

Abstract
Genome-wide association studies have established that human REL is a susceptibility gene for lymphoid cancers and inflammatory diseases. REL is the hematopoietic member of the nuclear factor-κB (NF-κB) family and is frequently amplified in human lymphomas. However, the mechanism through which REL and its encoded protein c-Rel effect human lymphoma is largely unknown. Using both loss-of-function and gain-of-function approaches, we studied the roles of REL gene in human Jurkat leukemia cells. Compared with control Jurkat cells, REL knockout cells exhibited significant defects in cell growth and mitochondrial respiration. Genome-wide transcriptome analyses revealed that T cells lacking REL had selective defects in the expression of inflammatory and metabolic genes including c-Myc. We found that c-Rel controlled the expression of c-Myc through its promotor, and expressing c-Myc in c-Rel-deficient lymphoma cells rescued their proliferative and metabolic defects. Thus, the human c-Rel-c-Myc axis controls lymphoma growth and metabolism and could be a therapeutic target for lymphoma.
Keywords: immunometabolism, mitochondria, NF-κB, lymphoma, CRISPR-Cas9
Introduction
Metabolic reprogramming is a recurrent event in T cells throughout their life cycle, and is essential for their normal development and function[1]. Abnormal metabolic adaptation underpins a number of human diseases, including cancer[2]. Metabolic adaptation of T cells helps them survive in nutrient-restricted regions of the body and fulfill their functions in immune defense and surveillance; similarly, metabolic adaptation of cancer cells helps them survive the nutrient-deprived tumor microenvironment, making them more resilient and thus more difficult to combat[3]. In particular, cancer cells are characterized by increased energy demands and aberrant metabolism associated with rapid cell growth. Despite extensive research investigating the role of metabolism in driving cancer cell proliferation, the regulators governing cancer cell metabolism remain only partially characterized.
The nuclear factor-κB (NF-κB) family of dimeric transcription factors is comprised of five members: NF-κB1 (p105/p50), NF-κB2 (p100/p52), RelA/p65, RelB, and c-Rel[4]. They are key regulators of innate and adaptive immunity and also play pivotal roles in general cellular processes including cellular proliferation, differentiation, stress responses, and survival[5]. Unlike other NF-κB family members that are constitutively expressed in multiple cell types, c-Rel is expressed primarily in leukocytes[6]. c-Rel plays a crucial role in the development of regulatory T cells, inflammatory Th17 cells, and myeloid-derived suppressor cells through transcriptional regulation of forkhead box P3 (Foxp3), retinoic acid-related orphan receptor γt (RORγT), and CCAAT enhancer binding protein beta (Cebpb), respectively[6–8]. In addition, polymorphisms at the REL locus are associated with several autoimmune diseases and cancer [9, 10]. Moreover, REL gene amplifications and increased c-Rel expression have been found in a variety of B and T cell malignancies[9]. Despite these discoveries, the mechanisms by which c-Rel influences the disease course of lymphomas are largely unknown. Using CRISPR/Cas9 genome editing and metabolic profiling assays, we found that c-Rel controls human Jurkat T cell mitochondrial metabolism and proliferation via c-Myc.
Results
c-Rel is required for the growth of human Jurkat T cells.
To examine the role of c-Rel in human T cells, we generated REL knockout (KO) Jurkat T cells using a two-vector CRISPR/Cas9 system (Figure 1A). The main advantage of this two-vector system over most one-vector based systems is that the Cas9 endonuclease is expressed only when doxycycline is added to the culture medium. The level of Cas9 goes down following the withdrawal of doxycycline from the culture media, after gene editing has been completed. This helps to maintain the genomic stability of the knockout cells by preventing unintended mutations near the N-terminus of the c-Rel coding sequence, thereby eliminating the translated c-Rel protein in its entirety. We first transduced the wild-type Jurkat cells with the pCW-Cas9 vector, followed by one week of puromycin selection and one week of expansion. The selected cells were then transduced again with the pLX-single-guide RNA (sgRNA) vector followed by one week of blasticidin selection in the presence of doxycycline (Supplemental Figure 1A and 1B). One striking phenotype that we noticed immediately was that the polyclonal cells transduced with c-Rel-targeting sgRNA grew much slower than the cells transduced with the control AAVS1-targeting sgRNA (Figure 1B). After the clonal selection, three single-clonal c-Rel KO lines and three sgRNA control clones were successfully generated and used in subsequent comparisons (Figure 2A). Since the critical dependence of IL-2 induction by c-Rel has been well established in mice, we measured interleukin 2 (IL2) mRNA induction using RT-PCR for functional confirmation of successful c-Rel knockout. The results confirmed that all three c-Rel KO lines failed to induce IL2 expression upon phorbol myristate acetate (PMA)/ionomycin stimulation (which mimics T cell receptor engagement), while all three control cell lines created through the same processes, strongly expressed IL2 upon stimulation (Figure 2B).
Figure 1. c-Rel knockout polyclonal Jurkat cells show reduced cell growth.

A. a two-component lentiviral based CRISPR/Cas9 system was used to sequentially deliver Cas9 endonuclease and single-guide RNA (sgRNA) into cells. sgRNAs are constitutively expressed under the control of the U6 promoter, while Cas9 is expressed using a tet-on system in a doxycycline-dependent manner. B. cell growth of Jurkat cells treated with c-Rel-targeting sgRNA was measured against control sgRNA-treated and wild-type Jurkat cells. Values represent mean ± SEM, and are representative of 3 independent experiments. (n=3 biologically independent cultures in each group, P-values for control cells vs REL KO cells. ***, p<0.001).
Figure 2. c-Rel knockout single-clonal Jurkat cells show reduced cell growth.

A. Western blots of control and c-Rel knockout (KO) Jurkat clones. Each lane represents a single clone. B. control and c-Rel KO Jurkat cells were stimulated with or without PMA and ionomycin for 4 hours, followed by RT-PCR assay for human IL-2 (n=3 independent cultures of pooled clones). C. proliferation of control and c-Rel knockout Jurkat T cells in the culture (n=3 clones). D. Western blots of c-Rel KO Jurkat cell clones transduced with lentiviral control vector, or c-Rel-expressing vector. Each lane represents a single clone. E. cells were stimulated with PMA/ionomycin for 4 hours, followed by RT-PCR quantification of IL-2 (n=3 independent cultures of the indicated clones). F. proliferation of the indicated cells (n=3 clones). For all panels, data are expressed as mean ± SEM, and are representative of at least 3 independent experiments. *, p<0.05, **, p<0.01, ***, p<0.001.
The initial clonal selection process inevitably introduces selection bias in terms of growth rate, because non-proliferating or slow-proliferating cells will not emerge during the selection process. As a result, the growth rate differences we first observed before clonal selection growth were no longer evident when grown under standard culture medium containing 10% fetal bovine serum. However, upon placing the cells under lower serum concentration (2%) in the culture medium, all three c-Rel KO lines showed significantly lowered growth and survival rates (Figure 2C). To confirm that this difference was due to c-Rel deletion and not due to off-target effects of the CRISPR/Cas9 system, we rescued c-Rel expression in the c-Rel KO cells using a lentiviral vector with REL under the control of the human Ubiquitin C (UbC) promoter and that also co-expressed GFP under the control of an internal ribosome entry site (IRES) element so that we could employ flow cytometry-based selection (Supplemental Figures 2A to 2C). Western Blotting confirmed successful re-expression of the c-Rel protein (Figure 2D). This ectopic expression of c-Rel functionally rescued c-Rel target IL2 expression (Figure 2E), and largely rescued the proliferation defect of the c-Rel KO cells (Figure 2F), suggesting that the proliferation defect is mostly attributable to c-Rel ablation. Taken together, these results indicate that c-Rel is required for cell growth in human leukemia T cells during serum starvation.
Genome-wide transcriptome analyses identify mitochondrial metabolism and the c-Myc pathway as c-Rel targets in Jurkat T cells.
To determine the pathways downstream of c-Rel that mediate its regulation of Jurkat T cell proliferation, we performed a high-throughput microarray study and analyzed the transcriptome of control and c-Rel KO Jurkat cells in response to 4 hours of PMA/ionomycin stimulation. By using Ingenuity Pathway analysis (IPA) for the genes that were downregulated by more than 1.5-fold in c-Rel KO cells, we found that “Function of blood cells” and “Quantity of leukocytes” were among the most significantly downregulated pathways in c-Rel KO cells (Figure 3A), supporting the notion that c-Rel controls proliferation and function in human Jurkat T cells. Among the 30 genes in the “Function of blood cell” pathway, we identified several cytokines/chemokines (CCL1, CSF2, CXCL8, IL2, IL23A, IL3), T cell signature receptor molecules (IL2RA and TNFRSF9), transcription factors (MYC and STAT3), and T cell effector molecules (GZMA) to be c-Rel target genes in human Jurkat T cells (Figure 3B). Furthermore, when using Gene Set Enrichment Analysis (GSEA) to perform pathway analysis on the same data set, we found that the gene signatures of “oxidative phosphorylation” and “Myc target genes” were significantly downregulated in c-Rel KO cells, indicating defects of mitochondrial metabolism and the c-Myc pathway in the Jurkat T cells (Figures 3C and 3D). Lastly, we confirmed by RT-PCR that the expression of c-Myc, several T cell signature genes (/L2, IL2RA, TNFRSF9, CD83, PDCD1), nuclear receptors (NR4A2 and NR4A3), and a lipid transporter (SLC27A2) were downregulated in the c-Rel KO cells (Figure 3E). In silico analysis of the promoters of MYC, TNFRSF9, SLC23A2, and NR4A2 identified potential c-Rel binding sites, indicating that c-Rel might regulate these genes by binding to their promoter regions (Supplementary Figures 3A to 3D).
Figure 3. Genome-wide transcriptome analysis identifies mitochondrial metabolism and the c-Myc pathway as c-Rel targets in human Jurkat T cells.
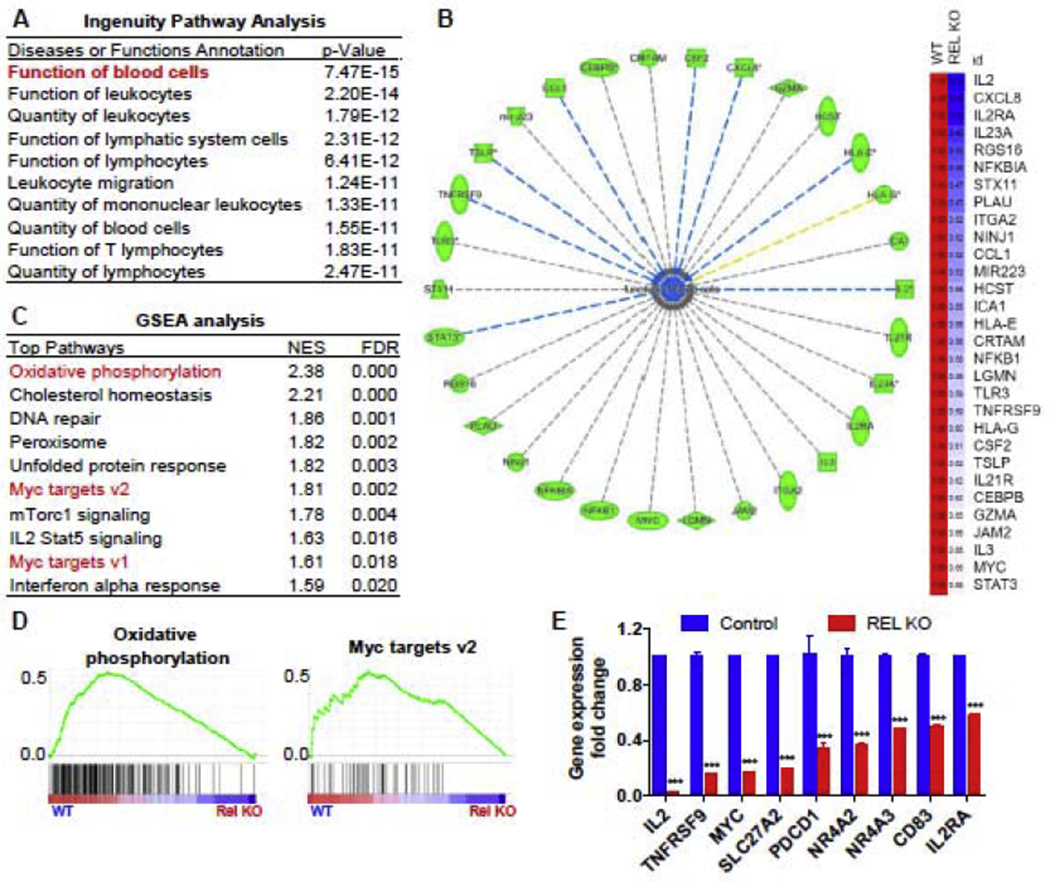
Control and c-Rel KO human Jurkat T cells (pooled clones in each group) were stimulated with PMA and ionomycin for 4 hours. Microarray (A-D) and RT-PCR (E) were performed afterwards. A. Ingenuity Pathway Analysis (IPA) showing the top reduced pathways in c-Rel KO cells. B. representative downregulated genes from the pathway “Function of blood cells” in panel A (left) and their relative gene expression levels compared with control (right). C. Gene Set Enrichment Analysis (GSEA) was performed and the top ten decreased gene signatures in c-Rel KO cells were shown. D. representative pathways from panel D are shown. E. RT-PCR confirmation of the genes that were identified from the microarray analysis (n=3 independent cultures of pooled clones). For all panels, data are expressed as mean ± SEM, and are representative of at least 2 independent experiments. ***, p<0.001.
c-Rel drives c-Myc gene expression in human Jurkat T cells.
Given the microarray results, we next tested the hypothesis that c-Rel is required for c-Myc gene expression in human Jurkat T cells, even without PMA/Ionomycin stimulation. Our results showed that the mRNA expression of MYC and its downstream target genes including SLC2A1 (also known as GLUT1; a glucose transporter), PPARGC1A (a master regulator of mitochondrial biogenesis), and TFAM (a mitochondrial transcription factor) were significantly downregulated in c-Rel KO cells under non-stimulated conditions (Figure 4A). Notably, the mRNA expression of IL2 and its receptor subunit IL2RA was not changed in c-Rel KO cells under non-stimulated conditions (Supplemental Figures 4A and 4B). In addition, the protein expression of c-Myc was also compromised in all three c-Rel KO clones (Figures 4B). Importantly, when c-Rel was reintroduced into the c-Rel KO cells (Supplemental Figures 2A to 2C), both protein and mRNA expression levels of c-Myc was restored, in the presence or absence of PMA/ionomycin stimulation, respectively (Figure 4C and 4D). To confirm that c-Rel could control c-Myc expression through transcriptional regulation at the c-Myc promoter, we employed a luciferase reporter assay. We cloned a 2kb genomic fragment containing the c-Myc promoter and inserted it upstream of a luciferase reporter gene (Supplemental Figure 5) and then tested the responsiveness of this reporter construct to c-Rel activation using control and c-Rel KO Jurkat cells. The results showed that the c-Myc promoter induced significant transcriptional activity upon PMA/ionomycin stimulation in the presence of c-Rel. However, this activity was significantly reduced in c-Rel KO cells (Figure 4E). Taken together, these results indicate that c-Rel might regulate the gene expression of c-Myc through binding to its promoter.
Figure 4. c-Rel drives c-Myc gene expression in human Jurkat T cells.

A. RT-PCR quantification of c-Myc and its target genes (SLC2A1, PPARGC1 A, and TFAM; n=3 independent cultures of pooled clones). B. Western blotting for c-Rel and c-Myc (Left panels) with quantification is shown on the right (n=3 clones). C. Western blots of c-Rel KO Jurkat cell clones transduced with GFP-expressing lentiviral control vector, or c-Rel- and GFP-expressing vector (Left panels), with quantification shown on the right (n=3 clones). D. c-Rel KO cells transduced with GFP and c-Rel KO cells transduced with c-Rel and GFP were stimulated with PMA/ionomycin for 4 hours, followed by RT-PCR quantification of c-Myc mRNA (n=3 independent cultures of the indicated clones). E. control and c-Rel KO Jurkat cells were transfected with a luciferase reporter plasmid containing either no promoter or a 2kb human c-Myc promoter, and stimulated overnight with PMA and ionomycin followed by luciferase assay (n=3 independent cultures of pooled clones). For all panels, data are expressed as mean ± SEM, and are representative of at least 3 independent experiments. *, p<0.05, **, p<0.01, ***, p<0.001.
c-Rel drives human Jurkat T cell proliferation and metabolism via c-Myc.
We next determined whether the observed c-Rel-dependent c-Myc induction was responsible for driving cell proliferation in our Jurkat cells. To test this hypothesis, we ectopically overexpressed c-Myc in c-Rel KO Jurkat T cells under the control of the UbC promoter, using a lentiviral vector that also co-expressed GFP under the control of an IRES element (Supplemental Figures 6A to 6C). The results showed that this ectopic MYC expression significantly increased c-Myc expression in c-Rel KO cells at both the protein and mRNA levels (Figure 5A and 5B). Moreover, c-Myc was rescued to a similar level to that seen in the control cells (Figure 5B). Importantly, restoring c-Myc expression partially reversed the proliferation defect of c-Rel KO cells under serum starvation (Figure 5C). Together, our results demonstrate that c-Rel controls proliferation in Jurat T cells partially via c-Myc.
FIGURE 5. c-Rel drives mitochondrial respiration and proliferation via c-Myc in human Jurkat T cells.
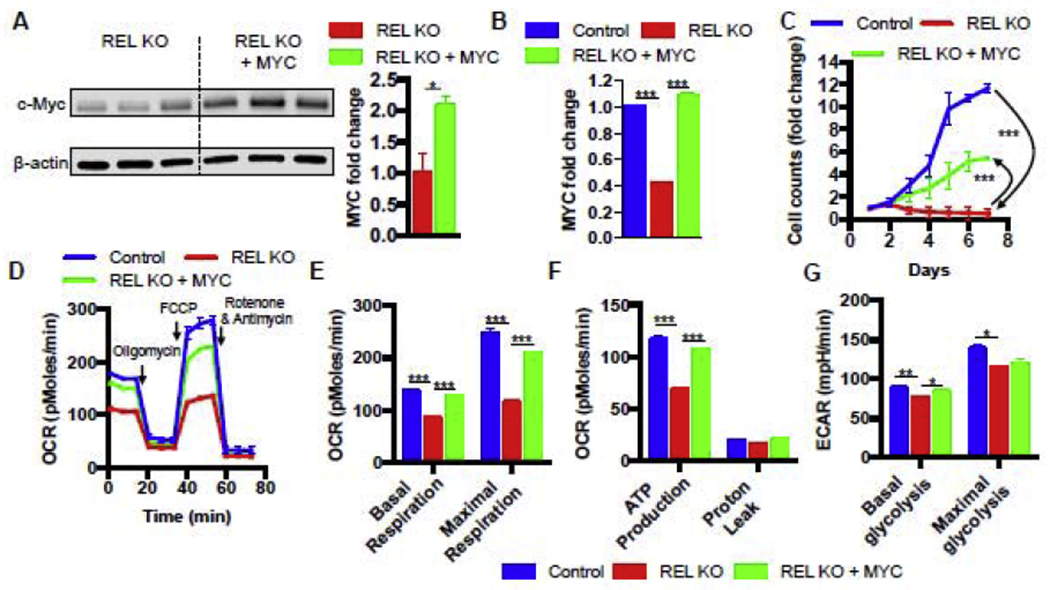
A. Western blots of c-Rel KO cells transduced with control vector or c-Myc-expressing vector with quantitation shown on the right (n=3 clones). B. RT-PCR of control Jurkat cells transduced with control vector, c-Rel KO cells transduced with control vector, and c-Rel KO cells transduced with c-Myc (n=3 independent cultures of pooled clones). C. proliferation of the indicated cells (n=3 clones). D. real-time recording of oxygen consumption rate (OCR) in a Seahorse mitochondrial stress test. Oligomycin, FCCP, Rotenone with Antimycin A were added sequentially to measure four mitochondrial parameters. E, F. quantitation of the four mitochondrial parameters from panel D (n=5 to 6 independent cultures of pooled clones). G. extracellular acidification rates (ECAR) were measured in real-time to gauge the basal and maximal glycolytic capacity using a Seahorse extracellular flux analyzer (n=6 independent cultures of pooled clones). For all panels, data are expressed as mean ± SEM, and are representative of at least 3 independent experiments. *, p<0.05, **, p<0.01, ***, p<0.001.
Since c-Myc serves as a master regulator of cellular metabolism, we hypothesized that c-Rel could control human Jurkat T cell metabolism via c-Myc regulation. Thus, we performed real-time metabolic profiling by simultaneously measuring glycolysis and mitochondrial respiration rates using a Seahorse XF extracellular flux analyzer. We found that before the electron transport chain inhibitors were added, the c-Rel KO cells showed significantly lower basal rate of oxygen consumption relative to the control cells (Figures 5D and 5E). Basal mitochondrial respiration is comprised of ATP production and proton leak. The addition of oligomycin (a mitochondrial ATP synthase inhibitor) suppressed oxygen consumption of both the c-Rel KO cells and the control cells to comparable levels, suggesting c-Rel KO cells have a defect in ATP production, but not proton leak (Figures 5D and 5F). After carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP) was added to uncouple electron transport from oxidative phosphorylation, allowing for maximal oxygen consumption, the control cells increased their oxygen consumption to a level significantly above the basal rate. However, the c-Rel KO cells merely increased their oxygen consumption to the basal level. These data revealed that c-Rel KO cells not only have reduced basal rates of mitochondrial respiration but also have significantly reduced maximal respiration (Figures 5D and 5E). Interestingly, overexpression of c-Myc in c-Rel KO cells almost completely rescued the mitochondrial respiration deficiency, indicating that c-Myc is the downstream target of c-Rel that controls mitochondrial metabolism. By contrast, c-Rel KO cells showed mild decrease of both basal and maximal glycolysis when compared with the control cells, while overexpression of c-Myc in c-Rel KO cells partially rescued the basal glycolysis defect (Figures 5G). Taken together, these results suggest that c-Rel controls human Jurkat T cell metabolism through c-Myc.
Discussion
Our data demonstrated that c-Rel is a critical factor for the proliferation and survival of Jurkat cells, and that a significant portion of the mechanism involves transcriptional upregulation of the proto-oncogene protein c-Myc. c-Myc has been proposed to function as a universal amplifier of already expressed genes in lymphocytes and tumor cells[11, 12]. Chromatin immunoprecipitation coupled to high-throughput sequencing (ChIP-Seq) studies suggest that instead of serving as an on-off switch for a new set of genes, c-Myc accumulates in the promoter regions of active genes and enhances the level of transcription within the cell’s existing gene expression program. This mode of function implies that a pre-established permissive chromatin environment is required at c-Myc target promoters. Interestingly, c-Rel has been shown to drive chromatin remodeling and establish a permissive chromatin environment at c-Rel-target promoters[13, 14]. Having confirmed that c-Myc itself is also a target of c-Rel, it is conceivable that their combined actions could form a synergistic mechanism to drive strong induction of their common targets.
It is possible that the stimuli used in this study, i.e., PMA and Ionomycin, might bypass TCR-induced signals seen under physiological conditions, as previously reported for primary murine T cells [15]. However, as we reported, our REL KO Jurkat T cells used in this study do produce significantly less IL-2 mRNA transcripts than WT control cells following activation with anti-CD3 and anti-CD28 [8]. Thus, in human Jurkat leukemia T cells, c-Rel controls IL-2 mRNA expression induced by either TCR or PMA/Ionomycin stimulation.
It has been reported that c-Myc-dependent growth during T cell activation requires c-Rel [16]. It was also previously shown that the proliferation defect of c-Rel knockout murine T cells is caused by a lack of IL-2 production upon T-cell receptor (TCR) activation[17]. It is thus conceivable that the growth and cellular metabolic defects observed in c-Rel KO Jurkat T cells might be due to impaired IL-2 signaling. However, in the proliferation assays and metabolic profiling assays reported here, control and c-Rel KO cells were not stimulated by PMA/Ionomycin. Under such conditions, we found that there were no differences in IL-2 or its receptor IL2RA expression between control and REL KO cells (Supplementary Figure 4A and 4B). Thus, we believe that it is the impaired c-Myc gene expression, rather than downregulated IL-2 signaling that contributed to the defects of spontaneous growth and mitochondrial respiration in c-Rel KO cells under non-stimulated condition. In other word, our data demonstrate that in cancerous human T cells, c-Rel may also control the intrinsic ability of these cells to survive and proliferate independent of IL-2 signaling. Future study is warranted to expand the current study on tumor cells to determine the relative contribution of IL-2 and c-Myc in mediating c-Rel’s effect on cellular metabolism and proliferation in primary human T cells.
Notably, while c-Rel KO Jurkat cells share many similarities with conditional c-Myc KO cells, such as compromised glycolysis as well as reduced cell growth and proliferation[18]. c-Rel and c-Myc are not complete substitutes for one another. For example, c-Myc overexpression in c-Rel KO cells almost fully rescued the defect in mitochondrial respiration but only partially reversed the proliferation defect. Based on our results we propose the following model: In human Jurkat T cells, c-Rel activates many target genes, including MYC. The increase in c-Myc, in turn, reinforces the gene expression shaped by c-Rel. In other words, c-Rel upregulates c-Myc to amplify many of c-Rel’s own targets. Without the presence of c-Rel, c-Myc alone is insufficient to boost the expression of these genes to the required levels, resulting in only a partial rescue. Thus, c-Rel and c-Myc work in a synergistic manner to drive cellular metabolism and promote cell proliferation (Supplemental Figure 7).
The reliance of cancer growth on altered metabolic pathways such as aerobic glycolysis, glutaminolysis, and fatty acid synthesis provides the basis for attacking cancer cells through metabolic interference. Targeting cancer metabolism has therefore become an area of intense interest and research focus. There has been some promising evidence supporting efficacy of this approach[19]. Given the critical importance of c-Rel in driving metabolic reprogramming to facilitate cell growth and proliferation under nutrient-deprived conditions, c-Rel could be a promising target for novel metabolism-based treatment strategies. Indeed, one study has shown that integrin β3-expressing human pancreatic carcinoma cells are resistant to EFGR tyrosine kinase inhibitor erlotinib and nutrient deprivation. However, this resistance can be overcome through inhibition of c-Rel using RNAi, and a combination of c-Rel shRNA and erlotinib successfully blocked tumor cell growth[20]. Therefore, similar approaches that interfere with mitochondrial respiration through c-Rel inhibition represent a potentially important avenue for developing novel combination cancer therapies.
Materials and Methods
Cells
Human Jurkat cells were obtained from ATCC and grown in RPMI 1640 supplemented with 10% or 2% fetal bovine serum (Hyclone), 2mM L-Glutamine (Gibco, Grand Island, NY), and penicillin/streptomycin (Sigma-Aldrich, St. Louis, MO).
Plasmid vectors
pCW-Cas9 and pLX-sgRNA were a gift from Eric Lander & David Sabatini (Addgene plasmid # 50661 & 50662). Lentiviral vectors used for c-Rel rescue and c-Myc overexpression were constructed based on the FUGW vector from the David Baltimore lab. pGL4.12[luc2CP] luciferase reporter vector was purchased from Promega (Madison, Wisconsin, USA).
Plasmid construction
pLX-sgRNA vectors
sgRNA sequences were ligated to U6 promoter sequence and sgRNA scaffold sequence using overlap-extension PCR, followed by restriction and ligation using the Xhol and Nhel restriction sites in the pLX-sgRNA plasmid. The sgRNA sequences used to generate the final c-Rel KO clones are: 1) AAAACGCATTCCCCTCTGCC (52 bp from 5’ in the reverse strand); 2) TAGAGATAATTGAACAACCC (45 bp from 5’ in the forward strand); and 3) TAATTGAACAACCCAGGCAG (51 bp from 5’ in the forward strand).
c-Rel and c-Myc rescue vectors
The backbone vector was first created by inserting an IRES element and the GFP coding sequence into the FUGW vector. Fluman c-Rel and c-Myc cDNA was then cloned into the vector using the following primer sets: 1) c-Rel forward primer: ATGGCCTCCGGTGCGTATAACCCG, c-Rel reverse primer: TTATACTTGAAAAAATTCATATGGAAAGGAG; 2) c-Myc forward primer: CTGGATTTTTTTCGGGTAGTGGAAAACC, c-Myc reverse primer: TTACGCACAAGAGTTCCGTAGCTG.
c-Myc promoter reporter vector
A 2kb genomic fragment containing the 5′ UTR and the proximal promoter of the c-Myc gene were amplified by PCR from human genomic DNA, and subcloned into the Hindi I l/Ncol sites of the pGL4.12[luc2P] luciferase reporter plasmid using the following primer set: forward primer: GGCCAAGCTTAAAAGGCACGGAAGTAATACTCCTCTCCTC; reverse primer: TCTTCCATGGTCTAAGCAGCTGCAAGGAGAGCCTTTC.
Lentivirus production and cell transduction
Lentiviruses used for generating the c-Rel KO, c-Rel KO c-Rel rescue, c-Rel KO c-Myc rescue, and the control cells were produced by co-transfecting the respective vectors with the 3rd generation packaging system into 80% confluent 293T cells using the FuGENE 6 (Promega, Madison, WI) transfection reagent according to the manufacturer’s instructions. Virus-containing supernatants were collected and filtered 36-48 h after transfection, and were either used immediately or stored at −80°C.
Jurkat cells were transduced in viral supernatants containing 10μg/mL polybrene at a density of 2x105 cells/mL, and were centrifuged under 3000 rpm, 30°C for 90 min, followed by replacement of fresh culture medium.
Generation of c-Rel KO cells using the CRISPR/Cas9 system
c-Rel KO Jurkat cells were generated using the CRISPR/Cas9 system described by Wang et al. The first vector (pCW-CAS9) carries the Cas9 endonuclease under the control of Tet-On tetracycline inducible expression system, as well as a constitutively expressed puromycin resistance gene. The second vector (pLX-SgRNA) carries a sgRNA driven by the U6 RNA polymerase III promoter, as well as a blasticidin resistance gene. 8 candidate sgRNA target sequences were generated based on complementarity to the N-terminus region of the c-Rel coding sequence. These target sequences were then fused to the sgRNA scaffold sequence and cloned into the pLX-sgRNA vector using the Xhol and Nhel restriction sites. The resulting vectors were used to produce lentiviruses in 293T cells by co-transfection with the 3rd generation packaging system. Jurkat cells were first transduced by lentivirus produced from the doxycycline-inducible Cas9 vector (pCW-Cas9) and selected under 1.5μg/mL puromycin for 3 days. The resulting cells were expanded and transduced by the c-Rel-targeting pLX-sgRNA lentiviruses and subsequently selected under 6pg/mL blasticidin in the presence of 750ng/mL doxycycline for one week. The cells were then expanded without doxycycline until sufficient numbers were obtained. c-Rel protein levels were analyzed by Western blotting.
Clonal selection to establish pure c-Rel KO lines
Jurkat cells were serial diluted to 0.5 cells / 200μL / well in 96-well plates and grew until density reaches confluency. The cells were then transferred to 12-well plates and expanded until sufficient numbers were obtained. c-Rel protein levels were analyzed by Western blotting.
PMA/lonomycin stimulation of Jurkat cells
1x106/mL Jurkat cells were stimulated in standard culture medium containing 10% fetal bovine serum with the addition of 10ng/mL PMA and 1 μM ionomycin for 4 hours before analyzed.
Western blotting
Cells were lysed in SDS and total protein concentration determined. 30 pg protein was loaded to each lane and separated by SDS-PAGE. After transferring to a nitrocellulose membrane, it was blocked with 5% milk in TBST and probed with the following primary antibodies, overnight at 4°C: c-Rel (#4727, rabbit polyclonal, 1:2000, Cell signaling), c-Myc(#9402, rabbit monoclonal, 1:1000, Cell Signaling), β-Actin (1:10000, mouse monoclonal, Sigma). HRP-conjugated secondary antibodies (anti-mouse or anti-rabbit IgG, 1:1000, GE healthcare) were incubated at room temperature for 2 hours. Detection was done using Amersham enhanced chemiluminescence (ECL).
Cell sorting by flow cytometry
Flow cytometric sorting was performed on Jurkat cells transduced with vectors carrying a GFP marker. Cells were analyzed on a BD FACS Aria II sorter. Data were analyzed with the FlowJo software.
Quantitative RT-PCR
Total RNA was extracted with RNeasy (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Reverse transcription was performed with oligo dT primers. RT-PCR was carried out in an Applied Biosystems 7500 system with Power SYBR Green PCR Master Mix (Applied Biosystems). Relative levels of gene expression were determined with GAPDH as the control. The primers used in the experiments are: 1) GAPDH-forward primer(F): GAGTCAACGGATTTGGTCGT, GAPDH-reverse primer (R): TTGATTTTGGAGGGATCTCG; 2) IL2-F: GTCACAAACAGTGCACCTAC, IL2-R: CCCTGGGTCTTAAGTGAAAG; 3) IL2RA-F: CAAGGGTCAGGAAGATGGATTC, IL2RA-R: CTTTGAATGTGGCGTGTGG; 4) TNFRSF9-F: ACTGCGTTGCTCTTCCTG, TNFRSF9-R: CAGCCATCTTCCTCTTGAGTAG; 5) CD83-F: TCCCTACACGGTCTCCTG, CD83-R: CGAAAGAACCATTTTGCCCC; 6) PDCD1-F: ATGCAGATCCCACAGGCGCC, PDCD1-R: TCAGAGGGGCCAAGAGCAGTG; 7) REL-F: GCAGGAATCAATCCATTCAA, REL-R: CAGGAGGAAGAGCAGTCGTC; 8) MYC-F: TTCGGGTAGTGGAAAACCAG, MYC-R: AGTAGAAATACGGCTGCACC; 9) SLC2A1-F: AAAGTGACAAGACACCCGAG, SLC2A1-R: TGTCAGGTTTGGAAGTCTCATC; 10) NR4A2-F: TCCTCGAAAACGCCTGTAAC, NR4A2-R:TGGAGTTAAGAAATCGGAGCTG; 11) NR4A3-F: AGTGTCTCAGTGTTGGAATGG, NR4A3-R: AGGAGAAGGTGGAGAGGG; 12) SLC27A2-F:CGAGAAAAGTTGGTGCTGTTG, SLC27A2-R: TGGGAACTCTGACGCAATATC; 13) PPARGC1A-F: ACCAAACCCACAGAGAACAG, PPARGC1A-R:GGTCAGAGGAAGAGATAAAGTTG; and 14) TFAM-F:GATTGAAGTTGGACGAAAGGATC, TFAM-R: CGATAGCTTCAGGTATTGAGACC.
In silico analysis of c-Rel binding sites
The promoter regions of targeted genes, defined as 1,500 base pairs upstream of the transcription start site, were retrieved from the NIH/NCBI Entrez Gene database (http://www.ncbi.nlm.nih.gov/gene). The promoter regions were then analyzed with PROMO (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3) to identify potential c-Rel binding sites.
Luciferase reporter assay
Control and c-Rel KO Jurkat cells were transfected with either promoter-less control plasmid or pGL4.12[luc2CP] c-Myc reporter plasmid by electroporation using a Gene Pulser II electroporator (Bio-Rad, Hercules, CA) at a concentration of 20μg/5x106 cells/mL. Cells were rested for 4 hours and stimulated with 10ng/ml PMA and 1pM ionomycin. 12 hours after stimulation, cells were lysed, and luciferase activities were measured using a luciferase assay kit (Promega, Madison, WI) and a GloMax 20/20 luminometer (Promega), according to the manufacturer’s protocols.
Seahorse metabolic analysis
Cells were harvested and resuspended at 5x106 cells/mL in appropriate Seahorse media. 40μL of cells were added to 140μL of appropriate seahorse media and plated in XF96 cell culture microplates that were precoated with 25μL of Cell tak reagent overnight according to manufacturer’s instructions (Corning Inc., Corning, NY, #354240). For examining mitochondrial function, a mito-stress kit was used with additions of the following reagents to cells in appropriate Seahorse media: 1 μM Oligomycin, 1μM fluoro-carbonyl cyanide phenylhydrazone (FCCP, an uncoupler of respiration and oxidative ATP synthesis), and pre-mixed 0.5 μM rotenone + antimycin A solution (Electron Transport Chain complex I and III inhibitors). Maximum respiratory capacity was calculated as the difference between the oxygen consumption rate (OCR) after addition of FCCP and the OCR after addition of rotenone/antimycin A. Spare respiratory capacity was calculated as the difference between maximal (post FCCP) and basal respiration. Plates were run on a Seahorse XF96 Extracellular Flux Analyzer according to the manufacturer’s protocols.
Statistical analyses
All statistics were performed using Prism 5 (GraphPad, La Jolla, CA). Data were expressed as mean ± standard error of mean (SEM) throughout the manuscript. For comparisons between two groups, two-tailed Studenťs t-test was used for evaluation of statistical significance. For comparisons across multiple groups, one-way ANOVA with Bonferroni post-test adjustment was used. Data shown are representative of two to three independent experiments. NS, not significant; *, p<0.05; **, p<0.01; ***, p<0.001.
Supplementary Material
Highlights.
REL knockout cells exhibited significant defects in cell growth and mitochondrial respiration.
Genome-wide transcriptome analyses revealed that T cells lacking REL had selective defects in the expression of inflammatory and metabolic genes including c-Myc.
c-Rel directly controls the expression of c-Myc through its promotor and expressing c-Myc in c-Rel-deficient lymphoma cells rescued their proliferative and metabolic defects.
The human c-Rel-c-Myc axis controls lymphoma growth and metabolism and could be a therapeutic target for lymphoma.
Acknowledgements:
We thank University of Pennsylvania Pancreatic Islet Cell Biology Core, Flow Cytometry & Cell Sorting Core, and Dr. Daniel P. Beiting from Penn Vet for technical support.
Funding: This work was supported in part by grants from the National Institutes of Health (NIH), USA (R01-AI099216, R01-AI121166, R01-AI136945 to Y.H.C.); X.L. was partially supported by NIH-T32-DK007780, J.R.G was supported by NIH-F32-DK116528.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: YHC is an inventor of the following patent: Chen, Y. H., R. Murali, J. Sun: Rel inhibitors and methods of use thereof. USA Patent Number US8609730B2, 2013. YHC is a member of the advisory boards of Amshenn Co., and Binde Co.
References
- 1.Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vazquez G, Yurchenko E, Raissi TC, van der Windt GJ, Viollet B, Pearce EL et al. : The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 2015, 42(1):41–54. [DOI] [PubMed] [Google Scholar]
- 2.Palm W, Thompson CB: Nutrient acquisition strategies of mammalian cells. Nature 2017, 546(7657):234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pavlova NN, Thompson CB: The Emerging Hallmarks of Cancer Metabolism. CellMetab 2016, 23(1):27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hayden MS, Ghosh S: Shared principles in NF-kappaB signaling. Cell 2008, 132(3):344–362. [DOI] [PubMed] [Google Scholar]
- 5.DiDonato JA, Mercurio F, Karin M: NF-kappaB and the link between inflammation and cancer. Immunol Rev 2012, 246(1):379–400. [DOI] [PubMed] [Google Scholar]
- 6.Ruan Q, Kameswaran V, Zhang Y, Zheng S, Sun J, Wang J, DeVirgiliis J, Liou HC, Beg AA, Chen YH: The Th17 immune response is controlled by the Rel-RORgamma-RORgamma T transcriptional axis. J Exp Med 2011, 208(11):2321–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruan Q, Kameswaran V, Tone Y, Li L, Liou HC, Greene MI, Tone M, Chen YH:Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity 2009, 31(6):932–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li T, Li X, Zamani A, Wang W, Lee C- N, Li M, Luo G, Eiler E, Sun H, Ghosh S: c-Rel is a myeloid checkpoint for cancer immunotherapy. Nature Cancer 2020:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilmore TD, Gerondakis S: The c-Rel Transcription Factor in Development and Disease. Genes Cancer 2011, 2(7):695–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Enciso-Mora V, Broderick P, Ma Y, Jarrett RF, Hjalgrim H, Hemminki K, van den Berg A, Olver B, Lloyd A, Dobbins SE et al. : A genome-wide association study of Hodgkin’s lymphoma identifies new susceptibility loci at 2p16.1 (REL), 8q24.21 and 10p14 (GATA3). Nat Genet 2010, 42(12):1126–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R et al. : c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 2012, 151(1):68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA: Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151(1):56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rao S, Gerondakis S, Woltring D, Shannon MF: c-Rel is required for chromatin remodeling across the IL-2 gene promoter. J Immunol 2003, 170(7):3724–3731. [DOI] [PubMed] [Google Scholar]
- 14.van Essen D, Zhu Y, Saccani S: A feed-forward circuit controlling inducible NF-kappaB target gene activation by promoter histone demethylation. Mol Cell 2010, 39(5):750–760. [DOI] [PubMed] [Google Scholar]
- 15.Kontgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D, Gerondakis S: Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev 1995, 9(16):1965–1977. [DOI] [PubMed] [Google Scholar]
- 16.Grumont R, Lock P, Mollinari M, Shannon FM, Moore A, Gerondakis S: The mitogen-induced increase in T cell size involves PKC and NFAT activation of Rel/NF-kappaB-dependent c-myc expression. Immunity 2004, 21(1):19–30. [DOI] [PubMed] [Google Scholar]
- 17.Liou HC, Jin Z, Tumang J, Andjelic S, Smith KA, Liou ML: c-Rel is crucial for lymphocyte proliferation but dispensable for T cell effector function. Int Immunol 1999, 11(3):361–371. [DOI] [PubMed] [Google Scholar]
- 18.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J et al. : The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35(6):871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG, Sotgia F, Lisanti MP: Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol 2017, 14(1):11–31. [DOI] [PubMed] [Google Scholar]
- 20.Seguin L, Kato S, Franovic A, Camargo MF, Lesperance J, Elliott KC, Yebra M, Mielgo A, Lowy AM, Husain H et al. : An integrin beta(3)-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat Cell Biol 2014, 16(5):457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.