

Abstract
This study developed a new rapid transcription activator-like effector nuclease (TALEN) preparation protocol by thoroughly redesigning the widely used Golden Gate TALEN and TAL Effector Kit 2.0. The new protocol can be used to prepare any custom 18-bp binding TALENs in just one day (about 12 h), more rapidly than CRISPR. This protocol used a set of linear monomers, a final TALE-FokI backbone plasmid, and a pipeline to assemble the ready-to-use TALEN expression plasmid, which were all newly developed for this study. The set of linear monomers can be easily produced and reproduced by high-fidelity polymerase chain reaction (PCR) amplification in a 96-well plate using a pair of universal primers. Most important of all, our rapid TALEN construction pipeline can easily obtain many positive colonies with high efficiency (over 80%). By preparing five pairs of TALENs targeting five NF-κB genes (RELA, RELB, CREL,NFKB1, and NFKB2) and editing these genes in different cell lines (293T, HepG2, and PANC1), this study demonstrated that the new protocol has high efficiency, reproducibility, reliability, and applicability. Moreover, this study showed that the fabricated TALEN has much higher editing efficiency than CRISPR. Finally, this study developed a dual-tagging system for simultaneously tagging target proteins and successfully edited cells with a streptavidin-binding peptide (SBP) or AviTag via homology-directed repair, which could have wide applications in protein (antigen) preparation, immunoprecipitation, and a transcription factor chromatin immunoprecipitation assay.
Introduction
A transcription activator-like effector (TALE) is a well-known second-generation genome-editing tool, which has a programmable DNA-binding domain (DBD) consisting of tandem repeats of 34 amino acid (aa) sequences (monomers) with repeat variable diresidues (RVDs).1 The TALE has been widely used to edit a genome by fusing with nuclease (TALEN) (i.e., TALE-FokI).2 Due to its high targeting efficiency,3 TALEN has already been used to produce the universal chimeric antigen receptor T (UCAR-T) cells for clinical cancer immunotherapy.4 However, with the advent of CRISPR in 2013, CRISPR rapidly replaced TALEN due to its simplicity. The Achillesʼ heel of TALEN is that it is time-consuming (5 days), labor-intensive, and a low-efficiency process of vector construction. Although some methods have been developed to overcome this key limitation,2b,5 they still did not make TALEN as easy as CRISPR due to its wide reapplication. Therefore, new TALEN preparation methods as easy as CRISPR are still in urgent need.
Attracted by its high specificity, we have ever tried to construct TALEs using a widely used TALE assembly kit, the Golden Gate TALEN and TAL Effector Kit 2.0 (Addgene Kit 2.0);2b however, we found that it was very difficult to obtain a usable TALE plasmid in 5 days. Therefore, to provide a TALE assembly protocol as convenient as CRISPR, we decided to improve the TALE assembly kit and pipeline. By fabricating a new set of linear monomers and a TALE-VP64/VPR backbone vector and designing a new assembly pipeline, we developed a new convenient and high-efficiency TALE preparation protocol.6 Using this protocol, variant TALE-VP64/VPR vectors could be easily prepared with high efficiency in 2 days, which is the same as CRISPR for activating various exogenous and endogenous genes.6 This TALE-VP64/VPR preparation protocol thus provides a new applicable alternative to the current various CRISPR/Cas9-sgRNA-based gene activators.
Although CRISPR is the current widely used genome-editing tool, the off-targeting still challenges its clinical application.7 By contrast, off-target activity appears to be less of an issue for TALEN. TALENs are often built with 34 aa sequences that are repeated 18 times. Due to the requirement of FokI dimerization for its activity, TALEN often edits a target site with a pair of TALEN proteins that orient oppositely. The binding sites of the two TALEN proteins often space 14–20 nucleotides. Therefore, TALEN has in fact a long (about 36 bp) DNA-binding site that is rarely found in genomes. Although some degeneracy exists in the RVD–DNA-binding code,8 TALEN rarely tolerates mismatch. For instance, a previous study revealed that TALEN showed no off-target activity in the human-induced pluripotent stem (iPS) cells.3 TALEN is known to have its relatively unconstrained target site requirements and a high degree of specificity. Therefore, TALEN has a significant advantage over CRISPR/Cas9 in clinical application and still serves as an indispensable alternative tool for genome editing.
Because the widest application of the TALE technique is TALEN as a genome-editing tool, in this study, based on our developed TALE assembly kit and pipeline, we constructed a ready-to-use TALE-FokI (TALEN) backbone plasmid and further simplified the TALEN assembly pipeline. We thus developed a new high-efficiency TALEN preparation protocol that is easier to use than CRISPR. Using this new TALEN preparation protocol, any custom TALEN expression vectors can be easily and rapidly prepared in just one day at very high efficiency. By preparing five pairs of TALENs targeting five genes of the NF-κB family and editing these genes in three types of human cell lines, we demonstrated that the constructed TALENs could more efficiently edit all genes in all cells than CRISPR. Moreover, in this study, we developed a dual-tagging system for simultaneously tagging the target protein and successfully edited cells with the streptavidin-binding peptide (SBP) or AviTag via homology-directed repair (HDR). This new HDR dual-tagging donor could have wide applications in protein (antigen) preparation, immunoprecipitation, and chromatin immunoprecipitation (ChIP) assays.
Results
Preparation of Monomers and TALEN Backbone Vectors
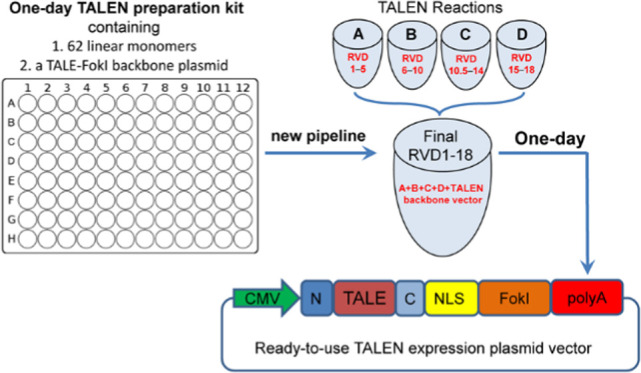
A new set of new linear monomers (Figure 1A) was prepared from plasmids of the Golden Gate TALEN and TAL Effector Kit 2.0 (Addgene) by polymerase chain reaction (PCR) amplification using a set of PCR primers (Table S1).6 Additionally, two new linking monomers, dsDNA10.5 and dsDNA17.5 (Figure 1A; Table S2), were designed. All of the linear monomers produced can then be reproduced by amplifying with a pair of universal primers (PCR-TAL-F/R; Table S1) in a 96-well PCR plate. This set of linear monomers can be used to construct any custom TALENs with 18-bp target sequences (Figure 1B). For preparing functional TALENs, a new TALEN (TALE-FokI) backbone plasmid was also constructed, which harbors a LacZ expression cassette to easily screen the final positive TALEN colony (white colony) with a blue-white screen (Figure 1C). A strong cytomegalovirus (CMV) promoter was used to enhance the expression of TALENs in mammalian cells (Figure 1C).
Figure 1.

Schematic of the new TALEN protocol. (A) Schematic of monomers in a 96-well PCR plate. There are a total of 60 base-determinant monomers and 2 linker monomers (dsDNA10.5 and dsDNA17.5). A TALEN backbone (TALE-FokI) plasmid was also constructed. (B) Structure of TALENs expression plasmid. (C) Structures of linear monomers and the TALEN backbone plasmid. All monomers can be reproduced by a 96-well PCR amplification using a pair of universal primers. CMV: cytomegalovirus promoter; N and C, nonrepetitive amino and carboxyl terminals of TALEN; BsmBI: the cutting site of type IIs restriction enzyme BsmBI used for inserting the custom TALEN DNA-binding domain; LacZ: LacZ expression cassette for a blue-white screen; NLS: nuclear localization signal; FokI: catalytic domain from the FokI endonuclease. (D) Schematic of DNA cut with TALEN (left and right TALENs), which can produce site-specific double-strand breaks (DSB) that can enhance DNA HDR. (E) Pipeline for constructing custom TALEN with linear monomers and the TALEN backbone plasmid. Time used in each step was given. The whole procedure can be completed in one day. TALENs ready to transfect mammalian cells can be obtained in one day. GG, Golden Gate.
Pipeline for Constructing TALEN with New Plasmid-Free Monomers
With the newly designed and produced linear monomers (PCR fragments) and TALEN backbone vector (Figure 1A–D), we designed a new pipeline to rapidly construct the custom TALEN in one day (Figure 1E). As with the Addgene kit 2.0, the pipeline also assembles a custom TALEN using the Golden Gate method, in which different cutting sites of two type IIs restriction enzymes, BsaI and BsmBI, are used to cut linear monomers, pentamers, and TALEN backbone vector. For constructing a custom TALEN, four digestion–ligation reactions were first built with monomers, BsaI, and T4 DNA ligase, which produced four circular pentamers. Then, another digestion–ligation reaction was built with four pentamers, TALEN backbone vector, BsmBI, and T4 DNA ligase, which produced the final TALEN vector that can be directly used to edit the genome. The final TALEN plasmid vector can be obtained by bacterial transformation, with or without colony PCR identification, and plasmid extraction. The positive final TALEN plasmid vector can be further rapidly confirmed by EcoRI digestion. In comparison with this pipeline of rapid TALEN plasmid preparation, the CRISPR/Cas9-sgRNA expression plasmid has to be prepared by at least 3 days (Figure S1).
Construction of TALENs Targeting NF-κB Genes
To verify the new TALEN assembly pipeline and gene editing function, we assembled TALENs targeting NF-κB following the assembling steps described in Methods, named RELA-left TALEN, RELA-right TALEN, RELB-left TALEN, RELB-right TALEN, CREL-left TALEN, CREL-right TALEN, NFKB1-left TALEN, NFKB1-right TALEN, NFKB2-left TALEN, and NFKB2-right TALEN. The final TALENs were transformed Escherichia coli (E. coli), and it was cultivated on solid agar. The results showed that many white spots were produced (Figure 2A). No blue spot was seen on the agar plate, indicating the high digestion–ligation efficiency of our protocol. The colony PCR detection of randomly picked colonies confirmed the insert size and also revealed that typically over 80% of the colonies were bona fide positive colonies (Figure 2B), indicating the high efficiency of constructing the custom TALEN with our method (Figure 1). The subsequent EcoRI digestion of extracted plasmids further confirmed the results of colony PCR, which indicates that typically 80% of the white spots are the correct and successfully assembled final TALENs (Figure 2B).
Figure 2.

Constructing custom TALENs with a new TALEN protocol. (A) TALEN plasmid-transformed E. coli colonies grew on an agar plate. There were few blue colonies in all plates. (B) Colony PCR detection of randomly picked white colonies. Eight colonies were detected for two RELA TALENs, and four colonies were detected for two TALENs of RELB, CREL, NFKB1, and NFKB2. The full-length PCR products are 2051 bp long. The ladder effect indicates a successful assembly. All colony PCR-detected colonies were cultivated to extract plasmids. The obtained plasmids were digested with EcoRI in which the positive colonies produced a full-length 3537-bp TALEN fragment, but the negative colonies (without inserted sequence) produced a 2143-bp band. (C) PCR and EcoRI detections of TALENs prepared with (up) or without (down) colony screening. (D) Two colonies were screened for each CRISPR/Cas9-sgRNA. M1: DL2000 DNA marker (Code No.3427A; Takara); M2: DL10000 DNA marker (code no. 3584A; Takara); and M3: 100-bp DNA ladder (code no. 3422A; Takara).
Due to the highly positive colonies, to further save time, we removed the colony screening step from the pipeline. After the bacteria were transformed by the TALEN plasmid, they were directly cultivated in 5 mL of liquid media containing antibiotics for 4 h (Figure 1E). The plasmids were then extracted from the whole 6 mL of bacteria. The bacteria and plasmids could also be confirmed by the colony PCR amplification and EcoRI digestion (Figure 2C). In this way, as many as over 25 μg of plasmid could be produced for each TALEN (Figure S2), which is sufficient for the transfection of as many as 6.25 × 107 cells (125 wells of a 24-well plate with 5 × 105 cells/well). This method saves the overnight culture time of bacteria on agar. In this way, the TALEN plasmid needed for cell transfection could be obtained in one day (Figure 1E).
With the TALEN expression vectors targeting NF-κB genes (Figure 2C), to compare the TALEN editing with the CRISPR/Cas9 editing, we also constructed Cas9/sgRNA expression vectors targeting the same five NF-κB genes, named RELA-sgRNA-Cas9, RELB-sgRNA-Cas9, CREL-sgRNA-Cas9, NFKB1-sgRNA-Cas9, and NFKB2-sgRNA-Cas9 (Figure 2D).
Investigation of Editing Efficiency with a Dual-Tagging Homologous Recombination
To check the reliability of our rapid TALEN preparing protocol and compare the editing effect of two editing tools, we inserted a dual-tagging DNA fragment at the end of the five genes of the NF-κB family by HDR. Five pairs of TALENs and five sgRNA-CRISPR/Cas9 were prepared targeting these genes. We prepared two kinds of linear HDR donors, SBP-IRES2-displaySBP and AviTag-IRES2-displayAviTag, in which the SBP/AviTag was fused to the C-terminal of the target gene for tagging the target protein, and the displaySBP/displayAviTag was used to display the SBP/AviTag on the cell surface for tagging cells (Figure 3A). If the HDR donors were successfully inserted into target genes, the dual tags could be expressed under the control of promoters of the target genes. The displayed SBP/AviTag could be detected using the fluorescently labeled streptavidin. The cells with the displayed SBP/AviTag could also be separated with streptavidin-coupled beads such as Dynabeads M-280 Streptavidin. The cell transfection with AviTag also needed a plasmid expressing pMy-BirA for biotinylating AviTag.
Figure 3.

Editing NF-κB RELA with TALEN and CRISPR. (A) Schematic HDR donor with two different dual-tagging systems and their tagging cells and target protein. SBP, streptavidin-binding peptide; SAV, streptavidin; HOM, homology arm; UTR, untranslated region; IRES2, internal ribosome entry site 2. (B–D) Editing NF-κB RELA with TALEN and CRISPR in three cell lines and detecting with near-infrared fluorescence (NIRF) imaging. The edited gene was repaired with a homologous donor with two different dual-tagging systems. (B) IRDye800CW-conjugated streptavidin-stained cells in wells. (C) Quantified NIRF intensity of the wells. (D) NIRF imaging of cells collected from wells by trypsinization. Up, NIRF imaging; down, bright field imaging. **p < 0.01 and *p < 0.05.
To evaluate the editing effects of two editing tools, we first cotransfected three different cell lines, 293T, HepG2, and PANC1, with TALEN/CRISPR targeting NF-κB RELA and two kinds of HDR donors. Then, the transfected cells were cultivated for 48 h and stained with IRDye800CW-conjugated streptavidin. The stained cells were imaged using a NIRF imager. The results indicated that the three cell lines were all successfully edited by both TALEN and CRISPR using two kinds of HDR donors (Figure 3B–D); however, the editing efficiency of RELA-TALEN was much higher than that of RELA-sgRNA-Cas9 in all cell lines (Figure 3B–D). Moreover, the HDR donor SBP-IRES2-displaySBP was superior to the AviTag-IRES2-displayAviTag (Figure 3B–D).
To further quantitatively evaluate the effects of the two editing tools and HDR donors, we performed three biologically replicated HepG2 and PANC1 transfections with TALEN/CRISPR targeting NF-κB RELA and two kinds of HDR donors. The transfected cells were stained with Alexa Fluor 488-conjugated Streptavidin and then quantitatively analyzed by flow cytometry. The results also indicated that the editing efficiency of TALEN was much higher than that of CRISPR/Cas9 (Figure 4A–C) and the HDR donor SBP-IRES2-displaySBP was better than the AviTag-IRES2-displayAviTag (Figure 4A–C). The cells were also quantitatively analyzed in another way, if flow cytometry was not available, in which the collected cells were dropped on slides and imaged using a fluorescence microscope in both the light field and fluorescence channel, and then the cells were counted using ImageJ (Figure 4D).
Figure 4.

Editing NF-κB RELA with TALEN and CRISPR. (A) Editing NF-κB RELA with TALEN and CRISPR in HepG2 cells and PANC1 cells and detecting with visible fluorescence. The edited gene was repaired with a homologous donor with two different dual-tagging systems. The cells were stained with Alexa Fluor 488-conjugated Streptavidin and imaged using a fluorescence microscope. The cells were imaged at 100× magnification. (B) Quantitative analysis of the Alexa Fluor 488-conjugated Streptavidin-stained cells by flow cytometry. (C) Statistical results of flow cytometry analysis. ***p < 0.001. (D) Statistical results of ImageJ analysis. ***p < 0.001.
Although the length of the SBP-IRES2-displaySBP (1076 bp) and the AviTag-IRES2-displayAviTag (1028 bp) is similar, a vector expressing biotin ligase pMy-BirA has to be cotransfected when using the AviTag-IRES2-displayAviTag. Subsequently, the more concise HDR donor SBP-IRES2-displaySBP was selected for further investigation. We transfected the HepG2 cells with the HDR donor SBP-IRES2-displaySBP and TALENs targeting five genes of the NF-κB family, respectively, to check the TALEN ability to edit different genes. The transfected cells were similarly stained with Alexa Fluor 488-conjugated Streptavidin and then quantitatively analyzed by flow cytometry. The results indicated that all five genes were efficiently edited by the TALENs that were prepared using the new pipeline (Figure 5).
Figure 5.

Editing five NF-κB genes with TALEN and CRISPR. (A) Editing five NF-κB genes with TALEN and CRISPR in HepG2 cells and detecting with visible fluorescence. The edited gene was repaired by the homologous donor of the SBP dual-tagging system. The cells were stained with Alexa Fluor 488-conjugated Streptavidin and imaged using a fluorescence microscope. The cells were imaged at 100× magnification. (B) Quantitative analysis of the Alexa Fluor 488-conjugated Streptavidin-stained cells by flow cytometry. The control cells only transfected with lipofectin showed no fluorescence (Figure S3).
To further confirm the TALEN editing and compare it with CRISPR, we detected the TALEN/CRISPR edited cells by qPCR that amplified the edited genes with a pair of primers annealing with the target gene and fused SBP coding sequences, respectively. The gDNA was extracted from the transfected cells, and the same amount of DNA was used as PCR templates. The results indicated that all five genes were successfully edited and repaired by the HDR donor (Figure 6A). The HDR donor was correctly fused to target genes (Figure 6B). TALEN showed a much higher editing efficiency than CRISPR in all five genes (Figure 6C).
Figure 6.
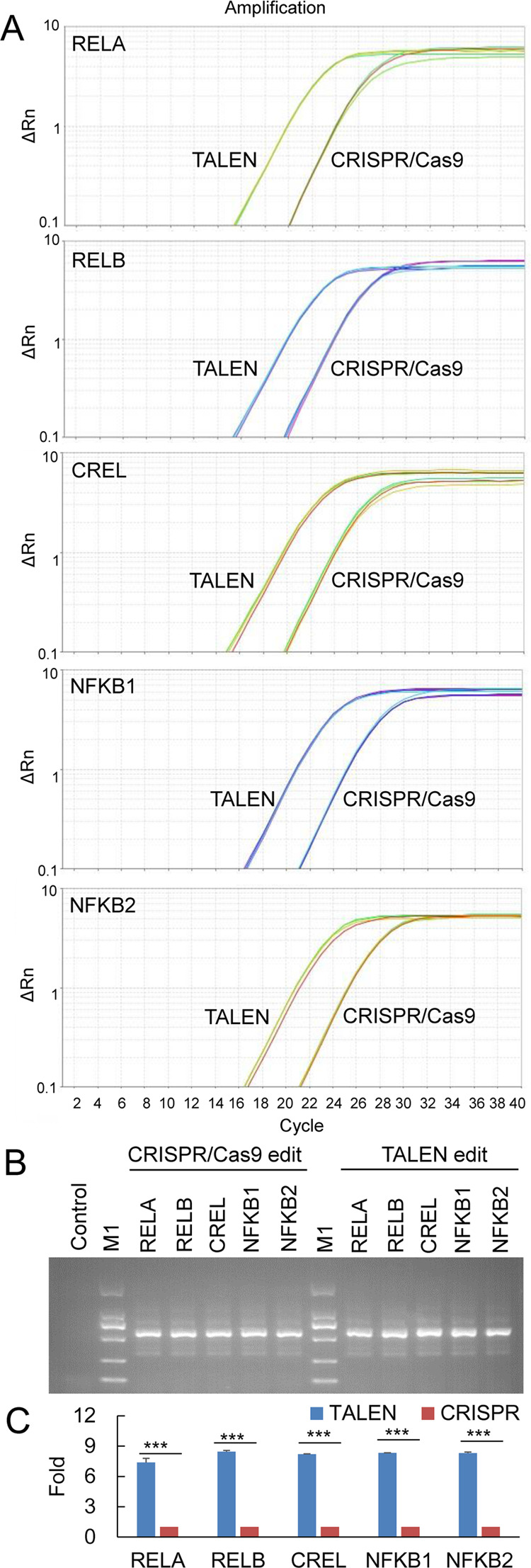
Editing five NF-κB genes with TALEN and CRISPR. (A) Editing five NF-κB genes with TALEN and CRISPR in HepG2 cells and detecting with qPCR. The edited gene was repaired by the homologous donor of the SBP dual-tagging system. The gDNA was prepared from the edited cells and detected by qPCR using a pair of primers, respectively, annealing with the target gene and inserted tag. (B) Visualization of the qPCR products by agarose gel electrophoresis. M1: DL2000 DNA marker (code no. 3427A; Takara). (C) Statistical results of qPCR detection. Fold, the fold of editing efficiency, calculated as (CtTALEN/CtCRISPR) × 10. ***p < 0.001.
Screening of Positive Cells and Validation of NF-κB Proteins
Using the HDR donors, a tag peptide SBP/AviTag was displayed on the surface of successfully edited cells. Not only are the displayed tags useful for easily detecting editing efficiency (as we did above) but also could be used to screen positive cells. We isolated the positive cells using the streptavidin-coupled magnetic beads (Dynabeads M-280 Streptavidin) (Figure 7A). The bead-captured cells were cultivated and again stained with Alexa Fluor 488-conjugated Streptavidin (Figure 7B) and analyzed by flow cytometry (Figure 7C). It was found that the positive cells were highly enriched (increased from 10 to 30%) after a single round of magnetic isolation. Of course, the positive cells can also be enriched by flow cytometry with an isolation function. Finally, to further validate the target protein tagging, we treated the enriched cells with TNFα. The nuclei were isolated from cells and detected by immunohistochemistry using Alexa Fluor 488-conjugated Streptavidin. The results indicated that the TNFα treatment induced a significant nuclei translocation of SBP-tagged NF-κB RELA (Figure 7D).
Figure 7.

Magnetic isolation of edited cells. The HepG2 cells edited by TALENs targeting five NF-κB genes and repaired by the homologous donor of the SBP dual-tagging system. (A) Cells isolated with streptavidin-coated magnetic beads. (B) and (C) Enriched cells were cultured for 48 h and stained with Alexa Fluor 488-conjugated Streptavidin. The stained cells were imaged using a fluorescence microscope (B) and quantitatively analyzed by flow cytometry (C). (D) Detection of NF-κB RELA tagging by staining nuclei of the edited cells with Alexa Fluor 488-conjugated Streptavidin. The stained cells were imaged using a fluorescence microscope (left) and quantitatively analyzed by flow cytometry (right). The edited cells were treated with TNFα for NF-κB RELA activation. The cells were imaged at 100× magnification.
Discussion
In this study, we edited the NF-κB RELA gene with both TALEN and CRISPR/Cas9 in three different cell lines, 293T, HepG2, and PANC1, and edited five different NF-κB genes, including RELA, RELB, CREL, NFKB1, and NFKB2, in HepG2 cells. The results indicated that TALEN showed much higher editing efficiency than CRISPR in all edited cells and genes. These results demonstrate that TALEN is a better gene-editing tool than CRISPR in regard to editing efficiency. However, TALEN was rapidly replaced by CRISPR due to the simplicity of the latter. TALEN lost due to its cumbersome, time-consuming, and low-efficiency construction process of the expression plasmid. In this study, we focused on addressing the problem. We ultimately established a new TALEN preparation protocol, which consists of a set of linear monomers, a final TALE-FokI (TALEN) backbone plasmid, and a one-day rapid TALEN construction pipeline. The set of linear monomers can be easily reproduced by high-fidelity PCR amplification in a 96-well plate using a pair of universal primers. Our rapid TALEN construction pipeline can obtain many usable colonies at high efficiency (over 80%). Moreover, using the pipeline to prepare the TALEN expression plasmid just in one day (Figure S2), we found that as many as over 25 μg of plasmid could be obtained for each TALEN. This amount of plasmid is sufficient for the transfection of as many as 6.25 × 107 cells, from which as many as 6 × 106 positive cells (i.e., successfully edited cells) can be obtained according to the editing efficiency of about 10%.
In this study, we used an HDR dual-tagging system to evaluate the editing efficiency of two editing tools, in which the successfully edited cells can be labeled by an SBP tag or AviTag displayed on the cell surface. The successfully edited cells were easily found by fluorescent streptavidin staining and quantitatively analyzed by flow cytometry. Moreover, it is worth emphasizing that we found that the fluorescent streptavidin (both IRDye800CW-conjugated streptavidin and Alexa Fluor 488-conjugated Streptavidin) can be directly added to the cells cultural media to stain cells. The stained cells can then be easily quantitatively analyzed and enriched by flow cytometry at the same time.
In this study, all studied genes were successfully edited in all detected cells by two editing tools, and TALEN showed much higher editing efficiency than CRISPR/Cas9. However, it seems that the total editing efficiency of both tools is not high. The TALEN only obtained about 10% editing efficiency and CRISPR only 3%. This low editing efficiency is related to our characterization of editing efficiency with HDR. In fact, two kinds of repair systems, nonhomologous end joining (NHEJ) and HDR, functioned in cells with the TALEN/CRISPR-mediated DNA DSB. Generally, the NHEJ is prior to the HDR; however, the NHEJ is not detected in our approach. The NHEJ has to be detected by other methods such as PCR amplification in combination with the T7 endonuclease I (T7EI) assay or sequencing. Anyway, NHEJ detection is beyond the interest of this study. This study only detected the cells successfully repaired by HDR. Therefore, the true editing efficiency should be higher than observed.
In this study, we developed a dual-tagging system, SBP-IRES2-displaySBP and AviTag-IRES2-displayAviTag, in which the SBP/AviTag was fused to the C-terminal of the target gene for tagging the target protein, and the displaySBP/displayAviTag was used to display the SBP/AviTag on the cell surface for tagging cells. We demonstrated that both HDR donors worked effectively in the studied cells. However, the SBP-IRES2-displaySBP is more concise and efficient. Moreover, a more plasmid expressing biotin ligase (pMy-BirA) has to be cotransfected when using the AviTag-IRES2-displayAviTag. We found that the more concise the HDR donor SBP-IRES2-displaySBP was the most ideal the HDR donor was for future applications. Nevertheless, the high binding affinity between biotin and streptavidin (Kd = 10–14 mol/L9) may make the AviTag-IRES2-displayAviTag a good choice in future applications. In this study, to avoid the construction of homology arm vectors using 700–800 nt,10 we used a shorter homologous arm of 35 nt,11 making it easier for us to prepare the HDR donor SBP/AviTag-IRES2-displaySBP/AviTag for other interested genes.
Currently, the widely used approach to isolate the successfully edited cells is drug screening, which is very time-consuming and very difficult to obtain the pure positive cell colonies. Long-term drug treatment also seriously affected cell growth. Our dual-tagging system provides a drug-free cell screening method. Our study revealed that the positive cell could be rapidly isolated from the edited cell pools without damaging cell viability. An easy single-round magnetic isolation increased the percentage of positive cells to 30%.
The target protein purification is important to various life science fields. The purified protein can be used as an antigen to produce antibodies that can interact with proteins in its natural state or structure, such as ChIP-grade antibodies. The purified proteins is essential for analyzing their structure by X-ray crystallography or cryo-electron microscopy. However, many proteins do not have their immunoprecipitation (IP)-grade antibodies. These proteins have to be produced by constructing a vector that expresses them in prokaryotic or eukaryotic cells, in which a tag such as the His tag has to be fused at N-terminals or C-terminals for purification. However, the proteins expressed in prokaryotic cells may have lost their natural modification. In our dual-tagging system, an SBP tag was fused to the C-terminal of the target protein, in which a GS linker was used. The SBP tag is a 38-residue peptide with higher binding affinities for streptavidin.12 The SBP tag can interact with streptavidin at a high binding affinity (Kd = 10–10 mol/L),13 which is higher than the binding affinity of most antibodies (Kd = 10–6–10–9 mol/L). The SBP tag is helpful for purification of the target protein. A cut site (such as thrombin) can also be placed before the GS linker, and the target protein can easily be cut down from streptavidin-coated magnetic beads to remove the GS linker and the SBP tag from the purified target protein. This kind of protein is more useful for analyzing the structure and antibody production.
Our study edited five genes of the NF-κB family, the SBP-tagged transcription factors (TFs) in cells can be used to enrich their binding DNA via ChIP without ChIP-grade antibodies. Many TFs do not have their commercially available ChIP-grade antibodies for identifying their target DNA and genes to clarify their function in gene regulation. Our dual-tagging system may provide a practicable antibody-free ChIP assay for these TF proteins. In this case, the cells can be transfected with a pair of TALENs targeting the interested TF and the HDR donor SBP-IRES2-displaySBP. The transfected cells can then be enriched with streptavidin-coated magnetic beads. The nuclei can be subsequently isolated from the enriched cells, which can then be used to prepare chromatin for precipitating the target TF and their complex DNA and other proteins using streptavidin-coated magnetic beads. The transfected cells can also be directly used to complete the ChIP assay without cell enrichment.
A limitation of the new TALEN protocol is that it can only be used to assemble TALENs binding an 18-bp sequence. This study focused on TALENs of this length because TALENs typically consist of 34 amino acid sequences repeated 18 times. With the constant T base, TALENs constructed with the new protocol can bind 19-bp target sequences in genomes. This is a frequently used length of TALEN with high specificity. This study revealed that different target genes could be effectively edited with the constructed 19-bp binding TALENs in various cells. In addition, using the same strategy to make linear monomers in this study, other monomers can be similarly manufactured for preparing longer TALENs if needed.
Conclusions
This study developed a new rapid TALEN preparation protocol, which can be used to prepare any ready-to-use custom TALEN expression plasmid vectors in just one day (about 12 h). The prepared TALEN expression plasmid vectors can express TALEN proteins that can bind an 18-bp binding site. This protocol used a set of linear monomers, a final TALE-FokI backbone plasmid, and a pipeline to assemble a ready-to-use TALEN expression plasmid, which were all newly developed in this study. By preparing five pairs of TALENs and editing five NF-κB genes in different cell lines, this study demonstrated that the new protocol has high efficiency, reproducibility, reliability, and applicability. This study also showed that TALEN has much higher editing efficiency than CRISPR. Additionally, this study also developed a dual-tagging system for simultaneously tagging target proteins and successfully edited cells, which has wide applications in protein (antigen) preparation, immunoprecipitation, and the TF ChIP-seq assay.
Experimental Section
Preparation of Linear TALEN Monomers
The linear TALEN monomers were prepared using the Golden Gate TALEN and TAL Effector Kit 2.0 (Addgene Kit 2.0; Addgene) by PCR amplification with primers and oligos listed in Tables S1 and S2 as we previously described.6 The prepared total of 62 linear TALEN monomers were arrayed in a 96-well plate as PCR templates. The monomers were reproduced by a 96-well PCR amplification using a pair of universal primers (PCR-TAL-F/R; Table S1). All PCR reactions (240 μL) contained 5 μL linear monomer, 10 μM PCR-TAL-F, 10 μM PCR-TAL-R, and 1× PrimeSTAR HS DNA polymerase (Takara). The PCR program was 96 °C for 3 min, 28 cycles of 96 °C for 20 s, 72 °C for 30 s, and 72 °C for 3 min. The PCR products were detected by 1% agarose gel electrophoresis. The PCR products were purified by phenol/chloroform extraction and precipitated with anhydrous ethanol. The PCR products were resuspended in water.
Construction of TALEN Backbone and sgRNA-CRISPR/Cas9 Plasmids
The FokI fragment was cloned from pTAL3-His (Addgene Kit 2.0) by PCR amplification, in which the EcoRI and NotI sites were introduced up- and down-stream. The PCR reaction (30 μL) consisted of 1 ng of pTAL3-His, 10 μM of FokI-F (Table S3), 10 μM of FokI-R (Table S3), and 1× PrimeSTAR HS DNA polymerase. The PCR program was 96 °C for 3 min, 28 cycles of 96 °C for 20 s, 58 °C for 20 s, 72 °C for 30 s, and 72 °C 3 min. The cloned fragment was ligated into pPIRES2-EGFP (Takara). The prepared plasmid was named pPIE-FokI (Supporting Information). The pPIE-FokI was verified by DNA sequencing. The TAL (LacZ) fragment was cut out from pTAL2 using the Golden Gate TALEN and TAL Effector Kit 2.0 with BamHI and ligated into PIE-FokI. The prepared plasmid was named as the TALEN backbone plasmid (Supporting Information).
A Cas9 and sgRNA coexpression plasmid was constructed for transfecting mammalian cells. A lac operon sequence with BbsI and BsaI (New England Biolabs) sites at both ends was cloned from the empty pEASY-Blunt-simple (Transgen, CB101–01) by PCR amplification with PrimeSTAR HS DNA polymerase using primers Lac-px-F and Lac-px-R (Table S3). This lac operon sequence was ligated into px459 (Addgene plasmid ID: 62988) to construct px459-lac (Supporting Information). The custom Cas9 and sgRNA coexpression plasmid can be constructed with px459-lac via a blue-white screen. The px459-lac was verified by sequencing.
Preparation of Homology Arm Cloning
Five genes of the NF-κB family, including RELA, RELB, CREL, NFKB1, and NFKB2 were chosen as targets. The SBP-IRES2-displaySBP and AviTag-IRES2-displayAviTag dual-tagging donors were synthesized (Supporting Information). Homology arms (only 35 nt) targeting various genes were designed as the terminals of the primers (Table S3) for amplifying dual-tagging donors by PCR. The PCR reaction (30 μL) contained 1 ng of SBP-IRES2-displaySBP or AviTag-IRES2-displayAviTag, 10 μM of hom-F (Table S3), 10 μM of hom-R (Table S3), and 1× PrimeSTAR HS DNA polymerase. The PCR program was 96 °C for 3 min, 28 cycles of 96 °C for 20 s, optimal annealing temperature for 20 s and 72 °C for 1.5 min, and 72 °C for 5 min. The PCR products were purified using a Gel Extraction Kit.
Design of TALEN and CRISPR Targets
A pair of TALENs (left and right TALENs) was designed for each gene by an online program TAL Effector Nucleotide Targeter 2.0 (https://tale-nt.cac.cornell.edu/) (Table S4 and Figure S4). The Cas9/sgRNA target sites were designed using the online program CHOPCHOP (https://chopchop.rc.fas.harvard.edu/) (Table S5 and Figure S4).
Construction of Custom 18-bp TALENs
All TALENs with an 18-bp binding site (Table S4) were prepared in two steps. First, Four Golden Gate reactions, including N1-N2-N3-N4-N5, N6-N7-N8-N9-N10, dsDNA10.5-N11-N12-N13-N14, and N15-N16-N17-dsDNA17.5-N18, were performed to produce four pentamers. Each reaction (20 μL) contained 10 U BsaI, 400 U T4 DNA ligase (New England Biolabs), 1× T4 DNA ligase buffer, 2 μg bovine serum albumin (BSA), and 200 ng each of five monomers. The reactions were incubated as follows: 3 cycles of 37 °C for 5 min and 16 °C for 10 min; 50 °C for 5 min, and 80 °C for 5 min. Into each reaction were then added 1 μL of Plasmid-Safe nuclease (10 U/μL; Epicenter Biotechnologies) and 1 μL of ATP (10 mM). The reactions were incubated at 37 °C for 15 min and 70 °C for 15 min. Second, a new Golden Gate reaction was performed to prepare the final TALEN vector. The reaction (20 μL) contained 75 ng TALEN backbone plasmid, 10 U BsmBI (New England Biolabs), 400 U T4 DNA ligase, 1× T4 DNA ligase buffer, and 200 ng each of four pentamers. The reactions were incubated as follows: 3 cycles of 37 °C for 5 min and 16 °C for 10 min; 50 °C for 5 min, and 80 °C for 5 min. The reaction (20 μL) was then used to transform the competent E. coli DH5α (Tiangen, China). Then, 800 μL of lysogeny broth (LB) media was added to the transformed DH5α (200 μL) and incubated at 37 °C for 1 h. The transformed DH5α (100 μL) was then cultured on agar containing 100 μg/mL of kanamycin overnight at 37 °C. The plate was imaged using a Bio-Rad gel imager (Gel Doc XR + imaging system). The colonies were then identified by colony PCR using primers TAL-F and TAL-R (Table S1). The colony PCR reaction (30 μL) contained 3 μL of colony suspension, 1× Premix Taq (Takara), 10 μM of TAL-F, and 10 μM of TAL-R. The PCR program was 96 °C for 10 min, 30 circles of 96 °C for 20 s, 55 °C for 20 s and 72 °C for 3 min, and 72 °C for 10 min. The PCR products (10 μL) were run with 1% agarose gel in 1× Tris-acetate–EDTA (TAE). The positive colonies were cultured in liquid LB media containing 100 μg/mL kanamycin and plasmids were extracted using a QIAprep Spin Miniprep Kit (Qiagen) following the manufacturer’s instructions. The plasmids were further confirmed by EcoRI digestion. The EcoRI reaction contained 1 μg TALEN plasmid, 15 U EcoRI, and 1× EcoRI buffer. The reaction was incubated at 37 °C for 30 min. The reaction was detected with 1% agarose gel in 1× TAE. The plasmids used for transfecting mammalian cells were extracted with the EndoFree Plasmid kit (CWBio, China) according to the manufacturer’s instruction.
For preparing the TALEN expression plasmid just in one day, at the end of the second Golden-gate cut-ligation reaction, a total of 20 μL of the reaction solution was then used to transform the competent E. coli DH5α. The transformed DH5α (200 μL) was added into 800 μL of LB media and incubated at 37 °C for 1 h. Then, a total of 1 mL of bacterial fluid was added into 5 mL of LB media containing 100 μg/mL of kanamycin and cultured at 37 °C for 4 h. The bacterial fluids were directly detected by PCR as described above. At the same time, plasmid DNA was extracted from the whole 6 mL of culture media using the EndoFree Plasmid kit. A total of 120 μL of DNA (containing over 25 μg of plasmid DNA) solution was obtained for each TALEN. The plasmid DNA was quantified using a NanoDrop. The extracted TALEN expression plasmids were also confirmed by EcoRI digestion.
Construction of sgRNA-CRISPR/Cas9 Targeting NF-κB
For preparing CRISPR/Cas9 vectors, to generate a single Cas9-sgRNA-puroR expressing vector, a modified pX459-lac expression vector expressing CMV-Cas9-puroR and sgRNA was linearized with BbsI digestion, and gel purified. A pair of oligos (Table S6) for each targeting site was annealed and ligated to the linearized pX459-lac. SgRNA was inserted into px459-lac for PCR validation and sequencing. We constructed five sgRNA-CRISPR/Cas9 plasmids targeting the NF-κB family, named RELA-sgRNA-Cas9, RELB-sgRNA-Cas9, CREL-sgRNA-Cas9, NFKB1-sgRNA-Cas9, and NFKB2-sgRNA-Cas9. To prepare the plasmids used to transfect mammalian cells, the competent E. coli DH5α was transformed with the constructed sgRNA-CRISPR/Cas9 plasmids, respectively. All plasmids were extracted from the cultures of the colony PCR-verified as positive colonies using the EndoFree Plasmid kits according to the manufacturer’s instruction. All plasmids were further verified by DNA sequencing.
Cell Culture and Transfection
293T, HepG2, and PANC1 cells (Shanghai institutes for biological sciences, Chinese Academy of Sciences; China) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (HyClone) containing 10% fetal bovine serum (FBS) (HyClone), 100 U/mL penicillin, and 100 μg/mL streptomycin in a humidified incubator with 5% CO2 at 37 °C. The cells were seeded in a 24-well plate with a density of 5 × 104 cells/well, and incubated for 24 h. The cells were then transfected with plasmids using Lipofectamine 2000 (Invitrogen). All vectors used for transfection were isolated using the EndoFree Plasmid kits. Before transfection, the medium was removed carefully. HepG2 and PANC1 cells were then rinsed with phosphate-buffered saline (PBS), but the 293T cells must not be rinsed with PBS. Subsequently, 500 μL of opti-MEM (Invitrogen) containing 2 μg of Lipofectamine 2000 and 800 ng of plasmid was added to each well of the 24-well plates. The cells were incubated for 4 h. The transfection media were removed, and the fresh complete medium was added. The cells were cultured for an additional 24 or 48 h.
Detection of the Efficiency of Gene Editing
293T, HepG2, and PANC1 cells cultivated in the 24-well plates were cotransfected with 400 ng of TALEN vectors (200 ng left TALEN and 200 ng right TALEN) and 400 ng of homology arm donor. 293T, HepG2, and PANC1 cells cultivated in the 24-well plates were cotransfected with 400 ng of sgRNA-CRISPR/Cas9 and 400 ng of homology arm donor. The cells were cultured for an additional 24 h. When the donor AviTag-IRES2-displayAviTag was used, the cells were cotransfected with an additional 100 ng pMy-BirA (Addgene).
One microliter of the IRDye800CW-conjugated streptavidin (1 mg/mL stock; Li-COR Bioscience) was added to the cells in the wells and cultivated for 4 h. The cells were rinsed with PBS and imaged using the Odyssey Infrared Imaging System (Li-COR Bioscience) at the channel of NIRF 800 nm. One microliter of the Alexa Fluor 488-conjugated Streptavidin (0.9 mg/mL stock; YEASEN, China) was added to the cells in wells and cultivated overnight. The cells were rinsed with PBS and photographed using a fluorescence microscope, IX51 with DP71 (Olympus), and quantitatively analyzed using a flow cytometer, Calibur (BD). At this point, the efficiency of gene editing was also determined by estimating the fraction of the fluorescent cells using a fluorescence microscope and ImageJ counting.
Homologous Recombination Validation by RT-qPCR
The HepG2 cells cultivated in the 24-well plates were cotransfected with TALEN vectors or CRISPR/Cas9-sgRNA together with a homology arm donor. The elution buffer of the EndoFree Plasmid kit was used as a blank control. At 72 h post-transfection, the genomic DNA (gDNA) was extracted using the gDNA extraction kit (Tiangen, China). The gDNA was detected by the quantitative PCR (qPCR). The PCR reaction (20 μL) consisted of 100 ng of gDNA, 10 μM of forward primer (RELA-F, RELB-F, CREL-F, NFKB1-F, or NFKB2-F), 10 μM of IRES2-R, and 2× SYBR Mix (Roche). The PCR program was 95 °C for 3 min, 40 cycles of 95 °C for 15 s, and 60 °C for 1 min. In this qPCR detection, a region around the TALEN/CRISPR cutting site was amplified using a pair of PCR primers (Table S7), one annealed with the target gene sequence and the other annealed with the inserted IRES2 coding sequence. The PCR products (3 μL) were detected with 1.5% agarose gel, including the addition of 3 μL of the quantitative DNA marker in one lane. The gel was exposed to 15 V/cm until all bands were clearly separated.
Screening of Positive Cells
HepG2 cotransfected with the TALEN vector and the homology arm donor or cotransfected with the sgRNA-CRISPR/Cas9 and the homology arm donor were collected by trypsinization. The cells were resuspended in 500 μL of magnetic bead sorting buffer and then mixed with 5 μL of Dynabeads M-280 Streptavidin beads (10 mg/mL; Invitrogen). The cells were incubated at room temperature for 30 min and gently shaken at an interval of 10 min to avoid precipitation of the magnetic beads. The cells were separated using a magnetic separator for 5–8 min, and the cells unbound to the magnetic beads were discarded. The bead-bound cells were washed three times with 1 mL of magnetic bead sorting buffer, and then cultured at 37 °C for 48 h to remove the magnetic beads.
Immunofluorescence Assay
The unedited HepG2 cells and the enriched edited HepG2 cells with RELA-TALEN were seeded in the wells and incubated for 24 h at 37 °C in 5% CO2. The cells were then stimulated with TNFα (Sigma) with a final concentration of 10 ng/mL for 30 min. The cells were stained with Alexa Fluor 488-conjugated Streptavidin and imaged using a fluorescence microscope. Then, the cells were washed with cold PBS and collected by trypsinization. The collected cells were washed with cold PBS, and partial cells were immediately quantitatively analyzed by flow cytometry. The remaining cells were solidified in 1% (w/v) formaldehyde (Sigma) for 10 min at room temperature. The crosslinking was terminated by adding glycine at a final concentration of 0.125 M. The cells were precipitated by centrifugation at 1500 rpm for 10 min at 4 °C. The cells were lysed with 500 μL of lysis buffer (10 mM Tris–HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, and 0.1% IGEPAL CA-630). The nuclei were collected by centrifugation at 1500 rpm for 10 min at 4 °C. The nuclei were permeabilized with 0.1% (v/v) Triton X-100 for 15 min and then incubated with Alexa Fluor 488-conjugated Streptavidin overnight at 37 °C. The nuclei were washed twice with PBS. Finally, the nuclei were imaged using a fluorescence microscope, IX51 with DP71 (Olympus), and analyzed by flow cytometry.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (grant 61971122).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c02396.
Primers for TALE monomer amplification and colony PCR (Table S1); synthetic double-stranded DNA (Table S2); primer vector and homology arm preparation and clone identification (Table S3); designed TALEN targets (Table S4); designed sgRNA targets (Table S5); oligonucleotide used to prepare target-specific regions (20 bp) of sgRNA (Table S6); primers for verifying homologous recombination (Table S7); vectors and their elements and sequences; HDR donor: SBP-IRES2-displaySBP; HDR donor: AviTag-IRES2-displayAviTag; pipeline for constructing custom CRISPR/Cas9-sgRNA expression vector (plasmid) (Figure S1); preparation of TALEN expression plasmid just in one day (Figure S2); editing five NF-κB genes with TALEN and CRISPR (control cells) (Figure S3); locations of binding sites of all TALENs and CRISPR/Cas9-sgRNA (Figure S4) (PDF)
Author Contributions
S.Z. and J.W. (co-first author) contributed equally to this work. S.Z. fabricated all monomers and all TALENs and CRISPR plasmids. J.W. (co-first author) cultured bacteria and cells and performed all detections together with S.Z. J.W. (the corresponding author) conceptualized, supervised, and funded the study. J.W. (the corresponding author) also wrote the paper.
The authors declare no competing financial interest.
Supplementary Material
References
- a Römer P.; Hahn S.; Jordan T.; Strauss T.; Bonas U.; Lahaye T. Plant pathogen recognition mediated by promoter activation of the pepper Bs3 resistance gene. Science 2007, 318, 645–648. 10.1126/science.1144958. [DOI] [PubMed] [Google Scholar]; b Kay S.; Hahn S.; Marois E.; Wieduwild R.; Bonas U. Detailed analysis of the DNA recognition motifs of the Xanthomonas type III effectors AvrBs3 and AvrBs3Deltarep16. Plant J. 2009, 59, 859–871. 10.1111/j.1365-313X.2009.03922.x. [DOI] [PubMed] [Google Scholar]; c Römer P.; Strauss T.; Hahn S.; Scholze H.; Morbitzer R.; Grau J.; Bonas U.; Lahaye T. Recognition of AvrBs3-Like Proteins Is Mediated by Specific Binding to Promoters of Matching Pepper Bs3 Alleles. Plant Physiol. 2009, 150, 1697–1712. 10.1104/pp.109.139931. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Boch J.; Bonas U. Xanthomonas AvrBs3 family-type III effectors: discovery and function. Annu. Rev. Phytopathol. 2010, 48, 419–436. 10.1146/annurev-phyto-080508-081936. [DOI] [PubMed] [Google Scholar]; e Boch J.; Scholze H.; Schornack S.; Landgraf A.; Hahn S.; Kay S.; Lahaye T.; Nickstadt A.; Bonas U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]; f Moscou M. J.; Bogdanove A. J. A simple cipher governs DNA recognition by TAL effectors. Science 2009, 326, 1501 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- a Geissler R.; Scholze H.; Hahn S.; Streubel J.; Bonas U.; Behrens S. E.; Boch J. Transcriptional activators of human genes with programmable DNA-specificity. PLoS One 2011, 6, e19509. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cermak T.; Doyle E. L.; Christian M.; Wang L.; Zhang Y.; Schmidt C.; Baller J. A.; Somia N. V.; Bogdanove A. J.; Voytas D. F. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011, 39, e82 10.1093/nar/gkr739. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Li T.; Huang S.; Zhao X.; Wright D. A.; Carpenter S.; Spalding M. H.; Weeks D. P.; Yang B. Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes. Nucleic Acids Res. 2011, 39, 6315–6325. 10.1093/nar/gkr188. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Wood A. J.; Lo T. W.; Zeitler B.; Pickle C. S.; Ralston E. J.; Lee A. H.; Amora R.; Miller J. C.; Leung E.; Meng X. D.; Zhang L.; Rebar E. J.; Gregory P. D.; Urnov F. D.; Meyer B. J. Targeted Genome Editing Across Species Using ZFNs and TALENs. Science 2011, 333, 307. 10.1126/science.1207773. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Christian M.; Cermak T.; Doyle E. L.; Schmidt C.; Zhang F.; Hummel A.; Bogdanove A. J.; Voytas D. F. Targeting DNA Double-Strand Breaks with TAL Effector Nucleases. Genetics 2010, 186, 757–U476. 10.1534/genetics.110.120717. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Hockemeyer D.; Wang H. Y.; Kiani S.; Lai C. S.; Gao Q.; Cassady J. P.; Cost G. J.; Zhang L.; Santiago Y.; Miller J. C.; Zeitler B.; Cherone J. M.; Meng X. D.; Hinkley S. J.; Rebar E. J.; Gregory P. D.; Urnov F. D.; Jaenisch R. Genetic engineering of human pluripotent cells using TALE nucleases. Nat. Biotechnol. 2011, 29, 731–734. 10.1038/nbt.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Tesson L.; Usal C.; Menoret S.; Leung E.; Niles B. J.; Remy S.; Santiago Y.; Vincent A. I.; Meng X. D.; Zhang L.; Gregory P. D.; Anegon I.; Cost G. J. Knockout rats generated by embryo microinjection of TALENs. Nat. Biotechnol. 2011, 29, 695–696. 10.1038/nbt.1940. [DOI] [PubMed] [Google Scholar]; h Sun N.; Liang J.; Abil Z.; Zhao H. M. Optimized TAL effector nucleases (TALENs) for use in treatment of sickle cell disease. Mol. BioSyst. 2012, 8, 1255–1263. 10.1039/c2mb05461b. [DOI] [PubMed] [Google Scholar]
- Park C. Y.; Kim J.; Kweon J.; Son J. S.; Lee J. S.; Yoo J. E.; Cho S. R.; Kim J. H.; Kim J. S.; Kim D. W. Targeted inversion and reversion of the blood coagulation factor 8 gene in human iPS cells using TALENs. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 9253–9258. 10.1073/pnas.1323941111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Qasim W.; Amrolia P. J.; Samarasinghe S.; Ghorashian S.; Zhan H.; Stafford S.; Butler K.; Ahsan G.; Gilmour K.; Adams S.; Pinner D.; Chiesa R.; Chatters S.; Swift S.; Goulden N.; Peggs K.; Thrasher A. J.; Veys P.; Pule M. First Clinical Application of Talen Engineered Universal CAR19 T Cells in B-ALL. Blood 2015, 126, 2046 10.1182/blood.V126.23.2046.2046. [DOI] [Google Scholar]; b Qasim W.; Zhan H.; Samarasinghe S.; Adams S.; Amrolia P.; Stafford S.; Butler K.; Rivat C.; Wright G.; Somana K.; Ghorashian S.; Pinner D.; Ahsan G.; Gilmour K.; Lucchini G.; Inglott S.; Mifsud W.; Chiesa R.; Peggs K. S.; Chan L.; Farzaneh F.; Thrasher A. J.; Vora A.; Pule M.; Veys P. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017, 9, eaaj2013 10.1126/scitranslmed.aaj2013. [DOI] [PubMed] [Google Scholar]
- a Sander J. D.; Cade L.; Khayter C.; Reyon D.; Peterson R. T.; Joung J. K.; Yeh J. R. J. Targeted gene disruption in somatic zebrafish cells using engineered TALENs. Nat. Biotechnol. 2011, 29, 697–698. 10.1038/nbt.1934. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Reyon D.; Tsai S. Q.; Khayter C.; Foden J. A.; Sander J. D.; Joung J. K. FLASH assembly of TALENs for high-throughput genome editing. Nat. Biotechnol. 2012, 30, 460–465. 10.1038/nbt.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. Y.; Chen H. T.; Wang J. K. Generate TALE/TALEN as Easily and Rapidly as Generating CRISPR. Mol. Ther.--Methods Clin. Dev. 2019, 13, 310–320. 10.1016/j.omtm.2019.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y.; Foden J. A.; Khayter C.; Maeder M. L.; Reyon D.; Joung J. K.; Sander J. D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanove A. J.; Voytas D. F. TAL effectors: customizable proteins for DNA targeting. Science 2011, 333, 1843–1846. 10.1126/science.1204094. [DOI] [PubMed] [Google Scholar]
- a Kim J.; Cantor A. B.; Orkin S. H.; Wang J. L. Use of in vivo biotinylation to study protein-protein and protein-DNA interactions in mouse embryonic stem cells. Nat. Protoc. 2009, 4, 506–517. 10.1038/nprot.2009.23. [DOI] [PubMed] [Google Scholar]; b Schatz P. J. Use of Peptide Libraries to Map the Substrate-Specificity of a Peptide-Modifying Enzyme - a 13 Residue Consensus Peptide Specifies Biotinylation in Escherichia coli. Nat. Biotechnol. 1993, 11, 1138–1143. 10.1038/nbt1093-1138. [DOI] [PubMed] [Google Scholar]
- Yao X.; Wang X.; Hu X.; Liu Z.; Liu J.; Zhou H.; Shen X.; Wei Y.; Huang Z.; Ying W.; Wang Y.; Nie Y. H.; Zhang C. C.; Li S.; Cheng L.; Wang Q.; Wu Y.; Huang P.; Sun Q.; Shi L.; Yang H. Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Res. 2017, 27, 801–814. 10.1038/cr.2017.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paix A.; Folkmann A.; Goldman D. H.; Kulaga H.; Grzelak M. J.; Rasoloson D.; Paidemarry S.; Green R.; Reed R. R.; Seydoux G. Precision genome editing using synthesis-dependent repair of Cas9-induced DNA breaks. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, E10745–E10754. 10.1073/pnas.1711979114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wilson D. S.; Keefe A. D.; Szostak J. W. The use of mRNA display to select high-affinity protein-binding peptides. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 3750–3755. 10.1073/pnas.061028198. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Keefe A. D.; Wilson D. S.; Seelig B.; Szostak J. W. One-step purification of recombinant proteins using a nanomolar-affinity streptavidin-binding peptide, the SBP-tag. Protein Expression Purif. 2001, 23, 440–446. 10.1006/prep.2001.1515. [DOI] [PubMed] [Google Scholar]
- a Barrette-Ng I. H.; Wu S. C.; Tjia W. M.; Wong S. L.; Ng K. K. S. The structure of the SBP-Tag-streptavidin complex reveals a novel helical scaffold bridging binding pockets on separate subunits. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 879–887. 10.1107/S0907444913002576. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wu S. C.; Wong S. L. Structure-Guided Design of an Engineered Streptavidin with Reusability to Purify Streptavidin-Binding Peptide Tagged Proteins or Biotinylated Proteins. PLoS One 2013, 8, e69530 10.1371/journal.pone.0069530. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.