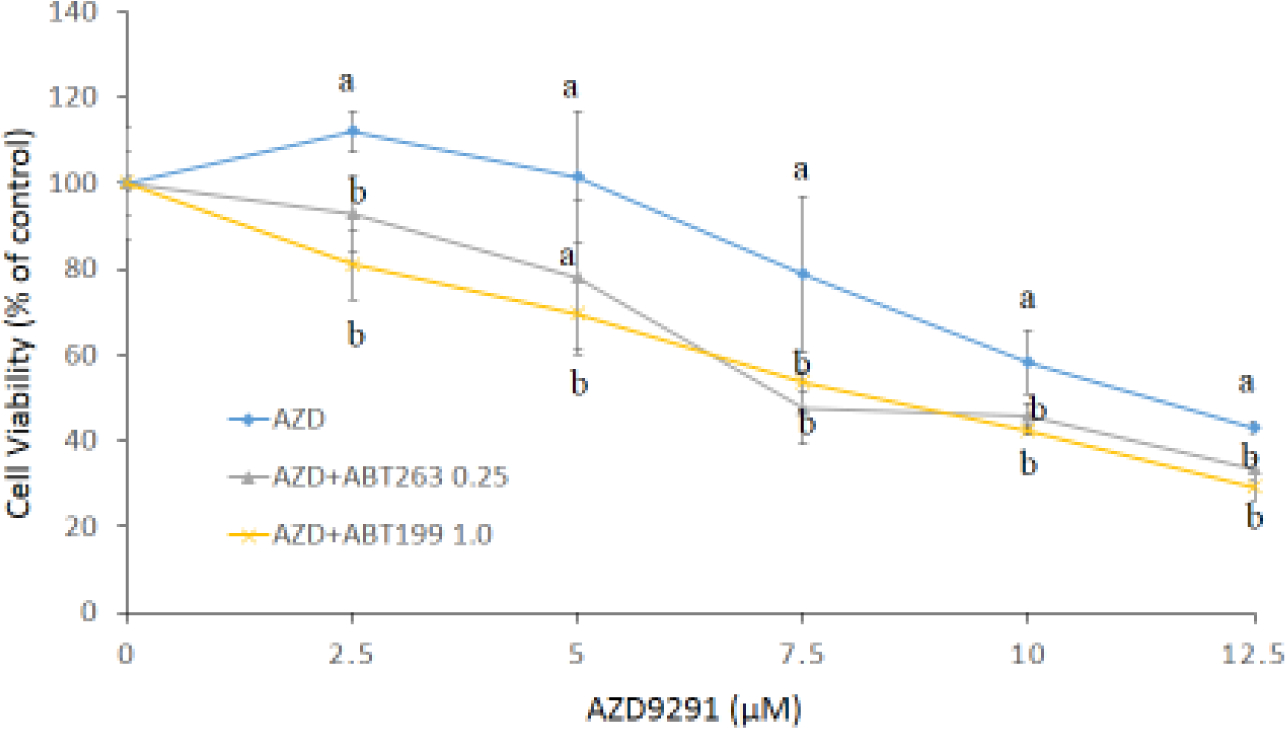
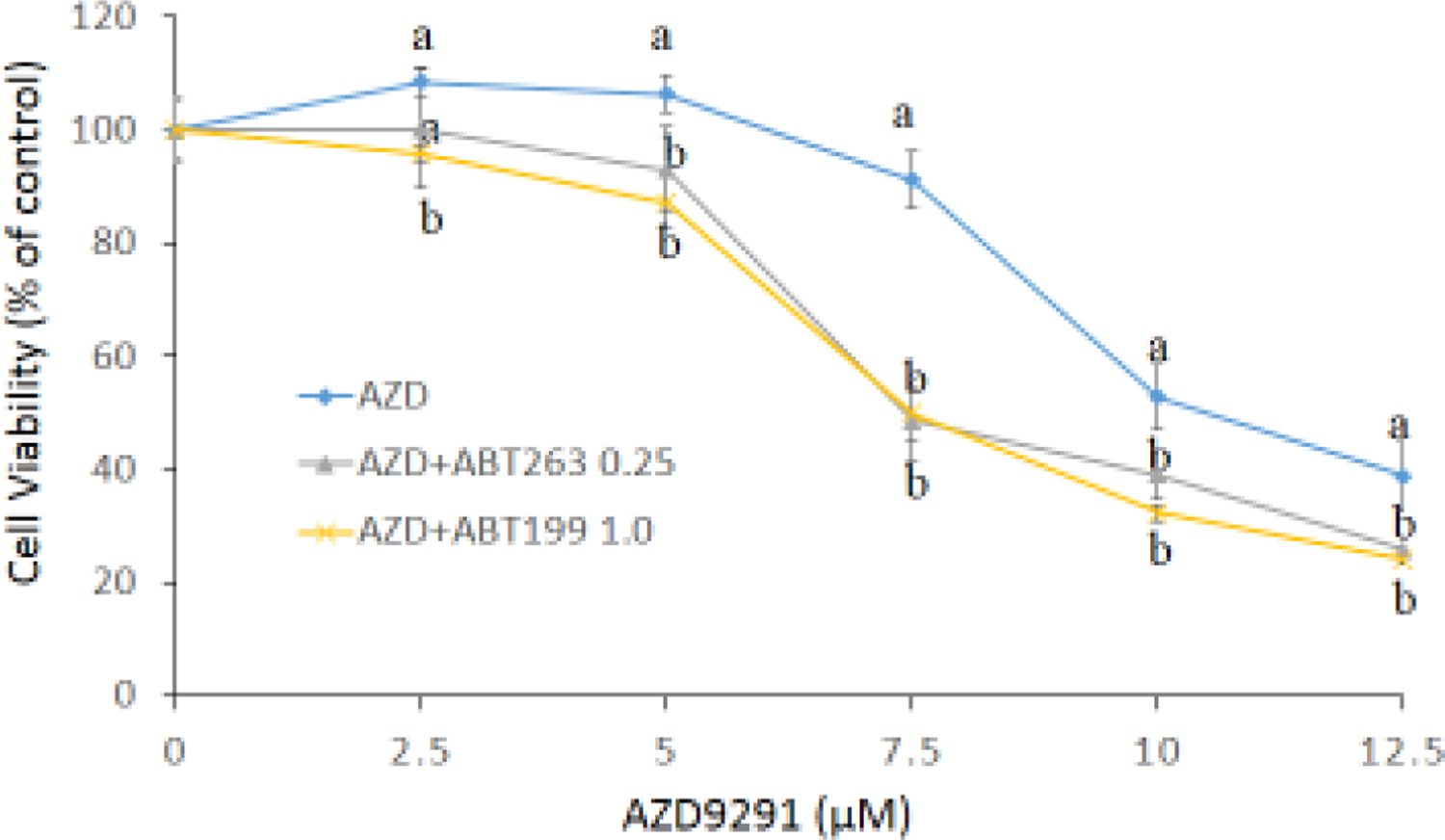
(a) protein and (b) mRNA expression levels in H1975 and H1975AR analyzed by Western blot and real-time PCR, respectively. * signified significant difference between H1975 and H1975AR (p<0.05). Cytotoxic effects of AZD alone and AZD+ABT263/ABT199 on the cell viability H1975AR at (c) 24 hrs and (d) 48 hrs were determined by the MTT assays. Data are expressed as the percentage by comparing to vehicle controls for AZD treatments or ABT263 (0.25 μM)/ABT199 (1 μM) for AZD+ABT263/ABT199 treatments. Values are represented as mean ± SD, n = 6. Mean values at the same concentration containing different letters indicate a significant difference among the treatment groups analyzed by a one-way ANOVA followed by a Fisher’s LSD test (p < 0.05). Values bearing the letter “a” indicate no significant differences compared with AZD treatments and those labeled “b” denote a significant difference when compared with AZD treatments.
Based on the MTT assays of ABT263 and ABT199 (data not shown), no significantly cytotoxic doses of ABT263 and ABT199, 0.25 μM and 1.0 μM respectively, were selected for the combination treatments. As shown in Figure 1c and 1d, the simultaneous treatments of AZD9291 (0–12.5 μM) and ABT263 (0.25 μM) or ABT199 (1.0 μM) showed synergistic cytotoxic effects on H1975AR as compared to AZD9291 alone at both 24 hrs and 48 hrs (p < 0.05, n = 6). The IC50 of AZD9291 at 48 hrs was 8.8±0.43 μM while it was significantly reduced to 6.6±0.41 μM with the combination of 0.25 μM ABT263 or 6.8±0.13 μM with the combination of 1.0 μM ABT199 (p < 0.05). Meanwhile, no synergistic effect of AZD9291+ABT263/ABT199 was found in H1975 (data not shown).
Flow cytometry analyses of apoptosis and cell cycle distributions in H1975AR treated by AZD9291 and/or ABT263/ABT199
Cell cycle analyses were conducted to further investigate the mechanism of the synergistic effects of AZD9291 and/or ABT263/ABT199 on the H1975AR apoptosis and cell cycle arrest. Table 1 and Figure 2 showed that AZD9291+ABT263/ABT199 co-treatments caused a significant increased proportion of apoptotic cells compared with AZD9291 or ABT263/ABT199 treatment alone. Table 1 showed that ABT263+AZD5 treatment group had the highest both early apoptotic (Annexin(+) 7-AAD(−), 14.8±3.01%) and late-stage apoptotic (Annexin(+) 7-AAD(+), 16.6±4.25%) cell populations (among all the treatment groups (p < 0.05) followed by ABT199+AZD5 that had significantly a higher late-stage apoptotic (Annexin(+) 7-AAD(+)) cell population (11.3±2.45%) compared with that of all other treatment groups (p < 0.05).
Table 1.
Effects of AZD9291 and/or ABT263/ABT199 on apoptosis of H1975AR
| Groups | Cell distribution (%) | |||
|---|---|---|---|---|
| Annexin(−) 7-AAD(+) | Annexin(+) 7-AAD(+) | Annexin(−) 7-AAD(−) | Annexin(+) 7-AAD(−) | |
| Control | 0.6±0.38a | 5.0±2.59a | 91.9±3.87a | 2.5±1.00a |
| AZD2.5 | 0.4±0.13a | 4.7±2.32a | 92.9±3.20a | 2.0±0.99a |
| AZD5 | 1.0±0.59a | 7.3±4.22a | 88.5±5.76a | 3.3±1.84a |
| ABT263 0.25 | 0.8±0.51a | 5.7±3.66a | 89.0±5.55a | 4.8±2.14a |
| ABT263+AZD2.5 | 1.0±0.97a,c | 8.4±4.03a,c | 84.3±7.05a,c | 6.3±3.86a,c |
| ABT263+AZD5 | 2.8±1.55a,c | 16.6±4.25b,d | 65.8±11.83b,d | 14.8±3.01b,d |
| ABT199 1.0 | 0.8±0.57a | 6.4±2.55a | 89.1±3.80a | 3.8±1.09a |
| ABT199+AZD2.5 | 0.5±0.29a,c | 5.0±2.93a,c | 90.5±4.45a,c | 3.5±1.73a,c |
| ABT199+AZD5 | 0.7±0.54a,c | 11.3±2.45b,d | 82.6±5.47b,d | 5.5±3.51a,c |
Data are represented as mean ± SD (n = 3). Mean values within the same column containing different superscript letters indicates significance among the treatment groups analyzed by one-way ANOVA followed by a Fisher’s LSD test (p < 0.05). Values bearing the letter “a” indicate no significant differences compared with control, those labeled “b” denote a significant difference (p < 0.05) when compared with the control, “c” denotes no significant difference when AZD9291+ABT263/ABT199 is compared with either AZD9291 or ABT263/ABT199 treatment alone at the corresponding concentration, and “d” denotes a significant difference (p < 0.05) when AZD9291+ABT263/ABT199 is compared with AZD9291 as well as ABT263/ABT199 treatment alone at the corresponding concentration.
Figure 2. Apoptosis analyses of H1975AR treated by AZD and/or ABT263/ABT199 by flow cytometry.
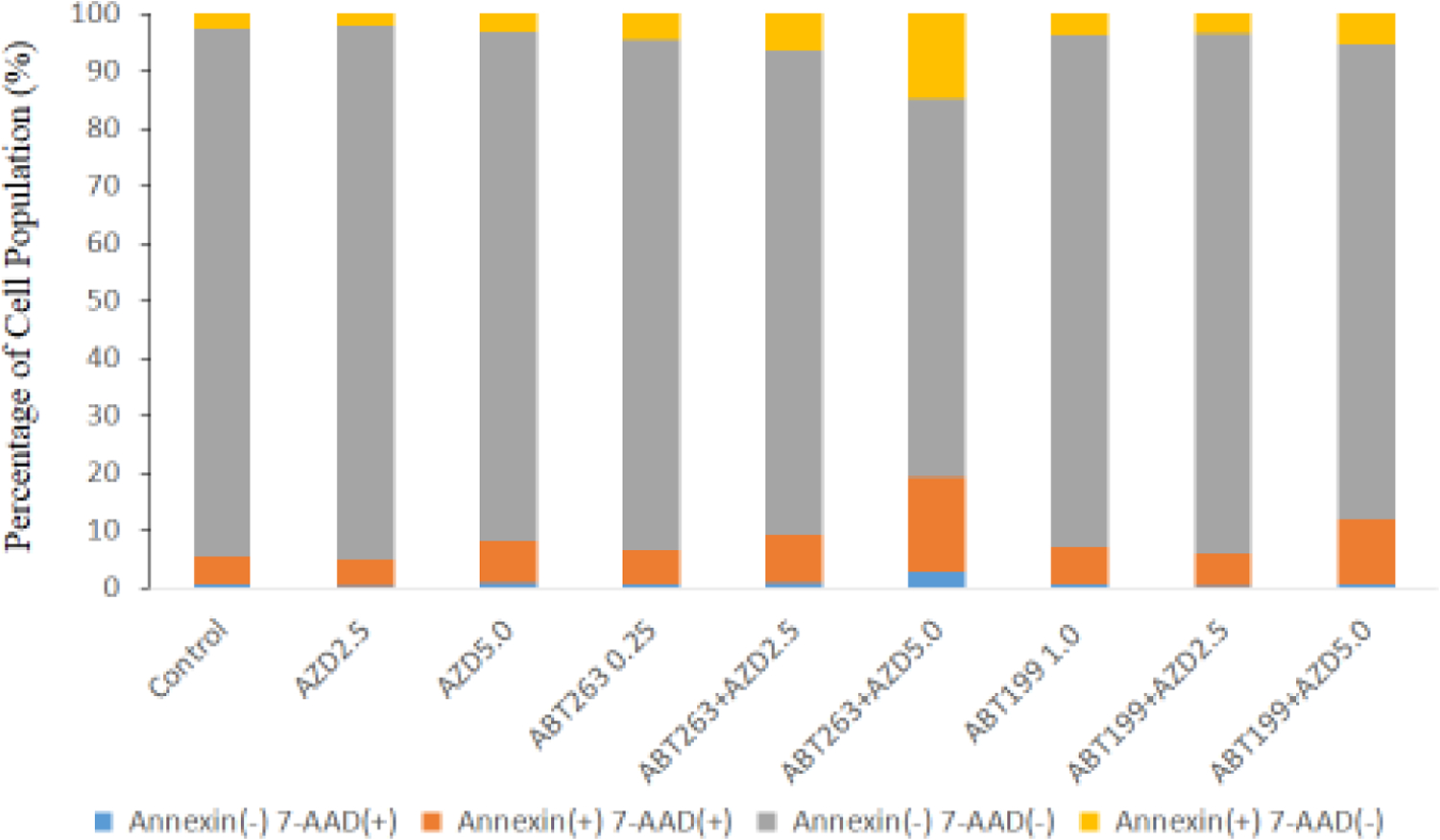
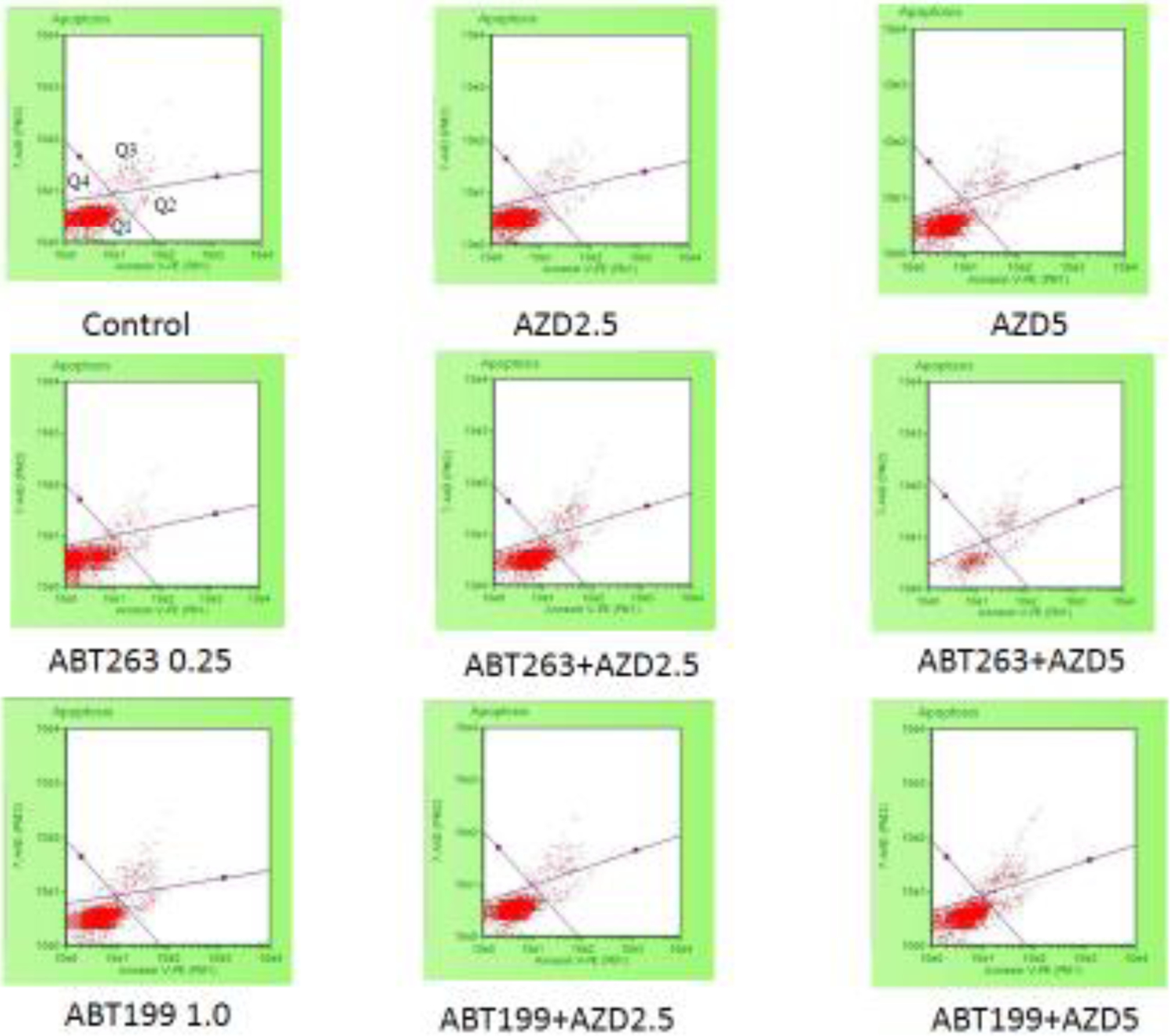
(a) Percentage of apoptosis analyzed by the Guava Nexin of H1975AR treated by AZD and/or ABT263/ABT199 for 48hrs and (b) Examples of H1975AR cells assayed using the Guava Nexin assay which allowed identification of the different apoptotic stages. As labeled in the “Control”, Q1 represents viable/non-apoptotic cells [Annexin V-PE (−) and 7-AAD (−)]; Q2 represents early-apoptotic cells [Annexin V-PE (+) and 7-AAD (−)]; Q3 represents late stage apoptotic/dead cells [Annexin V-PE(+) and 7-AAD (+)]; and Q4 represents nuclear debris [Annexin V-PE (−) and 7-AAD (+)]. Experiments performed in triplicate yielded similar results.
Table 2 and Figure 3 showed that AZD9291+ABT263/ABT199 co-treatments caused a significant accumulation of H1975AR in pre-G1 compared with ABT263/ABT199 or AZD9291 treatment alone, which further validated the synergistic effects of AZD9291+ABT263/ABT199 detected by the MTT and Nexin assays. AZD9291 treatment alone increased the numbers of cells in G0/G1 phase and reduced the number of cells in G2/M phase compared with control, which is consistent with the previous report of cell cycle arrest induced by AZD9291 treatment (Liu et al 2019). On the other hand, ABT263 and ABT199 significantly increased G2/M cell cycle arrest compared with the control. It is noticeable that ABT263+AZD5 co-treatments induced the highest pre-G1 cell cycle arrest (8.8±1.92%) compared with all other treatment groups while ABT199+AZD5 co-treatments significantly induced the highest G0/G1 cell cycle arrest (73.7±0.86%) in H1975AR compared with all other treatment groups.
Table 2.
Effects of AZD9291 and/or ABT263/ABT199 on cell cycles of H1975AR
| Groups | Cell Cycle (%) | |||
|---|---|---|---|---|
| pre-G1 | G0/G1 | S | G2/M | |
| Control | 3.1±0.25a | 65.5±2.31a | 4.0±1.19a | 23.6±0.54a |
| AZD2.5 | 4.6±1.15b | 68.2±1.67a | 3.0±0.07a | 22.0±3.62a |
| AZD5 | 4.3±0.36b | 69.3±0.77b | 3.9±0.21a | 19.8±2.66b |
| ABT263 0.25 | 5.6±0.31b | 60.3±0.82b | 3.0±0.38a | 28.2±0.38b |
| ABT263+AZD2.5 | 6.3±0.70b,c | 63.2±2.29a,c | 4.2±0.44a,d | 23.2±2.52a,c |
| ABT263+AZD5 | 8.8±1.92b,d | 62.9±1.14a,c | 3.9±1.12a,c | 22.0±2.29a,c |
| ABT199 1.0 | 4.0±0.44b | 64.1±0.72a | 3.0±0.55a | 27.9±1.86b |
| ABT199+AZD2.5 | 4.7±0.77b,c | 69.7±0.15b,c | 3.0±0.22a,c | 20.0±1.94b,c |
| ABT199+AZD5 | 6.5±1.70b,d | 73.7±0.86b,d | 3.0±0.54a,c | 14.9±1.13b,d |
Data are represented as mean ± SD (n = 3). Mean values within the same column containing different superscript letters indicates significance among the treatment groups analyzed by one-way ANOVA followed by a Fisher’s LSD test (p < 0.05). Values bearing the letter “a” indicate no significant differences compared with control, those labeled “b” denote a significant difference (p<0.05) when compared with the control, “c” denotes no significant difference when AZD9291+ABT263/ABT199 is compared with either AZD9291 or ABT263/ABT199 treatment alone at the corresponding concentration, and “d” denotes a significant difference (p<0.05) when AZD9291+ABT263/ABT199 is compared with AZD9291 as well as ABT263/ABT199 treatment alone at the corresponding concentration.
Figure 3. Cell cycle analyses of H1975AR treated by AZD and/or ABT263/ABT199 by flow cytometry.
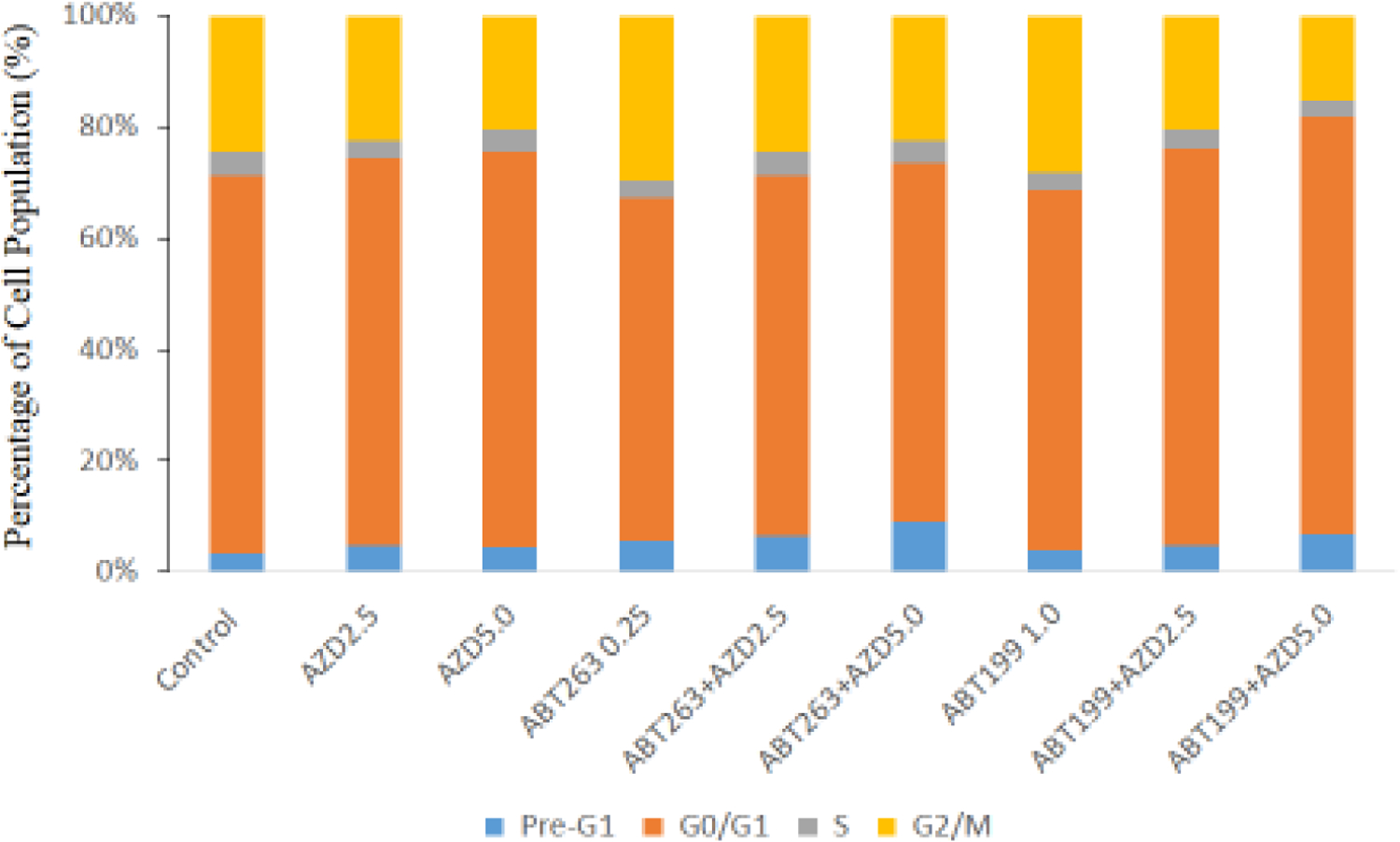
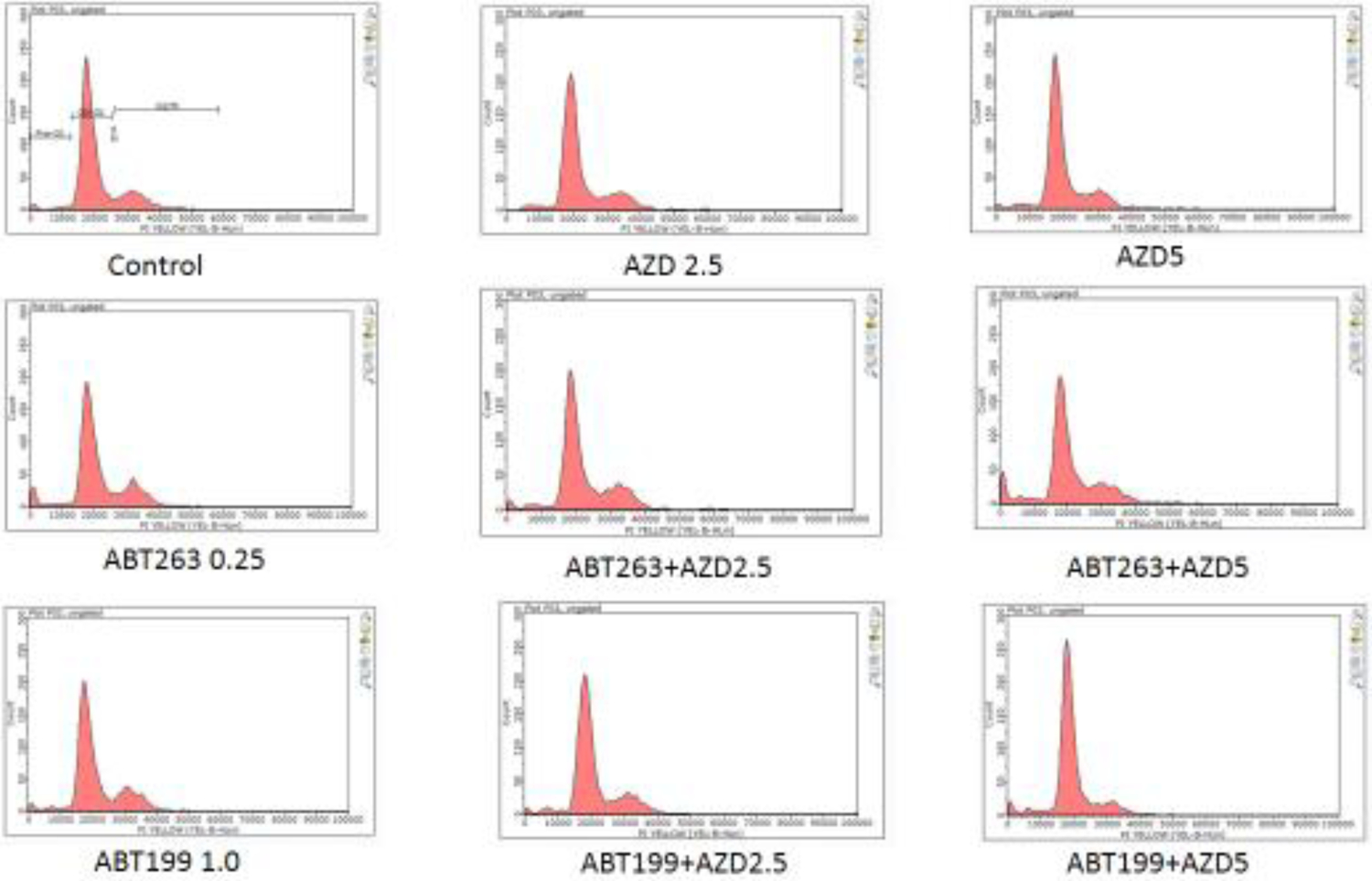
(a) Percentage of cell cycle arrest of H1975AR treated by AZD and/or ABT263/ABT199 and (b) Representative histograms of cell cycle analyses in AZD and/or ABT263/ABT199-treated H1975AR for 48 hrs were labeled with Guava Cell Cycle Reagent (Millipore) and analyzed by flow cytometry (pre-G1, G0/G1, S, and G2/M as labeled in “Control”). The y-axis shows the number of cells counted and the x-axis shows an increasing amount of Guava Cell Cycle Reagent incorporation/cell (left to right). Experiments performed in triplicate yielded similar results.
Protein analyses of EGFR signaling, Bcl-2, autophagy and other survival pathways in H1975AR after AZD9291 and/or ABT263/ABT199 treatments
Key molecules reported in our (Liu and Gao 2017; Liu and Gao 2019) or other previous studies (Froehlich et al 2019; Lin et al 2017; Zhan et al 2019) that played important roles in synergism between Bcl-2 inhibitors and EGFR-TKIs or other therapeutic agents were screened by multiscreen blot and Western blot (Figure 4), including p21, p-stat3, Bcl-XL, NF-κB, BIM, SQSTM1, Mcl-1, Twist1, and EGFR and other survival pathways. Among these selected molecules, p21 significantly decreased in AZD5+ABT263 compared with AZD5 (Figure 4a), while NF-κB and SQSTM1 significantly decreased in AZD5+ABT199 compared with AZD5 (Figure 4b).
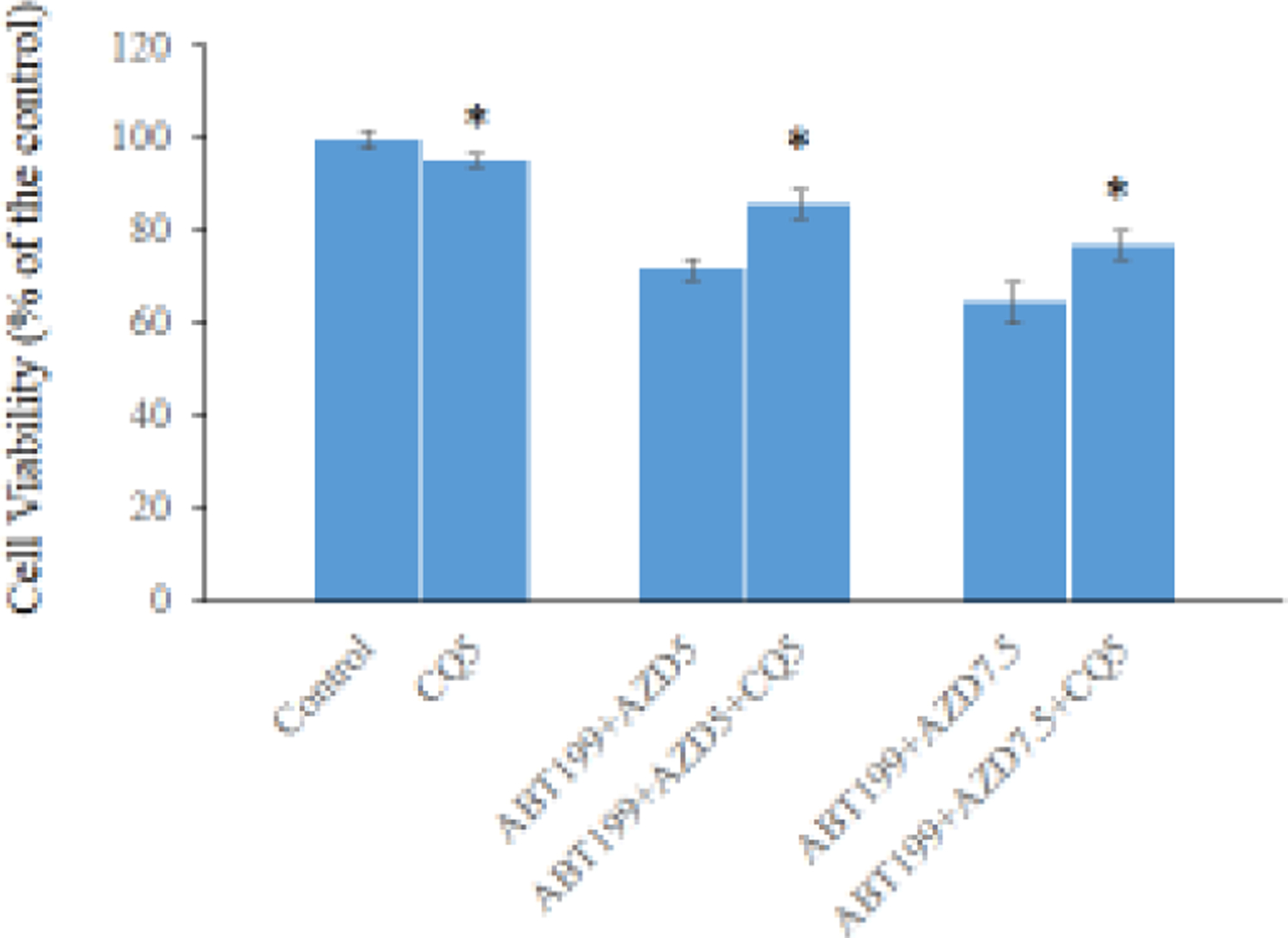
Figure 4. Multiscreen blot and Western blot analyses H1975AR after AZD and/or ABT263/ABT99 treatments and the effects of CQ on cell viabilities.
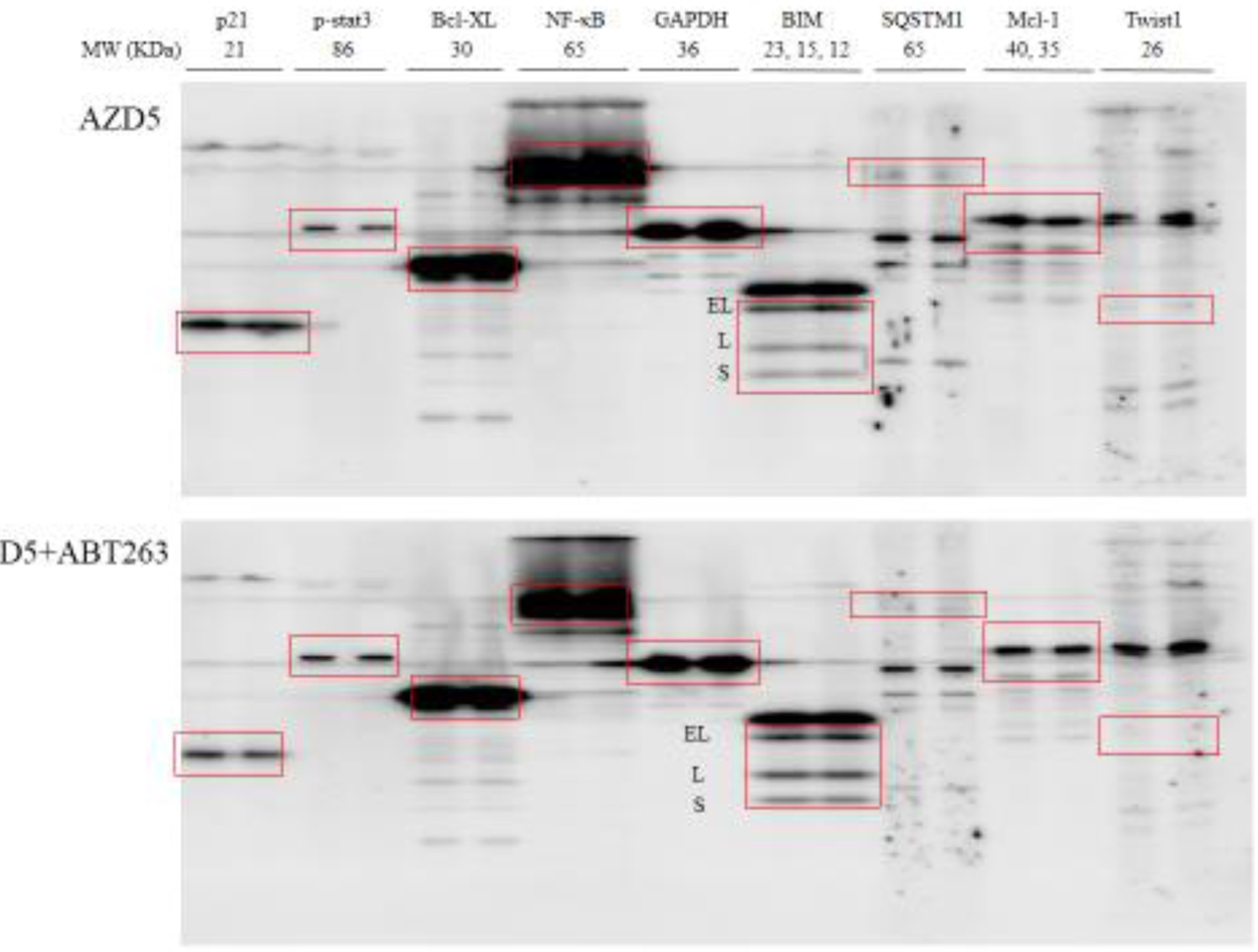
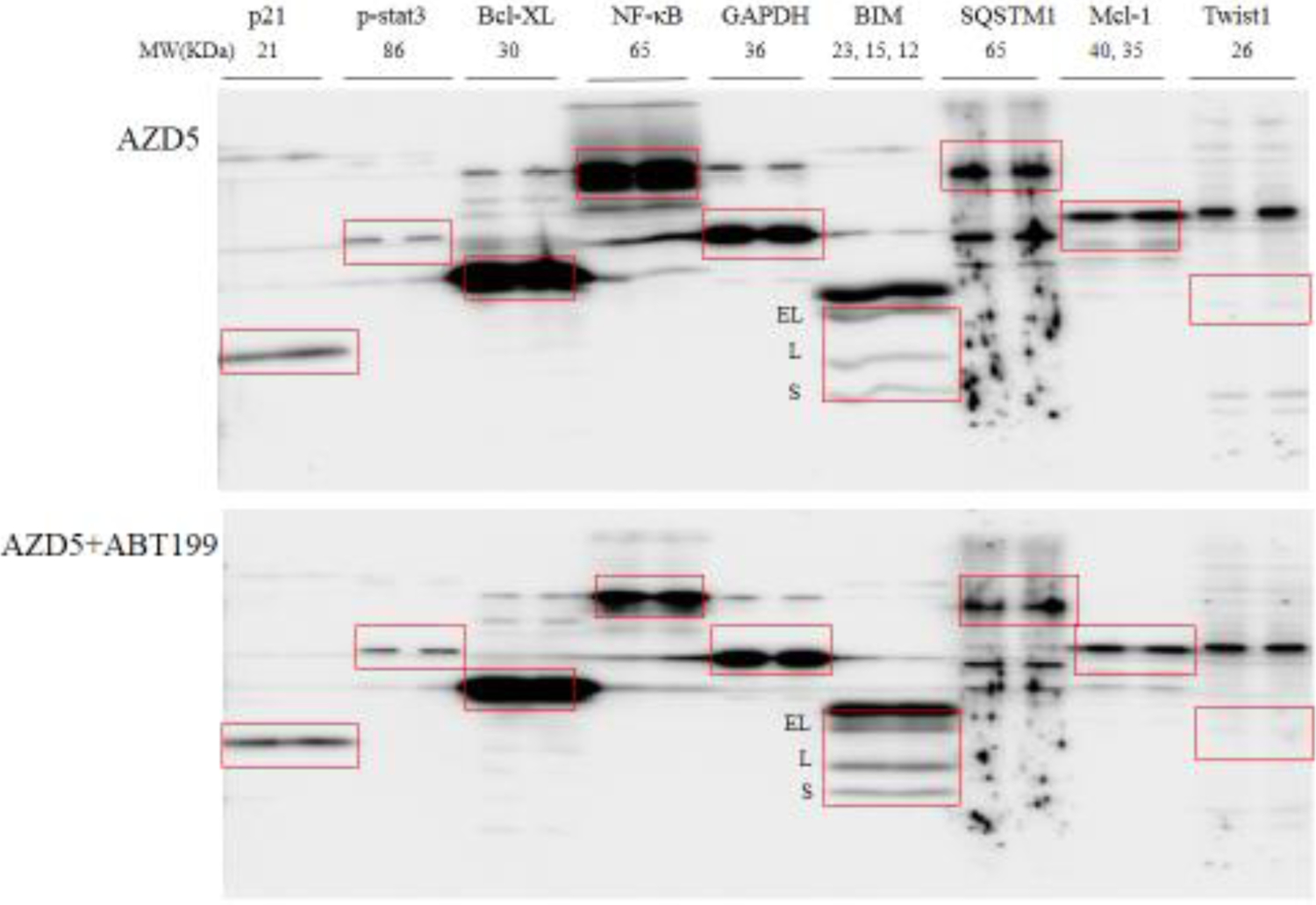
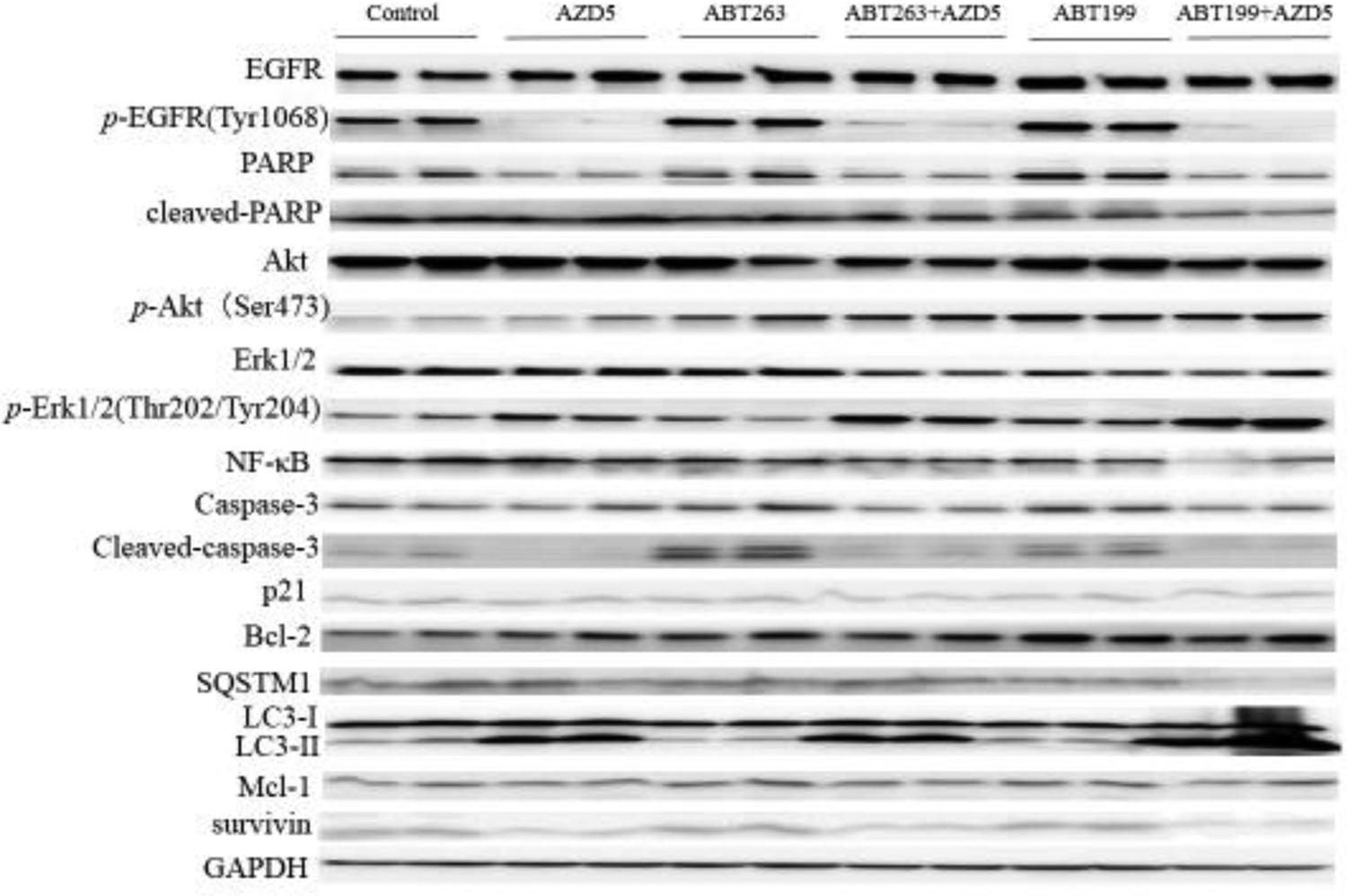
Multiscreen blots of protein profiles of H1975AR treated by (a) AZD5 and AZD5+ABT263 and (b) AZD5 and AZD5+ABT199. (c) Western blot analyses of EGFR, Bcl-2, autophagy, and other survival signaling pathways in H1975AR treated by AZD5, ABT263, ABT263+AZD5, ABT199, and ABT199+AZD5. (d) The effects of CQ on cell viabilities of ABT199+AZD9291 at 48 hrs were evaluated by comparing AZD5 for ABT199+AZD5 and ABT199+AZD5+CQ5 treatment groups, or by comparing AZD7.5 for ABT199+AZD7.5 and ABT199+AZD7.5+CQ5 treatment groups. * signified significant difference between with and without CQ in the corresponding treatment groups.
Further comprehensive Western blot analyses of all the treatment groups (Figure 4c) revealed that NF-κB were downregulated by combined treatments of AZD5+ABT263/ABT199 compared with AZD5, ABT263/ABT199, and the control, respectively. It is noticeable that AZD5+ABT199 treatment for 48 hrs caused an even more reduced expression of NF-κB compared with AZD5+ABT263 treatment for 48 hrs, which further confirmed the results of multiscreen blots. Meanwhile, downregulation of p21 was further confirmed in AZD5+ABT263 treatment group compared with AZD5, ABT263, and control, respectively. Whereas significant downregulations of survivin and SQSTM1 were observed specifically for AZD5+ABT199 compared with AZD5, ABT199, and the control, respectively. LC3-II was significantly higher in AZD5 and AZD5+ABT263/ABT199 and the highest in ABT199+AZD5 treatment group. Another important pro-apoptotic Bcl-2 family member Noxa was also examined by Western blot analyses as it was reported to a mechanism of synergism between ABT263 and EGFR inhibitor apigenin (Zhan et al 2019), but no change of Noxa was found in AZD5+ABT263/ABT199 compared with other treatment groups (data not shown).
To explore other mechanisms by which ABT263/ABT199 synergized with AZD9291 to overcome its drug resistance, phosphorylation status of EGFR and its downstream signaling pathways including EGFR, Akt, and Erk1/2 were examined by Western Blot. The results (Figure 4c) showed that the expression of EGFR did not change between different treatment groups while p-EGFR (Tyr1068) was downregulated in both AZD5 and AZD5+ABT263/ABT199 groups compared with control or ABT263/ABT199 treatment alone. Both p-Akt (Ser473) and p-Erk1/2 ((Thr202/Tyr204) were upregulated in AZD5+ABT263/ABT199 compared with control or AZD5 group. On the other hand, Akt and Erk1/2 were downregulated in AZD5+ABT263/ABT199 compared with control or AZD5 group. Similar with p-EGFR (Tyr1068), PARP was downregulated in both AZD5 and AZD5+ABT263/ABT199 groups compared with control or ABT263/ABT199 treatment alone while cleaved-PARP were downregulated in AZD5+ABT263/ABT199 compared with AZD5, ABT263/ABT199, and the control, respectively. No significant differences in the expressions of caspase-3 were found in AZD5+ABT263/ABT199 compared with the control or AZD5 group. However, compared with the control group, ABT263 and ABT199 treatment groups showed significantly increased expressions of cleaved-caspase-3. On the other hand, AZD5, ABT263+AZD5, and ABT199+AZD5 showed significantly decreased expressions of cleaved-caspase-3 compared with the control, AB263, and ABT199, respectively.
CQ inhibitory effect on cytotoxicity of ABT199+AZD9291 in H1975AR
The sequel of autophagy inhibitor CQ on the synergistic effects of AZD9291 and ABT199 was examined in H1975AR by MTT assay. Based on our cytotoxic data of CQ using MTT assay (data not shown) and the previous study (Tang et al 2017), 5 μM CQ was selected for autophagy inhibition to evaluate the consequence of autophagy induced by ABT199+AZD929 after the application of CQ in H1975AR. As shown in Figure 4d, 5 μM CQ could partially but significantly reverse ABT199+AZD9291-induced cytotoxicity in H1975AR. The cell viability was 71.8±2.04% in ABT199+AZD5 and 85.8±3.06% in ABT199+AZD5+CQ5 compared to AZD5, and 64.6±4.42% in ABT199+AZD7.5 and 77.1±3.18% in ABT199+AZD7.5+CQ5 compared to AZD7.5.
Discussion
MTT assay showed that ABT199+AZD5 displayed a similar synergistic effect on H1975AR compared with ABT263+AZD5. Flow cytometry analyses showed that ABT263+AZD5 had higher cell populations of early apoptotic and apoptotic in Nexin assay as well as higher cell population arrested at pre-G1 phase representing apoptotic cells than ABT199+AZD5. However, ABT199+AZD9291 induced more G0/G1 arrest in H1975AR. This contrast may be attributed to the different effects of ABT263 and ABT199 alone, in which ABT199 showed more G0/G1 while ABT263 had a tendency of more pre-G1. In addition, although the selected doses of ABT263 and ABT199 by itself had no significant cytotoxic effect, a relative higher potency and cytotoxicity of 0.25 μM ABT263 alone (91.3±2.21% as compared to the control) was found in H1975AR than 1 μM ABT199 alone (102.4±6.85% as compared to the control) at 48 hrs in MTT assays (data not shown). Nevertheless, 0.25 μM ABT263 and 1 μM ABT199 were taken as controls to calculate cell viabilities of ABT263+AZD5 and ABT199+AZD5 in MTT assays, respectively.
The clinical implementation of ABT263 was hampered due to concomitant inhibition of the anti-apoptotic protein Bcl-XL (Bate-Eya et al 2016). On the other hand, ABT199 showed less activity against Bcl-XL (Ki of 48 nM), Bcl-W (Ki of 245 nM) and Mcl-1 (Ki of >444 nM), and maintained its activity against Bcl-2 (Ki of <0.01 nM) (Bate-Eya et al 2016), which made ABT199 exhibited much less toxicity toward platelets that significantly limited the clinical applications of ABT263 in treating lymphoma (Bate-Eya et al 2016). Although the combination therapy of ABT263+AZD9291 in treating NSCLC patients of advanced and metastatic stage is undergoing clinical trials, the new combination of ABT199+AZD9291 presented in this study together with the new findings of ABT263+AZD9291 showed great clinical potential to treat NSCLC patients with EGFR-TKI resistance driven by significant Bcl-2 upregulation.
Western blot analyses revealed that the NF-κB is a common signaling pathway through which ABT263 or ABT199 synergized the cytotoxic effects of AZD9291 in H1975AR. NF-κB has been reported to play a key role in driving acquired resistance to another irreversible EGFR-TKI CNX-2006 and inhibition of NF-κB by either genetic or pharmacologic method could also efficiently overcome the drug resistance in CNX-2006 resistant H1975 cell line (Galvani et al 2015). The differences in binding activities (Ki) of ABT263 and ABT199 against different isoforms of Bcl-2 family proteins may explain the different inhibitory levels of p21, SQSTM1, and survivin by combined treatment of ABT263+AZD9291 or ABT199+AZD9291. Previous studies (Li et al 2014; Lin et al 2017) showed that p21 is a critical target of ABT263 or in combined use with other therapeutic agents while knockdown of p21 could significantly inhibited apoptosis in cancer cells caused by ABT263 treatments or ABT263 co-treatments (Li et al 2014; Lin et al 2017). Meanwhile, the previous study (Lin et al 2017) reported ABT263 (5–20 μM) caused apoptosis and autophagy mediated by G0/G1-phase arrest in human esophageal cancer cells in vitro. On the other hand, the results of our study demonstrated that ABT263 synergized the cytotoxic effects of AZD9291 by downregulating p21 compared to AZD9291, ABT263, and the control groups. Moreover, our qRT-PCR analyses revealed that p53, as a major transcriptional regulator of p21 (Bodzak et al 2008), was significantly downregulated in ABT263+AZD9291 compared with AZD9291 (data not shown), suggesting that the observed p21 downregulation could be associated with p53 reduction. The same RT-PCR array used in our previous study (Lv et al 2015) was also applied for screening of potential targets that play important roles in synergism observed in ABT263+AZD9291 in H1975AR, but no significant changes of signaling molecules were found in the array between ABT263+AZD9291 and AZD9291 treatment groups. These results indicated that unrecognized important molecular mechanisms other than p53/p21 pathways responsible for synergism between ABT263 and AZD9291 in H1975AR may warrant further research and exploration. It is noticeable that ABT263 treatment alone induced apparent G2/M arrest compared with the control in H1975AR, and ABT263+AZD9291 co-treatments also induced a relatively higher G2/M arrest compared AZD9291 treatment alone in H1975AR. Interestingly, the previous study (Lin et al 2017) showed that ABT263 at higher concentration range (1.25–20 μM) caused apoptosis and induced G0/G1 arrest with significant upregulation of p21 in human esophageal cancer cells, which suggest that p21, as an important regulator of cell cycle progression, may play quite different roles in cell cycle arrest induced by ABT263 at different concentrations and/or cell types.
SQSTM1/p62 plays a critical role in autophagy and the accumulation of SQSTM1 induced by AZD9291 could compromise the effectiveness of AZD9291 in NSCLC cells (Yang et al 2019). On the other hand, inhibition of NF-κB signaling via degradation of SQSTM1 could induce apoptosis and autophagy in cancer cells in vitro (Su et al 2015). Our study showed that ABT199+AZD9291 could significantly reduce the expression of NF-κB as well as SQSTM1 compared with all other treatment groups including the control, indicating that this may be a novel pathway with the potential to overcome the acquired resistance of AZD9291 in vitro. LC3-II, as a most important biomarker of autophagosome formations, have been significantly increased in AZD9291+ABT199 group compared with all other treatment groups, which was consistent with the degradation of SQSTM1, demonstrating that autophagy played an important role in synergism between AZD9291+ABT199 in H1975AR. On the other hand, AZD5 alone could also induce an upregulation of LC3-II compared with the control in H1975AR, which was consistent with the results of previous studies showing that AZD9291 induced autophagy with upregulation of LC3-II and downregulation of SQSTM1 in H1975 (Tang et al 2017; Zhang et al 2019). The cytotoxicity of AZD9291+ABT199 could be partially reversed by autophagy inhibitor CQ, suggesting that autophagy played an important role in synergism between AZD9291+ABT199. Survivin, as the smallest member of inhibitors of apoptosis (IAP) family of proteins involved in inhibition of apoptosis and regulation of cell cycle (Garg et al 2016), is most downregulated in ABT199+AZD5 treatment groups compared with other treatment or control groups. Survivin was reported to expressed highly at G2/M phase and declines rapidly in G1 phase of cell cycle (Jaiswal et al 2015), which was consistent with the observations of our flow cytometry analyses showing that ABT199+AZD5 treatment groups had highest G0/G1 and lowest G2/M cell populations compared with other treatment or control groups.
EGFR and its downstream signaling pathway were also examined, which revealed that p-EGFR (Tyr1068) was significantly downregulated in AZD5, AZD5+ABT263, and AZD5+ABT199 treatment groups to a similar extent compared with other treatment groups. Meanwhile, p-Erk1/2 (Thr202/Tyr204) was observed to be significantly upregulated in AZD5+ABT263/ABT199 groups compared with other treatment groups and no significant downregulation of p-Akt (Ser473) was observed in AZD5+ABT263/ABT199 groups compared with other treatment groups. The results indicated that inhibition of p-EGFR and its downstream pathways can be ruled out as a mechanism of synergistic effects between AZD5+ABT263/ABT199 on H1975AR. Degradation of PARP and the activation of cleaved-PARP were also important mechanisms underlying the synergism between Bcl-2 inhibitors and other therapeutic agents in cancer treatments revealed by previous studies (Lin et al 2017; Wang et al 2015). However, no significant difference of PARP degradation or activation of cleaved-PARP was found in AZD5+ABT263/ABT199 groups compared with AZD5 treatment group, demonstrating that PARP did not play a critical role for the observed synergistic cytotoxic effects in this study.
Meanwhile, no significant degradation of caspase-3 was found in AZD5+ABT263/ABT199 groups compared with AZD5 treatment group. However, cleaved-caspase-3 showed different changes among treatment groups. The higher cytotoxicity of ABT263 alone (0.25 μM) than that of ABT199 alone (1 μM) could induce a higher apoptosis in H1975AR with higher expression of cleaved-caspase-3 in ABT263 treatment group compared with ABT199 treatment group as well as the control. On the other hand, cleaved-caspase-3 significantly decreased in AZD5 compared with control in this study. This finding is consistent with the previous studies, suggesting that AZD5 could initiate an autophagy process that has an inverse relationship with activity of caspase-3 (Tsapras and Nezis 2017; Wu et al 2014). Meanwhile, cleaved-caspase-3 was significantly decreased in ABT263+AZD5 compared to ABT263 and the control, but was relatively higher than AZD5, indicating that the more pronounced cytotoxic effects of ABT263+AZD5 as compared to AZD5 may be caused by the combined mechanisms related to autophagy, apoptosis, and/or caspase-independent programmed cell death (Kögel and Prehn 2013; Qi and Liu 2006). Similarly, ABT199+AZD5 also showed a reduced expression of cleaved-caspase-3 compared with the control and ABT199, but a relatively lower expression compared to ABT263+AZD5, together with the findings of LC3-II, suggesting that autophagy-mediated cell death process is more likely to occur in ABT199+AZD5.
Although BIM (L) was found to be significantly upregulated in AZD5+ABT263/ABT199 compared with AZD5 revealed by multiscreen analyses, it was found that the strongest expression of BIM (EL, L, S) was also found in control groups by subsequent Western blot analyses (data not shown), which indicated that the synergism between ABT263/199 and AZD9291 was not achieved through activation of cytotoxic BIM family proteins. The expressions of other two anti-apoptotic proteins Bcl-2 and Mcl-1 were not found to be significantly downregulated in AZD5+ABT263/ABT199 treatment groups compared with control. Taken together, all these results demonstrated that the Bcl-2 family members were less likely to be responsible for the synergistic cytotoxic effects of AZD5+ABT263/ABT199 on H1975AR as shown in this study.
Conclusion
A novel combination therapy regimen ABT199/ABT263 and AZD9291 was firstly reported in our study to effectively overcome the acquired drug resistance of AZD9291 in H1975AR with significant upregulation of Bcl-2 compared with parental H1975. Flow cytometry analyses showed that ABT199+AZD9291 treatment caused a dramatically decreased G2/M cell population but increased G0/G1 of H1975AR, which could be closely associated with the significantly downregulation of survivin compared with ABT199, AZD9291, and control groups, considered as an important synergistic mechanism for ABT199+AZD9291. In contrast, a trend of upregulation of G2/M cell population was seen in ABT263 treatment groups compared with control and this trend was also observed in ABT263+AZD9291 compared with AZD9291 treatment groups, which could be associated with the downregulation of p53-dependent p21 expression as the synergistic mechanism of ABT263+AZD9291. ABT199 might also synergize with the cytotoxic effect of AZD9291 via enhanced autophagy evidenced by significantly increased expression of LC3-II and downregulation of SQSTM1 in H1975AR even compared with ABT263+AZD9291. In summary, our study demonstrated that 0.25 μM ABT263 or 1 μM ABT199 combined with AZD9291 showed similar efficiency to overcome the acquired resistance of AZD9291 in H975AR, though the synergism of ABT263 or ABT199 with AZD9291 against H1975AR was achieved by different molecular mechanisms.
Acknowledgement
This research was partially supported by the National Institute of Environmental Health Sciences of the National Institutes of Health under Award Number R15ES026789. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Bate-Eya LT, den Hartog IJ, van der Ploeg I et al. (2016) High efficacy of the BCL-2 inhibitor ABT199 (venetoclax) in BCL-2 high-expressing neuroblastoma cell lines and xenografts and rational for combination with MCL-1 inhibition. Oncotarget 7(19):27946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodzak E, Blough MD, Lee PW, Hill R (2008) p53 Binding to the p21 promoter is dependent on the nature of DNA damage. Cell Cycle 7(16):2535–2543 [DOI] [PubMed] [Google Scholar]
- Brown SP, Taygerly JP (2012) Small-molecule antagonists of Bcl-2 family proteins Annual Reports in Medicinal Chemistry. vol 47 Elsevier, p 253–266 [Google Scholar]
- Chen J, Jin S, Abraham V et al. (2011) The Bcl-2/Bcl-XL/Bcl-w inhibitor, navitoclax, enhances the activity of chemotherapeutic agents in vitro and in vivo. Mol Cancer Ther 10(12):2340–2349 [DOI] [PubMed] [Google Scholar]
- Cheong HT, Xu F, Choy CT et al. (2018) Upregulation of Bcl2 in NSCLC with acquired resistance to EGFR-TKI. Oncol Lett 15(1):901–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crombie J, Davids MS (2017) Venetoclax for the treatment of patients with chronic lymphocytic leukemia. Future Oncol 13(14):1223–1232 [DOI] [PubMed] [Google Scholar]
- Froehlich TC, Müller-Decker K, Braun JD et al. (2019) Combined inhibition of Bcl-2 and NFκB synergistically induces cell death in cutaneous T-cell lymphoma. Blood 134(5):445–455 [DOI] [PubMed] [Google Scholar]
- Galvani E, Sun J, Leon LG et al. (2015) NF-κB drives acquired resistance to a novel mutant-selective EGFR inhibitor. Oncotarget 6(40):42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg H, Suri P, Gupta JC, Talwar G, Dubey S (2016) Survivin: a unique target for tumor therapy. Cancer Cell Int 16(1):49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal PK, Goel A, Mittal R (2015) Survivin: A molecular biomarker in cancer. Indian J Med Res 141(4):389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kögel D, Prehn JH (2013) Caspase-independent cell death mechanisms Madame Curie Bioscience Database [Internet]. Landes Bioscience [Google Scholar]
- Li J, Chen Y, Wan J et al. (2014) ABT-263 enhances sorafenib-induced apoptosis associated with A kt activity and the expression of B ax and p21 (CIP1/WAF1) in human cancer cells. Br J Pharmacol 171(13):3182–3195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q-h, Que F-c, Gu C-p et al. (2017) ABT-263 induces G 1/G 0-phase arrest, apoptosis and autophagy in human esophageal cancer cells in vitro. Acta Pharmacol Sin 38(12):1632–1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Chen X, Shi L et al. (2019) The third-generation EGFR inhibitor AZD9291 overcomes primary resistance by continuously blocking ERK signaling in glioblastoma. J Exp Clin Cancer Res 38(1):219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Gao W (2017) Leptomycin B reduces primary and acquired resistance of gefitinib in lung cancer cells. Toxicol Appl Pharmacol 335:16–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Gao W (2019) Overcoming acquired resistance of gefitinib in lung cancer cells without T790M by AZD9291 or Twist1 knockdown in vitro and in vivo. Arch Toxicol 93:1555–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv T, Wang Q, Cromie M et al. (2015) Twist1-mediated 4E-BP1 regulation through mTOR in non-small cell lung cancer. Oncotarget 6(32):33006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR et al. (2005) An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435(7042):677. [DOI] [PubMed] [Google Scholar]
- Patel H, Pawara R, Ansari A, Surana S (2017) Recent updates on third generation EGFR inhibitors and emergence of fourth generation EGFR inhibitors to combat C797S resistance. Eur J Med Chem 142:32–47 [DOI] [PubMed] [Google Scholar]
- Qi R, Liu XY (2006) New advance in caspase-independent programmed cell death and its potential in cancer therapy. Int J Biomed Sci 2(3):211. [PMC free article] [PubMed] [Google Scholar]
- Santarpia M, Liguori A, Karachaliou N et al. (2017) Osimertinib in the treatment of non-small-cell lung cancer: design, development and place in therapy. Lung Cancer (Auckl) 8:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui WA, Ahad A, Ahsan H (2015) The mystery of BCL2 family: Bcl-2 proteins and apoptosis: an update. Arch Toxicol 89(3):289–317 [DOI] [PubMed] [Google Scholar]
- Su J, Liu F, Xia M et al. (2015) p62 participates in the inhibition of NF-κB signaling and apoptosis induced by sulfasalazine in human glioma U251 cells. Oncol Rep 34(1):235–243 [DOI] [PubMed] [Google Scholar]
- Suvarna V, Singh V, Murahari M (2019) Current overview on the clinical update of Bcl-2 anti-apoptotic inhibitors for cancer therapy. Eur J Pharmacol:172655. [DOI] [PubMed] [Google Scholar]
- Tang Z-H, Cao W-X, Su M-X, Chen X, Lu J-J (2017) Osimertinib induces autophagy and apoptosis via reactive oxygen species generation in non-small cell lung cancer cells. Toxicol Appl Pharmacol 321:18–26 [DOI] [PubMed] [Google Scholar]
- Tsapras P, Nezis IP (2017) Caspase involvement in autophagy. Cell Death Differ 24(8):1369–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Huang S-B, Yang M-C et al. (2015) Combining paclitaxel with ABT-263 has a synergistic effect on paclitaxel resistant prostate cancer cells. PLoS One 10(3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Che X, Zheng Q et al. (2014) Caspases: a molecular switch node in the crosstalk between autophagy and apoptosis. Int J Biol Sci 10(9):1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S-G, Shih J-Y (2018) Management of acquired resistance to EGFR TKI–targeted therapy in advanced non-small cell lung cancer. Mol Cancer 17(1):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Xie Y, Ni S, Liu H (2015) The latest therapeutic strategies after resistance to first generation epidermal growth factor receptor tyrosine kinase inhibitors (EGFR TKIs) in patients with non-small cell lung cancer (NSCLC). Ann Transl Med 3(7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Tang W, Li T, Zhang X, Sun Y (2019) Overcoming Resistance to AC0010, a Third Generation of EGFR Inhibitor, by Targeting c-MET and BCL-2. Neoplasia 21(1):41–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Ying S, Hu S et al. (2019) EGFR TKIs impair lysosome-dependent degradation of SQSTM1 to compromise the effectiveness in lung cancer. Signal Transduct Target Ther 4(1):1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda K, Imanishi N, Ichiki Y, Tanaka F (2019) Treatment of Non-small Cell Lung Cancer with EGFR-mutations. J UOEH 41(2):153–163 [DOI] [PubMed] [Google Scholar]
- Zhan Y, Wang Y, Qi M et al. (2019) BH3 mimetic ABT-263 enhances the anticancer effects of apigenin in tumor cells with activating EGFR mutation. Cell Biosci 9(1):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wang S, Wang L et al. (2015) Prognostic value of Bcl-2 expression in patients with non-small-cell lung cancer: A meta-analysis and systemic review. Onco Targets Ther 8:3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zhang M, Liu H, Yin W (2019) AZD9291 promotes autophagy and inhibits PI3K/Akt pathway in NSCLC cancer cells. J Cell Biochem 120(1):756–767 [DOI] [PubMed] [Google Scholar]
- Zhou Q, Wu L, Feng L et al. (2019) Safety and efficacy of abivertinib (AC0010), a third-generation EGFR tyrosine kinase inhibitor, in Chinese patients with EGFR-T790M positive non-small cell lung cancer (NCSLC). J Clin Oncol [Google Scholar]
- Zou M, Xia S, Zhuang L et al. (2013) Knockdown of the Bcl-2 gene increases sensitivity to EGFR tyrosine kinase inhibitors in the H1975 lung cancer cell line harboring T790M mutation. Int J Oncol 42(6):2094–2102 [DOI] [PubMed] [Google Scholar]