

Abstract
Introduction:
Natural, social, and constructed environments play a critical role in the development and exacerbation of respiratory diseases. However, less is known regarding the influence of these environmental/community risk factors on the health of individuals living with cystic fibrosis (CF), compared to other pulmonary disorders.
Areas Covered:
Here, we review current knowledge of environmental exposures related to CF, which suggest that environmental/community risk factors do interact with the respiratory tract to affect outcomes. Studies discussed in this review were identified in PubMed between March 2019 and March 2020. Although the limited data available do not suggest that avoiding other potentially detrimental exposures could improve outcome, additional research incorporating novel markers of environmental exposures and community characteristics obtained at localized levels is needed.
Expert Opinion:
As we outline, some environmental exposures and community characteristics are modifiable; if not by the individual, then by policy. We recommend a variety of strategies to advance understanding of environmental influences on CF disease progression.
Keywords: Cystic fibrosis, environment, geographic, geomarker, geocoding, climate, air pollution, socioeconomic status, stressors, secondhand smoke
1. Introduction and Definitions
1.1. Environmental influences on pulmonary outcomes
Environmental exposures (e.g., air pollution), are known to play an important role in the development and exacerbation of lung diseases and disorders including asthma and chronic obstructive pulmonary disease (COPD) (1) (2) (3) (4). The mechanisms by which air pollutants influence respiratory morbidity include both direct damage to the lung tissue upon exposure and indirect pathways via the production of reactive oxygen species and the induction of systemic inflammation (5, 6). Although there is a well-established link between air pollution and other environmental exposures on respiratory disorders, significantly less is known regarding how these environmental factors impact patients living with cystic fibrosis (CF) despite evidence that non-genetic influences explain 50% of the variation in CF lung function (7). Table 1 summarizes potential environmental exposures and the relevance to CF.
Table 1:
Examples of Environmental Exposures and Potential Relevance to CF Outcomes
| Exposure Domain† |
Variables* | Findings/Potential Relevance |
|---|---|---|
| Geographic | Ambient air pollutants/allergens and proxies:
|
CF-specific:
|
Climatology:
|
CF-specific:
|
|
| Community | Neighborhood characteristics/stressors:
|
CF-specific:
|
Household characteristics/stressors:
|
CF-specific: | |
Extent of greenspace:
|
Non-CF: | |
| Healthcare Access |
|
CF-specific: |
Not all variables are available at the same levels of geographic resolution and time.
It is plausible that exposures that accumulate over the lifespan of an individual change over time in response to stressors from the environment, thereby impacting respiratory health and well-being (Figure 1). Critical periods may include adolescence and early adulthood, when patients are at greatest risk of accelerated lung-function decline (25) (24). Increased knowledge surrounding the impact of environmental influences on lung function and other outcomes may lead to more precise environmental health interventions and policy recommendations, and accurate prognostic models for personalized care.
Figure 1: Conceptual framework outlining CF environmental health epidemiology*.
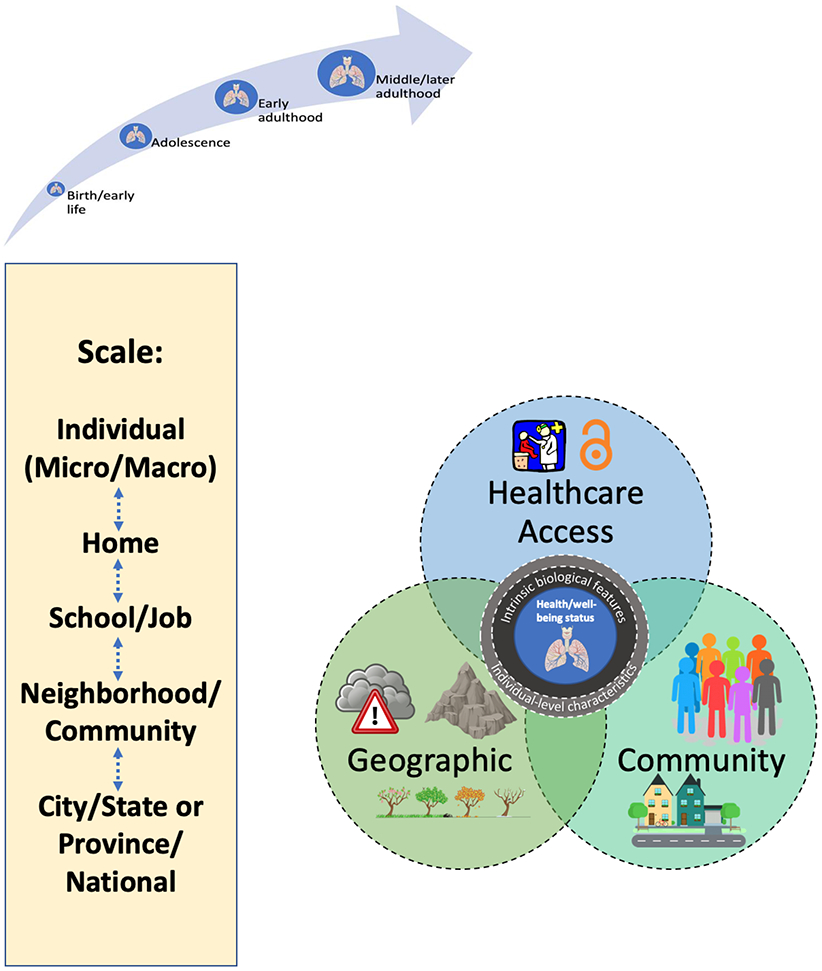
*See Section 2 for full description of key environmental domains (geographic, community and healthcare access) and exposures therein. Bi-directional arrows and dashed lines indicate reciprocal relationships.
1.2. Importance of location
Patients, caregivers and families who participated in a 2016 survey (Insight CF) conducted by the Cystic Fibrosis Foundation indicated that investigating how geographic and environmental factors impact lung infection and health outcomes was the third most important research priority after studying factors predicting survival and lung function decline (https://www.cff.org/Get-Involved/Connect/Help-Shape-CF-Research/ ). New and emerging technologies now enable researchers and others to geospatially map the location of individuals and characterize environmental exposures and identification of how these exposures worsen outcomes at the household level. These developments may allow for individual counseling of risk. Identifying geographic exposures that affect CF individuals more broadly may aid larger numbers of individuals through policy interventions and/or other recommendations. With the rapidly expanding knowledge of genetic modifiers, it becomes equally important to understand environmental risk factors in order to disentangle the interplay between genes and environment that leads to phenotypes.
Environmental factors are also not static for individuals owing to population mobility among other things. According to the 2016 Current Population Survey Annual and Economic Supplement (CPS ASEC), 11.2% of the U.S. population over the age of 1 year moved between 2015 and 2016. Although this is the lowest rate since tracking of this metric began in 1948, which is reflected in both local moves (within the same neighborhood or zip-code-tabulated centroid area) and non-local moves, the estimated moving rate has remained at roughly 15% since annual collection began in 2001. Data for individuals with CF are more limited, but a descriptive analysis of the US Cystic Fibrosis Foundation Data Registry from 2005-2015 suggests that, out of 37,391 patients with at least one recorded zip code, 17,528 (47%) moved at least once (with the caveat that patients have varying extent of follow up in the registry). It is unclear that less moving equates to improved long-term health consequences.
2. Environmental Exposure Domains and Relevance to CF
“Geomarkers” are measures that refer to any objective, contextual or geographic measure that influences or predicts the incidence of outcome or disease (26). The scale of geomarkers ranges from the individual patient to the national level, corresponding to degree of influence exerted by policy/decision making and components of the environment (Figure 1). The scale at which geomarkers are evaluated may yield different findings. Environmental features include inherent and manmade geographic characteristics (e.g., altitude and air pollution, respectively), community characteristics of environments (e.g., population density), and healthcare access (e.g., access to CF care center). In addition to intrinsic biologic features (e.g., sex) and behavioral characteristics (e.g., adherence to treatments), household-specific factors may impact CF respiratory health and well-being (the innermost circle), including secondhand smoke (SHS, described later in detail), indoor air pollution, and allergens. Studies discussed in this review were identified through semi-systematic searches of PubMed between 3/1/2019 and 3/12/2020, through the authors’ knowledge of the topic, and through citation review.
2.1. Geographic factors
2.1.1. Ambient air pollution and allergens
Outdoor air pollution is a complex mixture of solid and gaseous compounds including coarse, fine, and ultrafine particles (PM10, PM2.5, UFP respectively), ozone, oxides of nitrogen, and other toxicants. Major sources of outdoor air pollutants include industrial processes, biomass burning, and traffic. In contrast to other respiratory diseases, the evidence that outdoor air pollution influences CF morbidity is limited, but suggests that lung function, acquisition of respiratory pathogens, and exacerbation frequency are all impacted (27). Potential mechanisms may include increased airway inflammation, increased oxidative stress, and possibly decreased microbial clearance (28-31). Unlike asthma, there are few studies of the short-term effects of outdoor air pollution on exacerbation risk to inform CF patients regarding their outdoor exposure on poor air quality days as discussed below.
Exposure to particulate matter with an aerodynamic diameter of 2.5 microns or less (PM2.5), as measured by the U.S. Environmental Health Protection agency air quality monitoring stations, has been associated with earlier initial development of methicillin-resistant Staphylococcus aureus (MRSA) and Pseudomonas aeruginosa acquisition in U.S. children less than 6 years of age, but not methicillin-sensitive Staphylococcus aureus (MSSA), Stenotrophomonas maltophilia, or Achromobacter xylosoxidans (28, 32). However, the prevalence of P. aeruginosa, mucoid P. aeruginosa, or Burkholderia cepacia complex on respiratory cultures were not associated with higher PM2.5 in the CF Twin-Sibling Study after adjustment for other geographic factors (13).
A retrospective study conducted in Belgium demonstrated that the risk of CF exacerbations may be associated with short term increases in ozone, particulate matter with an aerodynamic diameter of 10 microns or less (PM10), and nitrogen dioxide; the finding for ozone was also observed in a separate study conducted in Sao Paulo, Brazil (9, 33). Annual exposure to air pollutants (PM10, PM2.5, and ozone) has also been reported to be associated with an increased exacerbation frequency in the CFF Data Registry (8). Although a decrease in FEV1 of 24mL for every increase in PM2.5 of 10 μg/m3 has been observed in the CFF Data Registry, this finding was not seen in the CF Twin-Sibling Study after adjustment for other geographic factors (8) (13). However, these findings are limited as pollution measured at local sites may not reflect individual exposures, either outdoors or indoors.
As noted, outdoor air pollutant concentrations arise from multiple sources, though evidence from epidemiologic studies of children with asthma suggest the composition of pollutants derived from traffic sources, including diesel exhaust particles, ultrafine particles in ambient air, nitrogen dioxide and elemental carbon, may confer specific risks to children who are exposed via residence near major roadways. Numerous association studies have been undertaken to examine relative contributions of traffic-related air pollution exposure on asthma severity (34). Similarly, CF exacerbation frequency has been associated with residential proximity to major arterial roadways in one study conducted in Los Angeles demonstrating a 6.7 greater odds of two or more exacerbations for every 1000 meters closer to a major roadway (10).
The presence of environmental allergens or sensitization to allergens has been reported to worsen severity of other respiratory diseases (e.g., asthma), but there are very limited data within CF to guide counselling regarding exposures (e.g. playing or exercising outside on high pollen count days). Within the CF Twin-Sibling Study, 14% self-reported environmental allergies (defined as allergies to pets, pollen, grasses, trees, dust mites and/or mold or seasonal allergies), which were associated with a higher risk of sinus disease (adjusted OR: 2.68), nasal polyps (adjusted OR: 1.74) and a more rapid decline in FEV1 (additional −1.1% predicted/year), but a later median age of acquisition of P. aeruginosa (6.6 years vs. 4.4 years of age) (35). The reported use of common allergy medications (i.e., anti-histamines and leukotriene inhibitors) did not alter the frequency of respiratory morbidities in this study.
2.1.2. Altitude
There are few studies examining the association between altitude (elevation) and CF phenotypes. Altitude may have effects on outcomes for individuals with pre-existing respiratory disease similar to air pollution exposure. For example, an observational study in Kyrgyzstan showed that individuals residing at lower altitudes (~750 m) had a lower prevalence of COPD (10.4%) than their counterparts in high-altitude areas (~2050 m; 36.7%), even after adjustment for indoor PM2.5 and smoking history.(36) As might be expected for CF, exercise capacity is decreased in higher altitudes where the partial pressure of inspired oxygen is reduced as outlined below, which is more pronounced in more severe lung disease and also non-specific to CF. However, it is unknown if there are any long-term consequences on lung function.
Data from the Dead Sea in Israel showed that exercise capacity (but not lung function) is increased by 7% as measured by peak oxygen consumption at very low altitude (396m below sea level) compared to sea level for individuals with CF (37). In contrast, hypoxemia has been observed during exercise at high altitudes (1500m above sea level) among individuals with CF, and this correlates with lower FEV1 (38). Hypoxemia has also been observed during rest at high altitudes (1800m above sea level) among 11-16 year old children with CF and moderate lung disease (mean FEV1: 64% predicted) (39).
While higher lung function (FEV1) and a reduced likelihood of mucoid P. aeruginosa, detection in respiratory cultures has been associated with higher altitude in the largely U.S. based CF Twin-Sibling Study, this finding did not remain significant in multivariable analyses including socioeconomic and other geographic factors (13). Altitude was also not associated with the presence of P. aeruginosa or B. cepacia complex on respiratory cultures examined in the same study.
2.1.3. Climatology
The majority of published work relating climate to outcomes in CF as outlined in this section is related to the acquisition and prevalence of respiratory pathogens, with a few studies examining lung function and one study focused on distal intestinal obstruction syndrome (DIOS). Potential associations with climate have been studied with both reference to long-term exposure (over years) and short-term exposure (generally seasonal). Overall, the data suggest that living in regions with warmer temperatures is associated with lower lung function in CF, largely (but not completely) mediated by higher prevalence of specific pathogens. However, the data are less clear on the specific climate factors (annual means or seasonal means) that result in earlier acquisition or higher prevalence of respiratory pathogens. It should also be emphasized that the observed associations between pathogens and climates may reflect differing behavior in different climates (e.g., the time spent outdoors), and not necessarily direct measures of temperature or humidity.
Higher mean annual ambient temperatures have been associated with lower lung function among patients with CF in the US and Australia patient registries, which have been replicated in a separate US-based dataset (−2.3 to −3.4 FEV1 percentile points per 10°F increase); this relationship persists after adjustment for some confounding demographic and socioeconomic factors, but it is possible that other unmeasured socioeconomic confounders exist (13) (40). It was found to be only partially mediated by P. aeruginosa, mucoid P. aeruginosa, and MRSA detection as the presence of these organisms accounted for 12 to 43% of the observed association between temperature and lung function (41). This relationship may be a function of human biology or CF-independent factors. An association between warmer temperatures and lower lung function has been reported in healthy individuals in the U.S.-based NHANES datasets with an effect size of lesser magnitude, −0.6 to −0.7 FEV1 percent predicted per 10°F increase (42). Of note, a higher prevalence of non-tuberculous mycobacteria has been reported in warmer climes in individuals with CF living in the U.S., but this did not mediate lung function (41).
Earlier P. aeruginosa acquisition in children less than 6 years of age may be more common in the Southern US (43). In addition, the rate of non-epidemic B. cepacia complex infection is 2.6-fold higher in patients residing in northern Australia, as compared to south of the Tropic of Capricorn (44). Both observations may be a function of temperature as in the warmest quartile regions of the US and Australia, the acquisition of P. aeruginosa may be 15 and 9 months earlier, respectively, than in the coldest quartile (13). Factors related to atmospheric moisture may play a role in acquisition and prevalence of infectious organisms as well. For example, higher rates of non-epidemic acquisition of B. cepacia complex have been correlated with higher rainfall in Australia in time series analyses (r=0.65-0.82) (44). For non-tuberculous mycobacteria, higher prevalence by center in the US was associated with higher annual water vapor pressure (r2=0.40), more so than individual behaviors, such as swimming in an indoor pool or observation of rusty/unclean tap water in the home (45).
In addition to climate effects by geographic regions, there may be seasonal influences on CF phenotypes. However, these effects may be region specific with seasonality serving as a proxy for accompanying changes in temperature and humidity. For example, the acquisition of P. aeruginosa has been reported to be 1.2-1.3-fold higher in the summer and fall among children less than 6 years of age in the CFF Data Registry (46), although over 60% of the Danish patient cohort followed from 1965-1990 contracted initial infection and chronic colonization with P. aeruginosa during the winter months (47), and no seasonal differences were seen in a smaller study of 120 newborns in Wisconsin (48). In terms of other CF-related respiratory pathogens, the highest rate of MRSA, A. xylosoxidans, H. influenzae acquisition were typically found to be in the winter months among U.S. children less than 6 years old (49). However, no seasonal variation was observed for S. maltophilia or S. aureus acquisition within the same population (46, 49).
Seasonal variation in respiratory viral infections has been observed in one study of 98 adults in the UK (Rhinovirus infections peaked in the fall, while non-rhinovirus infections peaked in the winter), but pulmonary exacerbations were not found to be associated with season (50). A prospective longitudinal study of 368 paired specimens from 33 children with CF showed that bacteria were more commonly detected in spring and summer, while viruses were more common in autumn and winter (51). Seasonal variation in lung function (Forced expiratory volume in 1 second: FEV1) has not been observed in the Danish or UK CF Registries (52). Within the US CFF Data Registry, FEV1 may be slightly higher on average (1.2 percentile points) in January compared to July, but this was not observed in the smaller largely US-based CF Twin-Sibling Study (13). Lastly, one single-center study in Australia found that higher temperatures in the prior week compared to the average for the season may be associated with admissions for DIOS and constipation (5°F and 7°F higher, respectively), but it is unknown whether living in warmer climates is associated with DIOS or constipation (53).
Seasonality was a strong indicator of Vitamin D levels in a study from a single pediatric center in the Northern US, which showed that Vitamin D levels were higher in summer, compared to winter (difference in means: 11.6 ng/mL)(54). A Danish pediatric study of 45 CF and 102 non-CF subjects affirmed the link between Vitamin D levels and seasonality, but there was no association between Vitamin D levels and demographic/clinical characteristics, including sex, BMI and FEV1 in the CF group; however, the CF group reported increased solar exposure, compared to the non-CF group (55).
2.1.4. Bodies of Water
A retrospective study conducted in Florida found that children with CF who lived within 500 meters of a body of water had a higher adjusted odds ratio (OR: 9.4; no CI reported, but estimated OR corresponded to logged OR and SE of 2.242 and 0.097, respectively) of acquiring non-tuberculous mycobacteria, although it is unclear whether the risk is due to aerosolized droplets with mycobacteria being carried on the wind versus visiting the body of water more frequently (56).
2.2. Community characteristics
2.2.1. Neighborhood characteristics
There are limited data regarding the role of population density on CF outcomes. A Wisconsin based study did not find any association between population density/urban location and the acquisition of P. aeruginosa in young children with CF (57). However, higher titers of Aspergillus fumigatus antibodies were found in CF patients living in low population density areas (rural) in one study from the UK in the mid-1990s (58). Population density has not been associated with lung function (FEV1), P. aeruginosa, mucoid P aeruginosa, or B. cepacia complex detection in the CF Twin-Sibling Study (13).
Material deprivation as measured by Area Deprivation Index and Rural-Urban Commuting Area scores has been associated with greater than 2-fold increase risk in having MRSA in CF for a single center in the Southeastern U.S. (40).
A study from England did not find an association between proximity to composting facilities and hospital admissions in CF with modelled concentrations of Aspergillus (59). Other community characteristics, such as extent of greenspace, have not been studied in CF.
2.2.2. Household characteristics
While outdoor air pollution has been demonstrated to have an effect on lung function and exacerbations, many children, particularly younger ones, spend much of their time indoors (60). While outdoor air pollution can penetrate the indoors, combustible indoor sources (SHS, gas or wood burning stoves, fireplaces, gas furnaces) also contribute to indoor concentrations of PM2.5 and NO2. The relationship between these sources of indoor air pollution and CF has not been reported; however, exposure to these pollutants have been associated with respiratory morbidity in children with asthma (61-63).
2.2.3. Active smoking
Active smoking has been examined in several studies of individuals with cystic fibrosis. The reported rates of ever smoking in adolescents is 21.1%, but only 3% for smoking regularly (64). The rate of regular smoking in adults with CF has been reported to by 8-11% in single center studies in the U.S. and U.K. (65) (66). In one study of adults, no difference was observed in decline in lung function (FEV1 or FVC) in active smokers versus non-smokers, but confounding may be present as sicker patients may choose not to smoke (66).
2.2.4. Secondhand smoke
Multiple studies have demonstrated the deleterious effects of secondhand tobacco smoke on lung function and nutritional status among individuals with CF, even after accounting for socioeconomic status (16, 67-70), although the effects have not been observed in every study (68). Tobacco smoke exposure has also been shown to produce cystic fibrosis transmembrane conductance regulator (CFTR) dysfunction in vitro and in vivo, with relevance to other respiratory diseases and potentially CF patients treated with CFTR modulators (71). In addition, SHS may increase airway inflammation and impair pathogen clearance in individuals with CF (72, 73), as evidenced by higher rates of MRSA seen in infants with CF who have parental smoke exposure (74). Caregiver-reported rates of SHS exposure for infants and children vary widely between studies (10% to 76%) (75). While this exposure occurs largely on a household level, local and state laws may impact additional exposure in public places.
Individuals with CF exposed to SHS have been shown to have lower lung function (9.8 FEV1 percentile points) in a retrospective cross-sectional study and these effects may be amplified by polymorphisms in specific modifier genes (i.e., TGFβ1)(16). In a study based on U.S. registry data, SHS was associated with 2.4% decrease in FEV1 in children after adjusting for socioeconomic factors (76). The effects may be dose-related as a decrease in FEV1 by 4% in children with CF has been reported for every 10 cigarettes smoked in the household each day (77). From a nutritional perspective, SHS has also been associated with reduced growth in infants and school age children with CF (68, 74).
Data from the Early Pseudomonas Infection Control study also showed that in utero tobacco exposure may be detrimental; maternal smoking during pregnancy was associated with lower FEV1 at 6-7 years of age (−4.4 percentile points) and lower height and weight percentiles (78, 79).
2.2.5. Indoor allergens
An allergen of particular importance in CF is Aspergillus as patients with CF are susceptible to allergic bronchopulmonary aspergillosis (ABPA). Limited data from the households of sixteen adult CF patients suggested that ABPA was associated with higher A. fumigatus DNA concentrations in the home (17). ABPA has also been associated with domestic pet ownership in one study of CF patients living in Germany (80). Within the CF Twin-Sibling Study, cat ownership was associated with a greater prevalence of nasal polyps and combined dog-cat ownership was associated with a greater rate of wheezing (81). There were no differences in lung function (FEV1) or the prevalence/age of acquisition for P. aeruginosa and MRSA between cat/dog owners and (potentially unexposed) non-cat/dog owners.
2.2.6. Siblings with cystic fibrosis
Given that CF is a genetic disorder, households may contain more than one family member with CF, usually siblings. One Israeli study matching siblings with CF to single CF patients found showed a lower FEV1 with a faster rate of decline decline rate, a higher prevalence of colonization by P. aeruginosa and Mycobacterium abscessus, and a higher likelihood of lung transplantation in siblings compared to singletons (82). Within families, studies have observed while younger siblings tend to be colonized with respiratory pathogens such as P. aeruginosa earlier in life than their older siblings, results are mixed as to whether lung function is higher in younger siblings compared to their older siblings (83, 84).
2.3. Healthcare access
Lack of healthcare access has important ramifications for health outcomes. Barriers to access can include time to travel and distance to care sites, availability of specific services within a geographic area, and financial coverage for healthcare services, all of which can be influenced by geography. In terms of services available, limited qualitative data from families living in rural Australia suggested that parents of children with CF sometimes had challenges in communicating with non-tertiary care centers regarding their children’s care (85). However, no differences in lung function (FEV1), BMI, or the prevalence of P. aeruginosa colonization were observed in a separate study conducted in Australia comparing those receiving care from academic CF centers versus other modes of outreach care (86).
Distance to CF care centers in the US was not associated with FEV1 in either the CFF Data Registry or the CF Twin-Sibling Study (13) (87). While moving residences has been associated with changes in lung function, there are many potential confounding factors. It is plausible that pediatric CF patients are primarily followed in accredited CF centers in the US; however, for other underserved geographic regions or chronic diseases, there may be no/remote access to a specialist. In any case, the longer distances to CF care centers may prevent quarterly follow up recommended by the CF Foundation. In the US, young adults who moved further away from CF care centers had an associated decrease in lung function, whereas for older patients, moving further away was associated with improved lung function (87). The negative association in young adults may not be related to the physical move but rather emerging independence and non-adherence to therapies and inadequate nutrition. As the authors acknowledged, the positive association found in older adults may be due to severity bias, as “healthier” adults who function more independently may choose to reside farther away from their designated CF care centers. Distance to CF care center was also not associated with the presence of P. aeruginosa, mucoid P aeruginosa, or B. cepacia complex on respiratory cultures in the CF Twin-Sibling Study (13). Access to public transportation routes could improve access to care but could also be detrimental if it increases exposure to traffic related air pollution (especially diesel exhaust emissions).
Variation in clinical management, either between care centers in a given country or between countries, may impact patient prognosis. Pulmonary exacerbation surveillance and treatment were shown to widely vary between US CF care centers (88). An Eastern European survey study on CF clinical management showed variable practice patterns and differences in patient severity according to region (89). The lack of high-quality patient registries has been noted as an obstacle to healthcare access (90); this may contribute to differences observed in mortality between Western and European countries (91).
3. Socioeconomic Status
Socioeconomic status, often measured as a combination of education, income and occupation, can determine an individual patient’s stressors and access to healthcare. As reflected by overlapping circles in the conceptual model (Figure 1), these domains are often intimately linked to socioeconomic status (individual-level characteristics including behavioral and socioeconomic as shown in inner circle of Figure 1), and deconstructing these relationships can be challenging. Environmental health exposures tend to be highly correlated with socioeconomic status (92). Although CF is a genetic disease, factors such as physical environment correspond to socioeconomic pathways, which, in turn, can create variability in disease progression and outcomes (93). The previously referenced study by Ong and colleagues in a CF pediatric cohort demonstrated that exposure to environmental tobacco smoke was increased in families with low socioeconomic status (67). They also found that the estimated impact of socioeconomic status on pulmonary and nutrition outcomes was similar regardless of whether or not they adjusted for exposure to environmental tobacco smoke, suggesting health disparity risks may be present that are independent exposure to environmental tobacco smoke. A UK CF registry study found that socioeconomic deprivation modified the association between employment chances and lung function (94).
The most commonly used proxy for individual-level socioeconomic status in CF linked to geography is insurance status as coverage for specific therapies may differ by state or country. Several association analyses using this measure, adjusted for age, genotype and other variables, have been performed. An single-center cohort study of 189 CF patients showed that median survival was 6.1 years in patients without insurance; while patients with Medicaid had the same median survival as those with private insurance (20.5 years) (95). Another single-center cross-sectional study of pediatric CF patients’ annualized data found that having Medicaid coverage corresponded to lower FEV1 (difference: 11.6% predicted), as well as more frequent and longer hospital stays to treat pulmonary exacerbations (per-patient difference: 0.80 hospitalization and 8.8 days, respectively) compared to patients covered by private insurance (96). In a larger study using the CFF Data Registry, patients with reported Medicaid coverage had a higher risk of death (hazard ratio: 3.65), lower FEV1 (difference: 9.2% predicted), higher likelihood of hospitalization for pulmonary exacerbation treatment (1.6 OR) and greater risk of growth measuring below the 5th percentile for weight and height (2.19 OR and 2.22 OR, respectively), compared to those who did not report using Medicaid insurance (97). Medicaid policies, which are determined on a state by state basis, can dictate what therapies and care are covered and, also eligibility for such insurance coverage. There are no published studies to date regarding whether variation in state policies affects outcomes in CF.
Different measures of individual and community socioeconomic status, including median household income by zip code, maternal education level and Medicaid/state insurance coverage, were linked to disease severity and practice patterns in a cross-sectional study derived from the Epidemiologic Study of CF (98). Pairwise correlations between socioeconomic measures were small to moderate (range in Spearman’s r: 0.19 to 0.34), implying that each measure may impact health in different ways. Each measure was studied individually, but all three measures were inversely associated with disease severity. Median household income by zip code was a more sensitive measure for healthcare utilization, e.g., patients residing in zip codes with low household income had more frequent intravenous antibiotic courses, compared to those in higher categories of median household income determined by zip code. However, this association was not detected with the other measures.
4. Useful Geomarker Tools and Methods for CF Environmental Health Studies
“Geocoding” refers to the process of translating addresses into a location on the earth’s surface (often latitude and longitude coordinates). Precise geolocation data (addresses) are not always available, and so patient location can often only be determined to the level of county or zip code. Measured and estimated geomarker values are often available at differing spatiotemporal resolutions (e.g., monthly estimations of greenspace at 12 km2, daily estimations of air pollution at 1 km2, or annual estimations of median household income at the census tract level). Estimating geomarker exposure for patients often requires harmonization of several heterogeneous data sources with geocoded location data, which themselves can be available at different spatiotemporal resolutions and extents. The error for estimating exposures must be carefully considered on a case-by-case basis for each geomarker depending on the precision of both the patient location and the geomarker data. Once geomarker exposures are estimated for individual patients, then associations between geomarkers and health outcomes can be tested for.
CF studies have largely focused on extent to which environmental exposures explain outcomes, primarily FEV1 and PE rates. Associations with quality of life have been largely understudied. The relative contributions of environmental exposures have been examined primarily through linear models of FEV1 decline, sometimes at zip code level, without adjustment for mortality or precise geomarker information. For example, studies have used data from Environmental Protection Agency monitors instead of exposure assessment modeling, limiting PM2.5 assessments to individuals within 20 miles of a monitor; these approaches have been shown to yield biased associations with health outcomes.(11) This highlights the importance of studying fine-scale (i.e., high spatial and/or temporal resolution at the individual level) rather than regional PM2.5 variations (99) (100) (101-103), which may lead to more informative prediction modeling for outcomes according to location. These models may also be used to characterize both acute and chronic exposures at the patient level. Sometimes, individuals share an unobserved heterogeneity within a region. For example, in some regions with better health services and less environmental problems, the mortality risk would be expected to be lower, but there may be isolated definable areas with specific high risk factors. By taking this heterogeneity into account, improved individualized predictions may be obtained.
5. Conclusions
The physical environment can have significant consequences for the health of individuals with CF. Epidemiologic research in CF, which has primarily been conducted using registry analyses, has shown that different measures of socioeconomic status, particularly access to care and community characteristics, have strong influences on morbidity and mortality. Recent studies have begun to examine the role of the physical environment on health outcomes in CF, but further work is necessary to determine the magnitude (effect size) of environmental factors and the cost/benefit ratio of preventing or ameliorating these exposures.
6. Expert Opinion
While the effect of environmental exposures on health outcomes has been known for centuries, to date most environmental factors have been studied at imprecise levels that inaccurately estimate both ambient and personal levels of exposure. Most of the environmental exposures that have been identified will require prospective studies with more precise measurements of exposures designed to confirm their associated effects. More recent tools, which include state-of-the-art environmental exposure models and fine-scale monitoring of air pollution and other exposures, offer increased geomarker precision. National CF registries across the world may be newly leveraged, given broader availability of geomarkers in recent years at more precise levels than previously available, as well as refined air sampling monitors and other technology developments to conduct these prospective studies. These studies should also be designed to account for modifier effects from other factors. For example, many environmental exposures that interact with the respiratory tract may be moderated by seasonality, temperature, and proximity to water.
In addition to advances in measuring environmental exposures more precisely, it is anticipated that combining more precise geomarkers with emerging physiologic biomarkers in CF (e.g., lung clearance index) may enhance environmental health decision making. Results from these studies can lead to pilot studies testing environmental health interventions and ultimately tailoring them to the individual CF patient (Figure 2).
Figure 2: Potential strategies to enhance CF environmental health decision making.
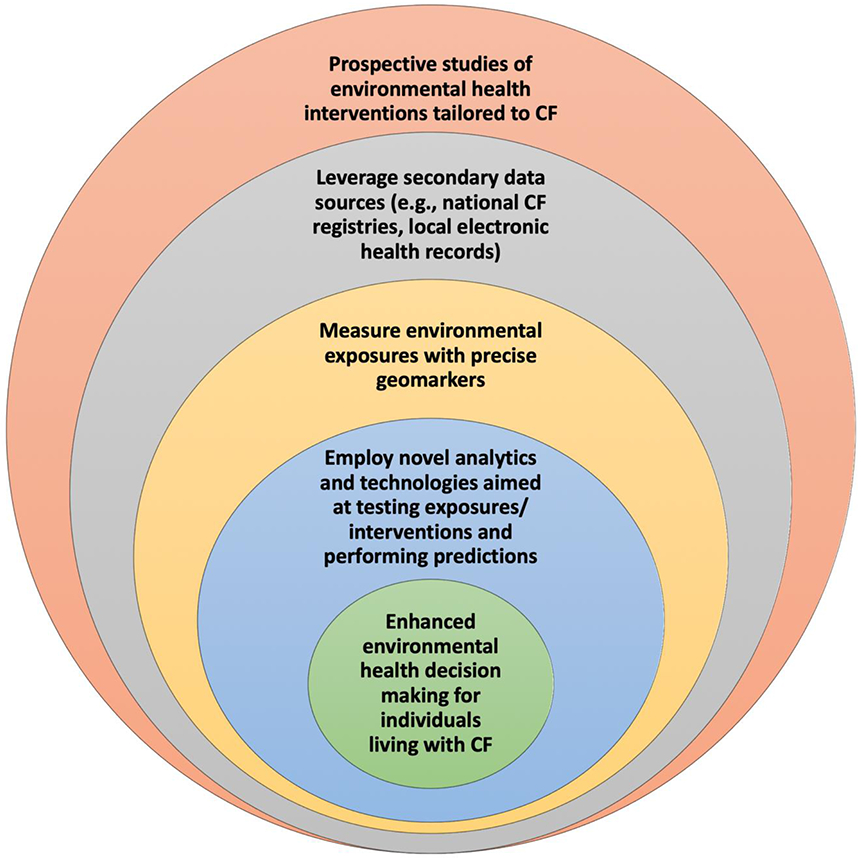
Interventions may take place on an individual or community level. For example, a modifiable exposure occurring on the household level is secondhand smoke. Other exposures could be modified on both an individual or community level. For example, if greenspace is ultimately found to be associated with outcomes in CF, interventions could occur on an individual level with residential relocation or on a community level through urban planning. In addition to preventative interventions, interventions that ameliorate exposures could also be found to be helpful in the future, potentially such as air purifiers. Other than exposure to secondhand smoke, published data do not necessarily support relocating to avoid or preventing other potentially detrimental exposures at this time.
Studies with clear associations between environmental exposures and health outcomes offer an opportunity to enact environmental health policies aimed at timely interventions and clinical decision-making strategies that can enhance the quality of life and longevity of individuals living with CF. As an example, U.S. Medicaid policies, which are determined on a state by state basis, can not only dictate what therapies and care are covered, but also eligibility for such insurance coverage. Likewise, policies and access to care may differ substantially for patients in living in adjacent countries with similar climatic features. A better understanding of the effects of environmental exposures may help advocate for more consistent policies across jurisdictions.
Looking towards the future, we would anticipate that there are more studies with more precise measurements of environmental exposure. Smart apps may be helpful in determining an individual’s proximity to fixed exposure sources (e.g., a coal-fired power plant). Additional modeling tools, such Bayesian modeling may help provide more nuanced probabilities for disease on the individual level. Ultimately, we would hope that personalized medicine also includes personalized information concerning environmental exposures to improve health care outcomes and quality of life.
Article Highlights Box:
The environment plays a critical role in the development and exacerbation of respiratory diseases.
However, less is known regarding the influence of these environmental/community risk factors on the health of individuals living with cystic fibrosis (CF), compared to other pulmonary disorders.
In this review, we summarize current knowledge regarding the risk of environmental exposures on outcomes in cystic fibrosis, including air pollution, secondhand smoke, climate, allergens, socioeconomic factors, community factors, and household factors.
Further work is necessary to determine the magnitude (effect size) of environmental factors and the cost/benefit ratio of preventing or ameliorating these exposures.
Advances in geocoding, more precise measurements of environmental exposure, and increased advocacy may improve outcomes in cystic fibrosis through changes in individual behavior and on a policy level.
Acknowledgments
Funding Details: This work was supported by the National Institutes of Health (Bethesda, MD, USA)(Grants R01 HL128475, R01 HL141286, and K23 ES029985). The funding sources had no involvement in the writing of the manuscript or the decision to submit. There are no conflicts of interest.
Financial and competing interests disclosure: This work was supported by the National Institutes of Health (Bethesda, MD, USA)(Grants R01 HL128475, R01 HL141286, and K23 ES029985). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
REFERENCES
- 1.Guarnieri M, Balmes JR. Outdoor air pollution and asthma. Lancet 383(9928), 1581–92 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mirabelli MC, Vaidyanathan A, Flanders WD, et al. Outdoor PM2.5, Ambient Air Temperature, and Asthma Symptoms in the Past 14 Days among Adults with Active Asthma. Environ Health Perspect 124(12), 1882–90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hansel NN, McCormack MC, Kim V. The Effects of Air Pollution and Temperature on COPD. COPD 13(3), 372–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heinrich J, Schikowski T. COPD Patients as Vulnerable Subpopulation for Exposure to Ambient Air Pollution. Curr Environ Health Rep 5(1), 70–6 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Schraufnagel DE, Balmes JR, Cowl CT, et al. Air Pollution and Noncommunicable Diseases: A Review by the Forum of International Respiratory Societies’ Environmental Committee, Part 2: Air Pollution and Organ Systems. Chest 155(2), 417–26 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schraufnagel DE, Balmes JR, Cowl CT, et al. Air Pollution and Noncommunicable Diseases: A Review by the Forum of International Respiratory Societies’ Environmental Committee, Part 1: The Damaging Effects of Air Pollution. Chest 155(2), 409–16 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.*.Collaco JM, Blackman SM, McGready J, et al. Quantification of the relative contribution of environmental and genetic factors to variation in cystic fibrosis lung function. The Journal of pediatrics 157(5), 802–7 e1–3 (2010).*A twin study attempting to derive the relative contributions of genetic and environmental factors to lung function.
- 8.***.Goss CH, Newsom SA, Schildcrout JS, et al. Effect of ambient air pollution on pulmonary exacerbations and lung function in cystic fibrosis. Am J Respir Crit Care Med 169(7), 816–21 (2004).***One of the first studies to examine the effects of an environmental exposure (ambient air pollution) to CF outcomes using geocoding.
- 9.Goeminne PC, Kicinski M, Vermeulen F, et al. Impact of air pollution on cystic fibrosis pulmonary exacerbations: a case-crossover analysis. Chest 143(4), 946–54 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Jassal MS, Yu AM, Bhatia R, et al. Effect of residential proximity to major roadways on cystic fibrosis exacerbations. Int J Environ Health Res 23(2), 119–31 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Miller KA, Siscovick DS, Sheppard L, et al. Long-term exposure to air pollution and incidence of cardiovascular events in women. N Engl J Med 356(5):447–58 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Margolis HG, Mann JK, Lurmann FW, et al. Altered pulmonary function in children with asthma associated with highway traffic near residence. Int J Environ Health Res 19(2), 139–55 (2009). [DOI] [PubMed] [Google Scholar]
- 13.***.Collaco JM, McGready J, Green DM, et al. Effect of temperature on cystic fibrosis lung disease and infections: a replicated cohort study. PloS one 6(11), e27784 (2011).***A replicated cohort study demonstrating an association between CF lung function and infectious pathogens with ambient temperature.
- 14.O’Connor GT, Quinton HB, Kneeland T, et al. Median household income and mortality rate in cystic fibrosis. Pediatrics 111(4 Pt 1), e333–9 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Auger KA, Kahn RS, Simmons JM, et al. Using Address Information to Identify Hardships Reported by Families of Children Hospitalized With Asthma. Acad Pediatr 17(1), 79–87 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collaco JM, Vanscoy L, Bremer L, et al. Interactions between secondhand smoke and genes that affect cystic fibrosis lung disease. JAMA 299(4), 417–24 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rocchi S, Richaud-Thiriez B, Barrera C, et al. Evaluation of mold exposure in cystic fibrosis patients’ dwellings and allergic bronchopulmonary risk. J Cyst Fibros 14(2), 242–7 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Feng X, Astell-Burt T. Is Neighborhood Green Space Protective against Associations between Child Asthma, Neighborhood Traffic Volume and Perceived Lack of Area Safety? Multilevel Analysis of 4447 Australian Children. Int J Environ Res Public Health 14(5), doi: 10.3390/ijerph14050543 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Douglas JA, Archer RS, Alexander SE. Ecological determinants of respiratory health: Examining associations between asthma emergency department visits, diesel particulate matter, and public parks and open space in Los Angeles, California. Prev Med Rep 14, 100855 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tischer C, Gascon M, Fernandez-Somoano A, et al. Urban green and grey space in relation to respiratory health in children. Eur Respir J 49(6), doi: 10.1183/13993003.02112-2015 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Gernes R, Brokamp C, Rice GE, et al. Using high-resolution residential greenspace measures in an urban environment to assess risks of allergy outcomes in children. Sci Total Environ 668, 760–7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsieh CJ, Yu PY, Tai CJ, et al. Association between the First Occurrence of Asthma and Residential Greenness in Children and Teenagers in Taiwan. Int J Environ Res Public Health 16(12), doi: 10.3390/ijerph16122076 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roberts JM, Wilcox PG, Quon BS. Evaluating Adult Cystic Fibrosis Care in BC: Disparities in Access to a Multidisciplinary Treatment Centre. Can Respir J 2016, 8901756 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szczesniak RD, Li D, Su W, et al. Phenotypes of Rapid Cystic Fibrosis Lung Disease Progression during Adolescence and Young Adulthood. Am J Respir Crit Care Med, doi: 10.1164/rccm.201612-2574OC (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vandenbranden SL, McMullen A, Schechter MS, et al. Lung function decline from adolescence to young adulthood in cystic fibrosis. Pediatr Pulmonol 47(2), 135–43 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beck AF, Huang B, Chundur R, Kahn RS. Housing code violation density associated with emergency department and hospital use by children with asthma. Health Aff (Millwood) 33(11), 1993–2002 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brugha R, Edmondson C, Davies JC. Outdoor air pollution and cystic fibrosis. Paediatric respiratory reviews, doi: 10.1016/j.prrv.2018.03.005 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Psoter KJ, De Roos AJ, Mayer JD, et al. Fine particulate matter exposure and initial Pseudomonas aeruginosa acquisition in cystic fibrosis. Annals of the American Thoracic Society 12(3), 385–91 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Qu F, Qin XQ, Cui YR, et al. Ozone stress down-regulates the expression of cystic fibrosis transmembrane conductance regulator in human bronchial epithelial cells. Chem Biol Interact 179(2–3), 219–26 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Ahmad S, Nichols DP, Strand M, et al. SERCA2 regulates non-CF and CF airway epithelial cell response to ozone. PloS one 6(11), e27451 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kunzi L, Krapf M, Daher N, et al. Toxicity of aged gasoline exhaust particles to normal and diseased airway epithelia. Scientific reports 5, 11801 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Psoter KJ, De Roos AJ, Wakefield J, et al. Air pollution exposure is associated with MRSA acquisition in young U.S. children with cystic fibrosis. BMC pulmonary medicine 17(1), 106 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Farhat SCL, Almeida MB, Silva-Filho L, et al. Ozone is associated with an increased risk of respiratory exacerbations in patients with cystic fibrosis. Chest 144(4), 1186–92 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng XY, Ding H, Jiang LN, et al. Association between Air Pollutants and Asthma Emergency Room Visits and Hospital Admissions in Time Series Studies: A Systematic Review and Meta-Analysis. PLoS One 10(9), e0138146 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collaco JM, Morrow CB, Green DM, et al. Environmental allergies and respiratory morbidities in cystic fibrosis. PediatrPulmonol 48(9), 857–64 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brakema EA, Tabyshova A, Kasteleyn MJ, et al. High COPD prevalence at high altitude: does household air pollution play a role? The European respiratory journal 53(2), doi: 10.1183/13993003.01193-2018 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Falk B, Nini A, Zigel L, et al. Effect of low altitude at the Dead Sea on exercise capacity and cardiopulmonary response to exercise in cystic fibrosis patients with moderate to severe lung disease. Pediatr Pulmonol 41(3):234–41 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Ryujin DT, Mannebach SC, Samuelson WM, Marshall BC. Oxygen saturation in adult cystic fibrosis patients during exercise at high altitude. Pediatr Pulmonol 32(6), 437–41 (2001). [DOI] [PubMed] [Google Scholar]
- 39.Oades PJ, Buchdahl RM, Bush A. Prediction of hypoxaemia at high altitude in children with cystic fibrosis. BMJ 308(6920), 15–8 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oates GR, Harris WT, Rowe SM, et al. Area Deprivation as a Risk Factor for Methicillin-resistant Staphylococcus aureus Infection in Pediatric Cystic Fibrosis. Pediatr Infect Dis J 38(11), e285–e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Collaco JM, Raraigh KS, Appel LJ, Cutting GR. Respiratory pathogens mediate the association between lung function and temperature in cystic fibrosis. Journal of cystic fibrosis. doi: 10.1016/j.jcf.2016.05.012 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Collaco JM, Appel LJ, McGready J, Cutting GR. The relationship of lung function with ambient temperature. PloS one 13(1), e0191409 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Psoter KJ, Rosenfeld M, De Roos AJ, et al. Differential geographical risk of initial Pseudomonas aeruginosa acquisition in young US children with cystic fibrosis. American journal of epidemiology. 179(12):1503–13 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Ramsay KA, Butler CA, Paynter S, et al. Factors influencing acquisition of Burkholderia cepacia complex organisms in patients with cystic fibrosis. Journal of clinical microbiology 51(12), 3975–80 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prevots DR, Adjemian J, Fernandez AG, et al. Environmental risks for nontuberculous mycobacteria. Individual exposures and climatic factors in the cystic fibrosis population. Annals of the American Thoracic Society 11(7), 1032–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Psoter KJ, De Roos AJ, Wakefield J, et al. Season is associated with Pseudomonas aeruginosa acquisition in young children with cystic fibrosis. Clinical microbiology and infection 19(11), E483–9 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Johansen HK, Hoiby N. Seasonal onset of initial colonisation and chronic infection with Pseudomonas aeruginosa in patients with cystic fibrosis in Denmark. Thorax 47(2), 109–11 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Farrell PM, Shen G, Splaingard M, et al. Acquisition of Pseudomonas aeruginosa in children with cystic fibrosis. Pediatrics 100(5), E2 (1997). [DOI] [PubMed] [Google Scholar]
- 49.Psoter KJ, De Roos AJ, Wakefield J, et al. Seasonality of acquisition of respiratory bacterial pathogens in young children with cystic fibrosis. BMC infectious diseases 17(1), 411 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Flight WG, Bright-Thomas RJ, Sarran C, et al. The effect of the weather on pulmonary exacerbations and viral infections among adults with cystic fibrosis. International journal of biometeorology 58(9), 1845–51 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Miro-Canis S, Capilla-Rubio S, Marzo-Checa L, et al. Multiplex PCR reveals that viruses are more frequent than bacteria in children with cystic fibrosis. J Clin Virol 86, 1–4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qvist T, Schluter DK, Rajabzadeh V, et al. Seasonal fluctuation of lung function in cystic fibrosis: A national register-based study in two northern European populations. Journal of cystic fibrosis, doi: 10.1016/j.jcf.2018.10.006 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ooi CY, Jeyaruban C, Lau J, et al. High ambient temperature and risk of intestinal obstruction in cystic fibrosis. Journal of paediatrics and child health 52(4), 430–5 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Lansing AH, McDonald C, Patel RA, et al. Vitamin D deficiency in pediatric patients with cystic fibrosis: associated risk factors in the northern United States. South Med J 108(3), 164–9 (2015). [DOI] [PubMed] [Google Scholar]
- 55.Oliveira MS, Matsunaga NY, Rodrigues MLE, et al. Lung disease and vitamin D levels in cystic fibrosis infants and preschoolers. Pediatr Pulmonol 54(5), 563–74 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Bouso JM, Burns JJ, Amin R, et al. Household proximity to water and nontuberculous mycobacteria in children with cystic fibrosis. Pediatr Pulmonol 52(3), 324–30 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Kosorok MR, Jalaluddin M, Farrell PM, et al. Comprehensive analysis of risk factors for acquisition of Pseudomonas aeruginosa in young children with cystic fibrosis. Pediatr Pulmonol 26(2), 81–8 (1998). [DOI] [PubMed] [Google Scholar]
- 58.Simmonds EJ, Littlewood JM, Hopwood V, Evans EG. Aspergillus fumigatus colonisation and population density of place of residence in cystic fibrosis. Archives of disease in childhood 70(2), 139–40 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roca-Barcelo A, Douglas P, Fecht D, et al. Risk of respiratory hospital admission associated with modelled concentrations of Aspergillus fumigatus from composting facilities in England. Environ Res. 2019, 108949 (2019). [DOI] [PubMed] [Google Scholar]
- 60.Klepeis NE, Nelson WC, Ott WR, et al. The National Human Activity Pattern Survey (NHAPS): a resource for assessing exposure to environmental pollutants. J Expo Anal Environ Epidemiol 11(3), 231–52 (2001). [DOI] [PubMed] [Google Scholar]
- 61.Hansel NN, Breysse PN, McCormack MC, et al. A longitudinal study of indoor nitrogen dioxide levels and respiratory symptoms in inner-city children with asthma. Environ Health Perspect 116(10), 1428–32 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Matsui EC. Environmental exposures and asthma morbidity in children living in urban neighborhoods. Allergy 69(5), 553–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McCormack MC, Breysse PN, Matsui EC, et al. In-home particle concentrations and childhood asthma morbidity. Environ Health Perspect 117(2), 294–8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Britto MT, Garrett JM, Dugliss MA, et al. Risky behavior in teens with cystic fibrosis or sickle cell disease: a multicenter study. Pediatrics 101(2), 250–6 (1998). [DOI] [PubMed] [Google Scholar]
- 65.Verma A, Clough D, McKenna D, et al. Smoking and cystic fibrosis. J R Soc Med 94 Suppl 40, 29–34 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stern RC, Byard PJ, Tomashefski JF, Jr., Doershuk CF. Recreational use of psychoactive drugs by patients with cystic fibrosis. J Pediatr 111(2), 293–9 (1987). [DOI] [PubMed] [Google Scholar]
- 67.Ong T, Schechter M, Yang J, et al. Socioeconomic Status, Smoke Exposure, and Health Outcomes in Young Children With Cystic Fibrosis. Pediatrics 139(2), doi: 10.1542/peds.2016-2730 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kovesi T, Corey M, Levison H. Passive smoking and lung function in cystic fibrosis. AmRevRespirDis 148(5), 1266–71 (1993). [DOI] [PubMed] [Google Scholar]
- 69.Rubin BK. Exposure of children with cystic fibrosis to environmental tobacco smoke. The New England journal of medicine 323(12), 782–8 (1990). [DOI] [PubMed] [Google Scholar]
- 70.Oates GR, Baker E, Rowe SM, et al. Tobacco smoke exposure and socioeconomic factors are independent predictors of pulmonary decline in pediatric cystic fibrosis. J Cyst Fibros, doi: 10.1016/j.jcf.2020.02.004 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cantin AM. Cystic Fibrosis Transmembrane Conductance Regulator. Implications in Cystic Fibrosis and Chronic Obstructive Pulmonary Disease. Annals of the American Thoracic Society 13 Suppl 2, S150–5 (2016). [DOI] [PubMed] [Google Scholar]
- 72.Ni I, Ji C, Vij N. Second-hand cigarette smoke impairs bacterial phagocytosis in macrophages by modulating CFTR dependent lipid-rafts. PloS one 10(3), e0121200 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kopp BT, Thompson R, Kim J, et al. Secondhand smoke alters arachidonic acid metabolism and inflammation in infants and children with cystic fibrosis. Thorax 74(3), 237–46 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kopp BT, Sarzynski L, Khalfoun S, et al. Detrimental effects of secondhand smoke exposure on infants with cystic fibrosis. Pediatr Pulmonol 50(1), 25–34 (2015). [DOI] [PubMed] [Google Scholar]
- 75.***.Kopp BT, Ortega-Garcia JA, Sadreameli SC, et al. The Impact of Secondhand Smoke Exposure on Children with Cystic Fibrosis: A Review. Int J Environ Res Public Health 13(10), doi: 10.3390/ijerph13101003 (2016).***Comprehensive review of the effects of secondhand smoke on CF outcomes.
- 76.Reid LEM, Pretsch U, Jones MC, et al. The acute medical unit model: A characterisation based upon the National Health Service in Scotland. PLoS One 13(10), e0204010 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Smyth A, O’Hea U, Williams G, et al. Passive smoking and impaired lung function in cystic fibrosis. Archives of disease in childhood 71(4), 353–4 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rosenfeld M, Emerson J, McNamara S, et al. Baseline characteristics and factors associated with nutritional and pulmonary status at enrollment in the cystic fibrosis EPIC observational cohort. PediatrPulmonol 45(9), 934–44 (2010). [DOI] [PubMed] [Google Scholar]
- 79.Sanders DB, Emerson J, Ren CL, et al. Early Childhood Risk Factors for Decreased FEV1 at Age Six to Seven Years in Young Children with Cystic Fibrosis. Annals of the American Thoracic Society 12(8), 1170–6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thronicke A, Heger N, Antweiler E, et al. Allergic bronchopulmonary aspergillosis is associated with pet ownership in cystic fibrosis. Pediatric allergy and immunology 27(6), 597–603 (2016). [DOI] [PubMed] [Google Scholar]
- 81.Morrow CB, Raraigh KS, Green DM, et al. Cat and dog exposure and respiratory morbidities in cystic fibrosis. The Journal of pediatrics 165(4), 830–5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lavie M, Shemer O, Sarouk I, et al. Several siblings with Cystic Fibrosis as a risk factor for poor outcome. Respir Med 109(1), 74–8 (2015). [DOI] [PubMed] [Google Scholar]
- 83.Katz SL, Strug LJ, Coates AL, Corey M. Disease severity in siblings with cystic fibrosis. Pediatr Pulmonol 37(5), 407–12 (2004). [DOI] [PubMed] [Google Scholar]
- 84.Slieker MG, van den Berg JM, Kouwenberg J, et al. Long-term effects of birth order and age at diagnosis in cystic fibrosis: a sibling cohort study. Pediatr Pulmonol 45(6), 601–7 (2010). [DOI] [PubMed] [Google Scholar]
- 85.Jessup M, Smyth W, Abernethy G, et al. Family-centred care for families living with cystic fibrosis in a rural setting: A qualitative study. Journal of clinical nursing 27(3–4), e590–e9 (2018). [DOI] [PubMed] [Google Scholar]
- 86.Weber HC, Robinson PF, Saxby N, et al. Do children with cystic fibrosis receiving outreach care have poorer clinical outcomes than those treated at a specialist cystic fibrosis centre? The Australian journal of rural health 25(1), 34–41 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Johnson B, Ngueyep R, Schechter MS, et al. Does distance to a cystic fibrosis center impact health outcomes? Pediatr Pulmonol 53(3), 284–92 (2018). [DOI] [PubMed] [Google Scholar]
- 88.Schechter MS. Reevaluating approaches to cystic fibrosis pulmonary exacerbations. Pediatr Pulmonol 53(S3), S51–S63 (2018). [DOI] [PubMed] [Google Scholar]
- 89.Walicka-Serzysko K, Peckova M, Noordhoek JJ, et al. Insights into the cystic fibrosis care in Eastern Europe: Results of survey. J Cyst Fibros 17(4), 475–7 (2018). [DOI] [PubMed] [Google Scholar]
- 90.Schwarz C, Hartl D. Cystic fibrosis in Europe: patients live longer but are we ready? Eur Respir J 46(1), 11–2 (2015). [DOI] [PubMed] [Google Scholar]
- 91.Burgel PR, Bellis G, Olesen HV, et al. Future trends in cystic fibrosis demography in 34 European countries. Eur Respir J 46(1), 133–41 (2015). [DOI] [PubMed] [Google Scholar]
- 92.Brulle RJ, Pellow DN. Environmental justice: human health and environmental inequalities. Annu Rev Public Health 27, 103–24 (2006). Epub 2006/03/15. [DOI] [PubMed] [Google Scholar]
- 93.Oates GR, Schechter MS. Socioeconomic status and health outcomes: cystic fibrosis as a model. Expert Rev Respir Med 10(9), 967–77 (2016). [DOI] [PubMed] [Google Scholar]
- 94.Taylor-Robinson DC, Smyth R, Diggle PJ, Whitehead M. A longitudinal study of the impact of social deprivation and disease severity on employment status in the UK cystic fibrosis population. PLoS One 8(8), e73322 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Curtis JR, Burke W, Kassner AW, Aitken ML. Absence of health insurance is associated with decreased life expectancy in patients with cystic fibrosis. Am J Respir Crit Care Med 155(6), 1921–4 (1997). [DOI] [PubMed] [Google Scholar]
- 96.Schechter MS, Margolis PA. Relationship between socioeconomic status and disease severity in cystic fibrosis. J Pediatr 132(2), 260–4 (1998). [DOI] [PubMed] [Google Scholar]
- 97.*.Schechter MS, Shelton BJ, Margolis PA, Fitzsimmons SC. The association of socioeconomic status with outcomes in cystic fibrosis patients in the United States. Am J Respir Crit Care Med 163(6), 1331–7 (2001).*Large study examining the effects of public insurance on CF health outcomes.
- 98.Schechter MS, McColley SA, Silva S, et al. Association of socioeconomic status with the use of chronic therapies and healthcare utilization in children with cystic fibrosis. J Pediatr 155(5), 634–9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Krieger N, Waterman P, Chen JT, et al. Zip code caveat: bias due to spatiotemporal mismatches between zip codes and US census-defined geographic areas--the Public Health Disparities Geocoding Project. Am J Public Health 92(7), 1100–2 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jerrett M, Burnett RT, Ma R, et al. Spatial analysis of air pollution and mortality in Los Angeles. Epidemiology 16(6), 727–36 (2005). [DOI] [PubMed] [Google Scholar]
- 101.Zandbergen PA, Chakraborty J. Improving environmental exposure analysis using cumulative distribution functions and individual geocoding. Int J Health Geogr 5:23, doi: 10.1186/1476-072X-5-23 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zandbergen PA. Influence of geocoding quality on environmental exposure assessment of children living near high traffic roads. BMC Public Health 7:37, doi: 10.1186/1471-2458-7-37 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zandbergen PA. Ensuring Confidentiality of Geocoded Health Data: Assessing Geographic Masking Strategies for Individual-Level Data. Adv Med 2014, 567049 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]