

Abstract
Notch 1 through 4 are transmembrane receptors that play a pivotal role in cell differentiation and function; this review addresses the role of Notch signaling in osteoclastogenesis and bone resorption. Notch receptors are activated following interactions with their ligands of the Jagged and Delta-like families. In the skeleton, Notch signaling controls osteoclast differentiation and bone-resorbing activity either directly acting on osteoclast precursors, or indirectly acting on cells of the osteoblast lineage and cells of the immune system. NOTCH1 inhibits osteoclastogenesis, whereas NOTCH2 enhances osteoclast differentiation and function by direct and indirect mechanisms. NOTCH3 induces the expression of RANKL in osteoblasts and osteocytes and as a result induces osteoclast differentiation. There is limited expression of NOTCH4 in skeletal cells. Selected congenital disorders and skeletal malignancies are associated with dysregulated Notch signaling and enhanced bone resorption. In conclusion, Notch signaling is a critical pathway that controls osteoblast and osteoclast differentiation and function and regulates skeletal homeostasis in health and disease.
Keywords: Notch, Jagged, tumor necrosis factor α, bone remodeling, inflammation, osteoclast, bone resorption
1. Introduction
Bone tissue continuously undergoes remodeling through the coordinated activity of cells of the osteoblast and the osteoclast lineages. A variety of molecules and signaling pathways control osteoclast differentiation and function either directly acting on osteoclast precursors or indirectly acting on osteoblasts, osteocytes and cells of the immune system that express osteoclastogenic factors under physiological conditions and in various disease states [1–5]. Notch signaling plays a critical role in cell fate determination and function and in the regulation of skeletal homeostasis [6–8]. Previous reviews on Notch signaling have described the function of Notch signaling in skeletal and non-skeletal cells, but have not addressed the specific function of each Notch receptor, an in particular the role of the receptors in osteoclastogenesis under physiological conditions and during inflammation. This is relevant since recent work has demonstrated distinct actions of each Notch receptor in bone remodeling and osteoclastogenesis. This review highlights recent insights into the role of Notch signaling in the regulation of osteoclastogenesis and bone resorption in physiological and selected pathological conditions affecting the skeleton.
2. Overview of Osteoclastogenesis
2.1. Regulation of Osteoclastogenesis
Osteoclasts are giant multinucleated cells that are responsible for bone resorption. Osteoclasts are derived from the differentiation and cell-cell fusion of myeloid progenitor cells that also have the potential to differentiate into monocytes, granulocytes and macrophages [1–4, 9, 10].
Macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor-κB (NF-κB) ligand (RANKL) are indispensable cytokines for osteoclast differentiation and survival. M-CSF and RANKL are produced by osteoblasts, osteocytes, stromal cells, and by cells of the immune system [1–4, 11]. M-CSF is secreted under physiological conditions, and it is not secreted by macrophages. M-CSF triggers downstream signaling through its receptor, CSF-1 receptor (CSF-1R), and promotes the differentiation of myeloid progenitors into osteoclast precursors by upregulating RANK expression. M-CSF signaling also is essential for cytoskeleton re-arrangement and for the survival of osteoclasts. RANKL-RANK signaling plays a critical role in osteoclast differentiation and bone-resorbing activity. RANKL binds to RANK, and facilitates the activation of the IκB kinase complex (IKK), the phosphatidylinositol 3-kinase (PI3K)-AKT signaling pathway, and mitogen-activated kinases (MAPKs) including p38, ERK and c-Jun N-terminal kinase (JNK) [5, 12]. This leads to the activation of the transcription factors NF-κB, c-Fos and c-Jun, which are required for the induction of nuclear factor of activated T cells c1 (NFATc1) at an early stage of osteoclastogenesis. NFATc1 is a transcription factor that is self-induced in a process of auto-amplification during osteoclastogenesis [12–16]. NFATc1 promotes the expression of genes required for osteoclast differentiation and function, such as tartrate-resistant acid phosphatase (TRAP), dendritic cell-specific transmembrane protein (DC-STAMP), v-ATPase V0 subunit d2 (ATP6V0D2), osteoclast-associated receptor (OSCAR), αvβ3 integrin receptor, and cathepsin K [17–28].
There are transcriptional repressors that act as ‘brakes’ of RANKL signaling and are constitutively expressed in osteoclast precursors. For osteoclastogenesis to proceed, RANKL needs to overcome the activity of the transcriptional repressors, including that of inhibitor of differentiation and DNA binding (IDs), v-maf musculoaponeurotic fibrosarcoma oncogene family, protein B (MAFB), interferon regulatory factor 8 (IRF8) and B cell lymphoma 6 (BCL6) [29–32]. These transcriptional repressors modulate the expression or the transcriptional activity of NFATc1. The constitutive expression of transcriptional repressors is downregulated at an early stage of osteoclastogenesis following the induction of B-lymphocyte-induced maturation protein-1 (BLIMP-1) by the actions of RANKL. The activity of RANKL is opposed by osteoprotegerin (OPG), which acts as a decoy receptor of RANKL decreasing its interaction with RANK [33–35] (Figure 1).
Figure 1.
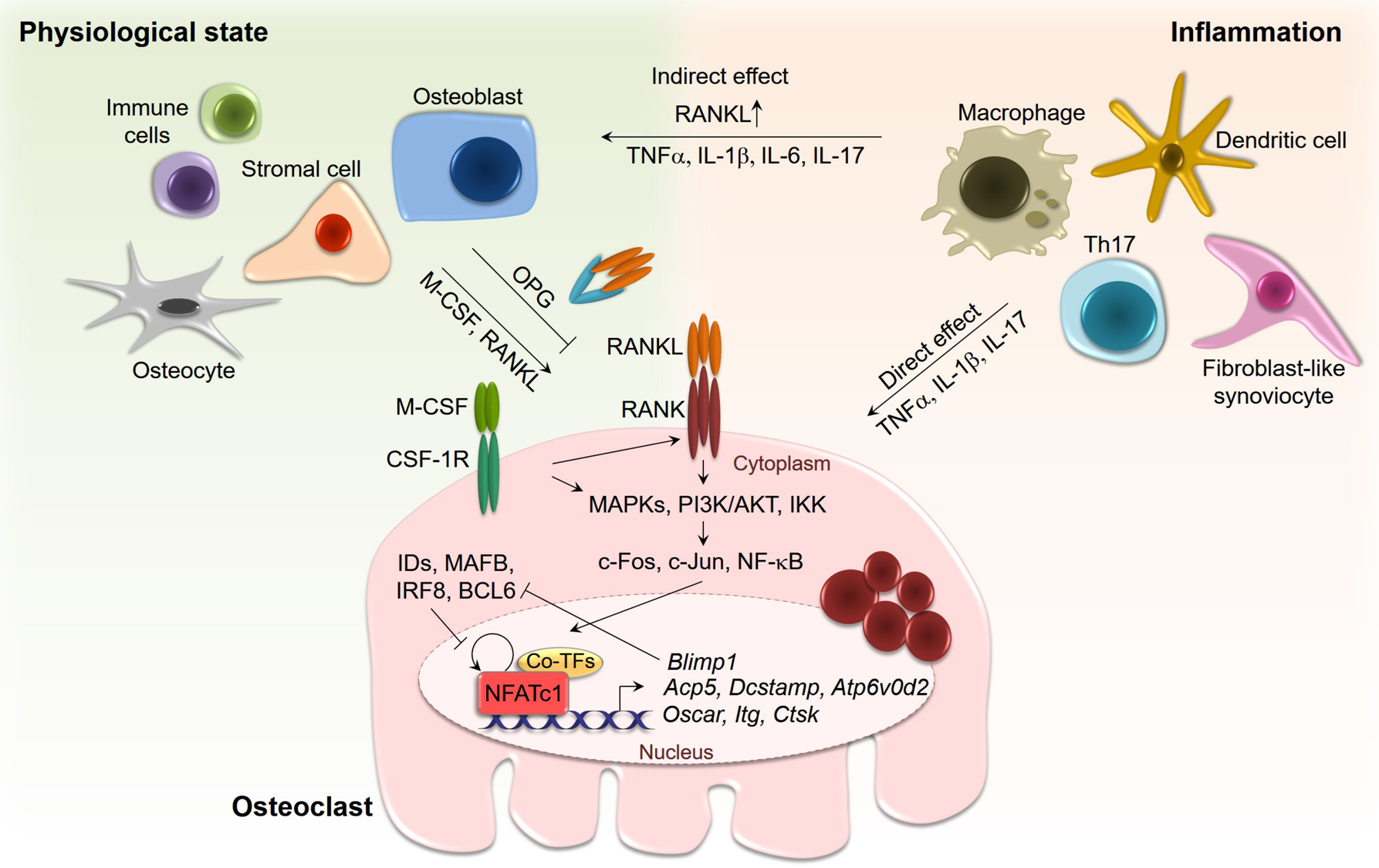
Regulation of osteoclastogenesis. M-CSF and RANKL, synthesized by osteoblasts, osteocytes, stromal cells and cells of the immune system, trigger osteoclast differentiation via NFATc1 induction. NFATc1 promotes gene expression required for osteoclast differentiation and function including Acp5 (encoding TRAP), Dcstamp, Atp6v0d2, Oscar, Itg (encoding integrin, αv and β3) and Ctsk (encoding cathepsin K). NFATc1 induces Blimp1 which downregulates transcriptional repressors of NFATc1. During inflammation, macrophages, dendritic cells, Th17 cells and synoviocytes secrete the proinflammatory and osteoclastogenic cytokines TNFα, IL-1β, IL-6, and IL-17 and these stimulate osteoclastogenesis directly and by inducing RANKL in osteoblasts.
2.2. Osteoclastogenesis and Inflammation
Osteoclast differentiation is excessively triggered under certain inflammatory conditions, such as infection, autoimmune disorders affecting the skeleton and fractures. Cells of the immune system, including macrophages, dendritic cells and T cells, as well as cells not forming part of the immune system, such as fibroblast-like synoviocytes secrete pro-inflammatory cytokines, including tumor necrosis factor α (TNFα), interleukin (IL)-1β, IL-6, and IL-17. These cytokines induce osteoclast differentiation by upregulating RANKL expression in osteoblasts and osteocytes, and through direct effects on osteoclast precursors that are either independent or dependent of RANKL signaling [3, 36, 37] (Figure 1).
TNFα is the main osteoclastogenic cytokine responsible for bone resorption during inflammation and in disease states characterized by an inflammatory process. TNFα induces the expression of IL-1β, IL-6 and TNFα itself. It can promote osteoclastogenesis independent of RANKL, although this has been an issue of controversy [38–41]. IL-1β alone does not induce osteoclast differentiation from osteoclast precursors although it has synergistic effects with RANKL and TNFα in the late stages of osteoclastogenesis and on bone-resorbing activity. As a result, IL-1β can cause bone destruction in a variety of diseases with an inflammatory component and affecting the skeleton [42–45]. IL-6 inhibits RANKL signaling in osteoclast precursors, however it increases osteoclastogenesis indirectly by upregulating RANKL production in osteoblasts [46, 47]. IL-17 enhances the sensitivity of osteoclast precursors to RANKL by augmenting RANK expression, and increases the expression of proinflammatory cytokines including TNFα, IL-1β and IL-6 [48–52].
3. Overview of Notch Signaling
3.1. Notch Receptors and Ligands
Notch receptors are single-pass type I transmembrane proteins that play a pivotal role in cell fate determination and function in a variety of cell lineages [6, 7]. In mammals, there are four Notch receptors (Notch1 through 4) and five classic ligands termed Jagged (JAG)1 and JAG2, and Delta-like (DLL)1, DLL3 and DLL4. Notch ligands also are transmembrane proteins and, when present in cells adjacent to those expressing Notch, their interactions with Notch result in signal activation. As a result, the Notch signaling pathway serves as a means of communication between neighboring cells.
Notch receptors have a complex structure; their extracellular domain consists of 29 to 36 epidermal growth factor (EGF) -like repeats and EGF repeats 11 and 12 interact with Notch ligands, although other EGF-like repeats modulate this interaction [53–55]. Notch receptors are cleaved (S1 cleavage) in the trans-Golgi network by a furin-like protease prior to their integration into the cell membrane as heterodimers [56] (Figure 2, top panel). At the junction of the extracellular and the transmembrane domain (TMD) rests the negative regulatory region (NRR), which consists of three Lin12-Notch repeats (LNR) that surround and protect the heterodimerization domain (HD) (Figure 2, lower panel). This is the site of cleavage (S2) required for Notch activation, and as a consequence plays a critical regulatory role in Notch signaling [57–60]. The intracellular domain of Notch (NICD) consists of a recombination signal-binding protein for Ig of κ region (RBPJκ)-association module (RAM) domain, nuclear localization sequences (NLS), and seven ankyrin (ANK) repeats; these domains are required to regulate transcription [61]. The C-terminus of Notch contains a proline (P)-, glutamic acid (E)-, serine (S)- and threonine (T)-rich (PEST) domain, which is the target of E3 ubiquitin ligases necessary for the proteasomal degradation of Notch [7, 62] (Figure 2).
Figure 2.
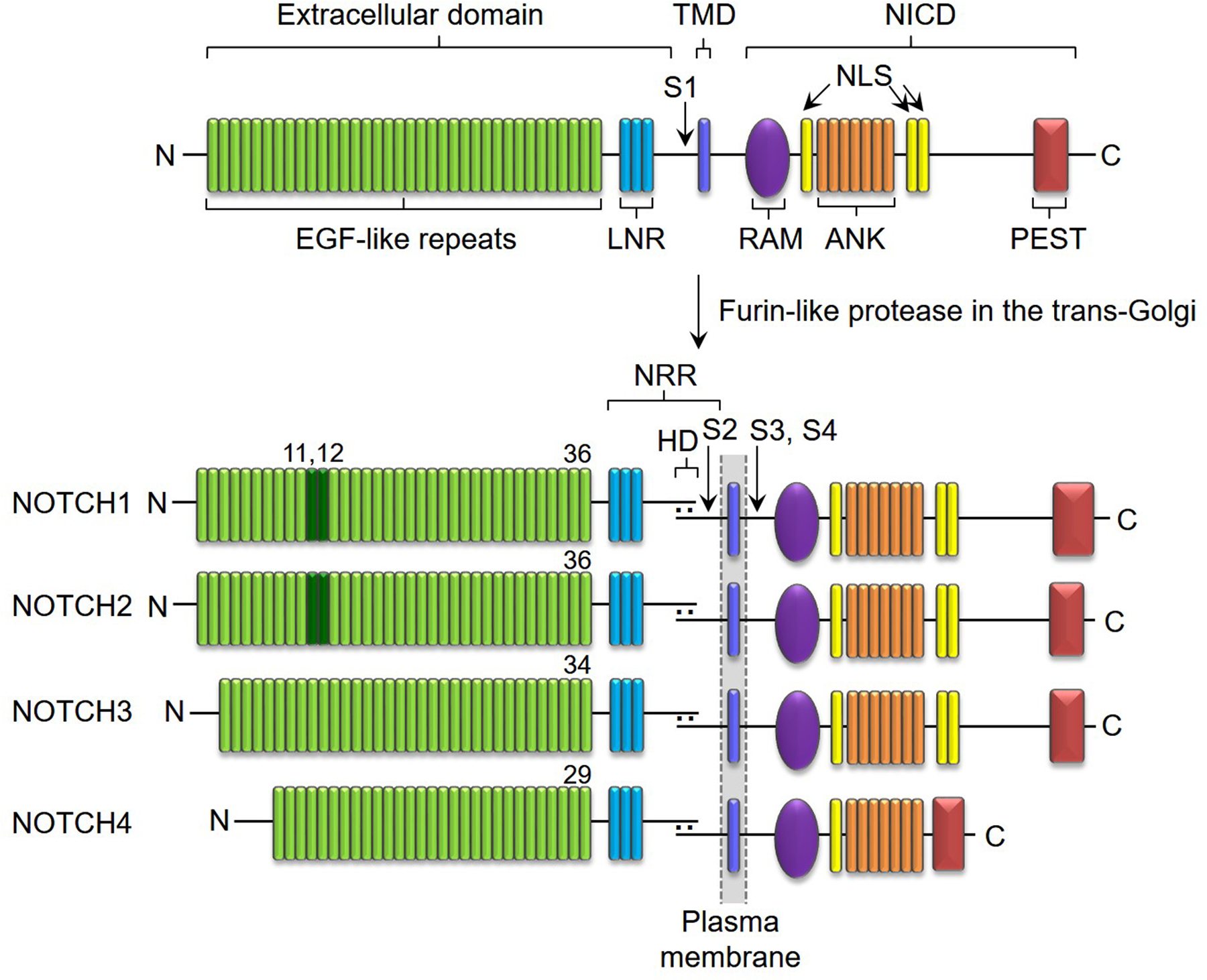
Domain structure of Notch receptors. Notch consists of an extracellular domain, TMD and NICD. The extracellular domain contains EGF-like repeats and the NRR. The NICD comprises RAM, NLS, ANK and PEST domains. Notch receptors are cleaved by a furin-like protease (S1) in the trans-Golgi network to form a heterodimer at the cell membrane. The NRR consists of LNR repeats and the HD with a cleavage site for ADAMs (S2). Cleavage sites for the γ-secretase complex are in the TMD (S3, S4). NOTCH1 and NOTCH2 have 36 EGF-like repeats; dark green (EGF11, 12) indicate the binding site for ligands. NOTCH3 has 34 and NOTCH4 has 29 EGF-like repeats. NOTCH1 and NOTCH2 have similar NICD. Reproduced in part with permission from Zanotti and Canalis Endocrine Reviews 37:223–253, 2016.
The structure and downstream signal transduction of Notch receptors are highly conserved; however, each Notch receptor has a distinct expression pattern and function, as demonstrated in studies of Notch gene inactivation in vivo. Whereas Notch1 null mice die during development due to widespread cellular death and hypomorphic Notch2 alleles cause perinatal death due to vascular and renal defects, Notch3 and Notch4 null mice develop normally and mutant adults are viable and fertile although Notch3 null mice have modest vascular alterations [63–67]. In the skeleton, NOTCH1, NOTCH2 and NOTCH3 and low levels of NOTCH4 are detected, and the most prevalent Notch ligand in skeletal cells is JAG1 [8, 68]. NOTCH 1 and NOTCH2 are expressed by osteoblasts, osteocytes and osteoclasts, whereas NOTCH3 is expressed by osteoblasts and osteocytes, and not by osteoclasts.
3.2. Activation and De-activation of Notch Signaling
Interactions of Notch with a Notch ligand present in an adjacent cell result in the endocytosis of the ligand and a pulling or hinge-like effect that unravels the NRR leaving the HD exposed to be cleaved by disintegrin and metalloprotease domain-containing proteins (ADAMs) (S2 cleavage) and the subsequent cleavage at the transmembrane domain by the γ-secretase complex (S3 and S4 cleavage) [69–71]. The “adjacent” cell expressing the Notch ligand required to activate Notch can be a skeletal cell or a cell in the bone marrow environment, and Notch activation by JAG1 in osteoblasts can regulate the hematopoietic stem cell niche [72]. Following endocytosis, the Notch ligand is recycled back to the cell surface, a process that involves ubiquitination by two E3 ubiquitin ligases [73–76]. Interactions of Notch with its ligands depend on a number of factors, including whether the ligand and Notch are in the same cell (cis) or in different adjacent cells (trans); cis interactions result in an inhibitory effect whereas trans interactions result in activation of Notch signaling. What determines or modulates the initial interaction of Notch with its ligand is not clear although does not seem to involve signals determining osteoclastogenesis, such as RANKL. Post-translational modifications of the Notch extracellular domain result in differential regulation of its interactions with its ligands. For instance, the glycosyltransferase EOGT transfers N-acetylglucosamine linked to Ser or Ther (O-GlcNAc) to select EGF repeats of Notch enhancing the binding of DLL1 and DLL4 but not the binding of JAG1 to Notch [77]. Fringe glycosyltransferases also modulate differential interactions of Notch receptors with its ligands, and lunatic and manic fringe enhance the binding of DLL1 to Notch and decrease the binding of JAG1 whereas radical fringe enhances the Notch response to both ligands [78–81]. Whether a ligand activates Notch signaling is dependent on its expression in a given cell system. In skeletal cells, JAG1 is expressed in osteoblasts, osteocytes and osteoclasts, and with the exception of DLL3 and DLL4, which are detected in osteocytes other Notch ligands are not detected in skeletal cells [82]. JAG1 is presumed to be the most critical Notch ligand in bone and its inactivation in progenitor cells phenocopies the loss of Notch function in osteoblasts [83].
The proteolytic cleavage of Notch leads to the release of the NICD. The NICD translocates to the nucleus, where it forms a complex with RBPJκ and mastermind-like (MAML) to regulate the transcription of target genes [71, 84, 85]. RBPJκ is also termed C promoter-binding factor 1 (CBF1), Suppressor of hairless, Lin-12 and Glp-1 (Lag-1) or CSL. It is important to note that RBPJκ, and not the NICD, binds to DNA and that under basal conditions RBPJκ associates with co-repressors to inhibit transcription. The translocation of the NICD to the nucleus, leads to the displacement of transcriptional inhibitors and the recruitment of activators of transcription so that the NICD, RBPJκ, MAML complex induces gene transcription [86]. This is considered the canonical Notch signaling pathway whereas Notch actions that are independent of RBPJκ are considered non-canonical. Targets of canonical Notch signaling include members of the Hairy and enhancer of split (HES) and HES with an YRPW motif (HEY) families of transcription factors [87–90]. The activation of Notch signaling is terminated by the actions of cyclin-dependent kinases (CDK) that phosphorylate the PEST domain of the NICD, resulting in the disassembly of the NICD, RBPJκ, MAML complex, the ubiquitination of the NICD by E3 ubiquitin ligases and the degradation of the NICD [91]. FBW7 is an F-box protein that serves as a substrate receptor of the E3 ligase complex and binds to the PEST domain of the NICD to trigger E3 ubiquitin ligase-dependent proteasome degradation [92, 93]. Figure 3 illustrates the cascade of events that lead to the activation of canonical Notch signaling and induction of its target genes.
Figure 3.
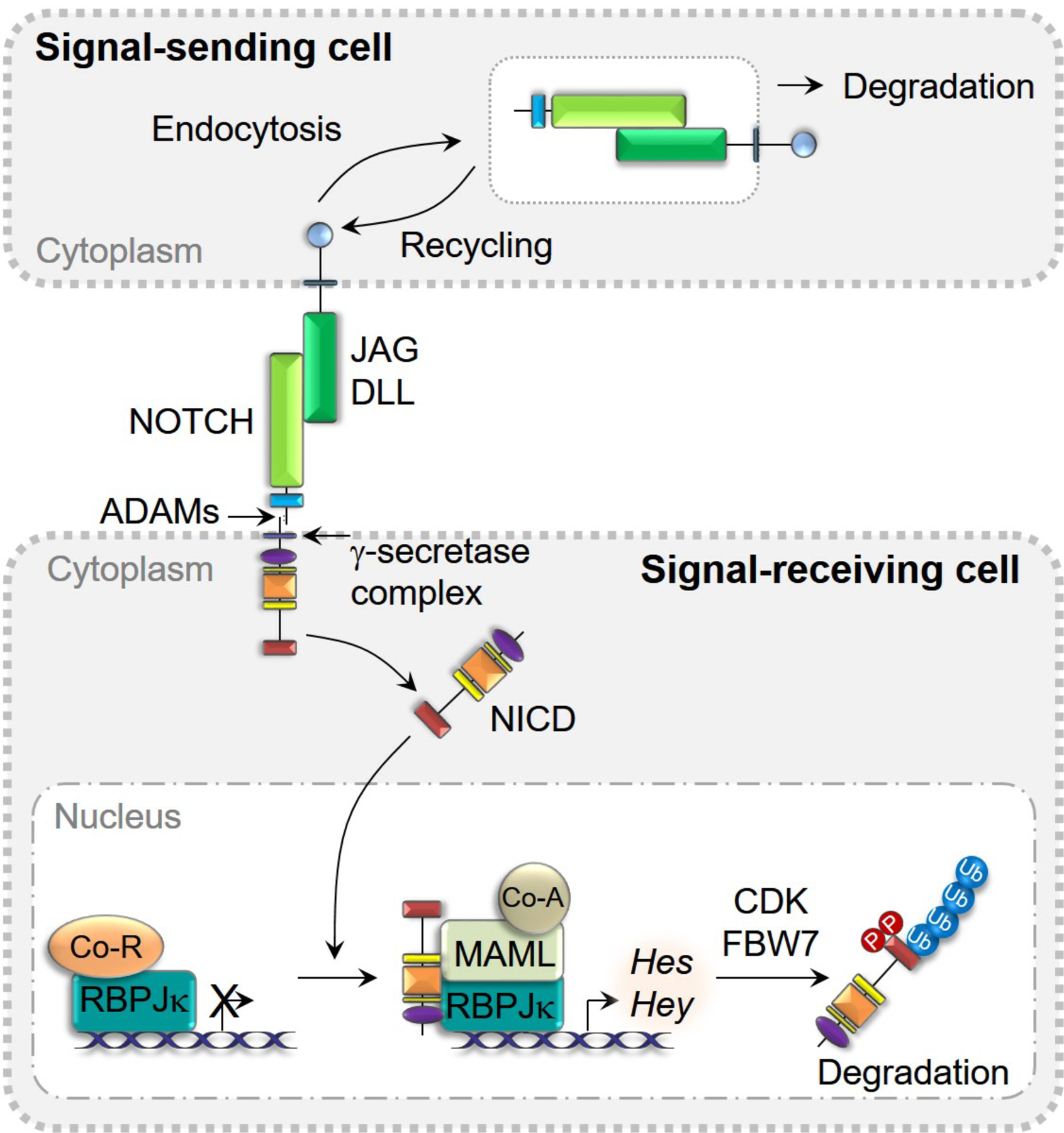
Activation and de-activation of Notch signaling. Binding of JAG or DLL to NOTCH receptors results in the cleavage of NOTCH and the release of NICD to the cytoplasm and the endocytosis and degradation of JAG and DLL in signal-sending cells. NICD translocates into nucleus and binds to RBPJκ, MAML, and co-activators of transcription to induce Hes and Hey. The phosphorylation of NICD by CDK results in the disassembly of the complex and FBW7 promotes NICD degradation via E3 ubiquitin ligase in the proteasome.
Although most of the activation of Notch depends on its interactions with ligands, a degree of basal activation has been reported under certain conditions, particularly for NOTCH3. Dysregulated activation of Notch signaling occurs in some malignancies and acute lymphoblastic leukemia and is associated with somatic mutations of the NRR that lead to the constitutive activation of Notch [6, 7, 94].
4. Notch Signaling and Osteoclast Differentiation and Function
4.1. Role of Notch Signaling in Osteoclastogenesis under Physiological Conditions
The role of Notch signaling in osteoclast differentiation has been studied by using in vitro cell culture systems and genetically modified mouse models. Results from this work have demonstrated unique actions of each Notch receptor on osteoclast differentiation and function (Figure 4). Although Notch gene misexpression can have developmental consequences, studies to determine the function of Notch in the skeleton have utilized conditional genetic mouse models that for the most part have an impact on the adult skeleton. NOTCH1 inhibits osteoclast differentiation by acting directly on osteoclast precursors and indirectly through its actions on osteoblasts. The genetic deletion of Notch1 in osteoclast precursors enhances osteoclastogenesis, whereas the overexpression of the NOTCH1 NICD (N1ICD) suppresses Nfatc1 transcription and osteoclast differentiation [95, 96]. Signal activation of NOTCH1 in the osteoblast lineage suppresses osteoblast differentiation and increases the level of OPG resulting in a pronounced inhibition of osteoclastogenesis [95, 97, 98]. Indeed, the overexpression of N1ICD in osteoblasts and in osteocytes causes an osteopetrotic phenotype [99, 100]. In osteocytes this is the result of an induction of OPG as well as a suppression of the Wnt antagonists dickkopf1 and sclerostin. As a consequence, Wnt signaling is enhanced resulting in an increase in cortical bone formation and a decrease in osteoclast number and bone resorption through the direct and indirect inhibitory effects of Wnt on osteoclastogenesis [101–104]. The effect of NOTCH1 in osteocytes is dependent on canonical activation of Notch signaling since the induction of OPG and Wnt antagonists and the osteopetrotic phenotype are reversed following the inactivation of Rbpjκ [105]. It is important to note that under basal conditions RBPJκ is dispensable for the function of osteoblasts, osteocytes and osteoclasts since the inactivation of Rbpjκ in these cell lineages in vivo does not result in a skeletal phenotype [96, 100, 105]. This would suggest that the genes transcriptionally inhibited by RBPJκ do not play a role in skeletal physiology and that RBPJκ is mostly relevant following the activation of Notch signaling. Although the inactivation of Rbpjκ in the myeloid lineage does not cause a skeletal phenotype, it amplifies the effects of TNFα and to a lesser extent RANKL on Nfatc1 transcription, osteoclastogenesis and bone resorption so that the activity of TNFα is comparable to that of RANKL [96, 106]. This has been interpreted to indicate that RBPJκ is an inhibitor of osteoclastogenesis. The mechanism by which RBPJκ suppresses Nfatc1 induction is by attenuating c-Fos activation and suppressing BLIMP-1; as a result, the transcriptional repressor IRF8 is not downregulated and osteoclastogenesis does not progress.
Figure 4.
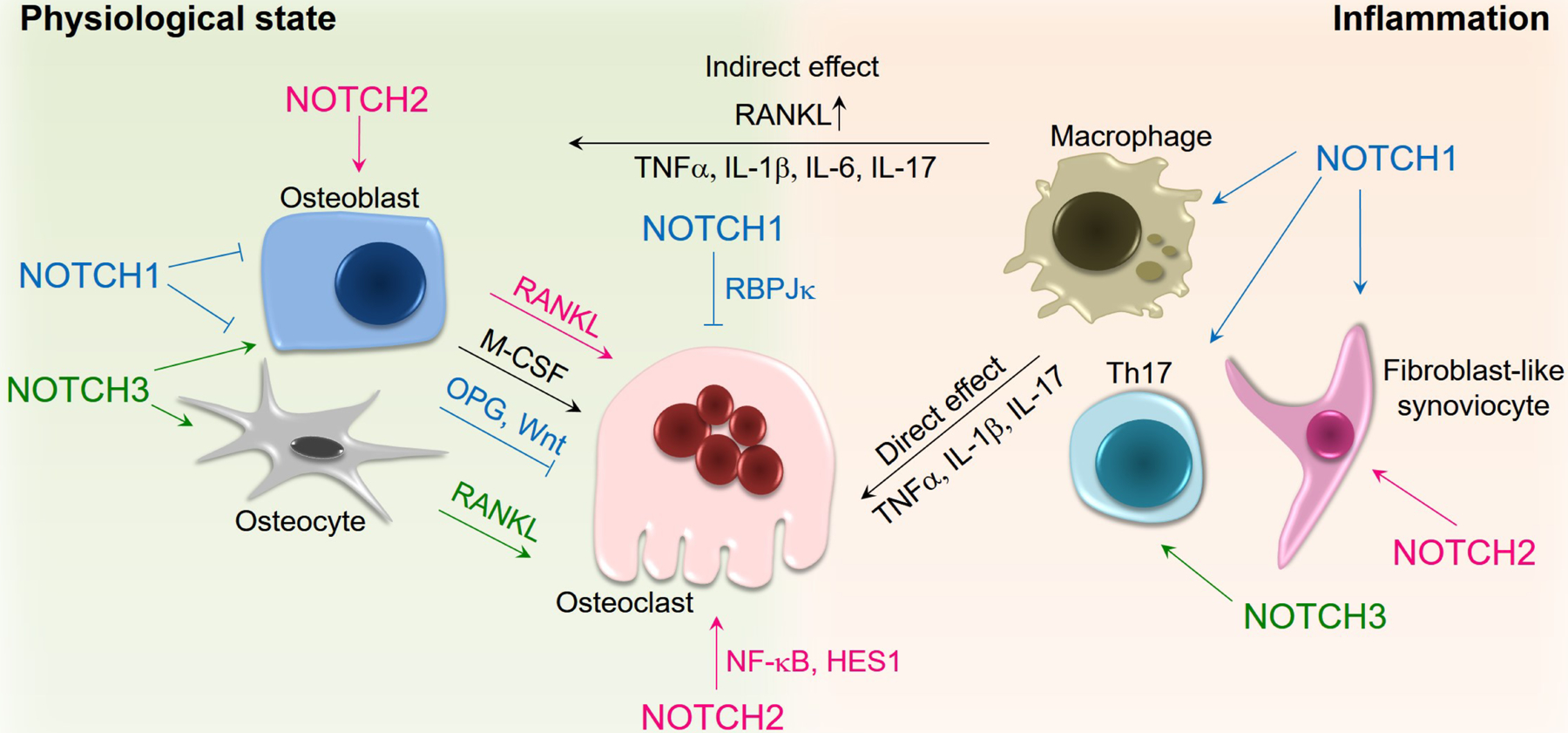
Notch signaling and osteoclastogenesis. NOTCH1 inhibits osteoclastogenesis through the RBPJκ canonical pathway and by enhancing OPG levels and Wnt signaling in osteoblasts and osteocytes. NOTCH2 induces osteoclastogenesis through NF-κB and HES1-dependent mechanisms and by increasing RANKL in osteoblasts. NOTCH3 promotes osteoclastogenesis by enhancing RANKL levels in osteoblasts and osteocytes. NOTCH1, NOTCH2 and NOTCH3 act on the cells of the immune system during inflammation leading to the induction of pro-inflammatory and osteoclastogenic cytokines that can promote osteoclast differentiation and function either directly on osteoclasts or indirectly through the release of RANKL by osteoblasts.
In contrast to the inhibitory effects of NOTCH1, NOTCH2 induces osteoclast differentiation by direct and indirect mechanisms. By acting on cells of the osteoblast lineage, NOTCH2 induces the expression of RANKL, and as a result enhances osteoclastogenesis. Importantly NOTCH2 is expressed in the myeloid lineage, where it promotes osteoclastogenesis directly. The effect requires the activation of the Notch receptor, since it is prevented by γ-secretase inhibitors and by antibodies directed to the NRR of NOTCH2 [107, 108]. Interactions of the NOTCH2 NICD (N2ICD) with the p65 subunit of NF-κB in osteoclast precursors lead to the transcription of Nfatc1 [109]. The inactivation of Notch2 in Lyz2 (LysM) expressing myeloid cells does not result in a skeletal phenotype [110]. However, the genetic deletion of Fbw7 in mature Ctsk expressing osteoclasts results in the stabilization of NOTCH2 and a gain-of-NOTCH2 function and causes enhanced osteoclastogenesis and osteopenia [93]. An analogous phenotype is observed in mutant mouse models expressing a truncated NOTCH2 protein that lacks the PEST domain resulting in the stabilization of NOTCH2 and a generalized NOTCH2 gain-of-function [111]. The lack of a phenotype in mice where Notch2 was deleted in the myeloid lineage might be related to inefficient Cre-mediated recombination or to low levels of Notch2 gene expression in undifferentiated cells [112]. This is possible since the levels of NOTCH2 increase substantially as cells of the myeloid lineage differentiate into mature osteoclasts in the presence of RANKL. Although some effects of NOTCH2 are due to direct interactions of the N2ICD with NF-κB on the Nfatc1 promoter, some are dependent on the induction of Notch target genes. Osteoclast precursors express Hes1 and low levels of Hes3 and Hes5 but do not express Hey1, Hey2 or HeyL transcripts [8]. Therefore, Hey genes cannot mediate the actions of Notch signaling in cells of the osteoclast lineage. The levels of HES1 increase during osteoclast differentiation in parallel to those of NOTCH2 [109, 111, 113]. Importantly, osteoclastogenesis is attenuated under conditions of Hes1 inactivation and the downregulation of Hes1 reverses the enhanced osteoclastogenesis caused by NOTCH2 demonstrating that HES1 is responsible for the effects of NOTCH2 on osteoclast differentiation [113].
NOTCH3 is not expressed by osteoclast precursors, but its activation in osteoblasts and osteocytes increases RANKL resulting in an induction of osteoclast differentiation by this indirect mechanism [114]. NOTCH4 is mostly detected in vascular cells and its levels in osteoclast precursors and in the osteoblast lineage are low and there is limited information on its role, if any, in skeletal physiology.
4.2. Role of Notch Signaling in Osteoclastogenesis under Inflammatory Conditions
Notch receptors and ligands are induced by pro-inflammatory cytokines in a variety of cell lineages, and Notch signaling plays a role in osteoclast differentiation during inflammation acting by direct and indirect mechanisms [115–117] (Figure 4). Overexpression of the N1ICD in osteoclast precursors suppresses TNFα-induced osteoclast formation and osteolysis in vivo, and the inactivation of Rbpjκ in myeloid cells enhances the osteoclastogenetic potential of TNFα [96]. This suggests that NOTCH1 canonical signaling has a direct inhibitory effect on TNFα-induced osteoclast differentiation. However, there is evidence indicating an indirect and stimulatory effect of NOTCH1 signaling on osteoclastogenesis and bone erosion in the context of certain inflammatory conditions, such as those occurring in joints affected by rheumatoid arthritis (RA). NOTCH1 is overexpressed and activated in fibroblast-like synoviocytes, Th17 cells and M1 macrophages, cells that play a pivotal role in the pathogenesis of RA [115, 116]. NOTCH1 signaling accelerates the production of the pro-inflammatory cytokines TNFα, IL-6 and IL-17 in synoviocytes and inhibition of Notch signaling using either a γ-secretase inhibitor or the transgenic delivery of NOTCH1-antisense constructs ameliorates arthritis severity and bone erosion in experimental models of RA [115–118]. Th17 cells secrete IL-17 in RA and periodontitis and NOTCH1 induces the transcription of Il17 in Th17 cells [119]. M1 macrophages produce pro-inflammatory cytokines, including TNFα, IL-6 and IL-1β so that M1 macrophages play a role in the pathogenesis of acute and chronic inflammatory conditions and the released cytokines induce osteolysis. In contrast, M2 macrophages synthesize anti-inflammatory cytokines, such as IL-4 and IL-10 and these are suppressed in RA [115, 120]. An imbalance between M1 and M2 macrophages is considered important in the pathogenesis of RA. The Notch inhibitor thapsigargin reduces TNFα-induced M1 macrophage polarization and attenuates inflammation and joint bone loss [121]. In the same context, microRNA-146a, which downregulates Notch1 transcripts, promotes M2 and decreases M1 macrophage polarization and ameliorates arthritis severity and bone erosion in a model of collagen-induced arthritis [122, 123].
NOTCH2 also plays a role in the bone loss associated with inflammation. Cells of the myeloid lineage expressing a truncated and stable NOTCH2 exhibit an enhanced osteoclastogenic response in the presence of TNFα [124]. TNFα induces Jag1 and Notch2 transcripts and NOTCH2 induces Hes1 as cells become differentiated into osteoclasts under the influence of TNFα. In this specific context, the effects of TNFα are independent of NF-κB and reversed following the inactivation of Hes1. In addition, a HES1-dependent increase in Il1b mRNA levels by TNFα is observed in osteoclasts harboring a NOTCH2 gain-of-function. Anti- JAG1 and anti-NOTCH2-NRR antibodies prevent TNFα-induced osteoclastogenesis in vitro, and osteolysis in vivo, confirming the NOTCH2 activation-dependency of the effects observed. In line with its actions in the myeloid lineage, TNFα enhances the expression of Notch2 and Hes1 in fibroblast-like synoviocytes from RA patients, and NOTCH2 contributes to the production of IL-6 by these cells [125].
Notch3 transcript levels are upregulated during the activation and differentiation of collagen II-specific Th1-Th17 expansion. The proliferation of Th17 cells is attenuated by a specific neutralizing antibody targeting NOTCH3, suggesting that NOTCH3 could have an indirect role in osteoclastogenesis by modulating the Th17 cell population [126].
5. Notch and Disorders of the Skeleton Associated with Altered Bone Resorption
5.1. Notch and Congenital Disorders of the Skeleton Associated with Altered Bone Resorption
Although there is a variety of congenital disorders of the skeleton that are associated with mutations in genes encoding various components of the Notch signaling pathway, most of them do not manifest alterations in osteoclastogenesis or bone resorption [7, 8]. Hajdu Cheney Syndrome (HCS) and Lateral Meningocele Syndrome (LMS) are possibly the only congenital disorders associated with mutations in Notch genes where bone resorption is known to be affected. HCS is a rare, inherited disease associated with nonsense mutations or deletions in exon 34 of NOTCH2 upstream of the PEST domain leading to the formation of a truncated and stable NOTCH2 protein and a NOTCH2 gain-of-function [127–130]. HCS is characterized by osteoporosis with fractures, acroosteolysis of the hands and feet, craniofacial developmental defects, spinal deformities and short stature [131–138]. Acroosteolysis is frequently present and accompanied by inflammation and lysis of the phalanges, which leads to short and broad digits. Platybasia and basilar invagination can result in severe neurological complications, including hydrocephalus and central respiratory arrest causing sudden death. Occasionally, polycystic kidneys, cardiac septal defects and valve abnormalities are present [138–140]. Iliac crest biopsies reveal the presence of cortical bone osteopenia, woven bone and increased osteoclast number and bone resorption [141]. We created a Notch2 mutant mouse model harboring a truncating mutation in exon 34 upstream of the PEST domain reproducing a mutation found in HCS [111]. The Notch2 mutant mouse model, termed Notch2tm1.1Ecan, exhibits pronounced cancellous and cortical bone osteopenia secondary to increased osteoclast number and bone resorption due to direct effects of NOTCH2 on cells of the myeloid lineage as well as the induction of RANKL by cells of the osteoblast lineage. The skeletal phenotype of Notch2tm1.1Ecan mice is congruent with the skeletal manifestations of HCS. Moreover, an alternate mouse model of HCS presents with increased number of osteoclasts and bone remodeling, confirming some of the phenotypic characteristics of Notch2tm1.1Ecan mice [142]. Although the mechanism of the inflammatory osteolysis of fingers and toes is poorly understood, Notch2tm1.1Ecan mice are sensitized to the osteolytic actions of TNFα in vivo and to the effect of TNFα on osteoclastogenesis [124].
Due to the limited number of subjects affected by HCS, there are no controlled trials on the management of the osteoporosis in this patient population. Bisphosphonates alone or in combination with teriparatide have been used, but evidence of benefit is scarce [140, 143, 144]. Because NOTCH2 induces RANKL and enhances osteoclastogenesis, a consideration is the use of the anti-RANKL antibody denosumab [145]. Although parathyroid hormone suppresses Notch signaling, the use of teriparatide in the treatment of HCS could pose risks since there is evidence of Notch activation in osteosarcoma in humans and prolonged activation of Notch in mice can cause osteosarcoma [68, 146, 147]. Moreover, since the mechanism responsible for the bone loss in HCS is increased bone resorption, the use of teriparatide would result in an increase in bone remodeling and possibly worsen the bone loss. NOTCH2 itself could become a future target in the treatment of HCS, and the skeletal phenotype of Notchtm1.1Ecan mouse mutants was reversed by anti-NOTCH2 NRR antibodies and ameliorated by the use of antisense oligonucleotides (ASOs) targeting Notch2 [108, 148, 149].
LMS is a rare congenital disorder characterized by craniofacial developmental abnormalities, intellectual disability, hypotonia, decreased muscle mass, syringomyelia, meningoceles and cardiac valve abnormalities [150]. Skeletal manifestations include short stature and scoliosis, increased density of the base of the skull, and increased bone remodeling and bone loss [151]. LMS is associated with point mutations or short deletions in exon 33 of NOTCH3, upstream of the PEST domain, leading to a truncated and stable NOTCH3 protein and a gain-of-function [152]. Our laboratory created a mouse model of LMS, where a tandem STOP codon was introduced into the mouse genome in exon 33 upstream of the PEST domain [114]. The mutant mouse, termed Notch3tm1.1Ecan, presents with osteopenia due to enhanced bone resorption secondary to increased osteoclast number due an induction of RANKL by cells of the osteoblast lineage including osteocytes. Because NOTCH3 is not detected in cells of the myeloid lineage, the effects on osteoclastogenesis are indirect and were documented in co-cultures of osteoblasts and bone marrow-derived macrophages. The enhanced osteoclastogenesis and osteopenic phenotype are reversed by the administration of anti-NOTCH3 NRR antibodies, selectively preventing the activation of NOTCH3; however, there is no information regarding possible therapeutic avenues in individuals with LMS [153].
5.2. Notch and Acquired Diseases of the Skeleton Associated with Altered Bone Resorption
A number of acquired skeletal disorders have been associated with dysregulated Notch signaling, including osteosarcoma, fractures, selected forms of arthritis and malignancies, particularly those presenting with bone metastases. The latter can cause lytic lesions of the skeleton or display enhanced bone resorption, which is the focus of this review. A more comprehensive review of acquired diseases of the skeleton associated with dysregulated Notch signaling was published recently [7, 8]. Although post-menopausal osteoporosis is associated with increased bone resorption, there is little evidence of dysregulated Notch signaling in osteoporosis. A SNP of the Notch ligand JAG1 is associated with bone mineral density [154]. Notch could play a role in the bone loss of aging since dysregulated gene expression of components of the Notch pathway is found in aging bone [155]. Aberrant Notch activation has been well established in hematological malignancies, where translocations that activate NOTCH1 and NOTCH2 as well as mutations that result in a truncated NOTCH1 or NOTCH2 receptor lacking a PEST domain are associated with leukemia, lymphoma and skeletal malignancies [94, 156–160]. NOTCH3 is constitutively active in carcinoma of the breast independent of ligand binding and NOTCH3 promotes carcinoma of the breast tumor growth in vitro and in vivo [161]. NOTCH3 also is expressed by myeloma cells and promotes cell growth and osteocytic invasion of myeloma cells [162]. Notch signaling plays a role in oncogenic transformation, in the epithelial-mesenchymal transition (EMT), on tumor invasiveness and in tumor angiogenesis favoring the metastatic potential of tumor cells [163, 164]. Because of these reasons, Notch plays an important function in tumor development and skeletal metastases in carcinoma of the breast and of the prostate, often altering the interactions between bone cells and metastatic cells.
Human bone marrow-derived osteoblasts induce the expression of NOTCH3 and its ligand JAG1 in human carcinoma of the breast cell lines. Inoculation of carcinoma of the breast cells, or their direct injection into the bone marrow of athymic mice, induces the formation of osteolytic bone metastases, and downregulation of NOTCH3 reduces their metastatic potential [165]. This indicates that NOTCH3 not only plays a role in the growth of breast cancer cells but also in their invasive potential to bone. The expression of JAG1 in mammary tumor cells correlates with tumor load and with the ability of tumors to form metastases in bone [166]. Tumor cells expressing JAG1 activate Notch signaling, which by inducing IL-6 in osteoblasts, enhances osteoclastogenesis and the formation of osteolytic bone metastases. The bone lysis results in a release from the bone matrix of transforming growth factor β, which upregulates JAG1 causing further activation of Notch signaling. This positive feedback loop favors the metastatic potential of tumor cells. Downregulation of JAG1 or the prevention of Notch activation decreases the osteolytic potential in experimental models of carcinoma of the breast.
Carcinoma of the prostate frequently metastasizes to bone, inducing osteoblastic woven bone formation and osteoclastic bone resorption [167]. NOTCH1, NOTCH3 and JAG1 are expressed by carcinoma of the prostate, and their levels are associated with high-grade tumors and their metastatic potential [168–172]. By inducing matrix metalloprotease 9 and of urokinase plasminogen activator receptor, NOTCH1 promotes prostate tumor invasiveness, which is associated with the presence of molecular markers of the EMT [173–175]. The observations suggest that Notch plays a role in EMT and tumor aggressiveness and in the invasive potential of carcinoma of the prostate.
Somatic mutations of NOTCH1 and dysregulated Notch signaling are often found in acute lymphoblastic T-cell leukemia and hypercalcemia. However, radiographic abnormalities of the skeleton including osteolytic lesions are uncommon and appear to be related to the secretion of parathyroid hormone related peptide by T-cells, and not to enhanced Notch signaling [176, 177]. NOTCH2 mutations resulting in a gain-of-NOTCH2 function are found in B-cell lymphomas and splenic marginal zone lymphomas, but do not seem to be associated with skeletal manifestations [159, 160, 178–180]. Chronic lymphoblastic leukemia is associated with osteolytic lesions and hypercalcemia, and precursor B-cell acute lymphoblastic leukemia is associated with bone loss and fractures, but the mechanism of the bone lytic lesions does not appear to involve dysregulated Notch signaling [181–184].
6. Ways to Control Notch Signal Activation
A variety of approaches have been developed to control Notch signaling including the use of biochemical inhibitors of Notch activation, small permeable molecules that prevent the formation of an NICD/RBPJκ/MAML ternary complex, antibodies to Notch receptors or to their ligands, and the use of antisense oligonucleotides targeting Notch genes. γ-secretase inhibitors block the cleavage of Notch receptors by interfering with the γ-secretase complex, but lack specificity since they target many substrates unrelated to Notch signal activation [185–187]. Thapsigargin is an inhibitor of the sarco/endoplasmic reticulum Ca2+-ATPase that precludes the proper folding of the Notch receptor leading to a decreased level of Notch receptors at the cell surface [188]. Synthetic small cell permeable molecules compete with the binding of MAML to the NICD/RBPJκ complex leading to suppression of Notch signaling [189]. These approaches result in a nonspecific inhibition of all Notch receptors. Since each Notch receptor has a distinct function, it is important to selectively target individual Notch receptors. To this end, antibodies directed against the NRR of NOTCH1, NOTCH2 and NOTCH3 have been developed and shown to have neutralizing activity specific to each receptor [148]. The targeting of the NRR prevents the cleavage and activation of Notch. In recent studies, the systemic administration of anti-NOTCH2 NRR and anti-NOTCH3 NRR antibodies were shown to reverse the osteopenic phenotype of HCS or LMS mutant mouse models [108, 153]. It is important to note that there may be safety concerns with the protracted suppression of a Notch receptor.
Potential future interventions to temper Notch signaling in skeletal disorders could include the administration of ASOs. These are short, synthetic, single-stranded oligodeoxynucleotides that bind target RNA by Watson-Crick base pairing resulting in RNA degradation by RNase H, but also in the inhibition of translation by blocking ribosomal subunits from attaching and/or running along the target transcript [190, 191]. The administration of ASOs has emerged as a novel therapeutic approach because ASOs can target and downregulate transcripts harboring specific mutations. The use of ASOs has been successful in the silencing of mutant genes in the central and peripheral nervous system, retina and liver, but there is limited information about their usefulness in gene silencing in the skeleton [192–201]. We demonstrated that the subcutaneous administration of Notch2 ASOs downregulated wild type and mutant Notch2 transcripts in bone extracts from Notch2tm1.1Ecan mice and ameliorated the osteopenic phenotype in this experimental model of HCS [149]. In vitro studies revealed that Notch2 ASOs inhibited the enhanced osteoclastogenesis observed in osteoclast precursors from Notch2tm1.1Ecan mice as well as the induction of RANKL expression in cells of the osteoblast lineage.
Conclusions
Notch signaling regulates osteoclast differentiation and function either by acting directly on osteoclast precursors or indirectly in cells of osteoblast lineage through the regulation of the RANKL-OPG axis. Each Notch receptor plays a distinct role in osteoclastogenesis and dysregulated Notch signaling is associated with selected congenital and acquired diseases. In conclusion, Notch signaling is a critical pathway that controls osteoblast and osteoclast differentiation and function and regulates skeletal homeostasis in health and disease.
Highlights.
Notch receptors determine cell fate and function, and regulate osteoclastogenesis
NOTCH1 inhibits osteoclastogenesis directly and by inducing osteoprotegerin in osteoblasts and osteocytes
NOTCH2 stimulates osteoclastogenesis directly and by inducing RANKL in osteoblasts, and NOTCH3 induces RANKL in osteoblasts and osteocytes
Dysregulation of Notch signaling is found in congenital and acquired diseases and can be associated with alterations in bone remodeling
Acknowledgments
This work was supported by grants from the National Institute of Arthritis, Musculoskeletal and Skin Diseases [Grants: AR063049, AR072987, AR076747] and a grant from the National Institute of Diabetes, Digestive and Kidney Diseases [Grant: DK045227].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
All authors report no conflict of interest.
References
- [1].Walsh MC, Kim N, Kadono Y, Rho J, Lee SY, Lorenzo J, Choi Y, Osteoimmunology: interplay between the immune system and bone metabolism, Annu. Rev. Immunol 24 (2006) 33–63. 10.1146/annurev.immunol.24.021605.090646 [DOI] [PubMed] [Google Scholar]
- [2].Walsh MC, Takegahara N, Kim H, Choi Y, Updating osteoimmunology: regulation of bone cells by innate and adaptive immunity, Nat Rev Rheumatol 14(3) (2018) 146–156. 10.1038/nrrheum.2017.213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tsukasaki M, Takayanagi H, Osteoimmunology: evolving concepts in bone-immune interactions in health and disease, Nat. Rev. Immunol 19(10) (2019) 626–642. 10.1038/s41577-019-0178-8 [DOI] [PubMed] [Google Scholar]
- [4].Takayanagi H, Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems, Nat. Rev. Immunol 7(4) (2007) 292–304. 10.1038/nri2062 [DOI] [PubMed] [Google Scholar]
- [5].Asagiri M, Takayanagi H, The molecular understanding of osteoclast differentiation, Bone 40(2) (2007) 251–264. 10.1016/j.bone.2006.09.023 [DOI] [PubMed] [Google Scholar]
- [6].Siebel C, Lendahl U, Notch Signaling in Development, Tissue Homeostasis, and Disease, Physiol. Rev 97(4) (2017) 1235–1294. 10.1152/physrev.00005.2017 [DOI] [PubMed] [Google Scholar]
- [7].Zanotti S, Canalis E, Notch Signaling and the Skeleton, Endocr. Rev 37(3) (2016) 223–253. 10.1210/er.2016-1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Canalis E, Notch in skeletal physiology and disease, Osteoporos.Int 29(12) (2018) 2611–2621. 10.1007/s00198-018-4694-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jacquin C, Gran DE, Lee SK, Lorenzo JA, Aguila HL, Identification of multiple osteoclast precursor populations in murine bone marrow, J. Bone Miner. Res 21(1) (2006) 67–77. 10.1359/JBMR.051007 [DOI] [PubMed] [Google Scholar]
- [10].Lorenzo J, Osteoclast precursor cells, Adv. Exp. Med. Biol 602 (2007) 77–82. 10.1007/978-0-387-72009-8_10 [DOI] [PubMed] [Google Scholar]
- [11].Manilay JO, Zouali M, Tight relationships between B lymphocytes and the skeletal system, Trends Mol. Med 20(7) (2014) 405–412. 10.1016/j.molmed.2014.03.003 [DOI] [PubMed] [Google Scholar]
- [12].Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, Wagner EF, Mak TW, Kodama T, Taniguchi T, Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts, Developmental cell 3(6) (2002) 889–901. 10.1016/s1534-5807(02)00369-6 [DOI] [PubMed] [Google Scholar]
- [13].Asagiri M, Sato K, Usami T, Ochi S, Nishina H, Yoshida H, Morita I, Wagner EF, Mak TW, Serfling E, Takayanagi H, Autoamplification of NFATc1 expression determines its essential role in bone homeostasis, J. Exp. Med 202(9) (2005) 1261–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gohda J, Akiyama T, Koga T, Takayanagi H, Tanaka S, Inoue J, RANK-mediated amplification of TRAF6 signaling leads to NFATc1 induction during osteoclastogenesis, EMBO J. 24(4) (2005) 790–799. 10.1038/sj.emboj.7600564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ikeda F, Nishimura R, Matsubara T, Hata K, Reddy SV, Yoneda T, Activation of NFAT signal in vivo leads to osteopenia associated with increased osteoclastogenesis and bone-resorbing activity, J. Immunol 177(4) (2006) 2384–2390. [DOI] [PubMed] [Google Scholar]
- [16].Takayanagi H, The role of NFAT in osteoclast formation, Ann. N.Y. Acad. Sci 1116 (2007) 227–237. [DOI] [PubMed] [Google Scholar]
- [17].Habermann B, Eberhardt C, Feld M, Zichner L, Kurth AA, Tartrate-resistant acid phosphatase 5b (TRAP 5b) as a marker of osteoclast activity in the early phase after cementless total hip replacement, Acta Orthop 78(2) (2007) 221–225. 10.1080/17453670710013717 [DOI] [PubMed] [Google Scholar]
- [18].Hayman AR, Tartrate-resistant acid phosphatase (TRAP) and the osteoclast/immune cell dichotomy, Autoimmunity 41(3) (2008) 218–223. 10.1080/08916930701694667 [DOI] [PubMed] [Google Scholar]
- [19].Miyamoto T, The dendritic cell-specific transmembrane protein DC-STAMP is essential for osteoclast fusion and osteoclast bone-resorbing activity, Modern rheumatology 16(6) (2006) 341–342. 10.1007/s10165-006-0524-0 [DOI] [PubMed] [Google Scholar]
- [20].Chiu YH, Ritchlin CT, DC-STAMP: A Key Regulator in Osteoclast Differentiation, J. Cell. Physiol 231(11) (2016) 2402–2407. 10.1002/jcp.25389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lee SH, Rho J, Jeong D, Sul JY, Kim T, Kim N, Kang JS, Miyamoto T, Suda T, Lee SK, Pignolo RJ, Koczon-Jaremko B, Lorenzo J, Choi Y, v-ATPase V0 subunit d2-deficient mice exhibit impaired osteoclast fusion and increased bone formation, Nature medicine 12(12) (2006) 1403–1409. 10.1038/nm1514 [DOI] [PubMed] [Google Scholar]
- [22].Nemeth K, Schoppet M, Al-Fakhri N, Helas S, Jessberger R, Hofbauer LC, Goettsch C, The role of osteoclast-associated receptor in osteoimmunology, J. Immunol 186(1) (2011) 13–18. 10.4049/jimmunol.1002483 [DOI] [PubMed] [Google Scholar]
- [23].Teitelbaum SL, Tanaka H, Mimura H, Inoue M, Shima M, Shioi A, Chiba M, Kitazawa S, Ross FP, Integrins and osteoclast polarization, Osteoporos.Int 7 Suppl 3 (1997) S54–56. 10.1007/BF03194343 [DOI] [PubMed] [Google Scholar]
- [24].Duong LT, Rodan GA, The role of integrins in osteoclast function, J. Bone Miner. Metab 17(1) (1999) 1–6. 10.1007/s007740050055 [DOI] [PubMed] [Google Scholar]
- [25].Nakamura I, Duong LT, Rodan SB, Rodan GA, Involvement of alpha(v)beta3 integrins in osteoclast function, J. Bone Miner. Metab 25(6) (2007) 337–344. 10.1007/s00774-007-0773-9 [DOI] [PubMed] [Google Scholar]
- [26].Teitelbaum SL, Osteoclasts: what do they do and how do they do it?, Am. J. Pathol 170(2) (2007) 427–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wilson SR, Peters C, Saftig P, Bromme D, Cathepsin K activity-dependent regulation of osteoclast actin ring formation and bone resorption, J. Biol. Chem 284(4) (2009) 2584–2592. 10.1074/jbc.M805280200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pang M, Rodriguez-Gonzalez M, Hernandez M, Recinos CC, Seldeen KL, Troen BR, AP-1 and Mitf interact with NFATc1 to stimulate cathepsin K promoter activity in osteoclast precursors, J. Cell. Biochem 120(8) (2019) 12382–12392. 10.1002/jcb.28504 [DOI] [PubMed] [Google Scholar]
- [29].Zhao B, Ivashkiv LB, Negative regulation of osteoclastogenesis and bone resorption by cytokines and transcriptional repressors, Arthritis Res Ther 13(4) (2011) 234 10.1186/ar3379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Miyauchi Y, Ninomiya K, Miyamoto H, Sakamoto A, Iwasaki R, Hoshi H, Miyamoto K, Hao W, Yoshida S, Morioka H, Chiba K, Kato S, Tokuhisa T, Saitou M, Toyama Y, Suda T, Miyamoto T, The Blimp1-Bcl6 axis is critical to regulate osteoclast differentiation and bone homeostasis, J. Exp. Med 207(4) (2010) 751–762. 10.1084/jem.20091957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nishikawa K, Nakashima T, Hayashi M, Fukunaga T, Kato S, Kodama T, Takahashi S, Calame K, Takayanagi H, Blimp1-mediated repression of negative regulators is required for osteoclast differentiation, Proc. Natl. Acad. Sci. U. S. A 107(7) (2010) 3117–3122. 10.1073/pnas.0912779107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhao B, Takami M, Yamada A, Wang X, Koga T, Hu X, Tamura T, Ozato K, Choi Y, Ivashkiv LB, Takayanagi H, Kamijo R, Interferon regulatory factor-8 regulates bone metabolism by suppressing osteoclastogenesis, Nature medicine 15(9) (2009) 1066–1071. 10.1038/nm.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, Shimamoto G, DeRose M, Elliott R, Colombero A, Tan HL, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Sander S, Van G, Tarpley J, Derby P, Lee R, Boyle WJ, Osteoprotegerin: a novel secreted protein involved in the regulation of bone density, Cell 89(2) (1997) 309–319. S0092–8674(00)80209–3 [pii] [DOI] [PubMed] [Google Scholar]
- [34].Gori F, Hofbauer LC, Dunstan CR, Spelsberg TC, Khosla S, Riggs BL, The expression of osteoprotegerin and RANK ligand and the support of osteoclast formation by stromal-osteoblast lineage cells is developmentally regulated, Endocrinology 141(12) (2000) 4768–4776. [DOI] [PubMed] [Google Scholar]
- [35].Min H, Morony S, Sarosi I, Dunstan CR, Capparelli C, Scully S, Van G, Kaufman S, Kostenuik PJ, Lacey DL, Boyle WJ, Simonet WS, Osteoprotegerin reverses osteoporosis by inhibiting endosteal osteoclasts and prevents vascular calcification by blocking a process resembling osteoclastogenesis, J. Exp. Med 192(4) (2000) 463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kwan Tat S, Padrines M, Theoleyre S, Heymann D, Fortun Y, IL-6, RANKL, TNF-alpha/IL-1: interrelations in bone resorption pathophysiology, Cytokine Growth Factor Rev. 15(1) (2004) 49–60. [DOI] [PubMed] [Google Scholar]
- [37].Amarasekara DS, Yu J, Rho J, Bone Loss Triggered by the Cytokine Network in Inflammatory Autoimmune Diseases, Journal of immunology research 2015 (2015) 832127 10.1155/2015/832127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Azuma Y, Kaji K, Katogi R, Takeshita S, Kudo A, Tumor necrosis factor-alpha induces differentiation of and bone resorption by osteoclasts, J. Biol. Chem 275(7) (2000) 4858–4864 [DOI] [PubMed] [Google Scholar]
- [39].Kobayashi K, Takahashi N, Jimi E, Udagawa N, Takami M, Kotake S, Nakagawa N, Kinosaki M, Yamaguchi K, Shima N, Yasuda H, Morinaga T, Higashio K, Martin TJ, Suda T, Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction, J. Exp. Med 191(2) (2000) 275–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kim N, Kadono Y, Takami M, Lee J, Lee SH, Okada F, Kim JH, Kobayashi T, Odgren PR, Nakano H, Yeh WC, Lee SK, Lorenzo JA, Choi Y, Osteoclast differentiation independent of the TRANCE-RANK-TRAF6 axis, J. Exp. Med 202(5) (2005) 589–595. 10.1084/jem.20050978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tanaka S, RANKL-Independent Osteoclastogenesis: A Long-Standing Controversy, J. Bone Miner. Res 32(3) (2017) 431–433. 10.1002/jbmr.3092 [DOI] [PubMed] [Google Scholar]
- [42].Lee ZH, Lee SE, Kim CW, Lee SH, Kim SW, Kwack K, Walsh K, Kim HH, IL-1alpha stimulation of osteoclast survival through the PI 3-kinase/Akt and ERK pathways, J Biochem 131(1) (2002) 161–166. 10.1093/oxfordjournals.jbchem.a003071 [DOI] [PubMed] [Google Scholar]
- [43].Tanabe N, Maeno M, Suzuki N, Fujisaki K, Tanaka H, Ogiso B, Ito K, IL-1 alpha stimulates the formation of osteoclast-like cells by increasing M-CSF and PGE2 production and decreasing OPG production by osteoblasts, Life Sci. 77(6) (2005) 615–626. 10.1016/j.lfs.2004.10.079 [DOI] [PubMed] [Google Scholar]
- [44].Kim JH, Jin HM, Kim K, Song I, Youn BU, Matsuo K, Kim N, The mechanism of osteoclast differentiation induced by IL-1, J. Immunol 183(3) (2009) 1862–1870. 10.4049/jimmunol.0803007 [DOI] [PubMed] [Google Scholar]
- [45].Shiratori T, Kyumoto-Nakamura Y, Kukita A, Uehara N, Zhang J, Koda K, Kamiya M, Badawy T, Tomoda E, Xu X, Yamaza T, Urano Y, Koyano K, Kukita T, IL-1beta Induces Pathologically Activated Osteoclasts Bearing Extremely High Levels of Resorbing Activity: A Possible Pathological Subpopulation of Osteoclasts, Accompanied by Suppressed Expression of Kindlin-3 and Talin-1, J. Immunol 200(1) (2018) 218–228. 10.4049/jimmunol.1602035 [DOI] [PubMed] [Google Scholar]
- [46].Palmqvist P, Persson E, Conaway HH, Lerner UH, IL-6, leukemia inhibitory factor, and oncostatin M stimulate bone resorption and regulate the expression of receptor activator of NF-kappa B ligand, osteoprotegerin, and receptor activator of NF-kappa B in mouse calvariae, J. Immunol 169(6) (2002) 3353–3362. 10.4049/jimmunol.169.6.3353 [DOI] [PubMed] [Google Scholar]
- [47].Yoshitake F, Itoh S, Narita H, Ishihara K, Ebisu S, Interleukin-6 directly inhibits osteoclast differentiation by suppressing receptor activator of NF-kappaB signaling pathways, J. Biol. Chem 283(17) (2008) 11535–11540. [DOI] [PubMed] [Google Scholar]
- [48].Adamopoulos IE, Bowman EP, Immune regulation of bone loss by Th17 cells, Arthritis Res Ther 10(5) (2008) 225 10.1186/ar2502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Moseley TA, Haudenschild DR, Rose L, Reddi AH, Interleukin-17 family and IL-17 receptors, Cytokine Growth Factor Rev. 14(2) (2003) 155–174. 10.1016/s1359-6101(03)00002-9 [DOI] [PubMed] [Google Scholar]
- [50].Lubberts E, van den Bersselaar L, Oppers-Walgreen B, Schwarzenberger P, Coenen-de Roo CJ, Kolls JK, Joosten LA, van den Berg WB, IL-17 promotes bone erosion in murine collagen-induced arthritis through loss of the receptor activator of NF-kappa B ligand/osteoprotegerin balance, J. Immunol 170(5) (2003) 2655–2662. 10.4049/jimmunol.170.5.2655 [DOI] [PubMed] [Google Scholar]
- [51].Adamopoulos IE, Chao CC, Geissler R, Laface D, Blumenschein W, Iwakura Y, McClanahan T, Bowman EP, Interleukin-17A upregulates receptor activator of NF-kappaB on osteoclast precursors, Arthritis Res Ther 12(1) (2010) R29 10.1186/ar2936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Onishi RM, Gaffen SL, Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease, Immunology 129(3) (2010) 311–321. 10.1111/j.1365-2567.2009.03240.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kelley MR, Kidd S, Deutsch WA, Young MW, Mutations altering the structure of epidermal growth factor-like coding sequences at the Drosophila Notch locus, Cell 51(4) (1987) 539–548. 10.1016/0092-8674(87)90123-1 [DOI] [PubMed] [Google Scholar]
- [54].de Celis JF, Bray SJ, The Abruptex domain of Notch regulates negative interactions between Notch, its ligands and Fringe, Development 127(6) (2000) 1291–1302. [DOI] [PubMed] [Google Scholar]
- [55].Xu A, Lei L, Irvine KD, Regions of Drosophila Notch that contribute to ligand binding and the modulatory influence of Fringe, J. Biol. Chem 280(34) (2005) 30158–30165. 10.1074/jbc.M505569200 [DOI] [PubMed] [Google Scholar]
- [56].Logeat F, Bessia C, Brou C, LeBail O, Jarriault S, Seidah NG, Israel A, The Notch1 receptor is cleaved constitutively by a furin-like convertase, Proc. Natl. Acad. Sci. U. S. A 95(14) (1998) 8108–8112. 10.1073/pnas.95.14.8108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Gordon WR, Vardar-Ulu D, Histen G, Sanchez-Irizarry C, Aster JC, Blacklow SC, Structural basis for autoinhibition of Notch, Nat. Struct. Mol. Biol 14(4) (2007) 295–300. 10.1038/nsmb1227 [DOI] [PubMed] [Google Scholar]
- [58].Gordon WR, Roy M, Vardar-Ulu D, Garfinkel M, Mansour MR, Aster JC, Blacklow SC, Structure of the Notch1-negative regulatory region: implications for normal activation and pathogenic signaling in T-ALL, Blood 113(18) (2009) 4381–4390. 10.1182/blood-2008-08-174748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Gordon WR, Zimmerman B, He L, Miles LJ, Huang J, Tiyanont K, McArthur DG, Aster JC, Perrimon N, Loparo JJ, Blacklow SC, Mechanical Allostery: Evidence for a Force Requirement in the Proteolytic Activation of Notch, Developmental cell 33(6) (2015) 729–736. 10.1016/j.devcel.2015.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Sanchez-Irizarry C, Carpenter AC, Weng AP, Pear WS, Aster JC, Blacklow SC, Notch subunit heterodimerization and prevention of ligand-independent proteolytic activation depend, respectively, on a novel domain and the LNR repeats, Mol. Cell. Biol 24(21) (2004) 9265–9273. 10.1128/mcb.24.21.9265-9273.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Aster JC, Xu L, Karnell FG, Patriub V, Pui JC, Pear WS, Essential roles for ankyrin repeat and transactivation domains in induction of T-cell leukemia by notch1, Mol. Cell Biol 20(20) (2000) 7505–7515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Rogers S, Wells R, Rechsteiner M, Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis, Science (New York, N.Y.) 234(4774) (1986) 364–368. [DOI] [PubMed] [Google Scholar]
- [63].Swiatek PJ, Lindsell CE, del Amo FF, Weinmaster G, Gridley T, Notch1 is essential for postimplantation development in mice, Genes Dev. 8(6) (1994) 707–719. [DOI] [PubMed] [Google Scholar]
- [64].McCright B, Gao X, Shen L, Lozier J, Lan Y, Maguire M, Herzlinger D, Weinmaster G, Jiang R, Gridley T, Defects in development of the kidney, heart and eye vasculature in mice homozygous for a hypomorphic Notch2 mutation, Development 128(4) (2001) 491–502. [DOI] [PubMed] [Google Scholar]
- [65].Domenga V, Fardoux P, Lacombe P, Monet M, Maciazek J, Krebs LT, Klonjkowski B, Berrou E, Mericskay M, Li Z, Tournier-Lasserve E, Gridley T, Joutel A, Notch3 is required for arterial identity and maturation of vascular smooth muscle cells, Genes Dev. 18(22) (2004) 2730–2735. 10.1101/gad.308904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Krebs LT, Xue Y, Norton CR, Sundberg JP, Beatus P, Lendahl U, Joutel A, Gridley T, Characterization of Notch3-deficient mice: normal embryonic development and absence of genetic interactions with a Notch1 mutation, Genesis 37(3) (2003) 139–143. 10.1002/gene.10241 [DOI] [PubMed] [Google Scholar]
- [67].Krebs LT, Xue Y, Norton CR, Shutter JR, Maguire M, Sundberg JP, Gallahan D, Closson V, Kitajewski J, Callahan R, Smith GH, Stark KL, Gridley T, Notch signaling is essential for vascular morphogenesis in mice, Genes Dev. 14(11) (2000) 1343–1352. [PMC free article] [PubMed] [Google Scholar]
- [68].Zanotti S, Canalis E, Parathyroid hormone inhibits Notch signaling in osteoblasts and osteocytes, Bone 103 (2017) 159–167. 10.1016/j.bone.2017.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Ehebauer M, Hayward P, Martinez-Arias A, Notch signaling pathway, Sci. STKE 2006(364) (2006) cm7. [DOI] [PubMed] [Google Scholar]
- [70].Sato T, Diehl TS, Narayanan S, Funamoto S, Ihara Y, De SB, Steiner H, Haass C, Wolfe MS, Active gamma-secretase complexes contain only one of each component, J. Biol. Chem 282(47) (2007) 33985–33993. [DOI] [PubMed] [Google Scholar]
- [71].Song W, Nadeau P, Yuan M, Yang X, Shen J, Yankner BA, Proteolytic release and nuclear translocation of Notch-1 are induced by presenilin-1 and impaired by pathogenic presenilin-1 mutations, Proc. Natl. Acad. Sci. U. S. A 96(12) (1999) 6959–6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, Martin RP, Schipani E, Divieti P, Bringhurst FR, Milner LA, Kronenberg HM, Scadden DT, Osteoblastic cells regulate the haematopoietic stem cell niche, Nature 425(6960) (2003) 841–846. 10.1038/nature02040 [DOI] [PubMed] [Google Scholar]
- [73].Yamamoto S, Charng WL, Bellen HJ, Endocytosis and intracellular trafficking of Notch and its ligands, Curr Top Dev Biol 92 (2010) 165–200. 10.1016/S0070-2153(10)92005-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Lai EC, Deblandre GA, Kintner C, Rubin GM, Drosophila neuralized is a ubiquitin ligase that promotes the internalization and degradation of delta, Developmental cell 1(6) (2001) 783–794. 10.1016/s1534-5807(01)00092-2 [DOI] [PubMed] [Google Scholar]
- [75].Le Borgne R, Schweisguth F, Notch signaling: endocytosis makes delta signal better, Curr. Biol 13(7) (2003) R273–275. 10.1016/s0960-9822(03)00199-4 [DOI] [PubMed] [Google Scholar]
- [76].Deblandre GA, Lai EC, Kintner C, Xenopus neuralized is a ubiquitin ligase that interacts with XDelta1 and regulates Notch signaling, Developmental cell 1(6) (2001) 795–806. 10.1016/s1534-5807(01)00091-0 [DOI] [PubMed] [Google Scholar]
- [77].Sawaguchi S, Varshney S, Ogawa M, Sakaidani Y, Yagi H, Takeshita K, Murohara T, Kato K, Sundaram S, Stanley P, Okajima T, O-GlcNAc on NOTCH1 EGF repeats regulates ligand-induced Notch signaling and vascular development in mammals, eLife 6 (2017) e24419 10.7554/eLife.24419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Hicks C, Johnston SH, diSibio G, Collazo A, Vogt TF, Weinmaster G, Fringe differentially modulates Jagged1 and Delta1 signalling through Notch1 and Notch2, Nat. Cell Biol 2(8) (2000) 515–520. 10.1038/35019553 [DOI] [PubMed] [Google Scholar]
- [79].Lei L, Xu A, Panin VM, Irvine KD, An O-fucose site in the ligand binding domain inhibits Notch activation, Development 130(26) (2003) 6411–6421. 10.1242/dev.00883 [DOI] [PubMed] [Google Scholar]
- [80].Bruckner K, Perez L, Clausen H, Cohen S, Glycosyltransferase activity of Fringe modulates Notch-Delta interactions, Nature 406(6794) (2000) 411–415. 10.1038/35019075 [DOI] [PubMed] [Google Scholar]
- [81].LeBon L, Lee TV, Sprinzak D, Jafar-Nejad H, Elowitz MB, Fringe proteins modulate Notch-ligand cis and trans interactions to specify signaling states, eLife 3 (2014) e02950 10.7554/eLife.02950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Canalis E, Notch in skeletal physiology and disease, Osteoporos. Int 29(12) (2018) 2611–2621. 10.1007/s00198-018-4694-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Lawal RA, Zhou X, Batey K, Hoffman CM, Georger MA, Radtke F, Hilton MJ, Xing L, Frisch BJ, Calvi LM, The Notch Ligand Jagged1 Regulates the Osteoblastic Lineage by Maintaining the Osteoprogenitor Pool, J. Bone Miner. Res 32(6) (2017) 1320–1331. 10.1002/jbmr.3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Kovall RA, Structures of CSL, Notch and Mastermind proteins: piecing together an active transcription complex, Curr. Opin. Struct. Biol 17(1) (2007) 117–127. [DOI] [PubMed] [Google Scholar]
- [85].Kovall RA, More complicated than it looks: assembly of Notch pathway transcription complexes, Oncogene 27(38) (2008) 5099–5109. [DOI] [PubMed] [Google Scholar]
- [86].Artavanis-Tsakonas S, Rand MD, Lake RJ, Notch signaling: cell fate control and signal integration in development, Science (New York, N.Y.) 284(5415) (1999) 770–776. [DOI] [PubMed] [Google Scholar]
- [87].Ohtsuka T, Ishibashi M, Gradwohl G, Nakanishi S, Guillemot F, Kageyama R, Hes1 and Hes5 as notch effectors in mammalian neuronal differentiation, EMBO J. 18(8) (1999) 2196–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Iso T, Sartorelli V, Poizat C, Iezzi S, Wu HY, Chung G, Kedes L, Hamamori Y, HERP, a novel heterodimer partner of HES/E(spl) in Notch signaling, Mol. Cell. Biol 21(17) (2001) 6080–6089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Katoh M, Katoh M, Identification and characterization of human HES2, HES3, and HES5 genes in silico, Int. J. Oncol 25(2) (2004) 529–534. [PubMed] [Google Scholar]
- [90].Iso T, Kedes L, Hamamori Y, HES and HERP families: multiple effectors of the Notch signaling pathway, J. Cell. Physiol 194(3) (2003) 237–255. 10.1002/jcp.10208 [DOI] [PubMed] [Google Scholar]
- [91].Fryer CJ, White JB, Jones KA, Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover, Mol. Cell 16(4) (2004) 509–520. [DOI] [PubMed] [Google Scholar]
- [92].Thompson BJ, Buonamici S, Sulis ML, Palomero T, Vilimas T, Basso G, Ferrando A, Aifantis I, The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia, J. Exp. Med 204(8) (2007) 1825–1835. 10.1084/jem.20070872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Fukushima H, Shimizu K, Watahiki A, Hoshikawa S, Kosho T, Oba D, Sakano S, Arakaki M, Yamada A, Nagashima K, Okabe K, Fukumoto S, Jimi E, Bigas A, Nakayama KI, Nakayama K, Aoki Y, Wei W, Inuzuka H, NOTCH2 Hajdu-Cheney Mutations Escape SCF(FBW7)-Dependent Proteolysis to Promote Osteoporosis, Mol. Cell 68(4) (2017) 645–658 e645. 10.1016/j.molcel.2017.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Chiang MY, Xu ML, Histen G, Shestova O, Roy M, Nam Y, Blacklow SC, Sacks DB, Pear WS, Aster JC, Identification of a conserved negative regulatory sequence that influences the leukemogenic activity of NOTCH1, Mol. Cell. Biol 26(16) (2006) 6261–6271. 10.1128/mcb.02478-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Bai S, Kopan R, Zou W, Hilton MJ, Ong CT, Long F, Ross FP, Teitelbaum SL, NOTCH1 regulates osteoclastogenesis directly in osteoclast precursors and indirectly via osteoblast lineage cells, J. Biol. Chem 283(10) (2008) 6509–6518. [DOI] [PubMed] [Google Scholar]
- [96].Zhao B, Grimes SN, Li S, Hu X, Ivashkiv LB, TNF-induced osteoclastogenesis and inflammatory bone resorption are inhibited by transcription factor RBP-J, J. Exp. Med 209(2) (2012) 319–334. 10.1084/jem.20111566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Engin F, Yao Z, Yang T, Zhou G, Bertin T, Jiang MM, Chen Y, Wang L, Zheng H, Sutton RE, Boyce BF, Lee B, Dimorphic effects of Notch signaling in bone homeostasis, Nat. Med 14(3) (2008) 299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Hilton MJ, Tu X, Wu X, Bai S, Zhao H, Kobayashi T, Kronenberg HM, Teitelbaum SL, Ross FP, Kopan R, Long F, Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation, Nature medicine 14(3) (2008) 306–314. 10.1038/nm1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Canalis E, Adams DJ, Boskey A, Parker K, Kranz L, Zanotti S, Notch Signaling in Osteocytes Differentially Regulates Cancellous and Cortical Bone Remodeling, J. Biol. Chem 288(35) (2013) 25614–25625. M113.470492 [pii]; 10.1074/jbc.M113.470492 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Tao J, Chen S, Yang T, Dawson B, Munivez E, Bertin T, Lee B, Osteosclerosis owing to Notch gain of function is solely Rbpj-dependent, J. Bone Miner. Res 25(10) (2010) 2175–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Canalis E, Wnt signalling in osteoporosis: mechanisms and novel therapeutic approaches, Nat. Rev. Endocrinol 9(10) (2013) 575–583. nrendo.2013.154 [pii]; 10.1038/nrendo.2013.154 [doi] [DOI] [PubMed] [Google Scholar]
- [102].Wei W, Zeve D, Suh JM, Wang X, Du Y, Zerwekh JE, Dechow PC, Graff JM, Wan Y, Biphasic and dosage-dependent regulation of osteoclastogenesis by beta-catenin, Mol. Cell Biol 31(23) (2011) 4706–4719. MCB.05980–11 [pii]; 10.1128/MCB.05980-11 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Chen J, Long F, beta-catenin promotes bone formation and suppresses bone resorption in postnatal growing mice, J. Bone Miner. Res 28(5) (2013) 1160–1169. 10.1002/jbmr.1834 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Otero K, Shinohara M, Zhao H, Cella M, Gilfillan S, Colucci A, Faccio R, Ross FP, Teitelbaum SL, Takayanagi H, Colonna M, TREM2 and beta-catenin regulate bone homeostasis by controlling the rate of osteoclastogenesis, J. Immunol 188(6) (2012) 2612–2621. jimmunol.1102836 [pii]; 10.4049/jimmunol.1102836 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Canalis E, Bridgewater D, Schilling L, Zanotti S, Canonical Notch activation in osteocytes causes osteopetrosis, American journal of physiology. Endocrinology and metabolism 310(2) (2016) E171–182. 10.1152/ajpendo.00395.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Swarnkar G, Karuppaiah K, Mbalaviele G, Chen TH, Abu-Amer Y, Osteopetrosis in TAK1-deficient mice owing to defective NF-kappaB and NOTCH signaling, Proc. Natl. Acad. Sci. U. S. A 112(1) (2015) 154–159. 10.1073/pnas.1415213112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Ashley JW, Ahn J, Hankenson KD, Notch signaling promotes osteoclast maturation and resorptive activity, J. Cell. Biochem 116(11) (2015) 2598–2609. 10.1002/jcb.25205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Canalis E, Sanjay A, Yu J, Zanotti S, An Antibody to Notch2 Reverses the Osteopenic Phenotype of Hajdu-Cheney Mutant Male Mice, Endocrinology 158(4) (2017) 730–742. 10.1210/en.2016-1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Fukushima H, Nakao A, Okamoto F, Shin M, Kajiya H, Sakano S, Bigas A, Jimi E, Okabe K, The association of Notch2 and NF-kappaB accelerates RANKL-induced osteoclastogenesis, Mol. Cell Biol 28(20) (2008) 6402–6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Yorgan T, Vollersen N, Riedel C, Jeschke A, Peters S, Busse B, Amling M, Schinke T, Osteoblast-specific Notch2 inactivation causes increased trabecular bone mass at specific sites of the appendicular skeleton, Bone 87 (2016) 136–146. 10.1016/j.bone.2016.04.012 [DOI] [PubMed] [Google Scholar]
- [111].Canalis E, Schilling L, Yee SP, Lee SK, Zanotti S, Hajdu Cheney Mouse Mutants Exhibit Osteopenia, Increased Osteoclastogenesis and Bone Resorption, J. Biol. Chem 291 (2016) 1538–1551. 10.1074/jbc.M115.685453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Aliprantis AO, Ueki Y, Sulyanto R, Park A, Sigrist KS, Sharma SM, Ostrowski MC, Olsen BR, Glimcher LH, NFATc1 in mice represses osteoprotegerin during osteoclastogenesis and dissociates systemic osteopenia from inflammation in cherubism, J. Clin. Invest 118(11) (2008) 3775–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Fujiwara T, Zhou J, Ye S, Zhao H, RNA-binding protein Musashi2 induced by RANKL is critical for osteoclast survival, Cell Death Dis 7 (2016) e2300 10.1038/cddis.2016.213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Canalis E, Yu J, Schilling L, Yee SP, Zanotti S, The lateral meningocele syndrome mutation causes marked osteopenia in mice, J. Biol. Chem 293(36) (2018) 14165–14177. 10.1074/jbc.RA118.004242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Keewan E, Naser SA, The Role of Notch Signaling in Macrophages during Inflammation and Infection: Implication in Rheumatoid Arthritis?, Cells 9(1) (2020) 10.3390/cells9010111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Duan L, Ren Y, Role of notch signaling in osteoimmunology--from the standpoint of osteoclast differentiation, European journal of orthodontics 35(2) (2013) 175–182. 10.1093/ejo/cjs002 [DOI] [PubMed] [Google Scholar]
- [117].Shang Y, Smith S, Hu X, Role of Notch signaling in regulating innate immunity and inflammation in health and disease, Protein Cell 7(3) (2016) 159–174. 10.1007/s13238-016-0250-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Park JS, Kim SH, Kim K, Jin CH, Choi KY, Jang J, Choi Y, Gwon AR, Baik SH, Yun UJ, Chae SY, Lee S, Kang YM, Lee KC, Arumugam TV, Mattson MP, Park JH, Jo DG, Inhibition of notch signalling ameliorates experimental inflammatory arthritis, Ann. Rheum. Dis 74(1) (2015) 267–274. 10.1136/annrheumdis-2013-203467 [DOI] [PubMed] [Google Scholar]
- [119].Keerthivasan S, Suleiman R, Lawlor R, Roderick J, Bates T, Minter L, Anguita J, Juncadella I, Nickoloff BJ, Le Poole IC, Miele L, Osborne BA, Notch signaling regulates mouse and human Th17 differentiation, J. Immunol 187(2) (2011) 692–701. 10.4049/jimmunol.1003658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Gu Q, Yang H, Shi Q,Macrophages and bone inflammation, J Orthop Translat 10 (2017) 86–93. 10.1016/j.jot.2017.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Sun W, Zhang H, Wang H, Chiu YG, Wang M, Ritchlin CT, Kiernan A, Boyce BF, Xing L, Targeting Notch-Activated M1 Macrophages Attenuates Joint Tissue Damage in a Mouse Model of Inflammatory Arthritis, J. Bone Miner. Res 32(7) (2017) 1469–1480. 10.1002/jbmr.3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].He Y, Sun X, Huang C, Long XR, Lin X, Zhang L, Lv XW, Li J, MiR-146a regulates IL-6 production in lipopolysaccharide-induced RAW264.7 macrophage cells by inhibiting Notch1, Inflammation 37(1) (2014) 71–82. 10.1007/s10753-013-9713-0 [DOI] [PubMed] [Google Scholar]
- [123].Ammari M, Presumey J, Ponsolles C, Roussignol G, Roubert C, Escriou V, Toupet K, Mausset-Bonnefont AL, Cren M, Robin M, Georgel P, Nehmar R, Taams L, Grun J, Grutzkau A, Haupl T, Pers YM, Jorgensen C, Duroux-Richard I, Courties G, Apparailly F, Delivery of miR-146a to Ly6C(high) Monocytes Inhibits Pathogenic Bone Erosion in Inflammatory Arthritis, Theranostics 8(21) (2018) 5972–5985. 10.7150/thno.29313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Yu J, Canalis E, The Hajdu Cheney mutation sensitizes mice to the osteolytic actions of tumor necrosis factor alpha, J. Biol. Chem 294(39) (2019) 14203–14214. 10.1074/jbc.RA119.009824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Jiao Z, Wang W, Ma J, Wang S, Su Z, Xu H, Notch signaling mediates TNF-alpha-induced L-6 production in cultured fibroblast-like synoviocytes from rheumatoid arthritis, Clin. Dev. Immunol 2012 (2012) 350209 10.1155/2012/350209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Jiao Z, Wang W, Xu H, Wang S, Guo M, Chen Y, Gao J, Engagement of activated Notch signalling in collagen II-specific T helper type 1 (Th1)- and Th17-type expansion involving Notch3 and Delta-like1, Clin. Exp. Immunol 164(1) (2011) 66–71. 10.1111/j.1365-2249.2010.04310.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Isidor B, Lindenbaum P, Pichon O, Bezieau S, Dina C, Jacquemont S, Martin-Coignard D, Thauvin-Robinet C, Le MM, Mandel JL, David A, Faivre L, Cormier-Daire V, Redon R, Le CC, Truncating mutations in the last exon of NOTCH2 cause a rare skeletal disorder with osteoporosis, Nat. Genet 43(4) (2011) 306–308. [DOI] [PubMed] [Google Scholar]
- [128].Majewski J, Schwartzentruber JA, Caqueret A, Patry L, Marcadier J, Fryns JP, Boycott KM, Ste-Marie LG, McKiernan FE, Marik I, Van EH, Michaud JL, Samuels ME, Mutations in NOTCH2 in families with Hajdu-Cheney syndrome, Hum. Mutat 32(10) (2011) 1114–1117. 10.1002/humu.21546 [doi] [DOI] [PubMed] [Google Scholar]
- [129].Simpson MA, Irving MD, Asilmaz E, Gray MJ, Dafou D, Elmslie FV, Mansour S, Holder SE, Brain CE, Burton BK, Kim KH, Pauli RM, Aftimos S, Stewart H, Kim CA, Holder-Espinasse M, Robertson SP, Drake WM, Trembath RC, Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss, Nat. Genet 43(4) (2011) 303–305. [DOI] [PubMed] [Google Scholar]
- [130].Zhao W, Petit E, Gafni RI, Collins MT, Robey PG, Seton M, Miller KK, Mannstadt M, Mutations in NOTCH2 in patients with Hajdu-Cheney syndrome, Osteoporos. Int 24(8) (2013) 2275–2281. 10.1007/s00198-013-2298-5 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Brennan AM, Pauli RM, Hajdu--Cheney syndrome: evolution of phenotype and clinical problems, Am. J. Med. Genet 100(4) (2001) 292–310. [DOI] [PubMed] [Google Scholar]
- [132].Cheney WD, Acro-Osteolysis, Am. J. Roentgenol. Radium. Ther. Nucl. Med 94 (1965) 595–607. [PubMed] [Google Scholar]
- [133].Currarino G, Hajdu-Cheney syndrome associated with serpentine fibulae and polycystic kidney disease, Pediatr. Radiol 39(1) (2009) 47–52. 10.1007/s00247-008-0992-9 [doi] [DOI] [PubMed] [Google Scholar]
- [134].Descartes M, Rojnueangnit K, Cole L, Sutton A, Morgan SL, Patry L, Samuels ME, Hajdu-Cheney syndrome: phenotypical progression with de-novo NOTCH2 mutation, Clin. Dysmorphol 23(3) (2014) 88–94. 10.1097/mcd.0000000000000034 [DOI] [PubMed] [Google Scholar]
- [135].Gray MJ, Kim CA, Bertola DR, Arantes PR, Stewart H, Simpson MA, Irving MD, Robertson SP, Serpentine fibula polycystic kidney syndrome is part of the phenotypic spectrum of Hajdu-Cheney syndrome, Eur. J. Hum. Genet 20(1) (2012) 122–124. ejhg2011125 [pii]; 10.1038/ejhg.2011.125 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Hajdu N, Kauntze R, Cranio-skeletal dysplasia, Br. J. Radiol 21(241) (1948) 42–48. [DOI] [PubMed] [Google Scholar]
- [137].Silverman FN, Dorst JP, Hajdu N, Acroosteolysis (Hajdu-Cheney syndrome), Birth Defects Orig. Artic. Ser 10(12) (1974) 106–123. [PubMed] [Google Scholar]
- [138].Canalis E, Clinical and experimental aspects of notch receptor signaling: Hajdu-Cheney syndrome and related disorders, Metabolism 80 (2018) 48–56. 10.1016/j.metabol.2017.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Kaler SG, Geggel RL, Sadeghi-Nejad A, Hajdu-Cheney syndrome associated with severe cardiac valvular and conduction disease., Dysmorph. Clin. Genet 4 (1990) 43–47. [Google Scholar]
- [140].Sargin G, Cildag S, Senturk T, Hajdu-Cheney syndrome with ventricular septal defect, Kaohsiung J. Med. Sci 29(6) (2013) 343–344. 10.1016/j.kjms.2012.10.009 [DOI] [PubMed] [Google Scholar]
- [141].Sakka S, Gafni RI, Davies JH, Clarke B, Tebben P, Samuels M, Saraff V, Klaushofer K, Fratzl-Zelman N, Roschger P, Rauch F, Hogler W, Bone Structural Characteristics and Response to Bisphosphonate Treatment in Children With Hajdu-Cheney Syndrome, J. Clin. Endocrinol. Metab 102(11) (2017) 4163–4172. 10.1210/jc.2017-01102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Vollersen N, Hermans-Borgmeyer I, Cornils K, Fehse B, Rolvien T, Triviai I, Jeschke A, Oheim R, Amling M, Schinke T, Yorgan TA, High Bone Turnover in Mice Carrying a Pathogenic Notch2 Mutation Causing Hajdu-Cheney Syndrome, J. Bone Miner. Res 33(1) (2018) 70–83. 10.1002/jbmr.3283 [DOI] [PubMed] [Google Scholar]
- [143].Galli-Tsinopoulou A, Kyrgios I, Giza S, Giannopoulou EM, Maggana I, Laliotis N, Two-year cyclic infusion of pamidronate improves bone mass density and eliminates risk of fractures in a girl with osteoporosis due to Hajdu-Cheney syndrome, Minerva Endocrinol. 37(3) (2012) 283–289. R07121871 [pii] [PubMed] [Google Scholar]
- [144].McKiernan FE, Integrated anti-remodeling and anabolic therapy for the osteoporosis of Hajdu-Cheney syndrome: 2-year follow-up, Osteoporos. Int 19(3) (2008) 379–380. 10.1007/s00198-007-0461-6 [doi] [DOI] [PubMed] [Google Scholar]
- [145].Adami G, Rossini M, Gatti D, Orsolini G, Idolazzi L, Viapiana O, Scarpa A, Canalis E, Hajdu Cheney Syndrome; report of a novel NOTCH2 mutation and treatment with denosumab, Bone 92 (2016) 150–156. 10.1016/j.bone.2016.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Engin F, Bertin T, Ma O, Jiang MM, Wang L, Sutton RE, Donehower LA, Lee B, Notch signaling contributes to the pathogenesis of human osteosarcomas, Hum. Mol. Genet 18(8) (2009) 1464–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Tao J, Jiang MM, Jiang L, Salvo JS, Zeng HC, Dawson B, Bertin TK, Rao PH, Chen R, Donehower LA, Gannon F, Lee BH, Notch activation as a driver of osteogenic sarcoma, Cancer Cell 26(3) (2014) 390–401. 10.1016/j.ccr.2014.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, de Leon GP, Chen Y, Finkle D, Venook R, Wu X, Ridgway J, Schahin-Reed D, Dow GJ, Shelton A, Stawicki S, Watts RJ, Zhang J, Choy R, Howard P, Kadyk L, Yan M, Zha J, Callahan CA, Hymowitz SG, Siebel CW, Therapeutic antibody targeting of individual Notch receptors, Nature 464(7291) (2010) 1052–1057. 10.1038/nature08878 [DOI] [PubMed] [Google Scholar]
- [149].Canalis E, Grossman TR, Carrer M, Schilling L, Yu J, Antisense oligonucleotides targeting Notch2 ameliorate the osteopenic phenotype in a mouse model of Hajdu-Cheney syndrome, J. Biol. Chem 295(12) (2020) 3952–3964. 10.1074/jbc.RA119.011440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Lehman RA, Stears JC, Wesenberg RL, Nusbaum ED, Familial osteosclerosis with abnormalities of the nervous system and meninges, J. Pediatr 90(1) (1977) 49–54. [DOI] [PubMed] [Google Scholar]
- [151].Gripp KW, Scott CI Jr., Hughes HE, Wallerstein R, Nicholson L, States L, Bason LD, Kaplan P, Zderic SA, Duhaime AC, Miller F, Magnusson MR, Zackai EH, Lateral meningocele syndrome: three new patients and review of the literature, Am. J. Med. Genet 70(3) (1997) 229–239. [PubMed] [Google Scholar]
- [152].Gripp KW, Robbins KM, Sobreira NL, Witmer PD, Bird LM, Avela K, Makitie O, Alves D, Hogue JS, Zackai EH, Doheny KF, Stabley DL, Sol-Church K, Truncating mutations in the last exon of NOTCH3 cause lateral meningocele syndrome, Am. J. Med. Genet. A 167A(2) (2015) 271–281. 10.1002/ajmg.a.36863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Yu J, Siebel CW, Schilling L, Canalis E, An antibody to Notch3 reverses the skeletal phenotype of lateral meningocele syndrome in male mice, J. Cell. Physiol 235(1) (2020) 210–220. 10.1002/jcp.28960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Kung AW, Xiao SM, Cherny S, Li GH, Gao Y, Tso G, Lau KS, Luk KD, Liu JM, Cui B, Zhang MJ, Zhang ZL, He JW, Yue H, Xia WB, Luo LM, He SL, Kiel DP, Karasik D, Hsu YH, Cupples LA, Demissie S, Styrkarsdottir U, Halldorsson BV, Sigurdsson G, Thorsteinsdottir U, Stefansson K, Richards JB, Zhai G, Soranzo N, Valdes A, Spector TD, Sham PC, Association of JAG1 with bone mineral density and osteoporotic fractures: a genome-wide association study and follow-up replication studies, Am. J. Hum. Genet 86(2) (2010) 229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Farr JN, Roforth MM, Fujita K, Nicks KM, Cunningham JM, Atkinson EJ, Therneau TM, McCready LK, Peterson JM, Drake MT, Monroe DG, Khosla S, Effects of Age and Estrogen on Skeletal Gene Expression in Humans as Assessed by RNA Sequencing, PLoS One 10(9) (2015) e0138347 10.1371/journal.pone.0138347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Weng AP, Ferrando AA, Lee W, Morris J.P.t., Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC, Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia, Science (New York, N.Y.) 306(5694) (2004) 269–271. 10.1126/science.1102160 [DOI] [PubMed] [Google Scholar]
- [157].Robinson DR, Kalyana-Sundaram S, Wu YM, Shankar S, Cao X, Ateeq B, Asangani IA, Iyer M, Maher CA, Grasso CS, Lonigro RJ, Quist M, Siddiqui J, Mehra R, Jing X, Giordano TJ, Sabel MS, Kleer CG, Palanisamy N, Natrajan R, Lambros MB, Reis-Filho JS, Kumar-Sinha C, Chinnaiyan AM, Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer, Nature medicine 17(12) (2011) 1646–1651. 10.1038/nm.2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Pear WS, Aster JC, T cell acute lymphoblastic leukemia/lymphoma: a human cancer commonly associated with aberrant NOTCH1 signaling, Curr Opin Hematol 11(6) (2004) 426–433. [DOI] [PubMed] [Google Scholar]
- [159].Rossi D, Trifonov V, Fangazio M, Bruscaggin A, Rasi S, Spina V, Monti S, Vaisitti T, Arruga F, Fama R, Ciardullo C, Greco M, Cresta S, Piranda D, Holmes A, Fabbri G, Messina M, Rinaldi A, Wang J, Agostinelli C, Piccaluga PP, Lucioni M, Tabbo F, Serra R, Franceschetti S, Deambrogi C, Daniele G, Gattei V, Marasca R, Facchetti F, Arcaini L, Inghirami G, Bertoni F, Pileri SA, Deaglio S, Foa R, Dalla-Favera R, Pasqualucci L, Rabadan R, Gaidano G, The coding genome of splenic marginal zone lymphoma: activation of NOTCH2 and other pathways regulating marginal zone development, J. Exp. Med 209(9) (2012) 1537–1551. 10.1084/jem.20120904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Lee SY, Kumano K, Nakazaki K, Sanada M, Matsumoto A, Yamamoto G, Nannya Y, Suzuki R, Ota S, Ota Y, Izutsu K, Sakata-Yanagimoto M, Hangaishi A, Yagita H, Fukayama M, Seto M, Kurokawa M, Ogawa S, Chiba S, Gain-of-function mutations and copy number increases of Notch2 in diffuse large B-cell lymphoma, Cancer Sci. 100(5) (2009) 920–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Choy L, Hagenbeek TJ, Solon M, French D, Finkle D, Shelton A, Venook R, Brauer MJ, Siebel CW, Constitutive NOTCH3 Signaling Promotes the Growth of Basal Breast Cancers, Cancer Res. 77(6) (2017) 1439–1452. 10.1158/0008-5472.CAN-16-1022 [DOI] [PubMed] [Google Scholar]
- [162].Delgado-Calle J, Anderson J, Cregor MD, Hiasa M, Chirgwin JM, Carlesso N, Yoneda T, Mohammad KS, Plotkin LI, Roodman GD, Bellido T, Bidirectional Notch Signaling and Osteocyte-Derived Factors in the Bone Marrow Microenvironment Promote Tumor Cell Proliferation and Bone Destruction in Multiple Myeloma, Cancer Res. 76(5) (2016) 1089–1100. 10.1158/0008-5472.CAN-15-1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Timmerman LA, Grego-Bessa J, Raya A, Bertran E, Perez-Pomares JM, Diez J, Aranda S, Palomo S, McCormick F, Izpisua-Belmonte JC, de la Pompa JL, Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation, Genes Dev. 18(1) (2004) 99–115. 10.1101/gad.276304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Sethi N, Kang Y, Notch signalling in cancer progression and bone metastasis, Br. J. Cancer 105(12) (2011) 1805–1810. 10.1038/bjc.2011.497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Zhang Z, Wang H, Ikeda S, Fahey F, Bielenberg D, Smits P, Hauschka PV, Notch3 in human breast cancer cell lines regulates osteoblast-cancer cell interactions and osteolytic bone metastasis, Am. J. Pathol 177(3) (2010) 1459–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Sethi N, Dai X, Winter CG, Kang Y, Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells, Cancer Cell 19(2) (2011) 192–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Koutsilieris M, Osteoblastic metastasis in advanced prostate cancer, Anticancer Res. 13(2) (1993) 443–449. [PubMed] [Google Scholar]
- [168].Zayzafoon M, Abdulkadir SA, McDonald JM, Notch signaling and ERK activation are important for the osteomimetic properties of prostate cancer bone metastatic cell lines, J. Biol. Chem 279(5) (2004) 3662–3670. [DOI] [PubMed] [Google Scholar]
- [169].Santagata S, Demichelis F, Riva A, Varambally S, Hofer MD, Kutok JL, Kim R, Tang J, Montie JE, Chinnaiyan AM, Rubin MA, Aster JC, JAGGED1 expression is associated with prostate cancer metastasis and recurrence, Cancer Res. 64(19) (2004) 6854–6857. 10.1158/0008-5472.CAN-04-2500 [DOI] [PubMed] [Google Scholar]
- [170].Carvalho FL, Simons BW, Eberhart CG, Berman DM, Notch signaling in prostate cancer: a moving target, Prostate 74(9) (2014) 933–945. 10.1002/pros.22811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Ross AE, Marchionni L, Vuica-Ross M, Cheadle C, Fan J, Berman DM, Schaeffer EM, Gene expression pathways of high grade localized prostate cancer, Prostate 71(14) (2011) 1568–1577. 10.1002/pros.21373 [DOI] [PubMed] [Google Scholar]
- [172].Ganguly SS, Hostetter G, Tang L, Frank SB, Saboda K, Mehra R, Wang L, Li X, Keller ET, Miranti CK, Notch3 promotes prostate cancer-induced bone lesion development via MMP-3, Oncogene 39(1) (2020) 204–218. 10.1038/s41388-019-0977-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Emadi Baygi M, Soheili ZS, Schmitz I, Sameie S, Schulz WA, Snail regulates cell survival and inhibits cellular senescence in human metastatic prostate cancer cell lines, Cell Biol. Toxicol 26(6) (2010) 553–567. 10.1007/s10565-010-9163-5 [DOI] [PubMed] [Google Scholar]
- [174].Sethi S, Macoska J, Chen W, Sarkar FH, Molecular signature of epithelial-mesenchymal transition (EMT) in human prostate cancer bone metastasis, Am J Transl Res 3(1) (2010) 90–99. [PMC free article] [PubMed] [Google Scholar]
- [175].Bin Hafeez B, Adhami VM, Asim M, Siddiqui IA, Bhat KM, Zhong W, Saleem M, Din M, Setaluri V, Mukhtar H, Targeted knockdown of Notch1 inhibits invasion of human prostate cancer cells concomitant with inhibition of matrix metalloproteinase-9 and urokinase plasminogen activator, Clin. Cancer. Res 15(2) (2009) 452–459. 10.1158/1078-0432.CCR-08-1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Prager D, Rosenblatt JD, Ejima E, Hypercalcemia, parathyroid hormone-related protein expression and human T-cell leukemia virus infection, Leuk. Lymphoma 14(5–6) (1994) 395–400. 10.3109/10428199409049695 [DOI] [PubMed] [Google Scholar]
- [177].Aoki J, Sato N, Oya N, Inoue T, Endo K, Adult T-cell leukemia, Semin Musculoskelet Radiol 5(2) (2001) 95–98. [DOI] [PubMed] [Google Scholar]
- [178].Zhang X, Shi Y, Weng Y, Lai Q, Luo T, Zhao J, Ren G, Li W, Pan H, Ke Y, Zhang W, He Q, Wang Q, Zhou R, The Truncate Mutation of Notch2 Enhances Cell Proliferation through Activating the NF-kappaB Signal Pathway in the Diffuse Large B-Cell Lymphomas, PLoS One 9(10) (2014) e108747 10.1371/journal.pone.0108747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Kiel MJ, Velusamy T, Betz BL, Zhao L, Weigelin HG, Chiang MY, Huebner-Chan DR, Bailey NG, Yang DT, Bhagat G, Miranda RN, Bahler DW, Medeiros LJ, Lim MS, Elenitoba-Johnson KS, Whole-genome sequencing identifies recurrent somatic NOTCH2 mutations in splenic marginal zone lymphoma, J. Exp. Med 209(9) (2012) 1553–1565. 10.1084/jem.20120910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Xing KH, Kahlon A, Skinnider BF, Connors JM, Gascoyne RD, Sehn LH, Savage KJ, Slack GW, Shenkier TN, Klasa R, Gerrie AS, Villa D, Outcomes in splenic marginal zone lymphoma: analysis of 107 patients treated in British Columbia, Br. J. Haematol 169(4) (2015) 520–527. 10.1111/bjh.13320 [DOI] [PubMed] [Google Scholar]
- [181].McMillan P, Mundy G, Mayer P, Hypercalcaemia and osteolytic bone lesions in chronic lymphocytic leukaemia, Br. Med. J 281(6248) (1980) 1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Mian M, Ceru S, Billio A, Rosanelli C, Cortelazzo S, Osteolytic bone lesions as a rare sign of progression of chronic lymphocytic leukemia without evidence of Richter syndrome, Leuk. Lymphoma 53(5) (2012) 993–995. 10.3109/10428194.2011.634044 [DOI] [PubMed] [Google Scholar]
- [183].Alos N, Grant RM, Ramsay T, Halton J, Cummings EA, Miettunen PM, Abish S, Atkinson S, Barr R, Cabral DA, Cairney E, Couch R, Dix DB, Fernandez CV, Hay J, Israels S, Laverdiere C, Lentle B, Lewis V, Matzinger M, Rodd C, Shenouda N, Stein R, Stephure D, Taback S, Wilson B, Williams K, Rauch F, Siminoski K, Ward LM, High incidence of vertebral fractures in children with acute lymphoblastic leukemia 12 months after the initiation of therapy, J. Clin. Oncol 30(22) (2012) 2760–2767. 10.1200/JCO.2011.40.4830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Halton J, Gaboury I, Grant R, Alos N, Cummings EA, Matzinger M, Shenouda N, Lentle B, Abish S, Atkinson S, Cairney E, Dix D, Israels S, Stephure D, Wilson B, Hay J, Moher D, Rauch F, Siminoski K, Ward LM, Advanced vertebral fracture among newly diagnosed children with acute lymphoblastic leukemia: results of the Canadian Steroid-Associated Osteoporosis in the Pediatric Population (STOPP) research program, J. Bone Miner. Res 24(7) (2009) 1326–1334. 10.1359/jbmr.090202 [doi]; 10.1359/jbmr.090202 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Ryeom SW, The cautionary tale of side effects of chronic Notch1 inhibition, J. Clin. Invest 121(2) (2011) 508–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R, A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain, Nature 398(6727) (1999) 518–522. 10.1038/19083 [DOI] [PubMed] [Google Scholar]
- [187].Duggan SP, McCarthy JV, Beyond gamma-secretase activity: The multifunctional nature of presenilins in cell signalling pathways, Cell. Signal 28(1) (2016) 1–11. 10.1016/j.cellsig.2015.10.006 [DOI] [PubMed] [Google Scholar]
- [188].Ilagan MX, Kopan R, Selective blockade of transport via SERCA inhibition: the answer for oncogenic forms of Notch?, Cancer Cell 23(3) (2013) 267–269. 10.1016/j.ccr.2013.02.020 [DOI] [PubMed] [Google Scholar]
- [189].Moellering RE, Cornejo M, Davis TN, Del BC, Aster JC, Blacklow SC, Kung AL, Gilliland DG, Verdine GL, Bradner JE, Direct inhibition of the NOTCH transcription factor complex, Nature 462(7270) (2009) 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Rinaldi C, Wood MJA, Antisense oligonucleotides: the next frontier for treatment of neurological disorders, Nature reviews. Neurology 14(1) (2018) 9–21. 10.1038/nrneurol.2017.148 [DOI] [PubMed] [Google Scholar]
- [191].Bennett CF, Baker BF, Pham N, Swayze E, Geary RS, Pharmacology of Antisense Drugs, Annu. Rev. Pharmacol. Toxicol 57 (2017) 81–105. 10.1146/annurev-pharmtox-010716-104846 [DOI] [PubMed] [Google Scholar]
- [192].Carroll JB, Warby SC, Southwell AL, Doty CN, Greenlee S, Skotte N, Hung G, Bennett CF, Freier SM, Hayden MR, Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene / allele-specific silencing of mutant huntingtin, Mol. Ther 19(12) (2011) 2178–2185. 10.1038/mt.2011.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Limmroth V, Barkhof F, Desem N, Diamond MP, Tachas G, Group ATLS, CD49d antisense drug ATL1102 reduces disease activity in patients with relapsing-remitting MS, Neurology 83(20) (2014) 1780–1788. 10.1212/WNL.0000000000000926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Murray SF, Jazayeri A, Matthes MT, Yasumura D, Yang H, Peralta R, Watt A, Freier S, Hung G, Adamson PS, Guo S, Monia BP, LaVail MM, McCaleb ML, Allele-Specific Inhibition of Rhodopsin With an Antisense Oligonucleotide Slows Photoreceptor Cell Degeneration, Invest Ophthalmol Vis Sci 56(11) (2015) 6362–6375. 10.1167/iovs.15-16400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Shy ME, Antisense oligonucleotides offer hope to patients with Charcot-Marie-Tooth disease type 1A, J. Clin. Invest 128(1) (2018) 110–112. 10.1172/JCI98617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].McCampbell A, Cole T, Wegener AJ, Tomassy GS, Setnicka A, Farley BJ, Schoch KM, Hoye ML, Shabsovich M, Sun L, Luo Y, Zhang M, Comfort N, Wang B, Amacker J, Thankamony S, Salzman DW, Cudkowicz M, Graham DL, Bennett CF, Kordasiewicz HB, Swayze EE, Miller TM, Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models, J. Clin. Invest 128(8) (2018) 3558–3567. 10.1172/JCI99081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Zhao HT, Damle S, Ikeda-Lee K, Kuntz S, Li J, Mohan A, Kim A, Hung G, Scheideler MA, Scherer SS, Svaren J, Swayze EE, Kordasiewicz HB, PMP22 antisense oligonucleotides reverse Charcot-Marie-Tooth disease type 1A features in rodent models, J. Clin. Invest 128(1) (2018) 359–368. 10.1172/JCI96499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Zhu C, Kim K, Wang X, Bartolome A, Salomao M, Dongiovanni P, Meroni M, Graham MJ, Yates KP, Diehl AM, Schwabe RF, Tabas I, Valenti L, Lavine JE, Pajvani UB, Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis, Sci Transl Med 10(468) (2018) 10.1126/scitranslmed.aat0344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Crooke ST, Witztum JL, Bennett CF, Baker BF, RNA-Targeted Therapeutics, Cell metabolism 29(2) (2019) 501 10.1016/j.cmet.2019.01.001 [DOI] [PubMed] [Google Scholar]
- [200].Wang FS, Ko JY, Lin CL, Wu HL, Ke HJ, Tai PJ, Knocking down dickkopf-1 alleviates estrogen deficiency induction of bone loss. A histomorphological study in ovariectomized rats, Bone 40(2) (2007) 485–492. 10.1016/j.bone.2006.09.004 [DOI] [PubMed] [Google Scholar]
- [201].Wang FS, Wu RW, Ko JY, Tai MH, Ke HC, Yeh DW, Wu SL, Chen MW, Heat shock protein 60 protects skeletal tissue against glucocorticoid-induced bone mass loss by regulating osteoblast survival, Bone 49(5) (2011) 1080–1089. 10.1016/j.bone.2011.08.006 [DOI] [PubMed] [Google Scholar]