

SUMMARY
Oxidative stress plays a critical role in liver tissue damage and in hepatocellular carcinoma (HCC) initiation and progression. However, the mechanisms that regulate autophagy and metabolic reprogramming during reactive oxygen species (ROS) generation, and how ROS promote tumorigenesis, still need to be fully understood. We show that protein kinase C (PKC)λ/ι loss in hepatocytes promotes autophagy and oxidative phosphorylation. This results in ROS generation, which through NRF2 drives HCC through cell autonomous and non-autonomous mechanisms. Although PKCλ/ι promotes tumorigenesis in oncogene-driven cancer models, emerging evidence demonstrate that it is a tumor suppressor in more complex carcinogenic processes. Consistently, PKCλ/ι levels negatively correlate with HCC histological tumor grade, establishing this kinase as a tumor suppressor in liver cancer.
Graphical Abstract
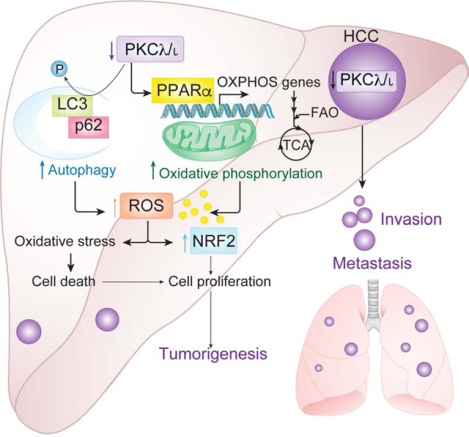
In Brief:
Yotaro et al. define a tumor suppressor role for PKCλ/ι in hepatocellular carcinoma (HCC). PKCλ/ι loss induces oxidative phosphorylation, reactive oxygen species production, and autophagy. These events activate an NRF2 transcriptional program that supports HCC progression.
INTRODUCTION
Liver cancer results from chronic tissue damage associated with oxidative stress caused by the generation of ROS (El-Serag and Rudolph, 2007). Obesity-driven steatosis is growing in importance as HCC risk factor (El-Serag, 2011; Starley et al., 2010). The heterogeneous nature of HCC makes the design of oncogene-targeted therapies highly challenging (Schulze et al., 2015; Totoki et al., 2014). Our understanding of the signaling and metabolic pathways that lead to ROS generation and HCC is fragmentary. Identifying these crucial elements in the etiopathogenesis of HCC is urgent due to the lack of effective therapies.
Autophagy and metabolic reprogramming are currently considered vulnerabilities in different types of cancer (Kimmelman and White, 2017; Pavlova and Thompson, 2016; Vander Heiden and DeBerardinis, 2017). Obesity is associated with autophagy inhibition (Yang et al., 2010), which has been postulated to be a tumor suppressor at least during cancer initiation (Takamura et al., 2011). Autophagy inhibition leads to the accumulation of the signaling adaptor and autophagy chaperone p62 (encoded by the SQSTM1 gene) (Moscat and Diaz-Meco, 2009; Moscat et al., 2016), which results in the activation of key pro-tumorigenic signaling pathways, including mTORC1, MYC and NRF2 (Duran et al., 2011; Duran et al., 2008; Moscat et al., 2016; Umemura et al., 2016).. Therefore, the upregulation of p62 appears as a critical step in liver cancer initiation during autophagy inhibition. However, the involvement of NRF2 downstream of autophagy inhibition is more intricate, likely due to its dual pro- and anti-tumorigenic function, and its pleiotropic implication in virtually all of the hallmarks of cancer (Rojo de la Vega et al., 2018). Germane to this question is that autophagy is an essential mechanism for the supply of nutrients for oxidative phosphorylation, which critically contributes to tumor maintenance and progression (Guo et al., 2016; Rabinowitz and White, 2010), and is a powerful generator of ROS (Murphy, 2009). Increased ROS is the best characterized route for NRF2 activation through the oxidation of its repressor KEAP1 that makes it unable to bind NRF2 (McMahon et al., 2003; Rojo de la Vega et al., 2018). This scenario is further complicated by the fact that p62 itself is transcriptionally upregulated by NRF2, which significantly confounds the identification of the initial trigger of this pathway (Jain et al., 2010). Altogether, these evidences demonstrate a fragmentary view of the link between liver cancer and autophagy activation and function.
Among the critical modules present in p62 that govern its involvement in the different signaling pathways, the PB1 domain is particularly interesting because it drives the binding of p62 to other PB1 domain-containing signaling proteins, such as the atypical protein kinase Cs (aPKCs; PKCζ and PKCλ/ι)(Moscat and Diaz-Meco, 2009; Moscat et al., 2016). The implication of the aPKCs in cancer seems dual (Reina-Campos et al., 2019a). Thus, PKCλ/ι has been proposed to be a pro-tumorigenic kinase in situations of oncogene-induced stress (Reina-Campos et al., 2019a). However, evidence in genetically engineered mouse models demonstrate that PKCλ/ι is actually a tumor suppressor in the context of changes in lineage plasticity associated with therapy resistance, as found in neuroendocrine prostate cancer (Reina-Campos et al., 2019b). Also, the sole genetic inactivation of PKCλ/ι in the intestinal epithelium in the absence of any other stress or oncogenic hit, results in the spontaneous generation of aggressive serrated cancer similar to the human disease under conditions of immunosuppression (Nakanishi et al., 2018). Therefore, it is clear that the effects of PKCλ/ι are context-dependent and need to be clarified in each type of cancer. Based on all these previous contradictory and dual roles of autophagy and PKCλ/ι in tumorigenesis, in this study we addressed the precise function and mechanisms of action of PKCλ/ι in HCC in vivo and analyzed its impact on human liver cancer.
RESULTS
Hepatocyte-specific deletion of PKCλ/ι promotes HCC
To explore PKC λ/ι protein expression in mouse hepatic tumors, we subjected 2 weeks-old wild-type mice to an endogenous HCC protocol consisting of the injection of 25 mg/kg of the hepatic carcinogen diethylnitrosamine (DEN), followed by feeding with a high-fat diet (HFD) as a tumor promoter. This is a well-established model that recapitulates liver cancer promotion under conditions of obesity (Park et al., 2010). At 36 weeks of age, DEN/HFD-treated mice developed multiple hepatic tumors of different histological grade, ranging from benign adenoma to high-grade tumors (Figure S1A). PKCλ/ι expression was highest in adenomas and gradually decreased as tumors became more de-differentiated (Figure S1A). This negative correlation between tumor histological grade and PKCλ/ι expression suggests that the reduced expression of PKCλ/ι might contribute to the aggressiveness of cancer cells. To establish the function of PKCλ/ι in liver physiology and cancer, we crossed Prkcif/f and Alb-Cre mice to generate a mouse line with the selective deletion of PKCλ/ι in hepatocytes (Prkcif/f;Alb-Cre). These mice were born at the expected Mendelian ratios and, when analyzed at 30 weeks of age, exhibited livers with a rough surface and blunt edge, indicative of chronic liver injury (Figures 1A and 1B). Prkcif/f;Alb-Cre mice displayed elevated serum alanine aminotransferase (ALT), reflecting hepatic damage (Figure 1C). Liver histology demonstrated disseminated cell necrosis, characterized by cytoplasmic eosinophilia and swelling, and washed-out nuclei (Figure 1D). There were no signs of steatosis in the mutant livers, but histological analysis revealed cell death including hepatocyte apoptosis (Figure S1B). Fibrosis was detected in the Prkcif/f;Alb-Cre mice (Figure S1C), as well as increased hepatocyte proliferation (Figure S1D). Older Prkcif/f;Alb-Cre mice (age range 33–52 weeks) had a similar histology with evidence of liver damage, cell death, cellular infiltrates (Figure S1E), fibrosis (Figure S1F), and increased hepatocyte proliferation (Figure S1G). None of the Prkcif/f;Alb-Cre mice develop any type of tumors during the time of the experiment while fed with chow diet (RD). However, the increased cell death and proliferation in these mutant livers suggested a microenvironment conducive to cancer. To test that possibility, Prkcif/f;Alb-Cre mice were subjected to the DEN/HFD protocol (Figure 1E). Similar to the RD-fed mice, those under the DEN/HFD showed increased ALT in serum (Figure 1F). However, in contrast to RD-fed mice, both male and female DEN/HFD-treated Prkcif/f;Alb-Cre mice displayed enhanced hepatocarcinogenesis (Figures 1G–1I); they also showed increased apoptosis, were slightly more steatotic, and had increased fibrosis and cell proliferation (Figure S1H). HCCs were present in the livers of 88% of the male and 43% of the female Prkcif/f;Alb-Cre mice, consistent with the more aggressive nature of HCC in males (Naugler et al., 2007). In contrast, all of the Prkcif/f mice displayed only benign hepatic adenomas (Figure 1J). HCCs in Prkcif/f;Alb-Cre mice exhibited a wide range of histological grades, from well to moderately differentiated HCCs of trabecular and pseudograndular type, and poorly differentiated HCCs, similar to those in human clinical HCCs (Figure 1K). In addition, 63% of the male and 14% of the female Prkcif/f;Alb-Cre mice showed lung metastases (Figures 1J and 1L). These findings indicate that the PKCλ/ι function is critical to suppress liver tumor progression, consistent with the negative correlation found between tumor histological grade and PKCλ/ι expression (Figure S1A). Tumors in the Prkcif/f;Alb-Cre mouse livers were completely devoid of PKCλ/ι as was the surrounding non-tumor liver tissue (Figure S1I), and the lung metastases (Figure S1J).
Figure 1. Hepatocyte-specific PKCλ/ι Ablation Promotes Hepatocarcinogenesis.
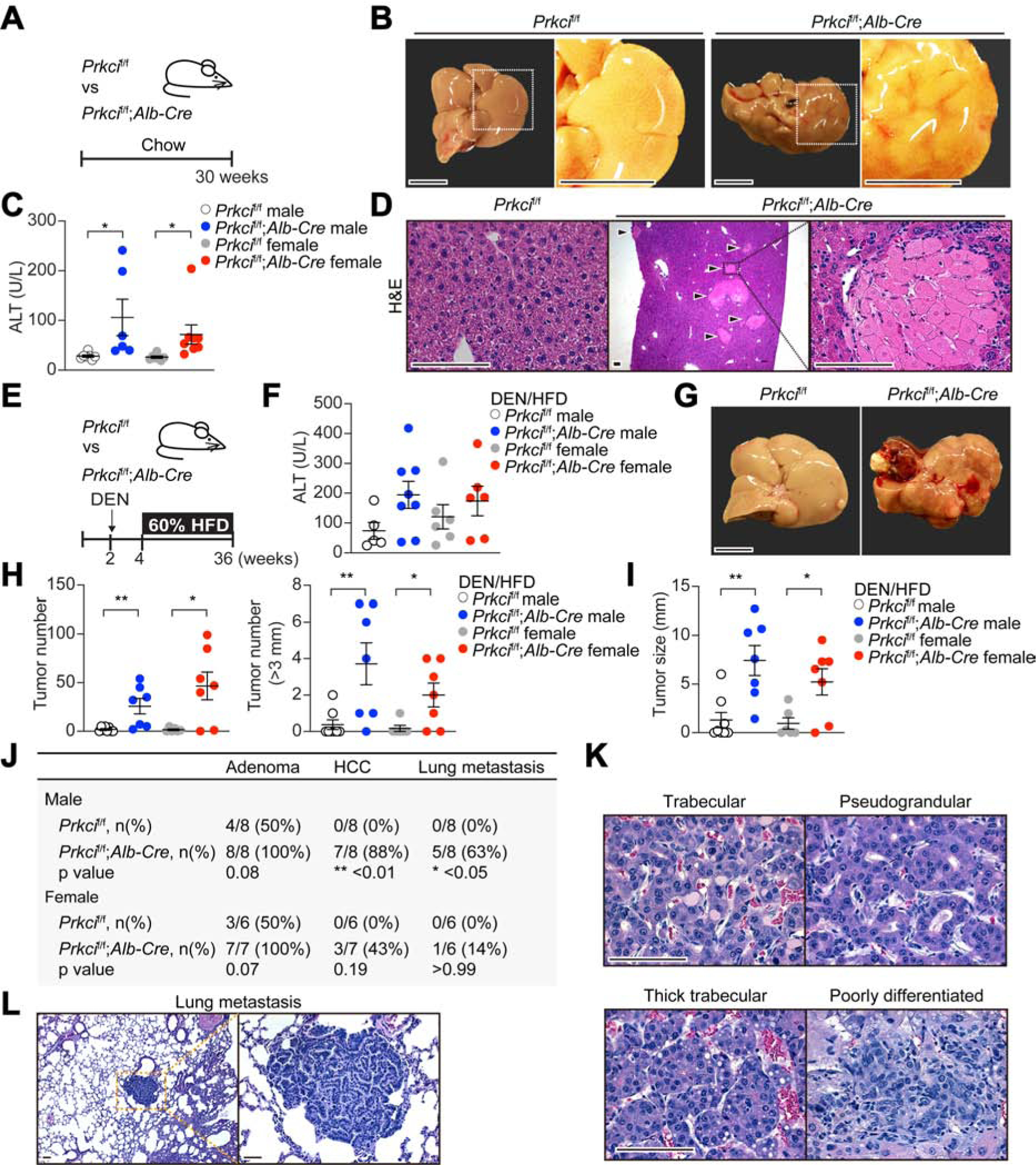
(A) Schematic representation of Prkcif/f;Alb-Cre mice examination.
(B) Images of livers from Prkcif/f and Prkcif/f;Alb-Cre mice. Scale bar, 1 cm.
(C) Serum ALT levels in Prkcif/f (n = 7 males and 6 females) and Prkcif/f;Alb-Cre mice (n = 7 males and 8 females).
(D) H&E staining of Prkcif/f and Prkcif/f;Alb-Cre livers. Arrow heads indicate necrotic foci. Scale bar, 100 μm.
(E) Schematic representation of DEN/HFD-induced HCC model.
(F) Serum ALT levels in Prkcif/f (n = 5 males and 8 females) and Prkcif/f;Alb-Cre (n = 6 males and 6 females) mice treated as in (E).
(G) Images of livers from Prkcif/f and Prkcif/f;Alb-Cre mice treated as in (E). Scale bar, 1 cm.
(H and I) Tumor incidence (H), and maximal tumor diameters (I) in Prkcif/f (n = 8 males and 7 females) and Prkcif/f;Alb-Cre (n = 6 males and 7 females) livers.
(J) Frequencies of liver adenoma, HCC and lung metastasis in Prkcif/f and Prkcif/f;Alb-Cre mice.
(K and L) H&E staining of tumor sections from Prkcif/f;Alb-Cre livers showing different tumor histological grades (K), and lung metastasis (L) from Prkcif/f;Alb-Cre animals. Scale bar, 100 μm.
Mean ± SEM. *p < 0.05, **p < 0.01. See also Figures S1 and S2.
As compared to that of Prkcif/f, Prkcif/f;Alb-Cre mice treated with DEN/HFD did not shown increased serum total bilirubin (Figure S2A), suggesting lack of significant dysregulation of the cholangiocellular compartment. Consistently, very few ductular reactions were observed in the mutant livers in both feeding conditions (Figure S2B). Furthermore, IHC analysis demonstrated positive staining for the hepatocyte lineage marker, HNF4α, and negative staining for the cholangiocyte lineage marker, KRT19, which is consistent with the notion that the Prkcif/f;Alb-Cre tumors are HCC and not intrahepatic cholangiocarcinomas (ICC) (Figure S2C). Analysis of the immunological microenvironment of Prkcif/f;Alb-Cre mice shows no differences in immune populations of B cells (B220), T cells (CD3), Ly6G or F4/80 as compared to controls (Figures S2D and S2E). The presence of ectopic lymphoid-like structures (ELS) is a poor prognostic factor in HCC and is often associated to chronic inflammation (Finkin et al., 2015). However, no proper ELS were detected but only small immune aggregates of F4/80+ macrophages and Kupffer cells (Figures S2F and S2G).
Hepatocyte-specific deletion of PKCλ/ι increases oxidative phosphorylation, oxidative stress, and the upregulation of NRF2 in vivo
Gene Set Enrichment Analysis (GSEA) of RNA-seq of livers of both genotypes showed enrichment of signatures corresponding to oxidative phosphorylation (OXPHOS), fatty acid metabolism and oxidation (FAO) and ROS in the livers of Prkcif/f;Alb-Cre mice fed with either RD (Figures 2A–2E) or HFD (Figures S3A–S3E). An epithelial-mesenchymal transition (EMT) signature was also upregulated in the Prkcif/f;Alb-Cre livers, consistent with their increased tumorigenesis (Figures 2A and S3A). Quantitative RT-PCR confirmed the upregulation of transcripts representative of oxidative metabolism in these samples (Figure 2F). The Prkcif/f;Alb-Cre mice showed increased ketone body levels in plasma (Figure 2G) and reduced triacylglycerol in liver (Figure 2H). Consistently, the oxygen consumption rate (OCR) of hepatocytes isolated from the Prkcif/f;Alb-Cre livers was increased (Figures 2I and 2J). Ingenuity Pathway Analysis (IPA) and NextBio to find “regulatory motifs” led to the identification of PPARα as a potential link between PKCλ/ι deficiency in hepatocytes and the activation of the OXPHOS metabolic program (Figures 2K, 2L, S3F and S3G).
Figure 2. Selective PKCλ/ι Deficiency in Hepatocytes Results in Metabolic Reprogramming.
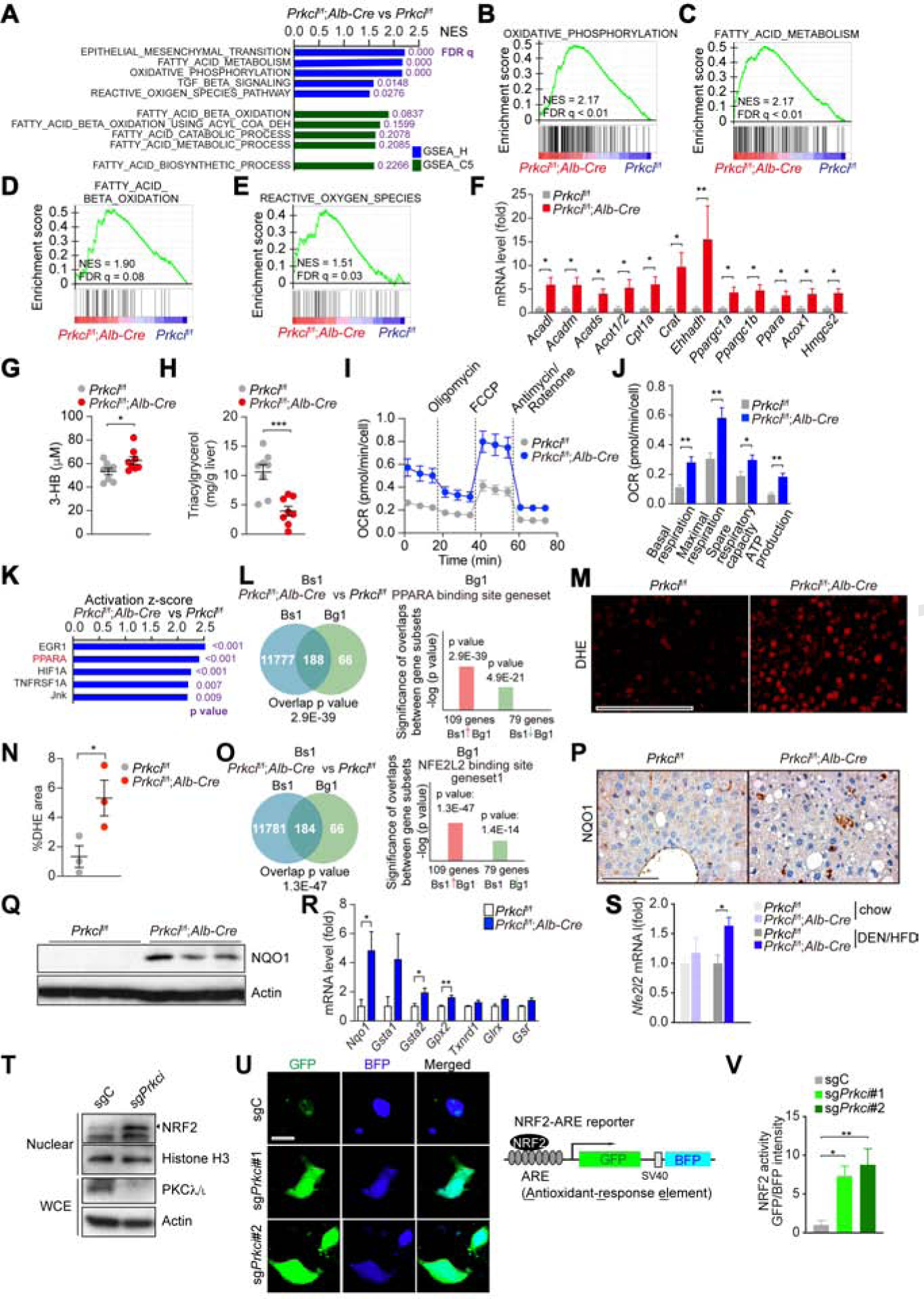
(A) Top 5 GSEA results of Prkcif/f;Alb-Cre vs Prkcif/f livers (n = 3) using compilation H and C5 (MSigDb).
(B-E) GSEA of the indicated genesets.
(F) qPCR of FAO/OXPHOS-related genes in Prkcif/f (n = 6) and Prkcif/f;Alb-Cre (n = 8) livers.
(G and H) Plasma ketone body (G) and hepatic triacylglycerol (H) in Prkcif/f and Prkcif/f;Alb-Cre mice (n = 8) after 12 h fasting followed by 2 h of refeeding.
(I and J) OCR in primary hepatocytes isolated from Prkcif/f and Prkcif/f;Alb-Cre mice (n = 5).
(K) Upstream Regulator Analysis by IPA in Prkcif/f;Alb-Cre livers vs Prkcif/f livers (n = 3).
(L) NextBio analysis of gene overlap between genes upregulated in Prkcif/f;Alb-Cre livers vs Prkcif/f livers (n = 3, Bioset1, Bs1) and PPARα binding gene set (Biogroup 1, Bg1).
(M and N) Images of DHE assay in Prkcif/f and Prkcif/f;Alb-Cre livers (M) and positive area quantification (N) (n = 4). Scale bar, 100 μm.
(O) NextBio analysis of gene overlap between genes upregulated in Prkcif/f;Alb-Cre livers vs Prkcif/f livers (n = 3, Bioset1, Bs1) and NRF2 binding gene set (Biogroup 1, Bg1).
(P and Q) NQO1 IHC (P) and immunoblot (Q) in Prkcif/f and Prkcif/f;Alb-Cre livers. Scale bar, 100 μm.
(R) qPCR of NRF2 target genes in Prkcif/f (n = 6) and Prkcif/f;Alb-Cre (n = 8) livers.
(S) Nfe2l2 expression levels (RNA-seq, FPKM) in Prkcif/f (n = 3 for chow diet and DEN/HFD) and Prkcif/f;Alb-Cre (n = 3 and n = 4 for chow and DEN/HFD, respectively) livers.
(T) Immunoblots of indicated proteins in sgPrkci or sgC cells.
(U and V) IF images left) and design of the NRF2 reporter (right) (U). Scale bar, 10 μm. Quantification of NRF2 activity (n > 10 cells) (V).
Mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S3.
The enrichment in the ROS signature in the GSEA of Prkcif/f;Alb-Cre livers (Figures 2A, 2E, S3A and S3E) is in agreement with the generation of ROS associated with augmented OXPHOS (Murphy, 2009). Dihydroethidium (DHE) staining of the Prkcif/f;Alb-Cre liver tissues under both treatment conditions demonstrate increased levels of ROS in vivo (Figures 2M, 2N, S3H and S3I), which would lead to the activation of NRF2 (Rojo de la Vega et al., 2018). NextBio analysis demonstrated the upregulation of a gene signature corresponding to “NFE2L2 biding site geneset” in the Prkcif/f;Alb-Cre livers (Figure 2O). Prkcif/f;Alb-Cre livers showed increased expression of Nqo1, a bona fide target of NRF2, and of several other NRF2 targets (Figures 2R–2R and S3J). Double IHC staining demonstrated that NQO1 was upregulated in hepatocytes (Figure S3K). Also, Nfe2l2 mRNA and its target genes were synergistically upregulated by HFD feeding in Prkcif/f;Alb-Cre livers (Figures 2S and S3L). PKCλ/ι KO by CRISPR/Cas9 in the mouse hepatocyte cell line, BNL CL.2 (sgPrkci), resulted in increased nuclear levels of NRF2 as well in high NRF2 activity (Figures 2T–2V). These results establish that the loss of PKCλ/ι in hepatocytes results in the activation of an OXPHOS gene expression program that correlates with increased ROS accumulation and NRF2 activity.
Cell-autonomous role of PKCλ/ι in the regulation of OXPHOS
The upregulation of OXPHOS genes in sgPrkci cells demonstrated the cell-autonomous role of PKCλ/ι as a negative regulator of OXPHOS (Figure 3A). CPT1 protein levels, a rate-limiting enzyme in FAO, was also increased in sgPrkci cells (Figure 3B). As a consequence of OXPHOS upregulation, sgPrkci cells consumed less glucose and produced less lactate, with no differences in glutamine consumption (Figures S4A–S4C). Importantly, sgPrkci cells displayed increased OCR, which was abolished by the PPARα inhibitor GW6471 (Figures 3C and 3D). The OCR was also increased in a PPARα-dependent manner in PKCλ/ι -deficient human hepatoma HepG2 cells (Figures S4D–S4F).
Figure 3. Loss of PKCλ/ι in Hepatocytes Enhances Oxidative Phosphorylation.
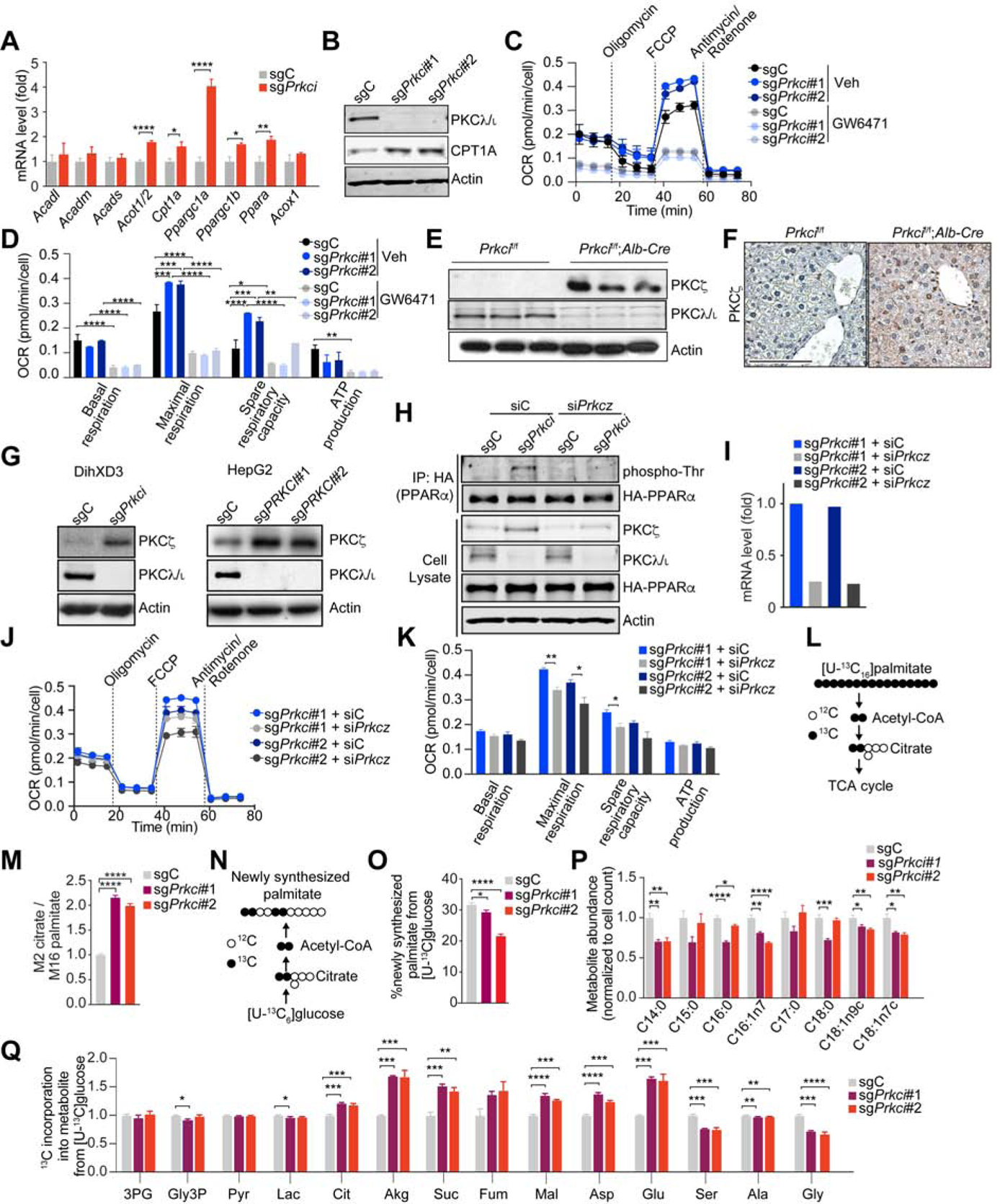
(A) qPCR of FAO/OXPHOS-related genes in sgPrkci (n=6) or sgC (n = 5) cells.
(B) Immunoblot of indicated proteins in sgPrkci (#1 and #2) or sgC cells.
(C and D) OCR measurement in sgPrkci (#1 and #2) and sgC cells (n = 3 – 5) treated with GW6471 (10 μM) or DMSO (Veh) for 24 h.
(E) Immunoblots of indicated proteins in Prkcif/f and Prkcif/f;Alb-Cre livers from 30-week-old mice (n = 3).
(F) IHC for PKCζ in Prkcif/f and Prkcif/f;Alb-Cre livers.
(G) Immunoblot of PKCζ in sgPrkci or sgC DihXD3 and HepG2 cells.
(H) Immunoprecipitation of HA-tagged human PPARα in BNL CL.2 cells treated with Calyculin A (100 nM, 2 h) and immunoblot for the indicated proteins.
followed by immunoblot of phospho-Threonine.
(I) qPCR of Prkcz in the indicated cells lines.
(J and K) OCR measurement in cell lines form (I) (n = 3).
(L) Schematic depicting 13C carbon labeling on citrate from [U-13C]palmitate via fatty acid oxidation.
(M) Fatty acid oxidation over 6 h depicted as M2 labeling on citrate relative to M16 labeling on intracellular palmitate from [U-13C16]palmitate trace in sgPrkci (#1 and #2) and sgC cells (n = 3).
(N) Schematic depicting 13C carbon labeling on palmitate from [U-13C]glucose via de novo lipogenesis.
(O) Percent of newly synthesized palmitate from [U-13C6]glucose over 24 h in sgPrkci (#1 and #2) and sgC cells (n = 3).
(P) Intracellular metabolite abundance in sgPrkci (#1 and #2) BNL CL.2 cells relative to control (sgC) (n = 3)
(Q) 13C incorporation into intracellular metabolites from [U-13C6]glucose over 24h in sgPrkci (#1 and #2) and sgC cells (n = 3).
Mean ± SEM with exception of (H), in which mean ± 95% confidence interval. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. See also Figure S4.
A critical question is to determine the mechanism whereby PKC λ/ι regulates PPARα activity. Interestingly, PKCζ was previously reported as an upstream activator of PPARα by direct phosphorylation (Burns and Vanden Heuvel, 2007; Gray et al., 2005).Therefore, loss of PKCλ/ι might result in the upregulation of PKCζ. Indeed, Prkcif/f;Alb-Cre livers showed increased PKCζ expression in PKCλ/ι -deficient hepatocytes (Figures 3E and 3F). Similarly, deletion of Prkci in DihXD3 cells, a murine HCC cell line or in HepG2 cells resulted in increased PKCζ levels (Figure 3G). We next transfected an HA-tagged PPARα construct into sgC or sgPrkci cells, after which PKCζ (encoded by the Prkcz gene) was knocked down by siRNA. HA-PPARα was immunoprecipitated and analyzed by IB with an anti-phospho-threonine antibody. While PPARα phosphorylation was increased in sgPrkci cells proficient for PKCζ, it was completely abolished in PKCζ-deficient sgPrkci cells (Figure 3H), consistent with a model whereby PKCλ/ι controls PPARα phosphorylation through PKCζ. Of note, Prkcif/f;Alb-Cre mice were born at the expected Mendelian ratios, developed normally, and did not show alterations in the liver, or in ALT, ALP or TBil serum levels (Figures S4G–S4J). Likewise, when subjected to the DEN/HFD protocol, Prkcif/f;Alb-Cre mice displayed similar steatosis and tumorigenesis as control mice (Figures S4K–S4P). These results demonstrate that although PKCζ is not involved in HCC under conditions of PKCλ/ι proficiency in hepatocytes, its upregulation upon PKCλ/ι loss unveils its role in the activation of PPARα. Interestingly, PKCζ knockdown reduced the increased OCR in sgPrkci BNL CL.2 cells (Figures 3I–3K), which established that PKCζ is a functionally relevant step downstream of PKCλ/ι in the regulation of PPARα-driven OXPHOS.
How PKC λ/ι deficiency promotes OXPHOS was next investigated using isotopic tracing strategies to quantify substrate utilization for the TCA cycle. Thus, sgC and sgPrkci BNL CL.2 cells were incubated with [U-13C16]palmitate and the carbon labeling of citrate served to quantify FAO (Figure 3L). Consistent with increased FAO, sgPrkci cells showed higher levels of carbon labeling into citrate (Figure 3M), indicating that the loss of PKCλ/ι promotes carbon flux from fatty acids for OXPHOS. By incubating cells with [U-13C]glucose, we observed decreased de novo lipogenesis (Figures 3N and 3O), while the contribution of glucose to the lipogenic acetyl-CoA pool was not affected (Figure S4Q). Fatty acid levels were decreased in sgPrkci cells (Figures 3P). Furthermore, while the relative contribution of [U-12C6]glucose into palmitate was decreased (Figures 3O and S4R), that into the TCA cycle was increased in sgPrkci cells (Figure 3Q). These metabolic studies indicate that the loss of PKCλ/ι induces the reprogramming of central carbon metabolism to promote the flux of carbons from glucose into oxidative pathways.
PKC λ/ι deficiency in hepatocytes results in autophagy activation
Autophagy is a source of substrates for OXPHOS and FAO (Guo et al., 2016; Rabinowitz and White, 2010). Liver extracts from Prkcif/f;Alb-Cre displayed increased levels of LC3 and its lipidated form LC3-II, as compared to controls (Figure 4A); there was also a concomitant increase in the expression of genes coding for components of the autophagy and lysosome pathways (Figures 4B and 4C). Electron microscopy and morphometric analyses revealed an increased total number of autophagy vesicles resulting from higher abundance of both autophagosomes (APG) and autolysosomes (AUT) in sgPrkci hepatocytes as compared to sgC controls both under basal conditions and in response to nutrient deprivation (Figures 4D–4F). The increase in autophagic vacuoles with adequate maturation of APG in AUT is compatible with higher induction of autophagy in PKCλ/ι -deficient cells. Since autophagic flux is a combination of both autophagosome formation and clearance, we analyzed autophagosome biogenesis by comparing changes in LC3-II levels at 2 and 4 h of inhibitor treatment. This study revealed that sgPrkci hepatocytes had higher basal levels of LC3-II, which were further increased in the presence of the lysosomal protease inhibitor (PI), indicating enhanced autophagic flux (Figures 4G and 4H). This increase was mainly due to higher rates of autophagosome formation coupled with their efficient clearance through lysosomal degradation (Figure 4I). We next examined autophagic flux by using a tandem fluorescent-tagged LC3 reporter (mCherry-GFP-LC3). As compared to sgC, sgPrkci cells displayed higher overall abundance of LC3-positive puncta and increased LC3 flux (greater amount of AUT) under starvation conditions (Figures 4J and S5A). In contrast to the PKCζ dependency for PPARα activation, the knockdown of PKCζ did not affect the enhanced autophagy in sgPrkci cells (Figure S5B). These results establish that PKC λ/ι is a negative regulator of autophagy independently of PKCζ.
Figure 4. PKCλ/ι deficiency in hepatocytes Enhances Autophagy Flux.
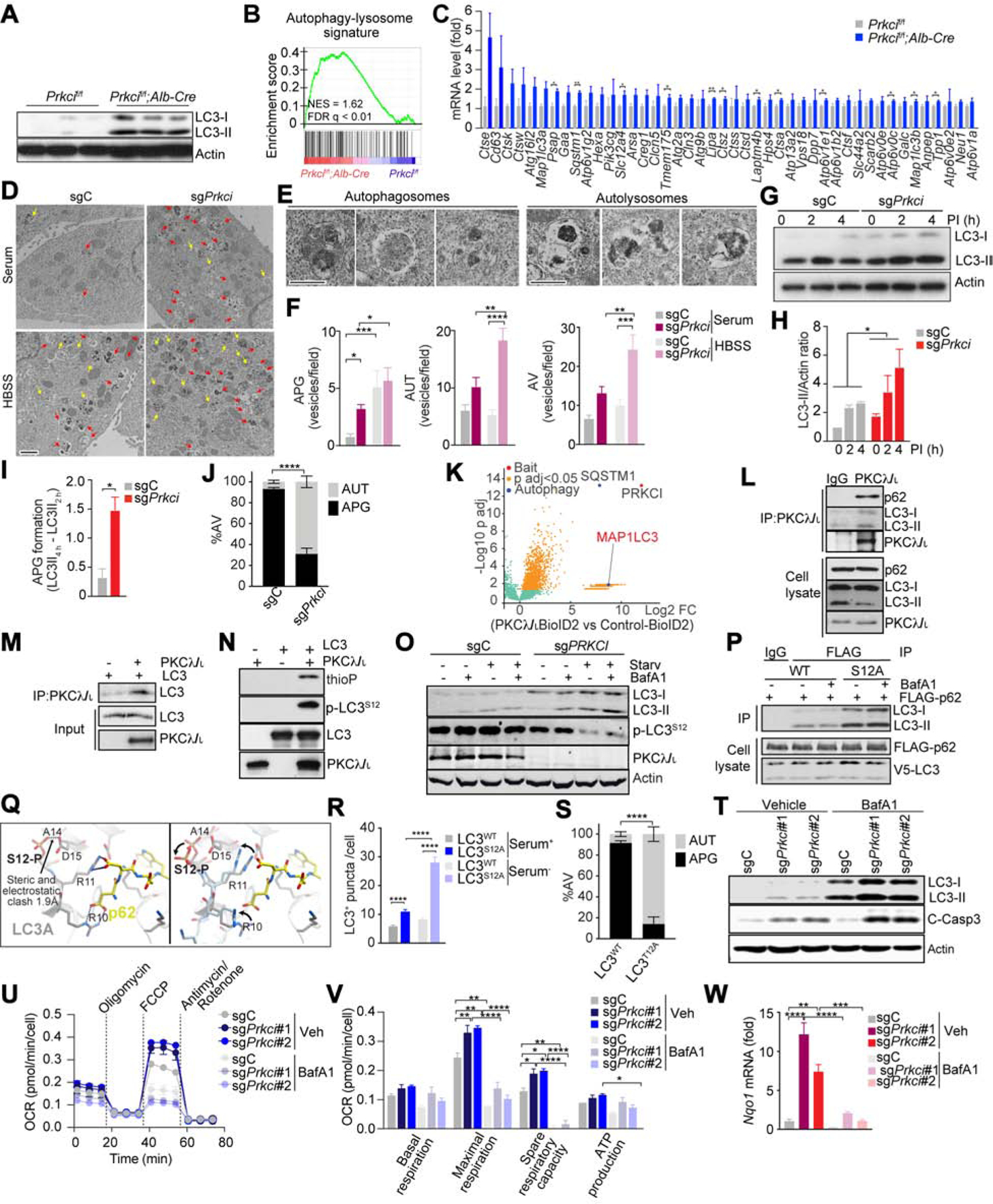
(A) Immunoblot for LC3 and actin in Prkcif/f and Prkcif/f;Alb-Cre livers (n = 3).
(B and C) GSEA of the autophagy associated geneset in Prkcif/f;Alb-Cre livers vs Prkcif/f livers (n = 3) (B), and expression of core genes in the signature (FPKM) (C).
(D) EM images of sgPrkci and sgC cells in serum supplemented media (Serum) or HBSS. Arrows: autophagosomes (yellow) and autolysosomes (red). Scale bar, 1 μm.
(E-F) Images of autophagosomes (APG) and autolysosomes (AUT) (E), and quantification of APG, AUT and autophagic vacuoles (AV) (F). Scale bar, 0.5 μm.
(G-H) Immunoblot for LC3 in response to lysosomal inhibitors (PI) (G) and quantification of LC3-II intensity normalized to actin (n = 3) (H), in sgPrkci and sgC cells cultured in FBS-free media.
(I) Calculated speed of autophagosome formation (n = 3).
(J) Quantification of APG and AUT in sgPrkci and sgC cells in response to 4 h of FBS starvation using tandem fluorescent-tagged LC3B reporter plasmid (n > 20 cells).
(K) Volcano plot of biotinylated proteins in PKCλ/ι-BioID2 vs Empty-BioID2 HEK239T cells.
(L) Immunoprecipitation for endogenous PKCλ/ι followed by immunoblot for indicated endogenous proteins in HEK293T cells.
(M) Interaction between recombinant human PKCλ/ι and human LC3.
(N) In vitro kinase assay. Phosphorylated LC3 detection by immunoblot for thiophosphate ester (thioP) and phosphorylated Ser 12 in LC3 (p-LC3S12).
(O) Immunoblot of indicated proteins in sgC and sgPRKCI HEK293T cells in response to control (DMSO), bafilomycin A1 (BafA1, 100 nM), FBS and amino acid-depletion (Starv), and BafA1 and Starv treatment for 48 h.
(P) Immunoblot of cell lysate and FLAG-tagged immunoprecipitates of HEK293T cells transfected with FLAG-tagged p62 and V5-tagged LC3 in response to BafA1 (200 nM) treatment for 12 h.
(Q) Molecular structure of human LC3.
(R) Quantification of LC3 positive puncta in LC3 wild-type (WT) or S12A mutant (S12A) BNL CL.2 cells (n > 20 cells) cultured with or without serum for 4 h.
(S) Quantification of AUT in BNL CL.2 cells in response to 4 h of FBS starvation using tandem fluorescent-tagged LC3B wild-type (WT) or T12A mutant (T12A) reporter plasmid (n > 20 cells).
(T) Immunoblot for LC3 and cleaved caspase-3 in sgC and sgPrkci cells with or without BafA1 (30 nM).
(U and V) OCR measurement in sgPrkci (#1 and #2) and sgC cells (n = 3) treated with BafA1 (30 nM) or control (Veh).
(W) qPCR for Nqo1 mRNA expression in sgC and sgPrkci cells with or without BafA1 (30 nM) (n = 3).
Mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. See also Figure S5.
To investigate the mechanism in autophagy, we carried out an unbiassed approach to identify interactors of PKCλ/ι using the proximity-dependent biotin method (BioID2) (Figure 4K) (Reina-Campos et al., 2019b). The autophagy proteins identified included p62/SQSTM1 and MAP1LC3 (Figure 4K). Immunoprecipitation of ectopically expressed and endogenous proteins confirmed that LC3 along with p62 was a bona fide binding partner of PKCλ/ι (Figures 4L and S5C). Since p62 also binds LC3 through its LIR domain, the interaction of PKCλ/ι with LC3 could be mediated by p62. A mutation in the PB1 domain of PKC λ/ι that abolishes its binding to p62 (Moscat et al., 2006), does not affect the interaction of PKCλ/ι with LC3 (Figures S5D and S5E). This result indicates that PKCλ/ι binds directly LC3 and independently of p62. A direct PKCλ/ι -LC3 interaction was demonstrated by binding assay of recombinant proteins (Figure 4M). The catalytic domain of PKCλ/ι was necessary and sufficient for its interaction with LC3 (Figures S5D, S5F and S5G), which suggested that LC3 might be a substrate of PKCλ/ι kinase activity. In vitro assays demonstrated that LC3 is directly phosphorylated by PKCλ/ι (Figure 4N), and mass spectrometry analysis identified serine (S) 12 as the unique phosphorylation site in LC3 (Figure S5H). ScanSite v4.0 software also predicted S12 as an aPKC phosphorylation site in LC3 (Figure S5I). This site is conserved across species in both LC3 isoforms, being serine in LC3A and threonine in LC3B (Figure S5J). S12 also conforms to a previously identified consensus substrate recognition pattern for aPKCs (Wang et al., 2012) (Figure S5J). In vitro phosphorylation followed by IB with an anti-phospho-S12-LC3 antibody independently established that S12 is a direct bona fide phosphorylation site for PKCλ/ι in LC3 (Figure 4N). This phosphorylation was abolished when the enzymatic assay was performed with a kinase-dead PKCλ/ι mutant (Figure S5K). Importantly, LC3 S12 phosphorylation was reduced, concomitant with increased autophagy, in PKCλ/ι -deficient cells (Figure 4O). The interaction of p62 with a S12A non-phosphorylatable LC3 mutant was stronger than with LC3 WT (Figure 4P), consistent with the negative effect of S12 phosphorylation on LC3 function. Modeling the structure of LC3A, based on the reported structural data of LC3B in complex with a p62 peptide, shows that S12 phosphorylation creates a destabilizing clash with aspartic acid (D) 15 (Figure 4Q). Molecular dynamics simulations suggested that helix anchored D15 is rigidly positioned, forcing phosphorylated S12 to rotate away from the protein (Figure 4Q). This motion causes twisting of the upstream loop, disrupting the positioning of both arginine (R) 10 and R11 for p62 binding, which could explain the reduced p62-LC3 binding upon phosphorylation. In agreement, hepatocytes expressing the LC3S12A mutant formed more autophagy vesicles compared to control and enhanced autophagy flux as determined by the tandem fluorescent-tagged-LC3T12 reporter (Figures 4R, 4S, S5L and S5M). These data demonstrated that PKC λ/ι deficiency in hepatocytes promotes two synergistic pathways. One results in increased autophagy that sustains OXPHOS, and the other transcriptionally reprograms hepatocytes to activate OXPHOS through PPARα. Indeed, treatment of sgPrkci cells with BafA1 reduces OCR to the levels found in sgC cells (Figures 4T–4V). In keeping with the existence of an autophagy-OXPHOS-ROS-NRF2 axis, BafA1 treatment also inhibited NRF2 activation (Figure 4W).
Increased OXPHOS, ROS and NRF2 are critical for hepatocyte proliferation, apoptosis and cancer cell invasion
Consistent with a cell-autonomous anti-tumorigenic function for PKCλ/ι, sgPrkci BNL CL.2 hepatocytes proliferated more than controls (Figure S6A). Similar results were obtained in HepG2 cells, which also showed enhanced sphere-forming activity (Figures S6B–S6D). Treatment of sgPrkci BNL CL.2 hepatocytes with etomoxir, an inhibitor of CPT1, or siRNA-induced knockdown of Cpt1a, resulted in growth inhibition to levels comparable to those of sgC cells (Figures 5A and 5B). Treatment with GW6471, an antagonist of PPARα, completely abrogated hepatocyte proliferation both in vitro and in vivo (Figures 5C–5E), which demonstrates the critical relevance of PPARα, and the subsequent upregulation of OXPHOS, for cell growth driven by PKCλ/ι deficiency. GW6471 treatment likewise abrogated the increased sphere-forming ability of sgPRKCI HepG2 cells (Figures 5F and 5G). Interestingly, the loss of PKCλ/ι in DihXD3 cells dramatically increased EMT (Figures S6E–S6H). Treatment with chloroquine (CQ), an autophagy inhibitor, or with GW6471 abolished the invasive phenotype of PKCλ/ι -deficient DihXD3 cells (Figures 5H–5K). To determine the in vivo relevance of the EMT phenotype caused by PKCλ/ι deficiency, PKCλ/ι -null or WT DihXD3 cells were transplanted via intrasplenic injection into NSG mice (Figure 5L). While both sgPrkci and sgC DihXD3 cells developed lung metastasis (Figures 5M and 5N), the metastatic potential of sgPrkci cells was significantly higher than that of sgC cells when corrected for the size of the primary tumor (Figures 5O–5R). PKCλ/ι expression was lost in the DihXD3 liver and lung metastasis (Figure 5S). Also, deletion of PRKCI in the human HCC cell line, SK-HEP-1, induced a similar invasive phenotype in vitro (Figures S6I–S6K), and increased lung metastasis in vivo (Figures S6L–S6Q). These results demonstrate that the loss of PKCλ/ι in hepatoma cells causes a cell-autonomous increase in cell proliferation, spheroid formation, and invasiveness through the upregulation of the OXPHOS metabolic program.
Figure 5. Loss of PKCλ/ι Enhances the Aggressive Phenotype of Hepatoma Cells.
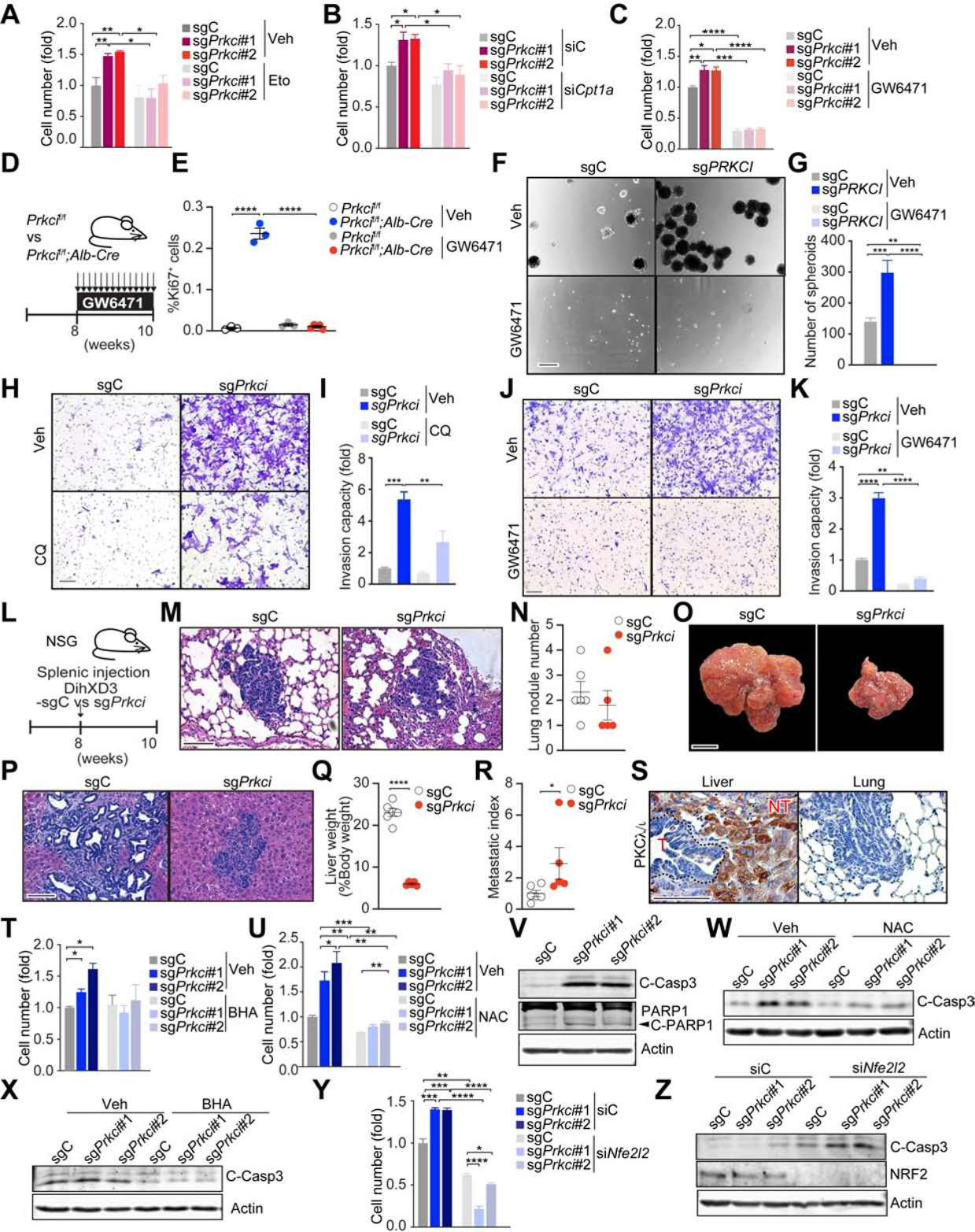
(A) Cell number of sgPrkci or sgC cells treated with etomoxir (Eto, 10 μm) or vehicle control (Veh) for 7 days (n = 3).
(B) Cell number of sgPrkci or sgC cells 7 days after siRNA-induced knockdown of Cpt1a (siCpt1a) or control siRNA (siC) (n = 3).
(C) Cell number of sgPrkci or sgC cells treated with GW6471 (5 μM) or Veh for 6 days (n = 3).
(D) Schematic representation of GW6471 treatment.
(E) Quantification of Ki-67 positive hepatocytes by IHC (n = 3–5) in mice treated as in (D).
(F and G) Sphere formation assay of sgPRKCI or sgC HepG2 cells treated with GW6471 (10 μM) or Veh. Images (F) and quantification of spheres (n = 4) (G).
(H and I) Invasion of sgPrkci or sgC DihXD3 cells treated with chloroquine (CQ, 5 μM) or Veh. Images (H) and quantification of invasive cells (n = 3) (I).
(J and K) Invasion of sgPrkci or sgC DihXD3 cells treated with GW6471 (10 μM) or Veh. Images (J) and quantification of invasive cells (n = 3) (K).
(L) Schematic representation of sgPrkci or sgC DihXD3 cells transplantation through splenic injection in male NSG mice (sgC, n = 6; sgPrkci, n = 5).
(M-N) H&E staining (M) and quantification of lung metastasis (N) in mice treated as in (L).
(O-R) Images (O), H&E staining (P), quantification of liver weight normalized to body weight (Q), and metastatic index (R) of livers from mice treated as in (L).
(S) IHC for PKCλ/ι in hepatic primary tumors and lung metastasis from the animal injected with sgC DihXD3 cells (T, tumor; NT, non-tumor).
(T and U) Cell number of sgPrkci or sgC BNL CL.2 cells treated with butylated hydroxyanisole (BHA, 100 μM) (T) or N-Acetylcysteine (NAC, 2 mM) (U) for 5 days (n = 3).
(V) Immunoblot for cleaved caspase-3 and cleaved PARP1 in sgPrkci or sgC BNL CL.2 cells.
(W and X) Immunoblot for cleaved caspase-3 in sgPrkci or sgC cells treated with NAC (2 mM) (W) or BHA (200 μM) (X) for 72 h.
(Y and Z) Cell number (Y) (n=3), and immunoblot for indicated proteins (Z), 7 days (Y) or 2 (Z) days after knockdown of Nfe2l2 in sgPrkci or sgC cells.
Mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Scale bar, 100 μm (F, H, J, M, P and S); 1 cm (O). See also Figure S6.
Treatment with the ROS scavengers N-acetylcysteine (NAC) or butylated hydroxyanisole (BHA) inhibited the increased proliferation of sgPrkci BNL CL.2 cells to levels similar to those of sgC cells (Figures 5T and 5U). Like in Prkcif/f;Alb-Cre livers, sgPrkci cells show increased apoptosis (Figure 5V). NAC or BHA inhibited caspase-3 activity in sgPrkci cells (Figures 5W and 5X), establishing that the enhanced proliferative capacity of PKCλ/ι -deficient hepatocytes can be accounted for by their higher levels of OXPHOS and ROS. An important consequence of high levels of cellular ROS is the activation of the NRF2 signaling pathway. The knockdown of Nfe2l2 reduced the increased proliferative capacity of sgPrkci cells (Figure 5Y). Therefore, NRF2 is a critical mediator of the hyper-proliferative phenotype of PKCλ/ι -deficient hepatocytes. Moreover, the loss of NRF2 resulted not only in decreased proliferation but also augmented caspase-3 activity (Figure 5Z), which likely contributes to the phenotype of the NRF2-deficient sgPrkci cells.
Cell non-autonomous tumor suppressor role of PKCλ/ι in HCC
The possibility that the loss of PKCλ/ι in hepatocytes might not only drive hepatic cell proliferation cell-autonomously but also create a pro-tumorigenic liver microenvironment was addressed in two in vivo experimental models. First, we transplanted via intrasplenic injection syngeneic mouse DihXD3 HCC cells, into either Prkcif/f or Prkcif/f;Alb-Cre mice (Figure 6A). To establish the role of NRF2 in this process, we also crossed Prkcif/f;Alb-Cre mice with Nf2l2 KO mice to generated Prkcif/f;Alb-Cre;Nf2l2+/−. Importantly, Prkcif/f;Alb-Cre mice showed significantly higher tumor burden and developed massive bloody ascites (a characteristic of cancerous peritonitis or rupture of HCC) than Prkcif/f control mice, which was completely reverted in Prkcif/f;Alb-Cre;Nf2l2+/− mice (Figures 6B–6E). The second model consisted of the direct implantation of the same HCC cell line into Prkcif/f or Prkcif/f;Alb-Cre mice liver that had been pre-conditioned by HFD feeding for 4 weeks (Figure 6F). In agreement with our hypothesis, DihXD3 cells produced more primary tumors in the liver and also metastatic lesions in the lung in the HFD-treated Prkcif/f;Alb-Cre mice than in equally treated Prkcif/f mice (Figures 6G–6J). These results strongly suggest that PKC λ/ι -deficient hepatocytes are not only more pro-tumorigenic cell-autonomously but also create a liver microenvironment that promotes higher levels of malignancy in a NRF2-dependnet manner.
Figure 6. PKCλ/ι Deficiency in Non-tumorous Liver Tissue Generates a Pro-tumorigenic Microenvironment.
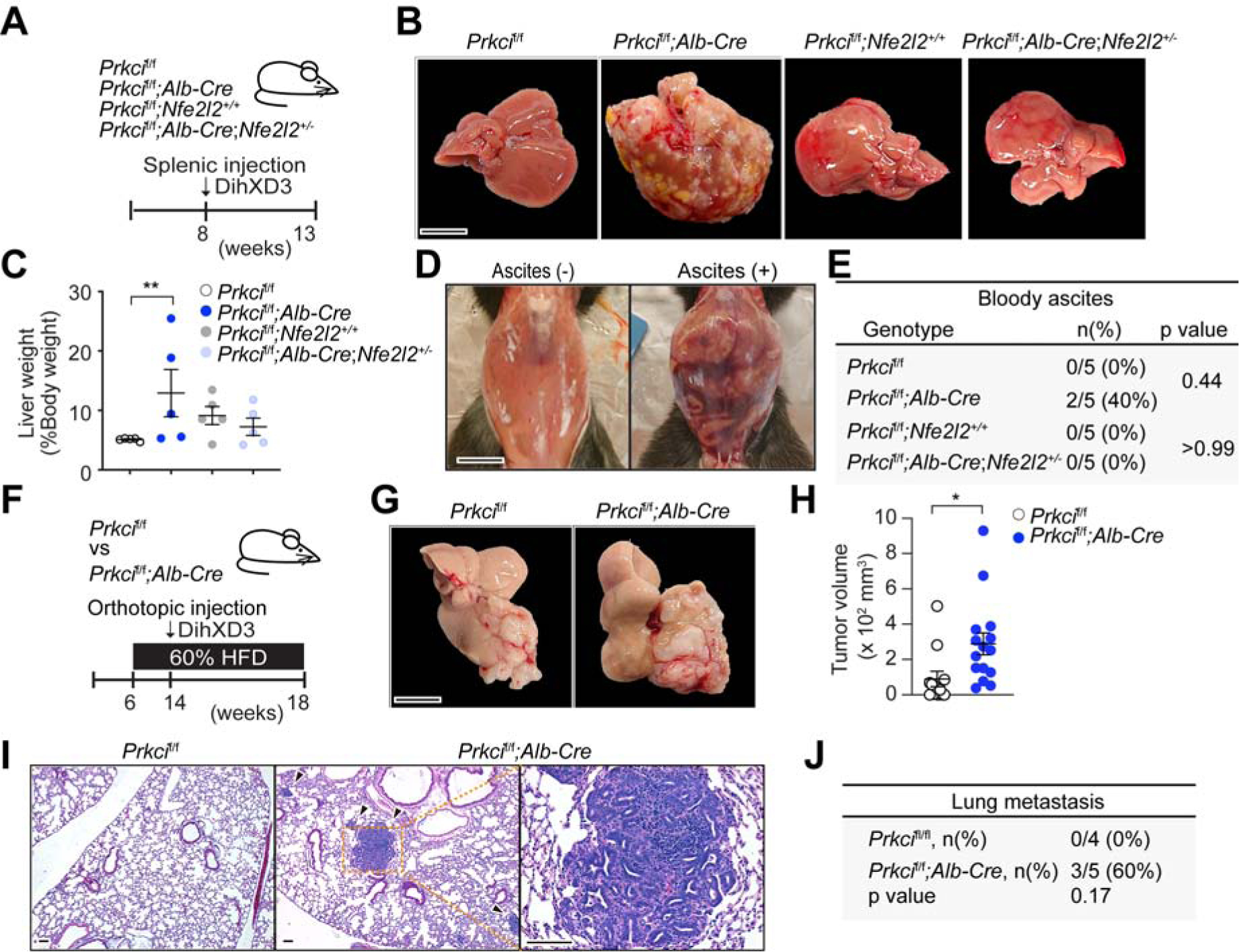
(A) Schematic representation of splenic injection of DihXD3.
(B-E) Images of transplanted livers (B), quantification of liver weight normalized to body weight (n = 5)
(C) and images (D), and incidence of bloody ascites (E) of mice treated as in (A).
(F) Schematic of orthotopic implantation of DihXD3 cells into livers of Prkcif/f and Prkcif/f;Alb-Cre male mice.
(G-J) Liver images (G), hepatic tumor volume (Prkcif/f, n = 12; Prkcif/f;Alb-Cre, n = 15 tumors) (H), H&E staining (I) and incidence (J) of lung metastasis (Prkcif/f, n = 4; Prkcif/f;Alb-Cre, n = 5 animals) of mice treated as in (F).
Mean ± SEM. *p < 0.05, **p < 0.01. Scale bar, 1 cm (B and G); 100 μm (I).
Human Relevance of PKCλ/ι in liver cancer
The negative correlation between tumor histological grade and PKCλ/ι expression observed in murine liver tumors (Figure S1A) was also seen in human clinical specimens of HCC that were analyzed by IHC with a PKCλ/ι -specific antibody. This analysis included 271 resected samples and showed that there is a significant reduction of PKCλ/ι expression in poorly differentiated human HCC (Figures 7A and 7B). Also, the mouse studies reported in this work demonstrated that the loss of PKCλ/ι in hepatocytes creates a pro-tumorigenic liver microenvironment (Figure 6). To validate these observations in human patients, we first investigated whether low PKCλ/ι expression in the surrounding non-tumor liver tissue constitute a risk factor for aggressive tumor development. Surgically resected tissues from 139 HCC patients were studied. Interestingly, both univariate and multivariate logistic regression analyses demonstrated that low PKCλ/ι expression (weak or no staining signals) in the non-tumor liver tissue is a significant predictor of poorly differentiated HCC (Figure 7C). Furthermore, the analysis of the transcriptomic data of 82 patients who had undergone surgery (Hoshida et al., 2008) revealed that low PKCλ/ι expression in the non-tumor liver area was significantly associated with late recurrence (Figure 7D). These findings support the human relevance of our studies in mice and establish that the loss of PKCλ/ι in non-tumor hepatocytes is decisive to create a microenvironment conducive to liver cancer. Interestingly, the upregulation of the PPARα-OXPHOS-ROS-NRF2 axis and the activation of autophagy revealed in the PKCλ/ι mutant mouse studies reported herein was also validated in human patients. Thus, the in-silico analysis of RNA-seq data of HCC patients in the TCGA dataset demonstrated a significant negative correlation between PKCλ/ι expression in background liver tissues and gene signatures corresponding to a more oxidative microenvironment, autophagy, NRF2 and PPARα (Figures 7E–7H).
Figure 7. Low PKCλ/ι Expression in Human Liver Tissue is a Risk Factor for Late HCC Recurrence.
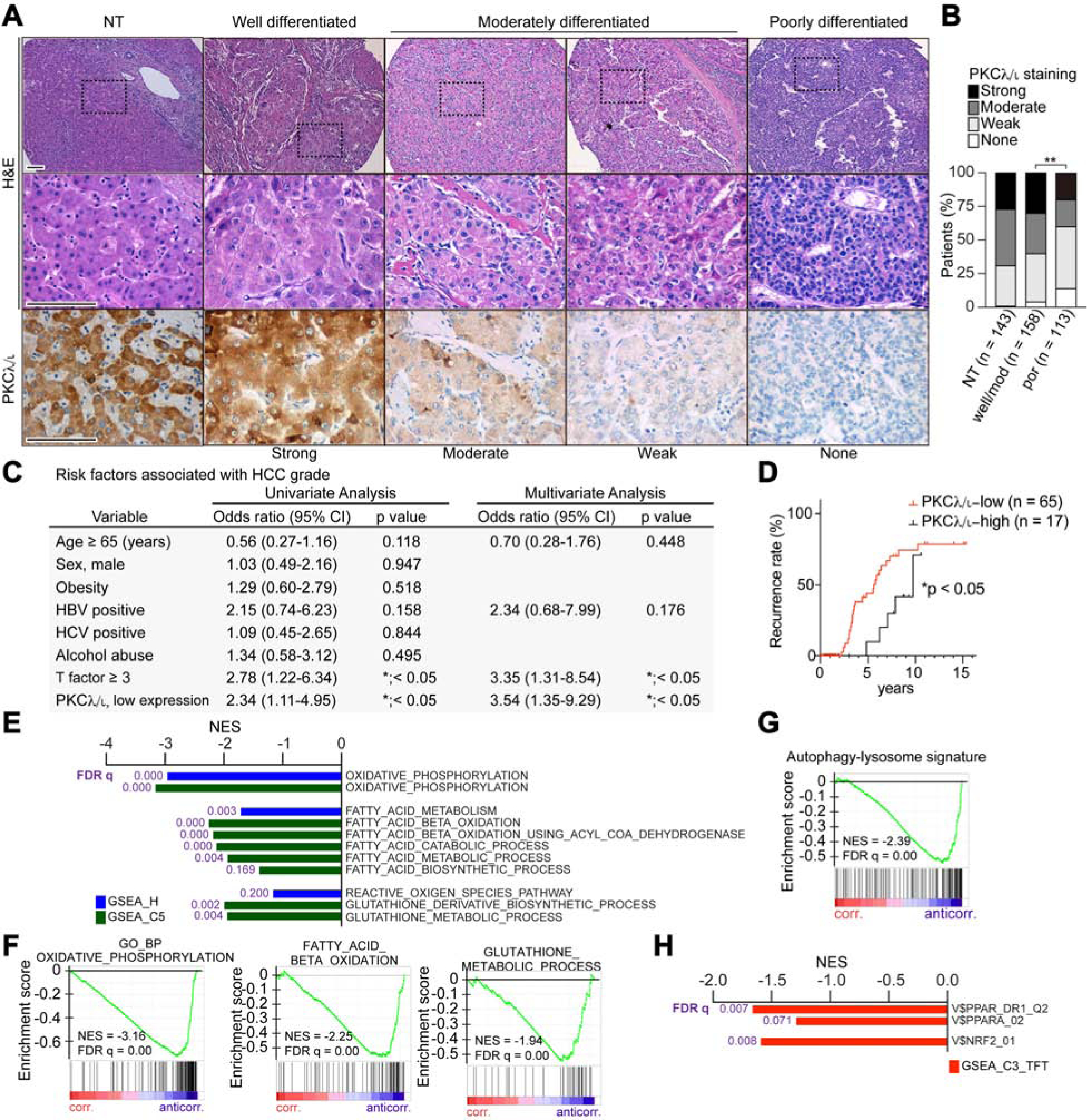
(A) H&E staining and IHC for PKCλ/ι in clinical HCC sections of different histological grade (n = 271 cases) and surrounding non-tumorous (NT, n = 143 cases) liver tissues. Scale bar, 100 μm.
(B) PKCλ/ι expression levels in HCC according to the IHC based classification; none, weak, moderate and strong (NT, n = 143; HCC, n = 271).
(C) Univariate and multivariate analyses to determine risk factors associated with HCC grade (odds ratio; poorly differentiated HCC vs HCC of well or moderately differentiated HCC), (n = 143).
(D) Kaplan-Meier curves of time to late recurrence in patients who underwent surgical resection of HCC. Patients were classified according to the PKCλ/ι expression level in surrounding non-tumorous liver tissues (n = 82).
(E) Negatively correlated pathways to Prkci gene expression in background liver tissues of HCC patients from TCGA dataset using compilation H and C5 (MSigDb).
(F-G) GSEA of the indicated genesets.
(H) Negative correlation between Prkci gene expression in background liver tissues of HCC patients from TCGA dataset with PPARα and NRF2 targets (C3, MSigDb).
*p < 0.05, **p < 0.01.
DISCUSSION
Here we show that the genetic inactivation of PKCλ/ι in hepatocytes results in metabolic changes that are critical for the progression of primary HCC tumors, the enhancement of its metastatic potential, and for creating a liver microenvironment conducive to cancer. Contrarily to previously reported pro-oncogenic roles of PKCλ/ι in leukemia and lung cancer (Reina-Campos et al., 2019a), its role in hepatocytes is to suppress tumorigenesis through the modulation of autophagy. Although autophagy inhibition in hepatocytes seems to drive hepatocarcinogenesis, in the context of PKCλ/ι deficiency, increased autophagy decisively contributes to the enhanced oxidative metabolism of PKCλ/ι-deficient hepatocytes and hepatoma cells. This metabolic reprogramming is essential for the upregulation of three major cancer hallmarks in PKCλ/ι-deficient transformed hepatocytes, namely, proliferation, spheroid formation, and EMT/invasion, which altogether drive the enhanced metastatic potential of these mutant hepatoma cells. Importantly, we also unveiled here the molecular mechanism whereby PKCλ/ι represses the autophagy machinery. This involves the phosphorylation of LC3 at S12 by PKCλ/ι, which impairs the LC3’s ability to interact with p62. This reveals the existence of a previously unappreciated p62-PKCλ/ι-LC3 ternary complex whereby PKCλ/ι binds p62, through their respective PB1 domains, to modulate the interaction of LC3 with p62’s LIR domain, which is inhibited by the constitutive phosphorylation of LC3 by PKCλ/ι. Therefore, our model establishes that during HCC progression, the reduction in the levels of PKCλ/ι in hepatocytes unleashes autophagy that sustains an oxidative metabolism that is required for tumor development. However, although the activation of autophagy is a required event in the tumorigenic pathways activated by PKCλ/ι deficiency, it might not be sufficient. To ensure the efficiency of this metabolic hallmark of HCC cells, the loss of PKCλ/ι also upregulates an oxidative metabolic program through the activation of PPARα. In this way, PKCλ/ι controls two critical arms in the oxidative program. On the one hand, it regulates the transcriptional machinery that provides the hepatocytes with the necessary oxidative network, while on the other hand, it also modulates the autophagy that sustains the oxidative function by providing the nutrients for oxidation.
This PKCλ/ι connection with PPARα and autophagy is essential to fulfill the metabolic requirements of hepatoma cells and for the generation of ROS, a “by-product” of an exacerbated oxidative metabolism (Murphy, 2009). ROS is responsible for the enhanced cell death of the PKCλ/ι-deficient hepatocytes and hepatoma cells, and most likely contributes to compensatory proliferation. The increased levels of ROS in this system serves to activate NRF2, which through the upregulation of ROS scavengers, keeps ROS levels below tolerable thresholds that allow proliferation without inducing insupportable levels of cell death. Under these conditions, the second role of NRF2 is to promote cell-autonomous proliferation by unleashing its pro-growth mechanisms. In this regard, NRF2 has been shown to virtually impact all of the hallmarks of cancer (Rojo de la Vega et al., 2018). However, in addition to these cell-autonomous dual effects as a pro-tumorigenic transcription factor, NRF2 is also critical for the cell non-autonomous role of PKCλ/ι in the liver tumor microenvironment. Therefore, NRF2 emerges as a critical component in the epithelium and the tumor microenvironment under conditions of enhanced autophagy.
STAR★ METHODS
RESOURCE AVAILABILITY
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jorge Moscat (jom4010@med.cornell.edu).
Data and Code Availability
The accession number for the RNA-seq data reported in this paper is GSE147801. The following data sets were analyzed: GSE10143 and UNSC Xena. The raw data of this study have been deposited in Mendeley Data and available at https://doi.org/10.17632/xm87k23hrh.1.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Prkcif/f and Alb-Cre mice were previously described (Nakanishi et al., 2018; Umemura et al., 2016). Alb-Cre mice were purchased from Jackson Laboratories. Nfe2l2−/− mice were kindly provided by Dr. Michael Karin, Ph.D. (Todoric et al., 2017). All these mouse strains were generated in a C57BL/6 background. NSG mice were purchased from the animal core at SBP Medical Discovery Institute. All mice were born and maintained under pathogen-free conditions. Animal handling and experimental procedures were approved by the Institutional Animal Care and Use Committee at SBP Medical Discovery Institute.
Human Samples
FFPE HCC tissue samples were obtained from Mayo Clinic (Minnesota, USA). Written informed consent was obtained from all patients with the protocol approved by the Ethics Committee of Mayo Clinic in accordance with the ethical guidelines for epidemiological research, as well as the principles expressed in the Declaration of Helsinki. De-identified samples were sent to SBP Medical Discovery Institute and used for histological analyses. The study was approved by the IRB Committee of SBP Medical Discovery Institute.
Cell Lines
HEK293T, Phoenix-GP, BNL CL.2, HepG2, and SK-HEP-1 cells were purchased from ATCC. All cells were negative for mycoplasm. BNL CL.2, DihXD3, HepG2, SK-HEP-1 and HEK293T cells were cultured in Dulbecco’s Modified Eagles Medium (DMEM, Corning) supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine in an atmosphere of 95% air and 5% CO2.
Establishment of DihXD3 Cells
Cell lines were derived from DEN-induced HCCs (Dih cells) of C57BL/6 female. Dih cells were injected subcutaneously in C57BL/6 males with low frequency of xenograft development. DihXD3 cells were derived from these developed xenografts.
METHODS DETAILS
Experiment Mouse Models
All genotyping was done by PCR. High Fat Calories (60%) Mouse Diet (F3282, Bio-Serv) was available ad libitum. Examinations of Prkcif/f;Alb-Cre and Prkczf/f;Alb-Cre mice were performed using both males and females, except those indicated respectively. Mice were fed regular chow and examined after 8 h of fasting at 30 weeks of age. Sex- and age-matched animals were allocated from each genotype into experimental groups. Animals for transplantation experiments were all males and were randomized between litters to prevent a bias towards age. An identification code was assigned to each animal and the investigators were not blinded to group allocation at the time of data collection and analysis. We subjected C57BL/6, Prkcif/f, Prkcif/f;Alb-Cre, Prkczf/f, and Prkczf/f;Alb-Cre mice to a well-established protocol to develop liver cancer under condition of obesity as previously described (Duran et al., 2016). Two-week-old mice were intraperitoneally injected with hepatic carcinogen diethylnitrosamine (DEN, 25 mg/kg) and two weeks later fed a 60% fat diet as a tumor promoter for 32 weeks. Animals were sacrificed at 36 weeks of age and examined after 6 h fast. For transplantation, sgPrkci (or sgC) DihXD3 cells or shPRKCI (or shC) SK-HEP-1 cells were trypsinized, washed two times in cold PBS, aliquoted to 2 million cells per dose in a 100 μl volume and injected into spleen of male 8-week NSG mice followed by splenectomy. Liver and lung tissues were examined on the 14th postoperative day. The number or the area of lung nodules was counted by H&E staining on the section that showed whole lobes. The burden of primary (hepatic) tumor was represented by ratio of liver weight over body weight. Metastatic capacity of cells was assessed as Metastatic index, defined as ratio of lung metastasis number (DihXD3 cells) or area (SK-HEP-1; it was hard to count the number of metastatic lesions because these cells developed disseminated lesions in lung) over the burden of primary tumor above. DihXD3 cells were trypsinized, washed two times in cold PBS, aliquoted to 2 million cells per dose in a 50 μl volume and injected into spleen of male 8-week Prkcif/f, Prkcif/f;Alb-Cre, Prkcif/f;Nfe2l2+/+, and Prkcif/f;Alb-Cre;Nfe2l2+/− mice followed by splenectomy. Liver tissues were examined on the 35th postoperative day. For the HCC orthotopic model, DihXD3 cells were trypsinized, washed two times in cold PBS, aliquoted to 1 million cells per dose in a 20 μl volume of 50% Matrigel and injected in the subcapsular region of the median and left lateral lobes of Prkcif/f and Prkcif/f;Alb-Cre mouse liver of 14 weeks of age. Each liver had totally 3 injection sites, which lead to 3 independent tumors. Animals were fed HFD for 8 weeks before and 4 weeks after the transplantation. Liver and lung tissues were examined on 28th postoperative day. Tumor volume was calculated for each tumor nodule using the following formula: 4/3 × 3.14 × (short diameter)2 × (long diameter). For GW6471 treatment experiment, GW6471 (10 mg/kg) or vehicle (4% DMSO, 2% Tween80 in saline) was daily administered intraperitoneally in Prkcif/f, Prkcif/f;Alb-Cre male mice at 8 weeks of age for 14 days, and livers were extracted 24 h after the last injection.
Cell Culture Experiments
To knockout PKCλ/ι in HEK293T, HepG2, BNL CL.2 and DihXD3 cells, 20-nucleotide single-guide RNA sequences targeting the human PRKCI (HEK293T and HepG2 cells) or mouse Prkci (BNL CL.2 and DihXD3 cells) gene were purchased from Synthego and transduced into cells with recombinant Streptococcus pyogenes Cas9 protein (Truecut Cas9 Protein v2, Thermo) using Neon Electroporation System (Invitrogen). Single clones were expanded and screened for PKCλ/ι expression by protein immunoblotting. To knockdown PRKCI gene in SK-HEP-1 cells, TRC lentiviral shRNA targeting human PRKCI (TRCN000006039) was obtained from Sigma-Aldrich. shRNA-encoding plasmids were cotransfected with psPAX2 (Addgene) and pMD2.G (Addgene) plasmids into HEK293T cells by using XtremeGene HP transfection reagent (Roche). Virus-containing supernatants were collected 48 h after transfection, filtered to eliminate cells, and then used to infect SK-HEP-1 cells in the presence of 8 μg/ml polybrene followed by puromycin selection (2 μg/ml). Knockdown of Prkcz, Cpt1a and Nfe2l2 genes in BNL CL.2 cells were achieved by siRNA transfection using Lipofectamine RNAiMAX Transfection Reagent (Invitrogen).
Transient overexpression was achieved by transfection using X-tremeGENE HP transfection reagent (Roche). Transfected cells were examined 48 h after transfection. Establishing stably FLAG-tagged LC3A (WT or S12A mutant) or mCherry-GFP-LC3B (WT or T12A mutant) expressing BNL CL.2 cells was achieved by retrovirus-mediated transduction. Retroviruses were prepared and used as previously described (Linares et al., 2015). Briefly, retroviruses were produced in Phoenix-GP cells using X-tremeGENE HP transfection reagent (Roche). Virus-containing supernatants were collected 48, 72 and 96 h after transfection, filtered to eliminate cells, and supplemented with 8 μg/ml polybrene. BNL CL.2 cells were infected with three rounds of viral supernatants and selected with hygromycin (75 μg/ml) or puromycin (3 μg/ml). For amino acid starvation with or without bafilomycin A1 (BafA1) treatment, HEK293T cells in culture dishes were rinsed with PBS and incubated in serum and amino acid-free RPMI media for 48 h with BafA1 (100 nM). Cell viability was determined by trypan blue exclusion assay at the indicated time.
Respiration Assays
The cellular oxygen consumption rate (OCR) of primary adipocytes was determined using an XFp Extracellular Flux Analyzer (Seahorse Bioscience). Prior to assay, 2 × 104 cells were seeded into XFp microplates. One hour prior to OCR measurement, culture medium was changed to XF basal medium containing 10 mM glucose and 1 mM pyruvate. L-glutamine (2 mM) was supplemented when indicated. The uncoupled and maximal OCRs were determined using 1 μM oligomycin and 1 μM carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), respectively. Complex-I-dependent respiration was inhibited with a mix of antimycin A and rotenone, 2 μM each. After OCR measurement, cells were stained with DAPI and imaged with the EVOS FL Auto Imaging System. Cell number was quantified in more than 8 random fields in each sample using Image J software (NIH). Oxygen consumption values were normalized to cell number per field. Cells were treated with GW6471 (10 μM) or bafilomycin A1 (BafA1, 30 nM) for 24 h prior to OCR measurement when indicated, and 10 μM GW6471 or 30 nM BafA1was also supplemented in the XF basal medium through the experiment. Prkcz knockdown BNL CL.2 cells were analyzed a day after siRNA transfection.
Isotopic Labeling
sgC and sgPrkci BNL CL.2 cells were cultured in DMEM medium containing 1% (v/v) delipidated FBS 24 h prior starting tracing experiments. Cells were then incubated for 24 h in [13C]glucose medium (DMEM (Sigma) medium supplemented with 1% (v/v) delipidated FBS, 6.25 mM [U-13C6] glucose, 18.75 mM [12C]glucose, and 4 mM [12C]glutamine) prior metabolite extraction. For analysis of fatty acid oxidation, cells were incubated for 6 h in DMEM medium containing 1% (v/v) delipidated FBS. [U-13C16]palmitate was noncovalently bound to fatty acid-free BSA and added to culture medium at 5% of the final volume (50 μM final concentration). Metabolite abundances were normalized to cell counts. Labeling (corrected for natural abundance using in-house software) is depicted as isotopologue distributions or as labeled fraction of metabolites (mole percent enrichment (MPE)). The contribution of [13C]glucose to lipogenic acetyl-CoA pool as well as fraction of newly synthesized palmitate was determined via isotopomer spectral analysis analysis (ISA) using INCA. Significance was considered as non-overlapping confidence intervals. Glucose, glutamine consumption, and lactate production was determined using the YSI2950 analyzer as described (Valencia et al., 2014).
Gas Chromatography-Mass Spectrometry (GC-MS) Sample Preparation and Analysis
Metabolites were extracted using a modified Bligh and Dyer method and analyzed as previously described in detail (Linares et al., 2017). Briefly, metabolites were extracted with 0.5 ml −20°C methanol, 0.2 ml 4°C cold water, and 0.5 ml −20 °C chloroform. The extracts were vortexed for 10 min at 4 °C and centrifuged at 16,000×g for 5 min at 4 °C. The upper aqueous phase was evaporated under vacuum at −4 °C, the lower organic phase under airflow. Derivatization for polar metabolites was performed using a Gerstel MPS with 15 μl of 2% (w/v) methoxyamine hydrochloride (Thermo Scientific) in pyridine (incubated for 60 min at 45 °C) and 15 μl N-tertbutyldimethylsilyl-N-methyltrifluoroacetamide (MTBSTFA) with 1% tert-butyldimethylchlorosilane (Regis Technologies) (incubated further for 30 min at 45 °C). Polar derivatives were analyzed by GC-MS using a DB-35MS column (30 m × 0.25 i.d. × 0.25 μm) installed in an Agilent 7890B gas chromatograph (GC) interfaced with an Agilent 5977B mass spectrometer (MS) operating under electron impact ionization at 70 eV. The MS source was held at 230°C, the quadrupole at 150°C, helium was used as carrier gas and the GC oven was held at 100°C for 1 min, increased to 300°C at 10°C/min, and held at 325°C for 3 min. The lower organic phase was derivatized to form fatty acid methyl esters (FAMES) using 500 μl 2% H2SO4 in MeOH and incubation at 50 °C for 2 h. FAMES were extracted via addition of 100 μl saturated salt solution and 500 μl hexane. FAMES were analyzed using a Select FAME column (100 m × 0.25 mm i.d.) installed in an Agilent 7890 A GC interfaced with an Agilent 5975C MS. Helium was used as carrier gas and the GC oven was held at 80 °C, increased by 20 °C/min to 170 °C, increased by 1 °C/min to 204 °C, then 20 °C/min to 250 °C and hold for 10 min.
LC3 Flux Analyses
sgPrkci and sgC BNL CL.2 cells were treated with lysosomal inhibitors (PI) (20 mM NH4Cl and 200 μM leupeptin) for 0, 2 and 4 h. After the incubation, cells were lysed and subjected to immunoblotting analyses. Amount of LC3-II was normalized to that of actin. Autophagosome formation as: LC3II4h - LC3II2h. We generated reporter cells stably expressing mCherry-GFP-LC3B, either WT or T12A mutant, reporter plasmid using retrovirus vector with selection by puromycin. After fixation followed by DAPI staining, cells were imaged with Zeiss LSM 710 NLO Confocal Microscope. The number of autophagosomes (shown as mCherry and GFP double positive puncta) and autolysosomes (shown as mCherry only positive puncta) were quantified in more than 20 cells using Image J software (NIH).
NRF2-ARE Reporter Assay
sgPrkci and sgC BNL CL.2 cells were transfected with pREP-8xARE-GFP-SV40-BFP plasmid (Addgene) and fixed with 4% PFA in PBS 48 h after transfection. Cells were imaged with Zeiss LSM 710 NLO Confocal Microscope, and NRF2-ARE transactivities were quantified as GFP intensity ratio to BFP intensity in more than 10 cells with BFP expression.
Primary Hepatocyte Isolation and Culture
Adult Prkcif/f and Prkcif/f;Alb-Cre mice were subjected to standard two-step collagenase perfusion for isolation of primary hepatocytes. Briefly, the liver was pre-perfused through the portal vein with perfusion buffer (0.5 mM EDTA, 137 mM NaCl, 2.7 mM KCl, 280 mM Na2HPO4, 10 mM HEPES, pH 7.65) and then perfused with digestion buffer (0.5 mg/ml Collagenase type IV, 14 mM CaCl2, 137 mM NaCl, 2.7 mM KCl, 280 mM Na2HPO4, 10 mM HEPES, pH 7.65). Digested liver was suspended in culture media (William’s E media supplemented with 2 mM glutamine, 100 nM dexamethasone and 10% FBS) and washed to eliminate dead cells and enrich hepatocytes. Viability of isolated hepatocytes was around 90% as determined by Trypan blue staining. Cells were plated on collagen-I-coated plates and subjected to Seahorse analysis 2 days after isolation.
Mouse Serum and Plasma Analysis
Mouse whole blood was collected from inferior vena cava. Serum was prepared by centrifugation of whole blood to remove clot after incubation at 4 °C overnight. Plasma was prepared by collecting whole blood in tubes treated with an anticoagulant (BD Microtainer K2EDTA, Becton Dickinson) followed by centrifugation to eliminate cells. Serum aminotransferase (ALT) levels were measured by using VetScan v2 Chemistry Analyzer (Abaxis). Plasma ketone body (β-hydroxybutyrate, 3-HB) levels were measured with fresh plasma from 36 weeks old mice by using β-hydroxybutyrate Colorimetric Assay Kit (Cayman).
Lipid Analysis
Total lipids were extracted from liver tissues by Folch method. Briefly, around 100 mg frozen liver tissue samples were homogenized in 1 ml of distilled water, 50 μl homogenates were further mixed with 1 ml chloroform-methanol (2:1 v/v) mixture and incubated at room temperature with shaking for 30 min to extract lipid. Samples were then mixed roughly with 0.2 ml H2O for 30 sec to separate phases. The lipid-containing organic phase (bottom) were collected and dried by nitrogen and finally reconstituted in 100 μl isopropanol. Triacylgrycerol concentration was determined spectrophotometrically (Wako Diagnostics). Liver tissue tryacylgrycerol leves was normalized to tissue weight.
Histological Analysis
Tissues from indicated mice were isolated, rinsed in cold PBS, fixed in 10% neutral buffered formalin for 24 h, dehydrated, and embedded in paraffin. Tissues were also embedded in Tissue Tek O.CT. compound and snap frozen in liquid nitrogen, then kept in −80C until examination. Tissue sections of 5 μm thickness were subjected to hematoxylin and eosin (H&E) and subsequent assays. Pathological characterization was assessed by a pathologist and a hepatologist independently. Frozen liver sections (5 μm) were stained with Oil red O (Sigma-Aldrich) to detect lipid accumulation. Sections were fixed in paraformaldehyde (PFA) and stained for 3 h in 0.5% Oil red O in propylene glycol, followed by 1 min incubations in 85% aqueous propylene glycol. After the slides were washed in distilled water, they were counterstained with hematoxylin. Fibrosis was assessed by Sirius Red staining; formalin-fixed paraffin-embedded (FFPE) liver sections were deparaffinized, rehydrated, and stained for 2 h in 0.1% Sirius red in saturated picric acid (Electron Microscopy Sciences), followed by rinse in 5% acetic acid. Frozen liver sections were stained with dihydroethidium (DHE) for intracellular ROS detection; sections were incubated in 2 μM DHE for 30 min at 37 °C and imaged with Zeiss LSM 710 NLO Confocal Microscope. Oil red O, Sirius red and DHE positive areas were quantified in more than 6 random fields on each slide using Image J software (NIH). For staining of immune cells in liver tissues, frozen sections were fixed with methanol, blocked, and incubated with primary antibody overnight at 4 °C, followed by incubation with fluorochrome-conjugated secondary antibody. For immunohistochemistry (IHC), FFPE sections were deparaffinized, rehydrated, and then treated for antigen retrieval. After blocking in avidin/biotin solutions (Vector Laboratories), tissues were incubated with primary antibody overnight at 4°C, followed by incubation with biotinylated secondary antibody. Endogenous peroxidase was quenched in 3% H2O2 in water at room temperature. Antibodies were visualized with avidin/biotin complex (Vectastain Elite, Vector Laboratories) using diaminobenzidine as the chromagen. For immunofluorescent (IF) observation, FFPE sections were deparaffinized, rehydrated, and then treated for antigen retrieval. After blocking sections were incubated with primary antibody overnight at 4 °C, followed by incubation with fluorochrome-conjugated secondary antibody, and imaged with Zeiss LSM 710 NLO Confocal Microscope.
Transmission electron microscopy and morphometric analysis.
BNL CL.2 cells (sgC,and sgPrkci) were fixed in glutaraldehyde. After embedding in Epon Araldite, sections were cut and examined using a Philips CM-100 Transmission Electron Microscope. Morphometric classification of autophagic vacuoles was done following the standard criteria as follows: autophagosomes were distinguished as double membrane vesicles with content of similar density as the surrounding cytosol and comprised often of recognizable cellular structures; autolysosomes were identified as single or partially double membrane vesicles of content of lower density than the surrounding cytosol and comprised of amorphous content or partially degraded cellular structures. In both cases, the limiting membrane had to be denuded of ribosomal particles. Morphometric analysis of autophagic compartments was done by double-blinded independent observers using a single-category allocation or each vesicle in at least 20 different micrographs per condition.
Flow Cytometry Analysis
Nonparenchymal cells (NPC) were extracted from livers according to previously described method (L’Hermitte et al., 2019) using percoll gradient density. Briefly, livers were harvested and perfused with 1× HBSS containing 10 mM HEPES to remove circulating blood cells. The liver was passed through a 10 μm cell strainer in RPMI 1640 supplemented with 2% FBS, 2 mM GlutaMAX, 100 U/ml penicillin, and 100 μg/ml streptomycin and 50 μM β-mercaptoethanol (Sigma-Aldrich). The liver cell suspension was collected, and parenchymal cells were separated from NPC by centrifugation for 2 min at 700 rpm. The supernatant containing the NPCs was collected and centrifuged for 10 min at 1500 rpm. The pellet was then resuspended in 40% percoll diluted in RPMI 1640 supplemented with 2% FBS, layered onto 80% percoll, and centrifuged for 15 min at room temperature at 2000 rpm. The cells concentrated at the interface were collected and washed in cold PBS. Red blood cells were removed with lysing buffer (BD Pharm Lyse) and live cells were counted using Trypan blue and then saturated with mouse Fc Block (purified anti-mouse CD16/CD32; 1:50; clone 2.4G2; BD Pharmingen) 30 min at 4°C before incubating with specific dyed antibodies.
Tumor Sphere Formation Assay
HepG2 cells (sgC,and sgPRKCI) were cultured in suspension of DMEM/F12 media (Gibco) supplemented with 2% B27 (Invitorgen), 1% Insulin-Transferrin-Selenium (Invitrogen), 2 ng/ml epidermal growth factor (Invitrogen), 2 ng/ml basic fibroblast growth factor (Invitrogen) and 1% methylcellulose (Sigma). Two thousand cells were plated into 24 well Ultra Low Attachment plates (Costar), and spheres with a diameter larger than 70 μm were counted after 10 days of culture. In the experiment involving GW6471 treatment, 50 μl of 1 μM GW6471 was supplemented to the culture media (500 μl) every 2 days throughout the experimental period.
Invasion Assay
DihXD3 cells (sgC,and sgPrkci) or SK-HEP-1 cells (shC and shPRKCI) were assayed for their ability to invade through a polyethylene terephthalate membrane inserts with 8 μm pores coated with extracellular matrix proteins in 24-well format (Matrigel Invasion Chambers, Corning). 1 × 105 cells (DihXD3) or 2 × 105 cells (SK-HEP-1) were seeded into the inner chamber in serum-free DMEM, and cells were challenged with 10% FBS-containing DEME in the outer chamber. After 22 h of incubation at 37 °C, 5% CO2, cells that invaded through the pores onto the bottom of the insert were fixed in cold methanol and stained with crystal violet. In the experiment involving chloroquine treatment, chloroquine (5 μM) was supplemented to both inner and outer chambers. In the experiment involving GW6471 treatment, cells were treated with GW6471 (10 μM) for 24 h prior to seeding, and GW6471 (10 μM) was supplemented to both inner and outer chambers.
Immunoblotting Analysis
Cells for protein analysis were lysed in RIPA buffer (20 mM Tris-HCl, 37 mM NaCl2, 2 mM EDTA, 1% Triton-X, 10% glycerol, 0.1% SDS, and 0.5% sodium deoxycholate) with phosphatase and protease inhibitors. For nuclear protein extraction, cells were incubated with buffer A (20 mM Tris-HCl at pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 1 mM PMSF) for 10 min on ice, centrifuged at 3,500 rpm, resuspended in buffer C (20 mM Tris-HCl at pH 7.9, 1.5 mM MgCl2, 0.42 M NaCl, 0.5 mM DTT, 0.2 mM PMSF, 0.2 mM EDTA), sonicated, and centrifuged at 14,000 rpm. Liver tissues were lysed using TissueLyser II (QIAGEN) in RIPA buffer with phosphatase and protease inhibitors. To extract nuclear fraction from tissues, liver tissues were homogenized in homogenize buffer (10 mM HEPES at pH 7.8, 10 mM KCl, 0.1 mM EDTA and 20% glycerol) using Dounce homogenizer, centrifuged at 1000 g, resuspended in nuclear fraction buffer (20 mM HEPES at pH 7.8, 0.4 M NaCl, 1mM EDTA, and 20% glycerol) with phosphatase and protease inhibitors for 15 min on ice, and centrifuged at 1,000 g. For immunoprecipitations, cells were lysed in σ3 buffer (100 mM NaCl, 25 mM Tris, 1% Triton-X, 10% glycerol, with phosphatase and protease inhibitors) and immunoprecipitated with 25 μl of 50% slurry of protein Glutathione-Sepharose 4B beads (Bioworld). Immunoprecipitates were washed three times with lysis buffer, once with high salt (500 mM NaCl), and once more with lysis buffer. Protein concentration in lysates were determined by using Protein Assay Kit (Bio-Rad). Cell extracts and immunoprecipitated proteins were denatured, subjected to SDS-PAGE, transferred to PVDF membranes (GE Healthcare). After blocking with 5% nonfat dry milk in Tris-buffered saline and 0.1% Tween (TBS-T), the membranes were incubated with the specific antibodies (as listed in Key Resources Table) overnight at 4 °C. After 2 h incubation with the appropriate horseradish peroxidase-conjugated antibodies, the immune complexes were detected by chemiluminescence (Thermo Scientific).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Mouse anti-β-actin | Sigma-Aldrich | Cat# A1978, RRID:AB_476692 |
| Mouse anti-CPT1A | Abcam | Cat# ab128568, RRID:AB_11141632 |
| Mouse anti-E-cadherin | BD Biosciences | Cat# 610181, RRID:AB_397580 |
| Mouse anti-FLAG® | Sigma-Aldrich | Cat# F3165, RRID:AB_259529 |
| Mouse anti-His-probe | Santa Cruz Biotechnology | Cat# sc-8036, RRID:AB_627727 |
| Mouse anti-HNF4α | Santa Cruz Biotechnology | Cat# sc-374229, RRID:AB_10989766 |
| Mouse anti-N-Cadherin | BD Biosciences | Cat# 610920, RRID:AB_2077527 |
| Mouse anti-NK1.1 | BD Pharmingen | Cat# 553165, RRID:AB_394677 |
| Mouse anti-NQO1 | Santa Cruz Biotechnology | Cat# sc-32793, RRID:AB_628036 |
| Mouse anti-NRF2 | Santa Cruz Biotechnology | Cat# sc-365949, RRID:AB_10917561 |
| Mouse anti-phospho-Threonine | Cell Signaling Technology | Cat# 9386, RRID:AB_331239 |
| Mouse anti-PKCλ | BD Biosciences | Cat# 610208, RRID:AB_397607 |
| Rabbit anti-Albumin [EPR12774] | Abcam | Cat# ab192603 |
| Rabbit anti MAP1LC3A | Cell Signaling Technology | Cat# 4599, RRID:AB_10548192 |
| Rabbit anti-Cleaved Caspase3 | Cell Signaling Technology | Cat# 9664, RRID:AB_2070042 |
| Rabbit anti-GFP | Cell Signaling Technology | Cat# 2956, RRID:AB_1196615 |
| Rabbit anti-HA-Tag | Cell Signaling Technology | Cat# 3724, RRID:AB_1549585 |
| Rabbit anti-Ki-67 | Abcam | Cat# ab16667, RRID:AB_302459 |
| Rabbit anti-PARP-1 | Santa Cruz Biotechnology | Cat# sc-7150, RRID:AB_2160738 |
| Rabbit anti-Phospho MAP1LC3A(S12) | Abgent | Cat# AP3301a, RRID:AB_2137560 |
| Rabbit anti-PKCz | Cell Signaling Technology | Cat# 9368, RRID:AB_10693777 |
| Rabbit anti-Snail | Cell Signaling Technology | Cat# 3879, RRID:AB_2255011 |
| Rabbit anti-SQSTM1/p62 | Thermo Fisher Scientific | Cat# PA5–20839, RRID:AB_11157045 |
| Rabbit anti-Thiophosphate ester | Abcam | Cat# ab92570, RRID:AB_10562142 |
| Rabbit anti-V5 tag | Thermo Fisher Scientific | Cat# PA1–993, RRID:AB_561893 |
| Rabbit anti-ZEB1 | Santa Cruz Biotechnology | Cat# sc-25388, RRID:AB_2217979 |
| Rat anti-B220 (CD45R) | Thermo Fisher Scientific | Cat# 14-0452-82, RRID:AB_467254 |
| Rat anti-B220 (CD45R) | BD Pharmingen | Cat# 552771, RRID:AB_394457 |
| Hamster anti-CD3 | D Biosciences | Cat# 550275, RRID:AB_393572 |
| Hamster anti-TCRβ chain | BD Pharmingen | Cat# 553174, RRID:AB_398534 |
| Armenian hamster anti-TCRβ chain | eBioscience | Cat# 45–5961, RRID:AB_925764 |
| Armenian hamster anti-CD11c | BD Pharmingen | Cat# 558079, RRID:AB_647251 |
| Rat anti-CD16/CD32 | BD Pharmingen | Cat# 553142, RRID:AB_394657 |
| Rat anti-CD4 | BD Horizon | Cat# 560468, RRID:AB_1645271 |
| Rat anti-CD8 | BD Pharmingen | Cat# 557654, RRID:AB_396769 |
| Rat anti-CD11b | eBioscience | Cat# 47–0112, RRID:AB_1603193 |
| Rat anti-CD45 | BD Horizon | Cat# 563891, RRID:AB_2734134 |
| Rat anti-F4/80 (IF) | Abcam | Cat# ab6640, RRID:AB_1140040 |
| Rat anti-F4/80 (FACS) | eBioscience | Cat# 17–4801, RRID:AB_469452 |
| Rat anti-Ly6C | BD Horizon | Cat# 560594, RRID:AB_1727559 |
| Rat anti-Ly6G (IF) | Bio X Cell | Cat# BE0075–1, RRID:AB_1107721 |
| Rat anti-Ly6G (FACS) | BD Pharmingen | Cat# 561236, RRID:AB_10611860 |
| Rat anti-KRT19 | DSHB | Cat# TROMA-III, RRID:AB_2133570 |
| Rat anti-MHC class II (I-A/I-E) | eBioscience | Cat# 11–5321, RRID:AB_465232 |
| Rat anti-PD-L1 | eBioscience | Cat# 12–5982, RRID:AB_466088 |
| Goat anti-Mouse IgG, secondary, HRP | Thermo Fisher Scientific | Cat# 31436, RRID:AB_228313 |
| Goat anti-Mouse IgG1, secondary, HRP | Thermo Fisher Scientific | Cat# PA1–74421, RRID:AB_10988195 |
| Goat anti-Rabbit IgG1, secondary, HRP | Thermo Fisher Scientific | Cat# 31461, RRID:AB_228347 |
| Goat anti-Rabbit IgG, secondary, Biotin | Agilent | Cat# E0432, RRID:AB_2313609 |
| Rat anti-Mouse IgG1, secondary, Biotin | BD Biosciences | Cat# 550331, RRID:AB_2296342 |
| Normal mouse IgG | Santa Cruz Biotechnology | Cat# sc-2025, RRID:AB_737182 |
| DH5α Competent Cells | Thermo Scientific | Cat# 18265017 |
| One Shot Stbl3 Chemically Competent | Thermo Scientific | Cat# C737303 |
| Human samples (Normal liver tissues, hepatocellular carcinomas) | Mayo Clinic, Minnesota, USA | N/A |
| 2-Mercaptoethanol | Gibco | Cat# 31350010 |
| [U-13C16]Palmitate | Cambridge Isotope | Cat# CLM-409-PK |
| [U-13C6]Glucose | Cambridge Isotope | Cat# CLM-1396-PK |
| Ampicillin | Fisher BioReagents | Cat# BP1760–25 |
| Antimycin A | Sigma-Aldrich | Cat# A8674 |
| B27 Supplement | Gibco | Cat# 17504001 |
| Bafilomycin A1 | Selleck Chemicals LLC | Cat# S1413 |
| bFGF recombinant protein | Gibco | Cat# 13256029 |
| Butylated hydroxyanisole (BHA) | Sigma-Aldrich | Cat# B1253 |
| Collagenase type IV | Sigma-Aldrich | Cat# C5138–1G |
| Crystal violet solution | Sigma-Aldrich | Cat# V5265 |
| DAPI | Life Technologies | Cat# D1306 |
| Dexamethasone | Sigma-Aldrich | Cat# D1756 |
| Dihydroethidium (DHE) | Sigma-Aldrich | Cat# D7008 |
| DMEM | Corning | Cat# 10–017CV |
| DMEM/F-12, GlutaMAX supplement | Gibco | Cat# 10565018 |
| ECL Western Blotting Substrate | Thermo Scientific | Cat# 32106 |
| Etomoxir | Sigma-Aldrich | Cat# E1905 |
| FCCP | Sigma-Aldrich | Cat# C2920 |
| GW6471 | Sigma-Aldrich | Cat# G5045 |
| HBSS (no calcium, no magnesium) | Gibco | Cat# 14175095 |
| HEPES | Gibco | Cat# 15630080 |
| High Fat Calories (60%) Mouse Diet | Bio-Serv | Cat# F3282 |
| His6_LC3/MAP1LC3A recombinant human protein | BostonBiochem | Cat# UL-430 |
| Hygromycin B Solution | Corning | Cat# 30–240-CR |
| Insulin-Transferrin-Selenium (ITS) | Gibco | Cat# 41400045 |
| L-Glutamine | Corning | Cat# 25–005-CI |
| Lipofectamine RNAiMAX Transfection Reagent | Invitrogen | Cat# 13778030 |
| lonomycin | Sigma-Aldrich | Cat# I0634 |
| Lysing Buffer | BD Pharm Lyse | Cat# 555899 |
| Matrigel®Growth Factor Reduced (GFR) Basement Membrane Matrix | Corning | Cat# 356230 |
| Methyl cellulose, 400 cP | Sigma-Aldrich | Cat# 0262 |
| MultiScribe Reverse Transcriptase | Invitrogen | Cat# 4311235 |
| Murine EGF | Gibco | Cat# PMG8045 |
| N-Acetyl Cystein (NAC) | Sigma-Aldrich | Cat# A7250 |
| Oil Red O 0.5% Solution in Propylene Glycol | Poly Scientific R&D Corp. | Cat# s1848 |
| Oligomycin | Sigma-Aldrich | Cat# 75351 |
| Opti-MEM Reduced Serum Medium | Gibco | Cat# 31985070 |
| PBS (no calcium, no magnesium) | Gibco | Cat# 10010–023 |
| Percoll | GE Healthcare | Cat# GE17-0891-01 |
| Phorbolmyristate acetate | Sigma-Aldrich | Cat# P8139 |
| PKCλ recombinant human protein | The Proteomics Core at SBP Medical Discovery Institute | N/A |
| Polybrene Infection Reagent | Sigma-Aldrich | Cat# TR-1003-G |
| Puromycin | Omega Scientific, inc. | Cat# PR-01 |
| Quick-RNA Miniprep Kit | Zymo Research | Cat# R1054 |
| QuickChange II Site-Directed Mutagenesis Kit | Agilent | Cat# 200523 |
| RNAlater Stabilization Solution | Invitrogen | Cat# AM7021 |
| Rotenone | Sigma-Aldrich | Cat# R8875 |
| RPMI 1640 Medium, GlutaMAX supplement | Gibco | Cat# 61870036 |
| Sirius Red, 0.1% in Saturated Picric Acid | Electron Microscopy Sciences | Cat# 50-300-77 |
| Tissue-Tek OCT compound | Sakura Finetek USA | Cat# 4583 |
| TRIzol | Thermo Fisher Scientific | Cat# 15596018 |
| Trypan Blue Solution, 0.4% | Gibco | Cat# 15250061 |
| Trypsin-EDTA (0.25%), phenol red | Gibco | Cat#25200056 |
| X-tremeGENE HP DNA Transfection Reagent | Roche | Cat# 6366236001 |
| Calyculin A | Sigma | Cat# C5552 |
| β-hydroxybutyrate Colorimetric Assay Kit | Cayman Chemical | Cat# 700190 |
| Matrigel invasion assay | Corning | Cat# 354480 |
| Mouse on Mouse (M.O.M.) Basic Kit | Vector | Cat# BMK-2202 |
| SeaHorse Seahorse XF24 Islet Capture FluxPak | Agilent | Cat# 101174–100 |
| Triacylglycerol quantification assay | Wako | Cat# 992–02892 |
| VECTASTAIN® Elite(R)ABC-HRP Kit | Vector | Cat# PK-6100 |
| VetScan Mammalian Liver Profile | Abaxis | Cat# 500–0040 |
| RNA-seq (Prkcif/f and Prkcif/fAlbCre livers) | This study; GEO | GSE147801 |
| RNA-seq (Prkcif/f and Prkcif/fAlbCre livers under DEN/HFD protocol) | This study; GEO | GSE147801 |
| Microarray Gene Expression (human liver tissues) | GEO | GSE10143 |
| RNA-seq (human liver tissues) | TCGA-LIHC | UNSC Xena |
| Raw Data | This study; Mendeley Data | https://doi.org/10.17632/xm87k23hrh.1 |
| BNL CL.2 | ATCC | Cat# TIB-73, RRID:CVCL_4383 |
| DihXD3 | This study | N/A |
| HEK293T | ATCC | Cat# CRL-3216, RRID:CVCL_0063 |
| HepG2 | ATCC | Cat# HB-8065, RRID:CVCL_0027 |
| Phoenix-GP | ATCC | Cat# CRL-3215, RRID:CVCL_H718 |
| Mouse: Alb-cre | The Jackson Laboratories | Stock No: 003574 |
| Mouse: C57BL/6 | The Animal Facility Core at SBP Medical Discovery Institute | N/A |
| Mouse: Nfe2l2−/− | Gift from Michael Karin, Ph.D. | N/A |
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) | The Animal Facility Core at SBP Medical Discovery Institute | N/A |
| Mouse: Prkcif/f | Leitges et al., 2001 | N/A |
| Primers, see Table S1 | IDT | N/A |
| gRNA targeting human PRKCI gene: GCCGCCGCCTGCGACCGTGT | Synthego | N/A |
| gRNA targeting mouse Prkci gene: AGTCCCTCAAAGGAGATGGA | Synthego | N/A |
| Mouse Cpt1a siRNA | Santa Cruz Biotechnology | Cat# sc-40377 |
| Mouse Nfe2l2 siRNA | Santa Cruz Biotechnology | Cat# sc-37049 |
| Mouse Prkci siRNA | Thermo Fisher | Cat# 150153 |
| pBABE-puro-mCherry-eGFP-LC3B | Addgene | Cat# 22418 |
| pBABE-puro-mCherry-eGFP-LC3B (T12A) | This paper | N/A |
| pBABE-puro-myc-BioID2 | Addgene | Cat# 80900 |
| pBABE-puro-myc-BioID2-PRKCI | This paper | N/A |
| pCMV-FLAG-p62 | Duran et al, 2011 | N/A |
| pCMV-FLAG-PKCλ | Reina-Campos et al., 2019b | N/A |
| pCMV-FLAG-PKCλ (1–250) | This paper | N/A |
| pCMV-FLAG-PKCλ (250–596) | This paper | N/A |
| pCMV-FLAG-PKCλ (D7276AA) | This paper | N/A |
| pCMV-FLAG-PKCλ (K274W) | This paper | N/A |
| pEGFP-LC3 | Addgene | Cat# 24920 |
| pCSF107mT-PPARα-HA | This paper | N/A |
| pLX304-V5-LC3A (S12A) | This paper | N/A |
| pLX304-V5-LC3A | DNAsu | HsCD00440693 |
| pREP-8xARE-GFP-SV40-BFP | Addgene | Cat# 134910 |
| psPAX2 | Addgene | Cat# 12260 |
| pMD2.G | Addgene | Cat# 12259 |
| pWZL-FLAG-LC3A | This paper | N/A |
| pWZL-FLAG-LC3A (S12A) | This paper | N/A |
| FlowJo | Tree Star Inc. | N/A |
| GenePattern | Broad Institute | https://cloud.genepattern.org/gp/pages/login.jsf |
| Graphpad Prism 8 | Graphpad | https://www.graphpad.com/scientificsoftware/prism/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Ingenuity Pathway Analysis | QIAGEN | https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/ |
| NextBio | Illumina | https://www.nextbio.com/b/authentication/login.nb |
| R | R Core Team | https://www.r-project.org/ |
| Scansite v4.00.022 | Koch Institute | https://scansite4.mit.edu/4.0/#home |
| UniProt | NIH | https://www.uniprot.org/ |
| UCSC Xena | UCSC | https://xenabrowser.net/datapages/ |
| EP-1 Econo Pump | Bio-Rad | Cat# 731–8140 |
| EVOS FL Auto Imaging System | Thermo Fisher Scientific | N/A |
| EVOS XL Core Cell Imaging System | Thermo Fisher Scientific | N/A |
| LSRFortessa 14-colors analyzer | BD Biosciences | N/A |
| NanoDrop 1000 spectrophotometer | Thermo Fisher Scientific | N/A |
| Neon Transfection System | Thermo Fisher Scientific | MPK1096 |
| Philips CM-100 Transmission Electron Microscope | Philips Electron Optics | N/A |
| TyssueLyser II | QIAGEN | Cat# 85300 |
| Zeiss LSM 710 NLO Confocal Microscope | Carl Zeiss Microscopy | N/A |
| VetScan v2 Chemistry Analyzer | Abaxis | N/A |
Gene-Expression Analyses
Total RNA was extracted by using TRIzol reagent (Invitrogen), and purified by using Quick-RNA Mini Prep Kit (Zymo Research) following the manufacturer’s protocols. After quantification using a Nanodrop 1000 spectrophotometer (Thermo Scientific, 1 mg of RNA was reverse transcribed using random primers and MultiScribe Reverse Transcriptase (Applied Biosystems). Gene expression was analyzed using the CFX96 Real Time PCR Detection System with SYBR Green Master Mix (Bio-Rad). Primer sequences are listed in Table S1. The amplification parameters were set at 95 °C for 30 sec, 58 °C for 30 sec, and 72 °C for 30 sec (40 cycles total). Gene expression values for each sample were normalized to the 18S rRNA. RNA-seq studies were performed in the Genomics Core at SBP Medical Discovery Institute. Briefly, total RNA was extracted from liver tissues from Prkcif/f and Prkcif/f;Alb-Cre animals which were fed with regular chow or subjected to DEN/HFD protocol. PolyA RNA was isolated using the NEBNext Poly(A) mRNA Magnetic Isolation Module and barcoded libraries were made using the NEBNext Ultra Directional RNA Library Prep Kit for Illumina (NEB). Libraries were pooled and single end sequenced (1×75) on the Illumina NextSeq 500 using the High output V2.5 kit (Illumina Inc.)
Bioinformatics Analysis
Sequencing FASTQ files from RNA-seq were uploaded to BaseSpace and processed with RNA-Seq Alignment App (Illumina Inc.) to obtain raw reads counts for each gene. Gene matrix files were modified to comply with .gct file format and used as input file for Gene Set Enrichment Analysis (GSEA). GSEA was performed using GSEA v20.0.4 module on GenePattern (https://cloud.genepattern.org/, Broad Institute) with 5,000 gene-set permutations using the gene ranking metric tTest or with Hallmark, C3, and C5 MSigDb collections as specified in each case. In order to explore autophagy associated genes signature, we customized “Autophagy-lysosome signature” as described (Perera et al., 2015). Differentially expressed genes using DESeq2 with q-value < 0.05 were considered to be significantly differentially expressed and uploaded to Ingenuity Pathway Analysis v.49309495 (IPA, QIAGEN) to be analyzed using the “Core Pathway Analysis”. Meta-analyses to identify overlapping and associated genes with publicly available data set were performed using the NextBio™ platform (www.nextbio.com, Illumina).
Bioinformatics Analysis of Clinical Data
Data for TCGA-LIHC was accessed through UNSC Xena (https://xena.ucsc.edu/). Raw gene expression datasets (GSE10143) was directly accessed through GEO website (NCBI). HCC patients (n = 82 from GSE10143) who underwent surgery were classified in two groups according to their PRKCI gene expression in surrounding non-tumorous tissues using maximally selected rank statistics. To examine correlation between PRKCI gene expression level and gene signatures, GSEA was performed using Pearson as the gene ranking metric.
In Vitro Kinase Assay
Recombinant human LC3 protein was incubated at 30 °C for 60 min in kinase-assay buffer (25 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 0.5 mM EGTA, 1 mM DTT and 100 μM ATP) in the presence of absence of recombinant PKCλ/ι protein or FLAG-tagged PKCλ/ι immunoprecipitates.. ATP analog-based phosphorylation detection was performed as previously described (Reina-Campos et al., 2019b). Briefly, 100 μM of ATPγS (Biolog) was added to the reaction, after which PNBM (Abcam) and EDTA were added to a final concentration of 2.5 mM and 20 mM, respectively, and incubated for 1 h at room temperature. Immunoblotting detection was performed with anti-thiophophate ester antibody or anti-phoshorylated LC3 (Ser 12). Protein digestion, TiO2-based phosphopeptide enrichment, electrospray ionization-liquid chromatography tandem mass spectrometry, and MS/MS analysis were performed in the Proteomics Core at SBP Medical Discovery Institute as described previously (Ma et al., 2013).
BioID2-Based Screening
Hek293T cells stably expressing myc-BioID2 or myc-BioID2-PKCλ/ι were grown for 48 h in the presence of 50 μM biotin. Cells were lysed via sonication in 8M urea, 50 mM ammonium bicarbonate, and extracted proteins were centrifuged at 14,000 × g to remove cellular debris and quantified by BCA assay (Thermo Scientific) according to the manufacturer’s specifications. A total of 700 μg of protein extract from each sample was used for affinity purification of biotinylated proteins. First, cysteine disulfide bonds were reduced with 5 mM tris(2-carboxyethyl)phosphine (TCEP) at 30°C for 60 min followed by cysteine alkylation with 15 mM iodoacetamide (IAA) in the dark at room temperature for 30 min. Affinity purification was carried out in a Bravo AssayMap platform (Agilent) using AssayMap streptavidin cartridges (Agilent). Briefly, cartridges were first primed with 50 mM ammonium bicarbonate, and then proteins were slowly loaded onto the streptavidin cartridge. Background contamination was removed by extensively washing the cartridges with 8 M urea, 50 mM ammonium bicarbonate. Finally, cartridges were washed with Rapid digestion buffer (Promega, Rapid digestion buffer kit) and proteins were subjected to on-cartridge digestion with mass spec grade Trypsin/Lys-C Rapid digestion enzyme (Promega) at 70 °C for 2 h. Digested peptides were then desalted in the Bravo platform using AssayMap C18 cartridges and the organic solvent was removed in a SpeedVac concentrator prior to LC-MS/MS analysis. Digested samples were analyzed on a Thermo Fisher Orbitrap Lumos mass spectrometer equipped with an Easy nLC 1200 ultra-high pressure liquid chromatography system. Samples were injected on an in-house packed C18 reverse phase column (25 cm × 75 μm packed with 1.7 μm, 130 Å pore size Bridged Ethylene Hybrid particles (Waters)). Peptides were separated by an organic gradient from 5% to 30% ACN in 0.1% formic acid over 130 minutes at a flow rate of 300 nl/min. The MS continuously acquired spectra in a data-dependent manner throughout the gradient, acquiring a full scan in the Orbitrap (at 120,000 resolution with an AGC target of 1e6 and a maximum injection time of 100 ms) followed by as many MS/MS scans as could be acquired on the most abundant ions in 3 s in the Orbitrap (at 15,000 resolution, HCD collision energy of 30%, AGC target of 1e5, a maximum injection time of 22 ms, and an isolation width of 1.3 m/z). Singly and unassigned charge states were rejected. Dynamic exclusion was enabled with a repeat count of 1, an exclusion duration of 20 s, and an exclusion mass width of ± 10 ppm. Raw mass spectrometry data were processed using the MaxQuant software package and Andromeda search engine (version 1.5.5.1). Peptides were generated from a tryptic digestion of up to two missed cleavages and were searched against the Uniprot human protein database (downloaded on August 13, 2015). Variable modifications were allowed for N-terminal protein acetylation, methionine oxidation, and lysine acetylation. A static modification was indicated for carbamidomethyl cysteine. All other settings were left using MaxQuant default settings. Peptide and protein identifications were filtered to a 1% false discovery rate (FDR). Statistical analysis was performed using the MSstats software package.
Protein Structure Analysis
The crystal structure of the human LC3A-p62 complex was downloaded from PDB:6EHR. Molecular graphics and analyses were performed with UCSF Chimera.
QUANTIFICATION AND STATISTICAL ANALYSIS
All the statistical tests were justified for every figure. All samples represent biological replicates. Data are presented as the mean ± SEM. Statistical analysis was performed using GraphPad Prism 8 or R software environment (http://www.r-project.org/). Significant differences between groups were determined using a Student’s t-test (two-tailed) when the data met the normal distribution tested by D’Agostino test. If the data did not meet this test, a Mann-Whitney U-test was used. Differences between more than 3 groups were determined using one-way ANOVA test (parametric) or Brown-Forsythe and Welch ANOVA tests (nonparametric) followed by Dunnett post hoc test. If the data did not meet this test, a Mann-Whitney test was used. Differences in Kaplan Meier plots were analyzed by Gehan-Breslow-Wilcoxon test. The chi-square test or Fisher’s exact test was used to determine the significance of differences between covariates. Logistic regression analysis was employed to estimate univariate and multivariate odds ratio and 95% confidence interval (CI). Values of p < 0.05 were considered as significantly different.
Supplementary Material
SIGNIFICANCE.
The heterogeneous and complex mutational landscape of hepatocellular carcinomas (HCC) is a barrier to oncogene-targeted therapy. The identification of key players of the non-oncogenic metabolic and stress vulnerabilities is a major gap in the field and would be instrumental in understanding the etiology of this type of cancer and opening avenues for new treatments. Here we identified PKCλ/ι as a critical suppressor of autophagy and oxidative metabolism. We show that the loss of PKCλ/ι results in increased ROS-induced NRF2 through an autophagy-OXPHOS mechanism, which is critical for the cell-autonomous and non-autonomous development of HCC. The finding that PKCλ/ι levels correlate negatively with the tumor grade establishes that the signals controlled by PKCλ/ι are potential therapeutic targets in HCC.
HIGHLIGHTS.
PKCλ/ι levels negatively correlate with HCC histological tumor grade
PKCλ/ι-deficient hepatocytes promote HCC linked to increased ROS and NRF2 activation
PKCλ/ι represses autophagy directly and inhibits OXPHOS to restrain ROS and NRF2
Increased ROS and NRF2 by PKCλ/ι deficiency accounts for increased HCC
ACKNOWLEDGMENTS
Research was supported by grants by NCI and NIDDK of the National Institutes of Health under awards numbers: R01DK108743, R01CA211794 to J.M; R01CA218254 to M.T.D.-M.; CA211794 and CA234128 to M.K.; and R01CA234245 to C.M.M. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank the personnel of the Histology, Flow Cytometry, Proteomics, Cell Imaging, Genomics, Animal Facility, and Viral Vectors Shared Resources at SBP Medical Discovery Institute for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Burns KA, and Vanden Heuvel JP (2007). Modulation of PPAR activity via phosphorylation. Biochim Biophys Acta 1771, 952–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, Hansen M, Moscat J, and Diaz-Meco MT (2011). p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell 44, 134–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran A, Hernandez ED, Reina-Campos M, Castilla EA, Subramaniam S, Raghunandan S, Roberts LR, Kisseleva T, Karin M, Diaz-Meco MT, and Moscat J (2016). p62/SQSTM1 by Binding to Vitamin D Receptor Inhibits Hepatic Stellate Cell Activity, Fibrosis, and Liver Cancer. Cancer Cell 30, 595–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, and Moscat J (2008). The Signaling Adaptor p62 Is an Important NF-[kappa]B Mediator in Tumorigenesis. Cancer Cell 13, 343–354. [DOI] [PubMed] [Google Scholar]
- El-Serag HB (2011). Hepatocellular carcinoma. N Engl J Med 365, 1118–1127. [DOI] [PubMed] [Google Scholar]
- El-Serag HB, and Rudolph KL (2007). Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 132, 2557–2576. [DOI] [PubMed] [Google Scholar]
- Finkin S, Yuan D, Stein I, Taniguchi K, Weber A, Unger K, Browning JL, Goossens N, Nakagawa S, Gunasekaran G, et al. (2015). Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat Immunol 16, 1235–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JP, Burns KA, Leas TL, Perdew GH, and Vanden Heuvel JP (2005). Regulation of peroxisome proliferator-activated receptor alpha by protein kinase C. Biochemistry 44, 10313–10321. [DOI] [PubMed] [Google Scholar]
- Guo JY, Teng X, Laddha SV, Ma S, Van Nostrand SC, Yang Y, Khor S, Chan CS, Rabinowitz JD, and White E (2016). Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev 30, 1704–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshida Y, Villanueva A, Kobayashi M, Peix J, Chiang DY, Camargo A, Gupta S, Moore J, Wrobel MJ, Lerner J, et al. (2008). Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med 359, 1995–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, McMahon M, Hayes JD, and Johansen T (2010). p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem 285, 22576–22591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmelman AC, and White E (2017). Autophagy and Tumor Metabolism. Cell Metab 25, 1037–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- L’Hermitte A, Pham S, Cadoux M, Couchy G, Caruso S, Anson M, Crain-Denoyelle AM, Celton-Morizur S, Yamagoe S, Zucman-Rossi J, et al. (2019). Lect2 Controls Inflammatory Monocytes to Constrain the Growth and Progression of Hepatocellular Carcinoma. Hepatology 69, 160–178. [DOI] [PubMed] [Google Scholar]
- Linares JF, Cordes T, Duran A, Reina-Campos M, Valencia T, Ahn CS, Castilla EA, Moscat J, Metallo CM, and Diaz-Meco MT (2017). ATF4-Induced Metabolic Reprograming Is a Synthetic Vulnerability of the p62-Deficient Tumor Stroma. Cell Metab 26, 817–829 e816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linares JF, Duran A, Reina-Campos M, Aza-Blanc P, Campos A, Moscat J, and Diaz-Meco MT (2015). Amino Acid Activation of mTORC1 by a PB1-Domain-Driven Kinase Complex Cascade. Cell Rep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Tao Y, Duran A, Llado V, Galvez A, Barger JF, Castilla EA, Chen J, Yajima T, Porollo A, et al. (2013). Control of Nutrient Stress-Induced Metabolic Reprogramming by PKCzeta in Tumorigenesis. Cell 152, 599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon M, Itoh K, Yamamoto M, and Hayes JD (2003). Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J Biol Chem 278, 21592–21600. [DOI] [PubMed] [Google Scholar]
- Moscat J, and Diaz-Meco MT (2009). p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 137, 1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscat J, Diaz-Meco MT, Albert A, and Campuzano S (2006). Cell Signaling and Function Organized by PB1 Domain Interactions. Mol Cell 23, 631–640. [DOI] [PubMed] [Google Scholar]
- Moscat J, Karin M, and Diaz-Meco MT (2016). p62 in Cancer: Signaling Adaptor Beyond Autophagy. Cell 167, 606–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP (2009). How mitochondria produce reactive oxygen species. Biochem J 417, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi Y, Duran A, L’Hermitte A, Shelton PM, Nakanishi N, Reina-Campos M, Huang J, Soldevila F, Baaten BJG, Tauriello DVF, et al. (2018). Simultaneous Loss of Both Atypical Protein Kinase C Genes in the Intestinal Epithelium Drives Serrated Intestinal Cancer by Impairing Immunosurveillance. Immunity 49, 1132–1147 e1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, and Karin M (2007). Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 317, 121–124. [DOI] [PubMed] [Google Scholar]
- Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, and Karin M (2010). Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 140, 197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, and Thompson CB (2016). The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR, et al. (2015). Transcriptional control of autophagy lysosome function drives pancreatic cancer metabolism. Nature 524, 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz JD, and White E (2010). Autophagy and metabolism. Science 330, 1344–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina-Campos M, Diaz-Meco MT, and Moscat J (2019a). The Dual Roles of the Atypical Protein Kinase Cs in Cancer. Cancer Cell 36, 218–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina-Campos M, Linares JF, Duran A, Cordes T, L’Hermitte A, Badur MG, Bhangoo MS, Thorson PK, Richards A, Rooslid T, et al. (2019b). Increased Serine and One-Carbon Pathway Metabolism by PKClambda/iota Deficiency Promotes Neuroendocrine Prostate Cancer. Cancer Cell 35, 385–400 e389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo de la Vega M, Chapman E, and Zhang DD (2018). NRF2 and the Hallmarks of Cancer. Cancer Cell 34, 21–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C, Shinde J, Soysouvanh F, et al. (2015). Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet 47, 505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starley BQ, Calcagno CJ, and Harrison SA (2010). Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology 51, 1820–1832. [DOI] [PubMed] [Google Scholar]
- Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, and Mizushima N (2011). Autophagy-deficient mice develop multiple liver tumors. Genes & development 25, 795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todoric J, Antonucci L, Di Caro G, Li N, Wu X, Lytle NK, Dhar D, Banerjee S, Fagman JB, Browne CD, et al. (2017). Stress-Activated NRF2-MDM2 Cascade Controls Neoplastic Progression in Pancreas. Cancer Cell 32, 824–839 e828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totoki Y, Tatsuno K, Covington KR, Ueda H, Creighton CJ, Kato M, Tsuji S, Donehower LA, Slagle BL, Nakamura H, et al. (2014). Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet 46, 1267–1273. [DOI] [PubMed] [Google Scholar]
- Umemura A, He F, Taniguchi K, Nakagawa H, Yamachika S, Font-Burgada J, Zhong Z, Subramaniam S, Raghunandan S, Duran A, et al. (2016). p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia T, Kim JY, Abu-Baker S, Moscat-Pardos J, Ahn CS, Reina-Campos M, Duran A, Castilla EA, Metallo CM, Diaz-Meco MT, and Moscat J (2014). Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell 26, 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, and DeBerardinis RJ (2017). Understanding the Intersections between Metabolism and Cancer Biology. Cell 168, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Shang Y, Yu J, and Zhang M (2012). Substrate recognition mechanism of atypical protein kinase Cs revealed by the structure of PKCiota in complex with a substrate peptide from Par-3. Structure 20, 791–801. [DOI] [PubMed] [Google Scholar]
- Yang L, Li P, Fu S, Calay ES, and Hotamisligil GS (2010). Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell metabolism 11, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-seq data reported in this paper is GSE147801. The following data sets were analyzed: GSE10143 and UNSC Xena. The raw data of this study have been deposited in Mendeley Data and available at https://doi.org/10.17632/xm87k23hrh.1.