

Abstract
Matching experimental ion mobility-mass spectrometry data to computationally-generated collision cross section (CCS) values enables more confident metabolite identifications. Here, we show for the first time that accurately predicting CCS values with simple models for the largest library of metabolite cross sections is indeed possible, achieving a root mean square error of 7.0 Å2 (median error of ~2%) using linear methods accesible to most researchers. A comparison on the performance of 2D vs. 3D molecular descriptors for the purposes of CCS prediction is also presented for the first time, enabling CCS prediction without a priori knowledge of the metabolite’s energy-minimized structure.
Metabolomics employs a variety of techniques, including nuclear magnetic resonance spectroscopy (NMR), high resolution mass spectrometry (MS) and tandem MS (MS/MS) in combination with liquid chromatography (LC) or gas chromatography (GC) to separate, detect and identify hundreds of small molecules in complex biological matrices.1 In this context, NMR and MS data are most frequently analysed by way of automated peak alignment, de-replication, and database matching, rather than manual approaches. While mass spectral database coverage is becoming increasingly more comprehensive,2 high quality experimental MS/MS fragmentation information may still be unavailable for many metabolites due to low precursor ion abundances, incomplete fragmentation, or precursor ion co-selection. In this scenario, metabolite identification becomes increasingly ambiguous, necessitating additional experiments to increase annotation confidence.3
Ion mobility (IM) coupled to MS is emerging as a robust platform to aid in metabolite identification.4–6 IM can be thought of as analogous to a gas-phase electrophoretic experiment, where ions in a buffer gas pass through a chamber in the presence of an electric field, and the migration time is measured. This migration time can then be transformed into a CCS value through appropriate regression or calibration procedures.7 IM CCS values, while partially correlated with mass-to-charge ratios (m/z), are also dependent on molecular shape, granting IM-MS instrumentation a high degree of orthogonality that can be exploited for distinguishing isobars.8 Moreover, CCS values have higher inter-laboratory robustness than chromatographic retention times,9 and are starting to be compiled in large scale databases for unknown metabolite annotation.10
CCS values for molecules as small as metabolites and as large as protein complexes have been predicted computationally for comparison to IM measurements.11–13 However, such predictions typically require 3-dimensional structures by way of solid state NMR or X-ray crystallography experimental data. When such data are lacking, computationally-optimized structures representative of the conformation(s) taken in the gas phase are calculated by molecular dynamics. This approach, useful to gain insight into the microscopic structure of an individual molecule, requires significant computational resources that are typically not scalable to metabolomics applications with hundreds of biospecimens and thousands of detected compounds.
As a faster, less resource-intensive alternative, a number of efforts regarding CCS prediction using machine learning have been reported in the literature. These include CCS predictions for phenolics using traveling wave ion mobility spectrometry,14 support vector machine regression models used to derive the MetCCS and LipidCCS databases,15,16 joint prediction of CCS values and chromatographic retention times for screening purposes,17 CCS prediction for pesticide libraries,18 and predictions using deep neural networks.19 Along these efforts, which we compare in more detail in ESI Table S1,† we developed an approach named CCS Prediction (CCSP) where over 3800 molecular descriptors were calculated using Dragon 7.0, the worldwide most used application for this type of calculations, from 2D SDF files. Molecular descriptors are mathematical representations of a molecule calculated by well-specified algorithms which transform molecular structures into numbers.20 Partial least squares (PLS) linear multivariate regression was used to develop models to predict CCS values from such molecular descriptors. CCSP yielded accurate CCS values that were within 2% of experimental measurements for depsipeptides and lipids using only their 2D structures.21 The excellent accuracy of such predictions was associated with the high chemical homogeneity of the training sets studied, which was viewed as one of the main limitations of the work, therefore only being useful for predicting CCS of structurally-similar species, and requiring some degree of a priori knowledge of the unknown compound chemical characteristics. Here, the prediction of CCS values for the largest and most diverse metabolite IM database is explored for the first time using linear methods, showing that CCS values can be well predicted without the need for complex multi-layer deep learning methods, molecular dynamics, or 3D molecular descriptors.
CCS values (DTΩN2) were sourced from the database recently reported by Baker et al.22 This database includes over 500 primary metabolites, secondary metabolites, and xenobiotic entries from the glycolysis and pentose phosphate pathways, the tricarboxylic acid cycle, terpenes, flavonoids, antibiotics, and aromatic hydrocarbons. Experimental CCS values for protonated and deprotonated species in both positive and negative ionization modes were included, as well as for sodiated species, although not all adducts are available for each entry. Database entries selected for inclusion in the CCS predictive model development had to meet the following criteria: (1) [M + H]+ and/or [M − H]− adducts only, (2) an SDF file had to be available in the PubChem database, (3) molecular weight, molecular formula, InChI Key, PubChem ID, and CAS number from the CCS database had to be in agreement with the PubChem files downloaded, and (4) in preliminary models the predicted CCS value had to be in the range of expected experimental CCS values. If one or more of the criteria were not met, the metabolite was excluded from the multivariate models developed. Downloading of the 2D structure files through PubChem eliminated the need for manually drawing each entry. When available, a 3D conformer file was also sourced for comparison purposes, however no additional molecular dynamics were employed to optimize these files. In some cases, a single entry from the CCS database could be represented by multiple PubChem entries. When this occurred, the most representative PubChem ID was selected (see list of PubChem IDs selected under ESI†).
Dragon 7.0 was used to calculate molecular descriptors for each SDF file downloaded.23 The SDF files were first separated into four categories based on adduct ion type: 2-dimensional [M + H]+ (n = 254), 2-dimensional [M − H]− (n = 295), 3-dimensional [M + H]+ (n = 238), and 3-dimensional [M − H]− (n = 271). It is important to note that, while CCS values from the database are for a specific adduct, the SDF files used to calcu late molecular descriptors were not manipulated to add a charge. Molecular descriptors that were constant within a category were excluded from model development, resulting in a final matrix of 1955 descriptors for the 2D entries and 3608 descriptors for the 3D entries. Many of the descriptors relied on weighting schemes for calculation; these weighting schemes, based on properties such mass, van der Waals volume, Sanderson electronegativity, ionization potential, polarizability, and intrinsic state, were applied directly by Dragon 7.0 when appropriate. When required, 2D hydrogens were added to structures with missing hydrogens, and atom coordinates were rounded. For disconnected molecules such as salts, the Dragon approach, a theoretical method new to Dragon 7.0, was applied to allow for the descriptors to be calculated. Overall, calculation of all molecular descriptors with Dragon 7.0 required less than 10 minutes for the whole metabolite database on a standard desktop workstation with 8 GB RAM.
Each adduct category was randomly split into calibration and test sets, with approximately 25% of each reserved for independent validation as follows: 2-dimensional [M + H]+ (ncalibration = 189), 2-dimensional [M − H]− (ncalibration = 217), 3-dimensional [M + H]+ (ncalibration = 177), and 3-dimensional [M − H]− (ncalibration = 200). In cases where very few entries for a given chemical class were present, such class was split 50% to have adequate representation in both the calibration and independent test sets. The widely accessible Matlab with the PLS toolbox24 was used for PLS regression between molecular descriptors (X-block) and database CCS values (Y-block) for each adduct category. Cross validation of the PLS models was performed using a Venetian blinds approach with 10 splits, and 1 object per split. In previous work, variable selection using genetic algorithm (GA)-PLS regression was critical for lowering the percent error associated with predicted CCS values. Following a similar strategy, GA was performed in Matlab reducing the number of molecular descriptors in the X-block as follows: 2-dimensional [M + H]+ (n = 49), 2-dimensional [M − H]− (n = 45), 3-dimensional [M + H]+ (n = 178), and 3-dimensional [M − H]− (n = 187). GA settings used are described under ESI (Table S2†), with the frequency of selection of various descriptors shown in Fig. S1.†
While previous work with lipids and depsipeptides only used 2D structures, 3D conformers for the majority of the metabolites under study here are now accessible in PubChem.25 Therefore, we now attempted a comparison of the accuracy of CCS predictions obtained from 2D structures vs. those from 3D conformers. Results showed only minimal differences in the CCS root mean square error in prediction (RMSEP) of the independent test set, and the root mean square error in cross validation (RMSECV) of the calibration set (Table 1) between 2D and 3D descriptors, suggesting that 2D descriptors are sufficient for CCS prediction with good accuracy.
Table 1.
Root mean square error in cross validation (RMSECV), root mean square error in prediction (RMSEP), and R2 in cross validation (CV) and prediction for each of the 4 best PLS models developed. Lower RMSE values indicate a better match to the experimentally-derived collision cross sections. The median percent error is reported
| RMSECV (Å2) | R2 CV | RMSEP (Å2) | R2 Pred | Median % error | |
|---|---|---|---|---|---|
| [M + H]+ 2D | 4.00 | 0.99 | 6.98 | 0.94 | 1.84 |
| [M + H]+ 3D | 3.87 | 0.98 | 5.63 | 0.92 | 2.29 |
| [M − H]− 2D | 3.62 | 0.99 | 5.44 | 0.98 | 1.47 |
| [M − H]− 3D | 3.54 | 0.99 | 5.09 | 0.96 | 1.95 |
For the [M − H]− 2D PLS model, the RMSEP was 5.44 Å2, while for the corresponding 3D PLS model the RMSEP was 5.09 Å2 (Table 1). Of the 78 independent test entries for the [M − H]− 2D model, 67 (86%) were predicted within ±5% and an additional 11 were within ±8.25% of the experimental CCS values. The median relative error was 1.47%. In comparison, the [M − H]− 3D model, which had 71 independent test entries, resulted in 62 metabolites within ±5% error (87%), an additional 8 within ± 10%, and only 1 with an error greater than 10% (−10.3%) with a median relative error of 1.95%. Experimental vs. predicted CCS values for 2D [M − H]−ions are shown in Fig. 1a, with the results for 3D [M − H]− ions shown in Fig. S2b,† and a breakdown of the observed accuracies presented in Fig. S3.†
Fig. 1.
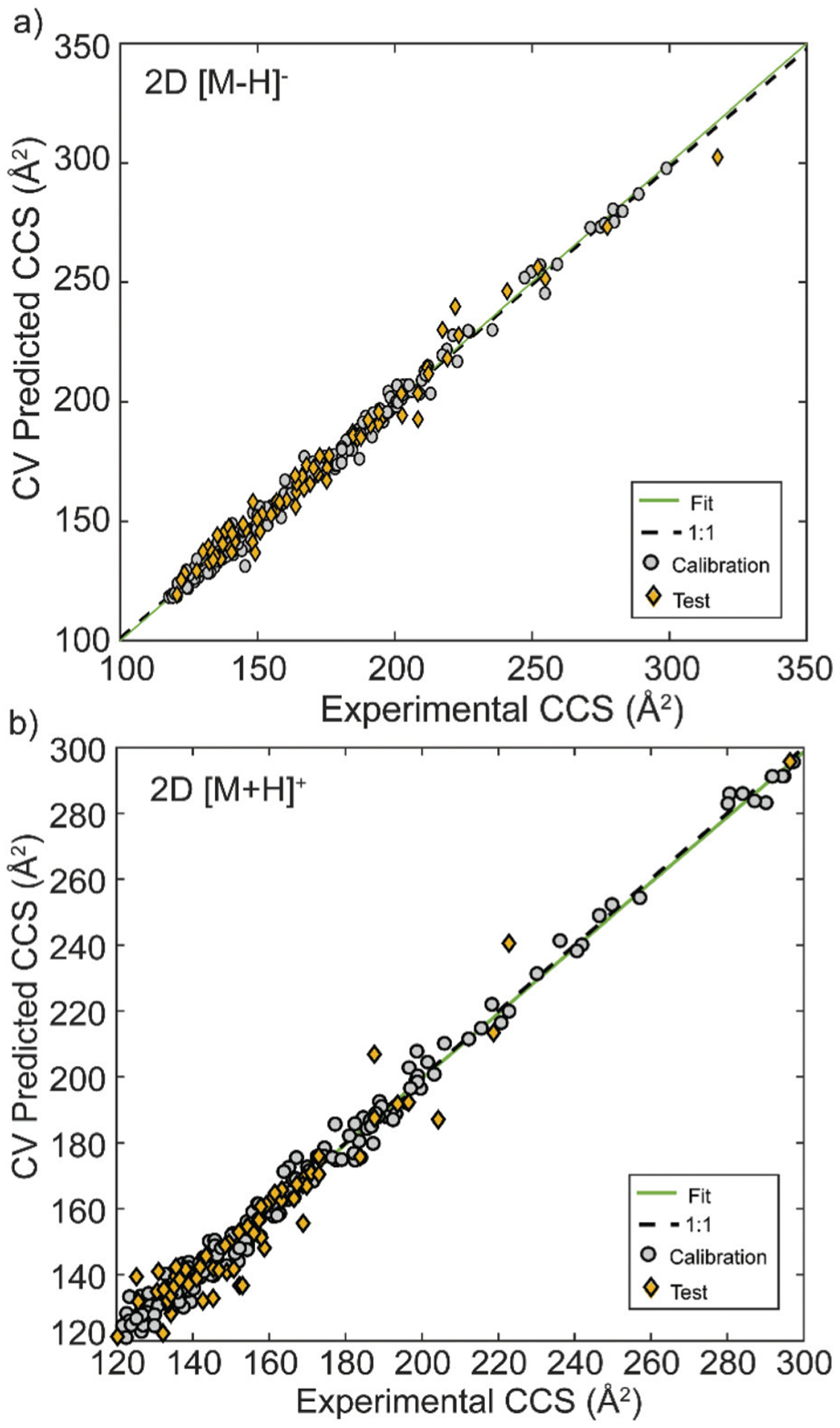
Measured vs. cross-validation (CV)-predicted CCS for metabolites developed using (a) 2D structural files to calculate molecular descriptors for [M − H]− adducts, and (b) 2D structural files to calculate molecular descriptors for [M + H]+ adducts. Optimal molecular descriptors were selected by a genetic algorithm.
For the predictive model developed for [M + H]+ ions, the RMSEP using 2D structures was 6.98 Å2, while the RMSEP using the 3D conformers was slightly lower at 5.63 Å2. For 2D structures, of the 64 independent test entries, 54 (81%) had predictions within ±5% error of the experimental measurement, an additional 11 were within ±10%, and the remaining 3 had an error lower than 12% (median relative error was 1.84%). With the 3D [M + H]+ model, 60 independent test entries were analysed and had a median relative error or 2.29%; 48 of these (80%) were within ±5% and 12 were within ±9%. Experimental vs. predicted CCS values for 2D [M + H]+ adducts are shown in Fig. 1b, 3D [M + H]+ are shown in Fig. S2a.† The model accuracy breakdown is shown in Fig. S3.† Results for PLS models without GA variable selection are provided in Fig. S4–S7,† for comparison purposes. Overall, these results indicated that accurate prediction of CCS values median relative errors better than 2% was possible for all [M + H]+ and [M − H]− ions of all metabolites considered using simple PLS models. These errors were slightly higher when 3D molecular descriptors were used. This is to be expected, as the 3D structures used to calculate those descriptors are not necessarily optimized. Also, the effect of charge localization on molecular structure (e.g. various protomers26) may be responsible for the observed CCS prediction error differences between 2D and 3D descriptors.
The molecular descriptors selected through GA were categorized into the blocks described within the molecular descriptor software, as indicated in ESI.†27 Two-dimensional atom pairs and 2D matrix-based descriptors were the top two most frequently used molecular descriptor blocks for all four models. Two-dimensional autocorrelations and edge adjacency indices were also frequently used blocks. The models lacking 3D descriptors used approximately half the number of molecular descriptor blocks than those developed with 3D information. This is expected considering the more simplistic nature of 2D models. Models for [M + H]+ species used one more molecular descriptor blocks than the corresponding 2 or 3D [M − H]− models.
The molecular descriptor blocks used for the 2D [M − H]− and [M + H]+ models were very similar. Blocks used for the 2D [M + H]+ model (14 blocks) and 2D [M − H]− model (13 blocks), shared 12 common descriptor blocks. Atom-type E-state indices were unique to the 2D [M − H]− model, while constitutional indices and information indices were unique to the 2D [M + H]+ model. These unique blocks, however, had few molecular descriptors of that category. Only one molecular descriptor fell within the atom-type E-state indices in the 2D [M − H]− model, with an extremely low variable importance in projection (VIP) score. While descriptors that fell within the constitutional indices and information indices blocks appeared infrequently in the 2D [M + H]+ model, they had several significant (greater than 1.4) VIP scores, leading to a higher weight when incorporated into the final model.
Three-dimensional models were more similar than 2D, where the 3D [M − H]− and 3D [M + H]+ model shared 27 descriptor blocks. The 3D [M + H]+ model included one extra block, ring descriptors, but this block was not significant for the model, containing only 2 molecular descriptors each with a VIP score lower than 1. A box and whisker plot for the descriptor blocks making up the 3D [M − H]− model is shown in Fig. 2, comparing the blocks by average VIP score of the individual molecular descriptors contained within them. Molecular descriptors falling within the matrix-based descriptors (2D and 3D), GETAWAY descriptors, walk and path counts, informational, constitutional, and connectivity indices blocks made up the highest VIP scoring descriptors consistently through the models. The importance of descriptors within these blocks indicates that the placement of atoms with respect to one another is critical to CCS prediction, as expected. ESI Table S3† presents a comparison of the specific descriptors in this work against those in our previous study and Table S4† gives the VIP score for the top 25 scoring molecular descriptors in each model. Of the descriptors chosen by GA in more than one model, none had consistently high VIP scores. This suggests that a few singular molecular descriptors on their own are not correlated with CCS, but instead it is the combination of molecular descriptors which provides strong predicting power. In other words, results indicate that the individual descriptors and what they represent are less critical than finding the best subset of molecular descriptors that combined can predict CCS accurately. Fig. S8 and S9† show correlation plots between these descriptors, suggesting that molecular descriptors from the same blocks are generally correlated with one another as expected, however not all are positively correlated with CCS. This further supports an aggregation of molecular descriptors being more important than any singular descriptors in CCS prediction.
Fig. 2.
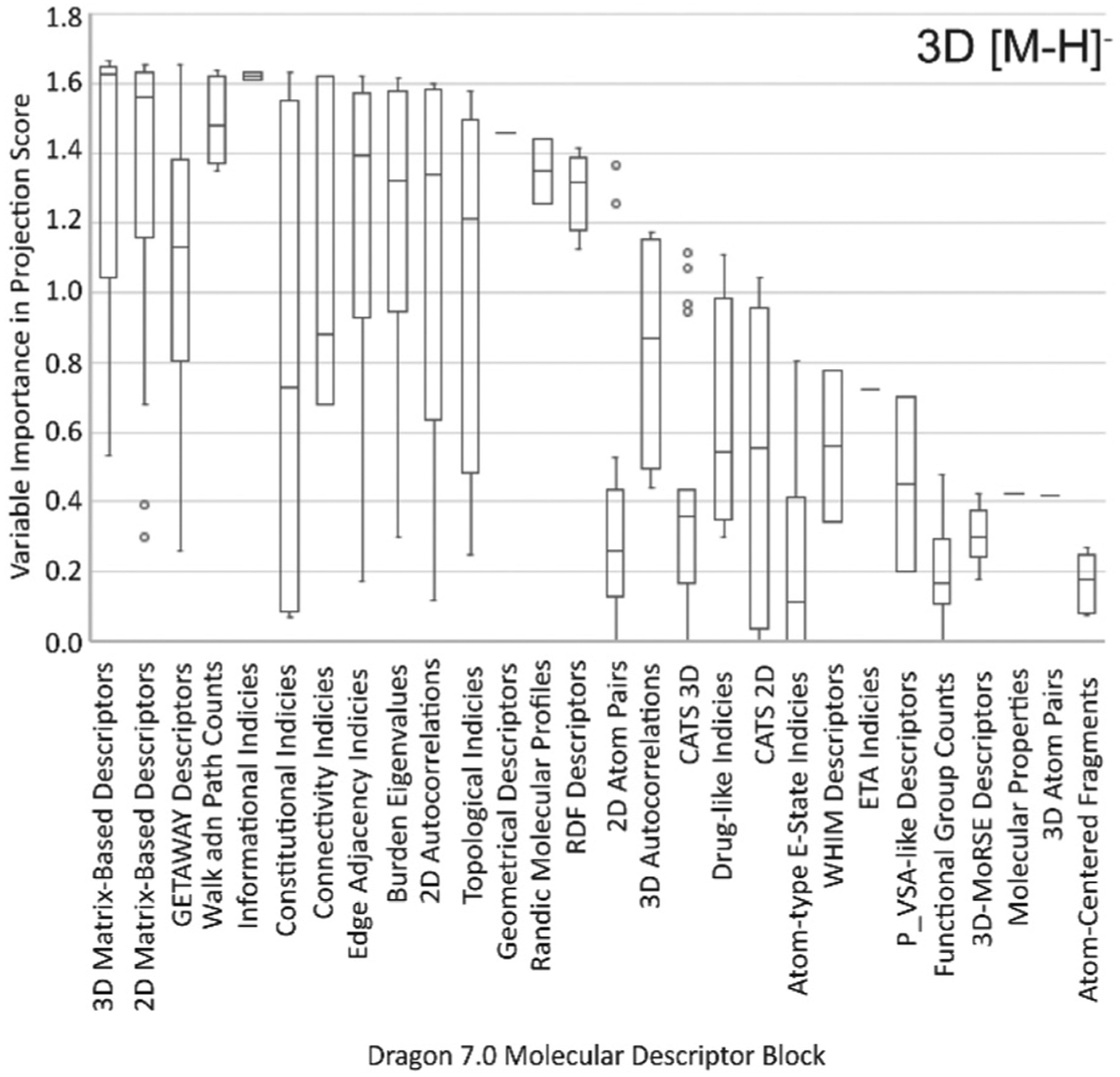
Each molecular descriptor used in the 3D [M − H]− model belongs to a block as defined by the Dragon 7.0 software. The box and whisker plot show the range of Variable Importance in Projection (VIP) scores given for each block. The higher the VIP score the more important the variable is to the multivariate model.
In comparison to our previous study with lipids and depsipeptides, the error for the training set used here was slightly higher due to the larger chemical diversity of the chemical species involved. In future work, even lower errors may be obtainable by developing chemical class-specific PLS models for more homogeneous training subsets or by increasing the overall training set size, following the continuing expansion of the Baker IM CCS database. Although it could be argued that with more chemically specific training subsets the risk of over-fitting may increase, use of some diagnostic fragment ions obtained by MS/MS to determine chemical class membership of the unknown metabolite in question may help guide the application of a given PLS model specific for that class. CCS prediction with CCSP did not rely on assigning a tentative metabolite class to the unknown, and thus has the advantage of identifying metabolites in a data independent fashion. Accurate CCS predictions were achievable even if the structure had not been energy minimized and only using 2D descriptors, thus removing the need for molecular dynamics optimization. In only a few cases 3D models produced predictions better than 2D models, but no obvious structural trend was observed for these species (Fig. S10 and S11†). Development of the models presented here did not require computing power in excess of a standard workstation for either molecular descriptor calculation or PLS regression within Matlab, allowing a quick approach to CCS prediction. However, it must be noted that even small molecules can exist as different isomers in the gas-phase, and that these isomers can sometimes be separated by ion mobility. In these cases, application of CCSP is likely to result in incorrect assignments, as only one CCS value is calculated per species.
Supplementary Material
Acknowledgements
FMF and ESB acknowledges support from NCI grant 1R01CA218664-01 and NIEHS grant P42 ES027704, respectively. MTSH acknowledges start up funding from NKU in support of this work.
Footnotes
Conflicts of interest
All authors declare no conflicts of interest with this work.
Electronic supplementary information (ESI) available: List of PubChem IDs included in each model, genetic algorithm parameters, and the results of models prior to genetic algorithm for variable selection. See DOI: 10.1039/d0an00198h
Notes and references
- 1.Want EJ, Wilson ID, Gika H, Theodoridis G, Plumb RS, Shockcor J, Holmes E and Nicholson JK, Nat. Protoc, 2010, 5, 1005–1018. [DOI] [PubMed] [Google Scholar]
- 2.Guijas C, Montenegro-Burke JR, Domingo-Almenara X, Palermo A, Warth B, Hermann G, Koellensperger G, Huan T, Uritboonthai W, Aisporna AE, Wolan DW, Spilker ME, Benton HP and Siuzdak G, Anal. Chem, 2018, 90, 3156–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Monge ME, Dodds JN, Baker ES, Edison AS and Fernandez FM, Annu. Rev. Anal. Chem, 2019, 12, 177–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paglia G and Astarita G, Nat. Protoc, 2017, 12, 797–813. [DOI] [PubMed] [Google Scholar]
- 5.Paglia G, Williams JP, Menikarachchi L, Thompson JW, Tyldesley-Worster R, Halldorsson S, Rolfsson O, Moseley A, Grant D, Langridge J, Palsson BO and Astarita G, Anal. Chem, 2014, 86, 3985–3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mairinger T, Causon TJ and Hann S, Curr. Opin. Chem. Biol, 2018, 42, 9–15. [DOI] [PubMed] [Google Scholar]
- 7.Gabelica V and Marklund E, Curr. Opin. Chem. Biol, 2018, 42, 51–59. [DOI] [PubMed] [Google Scholar]
- 8.Paglia G, Angel P, Williams JP, Richardson K, Olivos HJ, Thompson JW, Menikarachchi L, Lai S, Walsh C, Moseley A, Plumb RS, Grant DF, Palsson BO, Langridge J, Geromanos S and Astarita G, Anal. Chem, 2015, 87, 1137–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stow SM, Causon TJ, Zheng X, Kurulugama RT, Mairinger T, May JC, Rennie EE, Baker ES, Smith RD, McLean JA, Hann S and Fjeldsted JC, Anal. Chem, 2017, 89, 9048–9055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou ZW, Tu J and Zhu ZJ, Curr. Opin. Chem. Biol, 2018, 42, 34–41. [DOI] [PubMed] [Google Scholar]
- 11.Campuzano I, Bush MF, Robinson CV, Beaumont C, Richardson K, Kim H and Kim HI, Anal. Chem, 2012, 84, 1026–1033. [DOI] [PubMed] [Google Scholar]
- 12.Bleiholder C, Wyttenbach T and Bowers MT, Int. J. Mass Spectrom, 2011, 308, 1–10. [Google Scholar]
- 13.Valentine SJ, Counterman AE and Clemmer DE, J. Am. Soc. Mass Spectrom, 1999, 10, 1188–1211. [DOI] [PubMed] [Google Scholar]
- 14.Gonzales GB, Smagghe G, Coelus S, Adriaenssens D, De Winter K, Desmet T, Raes K and Van Camp J, Anal. Chim. Acta, 2016, 924, 68–76. [DOI] [PubMed] [Google Scholar]
- 15.Zhou ZW, Shen XT, Tu J and Zhu ZJ, Anal. Chem,2016, 88, 11084–11091. [DOI] [PubMed] [Google Scholar]
- 16.Zhou Z, Tu J, Xiong X, Shen X and Zhu ZJ, Anal. Chem, 2017, 89, 9559–9566. [DOI] [PubMed] [Google Scholar]
- 17.Mollerup CB, Mardal M, Dalsgaard PW, Linnet K and Barron LP, J. Chromatogr. A, 2018, 1542, 82–88. [DOI] [PubMed] [Google Scholar]
- 18.Bijlsma L, Bade R, Celma A, Mullin L, Cleland G, Stead S, Hernandez F and Sancho JV, Anal. Chem, 2017, 89, 6583–6589. [DOI] [PubMed] [Google Scholar]
- 19.Plante PL, Francovic-Fontaine E, May JC, McLean JA, Baker ES, Laviolette F, Marchand M and Corbeil J, Anal. Chem, 2019, 91, 5191–5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Consonni V and Todeschini R, Handbook of Molecular Descriptors, Wiley-VCH, Weinheim, New York, 2008. [Google Scholar]
- 21.Soper-Hopper MT, Petrov AS, Howard JN, Yu SS, Forsythe JG, Grover MA and Fernandez FM, Chem. Commun, 2017, 53, 7624–7627. [DOI] [PubMed] [Google Scholar]
- 22.Zheng X, Aly NA, Zhou Y, Dupuis KT, Bilbao A, Paurus VL, Orton DJ, Wilson R, Payne SH, Smith RD and Baker ES, Chem. Sci, 2017, 8, 7724–7736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hong HX, Xie Q, Ge WG, Qian F, Fang H, Shi LM, Su ZQ, Perkins R and Tong WD, J. Chem. Inf. Model, 2008, 48, 1337–1344. [DOI] [PubMed] [Google Scholar]
- 24.Wise BM, Gallagher N, Bro R, Shaver J, Windig W and Koch R, PLS Toolbox 3.5 for use with Matlab, Eigenvector Research Inc, Manson, WA, 2005. [Google Scholar]
- 25.Kim S, Thiessen PA, Bolton EE, Chen J, Fu G, Gindulyte A, Han L, He J, He S, Shoemaker BA, Wang J, Yu B, Zhang J and Bryant SH, Nucleic Acids Res, 2016, 44, D1202–D1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lalli PM, Iglesias BA, Toma HE, de Sa GF, Daroda RJ, Silva Filho JC, Szulejko JE, Araki K and Eberlin MN, J. Mass Spectrom, 2012, 47, 712–719. [DOI] [PubMed] [Google Scholar]
- 27.Todeschini R and Consonni V, Molecular Descriptors for Chemoinformatics, Wiley-VCH Verlag GmbH & Co; KGaA, 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.