

 This work is licensed under a
This work is licensed under a Abstract
The prevalence of non-alcoholic fatty liver disease (NAFLD) has increased dramatically worldwide and, subsequently, also the risk of developing non-alcoholic steatohepatitis (NASH), hepatic fibrosis, cirrhosis and cancer. Today, weight loss is the only available treatment, but administration of fibroblast growth factor 21 (FGF21) analogues have, in addition to weight loss, shown improvements on liver metabolic health but the mechanisms behind are not entirely clear. The aim of this study was to investigate the hepatic metabolic profile in response to FGF21 treatment. Diet-induced obese (DIO) mice were treated with s.c. administration of FGF21 or subjected to caloric restriction by switching from high fat diet (HFD) to chow to induce 20% weight loss and changes were compared to vehicle dosed DIO mice. Cumulative caloric intake was reduced by chow, while no differences were observed between FGF21 and vehicle dosed mice. The body weight loss in both treatment groups was associated with reduced body fat mass and hepatic triglycerides (TG), while hepatic cholesterol was slightly decreased by chow. Liver glycogen was decreased by FGF21 and increased by chow. The hepatic gene expression profiles suggest that FGF21 increased uptake of fatty acids and lipoproteins, channeled TGs toward the production of cholesterol and bile acid, reduced lipogenesis and increased hepatic glucose output. Furthermore, FGF21 appeared to reduce inflammation and regulate hepatic leptin receptor-a expression. In conclusion, FGF21 affected several metabolic pathways to reduce hepatic steatosis and improve hepatic health and markedly more genes than diet restriction (61 vs 16 out of 89 investigated genes).
Keywords: fibroblast growth factor 21 (FGF21), non-alcoholic fatty liver disease (NAFLD), lipid metabolism, glucose metabolism, inflammation
Introduction
Non-alcoholic fatty liver disease (NAFLD) is the most common liver disorder in the Western world (1, 2) with a global prevalence of 25% (3). It is defined as steatosis (fat accumulation) in at least 5% of hepatocytes (3). Obesity is a central risk factor for developing NAFLD (4), and it is estimated that 39% of all adults worldwide are overweight and 13% are obese (5). NASH is a further deterioration from the steatosis characteristic of NAFLD accompanied by inflammation and hepatocyte damage. Prevalence of NASH estimates among NAFLD patients range from 25 to 59% (3, 4). Liver fibrosis stage is the most important determinant of long-term outcome for NAFLD patients (6), and approximately 20% of NASH patients develop liver cirrhosis (4). Cirrhosis is the second most common cause of liver transplantation in the US (3), and patients with cirrhosis have an increased risk of developing liver cancer (4).
Currently, no pharmaceutical treatment for NASH is approved, but many compounds targeting various pathways are in the clinical pipeline, as reviewed by Rotman & Sanyal (7). Presently, the most effective method to alleviate NASH is body weight loss (4, 8), but to reverse fibrosis a body weight loss of at least 10% needs to be achieved (9). Fibroblast growth factor 21 (FGF21) is a hormone expressed primarily by the liver and adipose tissue. Treatment with FGF21 induces weight loss in rodents, non-human primates and early clinical trials (10, 11, 12). In diet-induced obese (DIO) mice, FGF21 lowers body weight by increasing energy expenditure (EE), whereas body weight loss in pigs (13) and in some non-human primates appears to be driven by a decrease in food intake (10). FGF21 treatment has multiple effects on the liver either by direct or indirect actions (11). For instance, FGF21 has been shown to reduce hepatic steatosis through increased fatty acid (FA) oxidation and decreased lipogenesis in rodents (11). Moreover, an FGF21 analogue has been shown to reduce inflammation and fibrogenesis in a NASH mouse model using leptin-deficient ob/ob mice and a methionine- and choline-deficient (MCD) diet (14). Likewise, in a phase 2 trial, a protracted FGF21 reduced liver fat, liver injury and fibrosis biomarkers in NASH patients (15). In addition to hepatic lipid regulation, FGF21 treatment also improved glycemic control in DIO mice (11, 16). However, this effect did not seem to translate as convincingly to humans (10, 12), while an increase in insulin sensitivity has been observed (17). Improved hepatic leptin signaling and leptin sensitivity have also been suggested to contribute to the effects by FGF21, as the leptin receptor (Lepr) expression was highly induced in DIO mice treated with FGF21 (11, 18). Leptin is an adipose-derived hormone, which plays an important role in control of appetite and energy homeostasis (19). LEBRB is considered to be the functional receptor for leptin’s effect on energy balance mainly through central regulation, while the effects of the shorter isoforms of the leptin receptor (isoform a, c, d and f) are largely unresolved (20). However, they have been suggested to function as leptin binding proteins (11, 21, 22, 23, 24, 25).
To obtain a broader mechanistic understanding of FGF21 in the liver, we aimed to provide a comprehensive analysis of simultaneous regulation of multiple hepatic metabolic pathways to drive new hypothesis and studies. In DIO mice, a 20% body weight loss was induced by FGF21 treatment or by caloric restriction (achieved by switching from high fat diet (HFD) to chow) for 18 days. Gene expression analysis in liver tissue was conducted by high-throughput qPCR investigating 89 genes within glucose- and lipid metabolism, inflammation and leptin signaling.
Materials and methods
Animals
Animals were used in compliance with internationally accepted principles for the care and use of laboratory animals and were approved by the internal Novo Nordisk Ethical Review Committee in Denmark. The study was conducted according to approved national regulations in Denmark. It was performed at Novo Nordisk, China, and was conducted under licenses granted by the Beijing Administration Office of Laboratory Animals (BAOLA). Animals were obtained from Jackson Laboratories and were housed and maintained in standard, temperature- and humidity-controlled environment and were subject to a 12 h light:12 h darkness cycle (lights on at 06:00 h). The animals had access to water and food ad libitum. During the treatment phase of the study, the mice were given either regular chow (no. 1324, Altromin, Brogaarden, Lynge, Denmark) or HFD (Research Diet #D12492, New Brunswick, NJ, USA).
Study design
Thirty male C57BL/6J DIO mice (approximately 20 weeks of age; 3.0 g) were put on a 60% kcal fat diet from 4 to 5 weeks of age. The mice were single-housed and acclimated to handling for 13 days prior to the start of treatment. Body weight (BW) and food intake were measured daily around 07:00 h by manual weighing. A magnetic resonance scan (Echo MRI) was conducted 6 days before study start, and mice were distributed into three groups such that statistical variations in BW, fat mass and fat/BW% were minimized between groups. At study start, the mice had a mean BW of 46.8 ± 3.0 g. All mice were dosed subcutaneously twice daily with either vehicle or native FGF21 (Novo Nordisk A/S, 0.6 mg/kg/day) for 18 days. The selected dose for FGF21 was based on internal data to obtain 20% body weight loss. Control and FGF21 treated mice were maintained on HFD, while mice in the diet restricted group were switched to standard chow ad libitum (~11% kcal fat). At day 18, without prior fasting, animals were subjected to a second MR scan (2 h after last dosing) and were killed by cervical dislocation under anesthesia by isoflurane inhalation (7 h after last dosing). Blood was sampled during anesthesia from the retro-orbital vein into EDTA-coated tubes, centrifuged (6000 g at 4°C for 5 min), and plasma isolated and stored at −80°C until analysis. Liver tissues were collected immediately, snap-frozen in liquid nitrogen and stored at −80°C.
Plasma and tissue analysis
Plasma leptin and insulin were measured by luminescent oxygen channeling (LOCI) assays (Perkin Elmer alphaLISA, kit AL521F). Frozen liver samples of 20–42 mg were used for the determination of lipid and glycogen as previously described (26). Note that, in this study, samples were homogenized with Tissuelyser II (Qiagen) at 30 Hz for 2× 45 s and analyzed on Cobas 6000 Analyzer (Roche Diagnostics). True TG was calculated as TG subtracted glycerol and glycogen as total glucose subtracted the free glucose. Due to the limited amount of tissue, the number of samples in each group was as follows: control = 9, chow = 10, and FGF21 = 8.
RNA purification
RNA extraction from frozen liver samples (10–30 mg) was conducted according to RNeasy Lipid Tissue Handbook 2009 (Qiagen), protocol ‘Purification of Total RNA Using the RNeasy Lipid Tissue Mini Kit’, with some modifications. TRIzolR Reagent (Invitrogen) was used for cell lysis, and homogenization was performed using Tissuelyser II (Qiagen) for 2× 2 min at 20 Hz. Phase separation was conducted with 1-Bromo-3-chloropropane (Sigma Life Science). DNase treatment was done on-columns after step 10, according to Appendix C of the protocol. RNA concentration was measured by NanoDrop-1000 (Thermo Scientific) and purity accessed from OD260/280. Integrity was tested on Agilent 2100 Bioanalyzer (Agilent Technologies) using the RNA 6000 Nano Kit. Samples with an RNA Integrity Number (RIN) ≥ 5 were used for downstream analysis (control: n = 10, OD260/280 = 2.0 ± 0.1, RIN = 7.8 ± 1.0; chow: n = 9, OD260/280 = 2.0 ± 0.0, RIN = 7.4 ± 1.2; FGF21: n = 7, OD260/280 = 2.1 ± 0.0, RIN = 5.9 ± 0.6. Values are mean ± s.d.).
cDNA synthesis
For each RNA sample two cDNA replicates were made separately from 1000 ng RNA using iScript™ cDNA Synthesis Kit (Bio-Rad), containing a blend of oligo(dT) and random hexamer primers, in a final reaction volume of 20 µL according to the manufacturer’s protocol. A negative control was made for each group by omitting RT in the reaction. Following manufacturer’s instructions, the reactions were incubated 5 min at 25°C, 30 min at 42°C and 5 min at 85°C. The cDNA was stored at −80°C until use.
Primer design
Selection of the genes of interest in the liver was focused on lipid and carbohydrate metabolic pathways, inflammation and leptin signaling and based on available RNA-sequencing data from in-house studies (Novo Nordisk A/S), on the presumption of differential gene expression between the groups. Gapdh, Ywhaz and Sdha were tested as reference genes, as they have been reported to be stably expressed in similar studies (27, 28, 29). Initially, 96 genes were selected. Primer sequences were obtained from the database PrimerBank (30) or designed using Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast). Primers were selected to flank an intron and make amplicons in the range of 75–200 nucleotides when possible and to have a narrow melting temperature between 60 and 63°C. Primers were purchased from Sigma-Aldrich. The sequences are listed in Supplementary Table 1 (see section on supplementary materials given at the end of this article).
High-throughput qPCR
High-throughput qPCR was conducted with the Biomark HD system (Fluidigm) on a 96.96 IFC chip. cDNA samples were pre-amplified with a pool of all assays to be analyzed. Fifteen PCR cycles of 5× diluted cDNA using TaqMan PreAmp Master Mix (Life Technologies) followed by Exonuclease I (New England BioLabs) cleanup was performed according to the manufacturer’s protocol (Fluidigm PN100-5875C1), apart from using primer concentrations of 250 nM. Samples were further 5× diluted and qPCR reactions were conducted using 2× SsoFast™ EvaGreen® Supermix with Low ROX (Bio-Rad Laboratories) according to manufacturer’s instructions (Fluidigm PN100-9792B1), except from final primer concentrations of 5 µM. A RT control from each group was included and standard curves were made from a cDNA pool of equal amounts of all pre-amplified and exonuclease treated samples. The thermal cycling profile used was GE 96.96 Fast PCR+Melt v2.pcl with melting curve analysis. Data were collected with Biomark HD Data Collection software.
qPCR data analysis
The efficiency for each assay was calculated from the log-linear portion of the standard curve and assays with efficiencies from 85 to 115% and R2 ≥ 0.98 were accepted. Data were analyzed in GenEx6 Pro (MultiD Analyses AB). Quantification cycle (Cq) values were corrected to assay efficiency and the reference genes were tested by the normalization algorithms GeNorm and Normfinder. Gapdh and Ywhaz were used for normalization, Cq values of cDNA replicates were averaged and fold changes (FC) were obtained by setting the expression of the treatment groups relative to the control group.
Follow-up by conventional qPCR
Two failed assays (Srebf1, Leprb) and three additional ones (Lepra, Lepr, Hmgcl) were subsequently analyzed using conventional qPCR on MX3005P (Agilent). For each 10 µL reaction, 1 µL 5× diluted cDNA, 5 µL 2× QuantiFast™ SYBR Green master mix (Qiagen) and 1 µM primer were mixed. Standard curves were made based on a cDNA pool of all samples in the study. Leprb was too low expressed to provide reliable analysis. Data were analyzed in GenEx6 Pro as described previously, apart from normalization to Gapdh, which was the most stable reference gene observed. The thermal profile was 95°C for 5 min, 40 cycles of 95°C for 10 s and 60°C for 30 s, ending with melting curve analysis (60–99°C). Data were collected using MxPro QPCR Software version 4.10.
Statistical analysis
Leptin and insulin plasma levels were log10 transformed and FCs log2 transformed to obtain normal distribution of residuals. One-way ANOVA followed by Tukey’s post hoc test was performed using either GenEx6 Pro or the statistical software R version 3.2.3. Unless otherwise stated, qPCR data are presented as mean treatment FC relative to the control group while other data are presented as means ± s.e.m. Since genes were specifically selected on the presumption of differential expression, multiple comparison analysis was not performed, and P < 0.05 was considered statistically significant. Graphs, figures and tables were created in Microsoft Excel and Microsoft PowerPoint.
Results
FGF21 reduced body weight to the same extent as switching diet from HFD to chow
In the FGF21 (0.6 mg/kg daily dosing) treated DIO mice, the caloric restriction induced by chow resulted in similar BW loss (Fig. 1A, chow 18.0% ± 1.2%, FGF21 21.3% ± 0.8%) and ~10.5% body fat mass reduction after 18 days (Fig. 1C, chow 10.5% ± 1.2%, FGF21 10.5% ± 1.4%). The cumulative caloric intake was reduced by chow, while no difference was observed between the FGF21-treated and control mice (Fig. 1B, control 244.4 ± 8.9 kcal, chow 172.0 ± 8.8 kcal, FGF21 239.3 ± 8.8 kcal).
Figure 1.

The effect of 18 days treatment of diet restriction (chow) and FGF21 in DIO mice. (A) Percentage of body weight loss, (B) the cumulative food intake (kcal), (C) percentage of body fat loss, (D) plasma leptin levels, (E) plasma insulin levels, (F) hepatic triglyceride levels, (G) hepatic cholesterol levels, and (H) hepatic glycogen level. Data are means ± s.e.m., with n = 8–10 in each group. ‘1’ indicates significant differences to the vehicle, ‘2’ significant differences to chow, and ‘3’ significant differences to FGF21 (*P < 0.05, **P < 0.01, ***P < 0.001). Statistical analyses were performed using one-way ANOVA with Tukey’s post hoc test. Group size, n = 10.
Plasma insulin and leptin
Plasma leptin and insulin were reduced to similar degree in mice treated with FGF21 and chow compared to the control group (Fig. 1D, control 70.1 ± 7.1 ng/mL, chow 11.5 ± 2.5 ng/mL, FGF21 16.2 ± 2.2 ng/mL; Fig. 1E, control 626.5 ± 192.0 pM, chow 191.1 ± 48.5 pM, FGF21 232.5 ± 65.0 pM).
Hepatic lipid and glycogen content
The hepatic triglyceride (TG) content was decreased to a similar degree by FGF21 and chow (Fig. 1F, control 136.5 ± 15.1 µM/g, chow 50.1 ± 9.2 µM/g, FGF21 35.0 ± 5.0 µM/g), while cholesterol was reduced by chow (Fig. 1G, control 9.9 ± 0.3 µM/g, chow 8.3 ± 0.3 µM/g, FGF21 9.3 ± 0.4 µM/g) compared to control mice. Glycogen content was decreased by FGF21 and increased by chow compared to control mice (Fig. 1H, control 253.5 ± 19.3 µM/g, chow 331.3 ± 15.2 µM/g, FGF21 173.3 ± 27.2 µM/g).
Hepatic gene expression
Of the 89 genes investigated in the liver, 70 showed significant differential regulation (Supplementary Table 2). The genes which are presented in the result section have focus on the impact of FGF21. Overall, treatment with FGF21 affected significant more genes than mice shifted diet from HFD to chow compared to vehicle-treated mice on HFD.
Regulation of transcription factors governing energy balance
Peroxisome proliferator-activated receptor alpha (Ppara), an important transcription factor (TF) inducing FA oxidation, was upregulated by FGF21 while slightly downregulated by chow (Table 1). FGF21 also upregulated the expression of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Ppargc1a, PGC-1a), a transcriptional coactivator that increases FA oxidation, gluconeogenesis and mitochondrial biosynthesis, and NAD-dependent protein deacetylase sirtuin-1 (Sirt1), an enzyme required for PGC-1a induction of gluconeogenic genes. Forkhead box protein A2 (Foxa2) and hepatocyte nuclear factor 4-alpha (Hnf4a) were both induced by FGF21 treatment but not by chow. Foxa2 is a central regulator of glucose, lipid and bile acid (BA) homeostasis (31, 32), while Hnf4a is another essential metabolic TF that induces gluconeogenesis (33) and BA metabolism (34, 35). Conversely, the farnesoid X receptor (Fxr) inhibitor of BA synthesis (36) was also upregulated by FGF21. Sterol regulatory element-binding protein 1 (Srebf1), encoding central activators of lipogenesis, cholesterol synthesis and uptake (37), as well as the glucose-induced lipogenic TF, carbohydrate-responsive element-binding protein (ChREBP, Mlxipl) (38), were upregulated by FGF21. Finally, the adipogenic TF peroxisome proliferator-activated receptor gamma (Pparg) was downregulated by FGF21.
Table 1.
Hepatic mRNA expression of transcriptional regulators involved in metabolism in DIO mice treated with diet restriction (chow) or FGF21 for 18 days.
| Transcript | Chow vs control | FGF21 vs control | FGF21 vs chow |
|---|---|---|---|
| Foxa2 | 1.4 | 3.0c | 2.1a |
| Fxr | 1.1 | 1.9c | 1.7c |
| Hnf4a | 1.1 | 2.4c | 2.2c |
| Mlxipl | 1.5 | 5.3c | 3.5b |
| Ppara | −1.2a | 1.6c | 1.9c |
| Pparg | −1.2 | −3.5c | −3.0c |
| Ppargc1a | 1.1 | 2.1c | 2.0c |
| Sirt1 | 1.2 | 2.1c | 1.8c |
| Srebf1 | 1.3 | 2.2c | 1.7a |
Values represent fold changes with ‘−’ indicating downregulation. Foxa2, Forkhead box protein A2; Fxr, farnesoid X receptor; Hnf4a, hepatocyte nuclear factor 4-alpha; Mlxipl, carbohydrate-responsive element-binding protein; Ppara, peroxisome proliferator-activated receptor alpha; Pparg, peroxisome proliferator-activated receptor gamma; Ppargc1a, peroxisome proliferator-activated receptor gamma co-activator 1a; Sirt1, sirtuin-1; Srebf1, sterol regulatory element-binding protein 1. Srebf1 was investigatied by conventional qPCR.
aP < 0.05; bP < 0.01; cP < 0.001. Statistical analyses were performed using one-way ANOVA with Tukey's post hoc test. Group size, n = 10.
Regulation of genes modulating carbohydrate metabolism
Genes downstream from the transcriptional regulators modulated by FGF21 generally changed expression profiles in the same direction as the corresponding TF. Glycogen phosphorylase (Pygl) was upregulated by FGF21 (Table 2), consistent with upregulation of the glucagon receptor (Gcgr) and reduced hepatic glycogen (Fig. 1H). Mitochondrial pyruvate carrier 2 (Mpc2), mediating pyruvate uptake into mitochondria, and pyruvate dehydrogenase E1 component subunit b (Pdhb), part of the pyruvate dehydrogenase complex (PDC), were both downregulated by FGF21. On the other hand, an inhibitor of the PDC, pyruvate dehydrogenase kinase isozyme 4 (Pdk4), was downregulated. The gluconeogenic phosphoenolpyruvate carboxykinase (Pepck) was upregulated (also in accordance with upregulated Gcgr), together with glucose 6-phosphatase 3 (G6pc3) and glucose transporter type 2 (Glut2). The glycolytic glucokinase (Gck) was also upregulated by FGF21, though to a lesser extent than G6pc3 (Fig. 2 and Table 2). FGF21 treatment also induced the expression of the insulin-sensitizing adiponectin receptor protein 2 (Adipor2) and the insulin receptor (Insr) compared to control and chow, while chow had significantly induced Insr compared to control (Fig. 4).
Table 2.
Hepatic mRNA expression of genes involved in glucose metabolism in DIO mice treated with diet restriction (chow) or FGF21 for 18 days.
| Transcript | Chow vs control | FGF21 vs control | FGF21 vs chow |
|---|---|---|---|
| Cox7a1 | −1.1 | −1.5a | −1.3 |
| G6pc3 | −1.0 | 2.7c | 2.8c |
| Gcgr | −1.1 | 2.1c | 2.3c |
| Gck | −1.1 | 1.4a | 1.6b |
| Glut2 | 1.1 | 1.5a | 1.3 |
| Gys2 | −1.2 | 1.1 | 1.3a |
| Mpc2 | −1.1 | −2.4b | −2.2a |
| Pdhb | −1.1 | −1.6b | −1.4a |
| Pdk4 | 1.0 | −1.6 | −1.7 |
| Pepck | 1.2 | 1.9b | 1.6a |
| Pklr | 1.1 | 1.1 | 1.0 |
| Pygl | 1.4 | 2.6c | 1.8a |
| Uqcrh | −1.0 | −1.6a | −1.5 |
| Usmg5 | −1.0 | −1.7a | −1.7a |
Values represent fold changes with ‘−’ indicating downregulation. Cox7a1, Cytochrome c oxidase subunit 7a1; G6pc3, glucose 6-phosphatase 3; Gcgr, glucagon receptor; Gck, glucokinase; Glut2, glucose transporter type 2; Gys2, glycogen synthase; Mpc2, mitochondrial pyruvate carrier 2; Pdhb, pyruvate dehydrogenase E1 component subunit beta; Pdk4, pyruvate dehydrogenase kinase isozyme 4; Pepck, phosphoenolpyruvate carboxykinase; Pklr, pyruvate kinase; Pygl, glycogen phosphorylase liver; Uqcrh, cytochrome b-c1 complex subunit 6; Usmg5, upregulated during skeletal muscle growth 5.
aP < 0.05; bP < 0.01; cP < 0.001. Statistical analyses were performed using one-way ANOVA with Tukey’s post hoc test. Group size, n = 10.
Figure 2.

Illustration of the hepatic mRNA expression of genes involved in glucose and lipid metabolism at day 18 in DIO mice treated with FGF21. Values in green and red colors represent significant (P < 0.05) fold changes in FGF21-treated mice compared to the control group. Green (↑) indicates upregulation by FGF21 and red (↓) means downregulation. G1P, Glucose 1-phosphate; G6P, glucose 6-phosphate; PEP, phosphoenolpyruvate; FA, fatty acid; TG, triglyceride. For gene names, see Tables 1–3. Statistical analyses were performed using one-way ANOVA with Tukey’s post hoc test. Group size, n = 10.
Figure 4.
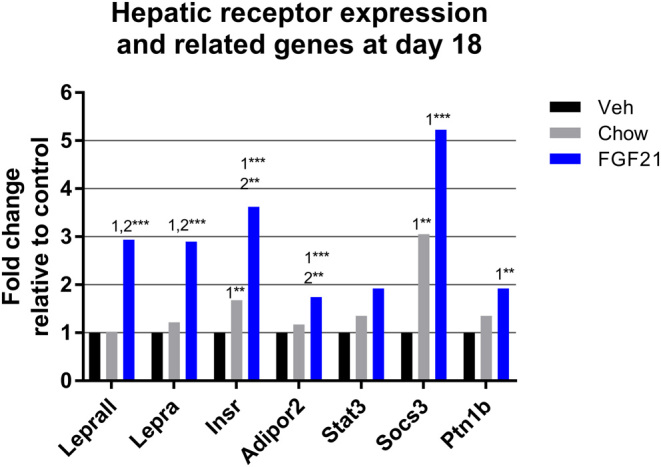
Hepatic mRNA expression of different receptors and related genes at day 18 in diet-induced obese (DIO) mice treated with diet restriction (chow) or FGF21. Lepr, leptin receptor all variants; Lepra, leptin receptor a; Insr, insulin receptor; Adipor2, adipornectin receptor 2; Stat3, signal transducer and activator of transcription 3; Socs3, suppressor of cytokine signaling-3; Ptp1b, tyrosine-protein phosphatase non-receptor type 1. Lepr and Lepra were measured by conventional qPCR. ‘1’ indicates significant differences to the vehicle, ‘2’ significant differences to chow, and ‘3’ significant differences to FGF21 (*P < 0.05, **P < 0.01, ***P < 0.001). Statistical analyses were performed using one-way ANOVA with Tukey’s post hoc test. Group size, n = 10.
Regulation of genes modulating lipid metabolism
FGF21 treatment regulated more genes than chow (Table 3). Chow induced the lipogenic fatty acid synthase (Fasn) and elongation of very long-chain fatty acids protein 3 (Elovl3), while the lipolytic lipoprotein lipase (Lpl) was downregulated. Fatty acid translocase (Cd36), facilitating uptake of oxidized low-density lipoprotein (oxLDL) and long-chain fatty acids (39), was downregulated. Microsomal triglyceride transfer protein (Mttp), controlling the rate-limiting process for very low-density lipoprotein (VLDL) secretion from the liver (38), was slightly upregulated. Finally, mice shifted to chow diet had increased expression levels of the rate-controlling enzyme of cholesterol biosynthesis, HMG-CoA reductase (Hmgcr) and the enzyme (cystein sulfinic acid decarboxylase (Csad)), catalyzing the synthesis of taurine, a component of bile salt. FGF21 treatment increased gene expression of LDL receptor (Ldlr) and high-density lipoprotein (HDL) receptor, scavenger receptor class B member 1 (Scarb1), the collective action of which would improve cholesterol clearance. On the other hand, the LDLR degrading proprotein convertase subtilisin/kexin type 9 (Pcsk9) was also upregulated by FGF21. The two lipogenic genes, stearoyl-CoA desaturase 1 (Scd1) and Elovl3, were markedly downregulated by FGF21 treatment, while, on the contrary, acetyl CoA carboxylase 1 (Acaca) was slightly upregulated. The FA oxidative carnitine palmitoyltransferase 1a (Cpt1a) was increased, and so was hepatic triacylglycerol lipase (Lipc) and adipose triglyceride lipase (Atgl), mediating degradation of TG from lipoproteins and lipid droplets (40). Also contributing to the hepatic FA pool was upregulation of fatty acid transport protein 2 (Fatp2). Cd36 was downregulated similar to chow, while Mttp was upregulated to a higher degree by FGF21. Decreased expression of the ketogenic HMG-CoA lyase (Hmgcl), together with increased expression of Hmgcr, and markedly downregulation of the HMGCR-inhibitor insulin-induced gene 2 (Insig2), suggest that the acetyl-CoA obtained from FA oxidation was used for cholesterol biosynthesis. Cholesterol 7-alpha-monooxygenase (Cyp7a1), mediating the rate-limiting step in BA synthesis, was upregulated, together with sterol 12-alpha-hydroxylase (Cyp8b1) and the cholesterol transporters Sterolin-1 (Abcg5) and sterolin-2 (Abcg8) (Fig. 2 and Table 3).
Table 3.
Hepatic mRNA expression of genes involved in lipid metabolism in DIO mice treated with diet restriction (chow) or FGF21 for 18 days.
| Transcript | Chow vs control | FGF21 vs control | FGF21 vs chow |
|---|---|---|---|
| Abcg5 | 1.1 | 2.6c | 2.3c |
| Abcg8 | 1.4 | 2.5b | 1.9a |
| Acaca | 1.3 | 1.6b | 1.2 |
| Atgl | 1.3 | 2.0c | 1.6b |
| Cd36 | −1.9a | −2.1b | −1.1 |
| Cpt1a | −1.1 | 2.2c | 2.5c |
| Csad | 2.9b | 1.6 | −1.8 |
| Cyp7a1 | 1.6 | 3.9c | 2.4b |
| Cyp8b1 | 1.4 | 2.5c | 1.7b |
| Dgat2 | −1.1 | 1.3 | 1.4b |
| Elovl3 | 1.8a | −3.6c | −6.5c |
| Fasn | 1.9a | 1.6 | −1.1 |
| Fatp2 | 1.1 | 2.2c | 2.0c |
| Gpam | 1.0 | 1.3 | 1.2 |
| Hmgcl | −1.4 | −2.c | −1.5a |
| Hmgcr | 1.7a | 3.8c | 2.2b |
| Insig2 | −1.2 | −5.8c | −5.0c |
| Ldlr | 1.1 | 3.1c | 2.8c |
| Lipc | 1.3 | 2.4c | 1.7b |
| Lpl | −1.5a | 1.3 | 1.9c |
| Mttp | 1.3a | 2.2c | 1.7c |
| Pcsk9 | 1.2 | 3.3c | 2.7c |
| Scarb1 | 1.1 | 3.4c | 3.2c |
| Scd1 | 1.7 | −5.3c | −9.3c |
Values represent fold changes with ‘−’ indicating downregulation. Abcg5, Sterolin-1; Abcg8, sterolin-2; Acaca, acetyl-CoA carboxylase 1; Atgl, adipose triglyceride lipase; Cd36, fatty acid translocase; Cpt1a, carnitine palmitoyltransferase 1a; Csad, cystein sulfinic acid decarboxylase; Cyp7a1, cholesterol 7-alpha-monooxygenase; Cyp8b1, sterol 12-alpha-hydroxylase; Dgat2, diacylglycerol O-acyltransferase 2; Elovl3, elongation of very long-chain fatty acids protein 3; Fasn, fatty acid synthase; Fatp2, fatty acid transport protein 2; Gpam, glycerol-3-phosphate acyltransferase; Hmgcl, HMG-CoA lyase; Hmgcr, HMG-CoA reductase; Insig2, insulin-induced gene 2; Ldlr, low-density lipoprotein receptor; Lipc, hepatic triacylglycerol lipase; Lpl, lipoprotein lipase; Mttp, microsomal triglyceride transfer protein; Pcsk9, proprotein convertase subtilisin/kexin type 9; Scarb1, scavenger receptor class B member 1; Scd1, stearoyl-CoA desaturase 1. Hmgcl was measured by conventional qPCR.
aP < 0.05; bP < 0.01; cP < 0.001. Statistical analyses were performed using one-way ANOVA with Tukey’s post hoc test. Group size, n = 10.
Regulation of Fgf21, Klb and Fgfrs
The mRNA expression of Fgf21 was significantly reduced by chow and FGF21 treatment, whereas FGF21 induced the expression of FGF receptor family Fgfr2 and Fgfr4 and Klb (Fig. 3).
Figure 3.
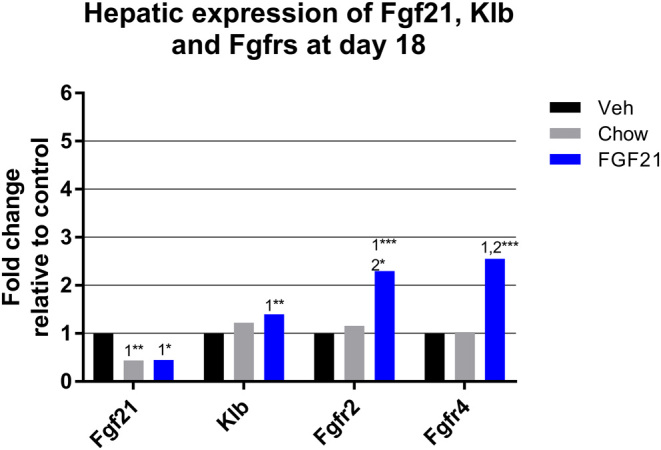
Hepatic mRNA expression of Fgf21 and Fgf receptors in DIO mice treated with diet restriction (chow) or FGF21. Fgf21, fibroblast growth factor 21; Klb, klotho-beta receptor subunit; Fgfr2, fibroblast growth factor receptor 2; Fgfr4, fibroblast growth factor receptor 4. ‘1’ indicates significant differences to the vehicle, ‘2’ significant differences to chow, and ‘3’ significant differences to FGF21 (*P < 0.05, **P < 0.01, ***P < 0.001). Statistical analyses were performed using one-way ANOVA with Tukey’s post hoc test. Group size, n = 10.
Regulation of leptin receptors
The functional isoform of the leptin receptor, Leprb, showed very low mRNA expression both on the Fluidigm platform and with conventional qPCR. Therefore, an assay targeting all leptin receptor transcript variants (Lepr) and one measuring only the short a-variant (Lepra) were run, both showing 2.9-fold upregulation by FGF21. Signal transducer and activator of transcription 3 (Stat3), a TF activated by LEPRB signaling, was not significantly regulated by the treatments, while inhibitors of leptin signalling, Socs3 and Ptp1b, were upregulated, primarily by FGF21 (Fig. 4).
Regulation of genes involved in inflammation and oxidative stress
Expression of the proinflammatory cytokine interleukin 18 (Il18) was 3.5-fold downregulated by FGF21, but not by chow, whereas another biomarker of inflammation, lipocalin 2 (Lcn2), was about five-fold downregulated by both treatments. The hepatokine fetuin-A (Ahsg), which mediates FA activation of toll-like receptor 4 (TLR4) (41), was induced by FGF21. Inhibitor of κB kinase epsilon (Ikbke), which recently was revealed to limit the chronic nature of inflammation (42), was 3.8-fold upregulated by FGF21 treatment. Mitochondrial uncoupling protein 2 (Ucp2), which reduces the generation of reactive oxidative species (ROS) (43), was upregulated by FGF21 compared to chow, while the endoplasmic reticulum (ER) chaperone 78-kDa glucose-regulated protein (Hspa5), a key regulator of protective responses to the accumulation of misfolded proteins, was upregulated by FGF21 compared to both chow and control (Fig. 5). Interleukin 1b (Il1b), macrophage migration inhibitory factor (Mif) and carbonyl reductase 1 (Cbr1) were not significantly regulated (Supplementary Table 2).
Figure 5.

Hepatic mRNA expression of inflammatory and anti-oxidative stress-related genes at day 18 in DIO mice treated with diet restriction (chow) or FGF21. Il18, interleukin 18; Lcn2, lipocalin-2; Ahsg, fetuin-A; Ikbke, inhibitor of nuclear kappa-B kinase subunit epsilon; Ucp2, mitochondrial uncoupling protein 2; Hspa5, 78-kDa glucose-regulated protein. ‘1’ indicates significant differences to the vehicle, ‘2’ significant differences to chow, and ‘3’ significant differences to FGF21 (*P < 0.05, **P < 0.01, ***P < 0.001). Statistical analyses were performed using one-way ANOVA with Tukey’s post hoc test. Group size, n = 10.
Discussion
In the present study, comparable (~20%) body weight loss was induced by FGF21 and by shifting diet from HFD to chow in DIO mice at day 18. Similar degree of BW loss allows the investigation of BW-independent actions of FGF21. The body weight loss was associated with reduction in body fat mass, liver fat, plasma leptin and insulin. For mice treated with FGF21, weight loss was independent of caloric intake, indicating an increase in EE as the primary driver for efficacy (11). To understand the effect of FGF21 in the liver, we investigated simultaneous regulation of multiple hepatic metabolic pathways. The presented gene expression profile demonstrated that the FGF21 treatment affected markedly more hepatic genes than mice shifted from HFD to chow (61 vs 16 out of 89). For the mice shifted from HFD to chow diet, the most regulated genes were found in the lipid metabolism related to reduced fat and cholesterol in the diet (44). We are aware that shifting from HFD to chow diet without including lean control mice makes it difficult to discuss whether the chow mice in the present study have normalized their gene regulation due to diet or weight loss. However, Siersbaek and colleagues have demonstrated a reversed hepatic gene expressing profile when changing from HFD to a chow diet, supporting normalization of the gene regulation profile by chow (44).
Within the hepatic carbohydrate metabolism, the gene expression profile suggested that FGF21 induce hepatic glucose mobilization. This was indicated by (1) upregulation of the several transcript factors (Ppargc1a, Foxa2, Hnf4a), (2), upregulation of genes in the gluconeogenesis (Pepck, G6pc3) and the glycogenolysis (Gcgr, Pygl), (3) reduced hepatic glycogen content and (4) downregulation of genes converting pyruvate into acetyl-CoA production (Pdhb, Mpc2) (Fig. 2 and Table 1). In addition, the increase in Glut2 expression by FGF21 suggested greater capacity for hepatic glucose flux. However, there are contradicting results showing a dose-dependent downregulation of G6pc3 by FGF21 in DIO mice while no effect on Pepck, suggesting a reduced hepatic glucose production derived from glycogenolysis (16). Coskun and colleagues showed a decreased Pepck expression indicating a decreased gluconeogenesis (11). In the present study, the three-fold upregulation of Foxa2, a TF for Pepck and G6pc3, together with the increased expression of Pygl and low hepatic glycogen levels highly suggest increased glycogenolysis and gluconeogenesis. Increased gluconeogenesis has previously been reported with FGF21 indicated by increased Foxa2 (11), PGC-1a (Ppargc1a) (45) and reduced PDC (46), supporting the present study result that FGF21 may induce hepatic glucose mobilization. The potentially enhanced hepatic glucose production is, however, not associated with increased blood glucose levels as FGF21 lowers blood glucose in other studies (11, 47). The enhanced hepatic glucose production may supply the brown adipose tissue (BAT) with fuel, secondary to increases in EE expenditure (48, 49). Villarroya and colleagues have previously demonstrated that FGF21 stimulate the glucose uptake in BAT and specifically induce the glucose oxidative activity (48). FGF21 has, moreover, been revealed to induce conversion of adipocytes from white adipose tissue to brown fat-like cells and increased the expression of thermogenic genes (49, 50). Thus, FGF21 may induce weight loss in DIO mice by directing glucose away from the liver toward BAT for heat production and increased energy expenditure.
Genes involved in hepatic lipid metabolism were also highly regulated by FGF21 compared to mice shifted to chow diet. The expression pattern induced by FGF21 suggests that hepatic TG was channeled toward the synthesis of acetyl-CoA, since lipolytic and FA oxidative genes were upregulated (Fig. 2). An exception to this pattern was the upregulation of the lipogenic Acaca which together with absent regulation of the FA oxidation inhibitor Acacb disagrees with previous publications (11, 16), and we do not have an explanation for this. However, Atgl was induced with two-fold upregulation which may be important for FGF21 to reduce hepatic steatosis, in part, by mobilizing FA from lipid droplets. ATGL has recently been reported as a major hepatic lipase; knockdown of Atgl in the liver of mice induced steatosis and overexpression of Atgl is associated with increased FA oxidation (51). Furthermore, downregulation of the ketogenic Hmgcl and upregulation of the rate-controlling enzyme in cholesterol synthesis Hmgcr suggest that the synthesized acetyl-CoA was channeled toward cholesterol production. These pathways were further supported by upregulation of cholesterol exporters and enzymes working in the synthesis of BAs pointing toward increased production of bile (Fig. 2). Previous, it has been shown that Abcg5 and Abcg8 were downregulated by hepatic overexpression of FGF21 (52), but this was measured in an acute study and may, therefore, differ from the present chronic study. On the other hand, overexpression of hepatic FGF21 and increased hepatic expression of Cyp7a1, Abcg5 and Abcg8 has likewise been shown in DIO mice associated with increased BA pool collected from liver, gallbladder, and small intestine and increased the fecal BA content (52). Unfortunately, neither BA nor cholesterol content in feces was measured in the present study, but Coskun et al. reported a 30% increase in fecal fat in mice treated with FGF21 (11). Thus, it may seem likely that increased cholesterol synthesis and subsequent catabolism (via synthesis of BAs and export into bile) may be contributing factors in the beneficial effects of FGF21 on steatosis. Particularly, the 5.8-fold reduced expression of Insig2 may be a central mechanism, as INSIG proteins act in a feedback mechanism to reduce the synthesis and uptake of cholesterol when endogenous levels are high (37). The role of INSIGs is to inhibit HMGCR expression, and reduced Insig2 expression will thereby circumvent the natural downregulation of cholesterol synthesis. Apart from being a way of dispatching energy via feces, BAs are increasingly being recognized as metabolic regulatory molecules. Stimulation of the BA receptor TGR5 increases EE in BAT as well as GLP-1 secretion in the intestinal L-cells (53). Also, in gastric bypass patients, increased BA has been suggested to contribute to the improved metabolic profile (54, 55). Thus, induction of cholesterol and bile metabolism may likely be important for FGF21 in the liver, but further investigations are needed.
SOCS3 and PTPB1 negative regulate several intracellular metabolic pathways such as growth hormone (GH) (56, 57), insulin (58, 59, 60), and leptin (20) signalling pathways. In the present study, the induction of Socs3 and Ptp1b may be mediated through adiponectin (25) as the Adipor2 was induced by FGF21 (61). Reduced GH response fits well with the reduced expression levels of Ghr and Igf (Supplementary Table 2) and has previously been found with FGF21 (56, 57). On the contrary, the increased hepatic Insr expression and reduced plasma insulin suggest increased hepatic insulin sensitivity (62). Within the liver, INSR stimulates lipogenesis and inhibits gluconeogenesis, but this was not evident in the present study despite upregulation of Insr. Thus, the increase in Insr may then be secondary to a dramatic decrease in plasma insulin which also was observed by chow. In addition, Emanuelli and colleagues showed that intact hepatic insulin signaling is required for most of the action of FGF21 on lipid metabolism (49). The LIRKO mice and FGF21 treated mice have overlapping phenotypes, indicating that hepatic lipid regulation by FGF21 is mediated by lack of insulin.
The leptin receptor transcripts and downstream signaling pathway were also investigated to determine hepatic leptin signaling, as leptin is important in body weight regulation through binding to LEPRB (20). In addition, the hepatic Lepr has been shown to be markedly increased by FGF21 in DIO mice and suggested to be associated with enhanced leptin signaling and sensitivity mice (11, 18). Within the liver, there is no clear evidence of leptin’s role, but it has been suggested to act as a profibrotic molecule by direct activation of LEPRB on hepatic stellate cells (25). In the present study, the Leprb was expressed at too low levels to be analyzed, which may be associated with the upregulation of Socs3 and Ptpb1 (63), respectively. This finding suggests that LEPRB was not a part of the hepatic improvement induced by FGF21. On the other hand, FGF21 induced same significant enhanced expression of all leptin transcript variants (Lepr) and the specific transcript Lepra that mimic the increased leptin receptor expression demonstrated by Coskun and colleagues (11, 18). Hepatic LEPRA is suggested to regulate leptin bioavailability via cleavage from the membrane to a soluble leptin receptor (SLR) (21, 64). Unfortunately, no data on SLR were available in the present study, but in-house data showed that FGF21 induce SLR dose dependently in plasma.
Obesity is characterized by a state of chronic low-grade inflammation, which is an important causative factor behind associated metabolic diseases (65). FGF21, but not chow, reduced the expression of the inflammatory cytokine Il18. To be active, pro-Il18 needs cleavage by the NLRP3 inflammasome that is central in metabolic triggered inflammation (65) and fibrosis development in the steatotic liver (66, 67). IKBKE negatively regulates the NLRP3 inflammasome (42), and the 3.8-fold induced expression of Ikbke seen by FGF21 in the present study, while not by chow, could then indicate that FGF21 protects against inflammatory processes induced by HFD. Oxidized low-density lipoproteins (oxLDLs) are also implicated in the inflammatory and fibrotic process in NASH through binding to the CD36 receptor (39). oxLDL is formed from LDL in response to oxidative stress (ROS), and the level of circulating LDL seems to be proportional to the amount of oxLDL (39). In the present study, the reduced expression of Cd36 together with increased expression of Ucp2 and Hspa5 (reduce oxidative stress), and increased expression of Ldlr (increase the hepatic clearance of LDL) by FGF21 may be protective mechanisms against hepatic inflammatory processes. In addition, increased expression of Adipor2 and reduced expression of Mpc2 by FGF21 and not by chow also support that FGF21 independent of weight loss protected the liver against inflammation and inflammatory processes (25, 68, 69, 70).
An important question is whether the findings in the DIO mice can be translated into humans. Weight loss observed in the DIO mice does not appear to correlate well in humans as only limited weight loss has been demonstrated in humans (10, 12). On the other hand, the pronounced effect of FGF21 in lowering hepatic fat have been observed in both DIO mice (11) and humans (10, 15, 71), indicating a more promising translation from the DIO mice to humans. Recently, Akero demonstrated impressive reduction in hepatic fat with a Fc conjugated FGF21 analogue in humans with a BMI > 25. The relative hepatic fat content was reduced to 70% after 12 weeks of treatment and was not driven by weight loss (71). Thus, the effect was driven by other mechanisms. In mice, it cannot be distinguished whether the increase in energy expenditure and/or weight loss have contributed to the reduction in hepatic fat. However, a lowering of plasma insulin has been observed in both the DIO mice and humans (12). This could indicate that insulin may contribute to the anti-steatotic effect through decreased hepatic lipogenesis and increased oxidation in both DIO mice and humans. In our study, the hepatic gene expression profile indicates a clear decrease in lipogenesis and increased lipid oxidation and may likely correspond to humans. Furthermore, the improved hepatic inflammatory response that appears observed in the DIO mice may also translate well into humans, as NASH patients treated with a protracted FGF21 analogue were protected against hepatic inflammation (15).
Another central question is whether the hepatic improvements by FGF21 are related directly or indirectly. The literature is controversial as direct signaling in hepatocytes in vitro (72) and liver in vivo (73) are observed, while the liver-specific KLB KO display unaltered FGF21 sensitivity (74). The direct effect on liver may depend on the pharmacological dose (72, 75), as FGF21 binds with higher affinity toward the FGFR1c/KLB complex compared to the FGFR3c/KLB complex (76). FGFR1c is not expressed by the liver, but FGF21 may mediate its hepatic effect through FGFR3c/KLB signaling (76, 77, 78). In the present study, Fgfr2, Fgfr4 and Klb were all upregulated by FGF21, suggesting a need for increased hepatic FGF signaling. FGF15’s suppressing effect on bile acids metabolism s dependent on FGFR4 and KLB expression in hepatocytes (77). The upregulation of Fgfr4 may be an endogenous compensatory mechanism to increase the binding sites for FGF15. However, as BA synthesis is increased due to upregulation of CYP7A1, a decrease in FGF15 activity is anticipated. Therefore, FGF21 may decrease inhibition of BA synthesis by antagonizing the effect of FGF15, which has also been reported previously in mice (79).
In conclusion, administration of FGF21 reduced BW and hepatic steatosis independent of caloric intake in DIO mice. In the present study, a panel of hepatic genes important for the multiple metabolic pathways was investigated to provide a broader mechanistic understanding of FGF21 and to drive new hypothesis and studies. We showed that FGF21 affected several metabolic pathways to reduce hepatic steatosis and to drive anti-inflammatory and anti-fibrogenic properties. The hepatic gene expression profile suggested that FGF21 increased uptake of fatty acids and lipoproteins, channeled TGs toward the production of cholesterol and bile acid, reduced lipogenesis and increased the secretion of VLDLc and glucose output. The increased hepatic BA synthesis and glucose mobilization do also suggest that FGF21 provide substrates derived from the liver for increasing EE. The increase of BA may likely be involved in the induction of EE through TGR5 activation where the glucose mobilization may be used as an energy source in BAT. This study was done in mice with high metabolic rate, which need to be taken into consideration when mode of actions of FGF21 are discussed in humans.
Supplementary Material
Declaration of interest
Birgitte Andersen, Helle Keinicke, Kirsten Raun, Linu Mary John and Marina Kjaergaard are full time employees at Novo Nordisk and stock owners through an employee offering program. The other authors have nothing to disclose.
Funding
This work was supported by Innovation Fund Denmark.
Acknowledgements
The authors express their thanks to all the animal technicians in the Animal Unit and the research associates Zhang Ning and Cao Zhiyou, Zach at the Novo Nordisk A/S Research Facility in Beijing and laboratory technicians and PhD students at the Faculty of Health and Medical Sciences for excellent assistance during this study. The authors are also grateful to Emma Henriksson, senior scientist in Liver Disease Research at Novo Nordisk A/S, for encouragements and highly motivated discussions.
References
- 1.Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, Hobbs HH. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology 2004. 40 1387–1395. ( 10.1002/hep.20466) [DOI] [PubMed] [Google Scholar]
- 2.Smits MM, Ioannou GN, Boyko EJ, Utzschneider KM. Non-alcoholic fatty liver disease as an independent manifestation of the metabolic syndrome: results of a US national survey in three ethnic groups. Journal of Gastroenterology and Hepatology 2013. 28 664–670. ( 10.1111/jgh.12106) [DOI] [PubMed] [Google Scholar]
- 3.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016. 64 73–84. ( 10.1002/hep.28431) [DOI] [PubMed] [Google Scholar]
- 4.Rinella ME. Nonalcoholic fatty liver disease a systematic review. JAMA 2015. 313 2263–2273. ( 10.1001/jama.2015.5370) [DOI] [PubMed] [Google Scholar]
- 5.WHO. Obesity and overweight. Geneva, Switzerland: World Health Organization, 2016. (available at: http://www.who.int/mediacentre/factsheets/fs311/en/) [Google Scholar]
- 6.Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, Mills PR, Keach JC, Lafferty HD, Stahler A, et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015. 149 389.e10–397.e10. ( 10.1053/j.gastro.2015.04.043) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rotman Y, Sanyal AJ. Current and upcoming pharmacotherapy for non-alcoholic fatty liver disease. Gut 2017. 66 180–190. ( 10.1136/gutjnl-2016-312431) [DOI] [PubMed] [Google Scholar]
- 8.Perry RJ, Samuel VT, Petersen KF, Shulman GI. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014. 510 84–91. ( 10.1038/nature13478) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, Torres-Gonzalez A, Gra-Oramas B, Gonzalez-Fabian L, Friedman SL, Diago M, Romero-Gomez M. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology 2015. 149 367.e5–378.e5; quiz e14. ( 10.1053/j.gastro.2015.04.005) [DOI] [PubMed] [Google Scholar]
- 10.Talukdar S, Zhou YJ, Li DM, Rossulek M, Dong J, Somayaji V, Weng Y, Clark R, Lanba A, Owen BM, et al. A long-acting FGF21 molecule, PF-05231023, decreases body weight and improves lipid profile in non-human primates and type 2 diabetic subjects. Cell Metabolism 2016. 23 427–440. ( 10.1016/j.cmet.2016.02.001) [DOI] [PubMed] [Google Scholar]
- 11.Coskun T, Bina HA, Schneider MA, Dunbar JD, Hu CC, Chen YY, Moller DE, Kharitonenkov A. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology 2008. 149 6018–6027. ( 10.1210/en.2008-0816) [DOI] [PubMed] [Google Scholar]
- 12.Gaich G, Chien JY, Fu HD, Glass LC, Deeg MA, Holland WL, Kharitonenkov A, Bumol T, Schilske HK, Moller DE. The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metabolism 2013. 18 333–340. ( 10.1016/j.cmet.2013.08.005) [DOI] [PubMed] [Google Scholar]
- 13.Christoffersen B, Straarup EM, Lykkegaard K, Fels JJ, Sass-Orum K, Zhang X, Raun K, Andersen B. FGF21 decreases food intake and body weight in obese Gottingen minipigs. Diabetes, Obesity and Metabolism 2019. 21 592–600. ( 10.1111/dom.13560) [DOI] [PubMed] [Google Scholar]
- 14.Lee JH, Kang YE, Chang JY, Park KC, Kim HW, Kim JT, Kim HJ, Yi HS, Shong M, Chung HK, et al. An engineered FGF21 variant, LY2405319, can prevent non-alcoholic steatohepatitis by enhancing hepatic mitochondrial function. American Journal of Translational Research 2016. 8 4750–4763. [PMC free article] [PubMed] [Google Scholar]
- 15.Sanyal A, Charles ED, Neuschwander-Tetri B, Loomba R, Harrison S, Abdelmalek M, Lawitz E, Halegoua-DeMarzio D, Dong Y, Noviello S, et al. BMS-986036 (pegylated FGF21) in patients with non-alcoholic steatohepatitis: a phase 2 study. Journal of Hepatology 2017. 66 S89–S90. ( 10.1016/S0168-8278(17)30443-9) [DOI] [Google Scholar]
- 16.Xu J, Lloyd DJ, Hale C, Stanislaus S, Chen M, Sivits G, Vonderfecht S, Hecht R, Li YS, Lindberg RA, et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 2009. 58 250–259. ( 10.2337/db08-0392) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Charles ED, Neuschwander-Tetri BA, Pablo Frias J, Kundu S, Luo Y, Tirucherai GS, Christian R. Pegbelfermin (BMS-986036), pegylated FGF21, in patients with obesity and type 2 diabetes: results from a randomized phase 2 study. Obesity 2019. 27 41–49. ( 10.1002/oby.22344) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coskun T, O'Farrell LS, Kauffman AL, Efanov A, Adams AC. Glucagon like peptide 1 (GLP1) in combination with fibroblast growth factor 21 (FGF21) enhances browning of white adipose tissue. Diabetologia 2013. 56 S34–S35. (available at: https://www.easd.org/virtualmeeting/home.html#!resources/glucagon-like-peptide-1-glp1-in-combination-with-fibroblast-growth-factor-21-fgf21-enhances-browning-of-white-adipose-tissue) [Google Scholar]
- 19.Friedman JM. Leptin, leptin receptors, and the control of body weight. Nutrition Reviews 1998. 56 s38–s46; discussion s54–s75. ( 10.1111/j.1753-4887.1998.tb01685.x) [DOI] [PubMed] [Google Scholar]
- 20.Myers MG, Cowley MA, Munzberg H. Mechanisms of leptin action and leptin resistance. Annual Review of Physiology 2008. 70 537–556. ( 10.1146/annurev.physiol.70.113006.100707) [DOI] [PubMed] [Google Scholar]
- 21.Cohen P, Yang GQ, Yu XX, Soukas AA, Wolfish CS, Friedman JM, Li C. Induction of leptin receptor expression in the liver by leptin and food deprivation. Journal of Biological Chemistry 2005. 280 10034–10039. ( 10.1074/jbc.M413684200) [DOI] [PubMed] [Google Scholar]
- 22.Huynh FK, Levi J, Denroche HC, Gray SL, Voshol PJ, Neumann UH, Speck M, Chua SC, Covey SD, Kieffer TJ. Disruption of hepatic leptin signaling protects mice from age- and diet-related glucose intolerance. Diabetes 2010. 59 3032–3040. ( 10.2337/db10-0074) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Polyzos SA, Kountouras J, Zavos C, Deretzi G. The potential adverse role of leptin resistance in nonalcoholic fatty liver disease: a hypothesis based on critical review of the literature. Journal of Clinical Gastroenterology 2011. 45 50–54. ( 10.1097/MCG.0b013e3181ec5c66) [DOI] [PubMed] [Google Scholar]
- 24.Marra F, Bertolani C. Adipokines in liver diseases. Hepatology 2009. 50 957–969. ( 10.1002/hep.23046) [DOI] [PubMed] [Google Scholar]
- 25.Saxena NK, Anania FA. Adipocytokines and hepatic fibrosis. Trends in Endocrinology and Metabolism 2015. 26 153–161. ( 10.1016/j.tem.2015.01.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kjærgaard M, Nilsson C, Rosendal A, Nielsen MO, Raun K. Maternal chocolate and sucrose soft drink intake induces hepatic steatosis in rat offspring associated with altered lipid gene expression profile. Acta Physiologica 2014. 210 142–153. ( 10.1111/apha.12138) [DOI] [PubMed] [Google Scholar]
- 27.Xu LY, Ma XR, Cui B, Li XY, Ning G, Wang S. Selection of reference genes for qRT-PCR in high fat diet-induced hepatic steatosis mice model. Molecular Biotechnology 2011. 48 255–262. ( 10.1007/s12033-010-9366-2) [DOI] [PubMed] [Google Scholar]
- 28.Svingen T, Letting H, Hadrup N, Hass U, Vinggaard AM. Selection of reference genes for quantitative RT-PCR (RT-qPCR) analysis of rat tissues under physiological and toxicological conditions. PeerJ 2015. 3 e855 ( 10.7717/peerj.855) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bruce KD, Sihota KK, Byrne CD, Cagampang FR. The housekeeping gene YWHAZ remains stable in a model of developmentally primed non-alcoholic fatty liver disease. Liver International 2012. 32 1315–1321. ( 10.1111/j.1478-3231.2012.02813.x) [DOI] [PubMed] [Google Scholar]
- 30.Spandidos A, Wang XW, Wang HJ, Dragnev S, Thurber T, Seed B. A comprehensive collection of experimentally validated primers for polymerase chain reaction quantitation of murine transcript abundance. BMC Genomics 2008. 9 633 ( 10.1186/1471-2164-9-633) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Newman JC, Verdin E. Ketone bodies as signaling metabolites. Trends in Endocrinology and Metabolism 2014. 25 42–52. ( 10.1016/j.tem.2013.09.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bochkis IM, Rubins NE, White P, Furth EE, Friedman JR, Kaestner KH. Hepatocyte-specific ablation of Foxa2 alters bile acid homeostasis and results in endoplasmic reticulum stress. Nature Medicine 2008. 14 828–836. ( 10.1038/nm.1853) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rhee J, Inoue Y, Yoon JC, Puigserver P, Fan ML, Gonzalez FJ, Spiegelman BM. Regulation of hepatic fasting response by PPAR gamma coactivator-1 alpha (PGC-1): requirement for hepatocyte nuclear factor 4 alpha in gluconeogenesis. PNAS 2003. 100 4012–4017. ( 10.1073/pnas.0730870100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. Hepatocyte nuclear factor 4 alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Molecular and Cellular Biology 2001. 21 1393–1403. ( 10.1128/MCB.21.4.1393-1403.2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Inoue Y, Yu AM, Yim SH, Ma XC, Krausz KW, Inoue J, Xiang CC, Brownstein MJ, Eggertsen G, Bjorkhem I, et al. Regulation of bile acid biosynthesis by hepatocyte nuclear factor 4 alpha. Journal of Lipid Research 2006. 47 215–227. ( 10.1194/jlr.M500430-JLR200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vallim TQD, Tarling EJ, Edwards PA. Pleiotropic roles of bile acids in metabolism. Cell Metabolism 2013. 17 657–669. ( 10.1016/j.cmet.2013.03.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell 2006. 124 35–46. ( 10.1016/j.cell.2005.12.022) [DOI] [PubMed] [Google Scholar]
- 38.Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. Journal of Clinical Investigation 2008. 118 829–838. ( 10.1172/JCI34275) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Houben T, Brandsma E, Walenbergh S, Hofker M, Shiri-Sverdlov R. Oxidized LDL at the crossroads of immunity in non-alcoholic steatohepatitis. Biochimica et Biophysica Acta: Molecular and Cell Biology of Lipids 2017. 1862 416–429. ( 10.1016/j.bbalip.2016.07.008) [DOI] [PubMed] [Google Scholar]
- 40.Sathyanarayan A, Mashek MT, Mashek DG. ATGL promotes autophagy/lipophagy via SIRT1 to control hepatic lipid droplet catabolism. Cell Reports 2017. 19 1–9. ( 10.1016/j.celrep.2017.03.026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, Ray S, Majumdar SS, Bhattacharya S. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nature Medicine 2012. 18 1279–1285. ( 10.1038/nm.2851) [DOI] [PubMed] [Google Scholar]
- 42.Patel MN, Bernard WG, Milev NB, Cawthorn WP, Figg N, Hart D, Prieur X, Virtue S, Hegyi K, Bonnafous S, et al. Hematopoietic IKBKE limits the chronicity of inflammasome priming and metaflammation. PNAS 2015. 112 506–511. ( 10.1073/pnas.1414536112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brand MD, Esteves TC. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metabolism 2005. 2 85–93. ( 10.1016/j.cmet.2005.06.002) [DOI] [PubMed] [Google Scholar]
- 44.Siersbaek M, Varticovski L, Yang S, Baek S, Nielsen R, Mandrup S, Hager GL, Chung JH, Grontved L. High fat diet-induced changes of mouse hepatic transcription and enhancer activity can be reversed by subsequent weight loss. Scientific Reports 2017. 7 40220 ( 10.1038/srep40220) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Potthoff MJ, Inagaki T, Satapati S, Ding XS, He TT, Goetz R, Mohammadi M, Finck BN, Mangelsdorf DJ, Kliewer SA, et al. FGF21 induces PGC-1 alpha and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. PNAS 2009. 106 10853–10858. ( 10.1073/pnas.0904187106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Samms RJ, Murphy M, Fowler MJ, Cooper S, Emmerson P, Coskun T, Adams AC, Kharitonenkov A, Ebling FJP, Tsintzas K. Dual effects of fibroblast growth factor 21 on hepatic energy metabolism. Journal of Endocrinology 2015. 227 37–47. ( 10.1530/JOE-15-0334) [DOI] [PubMed] [Google Scholar]
- 47.Laeger T, Baumeier C, Wilhelmi I, Wurfel J, Kamitz A, Schurmann A. FGF21 improves glucose homeostasis in an obese diabetes-prone mouse model independent of body fat changes. Diabetologia 2017. 60 2274–2284. ( 10.1007/s00125-017-4389-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Villarroya F, Hondares E, Diaz-Delfin J, Iglesias R, Giralt M. Brown adipose tissue, a sink of glucose disposal? Effects of FGF21 on glucose metabolism in brown adipose cells. Diabetologia 2013. 56 S35–S36. (available at: https://www.easd.org/virtualmeeting/home.html#!resources/brown-adipose-tissue-a-sink-of-glucose-disposal-effects-of-fgf21-on-glucose-metabolism-in-brown-adipose-cells--2) [Google Scholar]
- 49.Emanuelli B, Vienberg SG, Smyth G, Cheng C, Stanford KI, Arumugam M, Michael MD, Adams AC, Kharitonenkov A, Kahn CR. Interplay between FGF21 and insulin action in the liver regulates metabolism. Journal of Clinical Investigation 2014. 124 515–527. ( 10.1172/JCI67353) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fisher FM, Kleiner S, Douris N, Fox EC, Mepani RJ, Verdeguer F, Wu J, Kharitonenkov A, Flier JS, Maratos-Flier E, et al. FGF21 regulates PGC-1 alpha and browning of white adipose tissues in adaptive thermogenesis. Genes and Development 2012. 26 271–281. ( 10.1101/gad.177857.111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ong KT, Mashek MT, Bu SY, Greenberg AS, Mashek DG. Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning. Hepatology 2011. 53 116–126. ( 10.1002/hep.24006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang J, Gupte J, Gong Y, Weiszmann J, Zhang Y, Lee KJ, Richards WG, Li Y. Chronic over-expression of fibroblast growth factor 21 increases bile acid biosynthesis by opposing FGF15/19 action. EBioMedicine 2017. 15 173–183. ( 10.1016/j.ebiom.2016.12.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arab JP, Karpen SJ, Dawson PA, Arrese M, Trauner M. Bile acids and nonalcoholic fatty liver disease: molecular insights and therapeutic perspectives. Hepatology 2017. 65 350–362. ( 10.1002/hep.28709) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang W, Cheng Z, Wang Y, Dai Y, Zhang X, Hu S. Role of bile acids in bariatric surgery. Frontiers in Physiology 2019. 10 374 ( 10.3389/fphys.2019.00374) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Steinert RE, Peterli R, Keller S, Meyer-Gerspach AC, Drewe J, Peters T, Beglinger C. Bile acids and gut peptide secretion after bariatric surgery: a 1-year prospective randomized pilot trial. Obesity 2013. 21 E660–E668. ( 10.1002/oby.20522) [DOI] [PubMed] [Google Scholar]
- 56.Fazeli PK, Misra M, Goldstein M, Miller KK, Klibanski A. Fibroblast growth factor-21 may mediate growth hormone resistance in anorexia nervosa. Journal of Clinical Endocrinology and Metabolism 2010. 95 369–374. ( 10.1210/jc.2009-1730) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Inagaki T, Lin VY, Goetz R, Mohammadi M, Mangelsdorf DJ, Kliewer SA. Inhibition of growth hormone signaling by the fasting-induced hormone FGF21. Cell Metabolism 2008. 8 77–83. ( 10.1016/j.cmet.2008.05.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sachithanandan N, Fam BC, Fynch S, Dzamko N, Watt MJ, Wormald S, Honeyman J, Galic S, Proietto J, Andrikopoulos S, et al. Liver-specific suppressor of cytokine signaling-3 deletion in mice enhances hepatic insulin sensitivity and lipogenesis resulting in fatty liver and obesity. Hepatology 2010. 52 1632–1642. ( 10.1002/hep.23861) [DOI] [PubMed] [Google Scholar]
- 59.Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan CC, et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science 1999. 283 1544–1548. ( 10.1126/science.283.5407.1544) [DOI] [PubMed] [Google Scholar]
- 60.Ueki K, Kondo T, Kahn CR. Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Molecular and Cellular Biology 2004. 24 5434–5446. ( 10.1128/MCB.24.12.5434-5446.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Holland WL, Adams AC, Brozinick JT, Bui HH, Miyauchi Y, Kusminski CM, Bauer SM, Wade M, Singhal E, Cheng CC, et al. An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metabolism 2013. 17 790–797. ( 10.1016/j.cmet.2013.03.019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Camporez JP, Jornayvaz FR, Petersen MC, Pesta D, Guigni BA, Serr J, Zhang D, Kahn M, Samuel VT, Jurczak MJ, et al. Cellular mechanisms by which FGF21 improves insulin sensitivity in male mice. Endocrinology 2013. 154 3099–3109. ( 10.1210/en.2013-1191) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Myers MG, Leibel RL, Seeley RJ, Schwartz MW. Obesity and leptin resistance: distinguishing cause from effect. Trends in Endocrinology and Metabolism 2010. 21 643–651. ( 10.1016/j.tem.2010.08.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ge H, Huang L, Pourbahrami T, Li C. Generation of soluble leptin receptor by ectodomain shedding of membrane-spanning receptors in vitro and in vivo. Journal of Biological Chemistry 2002. 277 45898–45903. ( 10.1074/jbc.M205825200) [DOI] [PubMed] [Google Scholar]
- 65.Lackey DE, Olefsky JM. Regulation of metabolism by the innate immune system. Nature Reviews: Endocrinology 2016. 12 15–28. ( 10.1038/nrendo.2015.189) [DOI] [PubMed] [Google Scholar]
- 66.Watanabe A, Sohail MA, Gomes DA, Hashmi A, Nagata J, Sutterwala FS, Mahmood S, Jhandier MN, Shi Y, Flavell RA, et al. Inflammasome-mediated regulation of hepatic stellate cells. American Journal of Physiology: Gastrointestinal and Liver Physiology 2009. 296 G1248–G1257. ( 10.1152/ajpgi.90223.2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wree A, McGeough MD, Peña CA, Schlattjan M, Li H, Inzaugarat ME, Messer K, Canbay A, Hoffman HM, Feldstein AE. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. Journal of Molecular Medicine 2014. 92 1069–1082. ( 10.1007/s00109-014-1170-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ruan H, Dong LQ. Adiponectin signaling and function in insulin target tissues. Journal of Molecular Cell Biology 2016. 8 101–109. ( 10.1093/jmcb/mjw014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McCommis KS, Hodges WT, Brunt EM, Nalbantoglu I, McDonald WG, Holley C, Fujiwara H, Schaffer JE, Colca JR, Finck BN. Targeting the mitochondrial pyruvate carrier attenuates fibrosis in a mouse model of nonalcoholic steatohepatitis. Hepatology 2017. 65 1543–1556. ( 10.1002/hep.29025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Samuel VT, Shulman GI. Nonalcoholic fatty liver disease as a nexus of metabolic and hepatic diseases. Cell Metabolism 2018. 27 22–41. ( 10.1016/j.cmet.2017.08.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Akero. Press release: All AKR-001 dose groups met week 12 efficacy endpoints in NASH phase 2a BALANCED study, 2020. South San Francisco, CA, USA: Akero. (available at: https://ir.akerotx.com/news-releases/news-release-details/all-akr-001-dose-groups-met-week-12-efficacy-endpoints-nash) [Google Scholar]
- 72.Hansen AMK, Vienberg SG, Lykkegaard K, Zhao X, Tingqing G, Han D, Zhang X, Thogersen H, Sass-Orum K, Tagmose T, et al. Differential receptor selectivity of the FGF15/FGF19 orthologues determines distinct metabolic activities in db/db mice. Biochemical Journal 2018. 475 2985–2996. ( 10.1042/BCJ20180555) [DOI] [PubMed] [Google Scholar]
- 73.Fisher FM, Estall JL, Adams AC, Antonellis PJ, Bina HA, Flier JS, Kharitonenkov A, Spiegelman BM, Maratos-Flier E. Integrated regulation of hepatic metabolism by fibroblast growth factor 21 (FGF21) in vivo. Endocrinology 2011. 152 2996–3004. ( 10.1210/en.2011-0281) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lan T, Morgan DA, Rahmouni K, Sonoda J, Fu X, Burgess SC, Holland WL, Kliewer SA, Mangelsdorf DJ. FGF19, FGF21, and an FGFR1/beta-klotho-activating antibody act on the nervous system to regulate body weight and glycemia. Cell Metabolism 2017. 26 709.e3–718.e3. ( 10.1016/j.cmet.2017.09.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu J, Yang K, Yang J, Xiao W, Le Y, Yu F, Gu L, Lang S, Tian Q, Jin T, et al. Liver-derived fibroblast growth factor 21 mediates effects of glucagon-like peptide-1 in attenuating hepatic glucose output. EBioMedicine 2019. 41 73–84. ( 10.1016/j.ebiom.2019.02.037) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ogawa Y, Kurosu H, Yamamoto M, Nandi A, Rosenblatt KP, Goetz R, Eliseenkova AV, Mohammadi M, Kuro-o M. BetaKlotho is required for metabolic activity of fibroblast growth factor 21. PNAS 2007. 104 7432–7437. ( 10.1073/pnas.0701600104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kurosu H, Choi M, Ogawa Y, Dickson AS, Goetz R, Eliseenkova AV, Mohammadi M, Rosenblatt KP, Kliewer SA, Kuro-o M. Tissue-specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. Journal of Biological Chemistry 2007. 282 26687–26695. ( 10.1074/jbc.M704165200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Suzuki M, Uehara Y, Motomura-Matsuzaka K, Oki J, Koyama Y, Kimura M, Asada M, Komi-Kuramochi A, Oka S, Imamura T. betaKlotho is required for fibroblast growth factor (FGF) 21 signaling through FGF receptor (FGFR) 1c and FGFR3c. Molecular Endocrinology 2008. 22 1006–1014. ( 10.1210/me.2007-0313) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen MM, Hale C, Stanislaus S, Xu J, Veniant MM. FGF21 acts as a negative regulator of bile acid synthesis. Journal of Endocrinology 2018. 237 139–152. ( 10.1530/JOE-17-0727) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.