

Abstract
Background.
Potential adverse effects, such as functional impairment of islets, render conventional immunosuppressive drugs unsuitable for use in islet transplantation. In addition, as a single therapy, they cannot prolong islet allograft survival. Here, we investigated the utility of the mitogen-activated protein kinase inhibitor trametinib and asked whether it ameliorates acute rejection of transplanted islets without the need for conventional immunosuppressants.
Methods.
Islets from fully major histocompatibility complex-mismatched BALB/c mice were transplanted into streptozotocin-induced diabetic C57BL/6 mice via the portal vein. These mice received trametinib or vehicle (orally) for 28 days. Isolated islets from BALB/c mice were incubated in vitro with different concentrations of trametinib to determine viability and function.
Results.
Trametinib (0.1 and 0.3 mg/kg) prolonged graft survival significantly (P = 0.0007 and P = 0.005, respectively) when compared with vehicle. Histologic analyses revealed that cellular infiltration of the graft by lymphocytes was inhibited significantly on day 7 (P < 0.05). In addition, trametinib suppressed functional differentiation of naive CD4+ T cells in recipients. Expression of mRNA encoding inflammatory cytokines interleukin (IL)-2, tumor necrosis factor α, and interferon γ in recipients treated with trametinib was also inhibited (P < 0.001, P < 0.05, and P < 0.01, respectively). Trametinib also increased production of IL-4 and IL-10 (P < 0.05 and P = 0.20, respectively). In vitro, islets incubated with different concentrations of trametinib exhibited no harmful effects with respect to viability and function.
Conclusions.
Trametinib delayed islet graft rejection by inhibiting functional differentiation of naive CD4+ T cells and regulating inflammatory cytokines. Trametinib might be a promising candidate for maintenance immunosuppressive therapy after allogeneic islet transplantation.
INTRODUCTION
Clinical islet transplantation is a β-cell replacement therapy for patients with type 1 diabetes mellitus who experience severe hypoglycemic episodes.1,2 Recent clinical trials reveal that β-cell replacement therapy results in superior glycemic control, with fewer hypoglycemic episodes, than continuous insulin infusion or multiple daily injections of insulin.3,4 Although the islet transplantation procedure is less invasive than solid-organ pancreas transplantation,5,6 an islet transplant from a single donor has yet to achieve long-term insulin independence.7
Since the first islet transplantation in the 1970s, immunosuppressive regimens have been modified and improved continuously4,8; unfortunately, many immunosuppressive drugs used for clinical islet transplantation impair β cell growth, survival, and function.9–12 In addition, long-term immunosuppression increases the risk of infection, cancer, and organ impairment.13–15 These deleterious effects of immunosuppressants are significant obstacles to successful and long-lasting islet transplantation. The desirable immunological therapy for islet transplantation is, therefore, suppression of allorejection with no adverse effects on insulin secretion or survival of β cells.
The RAS/mitogen-activated protein kinase (MEK)/extracellular-regulated kinase (ERK) pathway plays a pivotal role in regulating cell survival, cell cycle progression, and differentiation.16 T-cell receptor ligation activates this pathway, and, once activated, phosphorylated ERK induces cytokine gene expression [particularly via activation of the interleukin (IL)-2 promoter], and T-cell proliferation.17–19 In addition, these cascades are involved in insulin signaling, which contributes to insulin resistance. Furthermore, MEK is a key therapeutic target; indeed, a number of MEK inhibitors have been developed, some of which have been approved for clinical cancer therapy.20
Previously, we demonstrated that MEK inhibitors selectively suppress alloreactivity of T cells while sparing herpesvirus-specific T cells and delaying onset of murine graft-versus-host disease after BMT.21,22 However, using an MEK inhibitor as a single drug therapy after organ transplantation did not prolong graft survival in a fully major histocompatibility complex (MHC)-mismatched rodent model.23,24 Therefore, the aim of the present report was to analyze the efficacy of a MEK inhibitor for allogeneic islet transplantation in a fully MHC-mismatched rodent model and to examine its effect on insulin secretion and cytotoxicity in vitro.
MATERIALS AND METHODS
Animals
Male C57BL/6 and BALB/c mice (8 wk old) were purchased from Shimizu Laboratory Supplies (Kyoto, Japan). All mouse experiments were conducted under pathogen-free conditions and in line with Institutional Animal Care protocols approved by Kyoto University (Q17-62 and 19-69).
Reagents
Trametinib was purchased from ChemScene (Monmouth Junction, NJ). For the in vitro experiments, the reagent was reconstituted in dimethyl sulfoxide (DMSO) to yield a final concentration of 10 mmol/L and stored at –80°C for up to 6 mo before addition to culture media. For the in vivo experiments, the drug was reconstituted in 200 μL vehicle (corn oil).
Pancreatic Islet Isolation and Culture
To isolate islets from BALB/c mice (8 wk old), the pancreas was inflated by injection of Hank’s Balanced Salt Solution containing 0.15 mg/mL collagenase P (Roche Diagnostics, IN) via the common bile duct. Next, the distended pancreas was excised and incubated at 37°C for 18 min. After the digested pancreas had dissociated, the tissue was washed with Hank’s Balanced Salt Solution. The islets in the dissociated pancreatic tissue were purified on discontinuous gradients (1.110, 1.103, 1.096, and 1.070 g/mL) of OptiPrep (Axis-Shield, Oslo, Norway) and ET Kyoto solution (Otsuka Pharmaceutical, Tokyo, Japan). Isolated islets were cultured (37°C/5% CO2/95% air humidified atmosphere) in RPMI1640 medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin.
Induction of Diabetes Mellitus in Recipient Mice
Diabetes mellitus was induced in 8-wk-old C57BL/6 mice via a single intraperitoneal injection of streptozotocin (STZ) (150 mg/kg body weight; STZ; Wako, Japan) 5–7 d before transplantation. Blood glucose was monitored by sampling via the tail vein and analyzed using an Accu-Chek monitor (Roche Diagnostics, Japan). Diabetes mellitus was considered to be established when 2 consecutive blood glucose measurements exceeded 450 mg/dL.
Islet Transplantation and Treatment Protocol
The donor islets were isolated from BALB/c mice. After culturing for 20 h, 600 donor islets were transplanted into the liver of diabetic C57BL6 mice (9–10 wk old) via the portal vein. The recipient mice received (orally) 0.1 mg/kg trametinib (Tra0.1), 0.3 mg/kg trametinib (Tra0.3), or vehicle once per day for 28 d, starting from the day of allogeneic islet transplantation. The dose of trametinib was determined based on previous studies.22,24 After transplantation, nonfasting blood glucose levels were monitored daily for the first 14 d and then 3 times per week thereafter until graft rejection. Engraftment was established when the blood glucose level fell below 200 mg/dL for 2 consecutive days. The day of graft rejection was defined as the first day on which blood glucose levels exceeded 350 mg/dL for 2 consecutive days.
Separately from the experiment designed to evaluate graft survival, islet grafts were retrieved from recipient mice to assess the immunological impact on islet grafts, expression of mRNA encoding intrahepatic inflammatory cytokines, and T-cell subpopulations.
Intraperitoneal Glucose Tolerance Tests
On day 7, recipient mice treated with trametinib or vehicle were subjected to an intraperitoneal glucose tolerance test (IPGTT) to evaluate islet graft function. Naive C57BL6 mice were used as controls. After 5 h of fasting, mice received an intraperitoneal injection of glucose solution (2 g of glucose/kg body weight) and blood glucose levels were checked after 0, 15, 30, 60, and 120 min. The area under the curve was calculated from the IPGTT data using the trapezoidal rule and the area above baseline.
Plasma C-peptide Measurements
During the IPGTT, blood samples were collected into heparin-coated capillary tubes at 0, 15, and 30 min after glucose administration. The plasma samples were obtained by centrifugation of blood samples at 1100 × g (15 min, 4°C). C-peptide was analyzed by LEBIS Mouse C-Peptide (U-type) ELISA kit (AKRCP-031, FUJIFILM Wako Shibayagi, Gunma, Japan) according to the manufacturer’s instructions. ΔC-peptide was calculated by subtracting the value for plasma C-peptide at 0 min from the value at each time point (0, 15, and 30 min).
Histopathological Analysis
Livers containing islet grafts were procured from recipient mice, fixed in 10% formalin neutral buffer solution, embedded in paraffin, and cut into 5 μm-thick sections. Hematoxylin and eosin (H&E) staining was preformed according to standard protocols. Immunohistochemical staining was performed as previously reported.25 Sections were incubated overnight at 4°C with the following primary antibodies: antiinsulin (1:2000; Proteintech, Tokyo, Japan), anti-CD4 (1:100; D7D2Z, Cell Signaling Technology), anti-CD8α (1:400; D4W2Z, Cell Signaling Technology), anti-CD31 (1:100; Cell Signaling Technology), and anti-F4/80 (1:50; Novus Biologicals), followed by incubation for 40 min with a biotinylated secondary antibody diluted 1:300 in PBS. Sections were washed with PBS before addition of avidin-biotin-peroxidase complex (1:100 in biotinylated secondary antibody; ABC-Elite, Vector Laboratories, Burlingame, CA) for 50 min. After washing in PBS, the color reaction was carried out using diaminobenzidine and nuclei were counterstained with hematoxylin. The number of positively stained lymphocytes that had infiltrated into the islet grafts was counted in all lobes of the liver (at the maximum cross section, which contained about 10 islets in total). The areas of liver sections that stained positively for insulin were measured and analyzed using NIH Image J software.
Preparation of Lymphocytes and Flow Cytometry
Lymphocytes from fresh livers and spleens were prepared essentially as previously reported.26 Briefly, excised liver from recipient mice was passed gently through a 200 μm steel mesh using a sterile syringe plunger. The liver cell suspension was collected, washed in PBS, and centrifuged for 10 min at 500 × g. The pellet was resuspended in 40% Percoll (GE Healthcare). The cell suspension was gently overlaid onto 70% Percoll and centrifuged for 25 min at 750 × g. Mononuclear cells were collected from the interface. Splenic cells were obtained from homogenized splenic tissue and filtered through a sieve. Contaminating red blood cells were lysed using red blood cells lysis buffer (BioLegend) and washed twice in PBS. Single cell suspensions obtained from the spleen and liver were subjected to flow cytometry analysis to detect surface antigens. The antibodies used to identify specific antigens were anti-CD3 (17A2), anti-CD4 (GK1.5), anti-CD8a (53-6.7), anti-CD44 (IM7), and anti-CD62L (MEL-14; all from BioLegend). Stained samples were assessed on a BD Accuri C6 Flow Cytometer (Becton Dickinson) and data analyzed using FlowJo Software (Tree Star).
Gene Expression Analyses
Total RNA was extracted from recipient liver samples using a PureLink RNA Mini Kit (Invitrogen, Tokyo, Japan), and cDNA was synthesized using a SuperScript VILO cDNA Synthesis Kit (Invitrogen), according to the manufacturer’s protocol. Expression of mRNA encoding IL-2, IL-4, IL-6, IL-10, tumor necrosis factor (TNF)-α, and interferon (IFN)-γ was measured by quantitative real-time PCR (q-PCR) using the StepOnePlus Real-Time PCR System (Applied Biosystems), along with TaqMan gene expression assays for IL-2 (Mm00434256_m1), IL-4 (Mm00445259_m1), IL-6 (Mm00446190_m1), IL-10 (Mm01288386_m1), TNF-α (Mm01168134_m1), and IFN-γ (Mm01168134_m1). 18S rRNA was used as an endogenous control (Mm03928990_g1). Fold changes in mRNA expression were calculated using the 2–ΔΔCt method. Each measurement was performed in triplicate.
Cell Viability Assay
To assess the viability of islet cells, Calcein-AM and propidium iodide (PI; Dojindo Laboratories, Kumamoto, Japan) were used as viability stains. Fresh solutions were prepared by diluting the stock in PBS to yield a final concentration of 1 μg/mL Calcein-AM and 10 μg/mL PI. The isolated islets were cultured for 24 h in the presence of trametinib (final concentration, 1 nmol/L–1 μM) or solvent (DMSO), and then incubated with the staining solution for 10 min at 37°C. The islets were then washed and assessed using fluorescence microscopy (BZ-X800 microscope; KEYENCE, Japan). Viable cells convert Calcein-AM to a green fluorescent product. Dead cells are permeable to PI, which is observed as red fluorescence. The viability of each islet was measured as a percentage of the total Calcein-AM positive area, subtracting the PI positive area from total Calcein-AM and PI positive areas. The areas were calculated using an image analyzer (KEYENCE, Japan).
Glucose-stimulated Insulin Secretion
The insulin secretory capacity of islets treated with trametinib was evaluated as described previously.27 Briefly, after culture for 24 h with trametinib (final concentration, 1 nmol/L–1 μM) or solvent (DMSO), 10 islet equivalents/Transwell were plated in 24-well Transwell microplates in RPMI1640 medium containing 3.3 mmol/L glucose and 0.1% FBS, and then incubated at 37°C for 60 min under a 5% CO2/95% air atmosphere to allow stabilization. Next, the Transwell was removed, placed in a second well containing RPMI1640 medium, 3.3 mmol/L glucose, and 0.1% FBS, and then incubated at 37°C for 60 min. Next, the Transwell was placed in a third well containing 20 mmol/L glucose for 60 min. After removal of the Transwell, the supernatants from the second and third wells were collected immediately, and then centrifuged at 800 × g for 1 min at 4°C. The insulin content of the supernatant was measured using a mouse insulin ELISA kit (AKRIN-011RU; FUJIFILM Wako Shibayagi, Gunma, Japan). The stimulation index was calculated by dividing the value for insulin secretion at the high-glucose concentration (3.3 mmol/L) by that at the low-glucose concentration (20 mmol/L).
Measurement of Total Insulin Content in Islets
After culture for 24 h with trametinib (final concentration, 1 nmol/L–1 μM) or solvent (DMSO), 10 islet equivalents per group were collected in microcentrifuge tubes and resuspended in acid ethanol (0.18 M HCl in 96% ethanol [v/v]). The islets were homogenized and incubated in 4°C for 12 h. Insulin in the supernatant was measured using a mouse insulin ELISA kit (AKRIN-011RU; FUJIFILM Wako Shibayagi, Gunma, Japan).
Statistical Analysis
Statistical analyses and data presentation were performed using Prism software (GraphPad). Data are expressed as the mean ± SD. Statistical analyses were performed using an unpaired Student’s t test (for 2 groups) or ANOVA followed by the Tukey-Kramer test (for groups of 3 or more). The log-rank test was used to compare graft survival between groups. Significance was defined as P < 0.05.
RESULTS
Trametinib Prolongs Survival of a Fully MHC-mismatched Islet Allograft
We examined the immunosuppressive effects of trametinib in mice harboring fully MHC-mismatched islet transplants. Six hundred BALB/c (H-2d haplotype) islets were transplanted into STZ-induced diabetic C57BL/6 (H-2b haplotype) mice via the portal vein. Vehicle-treated mice rejected the allografts with a median graft survival time of 12 d (n = 8; Figure 1A and B). By contrast, treatment with trametinib at a daily dose of 0.1 or 0.3 mg/kg for 28 d prolonged the median graft survival time to 32 (n = 8; P = 0.0007) and 30 d (n = 9; P = 0.005), respectively (Figure 1A and B). At the end of the administration period (d 28), 62.5% (5/8, 0.1 mg/kg) and 55.5% (5/9, 0.3 mg/kg) of recipient mice maintained normal blood glucose levels. None of the transplanted mice receiving vehicle or trametinib after islet transplantation died before graft rejection. The body weight of recipients treated with trametinib increased steadily during trametinib treatment (Figure 1C). In addition, we performed immunohistochemical staining of liver sections obtained from recipient mice on postoperative days 30 or 100 after completing trametinib treatment. Vascularization around islet grafts in recipient mice treated with trametinib (0.1 or 0.3 mg/kg) was successful (Figure S1, SDC, http://links.lww.com/TXD/A272). In addition, there were no changes in vascularization on days 30 and 100. By contrast, infiltration of islet grafts by macrophages was not observed, regardless of the number of days posttransplantation or the dose of trametinib (Figure S1, SDC, http://links.lww.com/TXD/A272). These results suggest that trametinib has no adverse effects on vascularization around islets grafts, and that it inhibits infiltration of the grafts by macrophages.
FIGURE 1.
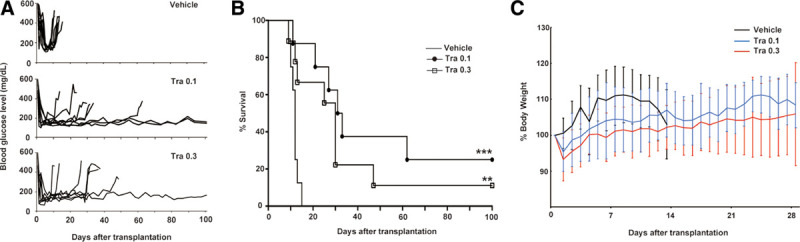
Six hundred islets isolated from BALB/c mice were transplanted into the liver of streptozotocin-induced diabetic C57/BL6 mice. Recipient mice received vehicle or trametinib (0.1 or 0.3 mg/kg) orally from the d of transplantation to d 28. Blood glucose levels (A) and islet graft survival (B) were monitored (n = 8 or 9). Graft survival was calculated using the Kaplan-Meier method and compared with vehicle treatment using a log-rank test (**P < 0.01, ***P < 0.001). (C) Preoperative and postoperative body weight were measured for 28 d or until graft rejection. Data were expressed as a percentage of preoperative body weight and the mean ± SD. Tra, trametinib.
Trametinib Preserves Graft Function and Glucose Responsiveness After Allogeneic Transplantation
To assess islet graft function, we performed an IPGTT on recipient mice at 7 d post–islet transplantation. Trametinib-treated groups had near-normal IPGTT profiles (Figure 2A) and a significantly smaller area under the curve for the IPGTT than vehicle-treated mice (Figure 2B; P < 0.0001 [0.1 mg/kg, n = 3]; P = 0.001 [0.3 mg/kg, n = 3]). During the IPGTT, plasma C-peptide levels at each time point (0, 15, and 30 min) were measured to assess insulin secretion. There was no significant difference in ΔPlasma C-peptide levels between trametinib-treated mice and naive mice (Figure 2C; P = 0.36, 2-way ANOVA, n = 3 per group). In addition, the glucose/C-peptide ratio (blood glucose level adjusted according to the plasma C-peptide level during IPGTT) showed near-normal profiles in trametinib-treated mice (Figure 2D; P < 0.001, 2-way ANOVA, n = 3 per group). These results indicate that trametinib preserves graft function and glucose responsiveness after allogeneic transplantation.
FIGURE 2.
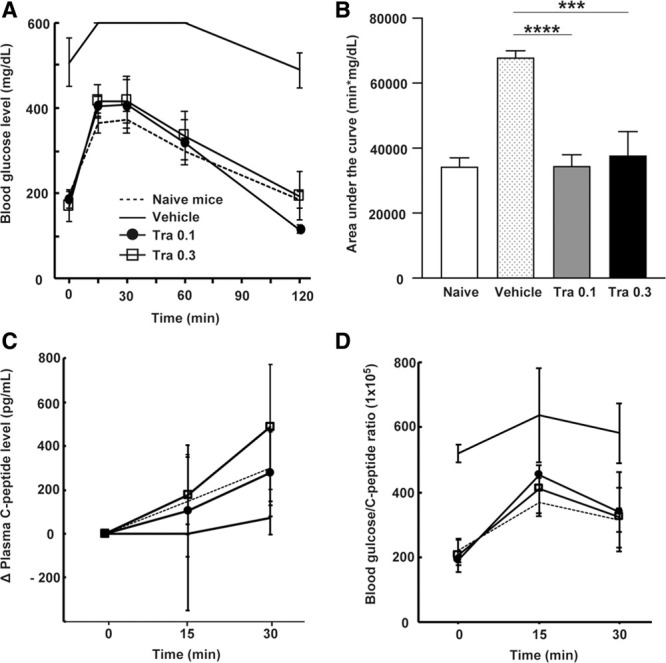
On d 7, an IPGTT was performed for recipient mice treated with vehicle or trametinib (0.1 or 0.3 mg/kg); also, blood glucose levels were measured and compared with those of naive mice (A). The area under the curve (AUC) was calculated (B); 1-way ANOVA followed by the Tukey-Kramer test (***P < 0.001 vs vehicle-treated mice, ****P < 0.0001 vs vehicle-treated mice). ΔPlasma C-peptide levels during IPGTT, expressed as pg/mL (C). Blood glucose/C-peptide ratio (D); 2-way ANOVA followed by the Tukey-Kramer test. Data are expressed as the mean ± SD of 3 independent experiments. IPGTT, intraperitoneal glucose tolerance test; Tra, trametinib.
Trametinib Suppresses Intragraft Infiltration by Lymphocytes
Due to antigen-specific rejection, the level of immune cell infiltration into the islet grafts peaks at around 7–10 d posttransplantation.28 Therefore, to assess the degree of the graft rejection, intrahepatic islet allografts transplanted into recipients treated with vehicle or with 0.1 or 0.3 mg/kg trametinib were procured at day 7 and evaluated histopathologically. H&E staining revealed severe cellular infiltration into and surrounding the islet grafts in the vehicle-treated group (Figure 3A; HE). Immunohistochemistry revealed that the immune response comprised mainly CD4+ and CD8+ cells (Figure 3A). Trametinib (0.3 mg/kg) significantly inhibited infiltration by CD4+ and CD8+ cells when compared with vehicle (Figure 3B; P < 0.05, n = 4/group). In addition, trametinib (0.3 mg/kg) preserved islet morphology (Figure 3A; insulin) and insulin-positive areas (Figure 3C; P < 0.05, n = 4/group). These results suggest that trametinib inhibits cellular infiltration associated with rejection.
FIGURE 3.
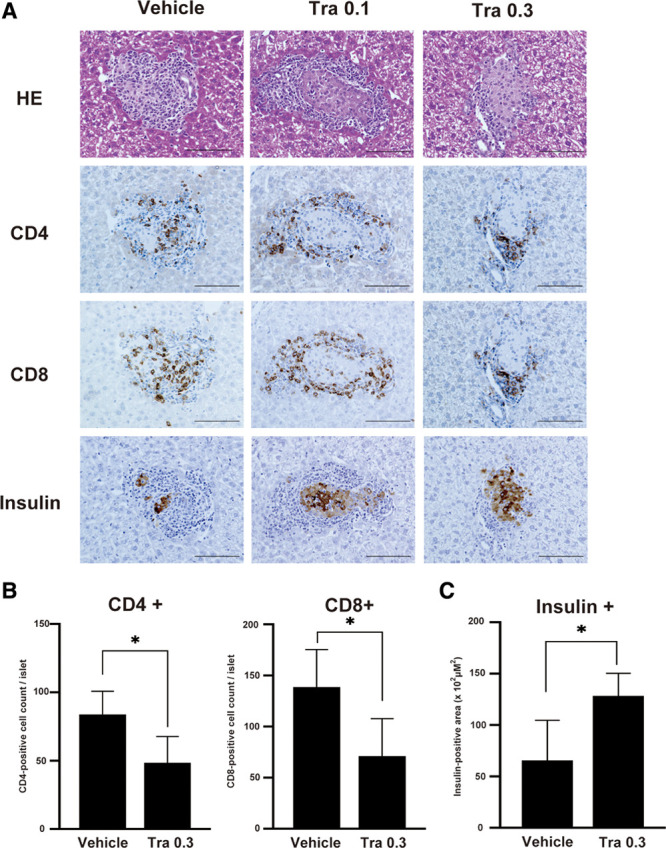
At 7 d posttransplantation, islet allografts from recipient livers were stained with HE and immunostained for expression of CD4, CD8α, and insulin. (A) Representative images of islet allograft recipients treated with vehicle or with 0.1 or 0.3 mg/kg trametinib (Tra; magnification, ×400; scale bar: 100 μm). (B) The number of CD4+ and CD8+ cells in the recipient liver were counted and compared between vehicle and Tra groups. (C) The insulin-positive area was measured and compared between groups. Data are expressed as the mean ± SD of 2 independent experiments (n = 4/group) and compared using an unpaired Student’s t test (*P < 0.05). HE, hematoxylin and eosin; Tra, trametinib.
Trametinib Suppresses Functional Differentiation of CD4+ T Cells In Vivo
We examined the mechanism by which trametinib impacts T-cell subpopulations after islet transplantation. T-cell subpopulations isolated from the liver and spleen of recipient mice on day 7, which were treated with vehicle or with 0.1 or 0.3 mg/kg trametinib, were analyzed by flow cytometry (n = 3/group). The ratio of CD8+ T cells to CD4+ T cells in the liver of trametinib-treated mice tended to be lower than that in the vehicle-treated group (Figure 4A; 1-way ANOVA; P = 0.26), indicating that trametinib suppresses infiltration of the liver by CD8+ T cells. Among the different CD4+ T-cell subpopulations, trametinib increased the percentage of naive T cells (CD62L+CD44–) in the liver in a dose-dependent manner; it also reduced the percentage of effector memory T cells (CD62L-CD44+) in the liver and spleen (Figure 4B; 1-way ANOVA; all P < 0.05). By contrast, trametinib had no effect on CD8+ T-cell subpopulations in the liver or spleen (Figure 4C). These results suggest that (at least in vivo) trametinib mainly suppresses functional differentiation of CD4+ naive T cells.
FIGURE 4.
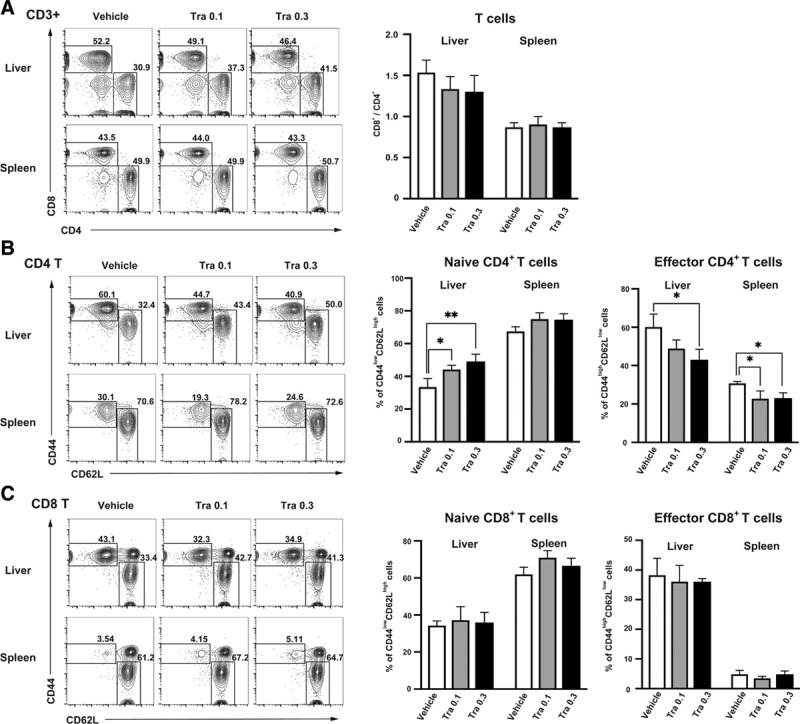
CD4/CD8 and naive/effector memory T-cell subsets of the liver and spleen were analyzed by flow cytometry at d 7 posttransplantation in the presence of 0.1 or 0.3 mg/kg trametinib (Tra) or vehicle. (A) Percentages of CD4+ and CD8+ T cells and the CD8+/CD4+ ratio. (B) Percentage of naive (CD62L+CD44–) and effector memory (CD62L–CD44+) T cells among the CD4+ T-cell and (C) CD8+ T-cell populations. Data are representative of 3 independent experiments (mean ± SD; n = 3/group; 1-way ANOVA followed by the Tukey-Kramer test [*P < 0.05 vs vehicle, ** P < 0.01 vs vehicle]).
Trametinib Inhibits Expression of mRNA Encoding Inflammatory Cytokines In Vivo
To investigate the effects of trametinib on inflammatory immune responses, which are related to T-cell activation and differentiation, we performed q-PCR on day 7 to measure expression of mRNA encoding inflammatory cytokines in the excised liver from recipient mice treated with vehicle or with 0.1 or 0.3 mg/kg trametinib (n = 3/group). Expression of mRNA encoding IL-2, TNF-α, or IFN-γ in the trametinib-treated group (0.1 mg/kg) was lower than that in the vehicle-treated group (Figure 5; P < 0.001, P < 0.05, and P < 0.01, respectively). However, trametinib (0.3 mg/kg) had no dose-dependent effect on suppression of inflammatory cytokines. As for the antiinflammatory cytokines IL-4 and IL-10, expression of mRNA tended to be higher in the trametinib-treated group (Figure 5; P < 0.05 [0.1 mg/kg] and P = 0.20 [0.3 mg/kg], respectively). These findings indicate that trametinib modulates Th1 and Th2 immune responses and regulates inflammatory cytokines in the recipient liver.
FIGURE 5.
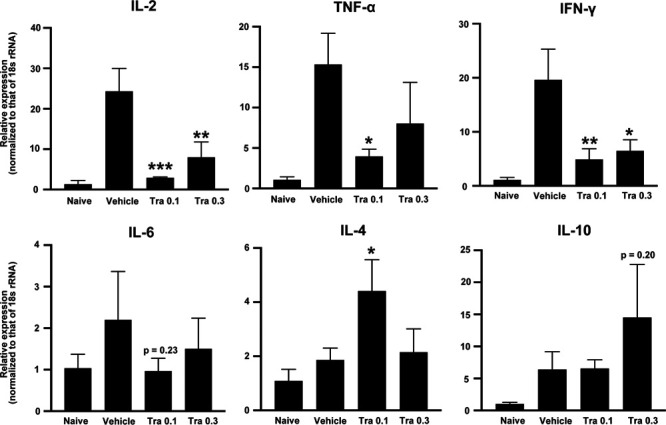
Q-PCR analysis of IL-2, IL-4, IL-6, IL-10, TNF-α, and IFN-γ mRNA expression in the liver of islet graft recipients treated with vehicle, or with 0.1 or 0.3 mg/kg trametinib (Tra 0.1 and Tra 0.3, respectively). Expression of each gene was normalized to that of 18s rRNA, and then quantified relative to that in naive mice. Data shown are from 3 independent experiments and are expressed as the mean ± SD (n = 3/group; *P < 0.05, **P < 0.01, ***P < 0.001, compared with the vehicle groups, 1-way ANOVA followed by the Tukey-Kramer test). IFN, interferon; IL, interleukin; Q-PCR, quantitative real-time PCR; TNF, tumor necrosis factor.
Trametinib Has No Adverse Effects on Islet Viability or Insulin Secretion In Vitro
We evaluated the effect of trametinib on islet viability and insulin secretion in vitro. Islet viability was assessed using Calcein-AM and PI staining (Figure 6A). We found no change in the viability of cultured islet cells exposed to 0, 1, 10, or 100 nmol/L, or to 1 μM, trametinib (Figure 6B; 93.7 ± 1.9%, 92.8 ± 3.8%, 94.5 ± 2.0%, 92.5 ± 1.7%, and 92.9 ± 3.5%, respectively; 1-way ANOVA; P = 0.79, n = 5/group). Next, we assessed the total insulin content and secretory capacity of islets cultured in the presence of each concentration of trametinib. Total insulin content and insulin release at low (3.3 mmol/L) or high (20 mmol/L) glucose levels showed no change at different concentrations (Figure 6C, D). In addition, there was no change in the stimulation index at different concentrations (0, 1, 10, or 100 nmol/L, or 1 μM) of trametinib (Figure 6E; 2.98 ± 0.31, 3.62 ± 1.17, 2.96 ± 1.00, 3.22 ± 1.84, and 3.29 ± 1.05, respectively; 1-way ANOVA; P = 0.95, n = 4/group). Thus, trametinib did not impair insulin secretion in vitro.
FIGURE 6.
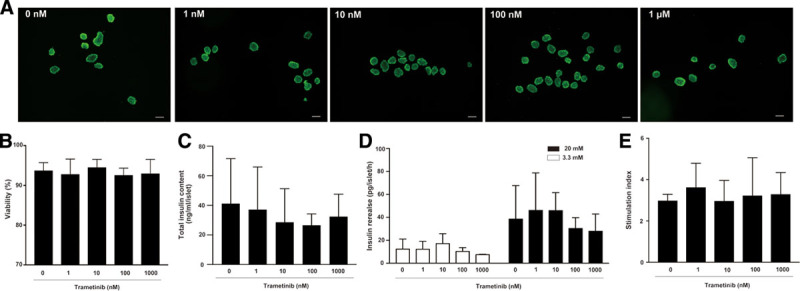
Islet cells isolated from BALB/c mice were incubated with 0, 1, 10, or 100 nmol/L, or 1 μM trametinib for 24 h. (A) Representative fluorescence microscopy images are shown for each concentration. Islet cells were stained with Calcein-AM (green: viable cells) and PI (red: dead cells). Original magnification, ×40; scale bar: 100 μm. (B) Percentage of viable islet cells in the presence of the indicated concentration of trametinib, calculated from an average of 40 islets (n = 5/group). (C) Total insulin content per islet after 24 h culture in the presence of each concentration of trametinib (n = 4/group). (D) Concentration of insulin detected in the presence of low (3.3 mmol/L) and high (20 mmol/L) glucose after 24 h of culture with the indicated concentrations of trametinib. (E) The stimulation index was calculated for each group (n = 4/group) by dividing the value for insulin secretion at high-glucose concentration by that at low-glucose concentration. Data are representative of 5 independent experiments (mean ± SD; 1-way ANOVA followed by the Tukey-Kramer test). PI, propidium iodide.
DISCUSSION
Here, we demonstrated that the MEK inhibitor trametinib prolongs graft survival after fully MHC-mismatched islet transplantation without the need for conventional immunosuppressants. This immunosuppressive effect is attributed mainly to inhibitory effects on naive CD4+ T cells; subsequent inhibition of the Th1 immune response induces an antiinflammatory environment in the recipient liver. We also confirmed that trametinib had no adverse effect on insulin secretion or survival of islet cells in vitro.
A previous study showed that conventional immunosuppressants such as tacrolimus, rapamycin, and mycophenolate mofetil, when used at doses that do not impair islet graft function, did not prolong islet allograft survival significantly in a fully MHC-mismatched mouse model.12 By contrast, we show here that treatment with trametinib alone prolonged allogeneic islet graft survival significantly, while at the same time preserving graft function moderately. These results are in contrast to those of a previous study showing that a combination of cyclosporine plus trametinib is needed to suppress acute rejection of fully MHC-mismatched lung transplants in rat models.24 Such differences between islet and organ transplantation may be attributed to the fact that primarily vascularized allografts induce direct, not indirect, T cell–mediated alloresponses associated with acute rejection.29 In the direct pathway, T cells recognize intact donor MHC molecules on transplanted cells, whereas in the indirect pathway donor peptides are first processed and presented by host antigen-presenting cells.30 Our recent study suggests that trametinib might suppress the indirect pathway but not the direct pathway.24 Therefore, we suggest that trametinib as a single agent can efficiently suppress acute rejection of a nonvascularized transplant such as an islet graft, which activates T cells via both direct and indirect pathways.
In our model, trametinib prolonged islet graft survival in 60% of recipient mice at the end of the administration period. However, graft acceptance was not maintained after treatment with trametinib ceased. Trametinib itself may not induce tolerance to allogeneic grafts. We reported previously that a MEK inhibitor, selumetinib, did not increase apoptosis of T cells stimulated with OKT3/CD28 in vitro.21 Thus, the immunosuppressive effect of trametinib depends on suppression rather than on deletion of proliferating T cells. Administration of trametinib over a longer time period may be required to achieve long-term graft engraftment.
We found here that trametinib suppressed both functional differentiation of naive CD4+ T cells and cellular infiltration into the graft in a dose-dependent manner. These findings are consistent with those of previous reports of BMT and lung transplantation models.22,24 However, in contrast to the other reports, we did not observe trametinib dose-dependent prolongation of graft survival time and suppression of inflammatory cytokines. This may be because trametinib increased the immunogenicity of the transplanted grafts. A previous study demonstrates that trametinib increases surface expression of MHC class I by a wide variety of cancer cells by inhibiting activation of mitogen-activated protein kinase signaling in a dose-dependent manner.31 Increased expression of MHC class I by cancer cells increases cytotoxic activity of antigen-specific T cells following treatment with trametinib. We suggest that trametinib has similar effects on donor islet grafts in a transplantation setting. The dose-dependent effects of trametinib suppress functional differentiation of recipient CD4+ T cells while increasing the antigenicity of the transplanted donor graft. Thus, setting the optimal dose of trametinib might be an important factor for achieving successful allogeneic transplantation.
In the present study, we show that trametinib suppressed production of inflammatory cytokines such as IL-2, TNF-α, and IFN-γ in the liver after transplantation (as determined by analysis of mRNA expression), suggesting inhibition of the Th1 immune response. Interestingly, trametinib also increased IL-4 and IL-10 production in vivo. These findings are consistent with those of a previous report showing that a low dose of MEK inhibitor, PD98059, inhibits production of IL-2, TNF-α, and IFN-γ by human T cells stimulated with an anti-CD3 monoclonal antibody and phorbol 12-myristate 13-acetate in vitro, while at the same time increasing production of IL-4 and IL-10.19 Trametinib might modulate Th1/Th2 immune responses. A switch from Th1 to Th2 responses might induce tolerance to allogeneic grafts with major histocompatibility antigen mismatches.32 In addition, we confirmed that trametinib inhibited inflammatory cytokines such as TNF-α, which is secreted mainly by macrophages.33 Inhibiting activation of mitogen-activated protein kinase pathways (including ERK1/2, p38, and c-Jun NH2-terminal kinase) suppresses inflammatory responses associated with instant blood-mediated inflammatory reactions during the peritransplant period.34–39 However, increased p38/c-Jun NH2-terminal kinase activation correlates more strongly with decreased islet survival than does ERK1/2 activation.39–41 Therefore, we conclude that trametinib does not suppress primary inflammation triggered by instant blood-mediated inflammatory reactions but rather attenuates alloreactive T cells and immune response related to graft rejection.
In vitro experiments show that the direct effects of conventional immunosuppressants (such as tacrolimus) on isolated human islets lead to a significant decrease in glucose-induced insulin secretion and to an increase in the percentage of apoptotic β cells.10 Here, we confirmed that various concentrations of trametinib did not exert cytotoxic effects or impair insulin secretion by isolated islets in vitro. These results indicated that trametinib had no direct toxic effects on cultured islets. In addition, MEK inhibitors inhibit β-cell apoptosis and preserve insulin secretion by human islets under hyperglycemic conditions and in the presence of IL-1β in vitro.38 In the present study, trametinib showed no dose-dependent effects on cultured islets in the absence of stress conditions (eg, proinflammatory cytokines). Therefore, MEK might not be fully activated before treatment with trametinib. Further studies are needed to assess the dose-dependent effects of trametinib on inflammatory responses, oxidative stress, and apoptotic responses of isolated islets under stress conditions.
In summary, we demonstrated for the first time that trametinib improves the outcome of fully MHC-mismatched islet transplantation without the need for conventional immunosuppressants. In addition, trametinib had no adverse effects on islet cell viability or insulin secretion. Trametinib might have beneficial effects after islet transplantation; these effects include preservation of graft function and vascularization around islet grafts, neither of which are observed after treatment with conventional immunosuppressants. Thus, trametinib may enable clinicians to reduce the dose of conventional immunosuppressants and avoid further adverse effects, thereby enabling maintenance of immunosuppressive therapy after allogenic islet transplantation.
ACKNOWLEDGMENTS
We thank the Center for Anatomical, Pathological, and Forensic Medical Research, Kyoto University Graduate School of Medicine, for preparing the microscope slides.
Footnotes
This work was supported by Grants-in-Aid for Scientific Research (T.A. and S.U., No. 18K08593) from the Japan Society for the Promotion of Science, Tokyo, Japan.
The authors declare no conflicts of interest.
S.T. designed the study protocol, performed the experiments, analyzed the data, and wrote the draft article. T.A. obtained the grant, designed the study protocol, participated in research, analyzed the data, and wrote the draft article. T.S. designed the study protocol, participated in research, analyzed the data, and wrote the draft article. K.Y., K.I., and N.F. participated in research. K.N., T.M., H.O., K.T., and S.S. helped writing of the draft article. S.U. supervised the research and revised the draft article.
REFERENCES
- 1.Shapiro AM, Ricordi C, Hering BJ, et al. International trial of the Edmonton protocol for islet transplantation. N Engl J Med. 2006; 355:1318–1330. doi:10.1056/NEJMoa061267 [DOI] [PubMed] [Google Scholar]
- 2.Hering BJ, Clarke WR, Bridges ND, et al. Clinical Islet Transplantation Consortium Phase 3 trial of transplantation of human islets in type 1 diabetes complicated by severe hypoglycemia. Diabetes Care. 2016; 39:1230–1240. doi:10.2337/dc15-1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holmes-Walker DJ, Gunton JE, Hawthorne W, et al. Islet transplantation provides superior glycemic control with less hypoglycemia compared with continuous subcutaneous insulin infusion or multiple daily insulin injections. Transplantation. 2017; 101:1268–1275. doi:10.1097/TP.0000000000001381 [DOI] [PubMed] [Google Scholar]
- 4.Schuetz C, Anazawa T, Cross SE, et al. IPITA YIC Young Investigator Committee β cell replacement therapy: the next 10 years. Transplantation. 2018; 102:215–229. doi:10.1097/TP.0000000000001937 [DOI] [PubMed] [Google Scholar]
- 5.Mittal S, Johnson P, Friend P. Pancreas transplantation: solid organ and islet. Cold Spring Harb Perspect Med. 2014; 4:a015610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wisel SA, Braun HJ, Stock PG. Current outcomes in islet versus solid organ pancreas transplant for β-cell replacement in type 1 diabetes. Curr Opin Organ Transplant. 2016; 21:399–404. doi:10.1097/MOT.0000000000000332 [DOI] [PubMed] [Google Scholar]
- 7.McCall M, Shapiro AM. Update on islet transplantation. Cold Spring Harb Perspect Med. 2012; 2:a007823 doi:10.1101/cshperspect.a007823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Najarian JS, Sutherland DE, Matas AJ, et al. Human islet transplantation: a preliminary report. Transplant Proc. 1977; 9:233–236 [PubMed] [Google Scholar]
- 9.Nir T, Melton DA, Dor Y. Recovery from diabetes in mice by beta cell regeneration. J Clin Invest. 2007; 117:2553–2561. doi:10.1172/JCI32959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bugliani M, Masini M, Liechti R, et al. The direct effects of tacrolimus and cyclosporin A on isolated human islets: a functional, survival and gene expression study. Islets. 2009; 1:106–110. doi:10.4161/isl.1.2.9142 [DOI] [PubMed] [Google Scholar]
- 11.Johnson JD, Ao Z, Ao P, et al. Different effects of FK506, rapamycin, and mycophenolate mofetil on glucose-stimulated insulin release and apoptosis in human islets. Cell Transplant. 2009; 18:833–845. doi:10.3727/096368909X471198 [DOI] [PubMed] [Google Scholar]
- 12.Marzorati S, Melzi R, Citro A, et al. Engraftment versus immunosuppression: cost-benefit analysis of immunosuppression after intrahepatic murine islet transplantation. Transplantation. 2014; 97:1019–1026. doi:10.1097/TP.0000000000000104 [DOI] [PubMed] [Google Scholar]
- 13.Gala-Lopez BL, Senior PA, Koh A, et al. Late cytomegalovirus transmission and impact of T-depletion in clinical islet transplantation. Am J Transplant. 2011; 11:2708–2714. doi:10.1111/j.1600-6143.2011.03724.x [DOI] [PubMed] [Google Scholar]
- 14.Ojo AO, Held PJ, Port FK, et al. Chronic renal failure after transplantation of a nonrenal organ. N Engl J Med. 2003; 349:931–940. doi:10.1056/NEJMoa021744 [DOI] [PubMed] [Google Scholar]
- 15.Ryan EA, Paty BW, Senior PA, et al. Risks and side effects of islet transplantation. Curr Diab Rep. 2004; 4:304–309. doi:10.1007/s11892-004-0083-8 [DOI] [PubMed] [Google Scholar]
- 16.Wu PK, Park JI. MEK1/2 inhibitors: molecular activity and resistance mechanisms. Semin Oncol. 2015; 42:849–862. doi:10.1053/j.seminoncol.2015.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seger R, Krebs EG. The MAPK signaling cascade. Faseb J. 1995; 9:726–735 [PubMed] [Google Scholar]
- 18.Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997; 15:707–747. doi:10.1146/annurev.immunol.15.1.707 [DOI] [PubMed] [Google Scholar]
- 19.Dumont FJ, Staruch MJ, Fischer P, et al. Inhibition of T cell activation by pharmacologic disruption of the MEK1/ERK MAP kinase or calcineurin signaling pathways results in differential modulation of cytokine production. J Immunol. 1998; 160:2579–2589 [PubMed] [Google Scholar]
- 20.Zhao Y, Adjei AA. The clinical development of MEK inhibitors. Nat Rev Clin Oncol. 2014; 11:385–400. doi:10.1038/nrclinonc.2014.83 [DOI] [PubMed] [Google Scholar]
- 21.Shindo T, Kim TK, Benjamin CL, et al. MEK inhibitors selectively suppress alloreactivity and graft-versus-host disease in a memory stage-dependent manner. Blood. 2013; 121:4617–4626. doi:10.1182/blood-2012-12-476218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Itamura H, Shindo T, Tawara I, et al. The MEK inhibitor trametinib separates murine graft-versus-host disease from graft-versus-tumor effects. JCI Insight. 2016; 1:e86331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang S, Guan Q, Diao H, et al. Prolongation of cardiac allograft survival by inhibition of ERK1/2 signaling in a mouse model. Transplantation. 2007; 83:323–332. doi:10.1097/01.tp.0000251374.49225.19 [DOI] [PubMed] [Google Scholar]
- 24.Takahagi A, Shindo T, Chen-Yoshikawa TF, et al. Trametinib attenuates delayed rejection and preserves thymic function in rat lung transplantation. Am J Respir Cell Mol Biol. 2019; 61:355–366. doi:10.1165/rcmb.2018-0188OC [DOI] [PubMed] [Google Scholar]
- 25.Toda Y, Kono K, Abiru H, et al. Application of tyramide signal amplification system to immunohistochemistry: a potent method to localize antigens that are not detectable by ordinary method. Pathol Int. 1999; 49:479–483. doi:10.1046/j.1440-1827.1999.00875.x [DOI] [PubMed] [Google Scholar]
- 26.Miyagi T, Takehara T, Tatsumi T, et al. CD1d-mediated stimulation of natural killer T cells selectively activates hepatic natural killer cells to eliminate experimentally disseminated hepatoma cells in murine liver. Int J Cancer. 2003; 106:81–89. doi:10.1002/ijc.11163 [DOI] [PubMed] [Google Scholar]
- 27.Ricordi C, Gray DW, Hering BJ, et al. Islet isolation assessment in man and large animals. Acta Diabetol Lat. 1990; 27:185–195. doi:10.1007/BF02581331 [DOI] [PubMed] [Google Scholar]
- 28.Evgenov NV, Medarova Z, Pratt J, et al. In vivo imaging of immune rejection in transplanted pancreatic islets. Diabetes. 2006; 55:2419–2428. doi:10.2337/db06-0484 [DOI] [PubMed] [Google Scholar]
- 29.Kant CD, Akiyama Y, Tanaka K, et al. Primary vascularization of allografts governs their immunogenicity and susceptibility to tolerogenesis. J Immunol. 2013; 191:1948–1956. doi:10.4049/jimmunol.1202092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marino J, Paster J, Benichou G. Allorecognition by T lymphocytes and allograft rejection. Front Immunol. 2016; 7:582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brea EJ, Oh CY, Manchado E, et al. Kinase regulation of human MHC class I molecule expression on cancer cells. Cancer Immunol Res. 2016; 4:936–947. doi:10.1158/2326-6066.CIR-16-0177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ricordi C, Strom TB. Clinical islet transplantation: advances and immunological challenges. Nat Rev Immunol. 2004; 4:259–268. doi:10.1038/nri1332 [DOI] [PubMed] [Google Scholar]
- 33.Kanak MA, Takita M, Kunnathodi F, et al. Inflammatory response in islet transplantation. Int J Endocrinol. 2014; 2014:451035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paraskevas S, Aikin R, Maysinger D, et al. Modulation of JNK and p38 stress activated protein kinases in isolated islets of Langerhans: insulin as an autocrine survival signal. Ann Surg. 2001; 233:124–133. doi:10.1097/00000658-200101000-00018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abdelli S, Ansite J, Roduit R, et al. Intracellular stress signaling pathways activated during human islet preparation and following acute cytokine exposure. Diabetes. 2004; 53:2815–2823. doi:10.2337/diabetes.53.11.2815 [DOI] [PubMed] [Google Scholar]
- 36.Arnette D, Gibson TB, Lawrence MC, et al. Regulation of ERK1 and ERK2 by glucose and peptide hormones in pancreatic beta cells. J Biol Chem. 2003; 278:32517–32525. doi:10.1074/jbc.M301174200 [DOI] [PubMed] [Google Scholar]
- 37.Lawrence MC, McGlynn K, Park BH, et al. ERK1/2-dependent activation of transcription factors required for acute and chronic effects of glucose on the insulin gene promoter. J Biol Chem. 2005; 280:26751–26759. doi:10.1074/jbc.M503158200 [DOI] [PubMed] [Google Scholar]
- 38.Fei H, Zhao B, Zhao S, et al. Requirements of calcium fluxes and ERK kinase activation for glucose- and interleukin-1beta-induced beta-cell apoptosis. Mol Cell Biochem. 2008; 315:75–84. doi:10.1007/s11010-008-9791-8 [DOI] [PubMed] [Google Scholar]
- 39.Jin SM, Kim KS, Lee SY, et al. The sequential combination of a JNK inhibitor and simvastatin protects porcine islets from peritransplant apoptosis and inflammation. Cell Transplant. 2011; 20:1139–1151. doi:10.3727/096368910X550170 [DOI] [PubMed] [Google Scholar]
- 40.Paraskevas S, Aikin R, Maysinger D, et al. Activation and expression of ERK, JNK, and p38 MAP-kinases in isolated islets of Langerhans: implications for cultured islet survival. FEBS Lett. 1999; 455:203–208. doi:10.1016/s0014-5793(99)00882-0 [DOI] [PubMed] [Google Scholar]
- 41.Noguchi H, Nakai Y, Ueda M, et al. Activation of c-Jun NH2-terminal kinase (JNK) pathway during islet transplantation and prevention of islet graft loss by intraportal injection of JNK inhibitor. Diabetologia. 2007; 50:612–619. doi:10.1007/s00125-006-0563-2 [DOI] [PubMed] [Google Scholar]